


Combination Strategies with HSP90 Inhibitors in Cancer Therapy: Mechanisms, Challenges, and Future Perspectives

 , , ,
, , , 
Abstract

1. Introduction
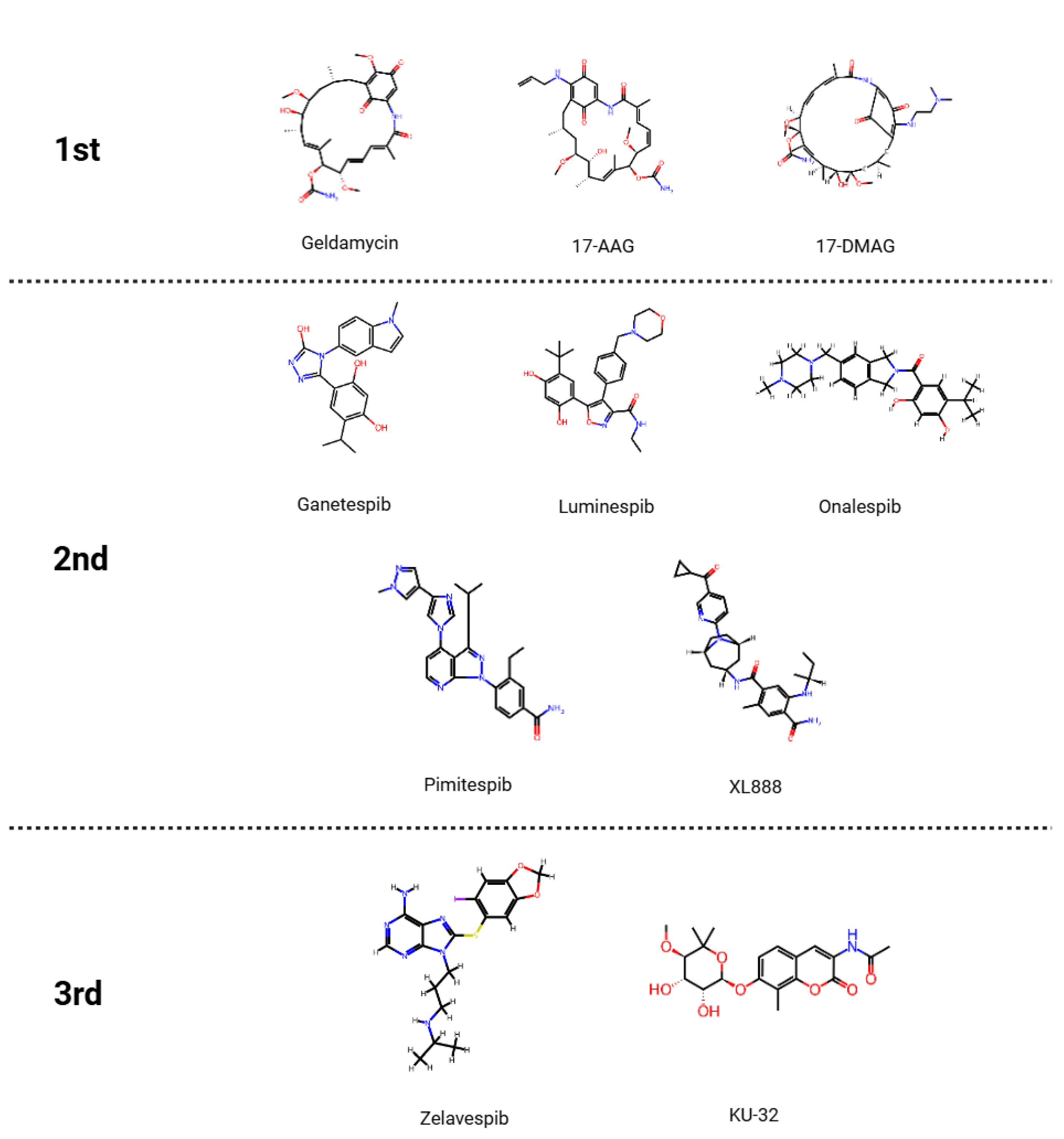
2. Classification and Mechanism of HSP90 Inhibitors
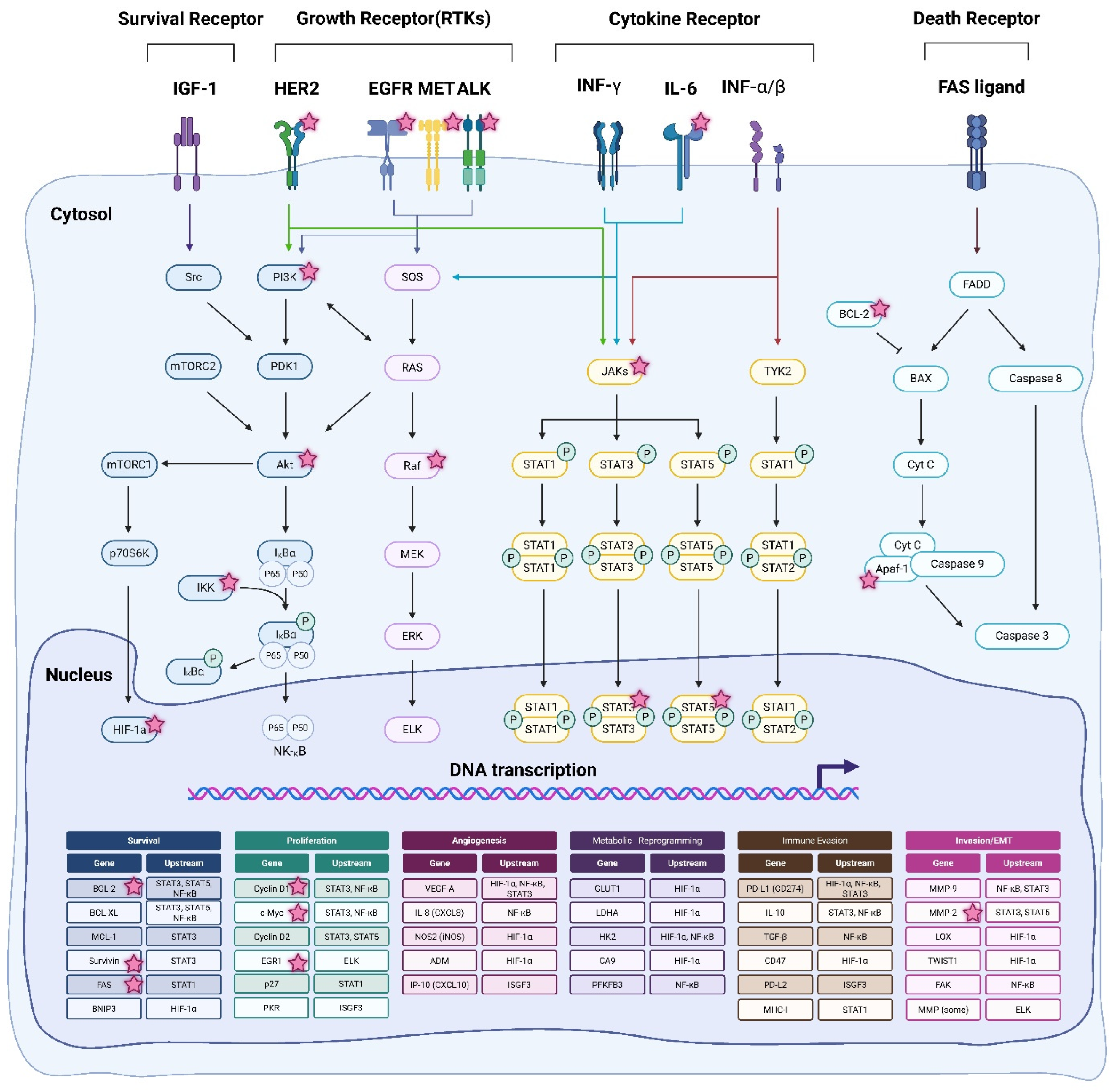
2.1. Mechanism of Action of HSP90 Inhibitors
2.2. Limitations of HSP90 Inhibitor Monotherapy
2.2.1. Drug Toxicity and Clinical Limitations
2.2.2. Mechanisms Underlying Resistance Development to HSP90 Inhibitor Monotherapy
2.3. Necessity and Clinical Significance of Combination Therapy
3. Mechanisms and Clinical Rationale for HSP90 Inhibitor Combination Therapy
3.1. Chemotherapeutic Agents
3.1.1. Taxane
3.1.2. Gemcitabine
3.1.3. Cisplatin
3.2. Targeted Therapeutic Agents
3.2.1. Membrane Receptor-Targeted Inhibitors
3.2.2. Intracellular Signaling Pathway Inhibitors
3.3. Immunotherapeutic Agents
3.3.1. PD-1 and PD-L1 Inhibitors
3.3.2. CTLA-4 Inhibitors
4. Toxicity Management, Optimization Strategies, and Future Research Directions for HSP90 Inhibitor Combination Therapy
4.1. How to Overcome Toxicity Management and Resistance When Using HSP90 Inhibitor Combination Therapy
4.2. Optimization of HSP90 Inhibitor Combination Therapy
4.3. Biomarker Limitations and Opportunities for Precise Application of HSP90 Inhibitor-Based Combination Therapy
4.4. Future Research Direction and Clinical Applicability
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| HSP90 | Heat Shock Protein 90 |
| EGFR | Epidermal Growth Factor Receptor |
| HER2 | Human Epidermal Growth Factor Receptor 2 |
| MET | Mesenchymal-Epithelial Transition factor |
| ALK | Anaplastic Lymphoma Kinase |
| RAF | Rapidly Accelerated Fibrosarcoma |
| MEK | Mitogen-Activated Protein Kinase Kinase (MAP2K) |
| ERK | Extracellular signal-Regulated Kinase |
| PI3K | Phosphoinositide 3-Kinase |
| AKT | AKT serine/threonine kinase (Protein Kinase B) |
| MAPK | Mitogen-Activated Protein Kinase |
| DNA | Deoxyribonucleic Acid |
| ATP | Adenosine Triphosphate |
| DLT | Dose-Limiting Toxicity |
| ORR | Objective Response Rate |
| PFS | Progression-Free Survival |
| HSF-1 | Heat Shock Factor 1 |
| IGF-1R | Insulin-like Growth Factor 1 Receptor |
| MDSCs | Myeloid-Derived Suppressor Cells |
| NSCLC | Non-Small Cell Lung Cancer |
| BRAF | v-Raf Murine Sarcoma Viral Oncogene Homolog B |
| PD-1 | Programmed Cell Death Protein 1 |
| PD-L1 | Programmed Death-Ligand 1 |
| Tregs | Regulatory T Cells |
| CDK1 | Cyclin-Dependent Kinase 1 |
| CDC25C | Cell Division Cycle 25C |
| RP2D | Recommended Phase 2 Dose |
| pCR | Pathological Complete Response |
| TNBC | Triple-Negative Breast Cancer |
| dC | Deoxycytidine |
| dFdCTP | Gemcitabine Triphosphate (active metabolite of gemcitabine) |
| DDR | DNA Damage Response |
| Chk1 | Checkpoint Kinase 1 |
| DCR | Disease Control Rate |
| OS | Overall Survival |
| SD | Stable Disease |
| PR | Partial Response |
| 5-FU | 5-Fluorouracil |
| CI | combination index |
| EC50 | Half Maximal Effective Concentration |
| DLBCL | Diffuse Large B-Cell Lymphoma |
| NPC | Nasopharyngeal Carcinoma |
| RTKs | Receptor Tyrosine Kinases |
| CBR | Clinical Benefit Rate |
| MTD | Maximum Tolerated Dose |
| FOXP3 | Forkhead Box P3 |
| CD8+ | Cluster of Differentiation 8 Positive |
| HS201-PDT | HS201 Photodynamic Therapy |
| CXCR3 | C-X-C Chemokine Receptor Type 3 |
| CTLA-4 | Cytotoxic T-Lymphocyte Associated Protein 4 |
| MD | Molecular Dynamics |
| ADMET | Absorption |
| Distribution | Metabolism |
| Excretion | and Toxicity |
| AI | Artificial Intelligence |
| 17-AAG | 17-N-allylamino-17-demethoxygeldanamycin |
| 17-DMAG | 17-dimethylaminoethylamino-17-demethoxygeldanamycin |
| IGF-1 | insulin-like growth factor 1 |
| INF-γ | Interferon-γ |
| INF-α/β | Interferon-α/β |
| IL-6 | Interleukin 6 |
| FAS ligand | Fas cell surface death receptor ligand |
| Src | SRC proto-oncogene, non-receptor tyrosine kinase |
| PDK1 | 3 Phosphoinositide Dependent Protein Kinase 1 |
| mTORC2 | Mechanistic Target Of Rapamycin Complex 2 |
| mTORC1 | Mechanistic Target Of Rapamycin Complex 1 |
| IκBα | Inhibitor of Nuclear Factor Kappa B Alpha |
| IKK | IκB Kinase (complex) |
| HIF 1α | Hypoxia Inducible Factor 1 Alpha |
| NF-κB | Nuclear Factor Kappa-Light-Chain-Enhancer of Activated B Cells |
| SOS | Son of Sevenless (guanine nucleotide exchange factor) |
| ELK | Ets-Like Kinase (transcription factor, e.g., ELK1) |
| JAKs | Janus Kinases (JAK1, JAK2, JAK3, TYK2) |
| TYK2 | Tyrosine Kinase 2 |
| BCL-2 | B-Cell Lymphoma 2 |
| BAX | Bcl-2-Associated X Protein |
| Cyt C | Cytochrome C (apoptogenic factor) |
| Apaf-1 | Apoptotic Protease Activating Factor 1 |
| FADD | Fas-Associated Death Domain protein |
| BCL-XL | B-Cell Lymphoma-Extra Large (also known as BCL2L1) |
| MCL-1 | Myeloid Cell Leukemia-1 |
| BNIP3 | BCL2/Adenovirus E1B 19kDa-Interacting Protein 3 |
| c-FLIP | Cellular FLICE-Like Inhibitory Protein |
| c-Myc | Cellular Myelocytomatosis Oncogene (transcription factor) |
| Pim-1 | Proto-Oncogene Serine/Threonine-Protein Kinase Pim-1 |
| c-FOS | Cellular FBJ Murine Osteosarcoma Oncogene (transcription factor) |
| EGR1 | Early Growth Response Protein 1 |
| PKR | Protein Kinase R (double-stranded RNA-activated kinase) |
| ISGF3 | Interferon-Stimulated Gene Factor 3 (STAT1–STAT2–IRF9 complex) |
| VEGF-A | Vascular Endothelial Growth Factor A |
