
Bridging the Gap in Breast Cancer Dormancy: Models, Mechanisms, and Translational Challenges

 , ,
, , 
Abstract
1. Introduction
2. Molecular and Cellular Mechanisms of Dormancy
2.1. Cell-Intrinsic Mechanisms: Molecular Pathways and Key Regulators
2.2. Epigenetic Modifications: Shaping the Dormancy Landscape
2.3. Metabolic Adaptations: Surviving in a Quiescent State
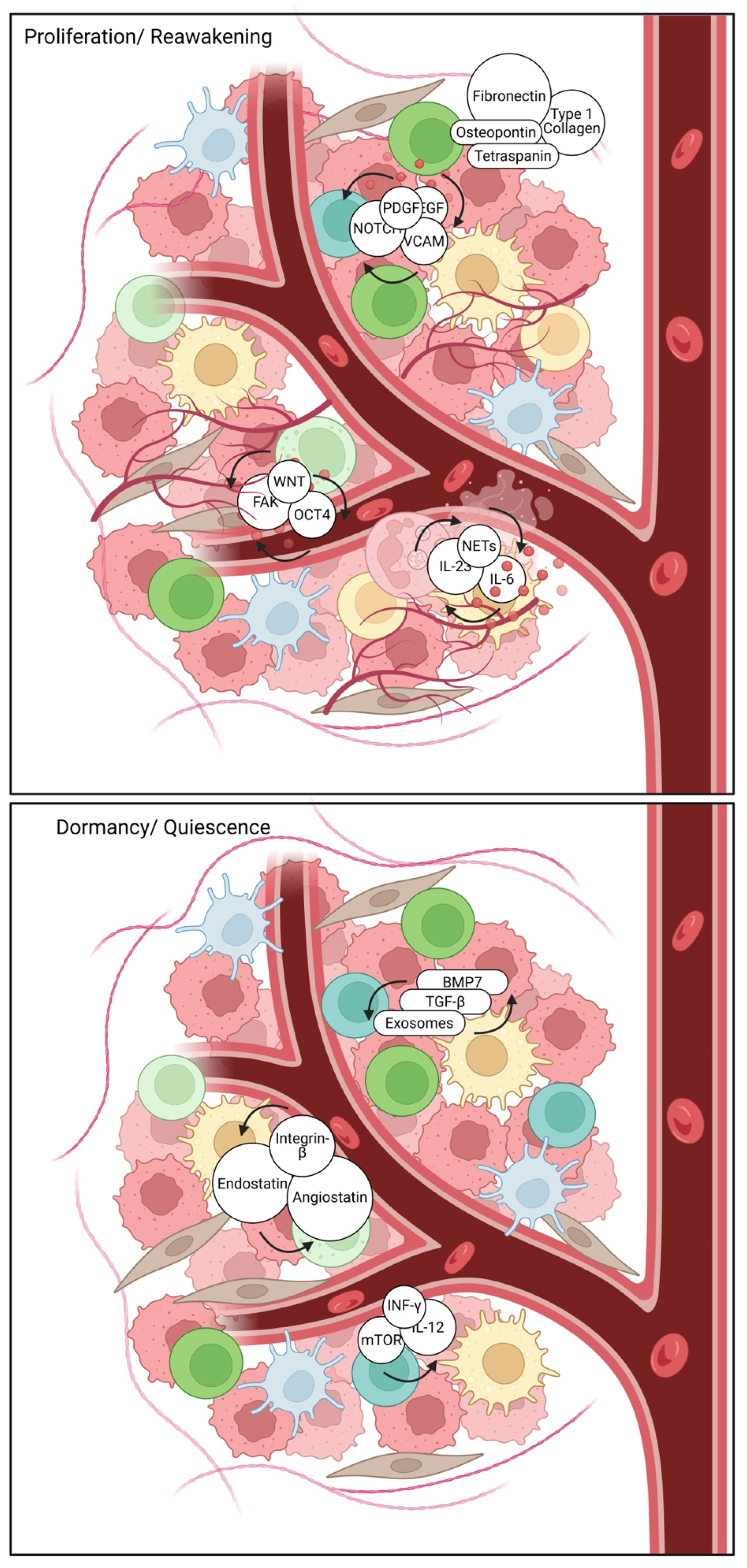
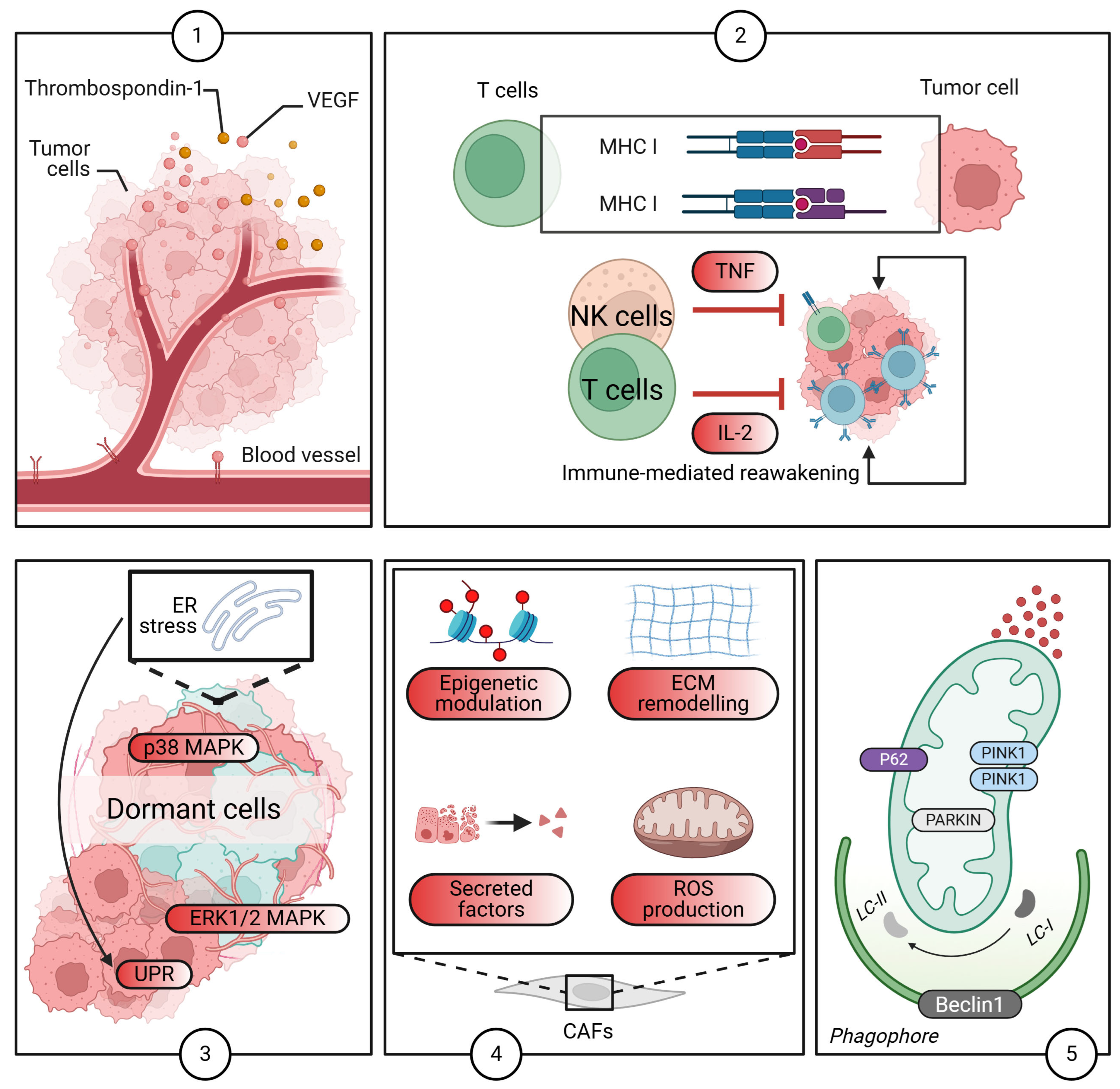
2.4. The TME: A Critical Regulator of Dormancy
2.5. The Role of Autophagy
2.6. Therapeutic Implications and Challenges
2.7. Current Immunotherapies and Dormant Cancer Cells: Challenges and Strategies
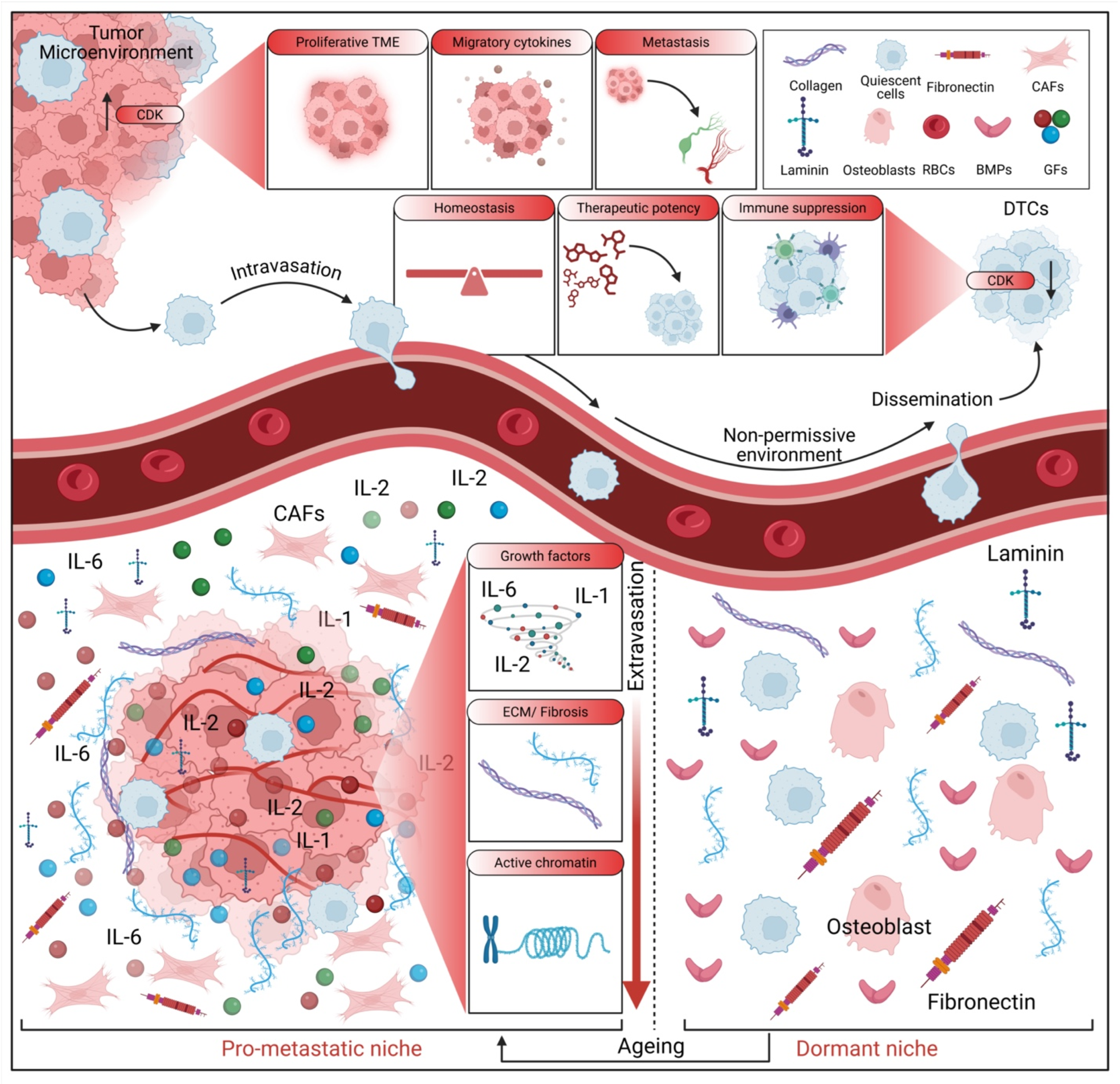
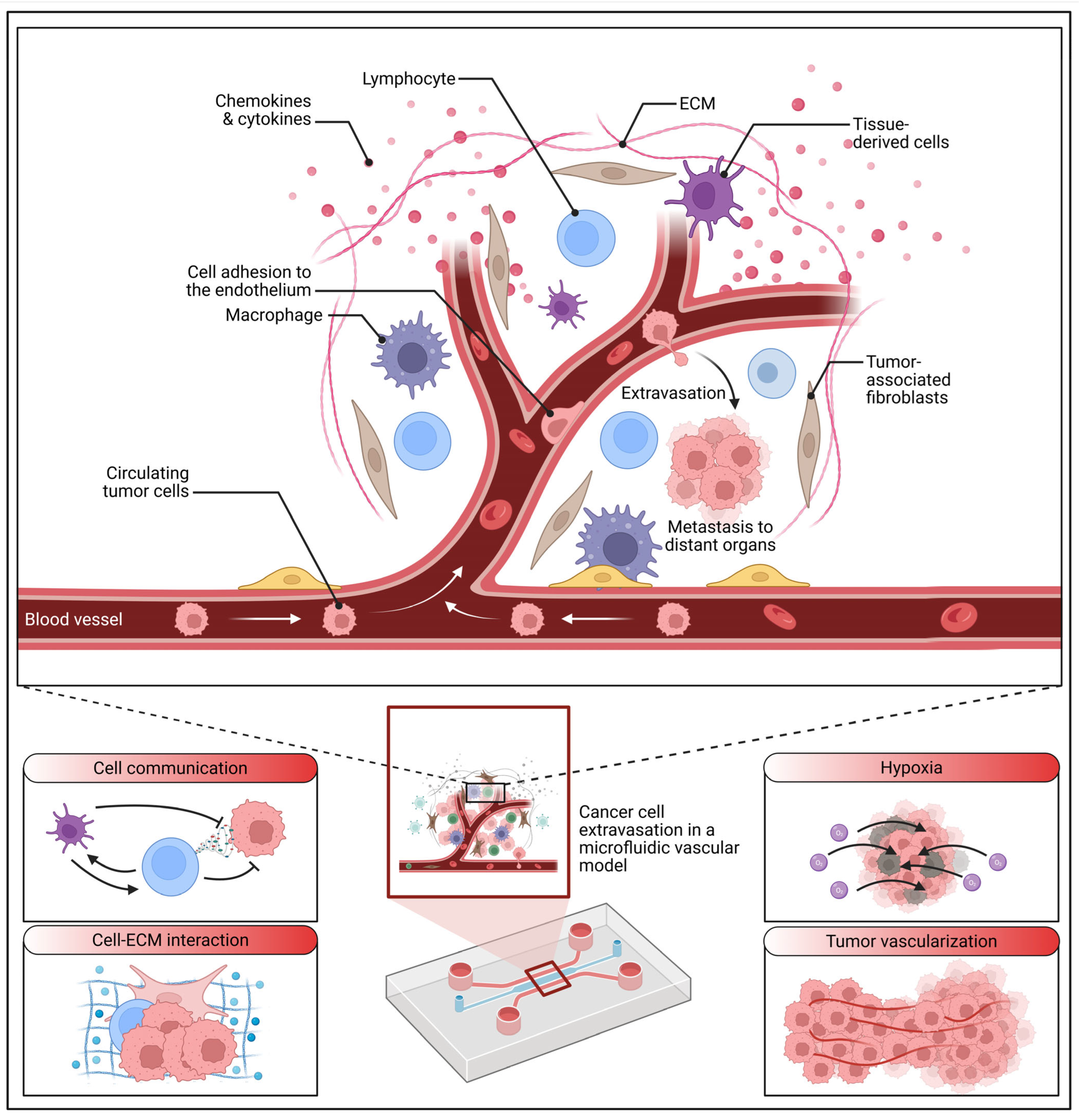
3. The Metastatic Niche and Microenvironmental Control
3.1. Cellular and Molecular Contributors to Metastatic Niche Formation
3.2. Emerging Concepts and Methodological Advances in Niche Research
3.3. Challenges in Targeting the Metastatic Niche
4. Experimental Models: Strengths and Limitations
4.1. In Vitro Models: 3D Cultures, Organoids, and Specialized Niches
4.1.1. Extracellular Vesicles and Niche Modeling
4.1.2. Modeling Organ-Specific Metastatic Niches
4.2. In Vivo Models: Mouse Models for Systemic Insights
4.3. Emerging Systems: Microfluidics and Advanced Methodologies for Niche Studies
4.4. Knowledge Gaps and Critical Analysis of Current Models
5. Clinical Translation and Biomarkers
5.1. Circulating Tumor DNA (ctDNA) as a Prognostic and Predictive Biomarker
5.2. Technological Advancements and Broader Biomarker Landscapes
5.3. Biomarkers in Guiding Treatment Strategies
5.4. Patient-Specific Variables Influencing Breast Cancer Dormancy
6. Future Directions and Research Priorities
6.1. Unraveling Molecular Mechanisms of Dormancy
6.2. Dissecting Microenvironmental Control of Dormancy
6.3. Advancing Experimental Models for Dormancy Research
6.4. Accelerating Clinical Translation
6.5. Fostering Interdisciplinary Approaches
6.6. Bridging the Translational Gap
7. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Elkholi, I.E.; Robert, A.; Malouf, C.; Kuasne, H.; Drapela, S.; Macleod, G.; Hébert, S.; Pacis, A.; Calderon, V.; Kleinman, C.L.; et al. Targeting the dependence on PIK3C3-mTORC1 signaling in dormancy-prone breast cancer cells blunts metastasis initiation. Cancer Res. 2025, 85, 2179–2198. [Google Scholar] [CrossRef] [PubMed]
- Louis, M.; Tapia, R.; Grabill, N.; Strom, P. Dormancy Leading to Late Recurrence in Breast Cancer: A Case of Hormone Receptor-Positive Supraclavicular Metastasis 10 Years After the Initial Treatment. Cureus 2024, 16, e71586. [Google Scholar] [CrossRef] [PubMed]
- Goddard, E.T.; Linde, M.H.; Srivastava, S.; Klug, G.; Shabaneh, T.B.; Iannone, S.; Grzelak, C.A.; Marsh, S.; Riggio, A.I.; Shor, R.E.; et al. Immune evasion of dormant disseminated tumor cells is due to their scarcity and can be overcome by T cell immunotherapies. Cancer Cell 2024, 42, 119–134.e112. [Google Scholar] [CrossRef] [PubMed]
- Heidrich, I.; Deitert, B.; Werner, S.; Pantel, K. Liquid biopsy for monitoring of tumor dormancy and early detection of disease recurrence in solid tumors. Cancer Metastasis Rev. 2023, 42, 161–182. [Google Scholar] [CrossRef]
- Bushnell, G.G.; Sharma, D.; Wilmot, H.C.; Zheng, M.; Fashina, T.D.; Hutchens, C.M.; Osipov, S.; Burness, M.; Wicha, M.S. Natural Killer Cell Regulation of Breast Cancer Stem Cells Mediates Metastatic Dormancy. Cancer Res. 2024, 84, 3337–3353. [Google Scholar] [CrossRef]
- Correia, A.L.; Guimaraes, J.C.; Auf der Maur, P.; De Silva, D.; Trefny, M.P.; Okamoto, R.; Bruno, S.; Schmidt, A.; Mertz, K.; Volkmann, K.; et al. Hepatic stellate cells suppress NK cell-sustained breast cancer dormancy. Nature 2021, 594, 566–571. [Google Scholar] [CrossRef]
- Nobre, A.R.; Dalla, E.; Yang, J.; Huang, X.; Wullkopf, L.; Risson, E.; Razghandi, P.; Anton, M.L.; Zheng, W.; Seoane, J.A.; et al. ZFP281 drives a mesenchymal-like dormancy program in early disseminated breast cancer cells that prevents metastatic outgrowth in the lung. Nat. Cancer 2022, 3, 1165–1180. [Google Scholar] [CrossRef]
- Parida, P.K.; Marquez-Palencia, M.; Nair, V.; Kaushik, A.K.; Kim, K.; Sudderth, J.; Quesada-Diaz, E.; Cajigas, A.; Vemireddy, V.; Gonzalez-Ericsson, P.I.; et al. Metabolic diversity within breast cancer brain-tropic cells determines metastatic fitness. Cell Metab. 2022, 34, 90–105.e107. [Google Scholar] [CrossRef]
- Montagner, M.; Sahai, E. In vitro Models of Breast Cancer Metastatic Dormancy. Front. Cell Dev. Biol. 2020, 8, 37. [Google Scholar] [CrossRef]
- Bakhshandeh, S.; Heras, U.; Taïeb, H.M.; Varadarajan, A.R.; Lissek, S.M.; Hücker, S.M.; Lu, X.; Garske, D.S.; Young, S.A.E.; Abaurrea, A.; et al. Dormancy-inducing 3D engineered matrix uncovers mechanosensitive and drug-protective FHL2-p21 signaling axis. Sci. Adv. 2024, 10, eadr3997. [Google Scholar] [CrossRef]
- Shboul, S.A.; DeLuca, V.J.; Dweiri, Y.A.; Saleh, T. Can 3D bioprinting solve the mystery of senescence in cancer therapy? Ageing Res. Rev. 2022, 81, 101732. [Google Scholar] [CrossRef] [PubMed]
- Kabak, E.C.; Foo, S.L.; Rafaeva, M.; Martin, I.; Bentires-Alj, M. Microenvironmental Regulation of Dormancy in Breast Cancer Metastasis: “An Ally that Changes Allegiances”. Adv. Exp. Med. Biol. 2025, 1464, 373–395. [Google Scholar] [CrossRef] [PubMed]
- Elkholi, I.E.; Rose, A.A.N.; Aguirre-Ghiso, J.A.; Côté, J.F. How can we integrate the biology of breast cancer cell dormancy into clinical practice? Cancer Cell 2024, 42, 1147–1151. [Google Scholar] [CrossRef] [PubMed]
- Leung, M.M.; Swanton, C.; McGranahan, N. Integrating model systems and genomic insights to decipher mechanisms of cancer metastasis. Nat. Rev. Genet. 2025, 26, 494–505. [Google Scholar] [CrossRef]
- Farino Reyes, C.J.; Pradhan, S.; Slater, J.H. The Influence of Ligand Density and Degradability on Hydrogel Induced Breast Cancer Dormancy and Reactivation. Adv. Healthc. Mater. 2021, 10, e2002227. [Google Scholar] [CrossRef]
- Farino Reyes, C.J.; Slater, J.H. Tuning Hydrogel Adhesivity and Degradability to Model the Influence of Premetastatic Niche Matrix Properties on Breast Cancer Dormancy and Reactivation. Adv. Biol. 2022, 6, e2200012. [Google Scholar] [CrossRef]
- Goodarzi, K.; Lane, R.; Rao, S.S. Varying the RGD concentration on a hyaluronic acid hydrogel influences dormancy versus proliferation in brain metastatic breast cancer cells. J. Biomed. Mater. Res. A 2024, 112, 710–720. [Google Scholar] [CrossRef]
- Kondapaneni, R.V.; Shevde, L.A.; Rao, S.S. A Biomimetic Hyaluronic Acid Hydrogel Models Mass Dormancy in Brain Metastatic Breast Cancer Spheroids. Adv. Biol. 2023, 7, e2200114. [Google Scholar] [CrossRef]
- Ovadia, E.M.; Pradhan, L.; Sawicki, L.A.; Cowart, J.E.; Huber, R.E.; Polson, S.W.; Chen, C.; van Golen, K.L.; Ross, K.E.; Wu, C.H.; et al. Understanding ER+ Breast Cancer Dormancy Using Bioinspired Synthetic Matrices for Long-Term 3D Culture and Insights into Late Recurrence. Adv. Biosyst. 2020, 4, e2000119. [Google Scholar] [CrossRef]
- Pradhan, L.; Moore, D.; Ovadia, E.M.; Swedzinski, S.L.; Cossette, T.; Sikes, R.A.; van Golen, K.; Kloxin, A.M. Dynamic bioinspired coculture model for probing ER+ breast cancer dormancy in the bone marrow niche. Sci. Adv. 2023, 9, eade3186. [Google Scholar] [CrossRef]
- Wieder, R. Bone Marrow Stroma Co-cultivation Model of Breast Cancer Dormancy. Methods Mol. Biol. 2024, 2811, 55–67. [Google Scholar] [CrossRef]
- Quinn, H.M.; Battista, L.; Scabia, V.; Brisken, C. Preclinical Mouse Intraductal Model (MIND) to Study Metastatic Dormancy in Estrogen Receptor-Positive Breast Cancer. Methods Mol. Biol. 2024, 2811, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Clements, M.E.; Holtslander, L.; Edwards, C.; Todd, V.; Dooyema, S.D.R.; Bullock, K.; Bergdorf, K.; Zahnow, C.A.; Connolly, R.M.; Johnson, R.W. HDAC inhibitors induce LIFR expression and promote a dormancy phenotype in breast cancer. Oncogene 2021, 40, 5314–5326. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhang, P.; Wu, Q.; Fang, H.; Wang, Y.; Xiao, Y.; Cong, M.; Wang, T.; He, Y.; Ma, C.; et al. Long non-coding RNA NR2F1-AS1 induces breast cancer lung metastatic dormancy by regulating NR2F1 and ΔNp63. Nat. Commun. 2021, 12, 5232. [Google Scholar] [CrossRef]
- Wang, J.; Ocadiz-Ruiz, R.; Hall, M.S.; Bushnell, G.G.; Orbach, S.M.; Decker, J.T.; Raghani, R.M.; Zhang, Y.; Morris, A.H.; Jeruss, J.S.; et al. A synthetic metastatic niche reveals antitumor neutrophils drive breast cancer metastatic dormancy in the lungs. Nat. Commun. 2023, 14, 4790. [Google Scholar] [CrossRef] [PubMed]
- Maurizi, A.; Ciocca, M.; Giuliani, C.; Di Carlo, I.; Teti, A. Role of Neural (N)-Cadherin in Breast Cancer Cell Stemness and Dormancy in the Bone Microenvironment. Cancers 2022, 14, 1317. [Google Scholar] [CrossRef]
- Sinha, G.; Ferrer, A.I.; Ayer, S.; El-Far, M.H.; Pamarthi, S.H.; Naaldijk, Y.; Barak, P.; Sandiford, O.A.; Bibber, B.M.; Yehia, G.; et al. Specific N-cadherin-dependent pathways drive human breast cancer dormancy in bone marrow. Life Sci. Alliance 2021, 4, e202000969. [Google Scholar] [CrossRef]
- Jiang, W.J.; Zhou, T.H.; Huang, H.J.; Li, L.S.; Tan, H.; Zhang, R.; Wang, Q.S.; Feng, Y.M. Breast Cancer Subtype-Specific Organotropism Is Dictated by FOXF2-Regulated Metastatic Dormancy and Recovery. Cancer Res. 2025, 85, 644–659. [Google Scholar] [CrossRef]
- Han, R.; Sun, X.; Wu, Y.; Yang, Y.H.; Wang, Q.C.; Zhang, X.T.; Ding, T.; Yang, J.T. Proteomic and Phosphoproteomic Profiling of Matrix Stiffness-Induced Stemness-Dormancy State Transition in Breast Cancer Cells. J. Proteome Res. 2024, 23, 4658–4673. [Google Scholar] [CrossRef]
- Kondapaneni, R.V.; Rao, S.S. Matrix stiffness and cluster size collectively regulate dormancy versus proliferation in brain metastatic breast cancer cell clusters. Biomater. Sci. 2020, 8, 6637–6646. [Google Scholar] [CrossRef]
- Drago-Garcia, D.; Giri, S.; Chatterjee, R.; Simoni-Nieves, A.; Abedrabbo, M.; Genna, A.; Rios, M.L.U.; Lindzen, M.; Sekar, A.; Gupta, N.; et al. Re-epithelialization of cancer cells increases autophagy and DNA damage: Implications for breast cancer dormancy and relapse. Sci. Signal. 2025, 18, eado3473. [Google Scholar] [CrossRef]
