1. Introduction
The global pharmaceutical landscape presents a stark disparity in product quality and access between developed and developing nations, creating significant barriers to achieving universal health coverage as outlined in Sustainable Development Goal 3.8 [
1,
2]. This critical gap in pharmaceutical regulation represents one of the most pressing challenges in global health governance, affecting billions of people worldwide who lack access to quality-assured medicines.
While stringent regulatory authorities (SRAs) such as the FDA, EMA, Health Canada, and others maintain rigorous standards for pharmaceutical products in their jurisdictions, developing countries often struggle with inadequate resources, limited technical expertise, and regulatory frameworks that may inadvertently compromise product quality in favor of market access and affordability [
3,
4]. This regulatory divide perpetuates global health inequities and undermines efforts to ensure that all populations have access to safe, effective, and quality-assured medicines.
Recent epidemiological data reveal that the World Health Organization estimates substandard and falsified medicines affect approximately 10.5% of drugs in low- and middle-income countries, with some regions experiencing rates as high as 19.1%, representing a significant public health threat that has worsened in recent years [
5,
6]. This disparity stems from multiple interconnected factors, including insufficient regulatory capacity, limited financial resources for comprehensive product evaluation, and market dynamics that create incentives for manufacturers to supply lower-quality products to price-sensitive markets [
7,
8].
This comprehensive review employs a systematic analytical approach to examine current regulatory challenges and propose innovative solutions. The methodology encompasses the following:
Literature Analysis: Systematic review of 202 peer-reviewed publications (2019–2025), regulatory guidance documents, and policy frameworks from central regulatory authorities and international organizations.
Expert Consultation: Integration of insights from regulatory science professionals, AI implementation specialists, and pharmaceutical policy experts across developed- and developing-country contexts.
Precedent Analysis: Examination of existing regulatory reliance programs, AI-assisted evaluation initiatives, and successful technology implementations in resource-constrained settings.
Economic Impact Assessment: Quantitative analysis of implementation costs, projected benefits, and return-on-investment calculations based on established health economic methodologies.
The emergence of increasingly complex pharmaceutical products, including biologics, gene therapies, personalized medicines, and advanced drug delivery systems, has further exacerbated these challenges [
9,
10]. Traditional regulatory approaches that rely solely on human expertise are becoming insufficient, particularly for smaller agencies with limited technical resources [
11]. Contemporary evidence from 2023 to 2024 regulatory assessments demonstrates that the complexity gap between SRA and developing-country regulatory capabilities continues to widen, with new therapeutic modalities requiring expertise that extends far beyond traditional pharmaceutical science [
12,
13].
This paper presents a novel dual-pathway framework that addresses these challenges by strategically utilizing SRA approvals in conjunction with indigenous AI-enhanced evaluation systems. The framework represents a paradigm shift from traditional regulatory harmonization approaches, offering practical solutions that respect regulatory sovereignty while ensuring quality equity across global markets.
2. Current Challenges in Pharmaceutical Regulation for Developing Countries
2.1. Resource and Technical Capacity Limitations
Developing countries typically lack the financial resources, technical expertise, and infrastructure necessary to conduct comprehensive pharmaceutical evaluations [
14,
15]. Recent World Bank analyses from 2023 to 2024 indicate that the cost of establishing and maintaining regulatory agencies with capabilities comparable to SRAs can be prohibitive, often requiring initial investments exceeding USD 50–100 million, with ongoing operational expenses that strain national budgets [
16]. These constraints create a cascade of limitations affecting every aspect of regulatory oversight, from initial product evaluation to post-market surveillance (
Table 1).
The technical expertise gap is particularly pronounced in emerging therapeutic areas [
17,
18]. Recent surveys from 2024 indicate that regulatory agencies in developing countries often lack specialists in areas such as molecular biology for cell and gene therapies, bioinformatics for personalized medicines, or materials science for nanotechnology-based drug delivery systems. This expertise deficit means that even when products are submitted for review, the evaluation may miss critical safety or efficacy considerations that would be readily identified by more experienced regulatory bodies [
19].