| IL-8 (CXCL8) | Interleukin-8, also known as C-X-C Motif Chemokine Ligand 8 |
| NOS2 (iNOS) | Nitric Oxide Synthase 2, inducible |
| ADM | Adrenomedullin |
| IP-10 (CXCL10) | Interferon Gamma-Induced Protein 10, or C-X-C Motif Chemokine Ligand 10 |
| GLUT1 | Glucose Transporter 1 (SLC2A1) |
| LDHA | Lactate Dehydrogenase A |
| HK2 | Hexokinase 2 |
| CA9 | Carbonic Anhydrase IX |
| PFKFB3 | 6-Phosphofructo-2-Kinase/Fructose-2,6-Bisphosphatase 3 |
| MMP-9 | Matrix Metallopeptidase 9 (gelatinase B) |
| MMP-2 | Matrix Metallopeptidase 2 (gelatinase A) |
| LOX | Lysyl Oxidase |
| TWIST1 | Twist-Related Protein 1 (transcription factor) |
| FAK | Focal Adhesion Kinase (PTK2) |
| MMP | Matrix Metallopeptidases (family of proteases) |
| MIA PaCa-2 | Human pancreatic carcinoma cell line (MIA PaCa-2) |
| PANC-1 | Human pancreatic carcinoma cell line (PANC-1) |
References
- Lim, K.S.; Lee, D.Y.; Han, S.; Bull, D.A.; Won, Y.-W. Targeted delivery of heat shock protein 90 inhibitors prevents growth of HER2-positive tumor. Biomaterials 2021, 273, 120817. [Google Scholar] [CrossRef]
- Trepel, J.; Mollapour, M.; Giaccone, G.; Neckers, L. Targeting the dynamic HSP90 complex in cancer. Nat. Rev. Cancer 2010, 10, 537–549. [Google Scholar] [CrossRef] [PubMed]
- Schopf, F.H.; Biebl, M.M.; Buchner, J. The HSP90 chaperone machinery. Nat. Rev. Mol. Cell Biol. 2017, 18, 345–360. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, J.; Carter, T.R.; Cohen, M.S.; Blagg, B.S.J. Old and new approaches to target the Hsp90 chaperone. Curr. Cancer Drug Targets 2020, 20, 253–270. [Google Scholar] [CrossRef]
- Jhaveri, K.; Modi, S. Ganetespib: Research and clinical development. OncoTargets Ther. 2015, 8, 1849–1858. [Google Scholar] [CrossRef]
- Birbo, B.; Madu, E.E.; Madu, C.O.; Jain, A.; Lu, Y. Role of HSP90 in Cancer. Int. J. Mol. Sci. 2021, 22, 10317. [Google Scholar] [CrossRef]
- Chatterjee, S.; Bhattacharya, S.; Socinski, M.A.; Burns, T.F. HSP90 inhibitors in lung cancer: Promise still unfulfilled. Clin. Adv. Hematol. Oncol. 2016, 14, 346–356. [Google Scholar]
- Park, H.K.; Yoon, N.G.; Lee, J.E.; Hu, S.; Yoon, S.; Kim, S.Y.; Hong, J.H.; Nam, D.; Chae, Y.C.; Park, J.B.; et al. Unleashing the full potential of Hsp90 inhibitors as cancer therapeutics through simultaneous inactivation of Hsp90, Grp94, and TRAP1. Exp. Mol. Med. 2020, 52, 79–91. [Google Scholar] [CrossRef]
- Yuno, A.; Lee, M.-J.; Lee, S.; Tomita, Y.; Rekhtman, D.; Moore, B.; Trepel, J.B. Clinical evaluation and biomarker profiling of Hsp90 inhibitors. Methods Mol. Biol. 2018, 1709, 423–441. [Google Scholar] [CrossRef]
- Kim, Y.S.; Alarcon, S.V.; Lee, S.; Lee, M.-J.; Giaccone, G.; Neckers, L.; Trepel, J.B. Update on Hsp90 inhibitors in clinical trial. Curr. Top. Med. Chem. 2009, 9, 1479–1492. [Google Scholar] [CrossRef]
- Gaspar, N.; Sharp, S.Y.; Pacey, S.; Jones, C.; Walton, M.; Vassal, G.; Eccles, S.; Pearson, A.; Workman, P. Acquired resistance to 17-allylamino-17-demethoxygeldanamycin (17-AAG, tanespimycin) in glioblastoma cells. Cancer Res. 2009, 69, 1966–1975. [Google Scholar] [CrossRef]
- Pearl, L.H. Review: The HSP90 molecular chaperone—An enigmatic ATPase. Biopolymers 2016, 105, 594–607. [Google Scholar] [CrossRef]
- Richter, K.; Moser, S.; Hagn, F.; Friedrich, R.; Hainzl, O.; Heller, M.; Schlee, S.; Kessler, H.; Reinstein, J.; Buchner, J. Intrinsic inhibition of the Hsp90 ATPase activity. J. Biol. Chem. 2006, 281, 11301–11311. [Google Scholar] [CrossRef]
- Ferraldeschi, R.; Welti, J.; Powers, M.V.; Yuan, W.; Smyth, T.; Seed, G.; Riisnaes, R.; Hedayat, S.; Wang, H.; Crespo, M.; et al. Second-generation HSP90 inhibitor onalespib blocks mRNA splicing of androgen receptor variant 7 in prostate cancer cells. Cancer Res. 2016, 76, 2731–2742. [Google Scholar] [CrossRef]
- Parimi, S.; Tsang, R.Y. Hsp90 inhibitors in oncology: Ready for prime time? Curr. Oncol. 2014, 21, e663–e667. [Google Scholar] [CrossRef] [PubMed]
- Zaarur, N.; Gabai, V.L.; Porco, J.A., Jr.; Calderwood, S.K.; Sherman, M.Y. Targeting heat shock response to sensitize cancer cells to proteasome and Hsp90 inhibitors. Cancer Res. 2006, 66, 1783–1791. [Google Scholar] [CrossRef] [PubMed]
- Miura, A.; Sootome, H.; Fujita, N.; Suzuki, T.; Fukushima, H.; Mizuarai, S.; Masuko, N.; Ito, K.; Hashimoto, A.; Uto, Y.; et al. TAS-119, a novel selective Aurora A and TRK inhibitor, exhibits antitumor efficacy in preclinical models with deregulated activation of the Myc, β-Catenin, and TRK pathways. Investig. New Drugs 2021, 39, 724–735. [Google Scholar] [CrossRef] [PubMed]
- Bussenius, J.; Blazey, C.M.; Aay, N.; Anand, N.K.; Arcalas, A.; Baik, T.; Bowles, O.J.; Buhr, C.A.; Costanzo, S.; Curtis, J.K.; et al. Discovery of XL888: A novel tropane-derived small molecule inhibitor of HSP90. Bioorg. Med. Chem. Lett. 2012, 22, 5396–5404. [Google Scholar] [CrossRef]
- Hoy, S.M. Pimitespib: First Approval. Drugs 2022, 82, 1413–1418. [Google Scholar] [CrossRef]
- Maiti, S.; Picard, D. Cytosolic Hsp90 Isoform-Specific Functions and Clinical Significance. Biomolecules 2022, 12, 1166. [Google Scholar] [CrossRef]
- Socinski, M.A.; Goldman, J.; El-Hariry, I.; Koczywas, M.; Vukovic, V.; Horn, L.; Paschold, E.; Salgia, R.; West, H.; Sequist, L.V.; et al. A multicenter phase II study of ganetespib monotherapy in patients with genotypically defined advanced non-small cell lung cancer. Clin. Cancer Res. 2013, 19, 3068–3077. [Google Scholar] [CrossRef]