- Aqbi, H.F.; Coleman, C.; Zarei, M.; Manjili, S.H.; Graham, L.; Koblinski, J.; Guo, C.; Xie, Y.; Guruli, G.; Bear, H.D.; et al. Local and distant tumor dormancy during early stage breast cancer are associated with the predominance of infiltrating T effector subsets. Breast Cancer Res. 2020, 22, 116. [Google Scholar] [CrossRef]
- Tallón de Lara, P.; Castañón, H.; Vermeer, M.; Núñez, N.; Silina, K.; Sobottka, B.; Urdinez, J.; Cecconi, V.; Yagita, H.; Movahedian Attar, F.; et al. CD39+PD-1+CD8+ T cells mediate metastatic dormancy in breast cancer. Nat. Commun. 2021, 12, 769. [Google Scholar] [CrossRef] [PubMed]
- Mehdizadeh, R.; Shariatpanahi, S.P.; Goliaei, B.; Rüegg, C. Targeting myeloid-derived suppressor cells in combination with tumor cell vaccination predicts anti-tumor immunity and breast cancer dormancy: An in silico experiment. Sci. Rep. 2023, 13, 5875. [Google Scholar] [CrossRef]
- Melzer, C.; Ohe, J.V.; Luo, T.; Hass, R. Spontaneous Fusion of MSC with Breast Cancer Cells Can Generate Tumor Dormancy. Int. J. Mol. Sci. 2021, 22, 5930. [Google Scholar] [CrossRef]
- Mohd Ali, N.; Yeap, S.K.; Ho, W.Y.; Boo, L.; Ky, H.; Satharasinghe, D.A.; Tan, S.W.; Cheong, S.K.; Huang, H.D.; Lan, K.C.; et al. Adipose MSCs Suppress MCF7 and MDA-MB-231 Breast Cancer Metastasis and EMT Pathways Leading to Dormancy via Exosomal-miRNAs Following Co-Culture Interaction. Pharmaceuticals 2020, 14, 8. [Google Scholar] [CrossRef]
- Sandiford, O.A.; Donnelly, R.J.; El-Far, M.H.; Burgmeyer, L.M.; Sinha, G.; Pamarthi, S.H.; Sherman, L.S.; Ferrer, A.I.; DeVore, D.E.; Patel, S.A.; et al. Mesenchymal Stem Cell-Secreted Extracellular Vesicles Instruct Stepwise Dedifferentiation of Breast Cancer Cells into Dormancy at the Bone Marrow Perivascular Region. Cancer Res. 2021, 81, 1567–1582. [Google Scholar] [CrossRef] [PubMed]
- Vishnubalaji, R.; Alajez, N.M. Disrupted Lipid Metabolism, Cytokine Signaling, and Dormancy: Hallmarks of Doxorubicin-Resistant Triple-Negative Breast Cancer Models. Cancers 2024, 16, 4273. [Google Scholar] [CrossRef] [PubMed]
- Sunderland, A.; Williams, J.; Andreou, T.; Rippaus, N.; Fife, C.; James, F.; Kartika, Y.D.; Speirs, V.; Carr, I.; Droop, A.; et al. Biglycan and reduced glycolysis are associated with breast cancer cell dormancy in the brain. Front. Oncol. 2023, 13, 1191980. [Google Scholar] [CrossRef]
- Ren, Q.; Khoo, W.H.; Corr, A.P.; Phan, T.G.; Croucher, P.I.; Stewart, S.A. Gene expression predicts dormant metastatic breast cancer cell phenotype. Breast Cancer Res. 2022, 24, 10. [Google Scholar] [CrossRef]
- Rosano, D.; Sofyali, E.; Dhiman, H.; Ghirardi, C.; Ivanoiu, D.; Heide, T.; Vingiani, A.; Bertolotti, A.; Pruneri, G.; Canale, E.; et al. Long-term Multimodal Recording Reveals Epigenetic Adaptation Routes in Dormant Breast Cancer Cells. Cancer Discov. 2024, 14, 866–889. [Google Scholar] [CrossRef]
- Bravo-Cordero, J.J.; O’Connor, R.S.; Lyssiotis, C.A. Metabolic vulnerabilities of dormant cancer cells. Nat. Rev. Cancer 2023, 23, 35–50. [Google Scholar]
- Fukuoka, M.; Ichikawa, Y.; Osako, T.; Fujita, T.; Baba, S.; Takeuchi, K.; Tsunoda, N.; Ebata, T.; Ueno, T.; Ohno, S.; et al. The ELEANOR noncoding RNA expression contributes to cancer dormancy and predicts late recurrence of estrogen receptor-positive breast cancer. Cancer Sci. 2022, 113, 2336–2351. [Google Scholar] [CrossRef] [PubMed]
- Iyer, D.P.; Khoei, H.H.; van der Weijden, V.A.; Kagawa, H.; Pradhan, S.J.; Novatchkova, M.; McCarthy, A.; Rayon, T.; Simon, C.S.; Dunkel, I.; et al. mTOR activity paces human blastocyst stage developmental progression. Cell 2024, 187, 6566–6583.e6522. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; He, M.; Zhao, L.; Qin, S.; Zhu, Z.; Du, X.; Zhou, B.; Yang, Y.; Liu, X.; Xia, G.; et al. HDAC6 regulates primordial follicle activation through mTOR signaling pathway. Cell Death Dis. 2021, 12, 559. [Google Scholar] [CrossRef]
- Chen, J.; Feng, W.; Sun, M.; Huang, W.; Wang, G.; Chen, X.; Yin, Y.; Chen, X.; Zhang, B.; Nie, Y.; et al. TGF-β1-Induced SOX18 Elevation Promotes Hepatocellular Carcinoma Progression and Metastasis Through Transcriptionally Upregulating PD-L1 and CXCL12. Gastroenterology 2024, 167, 264–280. [Google Scholar] [CrossRef]
- Chen, S.Y.; Kung, H.C.; Espinoza, B.; Washington, I.; Chen, K.; Wang, J.; Zlomke, H.; Loycano, M.; Wang, R.; Pickup, M.; et al. Targeting heterogeneous tumor microenvironments in pancreatic cancer mouse models of metastasis by TGF-β depletion. JCI Insight 2024, 9, e182766. [Google Scholar] [CrossRef]
- Ma, Z.; Sun, J.; Li, Z.; Huang, S.; Li, B. AMDHD1 acts as a tumor suppressor and contributes to activation of TGF-β signaling pathway in cholangiocarcinoma. Cell Death Differ. 2025, 32, 162–176. [Google Scholar] [CrossRef]
- Zahraeifard, S.; Xiao, Z.; So, J.Y.; Ahad, A.; Montoya, S.; Park, W.Y.; Sornapudi, T.; Andohkow, T.; Read, A.; Kedei, N.; et al. Loss of tumor suppressors promotes inflammatory tumor microenvironment and enhances LAG3+T cell mediated immune suppression. Nat. Commun. 2024, 15, 5873. [Google Scholar] [CrossRef]
- Boix, L.; López-Oliva, J.M.; Rhodes, A.C.; Bruix, J. Restoring miR122 in human stem-like hepatocarcinoma cells, prompts tumor dormancy through Smad-independent TGF-β pathway. Oncotarget 2016, 7, 71309–71329. [Google Scholar] [CrossRef]
- Singh, D.K.; Carcamo, S.; Farias, E.F.; Hasson, D.; Zheng, W.; Sun, D.; Huang, X.; Cheung, J.; Nobre, A.R.; Kale, N.; et al. 5-Azacytidine- and retinoic-acid-induced reprogramming of DCCs into dormancy suppresses metastasis via restored TGF-β-SMAD4 signaling. Cell Rep. 2023, 42, 112560. [Google Scholar] [CrossRef]
- Yumoto, K.; Eber, M.R.; Wang, J.; Cackowski, F.C.; Decker, A.M.; Lee, E.; Nobre, A.R.; Aguirre-Ghiso, J.A.; Jung, Y.; Taichman, R.S. Axl is required for TGF-β2-induced dormancy of prostate cancer cells in the bone marrow. Sci. Rep. 2016, 6, 36520. [Google Scholar] [CrossRef]
- Guereño, M.; Delgado Pastore, M.; Lugones, A.C.; Cercato, M.; Todaro, L.; Urtreger, A.; Peters, M.G. Glypican-3 (GPC3) inhibits metastasis development promoting dormancy in breast cancer cells by p38 MAPK pathway activation. Eur. J. Cell Biol. 2020, 99, 151096. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.; Ru, H.; Chen, Y.; Xu, J.; Wang, C.; Jin, Y. Gallic acid attenuates LPS-induced inflammation in Caco-2 cells by suppressing the activation of the NF-κB/MAPK signaling pathway. Acta Biochim. Biophys. Sin. 2024, 56, 905–915. [Google Scholar] [CrossRef]
- Deng, X.; Li, Y.; Chen, Y.; Hu, Q.; Zhang, W.; Chen, L.; Lu, X.; Zeng, J.; Ma, X.; Efferth, T. Paeoniflorin protects hepatocytes from APAP-induced damage through launching autophagy via the MAPK/mTOR signaling pathway. Cell. Mol. Biol. Lett. 2024, 29, 119. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Roy, A.M.; Tokumaru, Y.; Gandhi, S.; Asaoka, M.; Oshi, M.; Yan, L.; Ishikawa, T.; Takabe, K. NR2F1, a Tumor Dormancy Marker, Is Expressed Predominantly in Cancer-Associated Fibroblasts and Is Associated with Suppressed Breast Cancer Cell Proliferation. Cancers 2022, 14, 2962. [Google Scholar] [CrossRef] [PubMed]
- Lossaint, G.; Horvat, A.; Gire, V.; Bačević, K.; Mrouj, K.; Charrier-Savournin, F.; Georget, V.; Fisher, D.; Dulić, V. Reciprocal regulation of p21 and Chk1 controls the cyclin D1-RB pathway to mediate senescence onset after G2 arrest. J. Cell Sci. 2022, 135, jcs259114. [Google Scholar] [CrossRef]
- Francescangeli, F.; De Angelis, M.L.; Rossi, R.; Cuccu, A.; Giuliani, A.; De Maria, R.; Zeuner, A. Dormancy, stemness, and therapy resistance: Interconnected players in cancer evolution. Cancer Metastasis Rev. 2023, 42, 197–215. [Google Scholar] [CrossRef]
- Kumar, A.; Saha, L. Colorectal cancer cell dormancy: An insight into pathways. World J. Gastroenterol. 2024, 30, 3810–3817. [Google Scholar] [CrossRef]
- Llinas-Bertran, A.; Bellet-Ezquerra, M.; Seoane, J.A. Epigenetic Control of Cancer Cell Dormancy and Awakening in Endocrine Therapy Resistance. Cancer Discov. 2024, 14, 704–706. [Google Scholar] [CrossRef]
- Gomatou, G.; Syrigos, N.; Vathiotis, I.A.; Kotteas, E.A. Tumor Dormancy: Implications for Invasion and Metastasis. Int. J. Mol. Sci. 2021, 22, 4862. [Google Scholar] [CrossRef]
- Bai, J.; Jiang, P.; Ji, L.; Lam, W.K.J.; Zhou, Q.; Ma, M.L.; Ding, S.C.; Ramakrishnan, S.; Wan, C.W.; Yang, T.C.; et al. Histone modifications of circulating nucleosomes are associated with changes in cell-free DNA fragmentation patterns. Proc. Natl. Acad. Sci. USA 2024, 121, e2404058121. [Google Scholar] [CrossRef] [PubMed]
- Duan, X.; Xing, Z.; Qiao, L.; Qin, S.; Zhao, X.; Gong, Y.; Li, X. The role of histone post-translational modifications in cancer and cancer immunity: Functions, mechanisms and therapeutic implications. Front. Immunol. 2024, 15, 1495221. [Google Scholar] [CrossRef]
- Lee, K.; Barone, M.; Waterbury, A.L.; Jiang, H.; Nam, E.; DuBois-Coyne, S.E.; Whedon, S.D.; Wang, Z.A.; Caroli, J.; Neal, K.; et al. Uncoupling histone modification crosstalk by engineering lysine demethylase LSD1. Nat. Chem. Biol. 2025, 21, 227–237. [Google Scholar] [CrossRef]
- Simpson, B.; Tupper, C.; Al Aboud, N.M. Genetics, DNA Packaging. In StatPearls; StatPearls Publishing LLC.: Treasure Island FL, USA, 2025. [Google Scholar]
- Suraweera, A.; O’Byrne, K.J.; Richard, D.J. Epigenetic drugs in cancer therapy. Cancer Metastasis Rev. 2025, 44, 37. [Google Scholar] [CrossRef]
- Min, D.; Byun, J.; Lee, E.J.; Khan, A.A.; Liu, C.; Loudig, O.; Hu, W.; Zhao, Y.; Herlyn, M.; Tycko, B.; et al. Epigenetic Silencing of BMP6 by the SIN3A-HDAC1/2 Repressor Complex Drives Melanoma Metastasis via FAM83G/PAWS1. Mol. Cancer Res. 2022, 20, 217–230. [Google Scholar] [CrossRef]
- Smith, J.; Sen, S.; Weeks, R.J.; Eccles, M.R.; Chatterjee, A. Promoter DNA Hypermethylation and Paradoxical Gene Activation. Trends Cancer 2020, 6, 392–406. [Google Scholar] [CrossRef]
- Kim, H.; Wirasaputra, A.; Mohammadi, F.; Kundu, A.N.; Esteves, J.A.E.; Heiser, L.M.; Meyer, A.S.; Peyton, S.R. Live Cell Lineage Tracing of Dormant Cancer Cells. Adv. Healthc. Mater. 2023, 12, 2202275. [Google Scholar] [CrossRef]