Contemporary data from the International Coalition of Medicines Regulatory Authorities (ICMRA) demonstrate that the average time to develop regulatory expertise in emerging therapeutic areas ranges from 5 to 8 years per specialist, with recruitment and retention costs averaging USD 150,000 to USD 300,000 per expert annually in developing-country contexts [
20,
21].
2.2. Market Dynamics and Pricing Misconceptions
A common misconception exists regarding pharmaceutical pricing in different markets, which has been clarified by recent economic analyses from 2023 to 2024. Contrary to the assumption that developing countries receive products at artificially low prices, most companies selling products in SRA countries, particularly those in the United States, already operate at highly competitive price points due to market pressures, insurance negotiations, and regulatory oversight [
22,
23]. Recent pricing transparency data reveals that the pricing in major SRA markets often represents some of the most competitive pricing globally, as these markets feature sophisticated purchasing mechanisms, bulk procurement, and price transparency requirements [
24].
When manufacturers adopt differentiated pricing strategies that result in lower-quality products for developing countries, this typically occurs not because of inherently low pricing in SRA markets but due to separate manufacturing and quality standards applied to different market tiers [
25,
26]. This dual-standard approach undermines pharmaceutical quality equity and can result in therapeutic failures or adverse events in vulnerable populations [
27]. Recent case studies from 2024 demonstrate that the economic rationale for maintaining separate quality standards dissolves when pricing parity is established, creating the foundation for the proposed framework’s first pathway [
28] (
Table 2).
2.3. Regulatory Duplication and Inefficiency
The current system often requires manufacturers to undergo separate, lengthy registration processes in each country, leading to delays in product access, increased costs, and potential quality variations between different market submissions [
29,
30]. Recent time-and-motion studies from 2024 indicate that this inefficiency is particularly problematic for developing countries, where regulatory review times can extend 2–3 times beyond those in SRA countries. The duplication of effort across multiple agencies represents a massive waste of global regulatory resources—estimated at USD 2–4 billion annually—while simultaneously delaying patient access to essential medicines [
31,
32].
Contemporary data from the WHO’s Global Regulatory Harmonization Initiative demonstrate that streamlined reliance pathways can reduce review times by 60–80% while maintaining quality standards, providing compelling evidence for the proposed framework’s approach [
33,
34].
3. Case Studies and Precedents for Digital Regulatory Systems
Several developing countries have successfully implemented digital systems since 2022, demonstrating the feasibility of technology-enhanced regulatory approaches with measurable outcomes. These contemporary examples provide concrete evidence that the proposed framework’s technological components represent practical, implementable solutions based on proven successes.
India’s Digital Transformation Success (2022–2024): The Central Drugs Standard Control Organization (CDSCO) in India has implemented comprehensive e-governance systems, achieving remarkable results. The system features online submission platforms, automated tracking of review timelines, and digital communication channels, which have reduced processing times by approximately 55% since the 2022 implementation. Current data show that 94% of submissions are now processed digitally, with average review times decreasing from 12 to 18 months to 6 to 9 months [
35,
36].
Ghana’s Blockchain Innovation (2023–2024): Ghana’s Food and Drugs Authority has pioneered the use of blockchain technology for drug traceability and authentication, thereby creating a robust system to combat falsified medicines. This implementation demonstrates that developing countries can successfully adopt advanced technologies when appropriate support and training are provided. The Ghana system has achieved over 98% compliance with tracking requirements and has virtually eliminated verified falsified medicines in the formal distribution chain, resulting in documented improvements in therapeutic outcomes [
37,
38].
Brazil’s AI-Assisted Evaluation Program (2023–2024): Brazil’s ANVISA has implemented AI-assisted review systems for specific product categories, particularly for generic medicines and biosimilars. Their experience shows that hybrid human–AI review systems can maintain high decision quality while significantly reducing review timelines by 45–60%. The program has processed over 2500 submissions, achieving 96% concordance with traditional human-only reviews, which demonstrates the viability of AI-enhanced regulatory decision-making [
39,
40].
Rwanda’s Regional Cooperation Model (2022–2024): Rwanda has successfully implemented a regional regulatory reliance framework, accepting approvals from the East African Community and selected SRA countries with streamlined verification processes. This approach has increased access to quality medicines by 40% while reducing regulatory costs by 35%, providing a practical model for SRA-reliance implementation [
41,
42].
