
Tumour Microenvironment-Informed Radiotheranostics: Why and How Nuclear Medicine Could Advance Precision Oncology in the Decade Ahead

 , ,
, ,
Abstract
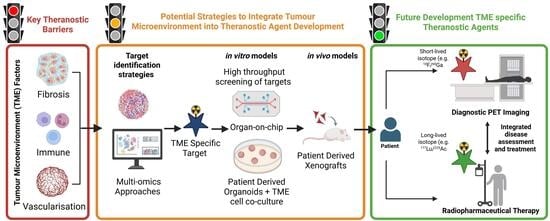
1. Introduction
2. The TME as a Barrier to Radiotheranostics
2.1. Fibrosis and Extracellular Matrix (ECM) Remodelling
2.2. Immune-Cell Infiltration and Immunosuppressive Niches
2.3. Vascular Abnormalities and Hypoxia
3. Barriers Imposed by the TME on Radiopharmaceutical Performance
3.1. Delivery and Distribution Limitations
3.2. Off-Target Uptake and Background Signal
3.3. Pharmacokinetics and Clearance
3.4. Radiation Dose and Toxicity Considerations
4. Radiotheranostics in the Context of the TME
4.1. Radiopharmaceuticals as Modulators of the TME
4.2. Targeting TME for Theranostics
5. Innovative Approaches to Develop TME-Responsive Theranostic Tools
5.1. Spatial and Single-Cell Omics for Target Discovery of Radiotheranostics
5.2. Organ-on-Chip Systems
5.3. Patient-Derived Organoids (PDOs)
5.4. Patient-Derived Xenografts (PDX)
6. Future Directions in Microenvironment-Driven Radiotheranostics
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CAF | Cancer-associated fibroblast |
| DNA | Deoxyribonucleic acid |
| ECM | Extracellular matrix |
| FGF | Fibroblast growth factor |
| LOX | Lysyl oxidase |
| LOXLs | Lysyl oxidase-like proteins |
| mCRPC | Metastatic castration-resistant prostate cancer |
| MMP | Matrix metalloproteinase |
| NET | Neuroendocrine tumour |
| OoC | Organ-on-chip |
| PD-1 | Programmed cell death protein-1 |
| PD-L1 | Programmed death-ligand 1 |
| PDGF | Platelet-derived growth factor |
| PDO | Patient-derived organoid |
| PDX | Patient-derived xenograft |
| PET | Positron emission tomography |
| PSMA | Prostate-specific membrane antigen |
| RPT | Radiopharmaceutical therapy |
| SPECT | Single-photon emission computed tomography |
| TAM | Tumour-associated macrophage |
| TME | Tumour microenvironment |
| Treg | Regulatory T cell |
| VEGF | Vascular endothelial growth factor |
References
- Balkwill, F.R.; Capasso, M.; Hagemann, T. The tumor microenvironment at a glance. J. Cell Sci. 2012, 125, 5591–5596. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.J.; Lei, K.F.; Han, F. Tumor microenvironment: Recent advances in various cancer treatments. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 3855–3864. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhou, H.; Ju, S.; Dong, X.; Zheng, C. The solid tumor microenvironment and related targeting strategies: A concise review. Front. Immunol. 2025, 16, 1563858. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Smolarska, A.; Kokoszka, Z.; Naliwajko, M.; Strupczewska, J.; Tondera, J.; Wiater, M.; Orzechowska, R. Cell-Based Therapies for Solid Tumors: Challenges and Advances. Int. J. Mol. Sci. 2025, 26, 5524. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Qian, J.; Meng, C.; Liu, Y.; Ding, Q.; Wu, H.; Li, P.; Ran, F.; Liu, G.-Q.; Wang, Y.; et al. TME-targeting theranostic agent uses NIR tracking for tumor diagnosis and surgical resection and acts as chemotherapeutic showing enhanced efficiency and minimal toxicity. Theranostics 2022, 12, 2535–2548. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Visvader, J.E. Cells of origin in cancer. Nature 2011, 469, 314–322. [Google Scholar] [CrossRef]
- Wu, Y.; Song, Y.; Wang, R.; Wang, T. Molecular mechanisms of tumor resistance to radiotherapy. Mol. Cancer 2023, 22, 96. [Google Scholar] [CrossRef] [PubMed]
- Rebucci, M.; Michiels, C. Molecular aspects of cancer cell resistance to chemotherapy. Biochem. Pharmacol. 2013, 85, 1219–1226. [Google Scholar] [CrossRef]
- Bodei, L.; Herrmann, K.; Schöder, H.; Scott, A.M.; Lewis, J.S. Radiotheranostics in oncology: Current challenges and emerging opportunities. Nat. Rev. Clin. Oncol. 2022, 19, 534–550. [Google Scholar] [CrossRef]
- Bilotta, M.T.; Antignani, A.; Fitzgerald, D.J. Managing the TME to improve the efficacy of cancer therapy. Front. Immunol. 2022, 13, 954992. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Tang, L.; Wei, F.; Wu, Y.; He, Y.; Shi, L.; Xiong, F.; Gong, Z.; Guo, C.; Li, X.; Deng, H.; et al. Role of metabolism in cancer cell radioresistance and radiosensitization methods. J. Exp. Clin. Cancer Res. 2018, 37, 87. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hynes, R.O.; Naba, A. Overview of the Matrisome—An Inventory of Extracellular Matrix Constituents and Functions. Cold Spring Harb. Perspect. Biol. 2012, 4, a004903. [Google Scholar] [CrossRef]
- Winkler, J.; Abisoye-Ogunniyan, A.; Metcalf, K.J.; Werb, Z. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat. Commun. 2020, 11, 5120. [Google Scholar] [CrossRef]
- Piersma, B.; Hayward, M.-K.; Weaver, V.M. Fibrosis and cancer: A strained relationship. Biochim. Biophys. Acta Rev. Cancer 2020, 1873, 188356. [Google Scholar] [CrossRef]
- Lavie, D.; Ben-Shmuel, A.; Erez, N.; Scherz-Shouval, R. Cancer-associated fibroblasts in the single-cell era. Nat. Cancer 2022, 3, 793–807. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Houthuijzen, J.M.; de Bruijn, R.; van der Burg, E.; Drenth, A.P.; Wientjens, E.; Filipovic, T.; Bullock, E.; Brambillasca, C.S.; Pulver, E.M.; Nieuwland, M.; et al. CD26-negative and CD26-positive tissue-resident fibroblasts contribute to functionally distinct CAF subpopulations in breast cancer. Nat. Commun. 2023, 14, 183. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Cox, T.R. The matrix in cancer. Nat. Rev. Cancer 2021, 21, 217–238. [Google Scholar] [CrossRef] [PubMed]
- Siddhartha, R.; Garg, M. Interplay Between Extracellular Matrix Remodeling and Angiogenesis in Tumor Ecosystem. Mol. Cancer Ther. 2023, 22, 291–305. [Google Scholar] [CrossRef] [PubMed]