- Zavareh, R.B.; Spangenberg, S.H.; Woods, A.; Martínez-Peña, F.; Lairson, L.L. HSP90 inhibition enhances cancer immunotherapy by modulating the surface expression of multiple immune checkpoint proteins. Cell Chem. Biol. 2021, 28, 158–168.e5. [Google Scholar] [CrossRef]
- Garcia-Carbonero, R.; Carnero, A.; Paz-Ares, L. Inhibition of HSP90 molecular chaperones: Moving into the clinic. Lancet Oncol. 2013, 14, e358–e369. [Google Scholar] [CrossRef]
- Akram, A.; Khalil, S.; Halim, S.A.; Younas, H.; Iqbal, S.; Mehar, S. Therapeutic uses of HSP90 inhibitors in non-small cell lung carcinoma (NSCLC). Curr. Drug Metab. 2018, 19, 335–341. [Google Scholar] [CrossRef]
- Gabai, V.L.; Yaglom, J.A.; Waldman, T.; Sherman, M.Y. Heat shock protein Hsp72 controls oncogene-induced senescence pathways in cancer cells. Mol. Cell. Biol. 2009, 29, 559–569. [Google Scholar] [CrossRef] [PubMed]
- Rao, A.; Taylor, J.L.; Chi-Sabins, N.; Kawabe, M.; Gooding, W.E.; Storkus, W.J. Combination therapy with HSP90 inhibitor 17-DMAG reconditions the tumor microenvironment to improve recruitment of therapeutic T cells. Cancer Res. 2012, 72, 3196–3206. [Google Scholar] [CrossRef] [PubMed]
- Song, K.H.; Oh, S.J.; Kim, S.; Cho, H.; Lee, H.J.; Song, J.S.; Chung, J.Y.; Cho, E.; Lee, J.; Jeon, S.; et al. HSP90A inhibition promotes anti-tumor immunity by reversing multi-modal resistance and stem-like property of immune-refractory tumors. Nat. Commun. 2020, 11, 562. [Google Scholar] [CrossRef] [PubMed]
- Neckers, L. Hsp90 Inhibitors as Novel Cancer Chemotherapeutic Agents. Trends Mol. Med. 2002, 8 (Suppl. 4), S55–S61. [Google Scholar] [CrossRef]
- Cools, R.; Vermeulen, K.; Vonck, E.; Baekelandt, V.; Varlow, C.; Narykina, V.; Cawthorne, C.; Van Laere, K.; Vanduffel, W.; Vasdev, N.; et al. In Vivo Visualization and Quantification of Brain Heat Shock Protein 90 with [11C]HSP990 in Healthy Aging and Neurodegeneration. J. Nucl. Med. 2025, 66, 940–947. [Google Scholar] [CrossRef]
- Shimomura, A.; Yamamoto, N.; Kondo, S.; Saijo, N.; Yamada, K.; Okamoto, I.; Hatake, K.; Satoh, T.; Watanabe, T.; Esaki, T.; et al. First-in-human phase I study of an oral HSP90 inhibitor, TAS-116, in patients with advanced solid tumors. Mol. Cancer Ther. 2019, 18, 531–540. [Google Scholar] [CrossRef]
- Travers, J.; Sharp, S.Y.; Workman, P. HSP90 inhibition: Two-pronged exploitation of cancer dependencies. Drug Discov. Today 2012, 17, 242–252. [Google Scholar] [CrossRef]
- Jim Sang, J.; Acquaviva, J.; Friedland, J.C.; Smith, D.L.; Sequeira, M.; Zhang, C.; Jiang, Q.; Xue, L.; Lovly, C.M.; Jimenez, J.P.; et al. Targeted inhibition of the molecular chaperone Hsp90 overcomes ALK inhibitor resistance in non-small cell lung cancer. Cancer Discov. 2013, 3, 430–443. [Google Scholar] [CrossRef]
- Haarberg, H.E.; Paraiso, K.H.T.; Wood, E.; Rebecca, V.W.; Sondak, V.K.; Koomen, J.M.; Smalley, K.S.M. Inhibition of Wee1, AKT, and CDK4 underlies the efficacy of the HSP90 inhibitor XL888 in an in vivo model of NRAS-mutant melanoma. Mol. Cancer Ther. 2013, 12, 901–912. [Google Scholar] [CrossRef]
- Altieri, D.C. Mitochondrial Hsp90 chaperones as novel molecular targets in prostate cancer. Future Oncol. 2010, 6, 487–489. [Google Scholar] [CrossRef]
- Jones, R.L.; Golčić, M. Recent advances in the systemic treatment of gastrointestinal stromal tumors. Cancer Biol. Med. 2023, 20, 701–705. [Google Scholar] [CrossRef] [PubMed]
- Phillips, M.J.; Alese, O.B.; Horvat, N.K.; Greene, E.; Gbolahan, O.B.; Coleman, K.; Doxie, D.B.; Parihar, V.; Mahdi, Z.K.; McCook Veal, A.; et al. XL888 and pembrolizumab modulate the immune landscape of colorectal tumors in a phase Ib II clinical trial. Oncoimmunology 2025, 14, 2475620. [Google Scholar] [CrossRef] [PubMed]
- Dernovšek, J.; Tomašič, T. Following the design path of isoform-selective Hsp90 inhibitors: Small differences, great opportunities. Pharmacol. Ther. 2023, 245, 108396. [Google Scholar] [CrossRef]
- Merfeld, T.; Peng, S.; Keegan, B.M.; Crowley, V.M.; Brackett, C.M.; Gutierrez, A.; McCann, N.R.; Reynolds, T.S.; Rhodes, M.C.; Byrd, K.M.; et al. Elucidation of novel TRAP1-Selective inhibitors that regulate mitochondrial processes. Eur. J. Med. Chem. 2023, 258, 115531. [Google Scholar] [CrossRef]
- Bricelj, A.; Steinebach, C.; Kuchta, R.; Gütschow, M.; Sosič, I. E3 Ligase Ligands in Successful PROTACs: An Overview of Syntheses and Linker Attachment Points. Front. Chem. 2021, 9, 707317. [Google Scholar] [CrossRef]
- Kelm, J.M.; Pandey, D.S.; Malin, E.; Kansou, H.; Arora, S.; Kumar, R.; Gavande, N.S. PROTAC’ing oncoproteins: Targeted protein degradation for cancer therapy. Mol. Cancer 2023, 22, 62. [Google Scholar] [CrossRef]
- Solomon, J.; Kendall, R.; Patel, S.; Yang, L.; Humbard, M.; Wittung-Stafshede, P.; Thiel, C.B. J-domain proteins form binary complexes with Hsp90 and ternary complexes with Hsp90–Hsp70: Insights into chaperone collaboration. J. Mol. Biol. 2023, 435, 168090. [Google Scholar] [CrossRef]
- Soga, S.; Akinaga, S.; Shiotsu, Y. Hsp90 inhibitors as anti-cancer agents, from basic discoveries to clinical development. Curr. Pharm. Des. 2013, 19, 366–376. [Google Scholar] [CrossRef] [PubMed]
- Sharp, S.Y.; Workman, P. Inhibitors of the Hsp90 molecular chaperone: Current status. Adv. Cancer Res. 2006, 95, 323–348. [Google Scholar] [CrossRef] [PubMed]