- Otani, M.; Zheng, L.; Kawakami, N. Genetic, Epigenetic, and Environmental Control of Seed Dormancy and Germination. Methods Mol. Biol. 2024, 2830, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Silva, H.G.; Sobral, R.; Alhinho, A.T.; Afonso, H.R.; Ribeiro, T.; Silva, P.M.A.; Bousbaa, H.; Morais-Cecílio, L.; Costa, M.M.R. Genetic and epigenetic control of dormancy transitions throughout the year in the monoecious cork oak. Physiol. Plant 2024, 176, e14620. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Chen, P.Y.; Zhong, S.; Dardick, C.; Callahan, A.; An, Y.Q.; van Knocker, S.; Yang, Y.; Zhong, G.Y.; Abbott, A.; et al. Thermal-responsive genetic and epigenetic regulation of DAM cluster controlling dormancy and chilling requirement in peach floral buds. Hortic. Res. 2020, 7, 114. [Google Scholar] [CrossRef]
- Ferrer-Diaz, A.I.; Sinha, G.; Petryna, A.; Gonzalez-Bermejo, R.; Kenfack, Y.; Adetayo, O.; Patel, S.A.; Hooda-Nehra, A.; Rameshwar, P. Revealing role of epigenetic modifiers and DNA oxidation in cell-autonomous regulation of Cancer stem cells. Cell Commun. Signal. 2024, 22, 119. [Google Scholar] [CrossRef]
- Robinson, N.J.; Parker, K.A.; Schiemann, W.P. Epigenetic plasticity in metastatic dormancy: Mechanisms and therapeutic implications. Ann. Transl. Med. 2020, 8, 903. [Google Scholar] [CrossRef]
- Guo, H. Metabolic reprogramming in dormant cancer cells. Front. Cell Dev. Biol. 2021, 9. [Google Scholar]
- Sharma, S. Reduced metabolic activity of dormant breast cancer cells and their susceptibility to re-awakening. J. Cell Sci. 2020, 133. [Google Scholar]
- Kim, K.J. Glycolytic adaptation is a hallmark of dormancy and resistance in a subset of breast cancer cells. Oncogene 2021, 40, 5440–5452. [Google Scholar]
- Ma, X. Hypoxia induces dormant breast cancer cell survival through glycolysis and fatty acid metabolism. Mol. Oncol. 2022, 16, 2119–2135. [Google Scholar]
- El-Gammal, Z.; Bakry, U.; El-Sayed, A.F.; Ahmed, T.A.; Oura, G.A.; Elshenawy, S.E.; El-Badri, N.; Romany, A.F.; Amer, K.; Elnagdy, T.; et al. Apolipoproteins have a major role in cellular tumor dormancy in triple negative breast cancer: In-silico study. Sci. Rep. 2024, 14, 23146. [Google Scholar] [CrossRef]
- Li, Y. Fatty acid oxidation fuels dormancy and resistance to chemotherapy in breast cancer. Cell Metab. 2022, 34, 1037–1051. [Google Scholar]
- Nakayama, T.; Sano, T.; Oshimo, Y.; Kawada, C.; Kasai, M.; Yamamoto, S.; Fukuhara, H.; Inoue, K.; Ogura, S.I. Enhanced lipid metabolism induces the sensitivity of dormant cancer cells to 5-aminolevulinic acid-based photodynamic therapy. Sci. Rep. 2021, 11, 7290. [Google Scholar] [CrossRef] [PubMed]
- Perego, M.; Tyurin, V.A.; Tyurina, Y.Y.; Yellets, J.; Nacarelli, T.; Lin, C.; Nefedova, Y.; Kossenkov, A.; Liu, Q.; Sreedhar, S.; et al. Reactivation of dormant tumor cells by modified lipids derived from stress-activated neutrophils. Sci. Transl. Med. 2020, 12, eabb5817. [Google Scholar] [CrossRef]
- Puente-Cobacho, B.; Esteo, C.; Altea-Manzano, P.; Garcia-Perez, J.L.; Quiles, J.L.; Sanchez-Rovira, P.; Martín-Salvago, M.D.; Molina-Jiménez, L.; Luque, R.J.; Fendt, S.M.; et al. De novo lipogenesis protects dormant breast cancer cells from ferroptosis and promotes metastasis. Redox Biol. 2025, 80, 103480. [Google Scholar] [CrossRef]
- Gómez-Banoy, N. Lipid droplet accumulation in dormant breast cancer cells promotes survival and resistance to therapy. Cancer Res. 2021, 81, 2686–2700. [Google Scholar]
- Park, S. Targeting lipid metabolism to eradicate dormant cancer cells. Trends Pharmacol. Sci. 2023, 44, 625–637. [Google Scholar]
- Dwyer, S.; Ruth, J.; Seidel, H.E.; Raz, A.A.; Chodosh, L.A. Autophagy is required for mammary tumor recurrence by promoting dormant tumor cell survival following therapy. Breast Cancer Res. 2024, 26, 143. [Google Scholar] [CrossRef]
- Tian, X.; He, Y.; Qi, L.; Liu, D.; Zhou, D.; Liu, Y.; Gong, W.; Han, Z.; Xia, Y.; Li, H.; et al. Autophagy Inhibition Contributes to Apoptosis of PLK4 Downregulation-induced Dormant Cells in Colorectal Cancer. Int. J. Biol. Sci. 2023, 19, 2817–2834. [Google Scholar] [CrossRef]
- Wang, Y. Autophagy blockade sensitizes dormant breast cancer cells to targeted therapies. Cancer Lett. 2023, 555. [Google Scholar]
- Kaur, J.; Debnath, J. Autophagy at the crossroads of catabolism and anabolism. Nat. Rev. Mol. Cell Biol. 2015, 16, 461–472. [Google Scholar] [CrossRef]
- Di Martino, J.S.; Nobre, A.R.; Mondal, C.; Taha, I.; Farias, E.F.; Fertig, E.J.; Naba, A.; Aguirre-Ghiso, J.A.; Bravo-Cordero, J.J. A tumor-derived type III collagen-rich ECM niche regulates tumor cell dormancy. Nat. Cancer 2022, 3, 90–107. [Google Scholar] [CrossRef]
- Dong, Y.; Bai, J.; Zhou, J. Developing a dormancy-associated ECM signature in TNBC that is linked to immunosuppressive tumor microenvironment and selective sensitivity to MAPK inhibitors. Heliyon 2024, 10, e32106. [Google Scholar] [CrossRef]
- Dai, J.; Cimino, P.J.; Gouin, K.H., 3rd; Grzelak, C.A.; Barrett, A.; Lim, A.R.; Long, A.; Weaver, S.; Saldin, L.T.; Uzamere, A.; et al. Astrocytic laminin-211 drives disseminated breast tumor cell dormancy in brain. Nat. Cancer 2022, 3, 25–42. [Google Scholar] [CrossRef]
- Kozlova, N.; Grossman, J.E.; Iwanicki, M.P.; Muranen, T. The Interplay of the Extracellular Matrix and Stromal Cells as a Drug Target in Stroma-Rich Cancers. Trends Pharmacol. Sci. 2020, 41, 183–198. [Google Scholar] [CrossRef]
- Cheng, S.-H.; Chiou, H.-Y.C.; Wang, J.-W.; Lin, M.-H. Reciprocal Regulation of Cancer-Associated Fibroblasts and Tumor Microenvironment in Gastrointestinal Cancer: Implications for Cancer Dormancy. Cancers 2023, 15, 2513. [Google Scholar] [CrossRef]
- Mun, J.-Y.; Leem, S.-H.; Lee, J.H.; Kim, H.S. Dual Relationship Between Stromal Cells and Immune Cells in the Tumor Microenvironment. Front. Immunol. 2022, 13, 864739. [Google Scholar] [CrossRef]
- Hen, O.; Barkan, D. Dormant disseminated tumor cells and cancer stem/progenitor-like cells: Similarities and opportunities. Semin. Cancer Biol. 2020, 60, 157–165. [Google Scholar] [CrossRef]
- Akkoc, Y.; Gozuacik, D. Autophagy and Hepatic Tumor Microenvironment Associated Dormancy. J. Gastrointest. Cancer 2021, 52, 1277–1293. [Google Scholar] [CrossRef]
- Jahangiri, L.; Ishola, T. Dormancy in Breast Cancer, the Role of Autophagy, lncRNAs, miRNAs and Exosomes. Int. J. Mol. Sci. 2022, 23, 5271. [Google Scholar] [CrossRef]
- Othman, A.; Winogradzki, M.; Lee, L.; Tandon, M.; Blank, A.; Pratap, J. Bone Metastatic Breast Cancer: Advances in Cell Signaling and Autophagy Related Mechanisms. Cancers 2021, 13, 4310. [Google Scholar] [CrossRef]
- Finnegan, R.M.; Elshazly, A.M.; Schoenlein, P.V.; Gewirtz, D.A. Therapeutic Potential for Targeting Autophagy in ER+ Breast Cancer. Cancers 2022, 14, 4289. [Google Scholar] [CrossRef]
- Wu, Q.; Sharma, D. Autophagy and Breast Cancer: Connected in Growth, Progression, and Therapy. Cells 2023, 12, 1156. [Google Scholar] [CrossRef]
- Niklaus, N.J.; Tokarchuk, I.; Zbinden, M.; Schläfli, A.M.; Maycotte, P.; Tschan, M.P. The Multifaceted Functions of Autophagy in Breast Cancer Development and Treatment. Cells 2021, 10, 1447. [Google Scholar] [CrossRef]
- Tyutyunyk-Massey, L.; Gewirtz, D.A. Roles of autophagy in breast cancer treatment: Target, bystander or benefactor. Semin. Cancer Biol. 2020, 66, 155–162. [Google Scholar] [CrossRef]
- Nicco, C.; Marine, T.; Julie, G.; Maryline, H.; Fanny, L.-T.; Ludivine, D.; François, D.; Frédéric, B.; and Tchenio, T. Mechanistic target of rapamycin (mTOR) regulates self-sustained quiescence, tumor indolence, and late clinical metastasis in a Beclin-1-dependent manner. Cell Cycle 2023, 22, 542–564. [Google Scholar] [CrossRef]
- Appiah, C.O.; Singh, M.; May, L.; Bakshi, I.; Vaidyanathan, A.; Dent, P.; Ginder, G.; Grant, S.; Bear, H.; Landry, J. The epigenetic regulation of cancer cell recovery from therapy exposure and its implications as a novel therapeutic strategy for preventing disease recurrence. Adv. Cancer Res. 2023, 158, 337–385. [Google Scholar] [CrossRef]
- Stojowska-Swędrzyńska, K.; Kuczyńska-Wiśnik, D.; Laskowska, E. New Strategies to Kill Metabolically-Dormant Cells Directly Bypassing the Need for Active Cellular Processes. Antibiotics 2023, 12, 1044. [Google Scholar] [CrossRef]
- Mehdizadeh, R.; Shariatpanahi, S.P.; Goliaei, B.; Peyvandi, S.; Rüegg, C. Dormant Tumor Cell Vaccination: A Mathematical Model of Immunological Dormancy in Triple-Negative Breast Cancer. Cancers 2021, 13, 245. [Google Scholar] [CrossRef]
- Agudo, J.; Aguirre-Ghiso, J.A.; Bhatia, M.; Chodosh, L.A.; Correia, A.L.; Klein, C.A. Targeting cancer cell dormancy. Nat. Rev. Cancer 2024, 24, 97–104. [Google Scholar] [CrossRef]
- Galeano Niño, J.L.; Wu, H.; LaCourse, K.D.; Kempchinsky, A.G.; Baryiames, A.; Barber, B.; Futran, N.; Houlton, J.; Sather, C.; Sicinska, E.; et al. Effect of the intratumoral microbiota on spatial and cellular heterogeneity in cancer. Nature 2022, 611, 810–817. [Google Scholar] [CrossRef]
- Yu, L.; Wei, J.; Liu, P. Attacking the PI3K/Akt/mTOR signaling pathway for targeted therapeutic treatment in human cancer. Semin. Cancer Biol. 2022, 85, 69–94. [Google Scholar] [CrossRef]
- Baldominos, P.; Barbera-Mourelle, A.; Barreiro, O.; Huang, Y.; Wight, A.; Cho, J.W.; Zhao, X.; Estivill, G.; Adam, I.; Sanchez, X.; et al. Quiescent cancer cells resist T cell attack by forming an immunosuppressive niche. Cell 2022, 185, 1694–1708.e1619. [Google Scholar] [CrossRef]
- Puttick, C.; Jones, T.P.; Leung, M.M.; Galvez-Cancino, F.; Liu, J.; Varas-Godoy, M.; Rowan, A.; Pich, O.; Martinez-Ruiz, C.; Bentham, R.; et al. MHC Hammer reveals genetic and non-genetic HLA disruption in cancer evolution. Nat. Genet. 2024, 56, 2121–2131. [Google Scholar] [CrossRef]
- Emens, L.A.; Cimino-Mathews, A. Quiescent cancer cells: Therapeutic targets to overcome immunotherapy resistance? Med 2022, 3, 358–360. [Google Scholar] [CrossRef]
- Au, L.; Hatipoglu, E.; Robert de Massy, M.; Litchfield, K.; Beattie, G.; Rowan, A.; Schnidrig, D.; Thompson, R.; Byrne, F.; Horswell, S.; et al. Determinants of anti-PD-1 response and resistance in clear cell renal cell carcinoma. Cancer Cell 2021, 39, 1497–1518.e1411. [Google Scholar] [CrossRef]