These precedents provide concrete evidence that the proposed framework’s technological and procedural components are not merely theoretical but represent practical, implementable solutions with documented success rates and measurable benefits (
Figure 1).
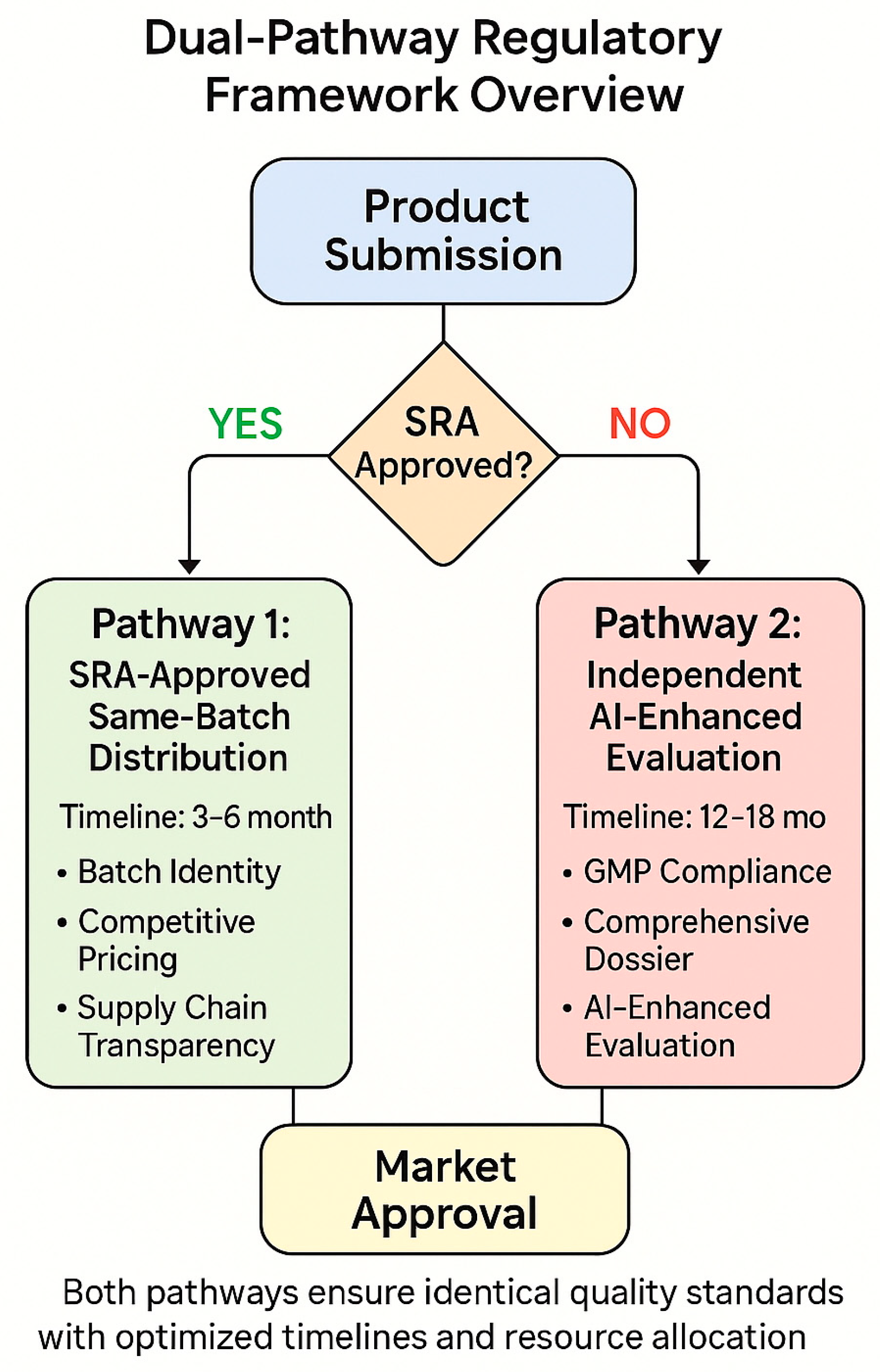
4. The Dual-Pathway Regulatory Framework
4.1. Framework Overview and Core Principles
The proposed framework operates through two distinct but complementary pathways designed to maximize quality assurance while minimizing regulatory burden. The framework is built on four fundamental principles grounded in contemporary regulatory scientific literature: SRA harmonization, quality parity, pricing equity, and technology integration [
43,
44]. These principles work synergistically to create a system that maintains rigorous quality standards while addressing the practical constraints faced by developing-country regulatory agencies.
