
α-Ketoglutarate Attenuates Oxidative Stress-Induced Neuronal Aging via Modulation of the mTOR Pathway
 ,
,
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Reversal of D-Gal-Induced Brain Aging in C57BL/6 Mice by AKG
2.2. AKG Attenuates Brain and Plasma Oxidative Stress in D-Gal-Induced Aging Mice
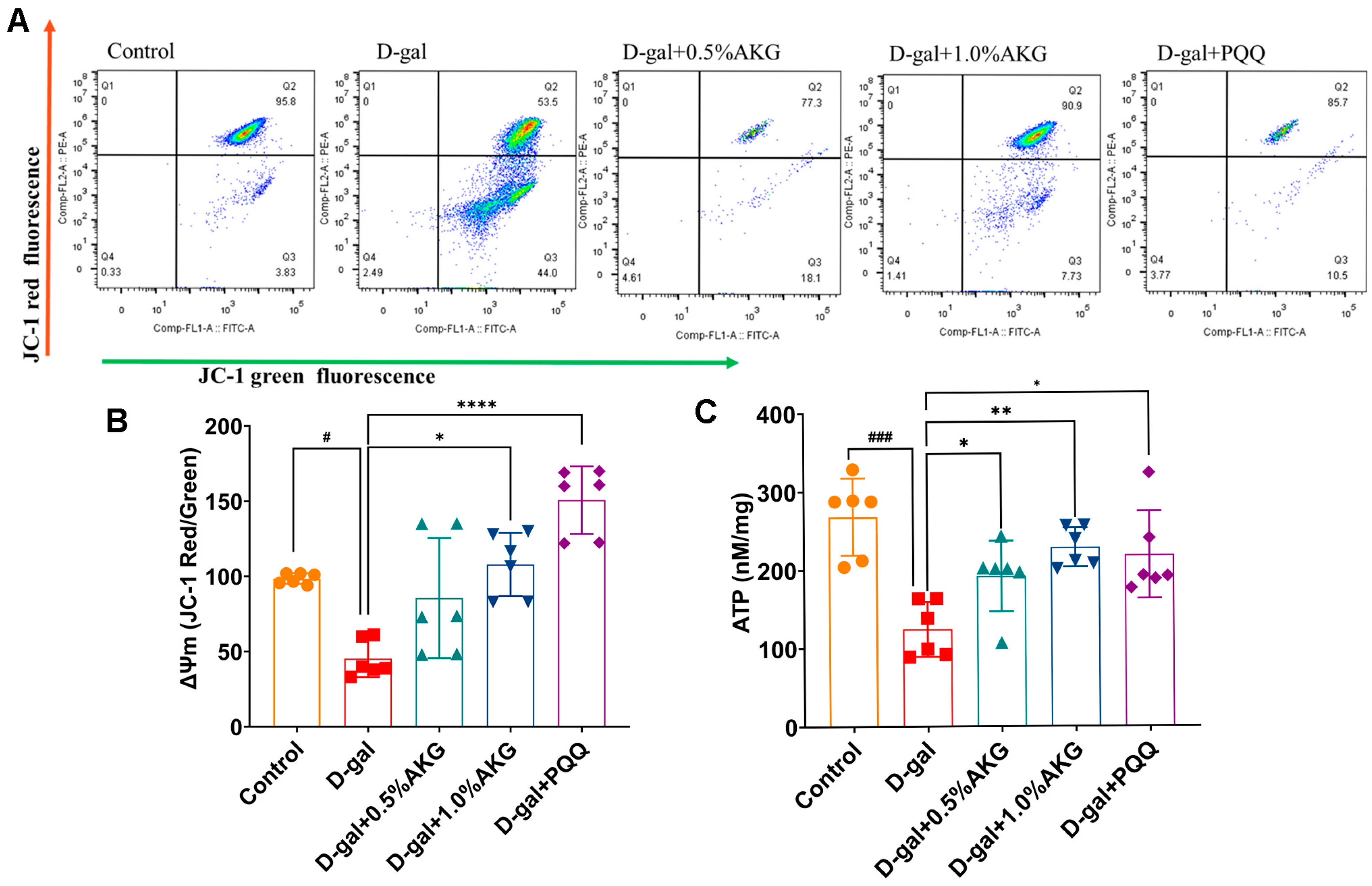
2.3. AKG Improves Mitochondrial Function in D-Gal-Induced Aging Mice
2.4. AKG Protects HT22 Cells Against H2O2-Induced Senescence and Promotes Cell Viability
2.5. AKG Alleviates Oxidative Stress and Promotes Mitochondrial Metabolism in H2O2-Induced HT22 Cells
2.6. AKG Regulates the mTOR and p53 Pathways, as Revealed by Proteomics and Verified by Western Blot
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Treatments
4.2. Measurement of Antioxidant Enzyme Activities
4.3. Malondialdehyde (MDA) Analysis
4.4. Determination of Protein Carbonyl Levels
4.5. Cell Viability Assay (CCK-8)
4.6. Intracellular ROS Detection
4.7. Mitochondrial Membrane Potential (Δψm) Assay
4.8. Measurement of Oxygen Consumption Rate (OCR)
4.9. Senescence-Associated β-Galactosidase (SA-β-Gal) Staining
4.10. Western Blotting
4.11. Quantitative Real-Time PCR Analysis of SASP Markers
4.12. Animal Model and Treatments
4.13. Morris Water Maze (MWM)
4.14. Rotarod Test
4.15. Passive Avoidance Test
4.16. Histomorphological Evaluations
4.17. Protein Extraction and DIA-Based Proteomics Analysis
4.18. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AKG | α-Ketoglutarate |
| TCA | Tricarboxylic acid |
| ROS | Reactive oxygen species |
| MMP | Mitochondrial membrane potential |
| SOD | Superoxide Dismutase |
| MDA | Malondialdehyde |
| PCO | Protein carbonyl |
| TBST | Tris-Buffered Saline with Tween® 20 |
| CCK-8 | Cell Viability Assay |
| OCR | Oxygen Consumption Rate |
| SA-β-Gal | Senescence-associated β-galactosidase |
| D-gal | D-galactose |
| MWM | Morris Water Maze |
| HE | Hematoxylin and eosin |
| DIA | Data-Independent Acquisition |
References
- Borghesan, M.; Hoogaars, W.M.H.; Varela-Eirin, M.; Talma, N.; Demaria, M. A Senescence-Centric View of Aging Implications for Longevity and Disease. Trends Cell Biol. 2020, 30, 777–791. [Google Scholar] [CrossRef] [PubMed]
- Harman, D. Free Radical Theory of Aging: An Update Increasing the Functional Life Span. Ann. N. Y. Acad. Sci. 2006, 1067, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.C.; Alhasawi, A.; Appanna, V.P.; Auger, C.; Appanna, V.D. Brain metabolism and Alzheimer’s disease: The prospect of a metabolite-based therapy. J. Nutr. Health Aging 2015, 19, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Wardlaw, J.M.; Smith, E.E.; Biessels, G.J.; Cordonnier, C.; Fazekas, F.; Frayne, R.; Lindley, R.I.; O’Brien, J.T.; Barkhof, F.; Benavente, O.R.; et al. Neuroimaging standards for research into small vessel disease and its contribution to ageing and neurodegeneration. Lancet Neurol. 2013, 12, 822–838. [Google Scholar] [CrossRef] [PubMed]