- Prakash, J.; Shaked, Y. The Interplay between Extracellular Matrix Remodeling and Cancer Therapeutics. Cancer Discov. 2024, 14, 1375–1388. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Dias, A.M.M.; Burgy, O.; Moreau, M.; Goncalves, V.; Pommerolle, L.; Douhard, R.; Courteau, A.; Helbling, A.; Guillemin, M.; Simonet, J.; et al. Collagen-targeted PET imaging for progressive experimental lung fibrosis quantification and monitoring of efficacy of anti-fibrotic therapies. Theranostics 2025, 15, 2092–2103. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Izquierdo-Garcia, D.; Désogère, P.; Le Fur, M.; Shuvaev, S.; Zhou, I.Y.; Ramsay, I.; Lanuti, M.; Catalano, O.A.; Catana, C.; Caravan, P. Biodistribution, dosimetry, and pharmacokinetics of 68Ga-CBP8: A type I collagen–targeted PET probe. J. Nucl. Med. 2023, 64, 775–781. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Zhao, Z.; Pan, L.; Wu, H.; Wang, S.; Tong, X.; Wu, S. Hyaluronidase: Structure, mechanism of action, diseases and therapeutic targets. Mol. Biomed. 2025, 6, 50. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Findlay, A.D.; Foot, J.S.; Buson, A.; Deodhar, M.; Jarnicki, A.G.; Hansbro, P.M.; Liu, G.; Schilter, H.; Turner, C.I.; Zhou, W.; et al. Identification and Optimization of Mechanism-Based Fluoroallylamine Inhibitors of Lysyl Oxidase-like 2/3. J. Med. Chem. 2019, 62, 9874–9889. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Younis, M.H.; Zhang, Y.; Cai, W.; Lan, X. Clinical summary of fibroblast activation protein inhibitor-based radiopharmaceuticals: Cancer and beyond. Eur. J. Nucl. Med. Mol. Imaging 2022, 49, 2844–2868. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jacobson, O.; Yan, X.; Niu, G.; Weiss, I.D.; Ma, Y.; Szajek, L.P.; Shen, B.; Kiesewetter, D.O.; Chen, X. PET imaging of tenascin-C with a radiolabeled single-stranded DNA aptamer. J. Nucl. Med. 2015, 56, 616–621. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Aloj, L.; D’Ambrosio, L.; Aurilio, M.; Morisco, A.; Frigeri, F.; Caraco’, C.; Di Gennaro, F.; Capobianco, G.; Giovannoni, L.; Menssen, H.D.; et al. Radioimmunotherapy with Tenarad, a 131I-labelled antibody fragment targeting the extra-domain A1 of tenascin-C, in patients with refractory Hodgkin’s lymphoma. Eur. J. Nucl. Med. Mol. Imaging 2014, 41, 867–877. [Google Scholar] [CrossRef] [PubMed]
- Jailkhani, N.; Ingram, J.R.; Rashidian, M.; Rickelt, S.; Tian, C.; Mak, H.; Jiang, Z.; Ploegh, H.L.; Hynes, R.O. Noninvasive imaging of tumor progression, metastasis, and fibrosis using a nanobody targeting the extracellular matrix. Proc. Natl. Acad. Sci. USA 2019, 116, 14181–14190. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Arnoldini, S.; Moscaroli, A.; Chabria, M.; Hilbert, M.; Hertig, S.; Schibli, R.; Béhé, M.; Vogel, V. Novel peptide probes to assess the tensional state of fibronectin fibers in cancer. Nat. Commun. 2017, 8, 1793. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yu, J.; Fu, L.; Wu, R.; Che, L.; Liu, G.; Ran, Q.; Xia, Z.; Liang, X.; Zhao, G. Immunocytes in the tumor microenvironment: Recent updates and interconnections. Front. Immunol. 2025, 16, 1517959. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Galdiero, M.R.; Garlanda, C.; Jaillon, S.; Marone, G.; Mantovani, A. Tumor associated macrophages and neutrophils in tumor progression. J. Cell. Physiol. 2013, 228, 1404–1412. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Zhang, B.; Li, B.; Wu, H.; Jiang, M. Cold and hot tumors: From molecular mechanisms to targeted therapy. Signal Transduct. Target. Ther. 2024, 9, 274. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Roerden, M.; Spranger, S. Cancer immune evasion, immunoediting and intratumour heterogeneity. Nat. Rev. Immunol. 2025, 25, 353–369. [Google Scholar] [CrossRef] [PubMed]
- Parihar, A.S.; Pant, N.; Heidari, P.; Fong, L.; Iravani, A. Approaches to Imaging Immune Activation Using PET. J. Nucl. Med. 2025, 66, 839–847. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Krutzek, F.; Kopka, K.; Stadlbauer, S. Development of Radiotracers for Imaging of the PD-1/PD-L1 Axis. Pharmaceuticals 2022, 15, 747. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bensch, F.; van der Veen, E.L.; Lub-de Hooge, M.N.; Jorritsma-Smit, A.; Boellaard, R.; Kok, I.C.; Oosting, S.F.; Schröder, C.P.; Hiltermann, T.J.N.; van der Wekken, A.J.; et al. 89Zr-atezolizumab imaging as a non-invasive approach to assess clinical response to PD-L1 blockade in cancer. Nat. Med. 2018, 24, 1852–1858. [Google Scholar] [CrossRef] [PubMed]
- De Groof, T.W.M.; Lauwers, Y.; De Pauw, T.; Saxena, M.; Vincke, C.; Van Craenenbroeck, J.; Chapon, C.; Le Grand, R.; Raes, G.; Naninck, T.; et al. Specific imaging of CD8 + T-Cell dynamics with a nanobody radiotracer against human CD8β. Eur. J. Nucl. Med. Mol. Imaging 2024, 52, 193–207. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Farwell, M.D.; Gamache, R.F.; Babazada, H.; Hellmann, M.D.; Harding, J.J.; Korn, R.; Mascioni, A.; Le, W.; Wilson, I.; Gordon, M.S.; et al. CD8-Targeted PET Imaging of Tumor-Infiltrating T Cells in Patients with Cancer: A Phase I First-in-Humans Study of 89Zr-Df-IAB22M2C, a Radiolabeled Anti-CD8 Minibody. J. Nucl. Med. 2022, 63, 720–726. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhou, H.; Wang, Y.; Xu, H.; Shen, X.; Zhang, T.; Zhou, X.; Zeng, Y.; Li, K.; Zhang, L.; Zhu, H.; et al. Noninvasive interrogation of CD8+ T cell effector function for monitoring early tumor responses to immunotherapy. J. Clin. Investig. 2022, 132, e161065. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Signore, A.; Galli, F.; Varani, M.; Campagna, G.; Bentivoglio, V.; Accardo, A.; Morelli, G.; Lauri, C. [68Ga]Ga-interleukin-2 for imaging activated T-lymphocytes: Biochemical characterization and phase I study in normal subjects. Eur. J. Nucl. Med. Mol. Imaging 2025, 53, 544–556. [Google Scholar] [CrossRef] [PubMed]
- Hackett, J.B.; Ramos, N.; Barr, S.; Bross, M.; Viola, N.T.; Gibson, H.M. Interferon gamma immunoPET imaging to evaluate response to immune checkpoint inhibitors. Front. Oncol. 2023, 13, 1285117. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Li, S.; Maes, A.; Vermassen, T.; Maes, J.; Sathekge, C.; Rottey, S.; Van de Wiele, C. PET and SPECT Imaging of Macrophages in the Tumor Stroma: An Update. J. Clin. Med. 2025, 14, 5075. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ren, J.; Xu, M.; Chen, J.; Ding, J.; Wang, P.; Huo, L.; Li, F.; Liu, Z. PET imaging facilitates antibody screening for synergistic radioimmunotherapy with a 177Lu-labeled αPD-L1 antibody. Theranostics 2021, 11, 304–315. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- LuLuna-Gutiérrez, M.; Azorín-Vega, E.; Oros-Pantoja, R.; Ocampo-García, B.; Cruz-Nova, P.; Jiménez-Mancilla, N.; Bravo-Villegas, G.; Santos-Cuevas, C.; Meléndez-Alafort, L.; Ferro-Flores, G. Lutetium-177 labeled iPD-L1 as a novel immunomodulator for cancer-targeted radiotherapy. EJNMMI Radiopharm. Chem. 2025, 10, 5. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Khatua, S.; Düring, M.; Lind, C.L.; Nielsen, J.R. 380TiP A phase I/II dose-escalation and expansion cohort study of intracerebroventricular radioimmunotherapy using 177Lu-DTPA-omburtamab in pediatric and adolescent patients with recurrent or refractory medulloblastoma. Ann. Oncol. 2021, 32, S528–S529. [Google Scholar] [CrossRef]