- Shiao, S.L.; Gouin, K.H., 3rd; Ing, N.; Ho, A.; Basho, R.; Shah, A.; Mebane, R.H.; Zitser, D.; Martinez, A.; Mevises, N.Y.; et al. Single-cell and spatial profiling identify three response trajectories to pembrolizumab and radiation therapy in triple negative breast cancer. Cancer Cell 2024, 42, 70–84.e78. [Google Scholar] [CrossRef]
- Singh, B.; Sarli, V.N.; Lucci, A. Sensitization of Resistant Breast Cancer Cells with a Jumonji Family Histone Demethylase Inhibitor. Cancers 2022, 14, 2631. [Google Scholar] [CrossRef]
- Singh, B.; Sarli, V.N.; Milligan, R.D.; Kinne, H.E.; Shamsnia, A.; Washburn, L.J.; Addanki, S.; Lucci, A. Sensitization of Resistant Cells with a BET Bromodomain Inhibitor in a Cell Culture Model of Deep Intrinsic Resistance in Breast Cancer. Cancers 2023, 15, 2036. [Google Scholar] [CrossRef]
- Usaite, I.; Biswas, D.; Dijkstra, K.; Watkins, T.B.; Pich, O.; Puttick, C.; Angelova, M.; Thakkar, K.; Hiley, C.; Birkbak, N.; et al. Quantifying the impact of immunotherapy on RNA dynamics in cancer. J. Immunother. Cancer 2023, 11, e007870. [Google Scholar] [CrossRef]
- Xiao, Y.; Cong, M.; Li, J.; He, D.; Wu, Q.; Tian, P.; Wang, Y.; Yang, S.; Liang, C.; Liang, Y.; et al. Cathepsin C promotes breast cancer lung metastasis by modulating neutrophil infiltration and neutrophil extracellular trap formation. Cancer Cell 2021, 39, 423–437.e427. [Google Scholar] [CrossRef]
- Yang, C.; Wang, Z.; Li, L.; Zhang, Z.; Jin, X.; Wu, P.; Sun, S.; Pan, J.; Su, K.; Jia, F.; et al. Aged neutrophils form mitochondria-dependent vital NETs to promote breast cancer lung metastasis. J. Immunother. Cancer 2021, 9, e002875. [Google Scholar] [CrossRef]
- He, X.Y.; Gao, Y.; Ng, D.; Michalopoulou, E.; George, S.; Adrover, J.M.; Sun, L.; Albrengues, J.; Daßler-Plenker, J.; Han, X.; et al. Chronic stress increases metastasis via neutrophil-mediated changes to the microenvironment. Cancer Cell 2024, 42, 474–486.e412. [Google Scholar] [CrossRef]
- Pan, J.; Zhang, L.; Wang, X.; Li, L.; Yang, C.; Wang, Z.; Su, K.; Hu, X.; Zhang, Y.; Ren, G.; et al. Chronic stress induces pulmonary epithelial cells to produce acetylcholine that remodels lung pre-metastatic niche of breast cancer by enhancing NETosis. J. Exp. Clin. Cancer Res. 2023, 42, 255. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, Y.; Zhong, J.; Li, M.; Zhou, Y.; Lin, Q.; Zong, S.; Luo, W.; Wang, J.; Wang, K.; et al. Tumor-derived Cav-1 promotes pre-metastatic niche formation and lung metastasis in breast cancer. Theranostics 2023, 13, 1684–1697. [Google Scholar] [CrossRef]
- Zheng, Z.; Li, Y.N.; Jia, S.; Zhu, M.; Cao, L.; Tao, M.; Jiang, J.; Zhan, S.; Chen, Y.; Gao, P.J.; et al. Lung mesenchymal stromal cells influenced by Th2 cytokines mobilize neutrophils and facilitate metastasis by producing complement C3. Nat. Commun. 2021, 12, 6202. [Google Scholar] [CrossRef]
- Yofe, I.; Shami, T.; Cohen, N.; Landsberger, T.; Sheban, F.; Stoler-Barak, L.; Yalin, A.; Phan, T.S.; Li, B.; Monteran, L.; et al. Spatial and Temporal Mapping of Breast Cancer Lung Metastases Identify TREM2 Macrophages as Regulators of the Metastatic Boundary. Cancer Discov. 2023, 13, 2610–2631. [Google Scholar] [CrossRef]
- Zou, Y.; Ye, F.; Kong, Y.; Hu, X.; Deng, X.; Xie, J.; Song, C.; Ou, X.; Wu, S.; Wu, L.; et al. The Single-Cell Landscape of Intratumoral Heterogeneity and The Immunosuppressive Microenvironment in Liver and Brain Metastases of Breast Cancer. Adv. Sci. 2023, 10, e2203699. [Google Scholar] [CrossRef]
- Morrissey, S.M.; Zhang, F.; Ding, C.; Montoya-Durango, D.E.; Hu, X.; Yang, C.; Wang, Z.; Yuan, F.; Fox, M.; Zhang, H.G.; et al. Tumor-derived exosomes drive immunosuppressive macrophages in a pre-metastatic niche through glycolytic dominant metabolic reprogramming. Cell Metab. 2021, 33, 2040–2058.e2010. [Google Scholar] [CrossRef]
- Jiang, X.; Wang, J.; Lin, L.; Du, L.; Ding, Y.; Zheng, F.; Xie, H.; Wang, Y.; Hu, M.; Liu, B.; et al. Macrophages promote pre-metastatic niche formation of breast cancer through aryl hydrocarbon receptor activity. Signal Transduct. Target. Ther. 2024, 9, 352. [Google Scholar] [CrossRef]
- Ferrucci, V.; Asadzadeh, F.; Collina, F.; Siciliano, R.; Boccia, A.; Marrone, L.; Spano, D.; Carotenuto, M.; Chiarolla, C.M.; De Martino, D.; et al. Prune-1 drives polarization of tumor-associated macrophages (TAMs) within the lung metastatic niche in triple-negative breast cancer. iScience 2021, 24, 101938. [Google Scholar] [CrossRef]
- Huang, Z.; Bu, D.; Yang, N.; Huang, W.; Zhang, L.; Li, X.; Ding, B.S. Integrated analyses of single-cell transcriptomics identify metastasis-associated myeloid subpopulations in breast cancer lung metastasis. Front. Immunol. 2023, 14, 1180402. [Google Scholar] [CrossRef] [PubMed]
- Gong, Z.; Li, Q.; Shi, J.; Wei, J.; Li, P.; Chang, C.H.; Shultz, L.D.; Ren, G. Lung fibroblasts facilitate pre-metastatic niche formation by remodeling the local immune microenvironment. Immunity 2022, 55, 1483–1500.e1489. [Google Scholar] [CrossRef] [PubMed]
- Shani, O.; Vorobyov, T.; Monteran, L.; Lavie, D.; Cohen, N.; Raz, Y.; Tsarfaty, G.; Avivi, C.; Barshack, I.; Erez, N. Fibroblast-Derived IL33 Facilitates Breast Cancer Metastasis by Modifying the Immune Microenvironment and Driving Type 2 Immunity. Cancer Res. 2020, 80, 5317–5329. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chen, S.; Wan, X.; Wang, R.; Luo, H.; Chang, C.; Dai, P.; Gan, Y.; Guo, Y.; Hou, Y.; et al. Tryptophan 2,3-dioxygenase-positive matrix fibroblasts fuel breast cancer lung metastasis via kynurenine-mediated ferroptosis resistance of metastatic cells and T cell dysfunction. Cancer Commun. 2024, 44, 1261–1286. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Hou, Y.; Zhou, X.; Lang, L.; Luo, H.; Sun, Y.; Wan, X.; Yuan, T.; Wang, R.; Liu, Y.; et al. Cancer-associated fibroblasts facilitate premetastatic niche formation through lncRNA SNHG5-mediated angiogenesis and vascular permeability in breast cancer. Theranostics 2022, 12, 7351–7370. [Google Scholar] [CrossRef]
- Yuan, X.; Qian, N.; Ling, S.; Li, Y.; Sun, W.; Li, J.; Du, R.; Zhong, G.; Liu, C.; Yu, G.; et al. Breast cancer exosomes contribute to pre-metastatic niche formation and promote bone metastasis of tumor cells. Theranostics 2021, 11, 1429–1445. [Google Scholar] [CrossRef]
- Roberts, B.K.; Li, D.I.; Somerville, C.; Matta, B.; Jha, V.; Steinke, A.; Brune, Z.; Blanc, L.; Soffer, S.Z.; Barnes, B.J. IRF5 suppresses metastasis through the regulation of tumor-derived extracellular vesicles and pre-metastatic niche formation. Sci. Rep. 2024, 14, 15557. [Google Scholar] [CrossRef]
- González-Callejo, P.; Gener, P.; Díaz-Riascos, Z.V.; Conti, S.; Cámara-Sánchez, P.; Riera, R.; Mancilla, S.; García-Gabilondo, M.; Peg, V.; Arango, D.; et al. Extracellular vesicles secreted by triple-negative breast cancer stem cells trigger premetastatic niche remodeling and metastatic growth in the lungs. Int. J. Cancer 2023, 152, 2153–2165. [Google Scholar] [CrossRef] [PubMed]
- Duan, S.; Nordmeier, S.; Byrnes, A.E.; Buxton, I.L.O. Extracellular Vesicle-Mediated Purinergic Signaling Contributes to Host Microenvironment Plasticity and Metastasis in Triple Negative Breast Cancer. Int. J. Mol. Sci. 2021, 22, 597. [Google Scholar] [CrossRef]
- Heo, W.; Lee, W.; Cheun, J.H.; Lee, E.S.; Li, S.; Kim, H.S.; Son, H.Y.; Kim, J.H.; Woo, Y.D.; Chung, D.H.; et al. Triple-Negative Breast Cancer-Derived Extracellular Vesicles Promote a Hepatic Premetastatic Niche via a Cascade of Microenvironment Remodeling. Mol. Cancer Res. 2023, 21, 726–740. [Google Scholar] [CrossRef]
- Ikari, A.; Ito, Y.; Taniguchi, K.; Shibata, M.A.; Kimura, K.; Iwamoto, M.; Lee, S.W. Role of CD44-Positive Extracellular Vesicles Derived from Highly Metastatic Mouse Mammary Carcinoma Cells in Pre-Metastatic Niche Formation. Int. J. Mol. Sci. 2024, 25, 9742. [Google Scholar] [CrossRef]
- Yu, P.; Han, Y.; Meng, L.; Tang, Z.; Jin, Z.; Zhang, Z.; Zhou, Y.; Luo, J.; Luo, J.; Han, C.; et al. The incorporation of acetylated LAP-TGF-β1 proteins into exosomes promotes TNBC cell dissemination in lung micro-metastasis. Mol. Cancer 2024, 23, 82. [Google Scholar] [CrossRef]
- Zhang, L.; Pan, J.; Wang, M.; Yang, J.; Zhu, S.; Li, L.; Hu, X.; Wang, Z.; Pang, L.; Li, P.; et al. Chronic Stress-Induced and Tumor Derived SP1+ Exosomes Polarizing IL-1β+ Neutrophils to Increase Lung Metastasis of Breast Cancer. Adv. Sci. 2025, 12, e2310266. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Q.; Zhang, R.; Lu, H.; Yue, X.M.; Huang, Y.F. Extracellular Vesicle-Packaged CDH11 and ITGA5 Induce the Premetastatic Niche for Bone Colonization of Breast Cancer Cells. Cancer Res. 2022, 82, 1560–1574. [Google Scholar] [CrossRef]
- Bado, I.L.; Zhang, W.; Hu, J.; Xu, Z.; Wang, H.; Sarkar, P.; Li, L.; Wan, Y.W.; Liu, J.; Wu, W.; et al. The bone microenvironment increases phenotypic plasticity of ER+ breast cancer cells. Dev. Cell 2021, 56, 1100–1117.e1109. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, T.; Zheng, B.; Lu, Y.; Liang, Y.; Xu, G.; Zhao, L.; Tao, Y.; Song, Q.; You, H.; et al. Lymphotoxin-β promotes breast cancer bone metastasis colonization and osteolytic outgrowth. Nat. Cell Biol. 2024, 26, 1597–1612. [Google Scholar] [CrossRef]
- Li, X.; Liang, Y.; Lian, C.; Peng, F.; Xiao, Y.; He, Y.; Ma, C.; Wang, Y.; Zhang, P.; Deng, Y.; et al. CST6 protein and peptides inhibit breast cancer bone metastasis by suppressing CTSB activity and osteoclastogenesis. Theranostics 2021, 11, 9821–9832. [Google Scholar] [CrossRef] [PubMed]
- Yue, Z.; Niu, X.; Yuan, Z.; Qin, Q.; Jiang, W.; He, L.; Gao, J.; Ding, Y.; Liu, Y.; Xu, Z.; et al. RSPO2 and RANKL signal through LGR4 to regulate osteoclastic premetastatic niche formation and bone metastasis. J. Clin. Invest. 2022, 132, e144579. [Google Scholar] [CrossRef]
- Yip, R.K.H.; Rimes, J.S.; Capaldo, B.D.; Vaillant, F.; Mouchemore, K.A.; Pal, B.; Chen, Y.; Surgenor, E.; Murphy, A.J.; Anderson, R.L.; et al. Mammary tumour cells remodel the bone marrow vascular microenvironment to support metastasis. Nat. Commun. 2021, 12, 6920. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Xu, Z.; Hao, X.; He, T.; Li, J.; Shen, Y.; Liu, K.; Gao, Y.; Liu, J.; Edwards, D.G.; et al. Bone Metastasis Initiation Is Coupled with Bone Remodeling through Osteogenic Differentiation of NG2+ Cells. Cancer Discov. 2023, 13, 474–495. [Google Scholar] [CrossRef]