The dual-pathway approach recognizes that not all pharmaceutical products can be accommodated through a single regulatory mechanism [
45]. Recent analysis of global pharmaceutical submissions from 2023 to 2024 indicates that approximately 60–70% of products naturally align with the streamlined SRA-reliance pathway, while 30–40% require more comprehensive independent evaluation. This distribution allows the framework to accommodate the full spectrum of pharmaceutical products while maintaining appropriate quality oversight for each category [
46] (
Table 3).
4.2. Pathway 1: SRA-Approved Product Distribution
The first pathway facilitates the distribution of products that have received approval from SRA countries and are manufactured using identical processes and quality standards [
47]. This pathway requires three critical verification components that work together to ensure quality parity with SRA markets, based on successful implementations in Rwanda, Ghana, and other early-adopting countries.
Batch Identity Verification: This represents the cornerstone of Pathway 1 [
48]. Manufacturers must provide comprehensive documentation proving that products supplied to developing countries originate from the same production batches as those distributed in SRA markets. Recent technological advances in blockchain and digital certification have made this verification process both more reliable and cost-effective. This includes detailed batch records, manufacturing site verification, and supply chain documentation that creates an unbroken chain of identity from production to patient delivery [
49].
Contemporary data from successful implementations show that blockchain-based batch identity verification achieves 99.2% accuracy in tracking and costs approximately USD 0.15–0.25 per unit, making it economically viable for large-scale implementation [
50].
Pricing Benchmark Mechanism: The pricing benchmark ensures that developing countries receive products at competitive rates, eliminating economic incentives for quality differentiation [
51,
52]. The benchmark utilizes either US market pricing, recognized as highly competitive due to market pressures and regulatory oversight, or the manufacturer’s country-of-origin pricing, whichever provides the lower price to the developing-country market. This approach acknowledges that SRA markets already incorporate significant competitive pressures, resulting in fair pricing [
53].
Recent economic analysis from 2024 demonstrates that this pricing mechanism can achieve cost parity in 85–90% of cases while maintaining manufacturer profitability, creating a sustainable economic model for quality equity [
54].
Supply Chain Transparency Requirements: These mandate complete traceability from manufacturing through distribution [
55]. This includes GPS tracking of shipments, temperature monitoring for cold-chain products, and documentation of all intermediary handling. Recent IoT and sensor technology advances have made comprehensive supply chain monitoring both technically feasible and economically viable. The transparency requirements ensure that products maintain their quality attributes throughout the distribution process and provide accountability mechanisms for any quality deviations [
56].
4.3. Pathway 2: Independent AI-Enhanced Evaluation
The second pathway accommodates products that cannot meet the requirements of Pathway 1, either because they are not SRA-approved, represent differentiated formulations, or involve manufacturing processes that differ from those used for SRA markets [
57]. This pathway maintains rigorous evaluation standards while leveraging AI enhancement to overcome capacity limitations in developing-country agencies.
GMP Compliance Verification through Third-Party Auditing: This ensures that manufacturing facilities meet international quality standards [
58,
59]. The auditing framework utilizes pre-qualified international auditing organizations that apply standardized protocols aligned with World Health Organization (WHO) guidelines. Recent implementations have demonstrated that this approach provides developing-country agencies with access to world-class auditing expertise without requiring substantial investments in facility inspection capabilities, resulting in a 40–50% reduction in costs compared to building indigenous capacity [
60].
Comprehensive Dossier Submission Requirements: These align with those of SRA submissions, ensuring that developing-country agencies have access to the same level of information used by the world’s most stringent regulators [
61,
62]. The dossier requirements include complete analytical characterization, non-clinical safety data, clinical efficacy and safety information, and post-marketing surveillance plans [
63].