- Kramer, K.; Pandit-Taskar, N.; Kushner, B.H.; Zanzonico, P.; Humm, J.L.; Tomlinson, U.; Donzelli, M.; Wolden, S.L.; Haque, S.; Dunkel, I.; et al. Phase 1 study of intraventricular 131I-omburtamab targeting B7H3 (CD276)-expressing CNS malignancies. J. Hematol. Oncol. 2022, 15, 165. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.; Liu, X.; Zhang, C.; Zhang, Q. Tumor hypoxia: From basic knowledge to therapeutic implications. Semin. Cancer Biol. 2023, 88, 172–186. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gasparini, G.; Longo, R.; Toi, M.; Ferrara, N. Angiogenic inhibitors: A new therapeutic strategy in oncology. Nat. Clin. Pract. Oncol. 2005, 2, 562–577. [Google Scholar] [CrossRef] [PubMed]
- Adams, R.H.; Alitalo, K. Molecular regulation of angiogenesis and lymphangiogenesis. Nat. Rev. Mol. Cell Biol. 2007, 8, 464–478. [Google Scholar] [CrossRef] [PubMed]
- Quaegebeur, A.; Carmeliet, P. Oxygen sensing: A common crossroad in cancer and neurodegeneration. In Diverse Effects of Hypoxia on Tumor Progression; Current Topics in Microbiology and Immunology; Springer: Berlin/Heidelberg, Germany, 2010; Volume 345, pp. 71–103. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Han, F.; Du, Y.; Shi, H.; Zhou, W. Hypoxic microenvironment in cancer: Molecular mechanisms and therapeutic interventions. Signal Transduct. Target. Ther. 2023, 8, 70. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Liu, Z.-L.; Chen, H.-H.; Zheng, L.-L.; Sun, L.-P.; Shi, L. Angiogenic signaling pathways and anti-angiogenic therapy for cancer. Signal Transduct. Target. Ther. 2023, 8, 198. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gilkes, D.M.; Semenza, G.L.; Wirtz, D. Hypoxia and the extracellular matrix: Drivers of tumour metastasis. Nat. Rev. Cancer 2014, 14, 430–439. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Beckers, C.; Pruschy, M.; Vetrugno, I. Tumor hypoxia and radiotherapy: A major driver of resistance even for novel radiotherapy modalities. Semin. Cancer Biol. 2024, 98, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.D.; Seano, G.; Jain, R.K. Normalizing Function of Tumor Vessels: Progress, Opportunities, and Challenges. Annu. Rev. Physiol. 2019, 81, 505–534. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Steiger, K.; Quigley, N.G.; Groll, T.; Richter, F.; Zierke, M.A.; Beer, A.J.; Weichert, W.; Schwaiger, M.; Kossatz, S.; Notni, J. There is a world beyond αvβ3-integrin: Multimeric ligands for imaging of the integrin subtypes αvβ6, αvβ8, αvβ3, and α5β1 by positron emission tomography. EJNMMI Res. 2021, 11, 106. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Masłowska, K.; Halik, P.K.; Tymecka, D.; Misicka, A.; Gniazdowska, E. The Role of VEGF Receptors as Molecular Target in Nuclear Medicine for Cancer Diagnosis and Combination Therapy. Cancers 2021, 13, 1072. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mittal, S.; Mallia, M.B. Molecular imaging of tumor hypoxia: Evolution of nitroimidazole radiopharmaceuticals and insights for future development. Bioorg. Chem. 2023, 139, 106687. [Google Scholar] [CrossRef] [PubMed]
- Shuch, B.; Pantuck, A.J.; Bernhard, J.-C.; Morris, M.A.; Master, V.; Scott, A.M.; van Praet, C.; Bailly, C.; Önal, B.; Aksoy, T.; et al. [89Zr]Zr-girentuximab for PET-CT imaging of clear-cell renal cell carcinoma: A prospective, open-label, multicentre, phase 3 trial. Lancet Oncol. 2024, 25, 1277–1287. [Google Scholar] [CrossRef] [PubMed]
- Muselaers, C.H.J.; Boers-Sonderen, M.J.; van Oostenbrugge, T.J.; Boerman, O.C.; Desar, I.M.E.; Stillebroer, A.B.; Mulder, S.F.; van Herpen, C.M.L.; Langenhuijsen, J.F.; Oosterwijk, E.; et al. Phase 2 Study of Lutetium 177-Labeled Anti-Carbonic Anhydrase IX Monoclonal Antibody Girentuximab in Patients with Advanced Renal Cell Carcinoma. Eur. Urol. 2016, 69, 767–770. [Google Scholar] [CrossRef] [PubMed]
- Chan, H.-W.; Kuo, D.-Y.; Shueng, P.-W.; Chuang, H.-Y. Visualizing the Tumor Microenvironment: Molecular Imaging Probes Target Extracellular Matrix, Vascular Networks, and Immunosuppressive Cells. Pharmaceuticals 2024, 17, 1663. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K. Normalization of Tumor Vasculature: An Emerging Concept in Antiangiogenic Therapy. Science 2005, 307, 58–62. [Google Scholar] [CrossRef]
- Tixier, F.; Groves, A.M.; Goh, V.; Hatt, M.; Ingrand, P.; Rest, C.C.L.; Visvikis, D. Correlation of Intra-Tumor 18F-FDG Uptake Heterogeneity Indices with Perfusion CT Derived Parameters in Colorectal Cancer. PLoS ONE 2014, 9, e99567. [Google Scholar] [CrossRef] [PubMed]
- Grkovski, M.; Schöder, H.; Lee, N.Y.; Carlin, S.D.; Beattie, B.J.; Riaz, N.; Leeman, J.E.; O’Donoghue, J.A.; Humm, J.L. Multiparametric Imaging of Tumor Hypoxia and Perfusion with 18F-Fluoromisonidazole Dynamic PET in Head and Neck Cancer. J. Nucl. Med. 2017, 58, 1072–1080. [Google Scholar] [CrossRef] [PubMed]
- Jochumsen, M.R.; Sörensen, J.; Tolbod, L.P.; Pedersen, B.G.; Frøkiær, J.; Borre, M.; Bouchelouche, K. Potential synergy between PSMA uptake and tumour blood flow for prediction of human prostate cancer aggressiveness. EJNMMI Res. 2021, 11, 12. [Google Scholar] [CrossRef] [PubMed]
- Verwer, E.E.; Boellaard, R.; van der Veldt, A.A. Positron emission tomography to assess hypoxia and perfusion in lung cancer. World J. Clin. Oncol. 2014, 5, 824–844. [Google Scholar] [CrossRef] [PubMed]
- Roncali, L.; Hindré, F.; Samarut, E.; Lacoeuille, F.; Rousseau, A.; Lemée, J.-M.; Garcion, E.; Chérel, M. Current landscape and future directions of targeted-alpha-therapy for glioblastoma treatment. Theranostics 2025, 15, 4861–4889. [Google Scholar] [CrossRef]
- van der Heide, C.D.; Dalm, S.U. Radionuclide imaging and therapy directed towards the tumor microenvironment: A multi-cancer approach for personalized medicine. Eur. J. Nucl. Med. Mol. Imaging 2022, 49, 4616–4641. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Miller, C.; Rousseau, J.; Ramogida, C.F.; Celler, A.; Rahmim, A.; Uribe, C.F. Implications of physics, chemistry and biology for dosimetry calculations using theranostic pairs. Theranostics 2022, 12, 232–259. [Google Scholar] [CrossRef]
- Parmar, A.; Jackson, S.; Sneiderman, C.; Stepniak, A.; Rajasundaram, D.; Frederico, S.; Edwards, B.; Nedrow, J.; Kohanbash, G. 1009 differences between alpha and beta-emitting radiopharmaceutical therapy on the tumor microenvironment using bulk RNA sequencing analysis in preclinical brain tumors. J. Immunother. Cancer 2024, 12, A1130. [Google Scholar] [CrossRef]
- IAEA. Guidance for Preclinical Studies with Radiopharmaceuticals. 2024. Available online: http://www.iaea.org/publications/14818/guidance-for-preclinical-studies-with-radiopharmaceuticals (accessed on 1 January 2026).
