




Ginsenoside Rg3 Adjunctively Increases the Efficacy of Gefitinib Against NSCLC by Regulating EGFR Copy Number

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. The Current Clinical Status of NSCLC
2.2. Clinical EGFR-TKI Therapy Does Not Have a Significant Advantage over First-Line Chemotherapeutic Agents
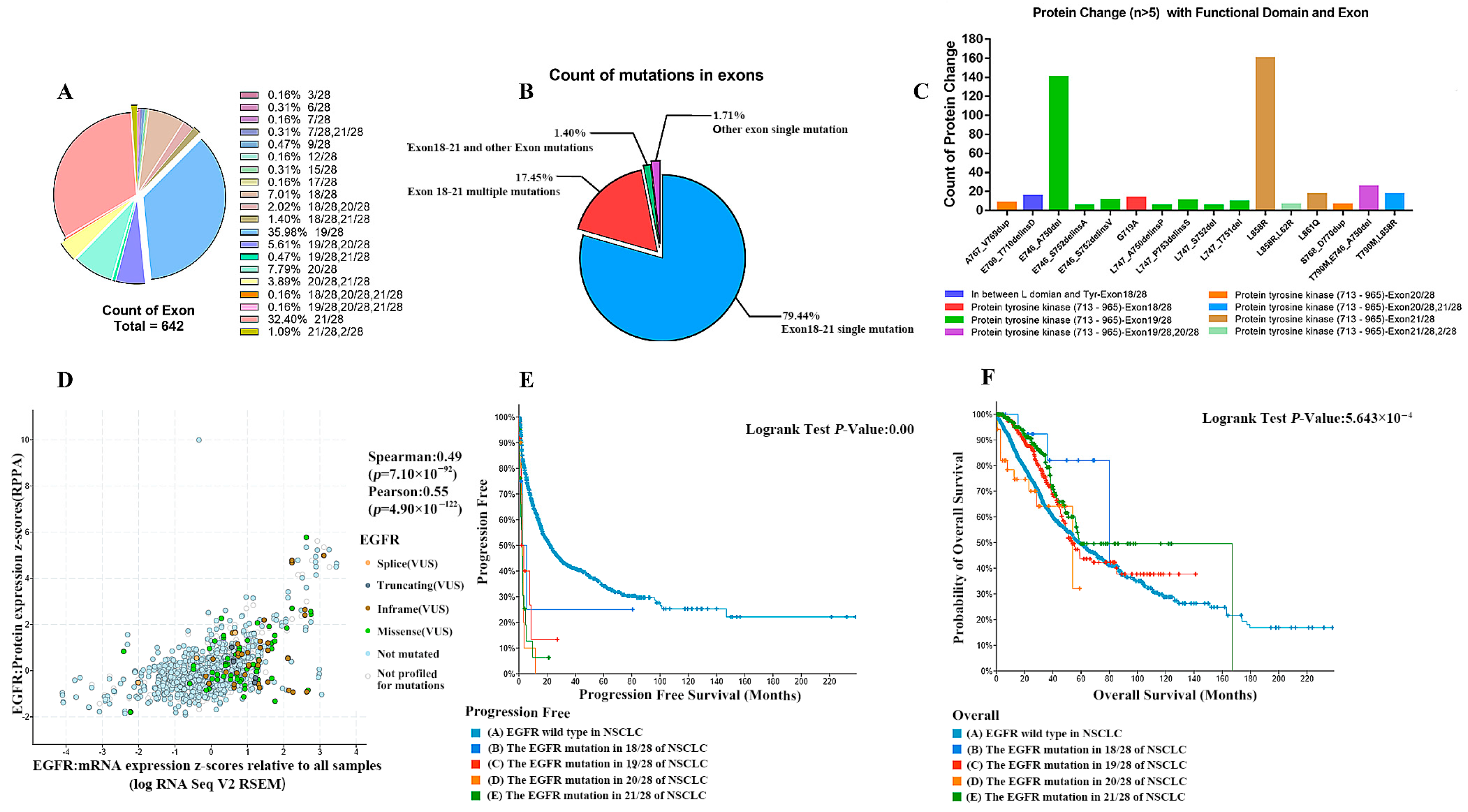
2.3. EGFR Abnormalities (Including Mutations, Structural Variants, and Copy Number Alterations) in NSCLC
2.4. A Higher Degree of Increase in EGFR Gene Copy Number Leads to a Worse Survival Prognosis of Patients
2.5. EGFR-TKI Agents Target May Not Be Effective in Patients with a Significant Increase in EGFR Copy Number
2.6. Ginsenoside Rg3 May Be More Sensitive for Patients with an EGFR Mutant Phenotype and EGFR May Be an Important Target for Its Action
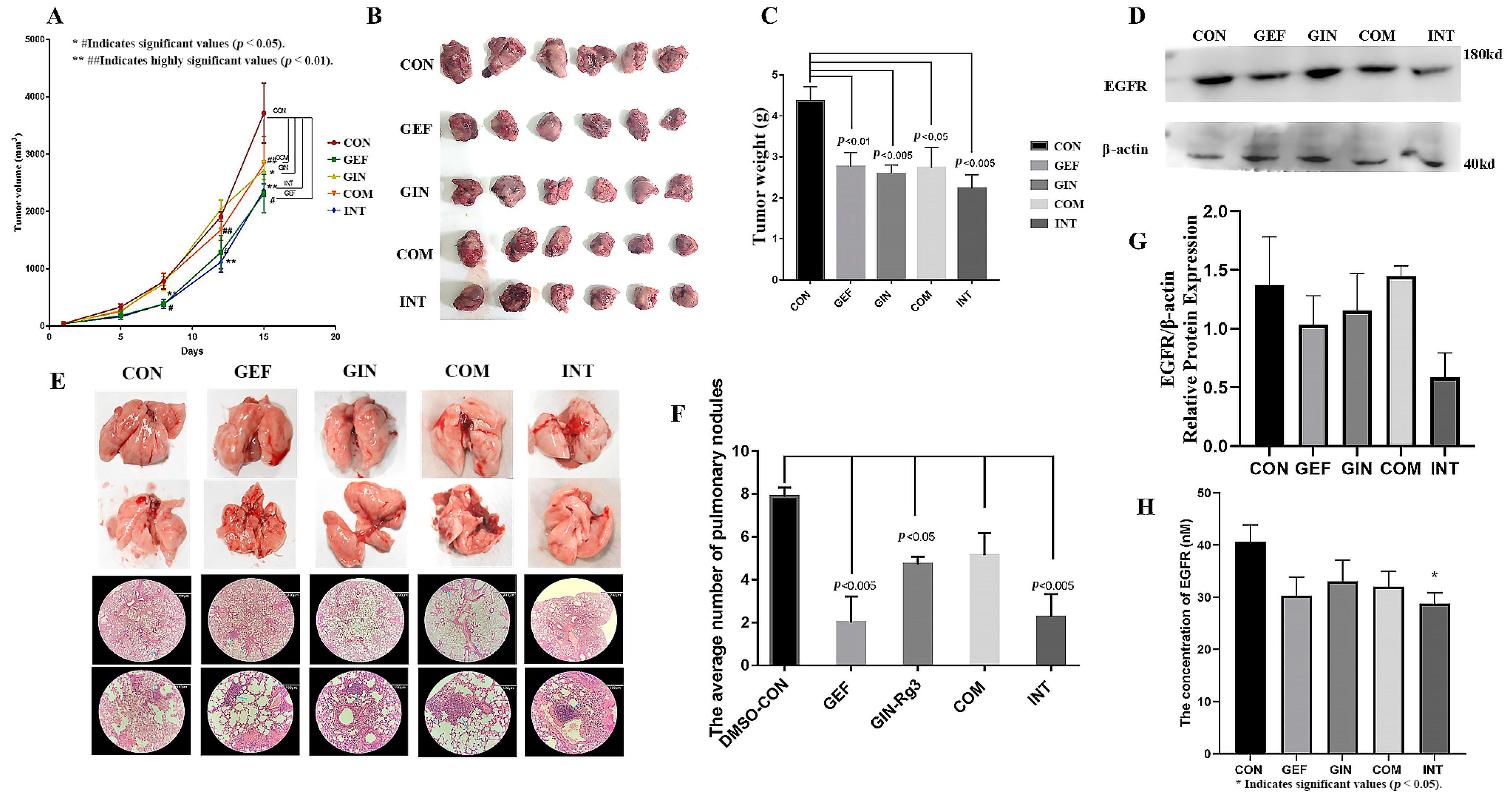
2.7. Cell Experiments Again Verified That the Combination of Ginsenosides and Gefitinib Was Superior to Gefitinib Alone
2.8. Animal Studies Demonstrate That the Addition of Ginsenosides Significantly Improves the Efficacy of Gefitinib Alone
3. Discussion
4. Materials and Methods
4.1. Build Database
4.2. Meta-Analysis
4.3. Reagents and Instruments
4.4. CCK8 Assay
4.5. Animal Studies
4.6. Histopathological Analysis
4.7. Quantitative Real-Time Polymerase Chain Reactions (RT-qPCR) Assay
4.8. Western Blotting
4.9. ELISA
4.10. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| EGFR | Epidermal Growth Factor Receptor |
| TKI | Tyrosine Kinase Inhibitors |
| NSCLC | Non-Small Cell Lung Cancer |
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Evison, M.; Barrett, E.; Cheng, A.; Mulla, A.; Walls, G.; Johnston, D.; McAleese, J.; Moore, K.; Hicks, J.; Blyth, K.; et al. Predicting the Risk of Disease Recurrence and Death Following Curative-intent Radiotherapy for Non-small Cell Lung Cancer: The Development and Validation of Two Scoring Systems From a Large Multicentre UK Cohort. Clin. Oncol. 2021, 33, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Jadhav, H.R. Targeting non-small cell lung cancer with small-molecule EGFR tyrosine kinase inhibitors. Drug Discov. Today 2018, 23, 745–753. [Google Scholar] [CrossRef] [PubMed]
- Voldborg, B.R.; Damstrup, L.; Spang-Thomsen, M.; Poulsen, H.S. Epidermal growth factor receptor (EGFR) and EGFR mutations, function and possible role in clinical trials. Ann. Oncol. 1997, 8, 1197–1206. [Google Scholar] [CrossRef] [PubMed]