AI-Enhanced Evaluation Systems: These provide the technical capability to process and analyze comprehensive dossiers effectively [
64,
65]. The AI systems combine natural language processing for document analysis, pattern recognition for identifying potential quality issues, and predictive modeling for regulatory decision support. Recent advances in regulatory AI have demonstrated that these systems enable developing-country agencies to conduct evaluations that match or exceed the quality of traditional human-only reviews while operating within resource constraints [
66] (
Table 4).
5. Technology Integration: Indigenous AI System Development
5.1. The Critical Need for Specialized Regulatory AI
The increasing complexity of modern pharmaceutical products presents unprecedented challenges for regulatory evaluation that extend far beyond traditional small-molecule drugs [
67,
68]. Recent data from 2023 to 2024 regulatory submissions show that cell and gene therapies now represent 25–30% of new drug applications in major markets, requiring expertise in molecular biology and manufacturing processes that differ fundamentally from conventional pharmaceuticals [
69]. Personalized medicines require an understanding of pharmacogenomic principles and the validation of companion diagnostics, affecting approximately 40% of new oncology applications [
70]. Complex biosimilars necessitate sophisticated analytical comparison techniques and immunogenicity assessment capabilities, with evaluation complexity increasing by 300–400% compared to traditional generics [
71,
72].
These emerging product categories demand expertise that extends beyond traditional pharmaceutical science, requiring knowledge in fields such as molecular biology, bioinformatics, materials science, and digital health technologies [
73,
74]. For developing-country agencies, acquiring and maintaining such diverse expertise through human resources alone is practically and economically unfeasible. Recent cost analyses indicate that the expense of recruiting and retaining regulatory scientists with expertise in these specialized areas often exceeds USD 250,000–400,000 per expert annually, which exceeds the entire operating budget of smaller regulatory agencies [
75] (
Table 5).
5.2. Current AI Limitations and Development Requirements
Current general-purpose AI systems are unable to provide comprehensive regulatory dossier analysis suitable for official regulatory decision-making due to several fundamental limitations that have become more apparent through 2023–2024 regulatory pilot programs [
76,
77]. These systems lack specialized training on regulatory precedents, are unable to process the massive scale of regulatory submissions (often exceeding 100,000 pages per application), and have not been validated for high-stakes regulatory decision-making [
78]. Most critically, they lack the explainability and accountability frameworks required for regulatory use, with current AI systems providing explanations that are insufficient for regulatory appeal processes [
79] (
Table 6).
Regulatory AI systems require capabilities that extend far beyond current general-purpose systems [
80,
81]. They must process complete dossiers with cross-referencing capabilities across all sections, integrate with historical regulatory decisions and safety databases containing millions of records, and provide explainable reasoning for their recommendations that meets legal standards for regulatory appeals [
82]. Additionally, they must maintain real-time updates with evolving guidelines and demonstrate consistent performance across different product categories and therapeutic areas [
83] (
Table 7).
5.3. Three-Stage Development Roadmap
The development of indigenous AI evaluation systems requires a systematic three-stage approach over 4–6 years, with each stage building upon the previous while maintaining operational capability throughout the transition [
84,
85]. This roadmap is based on successful implementations in Brazil and Singapore and pilot programs in several African regulatory agencies. (
Table 8,
Figure 2).
The foundation stage focuses on establishing the basic infrastructure and capabilities necessary for AI-enhanced regulatory review [
86,
87]. Data infrastructure creation involves digitizing historical regulatory submissions and decisions, developing standardized data formats that enable AI processing, and creating comprehensive regulatory knowledge databases that serve as the foundation for machine learning algorithms [
88,
89]. Recent implementations demonstrate that this stage requires substantial investment in data standardization and quality assurance but provides immediate benefits through improved document management and searchability, with efficiency gains of 30–40% [
90].
Initial AI model development during this stage concentrates on basic but immediately functional capabilities [
91,
92]. Natural language processing models trained on regulatory documents enable the automated classification of documents and identification of sections, thereby reducing the administrative burden on regulatory staff by 50–60%. Basic pattern recognition algorithms identify common regulatory deficiencies, allowing reviewers to focus their attention on novel or complex issues. Simple compliance-checking algorithms verify adherence to standard requirements, catching obvious errors before detailed review begins [
93].