- Zhang, S.; Wang, X.; Gao, X.; Chen, X.; Li, L.; Li, G.; Liu, C.; Miao, Y.; Wang, R.; Hu, K. Radiopharmaceuticals and their applications in medicine. Signal Transduct. Target. Ther. 2025, 10, 1. [Google Scholar] [CrossRef]
- Berton, C.; Klingler, S.; Prytuliak, S.; Holland, J.P. New tactics in the design of theranostic radiotracers. npj Imaging 2024, 2, 23. [Google Scholar] [CrossRef]
- O’Donoghue, J.; Zanzonico, P.; Humm, J.; Kesner, A. Dosimetry in Radiopharmaceutical Therapy. J. Nucl. Med. 2022, 63, 1467–1474. [Google Scholar] [CrossRef]
- Valkenburg, K.C.; de Groot, A.E.; Pienta, K.C. Targeting the tumour stroma to improve cancer therapy. Nat. Rev. Clin. Oncol. 2018, 15, 366–381. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Shea, A.G.; Idrissou, M.B.; Torres, A.I.; Chen, T.; Hernandez, R.; Morris, Z.S.; Sodji, Q.H. Immunological effects of radiopharmaceutical therapy. Front. Nucl. Med. 2024, 4, 1331364. [Google Scholar] [CrossRef]
- Núñez-Salinas, A.; Parra-Garretón, C.; Acuña, D.; Peñaloza, S.; Günther, G.; Bollo, S.; Arriagada, F.; Morales, J. Nanoradiopharmaceuticals: Design Principles, Radiolabeling Strategies, and Biomedicine Applications. Pharmaceutics 2025, 17, 912. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Dhoundiyal, S.; Srivastava, S.; Kumar, S.; Singh, G.; Ashique, S.; Pal, R.; Mishra, N.; Taghizadeh-Hesary, F. Radiopharmaceuticals: Navigating the frontier of precision medicine and therapeutic innovation. Eur. J. Med. Res. 2024, 29, 26. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, K. Development of Diagnostic and Therapeutic Probes with Controlled Pharmacokinetics for Use in Radiotheranostics. Chemical and Pharmaceutical Bulletin. Chem. Pharm. Bull. 2019, 67, 897–903. [Google Scholar] [CrossRef]
- Abbasi Gharibkandi, N.; Conlon, J.M.; Hosseinimehr, S.J. Strategies for improving stability and pharmacokinetic characteristics of radiolabeled peptides for imaging and therapy. Peptides 2020, 133, 170385. [Google Scholar] [CrossRef] [PubMed]
- Andrés, J.I.; Schmidt, M. Medicinal Chemistry strategies for PET tracer discovery. Drug Discov. Today Technol. 2017, 25, 11–17. [Google Scholar] [CrossRef]
- Crabbé, M.; Opsomer, T.; Vermeulen, K.; Ooms, M.; Segers, C. Targeted radiopharmaceuticals: An underexplored strategy for ovarian cancer. Theranostics 2024, 14, 6281–6300. [Google Scholar] [CrossRef]
- Park, E.A.; Graves, S.A.; Menda, Y. The Impact of Radiopharmaceutical Therapy on Renal Function. Semin. Nucl. Med. 2022, 52, 467–474. [Google Scholar] [CrossRef] [PubMed]
- Capala, J.; Graves, S.A.; Scott, A.; Sgouros, G.; James, S.S.; Zanzonico, P.; Zimmerman, B.E. Dosimetry for Radiopharmaceutical Therapy: Current Practices and Commercial Resources. J. Nucl. Med. 2021, 62, 3S–11S. [Google Scholar] [CrossRef] [PubMed]
- Pouget, J.-P.; Gabina, P.M.; Herrmann, K.; Deandreis, D.; Konijnenberg, M.; Taieb, D.; van Leeuwen, F.W.B.; Kurth, J.; Eberlein, U.; Lassmann, M.; et al. EANM expert opinion: How can lessons from radiobiology be applied to the design of clinical trials? Part I: Back to the basics of absorbed dose–response and threshold absorbed doses. Eur. J. Nucl. Med. Mol. Imaging 2025, 52, 1210–1222. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, J.P.B.; Rose, C.J.; Waterton, J.C.; Carano, R.A.D.; Parker, G.J.M.; Jackson, A. Imaging Intratumor Heterogeneity: Role in Therapy Response, Resistance, and Clinical Outcome. Clin. Cancer Res. 2015, 21, 249–257. [Google Scholar] [CrossRef]
- Dimou, K.; Roussakis, Y.; Zamboglou, C.; Stylianopoulos, T. The impact of tumor microenvironment and treatment schedule on the effectiveness of radiation therapy. PLoS ONE 2025, 20, e0331509. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.; Bae, S.; Cavinato, L.; Seifert, R.; Ryhiner, M.; Rominger, A.; Erlandsson, K.; Wilks, M.; Normandin, M.; El-Fakhri, G.; et al. Deciphering the effects of radiopharmaceutical therapy in the tumor microenvironment of prostate cancer: An in-silico exploration with spatial transcriptomics. Theranostics 2024, 14, 7122–7139. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Korde, A.; Patt, M.; Selivanova, S.V.; Scott, A.M.; Hesselmann, R.; Kiss, O.; Ramamoorthy, N.; Todde, S.; Rubow, S.M.; Gwaza, L.; et al. Position paper to facilitate patient access to radiopharmaceuticals: Considerations for a suitable pharmaceutical regulatory framework. EJNMMI Radiopharm. Chem. 2024, 9, 2. [Google Scholar] [CrossRef]
- Korde, A.; Mikolajczak, R.; Kolenc, P.; Bouziotis, P.; Westin, H.; Lauritzen, M.; Koole, M.; Herth, M.M.; Bardiès, M.; Martins, A.F.; et al. Practical considerations for navigating the regulatory landscape of non-clinical studies for clinical translation of radiopharmaceuticals. EJNMMI Radiopharm. Chem. 2022, 7, 18. [Google Scholar] [CrossRef]
- Knapp, F.F.; Dash, A. Translation of Radiopharmaceuticals from Bench to Bedside: Regulatory and Manufacturing Issues. In Radiopharmaceuticals for Therapy; Knapp, F.F., Dash, A., Eds.; Springer: New Delhi, India, 2016; pp. 323–343. [Google Scholar] [CrossRef]
- Sui, H.; Guo, F.; Liu, H.; Wang, R.; Li, L.; Wang, J.; Jia, C.; Xiang, J.; Liang, Y.; Chen, X.; et al. Safety, pharmacokinetics, and dosimetry of 177Lu-AB-3PRGD2 in patients with advanced integrin α v β 3-positive tumors: A first-in-human study. Acta Pharm. Sin. B 2025, 15, 669–680. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Knetsch, P.A.; Petrik, M.; Griessinger, C.M.; Rangger, C.; Fani, M.; Kesenheimer, C.; von Guggenberg, E.; Pichler, B.J.; Virgolini, I.; Decristoforo, C.; et al. [68Ga]NODAGA-RGD for imaging αvβ3 integrin expression. Eur. J. Nucl. Med. Mol. Imaging 2011, 38, 1303–1312. [Google Scholar] [CrossRef] [PubMed]