- Rybarczyk-Kasiuchnicz, A.; Ramlau, R.; Stencel, K. Treatment of Brain Metastases of Non-Small Cell Lung Carcinoma. Int. J. Mol. Sci. 2021, 22, 593. [Google Scholar] [CrossRef] [PubMed]
- Ayati, A.; Moghimi, S.; Salarinejad, S.; Safavi, M.; Pouramiri, B.; Foroumadi, A. A review on progression of epidermal growth factor receptor (EGFR) inhibitors as an efficient approach in cancer targeted therapy. Bioorgan. Chem. 2020, 99, 103811. [Google Scholar] [CrossRef] [PubMed]
- Sabbah, D.A.; Hajjo, R.; Sweidan, K. Review on Epidermal Growth Factor Receptor (EGFR) Structure, Signaling Pathways, Interactions, and Recent Updates of EGFR Inhibitors. Curr. Top. Med. Chem. 2020, 20, 815–834. [Google Scholar] [CrossRef] [PubMed]
- Chhouri, H.; Alexandre, D.; Grumolato, L. Mechanisms of Acquired Resistance and Tolerance to EGFR Targeted Therapy in Non-Small Cell Lung Cancer. Cancers 2023, 15, 504. [Google Scholar] [CrossRef] [PubMed]
- Okabe, T.; Okamoto, I.; Tamura, K.; Terashima, M.; Yoshida, T.; Satoh, T.; Takada, M.; Fukuoka, M.; Nakagawa, K. Differential Constitutive Activation of the Epidermal Growth Factor Receptor in Non–Small Cell Lung Cancer Cells Bearing EGFR Gene Mutation and Amplification. Cancer Res. 2007, 67, 2046–2053. [Google Scholar] [CrossRef] [PubMed]
- Normanno, N.; De Luca, A.; Bianco, C.; Strizzi, L.; Mancino, M.; Maiello, M.R.; Carotenuto, A.; De Feo, G.; Caponigro, F.; Salomon, D.S. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene 2006, 366, 2–16. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wu, C.; Chen, N.; Gu, H.; Yen, A.; Cao, L.; Wang, E.; Wang, L. PI3K/Akt/mTOR signaling pathway and targeted therapy for glioblastoma. Oncotarget 2016, 7, 33440–33450. [Google Scholar] [CrossRef] [PubMed]
- Jia, W.-T.; Xiang, S.; Zhang, J.-B.; Yuan, J.-Y.; Wang, Y.-Q.; Liang, S.-F.; Lin, W.-F.; Zhai, X.-F.; Shang, Y.; Ling, C.-Q.; et al. Jiedu recipe, a compound Chinese herbal medicine, suppresses hepatocellular carcinoma metastasis by inhibiting the release of tumor-derived exosomes in a hypoxic microenvironment. J. Integr. Med. 2024, 22, 696–708. [Google Scholar] [CrossRef] [PubMed]
- Chong, C.R.; Jänne, P.A. The quest to overcome resistance to EGFR-targeted therapies in cancer. Nat. Med. 2013, 19, 1389–1400. [Google Scholar] [CrossRef] [PubMed]