- Jhaveri, K.; Wang, R.; Teplinsky, E.; Chandarlapaty, S.; Solit, D.; Cadoo, K.; Speyer, J.; D’Andrea, G.; Adams, S.; Patil, S.; et al. A phase I trial of ganetespib in combination with paclitaxel and trastuzumab in patients with human epidermal growth factor receptor-2 (HER2)-positive metastatic breast cancer. Breast Cancer Res. 2017, 19, 89. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.; Kwon, J.; Hong, S.; Moon, A.-N.; Jeong, J.; Kwon, S.; Kim, J.-A.; Lee, M.; Lee, H.; Lee, J.H.; et al. Discovery of novel heat shock protein (Hsp90) inhibitors based on luminespib with potent antitumor activity. Bioorg. Med. Chem. Lett. 2020, 30, 127165. [Google Scholar] [CrossRef]
- Ramalingam, S.S.; Goss, G.; Rosell, R.; Schmid-Bindert, G.; Zaric, B.; Andric, Z.; Bondarenko, I.; Komov, D.; Ceric, T.; Khuri, F.; et al. A randomized phase II study of ganetespib, a heat shock protein 90 inhibitor, in combination with docetaxel in second-line therapy of advanced non-small cell lung cancer (GALAXY-1). Ann. Oncol. 2015, 26, 1741–1748. [Google Scholar] [CrossRef]
- Chandarlapaty, S.; Sawai, A.; Scaltriti, M.; Rodrik-Outmezguine, V.; Grbovic-Huezo, O.; Serra, V.; Majumder, P.K.; Baselga, J.; Rosen, N. AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell 2011, 19, 58–71. [Google Scholar] [CrossRef]
- Proia, D.A.; Kaufmann, G.F. Targeting Heat-Shock Protein 90 (HSP90) as a complementary strategy to immune checkpoint blockade for cancer therapy. Cancer Immunol. Res. 2015, 3, 583–589. [Google Scholar] [CrossRef]
- Miyajima, N.; Tsutsumi, S.; Sourbier, C.; Beebe, K.; Huang, Y.; Tatokoro, M.; Shinohara, N.; Nonomura, K.; Neckers, L.; Rivas, C. The HSP90 inhibitor ganetespib synergizes with the MET kinase inhibitor crizotinib in both crizotinib-sensitive and -resistant MET-driven tumor models. Cancer Res. 2013, 73, 7022–7033. [Google Scholar] [CrossRef]
- Prodromou, C.; Bjorklund, D.M. Advances towards understanding the mechanism of action of the Hsp90 complex. Biomolecules 2022, 12, 600. [Google Scholar] [CrossRef]
- Moser, C.; Lang, S.A.; Stoeltzing, O. Heat shock protein 90 (Hsp90) as a molecular target for therapy of gastrointestinal cancer. Anticancer Res. 2009, 29, 2031–2042. [Google Scholar]
- Youssef, M.E.; Cavalu, S.; Hasan, A.M.; Yahya, G.; Abd-Eldayem, M.A.; Saber, S. Role of Ganetespib, an HSP90 inhibitor, in cancer therapy: From molecular mechanisms to clinical practice. Int. J. Mol. Sci. 2023, 24, 5014. [Google Scholar] [CrossRef]
- Wei, H.; Zhang, Y.; Jia, Y.; Chen, X.; Niu, T.; Chatterjee, A.; He, P.; Hou, G. Heat shock protein 90: Biological functions, diseases, and therapeutic targets. MedComm 2024, 5, e470. [Google Scholar] [CrossRef]
- Cheng, X. A comprehensive review of HER2 in cancer biology and therapeutics. Genes 2024, 15, 903. [Google Scholar] [CrossRef] [PubMed]
- Bashraheel, S.S.; Kheraldine, H.; Khalaf, S.; Al Moustafa, A.E. Metformin and HER2 positive breast cancer: Mechanisms and therapeutic implications. Biomed. Pharmacother. 2023, 162, 114676. [Google Scholar] [CrossRef] [PubMed]
- Švec, X.; Štorkánová, H.; Špiritović, M.; Slabý, K.; Oreská, S.; Pekáčová, A.; Heřmánková, B.; Bubová, K.; Česák, P.; Khouri, H.; et al. Hsp90 as a myokine: Its association with systemic inflammation after exercise interventions in patients with myositis and healthy subjects. Int. J. Mol. Sci. 2022, 23, 11451. [Google Scholar] [CrossRef] [PubMed]
- Jego, G.; Hermetet, F.; Girodon, F.; Garrido, C. Chaperoning STAT3/5 by heat shock proteins: Interest of their targeting in cancer therapy. Cancers 2020, 12, 21. [Google Scholar] [CrossRef]
- Park, M.H.; Hong, J.T. Roles of NF-κB in cancer and inflammatory diseases and their therapeutic approaches. Cells 2016, 5, 15. [Google Scholar] [CrossRef]
- Kunachowicz, D.; Król Kulikowska, M.; Raczycka, W.; Sleziak, J.; Błażejewska, M.; Kulbacka, J. Heat shock proteins, a double-edged sword: Significance in cancer progression, chemotherapy resistance and novel therapeutic perspectives. Cancers 2024, 16, 1500. [Google Scholar] [CrossRef]
- Li, Y.; Dong, J.; Qin, J.J. Small molecule inhibitors targeting heat shock protein 90: An updated review. Eur. J. Med. Chem. 2024, 246, 116562. [Google Scholar] [CrossRef]
- Rahimi, A.; Baghernejadan, Z.; Hazrati, A.; Malekpour, K.; Samimi, L.N.; Najafi, A.; Falak, R.; Khorramdelazad, H. Combination therapy with immune checkpoint inhibitors in colorectal cancer: Challenges, resistance mechanisms, and the role of microbiota. Biomed. Pharmacother. 2025, 186, 118014. [Google Scholar] [CrossRef]
- Jhaveri, K.; Taldone, T.; Modi, S.; Chiosis, G. Advances in the clinical development of heat shock protein 90 inhibitors in cancers. Biochim. Biophys. Acta Mol. Cell Res. 2012, 1823, 742–755. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Goyal, S.; Jamal, S.; Singh, A.; Grover, A. Hsp90: Friends, clients and natural foes. Biochimie 2016, 127, 227–240. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Shu, H.; Xia, Q.; You, Q.; Wang, L. Recent developments of HSP90 inhibitors: An updated patent review (2020–present). Expert Opin. Ther. Pat. 2024, 34, 1–15. [Google Scholar] [CrossRef]
- Weaver, B.A. How Taxol/paclitaxel kills cancer cells. Mol. Biol. Cell 2014, 25, 2677–2681. [Google Scholar] [CrossRef] [PubMed]
- Jordan, M.A.; Wilson, L. Microtubules as a target for anticancer drugs. Nat. Rev. Cancer 2004, 4, 253–265. [Google Scholar] [CrossRef]