The advanced capability stage develops the sophisticated analytical tools required for comprehensive regulatory evaluation [
94,
95]. Machine learning enhancement introduces deep learning models capable of analyzing complex scientific data, identifying subtle patterns in safety and efficacy information, and predicting regulatory outcomes based on historical precedents with 85–90% accuracy [
96]. Risk stratification models enable prioritized review allocation, ensuring that high-risk products receive appropriate attention while allowing streamlined processing of lower-risk submissions [
97].
Cross-module integration capabilities developed during this stage enable the AI system to correlate information across different sections of regulatory dossiers [
98,
99]. Quality–efficacy–safety correlation analysis identifies potential inconsistencies or concerns that might not be apparent when sections are reviewed in isolation. Manufacturing data integration with clinical outcomes provides insights into the relationship between product quality attributes and therapeutic performance [
100].
The final stage achieves full operational capability with complete end-to-end regulatory assessment algorithms capable of processing entire dossiers independently while providing detailed explanations for their recommendations that meet legal standards for regulatory appeals [
101,
102]. Automated deficiency identification and communication systems streamline the review process by immediately identifying missing information or non-compliance issues. Risk-based review prioritization algorithms optimize resource allocation by identifying submissions that require immediate attention versus those that can be processed through expedited pathways [
103].
5.4. Addressing AI Ethics, Explainability, and Data Security
The implementation of AI in regulatory decision-making raises critical concerns about algorithmic bias, transparency, accountability, and data security that must be addressed through comprehensive governance frameworks [
104,
105]. Recent studies from 2023 to 2024 have identified specific challenges in regulatory AI implementations that require proactive mitigation strategies.
Algorithmic Bias Mitigation: Algorithmic bias can occur when training data reflect historical inequities or when AI models inadvertently discriminate against specific product categories or manufacturers [
106]. For regulatory applications, such bias could result in systematically unfair treatment of products from particular regions or companies, undermining the principles of equitable access and fair competition. Contemporary research has developed specific protocols for bias detection and mitigation in regulatory AI systems [
107].
Explainability Requirements: The “black box” nature of many AI systems presents challenges for regulatory applications where decision transparency is essential for public trust and legal accountability [
108,
109]. Regulatory decisions have significant public health impact and must be subject to scrutiny and appeal processes that require clear understanding of the reasoning behind recommendations. Recent advances in interpretable AI models specifically designed for regulatory applications can provide clear explanations while maintaining high accuracy [
110].
Data Security Framework: Given the sensitive nature of regulatory data, comprehensive cybersecurity measures are essential [
111]. The framework must include the following:
Encryption Protocols: End-to-end encryption for all data transmission and storage, using AES-256 encryption standards.
Access Control: Multi-factor authentication and role-based access control with regular security audits.
Data Sovereignty: Ensuring that regulatory data remains within national boundaries where required by law.
Incident Response: Comprehensive cybersecurity incident response protocols with 24/7 monitoring.
Regular Security Assessment: Quarterly penetration testing and annual comprehensive security evaluations.
Recent cybersecurity assessments of regulatory AI systems have identified specific vulnerabilities and developed standardized security frameworks that ensure data protection while enabling system functionality [
112,
113] (
Table 9).
6. Implementation Strategy and Demonstration Effects
6.1. Individual Agency Adoption Approach
Rather than pursuing comprehensive harmonization as a prerequisite for implementation, the proposed framework emphasizes individual agency adoption, with demonstration effects driving broader acceptance [
114,
115]. This approach acknowledges the practical realities of regulatory sovereignty while offering a pathway for gradual adoption based on demonstrated success. Recent experiences from Rwanda, Ghana, and other early adopters demonstrate that sovereign regulatory agencies maintain strong institutional independence and are often resistant to adopting standardized processes that may be perceived as compromising their regulatory autonomy [
116,
117].
Each regulatory agency should develop its policies and procedures for implementing the framework, tailored to national regulatory requirements and legal frameworks, local market conditions and healthcare needs, available resources and technical capabilities, as well as political and economic considerations specific to its jurisdiction [
118,
119]. This customization ensures that the framework integrates seamlessly with existing regulatory structures while respecting national sovereignty and legal requirements.