- Farrer, L.A.; Cupples, L.A.; Haines, J.L.; Hyman, B.; Kukull, W.A.; Mayeux, R.; Myers, R.H.; Pericak-Vance, M.A.; Risch, N.; Van Dujn, C.M. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta analysis. J. Am. Med. Assoc. 1997, 278, 1349–1356. [Google Scholar] [CrossRef]
- Stern, Y. Cognitive reserve in ageing and Alzheimer’s disease. Lancet Neurol. 2012, 11, 1006–1012. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.J.; Dan, X.L.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef] [PubMed]
- Finkel, T.; Holbrook, N.J. Oxidants, oxidative stress and the biology of ageing. Nature 2000, 408, 239–247. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Davalli, P.; Mitic, T.; Caporali, A.; Lauriola, A.; D’Arca, D. ROS, Cell Senescence, and Novel Molecular Mechanisms in Aging and Age-Related Diseases. Oxid. Med. Cell. Longev. 2016, 2016, 3565127. [Google Scholar] [CrossRef] [PubMed]
- Ton, A.L.Y.; Forbes, J.M.; Cooper, M.E. AGE, RAGE, and ROS in diabetic nephropathy. Semin. Nephrol. 2007, 27, 130–143. [Google Scholar] [CrossRef] [PubMed]
- Xiao, D.; Zeng, L.; Yao, K.; Kong, X.; Wu, G.; Yin, Y. The glutamine-alpha-ketoglutarate (AKG) metabolism and its nutritional implications. Amino Acids 2016, 48, 2067–2080. [Google Scholar] [CrossRef] [PubMed]
- Maus, A.; Peters, G.J. Glutamate and α-ketoglutarate: Key players in glioma metabolism. Amino Acids 2017, 49, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Legendre, F.; MacLean, A.; Appanna, V.P.; Appanna, V.D. Biochemical pathways to α-ketoglutarate, a multi-faceted metabolite. World J. Microbiol. Biotechnol. 2020, 36, 123. [Google Scholar] [CrossRef] [PubMed]
- Shahmirzadi, A.A.; Edgar, D.; Liao, C.-Y.; Hsu, Y.-M.; Lucanic, M.; Shahmirzadi, A.A.; Wiley, C.D.; Gan, G.; Kim, D.E.; Kasler, H.G.; et al. Alpha-Ketoglutarate, an Endogenous Metabolite, Extends Lifespan and Compresses Morbidity in Aging Mice. Cell Metab. 2020, 32, 447–456.e6. [Google Scholar] [CrossRef] [PubMed]
- Sandalova, E.; Goh, J.; Lim, Z.X.; Lim, Z.M.; Barardo, D.; Dorajoo, R.; Kennedy, B.K.; Maier, A.B. Alpha-ketoglutarate supplementation and BiologicaL agE in middle-aged adults (ABLE)-intervention study protocol. GeroScience 2023, 45, 2897–2907. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Liu, G.; Sun, W.; Jia, G.; Zhao, H.; Chen, X.; Wang, J. Dietary α-Ketoglutarate Alleviates Escherichia coli LPS-Induced Intestinal Barrier Injury by Modulating the Endoplasmic Reticulum-Mitochondrial System Pathway in Piglets. J. Nutr. 2024, 154, 2087–2096. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Lu, J.; Sun, W.; Jia, G.; Zhao, H.; Chen, X.; Wang, J. Alpha-ketoglutaric acid attenuates oxidative stress and modulates mitochondrial dynamics and autophagy of spleen in a piglet model of lipopolysaccharide-induced sepsis. Free Radic. Biol. Med. 2024, 214, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.Y.; Sabatini, D.M. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef] [PubMed]