- Proia, D.A.; Sang, J.; He, S.; Smith, D.L.; Sequeira, M.; Zhang, C.; Liu, Y.; Ye, S.; Zhou, D.; Blackman, R.K.; et al. Synergistic activity of the Hsp90 inhibitor ganetespib with taxanes in non-small cell lung cancer models. Investig. New Drugs 2012, 30, 2201–2209. [Google Scholar] [CrossRef]
- Solit, D.B.; Basso, A.D.; Olshen, A.B.; Scher, H.I.; Rosen, N. Inhibition of heat shock protein 90 function down-regulates Akt kinase and sensitizes tumors to Taxol. Cancer Res. 2003, 63, 2139–2144. [Google Scholar]
- Lang, J.E.; Forero-Torres, A.; Yee, D.; Yau, C.; Wolf, D.; Park, J.; Parker, B.A.; Chien, A.J.; Wallace, A.M.; Murthy, R.; et al. Safety and efficacy of HSP90 inhibitor ganetespib for neoadjuvant treatment of stage II/III breast cancer. NPJ Breast Cancer 2022, 8, 128. [Google Scholar] [CrossRef]
- Pillai, R.N.; Fennell, D.A.; Kovcin, V.; Ciuleanu, T.-E.; Ramlau, R.; Kowalski, D.; Schenker, M.; Yalcin, I.; Teofilovici, F.; Vukovic, V.; et al. Randomized Phase III study of ganetespib, a heat shock protein 90 inhibitor, with docetaxel versus docetaxel in advanced non-small-cell lung cancer (GALAXY-2). J. Clin. Oncol. 2020, 38, 613–622. [Google Scholar] [CrossRef]
- U.S. National Library of Medicine. Onalespib and Paclitaxel in Treating Patients with Advanced Triple Negative Breast Cancer. ClinicalTrials.gov Identifier: NCT02474173. 2020. Available online: https://clinicaltrials.gov/study/NCT02474173 (accessed on 11 June 2025).
- Seegobin, K.; Majeed, U.; Wiest, N.; Manochakian, R.; Lou, Y.; Zhao, Y. Immunotherapy in Non-Small Cell Lung Cancer with Actionable Mutations Other than EGFR. Front. Oncol. 2021, 11, 750657. [Google Scholar] [CrossRef]
- Mini, E.; Nobili, S.; Caciagli, B.; Landini, I.; Mazzei, T. Cellular pharmacology of gemcitabine. Ann. Oncol. 2006, 17 (Suppl. S5), v7–v12. [Google Scholar] [CrossRef] [PubMed]
- Cavalcante, L.S.; Monteiro, G. Gemcitabine: Metabolism and molecular mechanisms of action, sensitivity and chemoresistance in pancreatic cancer. Eur. J. Pharmacol. 2014, 741, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Pennisi, R.; Ascenzi, P.; Di Masi, A. Hsp90: A new player in DNA repair? Biomolecules 2015, 5, 2589–2618. [Google Scholar] [CrossRef] [PubMed]
- Ha, K.; Fiskus, W.; Rao, R.; Balusu, R.; Venkannagari, S.; Nalabothula, N.R.; Bhalla, K.N. Hsp90 inhibitor mediated disruption of chaperone association of ATR with Hsp90 sensitizes cancer cells to DNA damage. Mol. Cancer Ther. 2011, 10, 1194–1206. [Google Scholar] [CrossRef]
- Park, J.H.; Kim, W.S.; Kim, J.Y.; Park, M.; Nam, J.H.; Yun, C.W.; Kwon, Y.G.; Jo, I. Chk1 and Hsp90 cooperatively regulate phosphorylation of endothelial nitric oxide synthase at serine 1179. Free Radic. Biol. Med. 2011, 51, 2217–2226. [Google Scholar] [CrossRef]
- Orth, M.; Albrecht, V.; Seidl, K.; Kinzel, L.; Unger, K.; Hess, J.; Kreutzer, L.; Sun, N.; Stegen, B.; Nieto, A.; et al. Inhibition of HSP90 as a strategy to radiosensitize glioblastoma: Targeting the DNA damage response and beyond. Front. Oncol. 2021, 11, 612354. [Google Scholar] [CrossRef]
- Stecklein, S.R.; Kumaraswamy, E.; Behbod, F.; Wang, W.; Chaguturu, V.; Harlan Williams, L.M.; Jensen, R.A. BRCA1 and HSP90 cooperate in homologous and non homologous DNA double strand break repair and G2/M checkpoint activation. Proc. Natl. Acad. Sci. USA 2012, 109, 13650–13655. [Google Scholar] [CrossRef]
- Smyth, T.; Paraiso, K.H.T.; Hearn, K.; Rodriguez Lopez, A.M.; Munck, J.M.; Haarberg, H.E.; Sondak, V.K.; Thompson, N.T.; Azab, M.; Lyons, J.F.; et al. Inhibition of HSP90 by AT13387 delays the emergence of resistance to BRAF inhibitors and overcomes resistance to dual BRAF and MEK inhibition in melanoma models. Mol. Cancer Ther. 2014, 13, 2793–2804. [Google Scholar] [CrossRef]
- Koga, F.; Xu, W.; Karpova, T.S.; McNally, J.G.; Baron, R.; Neckers, L. Hsp90 inhibition transiently activates Src kinase and promotes Src-dependent Akt and Erk activation. Proc. Natl. Acad. Sci. USA 2006, 103, 11318–11322. [Google Scholar] [CrossRef]
- Pedersen, K.S.; Kim, G.P.; Foster, N.R.; Wang-Gillam, A.; Erlichman, C.; McWilliams, R.R. Phase II trial of gemcitabine and tanespimycin (17AAG) in metastatic pancreatic cancer: A Mayo Clinic Phase II Consortium study. Investig. New Drugs 2015, 33, 963–968. [Google Scholar] [CrossRef]
- Wahner Hendrickson, A.E.; Oberg, A.L.; Glaser, G.; Camoriano, J.K.; Peethambaram, P.P.; Colon-Otero, G.; Erlichman, C.; Ivy, S.P.; Kaufmann, S.H.; Karnitz, L.M.; et al. A phase II study of gemcitabine in combination with tanespimycin in advanced epithelial ovarian and primary peritoneal carcinoma. Gynecol. Oncol. 2012, 124, 210–215. [Google Scholar] [CrossRef]
- Daunys, S.; Matulis, D.; Petrikaitė, V. Synergistic activity of Hsp90 inhibitors and anticancer agents in pancreatic cancer cell cultures. Sci. Rep. 2019, 9, 16177. [Google Scholar] [CrossRef] [PubMed]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef]
- Galluzzi, L.; Senovilla, I.; Vitale, I.; Michels, J. Molecular mechanisms of cisplatin resistance. Oncogene 2012, 31, 1869–1883. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, L.; Issa, I.I.; Haraldsdóttir, H.; Hald, J.L.; Schmitz, A.; Due, H.; Dybkær, K. Hsp90 inhibition sensitizes DLBCL cells to cisplatin. Cancer Chemother. Pharmacol. 2022, 89, 431–440. [Google Scholar] [CrossRef]