- Baldassarri, I.; Tavakol, D.N.; Graney, P.L.; Chramiec, A.G.; Hibshoosh, H.; Vunjak-Novakovic, G. An engineered model of metastatic colonization of human bone marrow reveals breast cancer cell remodeling of the hematopoietic niche. Proc. Natl. Acad. Sci. USA 2024, 121, e2405257121. [Google Scholar] [CrossRef]
- Wirth, F.; Zoeller, C.; Lubosch, A.; Schroeder-Braunstein, J.; Wabnitz, G.; Nakchbandi, I.A. Insights into the metastatic bone marrow niche gained from fibronectin and β1 integrin transgenic mice. Neoplasia 2024, 58, 101058. [Google Scholar] [CrossRef]
- Ghiaur, G.; Valkenburg, K.C.; Esteb, C.; Ambinder, A.; Imus, P.H.; Pienta, K.J.; Jones, R.J. Bone marrow niche chemoprotection of metastatic solid tumors mediated by CYP3A4. Cancer 2023, 129, 1744–1751. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Gao, Z.; Yang, M.; Yang, C.; Yang, D.; Cui, W.; Wu, D.; Zhou, J. Unraveling the metastatic niche in breast cancer bone metastasis through single-cell RNA sequencing. Clin. Transl. Oncol. 2025, 27, 671–686. [Google Scholar] [CrossRef]
- Colombo, M.V.; Bersini, S.; Arrigoni, C.; Gilardi, M.; Sansoni, V.; Ragni, E.; Candiani, G.; Lombardi, G.; Moretti, M. Engineering the early bone metastatic niche through human vascularized immuno bone minitissues. Biofabrication 2021, 13, 035036. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Wang, N.; Wang, S.; Zhang, J.; Yang, B.; Wang, Z. Chronic psychological stress promotes breast cancer pre-metastatic niche formation by mobilizing splenic MDSCs via TAM/CXCL1 signaling. J. Exp. Clin. Cancer Res. 2023, 42, 129. [Google Scholar] [CrossRef]
- Hergueta-Redondo, M.; Sánchez-Redondo, S.; Hurtado, B.; Santos, V.; Pérez-Martínez, M.; Ximénez-Embún, P.; McDowell, S.A.C.; Mazariegos, M.S.; Mata, G.; Torres-Ruiz, R.; et al. The impact of a high fat diet and platelet activation on pre-metastatic niche formation. Nat. Commun. 2025, 16, 2897. [Google Scholar] [CrossRef]
- Theivanthiran, B.; Yarla, N.; Haykal, T.; Nguyen, Y.V.; Cao, L.; Ferreira, M.; Holtzhausen, A.; Al-Rohil, R.; Salama, A.K.S.; Beasley, G.M.; et al. Tumor-intrinsic NLRP3-HSP70-TLR4 axis drives premetastatic niche development and hyperprogression during anti-PD-1 immunotherapy. Sci. Transl. Med. 2022, 14, eabq7019. [Google Scholar] [CrossRef]
- Kim, S.; Oh, J.; Park, C.; Kim, M.; Jo, W.; Kim, C.S.; Cho, S.W.; Park, J. FAM3C in Cancer-Associated Adipocytes Promotes Breast Cancer Cell Survival and Metastasis. Cancer Res. 2024, 84, 545–559. [Google Scholar] [CrossRef] [PubMed]
- Kan, J.Y.; Lee, H.C.; Hou, M.F.; Tsai, H.P.; Jian, S.F.; Chang, C.Y.; Tsai, P.H.; Lin, Y.S.; Tsai, Y.M.; Wu, K.L.; et al. Metabolic shifts in lipid utilization and reciprocal interactions within the lung metastatic niche of triple-negative breast cancer revealed by spatial multi-omics. Cell Death Dis. 2024, 15, 899. [Google Scholar] [CrossRef]
- McGinnis, C.S.; Miao, Z.; Reticker-Flynn, N.E.; Winker, J.; Satpathy, A.T. The temporal progression of immune remodeling during metastasis. bioRxiv 2023. [Google Scholar] [CrossRef]
- McGinnis, C.S.; Miao, Z.; Superville, D.; Yao, W.; Goga, A.; Reticker-Flynn, N.E.; Winkler, J.; Satpathy, A.T. The temporal progression of lung immune remodeling during breast cancer metastasis. Cancer Cell 2024, 42, 1018–1031.e1016. [Google Scholar] [CrossRef]
- Zuo, Q.; Mogol, A.N.; Liu, Y.J.; Santaliz Casiano, A.; Chien, C.; Drnevich, J.; Imir, O.B.; Kulkoyluoglu-Cotul, E.; Park, N.H.; Shapiro, D.J.; et al. Targeting Metabolic Adaptations in the Breast Cancer-Liver Metastatic Niche Using Dietary Approaches to Improve Endocrine Therapy Efficacy. Mol. Cancer Res. 2022, 20, 923–937. [Google Scholar] [CrossRef]
- Gui, J.; Zahedi, F.; Ortiz, A.; Cho, C.; Katlinski, K.V.; Alicea-Torres, K.; Li, J.; Todd, L.; Zhang, H.; Beiting, D.P.; et al. Activation of p38α stress-activated protein kinase drives the formation of the pre-metastatic niche in the lungs. Nat. Cancer 2020, 1, 603–619. [Google Scholar] [CrossRef] [PubMed]
- Xia, C.; Bai, W.; Deng, T.; Li, T.; Zhang, L.; Lu, Z.; Zhang, Z.; Li, M.; He, Q. Sponge-like nano-system suppresses tumor recurrence and metastasis by restraining myeloid-derived suppressor cells-mediated immunosuppression and formation of pre-metastatic niche. Acta Biomater. 2023, 158, 708–724. [Google Scholar] [CrossRef] [PubMed]
- Orbach, S.M.; Brooks, M.D.; Zhang, Y.; Campit, S.E.; Bushnell, G.G.; Decker, J.T.; Rebernick, R.J.; Chandrasekaran, S.; Wicha, M.S.; Jeruss, J.S.; et al. Single-cell RNA-sequencing identifies anti-cancer immune phenotypes in the early lung metastatic niche during breast cancer. Clin. Exp. Metastasis 2022, 39, 865–881. [Google Scholar] [CrossRef] [PubMed]
- Orbach, S.M.; DeVaull, C.Y.; Bealer, E.J.; Ross, B.C.; Jeruss, J.S.; Shea, L.D. An engineered niche delineates metastatic potential of breast cancer. Bioeng. Transl. Med. 2024, 9, e10606. [Google Scholar] [CrossRef]
- Haq, A.T.A.; Yang, P.P.; Jin, C.; Shih, J.H.; Chen, L.M.; Tseng, H.Y.; Chen, Y.A.; Weng, Y.S.; Wang, L.H.; Snyder, M.P.; et al. Immunotherapeutic IL-6R and targeting the MCT-1/IL-6/CXCL7/PD-L1 circuit prevent relapse and metastasis of triple-negative breast cancer. Theranostics 2024, 14, 2167–2189. [Google Scholar] [CrossRef]
- Clark, A.M.; Heusey, H.L.; Griffith, L.G.; Lauffenburger, D.A.; Wells, A. IP-10 (CXCL10) Can Trigger Emergence of Dormant Breast Cancer Cells in a Metastatic Liver Microenvironment. Front. Oncol. 2021, 11, 676135. [Google Scholar] [CrossRef]
- Ruiu, R.; Cossu, C.; Iacoviello, A.; Conti, L.; Bolli, E.; Ponzone, L.; Magri, J.; Rumandla, A.; Calautti, E.; Cavallo, F. Cystine/glutamate antiporter xCT deficiency reduces metastasis without impairing immune system function in breast cancer mouse models. J. Exp. Clin. Cancer Res. 2023, 42, 254. [Google Scholar] [CrossRef]
- Pham, S.H.; Pratt, K.; Okolicsanyi, R.K.; Oikari, L.E.; Yu, C.; Peall, I.W.; Arif, K.T.; Chalmers, T.A.; Gyimesi, M.; Griffiths, L.R.; et al. Syndecan-1 and -4 influence Wnt signaling and cell migration in human breast cancers. Biochimie 2022, 198, 60–75. [Google Scholar] [CrossRef]
- Wang, X.; Wang, Z.; Liao, Q.; Yuan, P.; Mei, J.; Zhang, Y.; Wu, C.; Kang, X.; Zheng, S.; Yang, C.; et al. Spatially resolved atlas of breast cancer uncovers intercellular machinery of venular niche governing lymphocyte extravasation. Nat. Commun. 2025, 16, 3348. [Google Scholar] [CrossRef]
- Zhao, Z.; Zhang, Y.; Li, C.; Li, X.; Chu, Y.; Guo, Q.; Zhang, Y.; Xia, W.; Liu, P.; Chen, H.; et al. Microenvironment-tailored micelles restrain carcinoma-astrocyte crosstalk for brain metastasis. J. Control Release 2022, 349, 520–532. [Google Scholar] [CrossRef]
- Ahuja, S.; Lazar, I.M. Proteomic insights into breast cancer response to brain cell-secreted factors. Sci. Rep. 2024, 14, 19351. [Google Scholar] [CrossRef] [PubMed]
- Mondal, J.; Zhang, J.; Qing, F.; Li, S.; Kumar, D.; Huse, J.T.; Giancotti, F.G. Brd7 loss reawakens dormant metastasis initiating cells in lung by forging an immunosuppressive niche. Nat. Commun. 2025, 16, 1378. [Google Scholar] [CrossRef]
- Xia, J.; Ma, S.; Zhu, X.; Chen, C.; Zhang, R.; Cao, Z.; Chen, X.; Zhang, L.; Zhu, Y.; Zhang, S.; et al. Versatile ginsenoside Rg3 liposomes inhibit tumor metastasis by capturing circulating tumor cells and destroying metastatic niches. Sci. Adv. 2022, 8, eabj1262. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Ai, H.; Xi, Y.; Tan, J.; Qu, Y.; Xu, J.; Luo, F.; Dou, C. Osteoclast-derived apoptotic bodies inhibit naive CD8+ T cell activation via Siglec15, promoting breast cancer secondary metastasis. Cell Rep. Med. 2023, 4, 101165. [Google Scholar] [CrossRef] [PubMed]
- Crippa, M.; Bersini, S.; Gilardi, M.; Arrigoni, C.; Gamba, S.; Falanga, A.; Candrian, C.; Dubini, G.; Vanoni, M.; Moretti, M. A microphysiological early metastatic niche on a chip reveals how heterotypic cell interactions and inhibition of integrin subunit β(3) impact breast cancer cell extravasation. Lab. Chip 2021, 21, 1061–1072. [Google Scholar] [CrossRef]
- DeCastro, A.J.L.; Pranda, M.A.; Gray, K.M.; Merlo-Coyne, J.; Girma, N.; Hurwitz, M.; Zhang, Y.; Stroka, K.M. Morphological Phenotyping of Organotropic Brain- and Bone-Seeking Triple Negative Metastatic Breast Tumor Cells. Front. Cell Dev. Biol. 2022, 10, 790410. [Google Scholar] [CrossRef]
- Rimal, R.; Desai, P.; Marquez, A.B.; Sieg, K.; Marquardt, Y.; Singh, S. 3-D vascularized breast cancer model to study the role of osteoblast in formation of a pre-metastatic niche. Sci. Rep. 2021, 11, 21966. [Google Scholar] [CrossRef]
- Sterle, H.A.; Hildebrandt, X.; Valenzuela Álvarez, M.; Paulazo, M.A.; Gutierrez, L.M.; Klecha, A.J.; Cayrol, F.; Díaz Flaqué, M.C.; Rosemblit, C.; Barreiro Arcos, M.L.; et al. Thyroid status regulates the tumor microenvironment delineating breast cancer fate. Endocr. Relat. Cancer 2021, 28, 403–418. [Google Scholar] [CrossRef]
- Li, R.; Qi, Y.; Han, M.; Geng, B.; Wang, G.; Han, M. Computed tomography reveals microenvironment changes in premetastatic lung. Eur. Radiol. 2021, 31, 4340–4349. [Google Scholar] [CrossRef]
- Lu, Z.; Liu, H.; Ma, L.; Ren, K.; He, Z.; Li, M.; He, Q. Micellar nanoparticles inhibit breast cancer and pulmonary metastasis by modulating the recruitment and depletion of myeloid-derived suppressor cells. Nanoscale 2022, 14, 17315–17330. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ge, Z.; Toh, K.; Liu, X.; Dirisala, A.; Ke, W.; Wen, P.; Zhou, H.; Wang, Z.; Xiao, S.; et al. Enzymatically Transformable Polymersome-Based Nanotherapeutics to Eliminate Minimal Relapsable Cancer. Adv. Mater. 2021, 33, e2105254. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.H.; Park, J.; Cai, H.; Steinmetz, N.F. S100A9-Targeted Cowpea Mosaic Virus as a Prophylactic and Therapeutic Immunotherapy against Metastatic Breast Cancer and Melanoma. Adv. Sci. 2021, 8, e2101796. [Google Scholar] [CrossRef] [PubMed]
- Parida, S.; Siddharth, S.; Gatla, H.R.; Wu, S.; Wang, G.; Gabrielson, K.; Sears, C.L.; Ladle, B.H.; Sharma, D. Gut colonization with an obesity-associated enteropathogenic microbe modulates the premetastatic niches to promote breast cancer lung and liver metastasis. Front. Immunol. 2023, 14, 1194931. [Google Scholar] [CrossRef]