- Perluigi, M.; Di Domenico, F.; Butterfield, D.A. mTOR signaling in aging and neurodegeneration: At the crossroad between metabolism dysfunction and impairment of autophagy. Neurobiol. Dis. 2015, 84, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Crino, P.B. mTOR: A pathogenic signaling pathway in developmental brain malformations. Trends Mol. Med. 2011, 17, 734–742. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.Y.; Smit, D.J.; Juecker, M. The Role of PI3K/AKT/mTOR Signaling in Hepatocellular Carcinoma Metabolism. Int. J. Mol. Sci. 2023, 24, 2652. [Google Scholar] [CrossRef] [PubMed]
- Perl, A. Activation of mTOR (mechanistic target of rapamycin) in rheumatic diseases. Nat. Rev. Rheumatol. 2016, 12, 169–182. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Pang, Y.; Fan, X. Mitochondria in oxidative stress, inflammation and aging: From mechanisms to therapeutic advances. Sig. Transduct. Target. Ther. 2025, 10, 190. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Gao, Y.; Zhang, S.; Wang, Y.; Pei, X.; Chen, Y.; Zhang, J.; Zhang, Y.; Du, Y.; Hao, S.; et al. Mitochondrial dysfunction in the regulation of aging and aging-related diseases. Cell Commun. Signal. 2025, 23, 291. [Google Scholar] [CrossRef]
- Garcia-Dominguez, M. Pathological and Inflammatory Consequences of Aging. Biomolecules 2025, 15, 404. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.; Yang, M.Y.; Gaur, U.; Xu, H.L.; Yao, Y.F.; Li, D.Y. Alpha-Ketoglutarate: Physiological Functions and Applications. Biomol. Ther. 2016, 24, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.A.; Bhattacharjee, S.; Armin, S.M.; Qian, P.P.; Xin, W.; Li, H.Y.; Burritt, D.J.; Fujita, M.; Tran, L.S.P. Hydrogen peroxide priming modulates abiotic oxidative stress tolerance: Insights from ROS detoxification and scavenging. Front. Plant Sci. 2015, 6, 420. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.S.; Satsu, H.; Bae, M.J.; Zhao, Z.H.; Ogiwara, H.; Totsuka, M.; Shimizu, M. Anti-inflammatory effect of chlorogenic acid on the IL-8 production in Caco-2 cells and the dextran sulphate sodium-induced colitis symptoms in C57BL/6 mice. Food Chem. 2015, 168, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef] [PubMed]
- Jomova, K.; Raptova, R.; Alomar, S.Y.; Alwasel, S.H.; Nepovimova, E.; Kuca, K.; Valko, M. Reactive oxygen species, toxicity, oxidative stress, and antioxidants: Chronic diseases and aging. Arch. Toxicol. 2023, 97, 2499–2574. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.C.; Guan, K.L. mTOR: A pharmacologic target for autophagy regulation. J. Clin. Investig. 2015, 125, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Mossmann, D.; Park, S.; Hall, M.N. mTOR signalling and cellular metabolism are mutual determinants in cancer. Nat. Rev. Cancer 2018, 18, 744–757. [Google Scholar] [CrossRef] [PubMed]
- Zou, Z.L.; Tao, T.; Li, H.M.; Zhu, X. mTOR signaling pathway and mTOR inhibitors in cancer: Progress and challenge. Cell Biosci. 2020, 10, 31. [Google Scholar] [CrossRef] [PubMed]