- Kesler, M.; Levine, C.; Hershkovitz, D.; Mishani, E.; Menachem, Y.; Lerman, H.; Zohar, Y.; Shibolet, O.; Even-Sapir, E. 68Ga-Labeled Prostate-Specific Membrane Antigen Is a Novel PET/CT Tracer for Imaging of Hepatocellular Carcinoma: A Prospective Pilot Study. J. Nucl. Med. 2019, 60, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Long, Y.; Gai, Y.; Liu, Q.; Jiang, D.; Lan, X. [177Lu]Lu-PSMA-617 theranostic probe for hepatocellular carcinoma imaging and therapy. Eur. J. Nucl. Med. Mol. Imaging 2023, 50, 2342–2352. [Google Scholar] [CrossRef] [PubMed]
- Cole, E.L.; Kim, J.; Donnelly, D.J.; Smith, R.A.; Cohen, D.; Lafont, V.; Morin, P.E.; Huang, R.Y.-C.; Chow, P.L.; Hayes, W.; et al. Radiosynthesis and preclinical PET evaluation of 89Zr-nivolumab (BMS-936558) in healthy non-human primates. Bioorg. Med. Chem. 2017, 25, 5407–5414. [Google Scholar] [CrossRef]
- Frank, C.; Allen, K.J.H.; Dawicki, W.; Levin, M.C.; Dadachova, E. Fingolimod synergizes with anti-PD-1 radioimmunotherapy in an experimental multiple sclerosis model through enhanced lymph node retention and CD8+ T cell depletion. Front. Med. 2025, 12, 1593933. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bensch, F.; van der Veen, E.; Jorritsma, A.; Hooge, M.L.; Boellaard, R.; Oosting, S.; Schröder, C.; Hiltermann, J.; van der Wekken, A.; Groen, H.; et al. Abstract CT017: First-in-human PET imaging with the PD-L1 antibody 89Zr-atezolizumab. Cancer Res. 2017, 77, CT017. [Google Scholar] [CrossRef]
- Miedema, I.H.; Zwezerijnen, G.J.; van Dongen, G.A.; Vugts, D.J.; Huisman, M.C.; Hoekstra, O.S.; de Gruijl, T.D.; Verheul, H.M.; Menke, C.W.; van den Eertwegh, A.J. Abstract 1136: Tumor uptake and biodistribution of 89Zirconium-labeled ipilimumab in patients with metastatic melanoma during ipilimumab treatment. Cancer Res. 2019, 79, 1136. [Google Scholar] [CrossRef]
- Larimer, B.M.; Wehrenberg-Klee, E.; Dubois, F.; Mehta, A.; Kalomeris, T.; Flaherty, K.; Boland, G.; Mahmood, U. Granzyme B PET Imaging as a Predictive Biomarker of Immunotherapy Response. Cancer Res. 2017, 77, 2318–2327. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Shen, X.; Zhou, H.; Zhou, X.; Liu, Z.; Meng, X.; Zhang, L.; Song, Y.; Guo, R.; Wang, F.; Li, K.; et al. 68Ga-grazytracer PET for noninvasive assessment of response to immunotherapy in solid tumors and lymphomas: A phase 1/2 clinical trial. Nat. Commun. 2024, 15, 8791. [Google Scholar] [CrossRef] [PubMed]
- Gondry, O.; Xavier, C.; Raes, L.; Heemskerk, J.; Devoogdt, N.; Everaert, H.; Breckpot, K.; Lecocq, Q.; Decoster, L.; Fontaine, C.; et al. Phase I Study of [68Ga]Ga-Anti-CD206-sdAb for PET/CT Assessment of Protumorigenic Macrophage Presence in Solid Tumors (MMR Phase I). J. Nucl. Med. 2023, 64, 1378–1384. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Burvenich, I.J.G.; Parakh, S.; Lee, F.-T.; Guo, N.; Liu, Z.; Gan, H.K.; Rigopoulos, A.; O’Keefe, G.J.; Gong, S.J.; Goh, Y.W.; et al. Molecular imaging of T cell co-regulator factor B7-H3 with 89Zr-DS-5573a. Theranostics 2018, 8, 4199–4209. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kasten, B.B.; Ferrone, S.; Zinn, K.R.; Buchsbaum, D.J. B7-H3-targeted Radioimmunotherapy of Human Cancer. Curr. Med. Chem. 2020, 27, 4016–4038. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Rajendran, J.; Krohn, K. F18 Fluoromisonidazole for Imaging Tumor Hypoxia: Imaging the Microenvironment for Personalized Cancer Therapy. Semin. Nucl. Med. 2015, 45, 151–162. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Berg, T.J.; Pietras, A. Radiotherapy-induced remodeling of the tumor microenvironment by stromal cells. Semin. Cancer Biol. 2022, 86, 846–856. [Google Scholar] [CrossRef]
- Patel, R.B.; Hernandez, R.; Carlson, P.; Grudzinski, J.; Bates, A.M.; Jagodinsky, J.C.; Erbe, A.; Marsh, I.R.; Arthur, I.; Aluicio-Sarduy, E.; et al. Low-dose targeted radionuclide therapy renders immunologically cold tumors responsive to immune checkpoint blockade. Sci. Transl. Med. 2021, 13, eabb3631. [Google Scholar] [CrossRef]
- Strosberg, J.; Kunz, P.L.; Hendifar, A.; Yao, J.; Bushnell, D.; Kulke, M.H.; Baum, R.P.; Caplin, M.; Ruszniewski, P.; Deplassand, E.; et al. Impact of liver tumour burden, alkaline phosphatase elevation, and target lesion size on treatment outcomes with 177Lu-Dotatate: An analysis of the NETTER-1 study. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 2372–2382. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Halperin, D.; Myrehaug, S.; Herrmann, K.; Pavel, M.; Kunz, P.L.; Chasen, B.; Tafuto, S.; Lastoria, S.; Capdevila, J. [177Lu] Lu-DOTA-TATE plus long-acting octreotide versus high-dose long-acting octreotide for the treatment of newly diagnosed, advanced grade 2–3, well-differentiated, gastroenteropancreatic neuroendocrine tumours (NETTER-2): An open-label, randomised, phase 3 study. Lancet 2024, 403, 2807–2817. [Google Scholar] [PubMed]
- Zeng, Z.; Zhang, Z.; Xie, Q.; Tao, J.; Zhou, W.; Yuan, C.; Yang, Z.; Lu, M.; Yu, J. Predicting 177Lu-DOTATATE therapy response through immune microenvironment parameters in gastroenteropancreatic neuroendocrine tumors (GEP-NETs). Eur. J. Nucl. Med. Mol. Imaging 2026, 53, 940–953. [Google Scholar] [CrossRef]
- Wu, Y.; Pfeifer, A.K.; Myschetzky, R.; Garbyal, R.S.; Rasmussen, P.; Knigge, U.; Bzorek, M.; Kristensen, M.H.; Kjaer, A. Induction of Anti-Tumor Immune Responses by Peptide Receptor Radionuclide Therapy with 177Lu-DOTATATE in a Murine Model of a Human Neuroendocrine Tumor. Diagnostics 2013, 3, 344–355. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Seifert, R.; Alberts, I.L.; Afshar-Oromieh, A.; Rahbar, K. Prostate Cancer Theranostics: PSMA Targeted Therapy. PET Clin. 2021, 16, 391–396. [Google Scholar] [CrossRef] [PubMed]