- Harrison, P.T.; Vyse, S.; Huang, P.H. Rare epidermal growth factor receptor (EGFR) mutations in non-small cell lung cancer. Semin. Cancer Biol. 2020, 61, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Shan, Y.; Eastwood, M.P.; Zhang, X.; Kim, E.T.; Arkhipov, A.; Dror, R.O.; Jumper, J.; Kuriyan, J.; Shaw, D.E. Oncogenic Mutations Counteract Intrinsic Disorder in the EGFR Kinase and Promote Receptor Dimerization. Cell 2012, 149, 860–870. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Canepa, H.M.; Bailey, A.S.; Nakayama, S.; Yamaguchi, N.; Goldstein, M.A.; Huberman, M.S.; Costa, D.B. Compound EGFR Mutations and Response to EGFR Tyrosine Kinase Inhibitors. J. Thorac. Oncol. 2013, 8, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.K.; Wu, Y.L.; Ding, P.N.; Lord, S.J.; Inoue, A.; Zhou, C.; Mitsudomi, T.; Rosell, R.; Pavlakis, N.; Links, M.; et al. Impact of Specific Epidermal Growth Factor Receptor (EGFR) Mutations and Clinical Characteristics on Outcomes After Treatment with EGFR Tyrosine Kinase Inhibitors Versus Chemotherapy in EGFR -Mutant Lung Cancer: A Meta-Analysis. J. Clin. Oncol. 2015, 33, 1958–1965. [Google Scholar] [CrossRef] [PubMed]
- Ohsaki, Y.; Tanno, S.; Fujita, Y.; Toyoshima, E.; Fujiuchi, S.; Nishigaki, Y.; Ishida, S.; Nagase, A.; Miyokawa, N.; Hirata, S.; et al. Epidermal growth factor receptor expression correlates with poor prognosis in non-small cell lung cancer patients with p53 overexpression. Oncol. Rep. 2000, 7, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Gadgeel, S. (Eds.) Mechanisms of Resistance to EGFR Tyrosine Kinase Inhibitors and Therapeutic Approaches: An Update. Lung Cancer Pers. Med. 2016, 893, 137–153. [Google Scholar]
- Metro, G.; Crinò, L. The LUX-Lung clinical trial program of afatinib for non-small-cell lung cancer. Expert Rev. Anticancer Ther. 2011, 11, 673–682. [Google Scholar] [CrossRef] [PubMed]
- Malapelle, U.; Ricciuti, B.; Baglivo, S.; Pepe, F.; Pisapia, P.; Anastasi, P.; Tazza, M.; Sidoni, A.; Liberati, A.M.; Bellezza, G.; et al. Osimertinib. In Small Molecules in Oncology; Martens, U.M., Ed.; Springer International Publishing: Cham, Switzerland, 2018; pp. 257–276. [Google Scholar]
- Ramalingam, S.S.; Vansteenkiste, J.; Planchard, D.; Cho, B.C.; Gray, J.E.; Ohe, Y.; Zhou, C.; Reungwetwattana, T.; Cheng, Y.; Chewaskulyong, B.; et al. Overall Survival with Osimertinib in Untreated, EGFR-Mutated Advanced NSCLC. N. Engl. J. Med. 2020, 382, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Nishihara, S.; Yamaoka, T.; Ishikawa, F.; Higuchi, K.; Hasebe, Y.; Manabe, R.; Kishino, Y.; Kusumoto, S.; Ando, K.; Kuroda, Y.; et al. Mechanisms of EGFR-TKI-Induced Apoptosis and Strategies Targeting Apoptosis in EGFR-Mutated Non-Small Cell Lung Cancer. Genes 2022, 13, 2183. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Li, H.R.; Jin, C.; Jiang, J.H.; Ding, J.Y. Strategies to overcome acquired resistance to EGFR TKI in the treatment of non-small cell lung cancer. Clin. Transl. Oncol. 2019, 21, 1287–1301. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Z.; Chen, Q.; Zhong, X.; Chen, H.; Yu, R.; Tang, Y. Ginsenoside Rg3, a promising agent for NSCLC patients in the pandemic: A large-scale data mining and systemic biological analysis. J. Ginseng Res. 2023, 47, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Zhang, T.; Jing, S.; Zuo, P.; Li, T.; Wang, Y.; Xing, S.; Zhang, J.; Wei, Z. 20(S)-Ginsenoside Rg3 Inhibits Lung Cancer Cell Proliferation by Targeting EGFR-Mediated Ras/Raf/MEK/ERK Pathway. Am. J. Chin. Med. 2021, 49, 753–765. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Hou, W.; Wang, H.; Lin, H.S. Clinical Research on Shenyi Capsule Combined with Gefitinib for Advanced Non-Small Cell Lung Cancer: A Report of 50 Cases. J. Tradit. Chin. Med. 2012, 53, 933–935, 966. [Google Scholar]
- Jiang, Z.R.; Li, Q.X.; Qu, X. Effects of Shenyi Capsule Combined with Gefitinib on Pe-ripheral Serum MMP-9 in Patients with Advanced Non-small Cell Lung Cancer. World Chin. Med. 2018, 13, 1883–1886, 1890. [Google Scholar]
- Jiang, G.M.; Tan, Q.Q.; Liu, C.; Chen, X.; Xia, L.; Lu, D.; Kong, R.; Chen, Z.; Duan, Y.; Sun, J. Clinical efficacy and safety of Ginsenoside Rg3 combined with Osimertinib in treatment of the first-generation EGFR-TKI-resistant advanced non-small cell lung cancer. New Med. 2019, 50, 505–509. [Google Scholar]
- Li, Y. EGFR-TKI Combined with Ginsenoside Rg3 Compared with EGFR-TKI Alone in Patients with Advanced NSCLC: A Retrospective Study. Master’s Thesis, Third Military Medical University, Chongqing, China, 2016. [Google Scholar]
- Sozio, F.; Schioppa, T.; Sozzani, S.; Del Prete, A. Urethane-induced lung carcinogenesis. Methods Cell Biol. 2021, 163, 45–57. [Google Scholar] [PubMed]
- He, Q.; Sun, C.; Pan, Y. Whole-exome sequencing reveals Lewis lung carcinoma is a hypermutated Kras/Nras-mutant cancer with extensive regional mutation clusters in its genome. Sci. Rep. 2024, 14, 100. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Dong, X.; Jian, H.; Chen, J.; Chen, G.; Sun, Y.; Ji, Y.; Wang, Z.; Shi, J.; Lu, J.; et al. AENEAS: A Randomized Phase III Trial of Aumolertinib Versus Gefitinib as First-Line Therapy for Locally Advanced or MetastaticNon-Small-Cell Lung Cancer with EGFR Exon 19 Deletion or L858R Mutations. J. Clin. Oncol. 