- Rivera, V.M.M.; Juarez, J.R.T.; Melo, N.M.M.; Garcia, N.L.; Magana, J.J.; Martinez, J.C.; Gutierrez, G.E.J. Natural Autophagy Activators to Fight Age-Related Diseases. Cells 2024, 13, 1611. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Feng, W.; Shen, Q.; Yu, N.; Yu, K.; Wang, S.; Chen, Z.; Shioda, S.; Guo, Y. Rhizoma Coptidis and Berberine as a Natural Drug to Combat Aging and Aging-Related Diseases via Anti-Oxidation and AMPK Activation. Aging Dis. 2017, 8, 760–777. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Wang, T.; Wu, N.; Li, D.; Fan, X.; Xu, Z.; Mishra, S.K.; Yang, M. Alpha-ketoglutarate extends Drosophila lifespan by inhibiting mTOR and activating AMPK. Aging 2019, 11, 4183–4197. [Google Scholar] [CrossRef]
- Chiroma, S.M.; Moklas, M.A.M.; Taib, C.N.M.; Baharuldin, M.T.H.; Amon, Z. D-galactose and aluminium chloride induced rat model with cognitive impairments. Biomed. Pharmacother. 2018, 103, 1602–1608. [Google Scholar] [CrossRef] [PubMed]
- Shoji, H.; Takao, K.; Hattori, S.; Miyakawa, T. Age-related changes in behavior in C57BL/6J mice from young adulthood to middle age. Mol. Brain 2016, 9, 11. [Google Scholar] [CrossRef]
- Webster, S.J.; Bachstetter, A.D.; Nelson, P.T.; Schmitt, F.A.; Van Eldik, L.J. Using mice to model Alzheimer’s dementia: An overview of the clinical disease and the preclinical behavioral changes in 10 mouse models. Front. Genet. 2014, 5, 88. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Luo, H.; Chang, L.; Wang, S.; Geng, X.; Kang, L.; Zhong, Y.; Cao, Y.; Wang, R.; Yang, X.; et al. The NEDD8-activating enzyme inhibitor MLN4924 reduces ischemic brain injury in mice. Proc. Natl. Acad. Sci. USA 2022, 119, e2111896119. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guan, R.; Xue, Z.; Huang, K.; Zhao, Y.; He, G.; Dai, Y.; Liang, M.; Wen, Y.; Ye, X.; Liu, P.; et al. α-Ketoglutarate Attenuates Oxidative Stress-Induced Neuronal Aging via Modulation of the mTOR Pathway. Pharmaceuticals 2025, 18, 1080. https://doi.org/10.3390/ph18081080
Guan R, Xue Z, Huang K, Zhao Y, He G, Dai Y, Liang M, Wen Y, Ye X, Liu P, et al. α-Ketoglutarate Attenuates Oxidative Stress-Induced Neuronal Aging via Modulation of the mTOR Pathway. Pharmaceuticals. 2025; 18(8):1080. https://doi.org/10.3390/ph18081080
Chicago/Turabian StyleGuan, Ruoqing, Zhaoyun Xue, Kaikun Huang, Yanqing Zhao, Gongyun He, Yuxing Dai, Mo Liang, Yanzi Wen, Xueshi Ye, Peiqing Liu, and et al. 2025. "α-Ketoglutarate Attenuates Oxidative Stress-Induced Neuronal Aging via Modulation of the mTOR Pathway" Pharmaceuticals 18, no. 8: 1080. https://doi.org/10.3390/ph18081080
APA StyleGuan, R., Xue, Z., Huang, K., Zhao, Y., He, G., Dai, Y., Liang, M., Wen, Y., Ye, X., Liu, P., & Chen, J. (2025). α-Ketoglutarate Attenuates Oxidative Stress-Induced Neuronal Aging via Modulation of the mTOR Pathway. Pharmaceuticals, 18(8), 1080. https://doi.org/10.3390/ph18081080