- Kuo, P.H.; Benson, T.; Messmann, R.; Groaning, M. Why We Did What We Did: PSMA PET/CT Selection Criteria for the VISION Trial. J. Nucl. Med. 2022, 63, 816–818. [Google Scholar] [CrossRef]
- Sartor, O.; De Bono, J.; Chi, K.N.; Fizazi, K.; Herrmann, K.; Rahbar, K.; Tagawa, S.T.; Nordquist, L.T.; Vaishampayan, N.; El-Haddad, G.; et al. Lutetium-177–PSMA-617 for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2021, 385, 1091–1103. [Google Scholar] [CrossRef]
- Handke, A.; Lopes, L.; Kesch, C.; Darr, C.; Davicioni, E.; Shi, K.; Telli, T.; Fendler, W.P.; Herrmann, K.; Lückerath, K.; et al. Transcriptomic Profiling of the Tumor Immune Microenvironment Reveals Prognostic Markers in mCRPC Patients Treated with LuPSMA Therapy. Theranostics 2025, 15, 9447–9458. [Google Scholar] [CrossRef]
- Sandhu, K.; Chen, D.; Hennes, D.; Murphy, D.G.; Lawrentschuk, N.; Perera, M. PSMA Theranostics in Prostate Cancer and Beyond: Current and Future Perspectives. Cancers 2025, 17, 3717. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Minagawa, Y.; Shizukuishi, K.; Koike, I.; Horiuchi, C.; Watanuki, K.; Hata, M.; Omura, M.; Odagiri, K.; Tohnai, I.; Inoue, T.; et al. Assessment of tumor hypoxia by 62Cu-ATSM PET/CT as a predictor of response in head and neck cancer: A pilot study. Ann. Nucl. Med. 2011, 25, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Abida, W.; Cheng, M.L.; Armenia, J.; Middha, S.; Autio, K.A.; Vargas, H.A.; Rathkopf, D.; Morris, M.J.; Danila, D.C.; Slovin, S.F.; et al. Analysis of the Prevalence of Microsatellite Instability in Prostate Cancer and Response to Immune Checkpoint Blockade. JAMA Oncol. 2019, 5, 471. [Google Scholar] [CrossRef]
- Arbuznikova, D.; Eder, M.; Grosu, A.-L.; Meyer, P.T.; Gratzke, C.; Zamboglou, C.; Eder, A.-C. Towards Improving the Efficacy of PSMA-Targeting Radionuclide Therapy for Late-Stage Prostate Cancer—Combination Strategies. Curr. Oncol. Rep. 2023, 25, 1363–1374. [Google Scholar] [CrossRef] [PubMed]
- Handke, A.; Kesch, C.; Fendler, W.P.; Telli, T.; Liu, Y.; Hakansson, A.; Davicioni, E.; Hughes, J.; Song, H.; Lueckerath, K.; et al. Analysing the tumor transcriptome of prostate cancer to predict efficacy of Lu-PSMA therapy. J. Immunother. Cancer 2023, 11, e007354. [Google Scholar] [CrossRef]
- Kleinendorst, S.C.; Oosterwijk, E.; Bussink, J.; Westdorp, H.; Konijnenberg, M.W.; Heskamp, S. Combining Targeted Radionuclide Therapy and Immune Checkpoint Inhibition for Cancer Treatment. Clin. Cancer Res. 2022, 28, 3652–3657. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Reubi, J.C.; Schaer, J.C.; Waser, B.; Mengod, G. Expression and localization of somatostatin receptor SSTR1, SSTR2, and SSTR3 messenger RNAs in primary human tumors using in situ hybridization. Cancer Res. 1994, 54, 3455–3459. [Google Scholar] [PubMed]
- Tsourlakis, M.C.; Klein, F.; Kluth, M.; Quaas, A.; Graefen, M.; Haese, A.; Simon, R.; Sauter, G.; Schlomm, T.; Minner, S. PSMA expression is highly homogenous in primary prostate cancer. Appl. Immunohistochem. Mol. Morphol. 2015, 23, 449–455. [Google Scholar] [CrossRef]
- Xu, M.; Zhang, T.; Xia, R.; Wei, Y.; Wei, X. Targeting the tumor stroma for cancer therapy. Mol. Cancer 2022, 21, 208. [Google Scholar] [CrossRef]
- Rettig, W.J.; Garin-Chesa, P.; Beresford, H.R.; Oettgen, H.F.; Melamed, M.R.; Old, L.J. Cell-surface glycoproteins of human sarcomas: Differential expression in normal and malignant tissues and cultured cells. Proc. Natl. Acad. Sci. USA 1988, 85, 3110–3114. [Google Scholar] [CrossRef]
- Fitzgerald, A.A.; Weiner, L.M. The role of fibroblast activation protein in health and malignancy. Cancer Metastasis Rev. 2020, 39, 783–803. [Google Scholar] [CrossRef]
- Liu, F.; Qi, L.; Liu, B.; Liu, J.; Zhang, H.; Che, D.; Cao, J.; Shen, J.; Geng, J.; Bi, Y. Fibroblast activation protein overexpression and clinical implications in solid tumors: A meta-analysis. PLoS ONE 2015, 10, e0116683. [Google Scholar] [CrossRef]
- Wang, L.; Tang, G.; Hu, K.; Liu, X.; Zhou, W.; Li, H.; Huang, S.; Han, Y.; Chen, L.; Zhong, J.; et al. Comparison of 68Ga-FAPI and 18F-FDG PET/CT in the Evaluation of Advanced Lung Cancer. Radiology 2022, 303, 191–199. [Google Scholar] [CrossRef]
- Van Der Flier, A.; Sonnenberg, A. Function and interactions of integrins. Cell Tissue Res. 2001, 305, 285–298. [Google Scholar] [CrossRef]
- Campbell, I.D.; Humphries, M.J. Integrin structure, activation, and interactions. Cold Spring Harb. Perspect. Biol. 2011, 3, a004994. [Google Scholar] [CrossRef] [PubMed]
- Lewis, M.R. Radiolabeled RGD peptides move beyond cancer: PET imaging of delayed-type hypersensitivity reaction. J. Nucl. Med. 2005, 46, 2–4. [Google Scholar]
- Gao, S.; Wu, H.; Li, W.; Zhao, S.; Teng, X.; Lu, H.; Hu, X.; Wang, S.; Yu, J.; Yuan, S. A pilot study imaging integrin αvβ3 with RGD PET/CT in suspected lung cancer patients. Eur. J. Nucl. Med. Mol. Imaging 2015, 42, 2029–2037. [Google Scholar] [CrossRef]
- Reader, C.S.; Vallath, S.; Steele, C.W.; Haider, S.; Brentnall, A.; Desai, A.; Moore, K.M.; Jamieson, N.B.; Chang, D.; Bailey, P.; et al. The integrin αvβ6 drives pancreatic cancer through diverse mechanisms and represents an effective target for therapy. J. Pathol. 2019, 249, 332–342. [Google Scholar] [CrossRef] [PubMed]
- Mah, E.J.; Lefebvre, A.E.; McGahey, G.E.; Yee, A.F.; Digman, M.A. Collagen density modulates triple-negative breast cancer cell metabolism through adhesion-mediated contractility. Sci. Rep. 2018, 8, 17094. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.A.; Ghim, M.; Kukreja, G.; Neishabouri, A.; Zhang, Z.; Li, J.; Salarian, M.; Toczek, J.; Gona, K.; Hedayatyanfard, K. Collagen Hybridizing Peptide-Based Radiotracers for Molecular Imaging of Collagen Turnover in Pulmonary Fibrosis. J. Nucl. Med. 2025, 66, 425–433. [Google Scholar] [CrossRef]