- Kim, H.J.; Gong, M.K.; Yoon, C.Y.; Kang, J.; Yun, M.; Cho, N.H.; Rha, S.Y.; Choi, Y.D. Synergistic antitumor effects of combined treatment with HSP90 inhibitor and PI3K/mTOR dual inhibitor in cisplatin resistant human bladder cancer cells. Yonsei Med. J. 2020, 61, 587–596. [Google Scholar] [CrossRef]
- Feng, L.; Xu, X.; Zhao, K. NFYB potentiates STK33 activation to promote cisplatin resistance in diffuse large B-cell lymphoma via Hedgehog pathway. Leuk Res. 2021, 111, 106708. [Google Scholar] [CrossRef]
- Mortensen, A.C.L. Overcoming limitations of cisplatin therapy by additional treatment with the HSP90 inhibitor onalespib. Front. Oncol. 2020, 10, 532285. [Google Scholar] [CrossRef]
- Cho, W.C.; Wong, C.F. Potential benefits of combined treatment with Hsp90 inhibitor AUY922 and cisplatin for overcoming drug resistance in nasopharyngeal carcinoma. Am. J. Cancer Res. 2025, 15, 533–545. [Google Scholar] [CrossRef]
- Ewers, K.M.; Patil, S.; Kopp, W.; Thomale, J.; Quilitz, T.; Magerhans, A.; Wang, X.; Hessmann, E.; Dobbelstein, M. HSP90 inhibition synergizes with cisplatin to eliminate basal-like pancreatic ductal adenocarcinoma cells. Cancers. 2021, 13, 6163. [Google Scholar] [CrossRef] [PubMed]
- Huber, C.; Zinserling, J.; Benda, N.; Vetter, T.; Rueckbeil, M. Methodological Insights on Biomarker-Based Patient Selection: A Review of Scientific Advice Procedures at the European Medicines Agency. Clin. Pharmacol. Ther. 2025, 117, 1226–1235. [Google Scholar] [CrossRef] [PubMed]
- Roskoski , R., Jr. The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol. Res. 2014, 79, 34–74. [Google Scholar] [CrossRef] [PubMed]
- Arteaga, C.L.; Engelman, J.A. ERBB receptors: From oncogene discovery to basic science to mechanism-based cancer therapeutics. Cancer Cell 2014, 25, 282–303. [Google Scholar] [CrossRef]
- Yamaoka, T.; Kusumoto, S.; Ando, K.; Ohba, M.; Ohmori, T. Receptor tyrosine kinase-targeted cancer therapy. Int. J. Mol. Sci. 2018, 19, 3491. [Google Scholar] [CrossRef]
- Minuti, G.; D’Incecco, A.; Cappuzzo, F. Targeted therapy for NSCLC with driver mutations. Expert Opin. Biol. Ther. 2013, 13, 1401–1412. [Google Scholar] [CrossRef]
- Peron, M.; Bonvini, P.; Rosolen, A. Effect of inhibition of the ubiquitin proteasome system and Hsp90 on growth and survival of rhabdomyosarcoma cells in vitro. BMC Cancer 2012, 12, 233. [Google Scholar] [CrossRef]
- Huynh, T.K.; Ho, C.Y.; Tsai, C.H.; Wang, C.K.; Chen, Y.J.; Bau, D.T.; Tu, C.Y.; Li, T.S.; Huang, W.C. Proteasome inhibitors suppress ErbB family expression through HSP90-mediated lysosomal degradation. Int. J. Mol. Sci. 2019, 20, 4812. [Google Scholar] [CrossRef]
- Hsu, J.L.; Hung, M.C. The role of HER2, EGFR, and other receptor tyrosine kinases in breast cancer. Cancer Metastasis Rev. 2016, 35, 575–588. [Google Scholar] [CrossRef]
- Modi, S.; Stopeck, A.T.; Linden, H.; Solit, D.; Chandarlapaty, S.; Rosen, N.; D’Andrea, G.; Dickler, M.; Moynahan, M.E.; Sugarman, S.; et al. Hsp90 inhibition is effective in breast cancer: A Phase II trial of tanespimycin (17-AAG) plus trastuzumab in patients with HER2-positive metastatic breast cancer progressing on trastuzumab. Clin. Cancer Res. 2011, 17, 5132–5139. [Google Scholar] [CrossRef]
- Johnson, M.L.; Yu, H.A.; Hart, E.M.; Weitner, B.B.; Rademaker, A.W.; Patel, J.D.; Kris, M.G.; Riely, G.J. Phase I/II study of HSP90 inhibitor AUY922 and erlotinib for EGFR-mutant lung cancer with acquired resistance to EGFR tyrosine kinase inhibitors. J. Clin. Oncol. 2015, 33, 1666–1673. [Google Scholar] [CrossRef]
- Calderwood, S.K. Heat shock proteins and cancer: Intracellular chaperones or extracellular signalling ligands? Philos. Trans. R. Soc. Lond B Biol. Sci. 2018, 373, 20160524. [Google Scholar] [CrossRef]
- Bhullar, K.S.; Lagarón, N.O.; McGowan, E.M.; Parmar, I.; Jha, A.; Hubbard, B.P.; Rupasinghe, H.P.V. Kinase targeted cancer therapies: Progress, challenges and future directions. Mol. Cancer 2018, 17, 48. [Google Scholar] [CrossRef] [PubMed]
- Katayama, R.; Shaw, A.T.; Khan, T.M.; Mino-Kenudson, M.; Solomon, B.J.; Halmos, B.; Jessop, N.A.; Wain, J.C.; Yeo, A.T.; Benes, C.; et al. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung cancers. Sci. Transl. Med. 2012, 4, 120ra17. [Google Scholar] [CrossRef] [PubMed]
- Normant, E.; Paez, G.; West, K.A.; Lim, A.R.; Slocum, K.L.; Tunkey, C.; McDougall, J.; Wylie, A.A.; Robison, K.; Caliri, K.; et al. The Hsp90 inhibitor IPI504 rapidly lowers EML4 ALK levels and induces tumor regression in ALK driven NSCLC models. Oncogene 2011, 30, 2581–2586. [Google Scholar] [CrossRef]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.T.; Haarberg, H.E.; Wood, E.; Rebecca, V.W.; Chen, Y.A.; Xiang, Y.; Ribas, A.; Lo, R.S.; Weber, J.S.; Sondak, V.K.; et al. The HSP90 inhibitor XL888 overcomes BRAF inhibitor resistance mediated through diverse mechanisms. Clin. Cancer Res. 2012, 18, 2502–2514. [Google Scholar] [CrossRef]
- Ben-Hur, V.; Denichenko, P.; Siegfried, Z.; Maimon, A.; Krainer, A.; Davidson, B.; Karni, R. S6K1 alternative splicing modulates its oncogenic activity and regulates mTORC1. Cell Rep. 2013, 3, 103–115. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. A Study of AT13387 in Patients with Non-Small Cell Lung Cancer (NSCLC) Alone and in Combination with Crizotinib. 2016. Available online: https://clinicaltrials.gov/study/NCT01712217 (accessed on 17 June 2025).