- Chen, J.; Hou, S.; Liang, Q.; He, W.; Li, R.; Wang, H.; Zhu, Y.; Zhang, B.; Chen, L.; Dai, X.; et al. Localized Degradation of Neutrophil Extracellular Traps by Photoregulated Enzyme Delivery for Cancer Immunotherapy and Metastasis Suppression. ACS Nano. 2022, 16, 2585–2597. [Google Scholar] [CrossRef]
- Gallanis, G.T.; Sharif, G.M.; Schmidt, M.O.; Friedland, B.N.; Battina, R.; Rahhal, R.; Davis, J.E., Jr.; Khan, I.S.; Wellstein, A.; Riegel, A.T. Stromal Senescence following Treatment with the CDK4/6 Inhibitor Palbociclib Alters the Lung Metastatic Niche and Increases Metastasis of Drug-Resistant Mammary Cancer Cells. Cancers 2023, 15, 1908. [Google Scholar] [CrossRef]
- Chen, L.; Zheng, H.; Yu, X.; Liu, L.; Li, H.; Zhu, H.; Zhang, Z.; Lei, P.; Shen, G. Tumor-Secreted GRP78 Promotes the Establishment of a Pre-metastatic Niche in the Liver Microenvironment. Front. Immunol. 2020, 11, 584458. [Google Scholar] [CrossRef]
- Oskarsson, T. Stress-induced metastatic niches in breast cancer. Mol. Cell Oncol. 2020, 7, 1780105. [Google Scholar] [CrossRef]
- Santonja, Á.; Moya-García, A.A.; Ribelles, N.; Jiménez-Rodríguez, B.; Pajares, B.; Fernández-De Sousa, C.E.; Pérez-Ruiz, E.; Del Monte-Millán, M.; Ruiz-Borrego, M.; de la Haba, J.; et al. Role of germline variants in the metastasis of breast carcinomas. Oncotarget 2022, 13, 843–862. [Google Scholar] [CrossRef]
- Yao, Y.; Qian, R.; Gao, H.; Dai, Y.; Shi, Y.; An, P.; Xin, B.; Liu, Z.; Zhang, N.; Wan, Y.; et al. LSD1 deficiency in breast cancer cells promotes the formation of pre-metastatic niches. NPJ Precis. Oncol. 2024, 8, 260. [Google Scholar] [CrossRef]
- Corthay, A.; Bakacs, T.; Thangavelu, G.; Anderson, C.C. Tackling cancer cell dormancy: Insights from immune models, and transplantation. Semin. Cancer Biol. 2022, 78, 5–16. [Google Scholar] [CrossRef]
- Tamamouna, V.; Pavlou, E.; Neophytou, C.M.; Papageorgis, P.; Costeas, P. Regulation of Metastatic Tumor Dormancy and Emerging Opportunities for Therapeutic Intervention. Int. J. Mol. Sci. 2022, 23, 13931. [Google Scholar] [CrossRef] [PubMed]
- Fidelle, M.; Rauber, C.; Alves Costa Silva, C.; Tian, A.L.; Lahmar, I.; de La Varende, A.M.; Zhao, L.; Thelemaque, C.; Lebhar, I.; Messaoudene, M.; et al. A microbiota-modulated checkpoint directs immunosuppressive intestinal T cells into cancers. Science 2023, 380, eabo2296. [Google Scholar] [CrossRef] [PubMed]
- Koncina, E.; Nurmik, M.; Pozdeev, V.I.; Gilson, C.; Tsenkova, M.; Begaj, R.; Stang, S.; Gaigneaux, A.; Weindorfer, C.; Rodriguez, F.; et al. IL1R1+ cancer-associated fibroblasts drive tumor development and immunosuppression in colorectal cancer. Nat. Commun. 2023, 14, 4251. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Fang, W.; Zhou, S.; Zhu, D.; Chen, R.; Gao, X.; Li, Z.; Fu, Y.; Zhang, Y.; Yang, F.; et al. Single cell transcriptomic analyses implicate an immunosuppressive tumor microenvironment in pancreatic cancer liver metastasis. Nat. Commun. 2023, 14, 5123. [Google Scholar] [CrossRef]
- Yu, Y.; Cao, W.M.; Cheng, F.; Shi, Z.; Han, L.; Yi, J.; da Silva, E.M.; Dopeso, H.; Chen, H.; Yang, J.; et al. FOXK2 amplification promotes breast cancer development and chemoresistance. Cancer Lett. 2024, 597, 217074. [Google Scholar] [CrossRef]
- Nobre, A.R.; Risson, E.; Singh, D.K.; Di Martino, J.S.; Cheung, J.F.; Wang, J.; Johnson, J.; Russnes, H.G.; Bravo-Cordero, J.J.; Birbrair, A.; et al. Bone marrow NG2(+)/Nestin(+) mesenchymal stem cells drive DTC dormancy via TGFβ2. Nat. Cancer 2021, 2, 327–339. [Google Scholar] [CrossRef]
- De Angelis, M.L.; Francescangeli, F.; Aricò, E.; Verachi, P.; Zucchetti, M.; Matteo, C.; Petricci, E.; Pilozzi, E.; Orienti, I.; Boe, A.; et al. A nanoencapsulated oral formulation of fenretinide promotes local and metastatic breast cancer dormancy in HER2/neu transgenic mice. J. Exp. Clin. Cancer Res. 2024, 43, 296. [Google Scholar] [CrossRef] [PubMed]
- Hughes, R.; Chen, X.; Cowley, N.; Ottewell, P.D.; Hawkins, R.J.; Hunter, K.D.; Hobbs, J.K.; Brown, N.J.; Holen, I. Osteoblast-Derived Paracrine and Juxtacrine Signals Protect Disseminated Breast Cancer Cells from Stress. Cancers 2021, 13, 1366. [Google Scholar] [CrossRef]
- Ionkina, A.A.; Balderrama-Gutierrez, G.; Ibanez, K.J.; Phan, S.H.D.; Cortez, A.N.; Mortazavi, A.; Prescher, J.A. Transcriptome analysis of heterogeneity in mouse model of metastatic breast cancer. Breast Cancer Res. 2021, 23, 93. [Google Scholar] [CrossRef]
- De Martino, D.; Megino-Luque, C.; Bravo-Cordero, J.J. In Vivo Xenograft Model to Study Tumor Dormancy, Tumor Cell Dissemination and Metastasis. Methods Mol. Biol. 2024, 2811, 81–100. [Google Scholar] [CrossRef] [PubMed]
- Smit, D.J.; Hoffer, K.; Bettin, B.; Kriegs, M.; Cayrefourcq, L.; Schumacher, U.; Pantel, K.; Alix-Panabières, C.; Jücker, M. Analysis of the Plasticity of Circulating Tumor Cells Reveals Differentially Regulated Kinases During the Suspension-to-Adherent Transition. Cancer Med. 2024, 13, e70339. [Google Scholar] [CrossRef] [PubMed]
- Demicheli, R.; Desmedt, C.; Retsky, M.; Sotiriou, C.; Piccart, M.; Biganzoli, E. Late effects of adjuvant chemotherapy adumbrate dormancy complexity in breast cancer. Breast 2020, 52, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, P.; Ghoreyshi, Z.S.; George, J.T. How modulation of the tumor microenvironment drives cancer immune escape dynamics. Sci. Rep. 2025, 15, 7308. [Google Scholar] [CrossRef]
- Kordon, E.; Lanari, C.; Mando, P.; Novaro, V.; Rossi, M.; Simian, M. The BA-BCS 2021: An Initial “Trial” for Integrating Basic Science and Medical Progress on Breast Cancer in a Latin-American Country. J. Mammary Gland. Biol. Neoplasia 2021, 26, 227–234. [Google Scholar] [CrossRef]
- Koelsch, N.; Mirshahi, F.; Aqbi, H.F.; Seneshaw, M.; Idowu, M.O.; Olex, A.L.; Sanyal, A.J.; Manjili, M.H. Anti-Tumour Immunity Relies on Targeting Tissue Homeostasis Through Monocyte-Driven Responses Rather Than Direct Tumour Cytotoxicity. Liver Int. 2025, 45, e70110. [Google Scholar] [CrossRef]
- Hampsch, R.A.; Wells, J.D.; Traphagen, N.A.; McCleery, C.F.; Fields, J.L.; Shee, K.; Dillon, L.M.; Pooler, D.B.; Lewis, L.D.; Demidenko, E.; et al. AMPK Activation by Metformin Promotes Survival of Dormant ER+ Breast Cancer Cells. Clin. Cancer Res. 2020, 26, 3707–3719. [Google Scholar] [CrossRef]
- Roy, R.; Yang, J.; Shimura, T.; Merritt, L.; Alluin, J.; Man, E.; Daisy, C.; Aldakhlallah, R.; Dillon, D.; Pories, S.; et al. Escape from breast tumor dormancy: The convergence of obesity and menopause. Proc. Natl. Acad. Sci. USA 2022, 119, e2204758119. [Google Scholar] [CrossRef]
- Pascual, J.; Gil-Gil, M.; Proszek, P.; Zielinski, C.; Reay, A.; Ruiz-Borrego, M.; Cutts, R.; Ciruelos Gil, E.M.; Feber, A.; Muñoz-Mateu, M.; et al. Baseline Mutations and ctDNA Dynamics as Prognostic and Predictive Factors in ER-Positive/HER2-Negative Metastatic Breast Cancer Patients. Clin. Cancer Res. 2023, 29, 4166–4177. [Google Scholar] [CrossRef]
- Huebner, H.; Wimberger, P.; Laakmann, E.; Ruckhäberle, E.; Ruebner, M.; Lehle, S.; Uhrig, S.; Ziegler, P.; Link, T.; Hack, C.C.; et al. Cell-free tumor DNA analysis in advanced or metastatic breast cancer patients: Mutation frequencies, testing intention, and clinical impact. Precis. Clin. Med. 2025, 8, pbae034. [Google Scholar] [CrossRef]
- Fitzpatrick, A.; Iravani, M.; Mills, A.; Childs, L.; Alaguthurai, T.; Clifford, A.; Garcia-Murillas, I.; Van Laere, S.; Dirix, L.; Harries, M.; et al. Assessing CSF ctDNA to Improve Diagnostic Accuracy and Therapeutic Monitoring in Breast Cancer Leptomeningeal Metastasis. Clin. Cancer Res. 2022, 28, 1180–1191. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Rodríguez, A.; Fuentes-Antrás, J.; Lorca, V.; López de Sá, A.; Pérez-Segura, P.; Moreno, F.; García-Sáenz, J.A.; García-Barberán, V. Molecular Profiling of Endocrine Resistance in HR+/HER2-Metastatic Breast Cancer: Insights from Extracellular Vesicles-Derived DNA and ctDNA in Liquid Biopsies. Int. J. Mol. Sci. 2024, 25, 13045. [Google Scholar] [CrossRef]
- Liao, H.; Zhang, J.; Zheng, T.; Liu, X.; Zhong, J.; Shao, B.; Dong, X.; Wang, X.; Du, P.; King, B.L.; et al. Identification of mutation patterns and circulating tumour DNA-derived prognostic markers in advanced breast cancer patients. J. Transl. Med. 2022, 20, 211. [Google Scholar] [CrossRef]
- Zhou, Q.; Gampenrieder, S.P.; Frantal, S.; Rinnerthaler, G.; Singer, C.F.; Egle, D.; Pfeiler, G.; Bartsch, R.; Wette, V.; Pichler, A.; et al. Persistence of ctDNA in Patients with Breast Cancer During Neoadjuvant Treatment Is a Significant Predictor of Poor Tumor Response. Clin. Cancer Res. 2022, 28, 697–707. [Google Scholar] [CrossRef]
- Kindt, C.K.; Alves, C.L.; Ehmsen, S.; Kragh, A.; Reinert, T.; Vogsen, M.; Kodahl, A.R.; Rønlev, J.D.; Ardik, D.; Sørensen, A.L.; et al. Genomic alterations associated with resistance and circulating tumor DNA dynamics for early detection of progression on CDK4/6 inhibitor in advanced breast cancer. Int. J. Cancer 2024, 155, 2211–2222. [Google Scholar] [CrossRef] [PubMed]
- Migliaccio, I.; Romagnoli, D.; Galardi, F.; De Luca, F.; Biagioni, C.; Curigliano, G.; Criscitiello, C.; Minisini, A.M.; Moretti, E.; Risi, E.; et al. Mutational Analysis of Circulating Tumor DNA in Patients With Estrogen Receptor-Positive/Human Epidermal Growth Factor Receptor 2-Negative Advanced Breast Cancer Receiving Palbociclib: Results From the TREnd Trial. JCO Precis. Oncol. 2024, 8, e2300285. [Google Scholar] [CrossRef] [PubMed]
- Mouhanna, P.; Ståhlberg, A.; Andersson, D.; Albu-Kareem, A.; Elinder, E.; Eriksson, O.; Kavanagh, A.; Kovács, A.; Larsson, K.F.; Linderholm, B.; et al. Integration of personalised ultrasensitive ctDNA monitoring of patients with metastatic breast cancer to reduce imaging requirements. Int. J. Cancer 2025, 156, 1509–1517. [Google Scholar] [CrossRef]
- Scandino, R.; Nardone, A.; Casiraghi, N.; Galardi, F.; Genovese, M.; Romagnoli, D.; Paoli, M.; Biagioni, C.; Tonina, A.; Migliaccio, I.; et al. Enabling sensitive and precise detection of ctDNA through somatic copy number aberrations in breast cancer. NPJ Breast Cancer 2025, 11, 25. [Google Scholar] [CrossRef] [PubMed]