- Skinnider, M.A.; Courtine, G.; Bloch, J.; Squair, J.W. A clinical road map for single-cell omics. Cell 2025, 188, 3633–3647. [Google Scholar] [CrossRef] [PubMed]
- Seifert, R.; Seitzer, K.; Herrmann, K.; Kessel, K.; Schäfers, M.; Kleesiek, J.; Weckesser, M.; Boegemann, M.; Rahbar, K. Analysis of PSMA expression and outcome in patients with advanced Prostate Cancer receiving 177Lu-PSMA-617 Radioligand Therapy. Theranostics 2020, 10, 7812–7820. [Google Scholar] [CrossRef]
- Seifert, R.; Kessel, K.; Schlack, K.; Weckesser, M.; Kersting, D.; Seitzer, K.E.; Weber, M.; Bögemann, M.; Rahbar, K. Total tumor volume reduction and low PSMA expression in patients receiving Lu-PSMA therapy. Theranostics 2021, 11, 8143–8151. [Google Scholar] [CrossRef]
- Wang, J.; Seo, J.W.; Kare, A.J.; Schneider, M.; Pandrala, M.; Tumbale, S.K.; Raie, M.N.; Engudar, G.; Zhang, N.; Guo, Y.; et al. Spatial transcriptomic analysis drives PET imaging of tight junction protein expression in pancreatic cancer theranostics. Nat. Commun. 2024, 15, 10751. [Google Scholar] [CrossRef]
- Kraya, A.; Rathi, K.; Jin, R.; Kesherwani, V.; Resnick, A.C.; Storm, P.B.; Nabavizadeh, A. Evaluating the Potential of PSMA Targeting in CNS Tumors: Insights from Large-Scale Transcriptome Profiling. Cancers 2025, 17, 1239. [Google Scholar] [CrossRef] [PubMed]
- Mousavi Shaegh, S.A.; De Ferrari, F.; Zhang, Y.S.; Nabavinia, M.; Binth Mohammad, N.; Ryan, J.; Pourmand, A.; Laukaitis, E.; Banan Sadeghian, R.; Nadhman, A. A microfluidic optical platform for real-time monitoring of pH and oxygen in microfluidic bioreactors and organ-on-chip devices. Biomicrofluidics 2016, 10, 044111. Available online: https://pubs.aip.org/aip/bmf/article/10/4/044111/133942 (accessed on 1 January 2026). [CrossRef]
- Nasiri, R.; Sankaranthi, A.; Pratx, G. Organ-on-a-chip systems for modeling tumor and normal tissue microenvironments in radiotherapy research. Trends Biotechnol. 2025, 44, 333–350. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ingber, D.E. Human organs-on-chips for disease modelling, drug development and personalized medicine. Nat. Rev. Genet. 2022, 23, 467–491. [Google Scholar] [CrossRef]
- Abdollahi, H.; Fele-Paranj, A.; Rahmim, A. Model-Informed Radiopharmaceutical Therapy Optimization: A Study on the Impact of PBPK Model Parameters on Physical, Biological, and Statistical Measures in 177Lu-PSMA Therapy. Cancers 2024, 16, 3120. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wensink, G.E.; Elias, S.G.; Mullenders, J.; Koopman, M.; Boj, S.F.; Kranenburg, O.W.; Roodhart, J.M. Patient-derived organoids as a predictive biomarker for treatment response in cancer patients. npj Precis. Oncol. 2021, 5, 30. [Google Scholar] [CrossRef] [PubMed]
- Puca, L.; Bareja, R.; Prandi, D.; Shaw, R.; Benelli, M.; Karthaus, W.R.; Hess, J.; Sigouros, M.; Donoghue, A.; Kossai, M. Patient derived organoids to model rare prostate cancer phenotypes. Nat. Commun. 2018, 9, 2404. [Google Scholar] [CrossRef]
- Parikh, A.Y.; Masi, R.; Gasmi, B.; Hanada, K.; Parkhurst, M.; Gartner, J.; Sindiri, S.; Prickett, T.; Robbins, P.; Zacharakis, N.; et al. Using patient-derived tumor organoids from common epithelial cancers to analyze personalized T-cell responses to neoantigens. Cancer Immunol. Immunother. 2023, 72, 3149–3162. [Google Scholar] [CrossRef]
- Servant, R.; Garioni, M.; Vlajnic, T.; Blind, M.; Pueschel, H.; Müller, D.C.; Zellweger, T.; Templeton, A.J.; Garofoli, A.; Maletti, S.; et al. Prostate cancer patient-derived organoids: Detailed outcome from a prospective cohort of 81 clinical specimens. J. Pathol. 2021, 254, 543–555. [Google Scholar] [CrossRef]
- Kawasaki, K.; Toshimitsu, K.; Matano, M.; Fujita, M.; Fujii, M.; Togasaki, K.; Ebisudani, T.; Shimokawa, M.; Takano, A.; Takahashi, S. An organoid biobank of neuroendocrine neoplasms enables genotype-phenotype mapping. Cell 2020, 183, 1420–1435. [Google Scholar] [CrossRef]
- Raitanen, J.; Barta, B.; Fuchs, H.; Hacker, M.; Balber, T.; Georg, D.; Mitterhauser, M. Radiobiological assessment of targeted radionuclide therapy with [177Lu] Lu-PSMA-I&T in 2D vs. 3D Cell culture models. Int. J. Mol. Sci. 2023, 24, 17015. [Google Scholar]
- Shmuel, S.; Monette, S.; Ibrahim, D.; Pereira, P.M.R. PDX Models in Theranostic Applications: Generation and Screening for B Cell Lymphoma of Human Origin. Mol. Imaging Biol. 2024, 26, 569–576. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hidalgo, M.; Amant, F.; Biankin, A.V.; Budinská, E.; Byrne, A.T.; Caldas, C.; Clarke, R.B.; de Jong, S.; Jonkers, J.; Mælandsmo, G.M.; et al. Patient Derived Xenograft Models: An Emerging Platform for Translational Cancer Research. Cancer Discov. 2014, 4, 998–1013. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhou, H.; Qian, W.; Uckun, F.M.; Wang, L.; Wang, Y.A.; Chen, H.; Kooby, D.; Yu, Q.; Lipowska, M.; Staley, C.A.; et al. IGF1 Receptor Targeted Theranostic Nanoparticles for Targeted and Image-Guided Therapy of Pancreatic Cancer. ACS Nano 2015, 9, 7976–7991. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhang, Y.; Zhang, D.; An, S.; Liu, Q.; Liang, C.; Li, J.; Liu, P.; Wu, C.; Huang, G.; Wei, W.; et al. Development and Characterization of Nanobody-Derived CD47 Theranostic Pairs in Solid Tumors. Research 2023, 6, 0077. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Pereira, P.M.R.; Mandleywala, K.; Monette, S.; Lumish, M.; Tully, K.M.; Panikar, S.S.; Cornejo, M.; Mauguen, A.; Ragupathi, A.; Keltee, N.C.; et al. Caveolin-1 temporal modulation enhances antibody drug efficacy in heterogeneous gastric cancer. Nat. Commun. 