2022, 40, 3162–3171. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.Y.; Zhang, J.T.; Zeng, K.H.; Wu, Y.L. Perioperative targeted therapy for oncogene-driven NSCLC. Lung Cancer 2022, 172, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Ke, E.E.; Zhou, Q.; Wu, Y.L. Emerging paradigms in targeted treatments for Asian patients with NSCLC. Expert Opin. Pharmacother. 2015, 16, 1167–1176. [Google Scholar] [CrossRef] [PubMed]
- Goldman, J.W.; Bueno, A.M.; Dooms, C.; Jhaveri, K.; de Miguel, M.; Piha-Paul, S.A.; Unni, N.; Zick, A.; Mahipal, A.; Suga, J.M.; et al. Neratinib Efficacy in Patients with EGFR Exon 18-Mutant Non-Small-Cell Lung Cancer: Findings from the SUMMIT Basket Trial. Clin. Lung Cancer 2025, 26, 191–200.e1. [Google Scholar]
- Zhao, Y.; Yu, B.; Wang, Y.; Tan, S.; Xu, Q.; Wang, Z.; Zhou, K.; Liu, H.; Ren, Z.; Jiang, Z. Ang-1 and VEGF: Central regulators of angiogenesis. Mol. Cell Biochem. 2025, 480, 621–637. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Meng, P.; Terpstra, M.M.; van Rijk, A.; Tamminga, M.; Scherpen, F.; Ter Elst, A.; Alimohamed, M.Z.; Johansson, L.F.; Stigt, J.; et al. Clinical Value of EGFR Copy Number Gain Determined by Amplicon-Based Targeted Next Generation Sequencing in Patients with EGFR-Mutated NSCLC. Target. Oncol. 2021, 16, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Jin, R.; Li, M.; Lan, F.; Zhu, H.; Yu, Y.; Miao, D.; Wang, Q.; Zhou, Y.; Selvaggi, G.; et al. Potent antitumor activity of ensartinib in MET exon 14 skipping-mutated non-small cell lung cancer. Cancer Lett. 2023, 561, 216140. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Wu, W.W.; Yi, P. The Efficacy of Ginsenoside Rg3 Combined with First-line Chemotherapy in the Treatment of Advanced Non-Small Cell Lung Cancer in China: A Systematic Review and Meta-Analysis of Randomized Clinical Trials. Front. Pharmacol. 2020, 11, 630825. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Yang, Y.; Yang, Y.; Zhang, Y.; Yue, Z.; Pan, Z.; Ren, X. Ginsenoside Rg3 attenuates cisplatin resistance in lung cancer by downregulating PD-L1 and resuming immune. Biomed. Pharmacother. 2017, 96, 378–383. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, Y.; Niu, K.; Chen, X.; Xia, L.; Lu, D.; Kong, R.; Chen, Z.; Duan, Y.; Sun, J. Clinical benefit from EGFR-TKI plus ginsenoside Rg3 in patients with advanced non-small cell lung cancer harboring EGFR active mutation. Oncotarget 2016, 7, 70535–70545. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, J.; Yu, Z.; Ge, H.; Zhang, L.W.; Feng, L.X. Outcomes of concurrent versus sequential icotinib therapy and chemotherapy in advanced non-small cell lung cancer with sensitive EGFR mutations. Clin. Transl. Sci. 2021, 14, 890–897. [Google Scholar] [CrossRef] [PubMed]