- Martens, G.A.; Demol, J.; Dedeurwaerdere, F.; De Smet, K.; Wesolowski, J.; De Smet, D. Surveillance of Disease Progression in Metastatic Breast Cancer by Molecular Counting of Circulating Tumor DNA Using Plasma-SeqSensei Breast Cancer in Vitro Diagnostics Assay. J. Mol. Diagn. 2025, 27, 25–35. [Google Scholar] [CrossRef]
- Vannas, C.; Escobar, M.; Österlund, T.; Andersson, D.; Mouhanna, P.; Soomägi, A.; Molin, C.; Wennergren, D.; Fagman, H.; Ståhlberg, A. Treatment Monitoring of a Patient with Synchronous Metastatic Angiosarcoma and Breast Cancer Using ctDNA. Int. J. Mol. Sci. 2024, 25, 4023. [Google Scholar] [CrossRef]
- Gao, Y.; Zhao, H.; An, K.; Liu, Z.; Hai, L.; Li, R.; Zhou, Y.; Zhao, W.; Jia, Y.; Wu, N.; et al. Whole-genome bisulfite sequencing analysis of circulating tumour DNA for the detection and molecular classification of cancer. Clin. Transl. Med. 2022, 12, e1014. [Google Scholar] [CrossRef]
- Hai, L.; Li, L.; Liu, Z.; Tong, Z.; Sun, Y. Whole-genome circulating tumor DNA methylation landscape reveals sensitive biomarkers of breast cancer. MedComm 2022, 3, e134. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Zhang, Y.; Ren, Q.; Wang, X.; Zhu, J.; Yin, F.; Li, Z.; Zhang, M. Tetrahedral DNA nanostructure based biosensor for high-performance detection of circulating tumor DNA using all-carbon nanotube transistor. Biosens. Bioelectron. 2022, 197, 113785. [Google Scholar] [CrossRef]
- Sunami, K.; Bando, H.; Yatabe, Y.; Naito, Y.; Takahashi, H.; Tsuchihara, K.; Toyooka, S.; Mimori, K.; Kohsaka, S.; Uetake, H.; et al. Appropriate use of cancer comprehensive genome profiling assay using circulating tumor DNA. Cancer Sci. 2021, 112, 3911–3917. [Google Scholar] [CrossRef] [PubMed]
- Szostakowska-Rodzos, M.; Grzybowska, E.A.; Mysliwy, I.; Zub, R.; Jagiello-Gruszfeld, A.; Rubach, M.; Konieczna, A.; Fabisiewicz, A. The Combined Assessment of CTC and ESR1 Status in Liquid Biopsy Samples Enhances the Clinical Value of Prediction in Metastatic Breast Cancer. Int. J. Mol. Sci. 2025, 26, 2038. [Google Scholar] [CrossRef]
- Cani, A.K.; Dolce, E.M.; Darga, E.P.; Hu, K.; Liu, C.J.; Pierce, J.; Bradbury, K.; Kilgour, E.; Aung, K.; Schiavon, G.; et al. Serial monitoring of genomic alterations in circulating tumor cells of ER-positive/HER2-negative advanced breast cancer: Feasibility of precision oncology biomarker detection. Mol. Oncol. 2022, 16, 1969–1985. [Google Scholar] [CrossRef] [PubMed]
- Ruhen, O.; Mirzai, B.; Clark, M.E.; Nguyen, B.; Salomon, C.; Erber, W.; Meehan, K. Comparison of Circulating Tumour DNA and Extracellular Vesicle DNA by Low-Pass Whole-Genome Sequencing Reveals Molecular Drivers of Disease in a Breast Cancer Patient. Biomedicines 2020, 9, 14. [Google Scholar] [CrossRef]
- Weber, Z.T.; Collier, K.A.; Tallman, D.; Forman, J.; Shukla, S.; Asad, S.; Rhoades, J.; Freeman, S.; Parsons, H.A.; Williams, N.O.; et al. Modeling clonal structure over narrow time frames via circulating tumor DNA in metastatic breast cancer. Genome Med. 2021, 13, 89. [Google Scholar] [CrossRef]
- Yang, W.; You, N.; Jia, M.; Yeung, S.J.; Ou, W.; Yu, M.; Wang, Y.; Fu, X.; Zhang, Z.; Yang, J.; et al. Undetectable circulating tumor DNA levels correlate with low risk of recurrence/metastasis in postoperative pathologic stage I lung adenocarcinoma patients. Lung Cancer 2020, 146, 327–334. [Google Scholar] [CrossRef]
- Liu, X.; Davis, A.A.; Xie, F.; Gui, X.; Chen, Y.; Zhang, Q.; Gerratana, L.; Zhang, Y.; Shah, A.N.; Behdad, A.; et al. Cell-free DNA comparative analysis of the genomic landscape of first-line hormone receptor-positive metastatic breast cancer from the US and China. Breast Cancer Res. Treat. 2021, 190, 213–226. [Google Scholar] [CrossRef]
- Suppan, C.; Graf, R.; Jahn, S.; Zhou, Q.; Klocker, E.V.; Bartsch, R.; Terbuch, A.; Kashofer, K.; Regitnig, P.; Lindenmann, J.; et al. Sensitive and robust liquid biopsy-based detection of PIK3CA mutations in hormone-receptor-positive metastatic breast cancer patients. Br. J. Cancer 2022, 126, 456–463. [Google Scholar] [CrossRef]
- Fontana, E.; Rosen, E.; Lee, E.K.; Højgaard, M.; Mettu, N.B.; Lheureux, S.; Carneiro, B.A.; Cote, G.M.; Carter, L.; Plummer, R.; et al. Ataxia telangiectasia and Rad3-related (ATR) inhibitor camonsertib dose optimization in patients with biomarker-selected advanced solid tumors (TRESR study). J. Natl. Cancer Inst. 2024, 116, 1439–1449. [Google Scholar] [CrossRef] [PubMed]
- Prasath, V.; Boutrid, H.; Wesolowski, R.; Abdel-Rasoul, M.; Timmers, C.; Lustberg, M.; Layman, R.M.; Macrae, E.; Mrozek, E.; Shapiro, C.; et al. Phase II study of MEK inhibitor trametinib alone and in combination with AKT inhibitor GSK2141795/uprosertib in patients with metastatic triple negative breast cancer. Breast Cancer Res. Treat. 2025, 210, 179–189. [Google Scholar] [CrossRef]
- Hu, N.; Si, Y.; Yue, J.; Sun, T.; Wang, X.; Jia, Z.; Gao, S.; Li, Q.; Shao, Y.; Wang, J.; et al. Anlotinib has good efficacy and low toxicity: A phase II study of anlotinib in pre-treated HER-2 negative metastatic breast cancer. Cancer Biol. Med. 2021, 18, 849–859. [Google Scholar] [CrossRef] [PubMed]
- Brasó-Maristany, F.; Sansó, M.; Chic, N.; Martínez, D.; González-Farré, B.; Sanfeliu, E.; Ghiglione, L.; Carcelero, E.; Garcia-Corbacho, J.; Sánchez, M.; et al. Case Report: A Case Study Documenting the Activity of Atezolizumab in a PD-L1-Negative Triple-Negative Breast Cancer. Front. Oncol. 2021, 11, 710596. [Google Scholar] [CrossRef]
- Wang, J.; Liu, Y.; Liang, Y.; Zhang, Y.; Dong, H.; Zheng, T.; Yu, J.; Du, P.; Jia, S.; King, B.L.; et al. Clinicopathologic features, genomic profiles and outcomes of younger vs. older Chinese hormone receptor-positive (HR+)/HER2-negative (HER2-) metastatic breast cancer patients. Front. Oncol. 2023, 13, 1152575. [Google Scholar] [CrossRef] [PubMed]
- Kudo, R.; Safonov, A.; Jones, C.; Moiso, E.; Dry, J.R.; Shao, H.; Nag, S.; da Silva, E.M.; Yildirim, S.Y.; Li, Q.; et al. Long-term breast cancer response to CDK4/6 inhibition defined by TP53-mediated geroconversion. Cancer Cell 2024, 42, 1919–1935.e1919. [Google Scholar] [CrossRef]
- Cha, Y.J.; O’Connell, C.E.; Calhoun, B.C.; Felsheim, B.M.; Fernandez-Martinez, A.; Fan, C.; Brueffer, C.; Larsson, C.; Borg, Å.; Saal, L.H.; et al. Genomic Characteristics Related to Histology-Based Immune Features in Breast Cancer. Mod. Pathol. 2025, 38, 100736. [Google Scholar] [CrossRef]
- Ye, Y.; Wang, J.; Izban, M.G.; Ballard, B.R.; Barsky, S.H. Initiation of tumor dormancy by the lymphovascular embolus. Oncotarget 2024, 15, 726–740. [Google Scholar] [CrossRef]
- Dalla, E.; Papanicolaou, M.; Park, M.D.; Barth, N.; Hou, R.; Segura-Villalobos, D.; Valencia Salazar, L.; Sun, D.; Forrest, A.R.R.; Casanova-Acebes, M.; et al. Lung-resident alveolar macrophages regulate the timing of breast cancer metastasis. Cell 2024, 187, 6631–6648.e6620. [Google Scholar] [CrossRef]
- Lenart, N.A.; Rao, S.S. Cell-cell interactions mediating primary and metastatic breast cancer dormancy. Cancer Metastasis Rev. 2024, 44, 6. [Google Scholar] [CrossRef]
- Cackowski, F.C.; Korkaya, H. Breast Cancer Disseminated Tumor Cells: Do They Stay and Fight or Run and Hide? Cancer Res. 2024, 84, 3319–3321. [Google Scholar] [CrossRef]
- Garcia-Murillas, I.; Cutts, R.J.; Walsh-Crestani, G.; Phillips, E.; Hrebien, S.; Dunne, K.; Sidhu, K.; Daber, R.; Hubert, B.; Graybill, C.; et al. Longitudinal monitoring of circulating tumor DNA to detect relapse early and predict outcome in early breast cancer. Breast Cancer Res. Treat. 2025, 209, 493–502. [Google Scholar] [CrossRef]
- Zeng, Z.; Yi, Z.; Xu, B. The biological and technical challenges facing utilizing circulating tumor DNA in non-metastatic breast cancer patients. Cancer Lett. 2025, 616, 217574. [Google Scholar] [CrossRef]
- Gu, Y.; Bui, T.; Muller, W.J. Exploiting Mouse Models to Recapitulate Clinical Tumor Dormancy and Recurrence in Breast Cancer. Endocrinology 2022, 163, bqac055. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.K.; Brown, J.S.; Enderling, H.; Basanta, D.; Whelan, C.J. The Evolutionary Ecology of Dormancy in Nature and in Cancer. Front. Ecol. Evol. 2021, 9, 676802. [Google Scholar] [CrossRef]
- Pinheiro, I.; Torres-Padilla, M.E.; Almouzni, G. Epigenomics in the single cell era, an important read out for genome function and cell identity. Epigenomics 2021, 13, 981–984. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Qin, S.; Xia, L.; Ma, F.; Chen, L. Integrative single-cell sequencing analysis distinguishes survival-associated cells from the breast cancer microenvironment. Cancer Med. 2023, 12, 12896–12911. [Google Scholar] [CrossRef]
- Ors, A.; Mohammed, H.; Doe, A.; Haverlack, S.; Handu, M.; Mulqueen, R.M. Abstract 5288: Single-cell multiomics reveal divergent transcriptional and epigenetic cell states in breast cancer. Cancer Res. 2023, 83, 5288. [Google Scholar] [CrossRef]
- Gregorová, J.; Vychytilova-Faltejskova, P.; Kramářová, T.; Knechtová, Z.; Almáši, M.; Stork, M.; Pour, L.; Kohoutek, J.; Ševčíková, S. Proteomic analysis of the bone marrow microenvironment in extramedullary multiple myeloma patients. Neoplasma 2022, 69, 412–424. [Google Scholar] [CrossRef]
- Chen, L.; Zhu, X.; Han, B.; Ji, L.; Yao, L.; Wang, Z. High Expression of microRNA-223 Indicates a Good Prognosis in Triple-Negative Breast Cancer. Front. Oncol. 2021, 11, 630432. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Wang, Z.; Liu, L.; Yang, Z.; Liu, S.; Ma, Z.; Liu, Y.; Ma, Y.; Zhang, L.; Zhang, X.; et al. LncRNA Malat1 inhibition of TDP43 cleavage suppresses IRF3-initiated antiviral innate immunity. Proc. Natl. Acad. Sci. USA 2020, 117, 23695–23706. [Google Scholar] [CrossRef]
- Wan, G.; Feng, H.; Su, C.; Zhu, Y.; Zhang, L.; Zhang, Q.; Yu, L. A patent review of EZH2 inhibitors from 2017 and beyond. Expert. Opin. Ther. Pat. 2023, 33, 293–308. [Google Scholar] [CrossRef]
- Granata, V.; Crisafulli, L.; Nastasi, C.; Ficara, F.; Sobacchi, C. Bone Marrow Niches and Tumour Cells: Lights and Shadows of a Mutual Relationship. Front. Immunol. 2022, 13, 884024. [Google Scholar] [CrossRef] [PubMed]
- Pires, A.; Burnell, S.E.A.; Gallimore, A. Exploiting ECM remodelling to promote immune-mediated tumour destruction. Curr. Opin. Immunol. 2022, 13, 884024. [Google Scholar] [CrossRef]