2022, 13, 2526. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sheng, Y.; Xie, Z.; Wang, J.; Yang, X.; Yao, M.; Qian, W.; Zhang, L.; Chen, X.; Guo, S. Impact of Subcutaneous Versus Orthotopic Implantations on Patient-Derived Xenograft Transcriptomic Profiles. Cancer Res. Commun. 2025, 5, 871–880. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Liu, Y.; Wu, W.; Cai, C.; Zhang, H.; Shen, H.; Han, Y. Patient-derived xenograft models in cancer therapy: Technologies and applications. Signal Transduct. Target. Ther. 2023, 8, 160. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, G.J. Applications of patient-derived tumor xenograft models and tumor organoids. J. Hematol. Oncol. 2020, 13, 4. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- De Jong, M.; Maina, T. Of Mice and Humans: Are They the Same?—Implications in Cancer Translational Research. J. Nucl. Med. 2010, 51, 501–504. [Google Scholar] [CrossRef] [PubMed]
- Roll, W.; Schindler, P.; Masthoff, M.; Seifert, R.; Schlack, K.; Bögemann, M.; Stegger, L.; Weckesser, M.; Rahbar, K. Evaluation of 68Ga-PSMA-11 PET-MRI in Patients with Advanced Prostate Cancer Receiving 177Lu-PSMA-617 Therapy: A Radiomics Analysis. Cancers 2021, 13, 3849. [Google Scholar] [CrossRef]
- Seifert, R.; Telli, T.; Hadaschik, B.; Fendler, W.P.; Kuo, P.H.; Herrmann, K. Is 18F-FDG PET Needed to Assess 177Lu-PSMA Therapy Eligibility? A VISION-like, Single-Center Analysis. J. Nucl. Med. 2023, 64, 731–737. Available online: https://jnm.snmjournals.org/content/64/5/731.long (accessed on 1 January 2026). [CrossRef]
- Telli, T.; Lopes, L.; Karpinski, M.; Pabst, K.M.; Grünwald, V.; Shi, K.; Hadaschik, B.; Kesch, C.; Umutlu, L.; Herrmann, K.; et al. Prognostic value of [18F]FDG- and PSMA-PET in patients evaluated for [177Lu]Lu-PSMA therapy of mCRPC. Eur. J. Nucl. Med. Mol. Imaging 2025, 52, 3199–3210. [Google Scholar] [CrossRef] [PubMed]
- Avgoustakis, K.; Angelopoulou, A. Biomaterial-Based Responsive Nanomedicines for Targeting Solid Tumor Microenvironments. Pharmaceutics 2024, 16, 179. [Google Scholar] [CrossRef]
- Park, B.-N.; An, Y.-S.; Kim, S.-M.; Lee, S.-J.; Park, Y.-J.; Yoon, J.-K. 177Lu Anti-Angiogenic Radioimmunotherapy Targeting ATP Synthase in Gastric Cancer Model. Antibodies 2024, 13, 51. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Abyaneh, H.S.; Regenold, M.; McKee, T.D.; Allen, C.; Gauthier, M.A. Towards extracellular matrix normalization for improved treatment of solid tumors. Theranostics 2020, 10, 1960–1980. [Google Scholar] [CrossRef] [PubMed]
- Henke, E.; Nandigama, R.; Ergün, S. Extracellular Matrix in the Tumor Microenvironment and Its Impact on Cancer Therapy. Front. Mol. Biosci. 2020, 6, 160. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Example Radiopharmaceuticals | Precursor Format | Application | Refs. |
|---|---|---|---|---|
| Fibrosis- and ECM-related targets | ||||
| FAP | 68Ga-FAPI-04; 177Lu-FAP-2286 | Inhibitor | Theranostics | [24] |
| Collagen | 68Ga-CBP8 | Peptide | Imaging | [21] |
| Tenascin-C | 18F/64Cu-FB-tenascin-C aptamer; 131I-Tenarad | Aptamer, antibody | Theranostics | [25,26] |
| Fibronectin | 64Cu-NJB2; 131I-L19SIP | Antibody | Theranostics | [27] |
| Angiogenesis-related targets | ||||
| Integrin (e.g., αvβ3, αvβ6, αvβ8, α5β1) | 68Ga-NODAGA-RGD; 177Lu-AB-3PRGD2 | Peptide | Theranostics | [55,91,92] |
| VEGF | 89Zr-Bevacizumab; 177Lu-DOTA-VG76e | Antibody | Theranostics | [56] |
| PSMA | 68Ga-PSMA-11; 177Lu-PSMA-617 | Inhibitor | Theranostics | [93,94] |
| Immune-related targets | ||||
| PD-1 | 89Zr-nivolumab; 177Lu-αPD-1 | Antibody | Theranostics | [95,96] |
| PD-L1 | 89Zr-atezolizumab; 177Lu-DOTA-Y003 | Antibody | Theranostics | [35,42,97] |
| CTLA-4 | 89Zr-ipilimumab | Antibody | Imaging | [98] |
| CD8 | 89Zr-Df-IAB22M2C | Antibody | Imaging | [37] |
| Granzyme B | 68Ga-NOTA-GZP; 68Ga-grazytracer | Peptide | Imaging | [99,100] |
| IFN-γ | 89Zr-DFO-anti-IFNγ | Antibody | Imaging | [40] |
| IL-2R | 68Ga-interleukin-2 | Protein | Imaging | [39] |
| TAMs (e.g., CD206, TSPO) | 68Ga-Anti-CD206-sdAb | Antibody | Imaging | [41,101] |
| B7-H3 | 89Zr-DS-5573a; 124I/131I-8H9 | Antibody | Theranostics | [102,103] |
| Hypoxia-related targets | ||||
| / | 18F-FMISO | Nitroimidazole derivative | Imaging | [104] |
| CAIX | 89Zr/177Lu-girentuximab | Antibody | Theranostics | [58,59] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Share and Cite
Pandya, K.; Lin, Z.; Wadsak, M.; Wang, J.; Shi, K.; Seifert, R. Tumour Microenvironment-Informed Radiotheranostics: Why and How Nuclear Medicine Could Advance Precision Oncology in the Decade Ahead. Pharmaceuticals 2026, 19, 382. https://doi.org/10.3390/ph19030382
Pandya K, Lin Z, Wadsak M, Wang J, Shi K, Seifert R. Tumour Microenvironment-Informed Radiotheranostics: Why and How Nuclear Medicine Could Advance Precision Oncology in the Decade Ahead. Pharmaceuticals. 2026; 19(3):382. https://doi.org/10.3390/ph19030382
Chicago/Turabian StylePandya, Kalyani, Zhaoguo Lin, Magdalena Wadsak, Jiahui Wang, Kuangyu Shi, and Robert Seifert. 2026. "Tumour Microenvironment-Informed Radiotheranostics: Why and How Nuclear Medicine Could Advance Precision Oncology in the Decade Ahead" Pharmaceuticals 19, no. 3: 382. https://doi.org/10.3390/ph19030382
APA StylePandya, K., Lin, Z., Wadsak, M., Wang, J., Shi, K., & Seifert, R. (2026). Tumour Microenvironment-Informed Radiotheranostics: Why and How Nuclear Medicine Could Advance Precision Oncology in the Decade Ahead. Pharmaceuticals, 19(3), 382. https://doi.org/10.3390/ph19030382