- Eroglu, Z.; Chen, Y.A.; Smalley, I.; Li, J.; Markowitz, J.K.; Brohl, A.S.; Tetteh, L.; Taylor, H.; Sondak, V.K.; Khushalani, N.I.; et al. Combined BRAF, MEK, and heat-shock protein 90 inhibition in advanced BRAF V600-mutant melanoma. Cancer 2024, 130, 232–243. [Google Scholar] [CrossRef]
- Jhaveri, K.; Chandarlapaty, S.; Iyengar, N.; Morris, P.G.; Corben, A.D.; Patil, S.; Akram, M.; Towers, R.; Sakr, R.A.; King, T.A.; et al. Biomarkers That Predict Sensitivity to Heat Shock Protein 90 Inhibitors. Clin. Breast Cancer 2016, 16, 276–283. [Google Scholar] [CrossRef]
- Kim, J.Y.; Cho, T.M.; Park, J.M.; Kim, D.H.; Park, D.Y.; Yoon, J.H.; Jung, W.H.; Min, K.W.; Choi, S.Y.; Park, Y.M.; et al. A novel HSP90 inhibitor SL-145 suppresses metastatic triple-negative breast cancer without triggering the heat shock response. Oncogene 2022, 41, 3289–3297. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Hui, E.; Cheung, J.; Zhu, J.; Su, X.; Taylor, M.J.; Wallweber, H.A.; Sasmal, D.K.; Huang, J.; Kim, J.M.; Mellman, I.; et al. T cell costimulatory receptor CD28 is a primary target for PD-1 mediated inhibition. Science 2017, 355, 1428–1433. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer immune set point. Nat. Rev. Cancer 2017, 17, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Immune checkpoint blockade: A common denominator approach to cancer therapy. Cancer Cell 2015, 27, 450–461. [Google Scholar] [CrossRef]
- Wei, S.C.; Levine, J.H.; Cogdill, A.P.; Zhao, Y.; Anang, N.A.A.S.; Andrews, M.C.; Sharma, P.; Wang, J.; Wargo, J.A.; Pe’er, D.; et al. Distinct cellular mechanisms underlie anti-CTLA-4 and anti-PD-1 checkpoint blockade. Cell 2017, 170, 1120–1133.e17. [Google Scholar] [CrossRef]
- Socinski, M.A.; Jotte, R.M.; Cappuzzo, F.; Orlandi, F.; Stroyakovskiy, D.; Nogami, N.; Rodríguez-Abreu, D.; Moro-Sibilot, D.; Thomas, C.A.; Barlesi, F.; et al. Atezolizumab for first-line treatment of metastatic nonsquamous non–small-cell lung cancer. N. Engl. J. Med. 2018, 378, 2288–2301. [Google Scholar] [CrossRef]
- Mbofung, R.M.; McKenzie, J.A.; Malu, S.; Zhang, M.; Peng, W.; Liu, C.; Kuiatse, I.; Tieu, T.; Williams, L.; Devi, S.; et al. HSP90 inhibition enhances cancer immunotherapy by upregulating interferon response genes. Nat. Commun. 2017, 8, 451. [Google Scholar] [CrossRef]
- De Almeida, S.; Regimbeau, M.; Jego, G.; Garrido, C.; Girodon, F.; Hermetet, F. Heat shock proteins and PD-1/PD-L1 as potential therapeutic targets in myeloproliferative neoplasms. Cancers 2020, 12, 2592. [Google Scholar] [CrossRef]
- Tsuge, A.; Watanabe, S.; Kawazoe, A.; Togashi, Y.; Itahashi, K.; Masuda, M.; Sai, A.; Takei, S.; Muraoka, H.; Ohkubo, S.; et al. The HSP90 inhibitor pimitespib targets regulatory T cells in the tumor microenvironment. Cancer Immunol. Res. 2025, 13, 273–285. [Google Scholar] [CrossRef]
- Kawazoe, A.; Itahashi, K.; Yamamoto, N.; Kotani, D.; Kuboki, Y.; Taniguchi, H.; Harano, K.; Naito, Y.; Suzuki, M.; Fukutani, M.; et al. TAS-116 (pimitespib), an oral HSP90 inhibitor, in combination with nivolumab in patients with colorectal cancer and other solid tumors: An open-label, dose-finding, and expansion phase Ib trial (EPOC1704). Clin. Cancer Res. 2021, 27, 6709–6715. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, K.; Acharya, C.R.; Nagata, H.; Yang, X.; Hartman, Z.C.; Hobeika, A.; Hughes, P.F.; Haystead, T.A.J.; Morse, M.A.; Lyerly, H.K.; et al. Combination of a novel heat shock protein 90-targeted photodynamic therapy with PD-1/PD-L1 blockade induces potent systemic antitumor efficacy and abscopal effect against breast cancers. J. Immunother. Cancer 2022, 10, e004793. [Google Scholar] [CrossRef] [PubMed]
- Chambers, C.A.; Kuhns, M.S.; Egen, J.G.; Allison, J.P. CTLA-4-mediated inhibition in regulation of T cell responses: Mechanisms and manipulation in tumor immunotherapy. Annu. Rev. Immunol. 2001, 19, 565–594. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Albakova, Z.; Mangasarova, Y.; Sapozhnikov, A. Impaired heat shock protein expression in activated T cells in B-cell lymphoma. Biomedicines 2022, 10, 2747. [Google Scholar] [CrossRef]
- Alegre, M.-L.; Frauwirth, K.A.; Thompson, C.B. T-cell regulation by CD28 and CTLA-4. Nat. Rev. Immunol. 2001, 1, 220–228. [Google Scholar] [CrossRef]
- Pourmousa, M.; Jain, S.; Barnaeva, E.; Jin, W.; Hochuli, J.; Itkin, Z.; Maxfield, T.; Melo-Filho, C.; Thieme, A.; Wilson, K.; et al. AI-driven discovery of synergistic drug combinations against pancreatic cancer. Nat. Commun. 2025, 16, 4020. [Google Scholar] [CrossRef]
- Ayoub, N.M. Editorial: Novel combination therapies for the treatment of solid cancers. Front. Oncol. 2021, 11, 708943. [Google Scholar] [CrossRef]
- Gumusay, O.; Vitiello, P.P.; Wabl, C.; Corcoran, R.B.; Bardelli, A.; Rugo, H.S. Strategic combinations to prevent and overcome resistance to targeted therapies in oncology. Am. Soc. Clin. Oncol. Educ. Book 2020, 40, e292–e308. [Google Scholar] [CrossRef]
- Rached, L.; Laparra, A.; Sakkal, M.; Danlos, F.-X.; Barlesi, F.; Carbonnel, F.; De Martin, E.; Ducreux, M.; Even, C.; Le Pavec, J.; et al. Toxicity of immunotherapy combinations with chemotherapy across tumor indications: Current knowledge and practical recommendations. Cancer Treat. Rev. 2024, 127, 102751. [Google Scholar] [CrossRef]
- Williams, N.O.; Quiroga, D.; Johnson, C.; Brufsky, A.; Chambers, M.; Bhattacharya, S.; Patterson, M.; Sardesai, S.D.; Stover, D.; Lustberg, M.; et al. Phase Ib study of HSP90 inhibitor onalespib (AT13387) combined with paclitaxel in patients with advanced triple-negative breast cancer. Pharmaceutics 2023, 15, 17588359231217976. [Google Scholar] [CrossRef]
- Mall, C.; Sckisel, G.D.; Proia, D.A.; Mirsoian, A.; Grossenbacher, S.K.; Pai, C.-C.S.; Chen, M.; Monjazeb, A.M.; Kelly, K.; Blazar, B.R.; et al. Repeated PD-1/PD-L1 monoclonal antibody administration induces fatal xenogeneic hypersensitivity reactions in a murine 4T1 breast cancer model. Oncoimmunology 2015, 5, e1075114. [Google Scholar] [CrossRef] [PubMed]
- Duarte, D.; Vale, N. Evaluation of synergism in drug combinations and reference models for future orientations in oncology. Curr. Res. Pharmacol. Drug Discov. 2022, 3, 100110. [Google Scholar] [CrossRef]
- Ismail, J.; Shebaby, W.; Azar Atallah, S.; Taleb, R.I.; Kawrani, S.; Faour, W.; Mroueh, M. Combination of Cannabidiol with Cisplatin or Paclitaxel: Analysis Using the Chou–Talalay Method and Chemo-Sensitization Evaluation in Platinum-Resistant Ovarian Cancer Cells. Biomedicines 2025, 13, 520. [Google Scholar] [CrossRef] [PubMed]
- Flanary, V.L.; Fisher, J.L.; Wilk, E.J.; Howton, T.C.; Lasseigne, B.N. Computational advancements in cancer combination therapy prediction. JCO Precis. Oncol. 2023, 7, e2300261. [Google Scholar] [CrossRef] [PubMed]
- Hyun, S.Y.; Le, H.T.; Nguyen, C.-T.; Yong, Y.-S.; Boo, H.-J.; Lee, H.J.; Lee, J.-S.; Min, H.-Y.; Ann, J.; Chen, J.; et al. Development of a novel Hsp90 inhibitor NCT-50 as a potential anticancer agent for the treatment of non-small cell lung cancer. Sci. Rep. 2018, 8, 13924. [Google Scholar] [CrossRef]
- Muhammed, M.T.; Aki-Yalcin, E. Molecular docking: Principles, advances, and its applications in drug discovery. Lett. Drug Des. Discov. 2024, 21, 480–495. [Google Scholar] [CrossRef]
- Saouli, I.; Abrane, R.; Chahra, B.-H.; Boudiba, S. Insight into the structural and dynamic properties of novel HSP90 inhibitors through DFT calculations and molecular dynamics simulations. J. Mol. Model. 2024, 30, 420. [Google Scholar] [CrossRef]