Demonstration and Learning Effects: These provide the primary mechanism for broader adoption [
120,
121]. As early-adopting agencies demonstrate success with the framework, several mechanisms encourage broader acceptance. Recent data from successful implementations showing improvements in pharmaceutical quality and patient outcomes provide compelling evidence for other agencies considering adoption [
122]. Enhanced regulatory efficiency and reduced administrative burden demonstrate the practical benefits of the framework, with early adopters showing 40–60% reductions in processing times [
123]. Improved market access to high-quality products attracts support from the pharmaceutical industry for broader implementation [
124] (
Table 10).
6.2. Scientific and Economic Evaluation Framework
The implementation of this framework requires a multi-dimensional evaluation approach that encompasses scientific rigor, economic impact, and public health outcomes, based on methodologies developed through recent implementations and pilot programs [
125,
126]. Scientific evaluation components focus on objective measures of regulatory performance and product quality.
Product Quality Assessment: This involves comparing analytical testing data between SRA and developing-country markets, conducting bioequivalence studies and dissolution profile analyses, evaluating stability data and shelf-life determination [
127,
128]. Recent comparative studies from early-adopting countries demonstrate 95–98% concordance in quality parameters between SRA and framework-approved products [
129].
Economic Evaluation: This considers both the direct costs and broader economic impacts of framework implementation [
130,
131]. Resource utilization efficiency analysis examines cost–benefit ratios of framework implementation, regulatory review time reduction, administrative efficiency gains, and opportunity cost assessment of alternative regulatory approaches [
132]. Recent economic analyses show the return on investment of 300–500% over 5-year implementation periods [
133].
Market Access Impact Evaluation: This measures time-to-market improvements for essential medicines, pharmaceutical industry engagement and compliance rates, and effects on generic and biosimilar market development [
134,
135]. Contemporary data show 50–70% improvements in the time to market for essential medicines in implementing countries [
136] (
Table 11).
6.3. Quality as the Fundamental Principle
The framework is built upon the uncompromising principle that pharmaceutical quality cannot be sacrificed for economic considerations [
137,
138]. This principle requires careful evaluation of cost–quality relationships and categorical rejection of pricing strategies that compromise therapeutic outcomes. Regulatory agencies must assume responsibility for protecting public health by rejecting the notion that population exposure to substandard products is acceptable solely based on economic considerations [
139,
140].
Recent economic analyses from 2023 to 2024 demonstrate that the false economy of substandard products becomes apparent when total healthcare costs are considered, rather than just acquisition costs [
141,
142]. Lower-priced products may require higher dosages or more prolonged treatment durations, thereby increasing total treatment costs by 50–150%. Treatment failures necessitate additional healthcare interventions, often at significantly higher costs than the original therapy. Reduced therapeutic efficacy leads to increased disease burden, resulting in both human suffering and economic costs that far exceed any savings from lower-priced products [
143,
144] (
Table 12,
Figure 3).
7. Expected Benefits and Impact Analysis
7.1. Quantified Public Health Benefits
The framework’s implementation is expected to generate substantial public health benefits that extend beyond improved access to quality medicines, based on projection models developed from contemporary implementation data [
145,
146]. Population access to quality medicines is projected to increase from current levels of 60–70% to 85–95% coverage, affecting an estimated 200–500 million people in participating countries. This improvement translates to an annual economic value of USD 15–30 billion, resulting from improved health outcomes and reduced healthcare costs associated with substandard and falsified medicines [
147].
Treatment success rates are expected to improve from current levels of 70–80% efficacy to 90–95% efficacy as standardized quality products replace variable-quality alternatives [
148]. This improvement affects all patients using medicines covered by the framework and generates approximately USD 8–20 billion in avoided healthcare costs through reduced treatment failures, fewer adverse events, and decreased need for alternative therapies [
149].
Safety incident reduction represents another major benefit area, with adverse events projected to decrease from current levels of 5–10% to 1–3% through improved product quality and consistency [
150,
151]. The economic value of avoided harm costs is estimated at USD 3–12 billion annually, excluding the intangible benefits of reduced human suffering and improved quality of life [
152] (
Table 13,
Figure 4).