- Chang, Q.; Xu, J.; Qiang, H.; Teng, J.; Qian, J.; Lv, M.; Zhang, Y.; Lou, Y.; Zhao, Y.; Zhong, R.; et al. EGFR Tyrosine Kinase Inhibitor (TKI) Combined with Concurrent or Sequential Chemotherapy for Patients with Advanced Lung Cancer and Gradual Progression After First-Line EGFR-TKI Therapy: A Randomized Controlled Study. Clin. Lung Cancer 2021, 22, e395–e404. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Fang, D.; Liang, T.; Pang, H.; Nong, Y.; Tang, L.; Yang, Z.; Lu, C.; Han, X.; Zhao, S.; et al. Atractylodin may induce ferroptosis of human hepatocellular carcinoma cells. Ann. Transl. Med. 2021, 9, 1535. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zuo, L.; Guo, D.; Chai, X.; Xu, J.; Cui, Z.; Wang, Z.; Hou, C. Ginsenoside Rg3 regulates DNA damage in non-small cell lung cancer cells by activating VRK1/P53BP1 pathway. Biomed. Pharmacother. 2019, 120, 109483. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lv, X.; Song, Y.; Liu, T.; Zhang, D.; Ye, X.; Wang, Q.; Li, R.; Chen, J.; Zhang, S.; Yu, X.; et al. Ginsenoside Rg3 Adjunctively Increases the Efficacy of Gefitinib Against NSCLC by Regulating EGFR Copy Number. Pharmaceuticals 2025, 18, 1077. https://doi.org/10.3390/ph18071077
Lv X, Song Y, Liu T, Zhang D, Ye X, Wang Q, Li R, Chen J, Zhang S, Yu X, et al. Ginsenoside Rg3 Adjunctively Increases the Efficacy of Gefitinib Against NSCLC by Regulating EGFR Copy Number. Pharmaceuticals. 2025; 18(7):1077. https://doi.org/10.3390/ph18071077
Chicago/Turabian StyleLv, Xinyi, Yuehan Song, Tianhua Liu, Dingdan Zhang, Xinpeng Ye, Qingqing Wang, Rongrong Li, Jiayi Chen, Shujing Zhang, Xue Yu, and et al. 2025. "Ginsenoside Rg3 Adjunctively Increases the Efficacy of Gefitinib Against NSCLC by Regulating EGFR Copy Number" Pharmaceuticals 18, no. 7: 1077. https://doi.org/10.3390/ph18071077
APA StyleLv, X., Song, Y., Liu, T., Zhang, D., Ye, X., Wang, Q., Li, R., Chen, J., Zhang, S., Yu, X., & Hou, C. (2025). Ginsenoside Rg3 Adjunctively Increases the Efficacy of Gefitinib Against NSCLC by Regulating EGFR Copy Number. Pharmaceuticals, 18(7), 1077. https://doi.org/10.3390/ph18071077