- Ouassaf, M.; Bourougaa, L.; Al-Mijalli, S.H.; Abdallah, E.M.; Bhat, A.R.; Kawsar, S.M.A. Marine-derived compounds as potential inhibitors of Hsp90 for anticancer and antimicrobial drug development: A comprehensive in silico study. Molecules 2023, 28, 8074. [Google Scholar] [CrossRef]
- Díaz-Beyá, M.; García-Fortes, M.; Valls, R.; Artigas, L.; Gómez-Casares, M.T.; Montesinos, P.; Sánchez-Guijo, F.; Coma, M.; Vendranes, M.; Martínez-López, J. A systems biology- and machine learning-based study to unravel potential therapeutic mechanisms of midostaurin as a multitarget therapy on FLT3-mutated AML. BioMedInformatics 2022, 2, 375–397. [Google Scholar] [CrossRef]
- Dhiman, S.; Saha, M.; Ali, A.; Ali, A.; Gupta, G.D.; Asati, V. Structural aspects of triazole derivatives as HSP90 inhibitors for the treatment of cancer: In silico studies. J. Biomol. Struct. Dyn. 2023, 41, 4756–4769. [Google Scholar] [CrossRef]
- Bhowmick, S.; Malhotra, J.; Choudhary, R.; Shinde, O.D.; Metwally, A.S.M.; Patil, P.C. Machine Learning Aided De Novo Design Identifies Novel Benzimidazolone-Based Inhibitor-Modulators for Heat Shock Protein 90 (HSP90). ChemistrySelect 2024, 9, e202402147. [Google Scholar] [CrossRef]
- Zang, M.; Gan, H.; Zhou, X.; Wang, L.; Dong, H. Dual-Site targeting by peptide inhibitors of the N-terminal domain of Hsp90: Mechanism and design. J. Chem. Inf. Model. 2025, 65, 5113–5123. [Google Scholar] [CrossRef]
- Cheng, C.; Tang, X.; Woodley, D.T.; Chen, M.; Li, W. Previously unrecognized and potentially consequential challenges facing Hsp90 inhibitors in cancer clinical trials. Cell Stress Chaperones 2024, 29, 642–653. [Google Scholar] [CrossRef]
- Shin, S.-Y.; Centenera, M.M.; Hodgson, J.T.; Nguyen, E.V.; Butler, L.M.; Daly, R.J.; Nguyen, L.K. A Boolean-based machine learning framework identifies predictive biomarkers of HSP90-targeted therapy response in prostate cancer. Pharmaceuticals 2023, 10, 1094321. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, S.B.M.; Gülden, J.; Niederreiter, M.; Eichner, C.; Werner, J.; Mayer, B. Strong Hsp90α/β Protein Expression in Advanced Primary CRC Indicates Short Survival and Predicts Response to the Hsp90α/β-Specific Inhibitor Pimitespib. Cells 2025, 14, 836. [Google Scholar] [CrossRef] [PubMed]
- Wrona, A.; Dziadziuszko, R.; Jassem, J. Combining radiotherapy with targeted therapies in non-small cell lung cancer: Focus on anti-EGFR, anti-ALK and anti-angiogenic agents. Transl. Lung Cancer Res. 2021, 10, 2032–2047. [Google Scholar] [CrossRef] [PubMed]
- Marrugal, Á.; Ferrer, I.; Quintanal-Villalonga, Á.; Ojeda, L.; Pastor, M.D.; García-Luján, R.; Carnero, A.; Paz-Ares, L.; Molina-Pinelo, S. Inhibition of HSP90 in driver oncogene-defined lung adenocarcinoma cell lines: Key proteins underpinning therapeutic efficacy. Pharmaceuticals 2023, 14, 13830. [Google Scholar] [CrossRef]
- Liu, X.; Yu, J.; Li, Y.; Shi, H.; Jiao, X.; Liu, X.; Guo, D.; Li, Z.; Tian, Y.; Dai, F.; et al. Deciphering the tumor immune microenvironment of imatinib-resistance in advanced gastrointestinal stromal tumors at single-cell resolution. Cell Death Dis. 2024, 15, 190. [Google Scholar] [CrossRef]
- Rastogi, S.; Joshi, A.; Sato, N.; Lee, S.; Lee, M.-J.; Trepel, J.B.; Neckers, L. An update on the status of HSP90 inhibitors in cancer clinical trials. Cell Stress Chaperones 2024, 29, 519–539. [Google Scholar] [CrossRef]
- Ballard, J.L.; Wang, Z.; Li, W.; Shen, L.; Long, Q. Deep learning-based approaches for multi-omics data integration and analysis. BioData Min. 2024, 17, 38. [Google Scholar] [CrossRef]
- You, S.C.; Jung, S.; Swerdel, J.N.; Ryan, P.B.; Schuemie, M.J.; Suchard, M.A.; Lee, S.; Cho, J.; Hripcsak, G.; Park, R.W.; et al. Comparison of first-line dual combination treatments in hypertension: Real-world evidence from multinational heterogeneous cohorts. Korean Circ. J. 2020, 50, 52–68. [Google Scholar] [CrossRef]
- Sordo-Bahamonde, C.; Lorenzo-Herrero, S.; Gonzalez-Rodriguez, A.P.; Martínez-Pérez, A.; Rodrigo, J.P.; García-Pedrero, J.M.; Gonzalez, S. Chemo-Immunotherapy: A new trend in cancer treatment. Cancers 2023, 15, 2912. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Generation | Combination Therapy | Hsp90 Inhibitors | Types of Cancer | Preclinical and Clinical | NCT Number |
|---|---|---|---|---|---|
| I | Gemcitabine | Tanespimycin (17-AAG) | Metastatic pancreatic cancer (Stage IV) | Phase II | NCT00368590 |
| I | Gemcitabine | Tanespimycin (17-AAG) | Ovarian and peritoneal cancer | Phase II | NCT00387722 |
| I | Cisplatin | Tanespimycin (17-AAG) | Diffuse large B-cell lymphoma (DLBCL) | Preclinical | |
| I | Trastuzumab | Tanespimycin (17-AAG) | HER2-positive metastatic breast cancer | Phase II | NCT00027846 |
| II | Paclitaxel + Trastuzumab | Ganetespib | HER2-positive metastatic breast cancer | Phase I | NCT01273473 |
| II | Paclitaxel | Ganetespib | HER2-negative early-stage breast cancer | Phase II | NCT01042379 |
| II | Paclitaxel | Onalespib | Advanced triple-negative breast cancer (TNBC) | Phase I | NCT02474173 |
| II | Docetaxel | Ganetespib | Advanced non-small cell lung cancer (NSCLC) | Phase III | NCT01798485 |
| II | Gemcitabine | ICPD47 or ICPD62 | Pancreatic cancer cell lines (MIA PaCa-2, PANC-1) | Preclinical | |
| II | Cisplatin | Luminespib (AUY922) | Nasopharyngeal carcinoma (NPC) | Preclinical | |
| II | Cisplatin | Onalespib | Ovarian and peritoneal cancer | Preclinical | |
| II | Erlotinib | Luminespib (AUY922) | EGFR-mutant non-small cell lung cancer (NSCLC) | Phase I | NCT01259089 |
| II | paclitaxel + Trastuzumab | Ganetespib | HER2-positive metastatic breast cancer | Phase I | NCT00096391 |
| II | Crizotinib | Onalespib | ALK-positive non-small cell lung cancer (NSCLC) | Preclinical | |
| II | Crizotinib | Onalespib | ALK-positive non-small cell lung cancer (NSCLC) | Phase I/II | NCT01712217 |
| II | Vemurafenib (BRAF inhibitor) + Cobimetinib (MEK inhibitor) | XL888 | BRAF V600E-mutant melanoma | Phase I | NCT01657591 |
| II | Atezolizumab (PD-L1 inhibitor) | Pimitespib | Metastatic solid tumors | Preclinical | |
| II | Nivolumab (PD-1 inhibitor) | Pimitespib | Advanced solid tumors | Phase I | NCT02872116 |
| II | Clone 9H10 (CTLA-4 inhibitor) | Ganetespib | Melanoma | Preclinical |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, Y.; Lim, S.Y.; Kim, H.-O.; Ha, S.-J.; Park, J.-A.; Won, Y.-W.; Chae, S.; Lim, K.S. Combination Strategies with HSP90 Inhibitors in Cancer Therapy: Mechanisms, Challenges, and Future Perspectives. Pharmaceuticals 2025, 18, 1083. https://doi.org/10.3390/ph18081083
Kim Y, Lim SY, Kim H-O, Ha S-J, Park J-A, Won Y-W, Chae S, Lim KS. Combination Strategies with HSP90 Inhibitors in Cancer Therapy: Mechanisms, Challenges, and Future Perspectives. Pharmaceuticals. 2025; 18(8):1083. https://doi.org/10.3390/ph18081083
Chicago/Turabian StyleKim, Yeongbeom, Su Yeon Lim, Hyun-Ouk Kim, Suk-Jin Ha, Jeong-Ann Park, Young-Wook Won, Sehyun Chae, and Kwang Suk Lim. 2025. "Combination Strategies with HSP90 Inhibitors in Cancer Therapy: Mechanisms, Challenges, and Future Perspectives" Pharmaceuticals 18, no. 8: 1083. https://doi.org/10.3390/ph18081083
APA StyleKim, Y., Lim, S. Y., Kim, H.-O., Ha, S.-J., Park, J.-A., Won, Y.-W., Chae, S., & Lim, K. S. (2025). Combination Strategies with HSP90 Inhibitors in Cancer Therapy: Mechanisms, Challenges, and Future Perspectives. Pharmaceuticals, 18(8), 1083. https://doi.org/10.3390/ph18081083