- Lapp, E.A. Single-Cell Analysis Unveils the Role of the Tumor Immune Microenvironment and Notch Signaling in Dormant Minimal Residual Disease. Cancer Res. 2022, 82, 885–899. [Google Scholar]
- Wen, J.; Wang, S.; Guo, R.; Liu, D. CSF1R inhibitors are emerging immunotherapeutic drugs for cancer treatment. Eur. J. Med. Chem. 2022, 245, 114884. [Google Scholar] [CrossRef]
- Cai, D.; Gao, W.; Li, Z.; Zhang, Y.; Xiao, L.; Xiao, Y. Current Development of Nano-Drug Delivery to Target Macrophages. Adv. Cardiovasc. Dis. 2022, 10, 1203. [Google Scholar] [CrossRef]
- Manneken, J.D.; Currie, P.D. Macrophage-stem cell crosstalk: Regulation of the stem cell niche. Development 2023, 150, dev201510. [Google Scholar] [CrossRef]
- Kalot, R.; Mhanna, R.; Talhouk, R.S. Organ-on-a-chip platforms as novel advancements for studying heterogeneity, metastasis, and drug efficacy in breast cancer. Pharmacol. Ther. 2022, 237, 108156. [Google Scholar] [CrossRef]
- Brehm, M.A. Humanized mouse models for immuno-oncology research. Nat. Rev. Clin. Oncol. 2023, 20, 192–206. [Google Scholar] [CrossRef]
- Faragalla, H.; Plotkin, A.; Barnes, P.J.; Lu, F.I.; Kos, Z.; Mulligan, A.M.; Bane, A.; Mozes, S.N. Ki67 in Breast Cancer Assay: An Ad Hoc Testing Recommendation from the Canadian Association of Pathologists Task Force. Curr. Oncol. 2023, 30, 3079–3090. [Google Scholar] [CrossRef] [PubMed]
- Pozzi, L.; Kaur, J.S. Extracellular Fluid Flow Induces Shallow Quiescence Through Physical and Biochemical Cues. Front. Cell Dev. Biol. 2022, 10, 792719. [Google Scholar] [CrossRef]
- Baek, A.E. Metastatic dormancy needs STING. Sci. Signal. 2023, 16, eadi1372. [Google Scholar] [CrossRef]
- Bano, N.; Ansari, M.I.; Kainat, K.; Singh, V.K.; Sharma, P. Chloroquine synergizes doxorubicin efficacy in cervical cancer cells through flux impairment and down regulation of proteins involved in the fusion of autophagosomes to lysosomes. Biochem. Biophys. Res. Commun. 2023, 656, 131–138. [Google Scholar] [CrossRef]
- Omstead, A.; Paskewicz, M.; Gorbunova, A.; Zheng, P.; Salvitti, M.; Mansoor, R.; Reed, P.; Ballengee, S.; Wagner, P.; Jobe, B.; et al. CSF-1R inhibitor, pexidartinib, sensitizes esophageal adenocarcinoma to PD-1 immune checkpoint blockade in a rat model. Carcinogenesis 2022, 43, 842–850. [Google Scholar] [CrossRef] [PubMed]
- Ng, G.W. Clinical management and biology of tumor dormancy in breast cancer. Semin. Cancer Biol. 2022, 78, 49–62. [Google Scholar] [CrossRef]
- Borgen, E.; Rypdal, M.C.; Sosa, M.S.; Renolen, A.; Schlichting, E.; Lønning, P.E.; Synnestvedt, M.; Aguirre-Ghiso, J.A.; Naume, B. NR2F1 stratifies dormant disseminated tumor cells in breast cancer patients. Breast Cancer Res. 2018, 20, 120. [Google Scholar] [CrossRef]
- Ghobashi, A.H.; Kimani, J.W.; Ladaika, C.A.; O’Hagan, H.M. PTEN depletion reduces H3K27me3 levels to promote epithelial-to-mesenchymal transition in epithelial colorectal cancer cells. PLoS ONE 2024, 19, e0313769. [Google Scholar] [CrossRef]
- Yang, Q.; Zhou, Z.; Li, L.; Lu, R.; Hou, G.; Huang, C.; Huang, J.; Li, H.; Zhang, Y.; Li, J.; et al. The NEXT complex regulates H3K27me3 levels to affect cancer progression by degrading G4/U-rich lncRNAs. Nucleic Acids Res. 2025, 53, gkaf107. [Google Scholar] [CrossRef]
- Daly, A.J.; De Visscher, L.; Baetens, J.M.; De Baets, B. Quo vadis, agent-based modelling tools? Environ. Model. Softw. 2022, 157, 105514. [Google Scholar] [CrossRef]
- Tang, L.J.W.; Zaseela, A.; Toh, C.C.M.; Adine, C.; Aydar, A.O.; Iyer, N.G.; Fong, E.L.S. Engineering stromal heterogeneity in cancer. Adv. Drug Deliv. Rev. 2021, 175, 113817. [Google Scholar] [CrossRef]
- Wu, S.; Lv, X.; Li, Y.; Gao, X.; Ma, Z.; Fu, X.; Li, Y.H. Integrated Machine Learning and Single-Sample Gene Set Enrichment Analysis Identifies a TGF-Beta Signaling Pathway Derived Score in Headneck Squamous Cell Carcinoma. J. Oncol. 2022, 2022, 3140263. [Google Scholar] [CrossRef]
- Ho, H.Y.; Chung, K.K.; Kan, C.M.; Wong, S.C. Liquid Biopsy in the Clinical Management of Cancers. Int. J. Mol. Sci. 2024, 25, 8594. [Google Scholar] [CrossRef]
- Keup, C.; Kimmig, R.; Kasimir-Bauer, S. The Diversity of Liquid Biopsies and Their Potential in Breast Cancer Management. Cancers 2023, 15, 5463. [Google Scholar] [CrossRef] [PubMed]
- Medford, A.J.; Denault, E.N.; Moy, B.; Parsons, H.A.; Bardia, A. Circulating Tumor DNA in Breast Cancer: Current and Future Applications. Clin. Breast Cancer 2023, 23, 687–692. [Google Scholar] [CrossRef]
- Tay, T.K.Y.; Tan, P.H. Liquid Biopsy in Breast Cancer: A Focused Review. Arch. Pathol. Lab. Med. 2021, 145, 678–686. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Gao, H.; Guan, X.; Meng, J.; Ding, S.; Long, Q.; Yi, W. Circulating tumor DNA: From discovery to clinical application in breast cancer. Front. Immunol. 2024, 15, 1355887. [Google Scholar] [CrossRef]
- Bailey, C.; Black, J.R.M.; Reading, J.L.; Litchfield, K.; Turajlic, S.; McGranahan, N.; Jamal-Hanjani, M.; Swanton, C. Tracking Cancer Evolution through the Disease Course. Cancer Discov. 2021, 11, 916–932. [Google Scholar] [CrossRef]
- Chan, J.C.H.; Chow, J.C.H.; Ho, C.H.M.; Tsui, T.Y.M.; Cho, W.C. Clinical application of circulating tumor DNA in breast cancer. J. Cancer Res. Clin. Oncol. 2021, 147, 1431–1442. [Google Scholar] [CrossRef]
- Croessmann, S.; Park, B.H. Circulating tumor DNA in early-stage breast cancer: New directions and potential clinical applications. Clin. Adv. Hematol. Oncol. 2021, 19, 155–161. [Google Scholar] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Biomarker Type | Clinical Application/Focus | Specific Findings/Methodology | Citation |
|---|---|---|---|
| ctDNA | Prognostic and Predictive Value (General) | Baseline mutations and ctDNA dynamics as prognostic/predictive factors in ER+/HER2- metastatic BC. | [210] |
| ctDNA | Mutation Frequencies, Testing Intentions, Clinical Impact | Cell-free tumor DNA analysis in advanced/metastatic BC patients. | [211] |
| ctDNA (CSF) | Diagnostic and Therapeutic Monitoring | Improved diagnostic accuracy and therapeutic monitoring in BC leptomeningeal metastasis. | [212] |
| ctDNA (EV-derived) | Endocrine Resistance Profiling | Molecular profiling of endocrine resistance in HR+/HER2- metastatic BC using EV-derived DNA and ctDNA. | [213] |
| ctDNA | Prognostic Markers | Identification of mutation patterns and ctDNA-derived prognostic markers in advanced BC patients. | [214] |
| ctDNA | Predicting Treatment Response (Neoadjuvant) | The persistence of ctDNA during neoadjuvant treatment predicts poor tumor response. | [215] |
| ctDNA | Early Detection of Progression (CDK4/6 Inhibitors) | Genomic alterations and ctDNA dynamics linked to early detection of progression on CDK4/6 inhibitor therapy. | [216] |
| ctDNA | Mutational Analysis (Palbociclib) | Mutational analysis in ER+/HER2- advanced BC patients receiving palbociclib. | [217] |
| ctDNA | Reducing Imaging Requirements | Integration of personalized ultrasensitive ctDNA monitoring to potentially reduce imaging needs in metastatic BC. | [218] |
| ctDNA | Sensitive Detection Method | Methodological advancements for sensitive and precise ctDNA detection through somatic copy number aberrations. | [219] |
| ctDNA | Disease Progression Surveillance Assay | Discussion of Plasma-SeqSensei BC In vitro Diagnostics Assay for surveillance of disease progression. | [220] |
| ctDNA | Treatment Monitoring (Synchronous Cancers) | Treatment monitoring using ctDNA in a patient with synchronous metastatic angiosarcoma and BC. | [221] |
| ctDNA | Sensitive Biomarkers | Whole-genome circulating tumor DNA methylation landscape analysis revealing sensitive biomarkers. | [223] |
| ctDNA | High-Performance Detection | Development of a tetrahedral DNA nanostructure-based biosensor for high-performance ctDNA detection. | [224] |
| Circulating Tumor Cells (CTCs) | Serial Monitoring of Genomic Alterations | Serial monitoring of genomic alterations in circulating tumor cells of ER+/HER2- advanced BC. | [227] |
| ctDNA vs. Extracellular Vesicle DNA | Comparative Analysis | Comparative analyses between ctDNA and extracellular vesicle DNA. | [228] |
| ctDNA | Modeling Clonal Structure | Modeling clonal structure over narrow time frames via circulating tumor DNA. | [229] |
| Undetectable ctDNA | Recurrence/Metastasis Risk | Undetectable ctDNA levels correlate with a low risk of recurrence/metastasis in postoperative stage I lung adenocarcinoma. | [230] |
| Cell-free DNA (cfDNA) | Comparative Analysis | Comprehensive cell-free DNA comparative analysis. | [231] |
| Multi-omics | Clinicopathologic Features, Genomic Profiles, Outcomes | Examination of clinicopathologic features, genomic profiles, and outcomes concerning biomarkers. | [237] |
| PIK3CA Mutations | Specific Mutation Focus | Research focusing on PIK3CA mutations. | [232] |
| Biomarkers in Guiding Treatment | ATR Inhibitor (Camonsertib) | Dose optimization in biomarker-selected advanced solid tumors. | [233] |
| MEK/AKT Inhibitors (Trametinib/Uprosertib) | Phase II Study (mTNBC) | Phase II study in metastatic TNBC. | [234] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sabit, H.; Abdel-Ghany, S.; Albrahim, Y.; Wadan, A.-H.S.; Rashwan, S.; Arneth, R.; Arneth, B. Bridging the Gap in Breast Cancer Dormancy: Models, Mechanisms, and Translational Challenges. Pharmaceuticals 2025, 18, 961. https://doi.org/10.3390/ph18070961
Sabit H, Abdel-Ghany S, Albrahim Y, Wadan A-HS, Rashwan S, Arneth R, Arneth B. Bridging the Gap in Breast Cancer Dormancy: Models, Mechanisms, and Translational Challenges. Pharmaceuticals. 2025; 18(7):961. https://doi.org/10.3390/ph18070961
Chicago/Turabian StyleSabit, Hussein, Shaimaa Abdel-Ghany, Yasser Albrahim, Al-Hassan Soliman Wadan, Sanaa Rashwan, Rebekka Arneth, and Borros Arneth. 2025. "Bridging the Gap in Breast Cancer Dormancy: Models, Mechanisms, and Translational Challenges" Pharmaceuticals 18, no. 7: 961. https://doi.org/10.3390/ph18070961
APA StyleSabit, H., Abdel-Ghany, S., Albrahim, Y., Wadan, A.-H. S., Rashwan, S., Arneth, R., & Arneth, B. (2025). Bridging the Gap in Breast Cancer Dormancy: Models, Mechanisms, and Translational Challenges. Pharmaceuticals, 18(7), 961. https://doi.org/10.3390/ph18070961