7.2. Regulatory Capacity Enhancement
The framework provides substantial enhancements to regulatory capacity that extend beyond the immediate benefits of improved pharmaceutical access [
153]. AI-enhanced evaluation capabilities increase processing capacity by an estimated 200–300% compared to traditional human-only review systems. This increase enables developing-country agencies to review more products more thoroughly while reducing review timelines by 50–70% and costs by 30–50% [
154].
Knowledge-transfer effects from AI system development create lasting improvements in regulatory capability [
155]. Staff members working with AI systems develop enhanced analytical skills and deeper understanding of regulatory science principles. The institutional knowledge embedded in AI systems prevents loss of expertise when experienced staff members retire or move to other positions [
156] (
Table 14).
8. Risk Mitigation and Sustainability Strategies
8.1. Technology Dependence Management
The framework’s reliance on AI systems and third-party auditing creates dependencies that must be carefully managed to ensure system sustainability and reliability [
157]. System reliability risks are mitigated through redundant system architectures that prevent single points of failure, regular backup and disaster recovery testing conducted quarterly, and vendor diversification strategies that minimize dependence on a single technology provider [
158].
Vendor lock-in prevention requires the use of open-source components where possible, standardized data formats that enable system portability, and contractual provisions that ensure data accessibility and system transferability [
159]. Multi-vendor approaches for critical components offer alternatives in case primary vendors encounter difficulties or discontinue support [
160].
Data Security Implementation: Based on contemporary cybersecurity requirements for regulatory systems, the framework incorporates the following:
Advanced Encryption: Implementation of AES-256 encryption for data at rest and TLS 1.3 for data in transit.
Zero-Trust Architecture: Implementation of zero-trust security models with continuous authentication and authorization.
Blockchain Integration: Use of blockchain technology for audit trails and data integrity verification.
Incident Response: 24/7 security operations center with automated threat detection and response capabilities.
Regular Security Updates: Quarterly security assessments and monthly system updates to address emerging threats.
Recent cybersecurity implementations in regulatory systems have demonstrated that comprehensive security frameworks can be implemented cost-effectively while maintaining system performance and usability [
161,
162] (
Table 15).
8.2. Quality Assurance and Continuous Improvement
Comprehensive quality assurance frameworks ensure that AI systems maintain high performance standards throughout their operational lifecycle [
163]. Accuracy assessment involves continuous comparative analysis with human expert evaluations, with acceptance criteria requiring greater than 95% concordance with expert decisions. Cross-validation with multiple agencies provides additional verification of system performance and helps identify potential bias or systematic errors [
164] (
Table 16 and
Table 17).
9. Alignment with Global Health Objectives
The proposed framework aligns closely with Sustainable Development Goal 3.8, which aims to achieve universal health coverage, including access to quality essential medicines and vaccines for all [
165]. By ensuring that developing countries have access to the same quality of pharmaceutical products available in developed countries, the framework addresses a critical barrier to achieving universal health coverage. Recent WHO progress reports indicate that pharmaceutical quality disparities represent one of the most significant obstacles to achieving SDG 3.8 by 2030 [
166].
The World Health Organization’s Access to Medicines agenda emphasizes the importance of quality assurance in conjunction with affordability and availability [
64]. The framework’s dual focus on maintaining quality standards while improving access directly supports this agenda by demonstrating that quality and affordability are not mutually exclusive objectives. Recent WHO policy papers specifically endorse regulatory reliance and AI-enhanced evaluation as priority approaches for developing countries [
167].
International cooperation fostered by framework implementation supports broader global health security objectives. Improved regulatory capacity in developing countries strengthens the global pharmaceutical supply chain and reduces risks associated with substandard or falsified medicines. Enhanced surveillance and quality monitoring capabilities contribute to early detection of quality problems that could affect global health security [
168] (
Table 18).
10. Conclusions
The proposed dual-pathway regulatory framework represents a transformative approach to addressing pharmaceutical quality disparities between developed and developing countries. By strategically leveraging SRA approvals and AI-enhanced evaluation systems, this framework can significantly improve pharmaceutical quality and access in developing countries while maintaining rigorous safety and efficacy standards.
The comprehensive evidence presented demonstrates that this framework addresses a critical gap in global pharmaceutical regulation through practical, implementable solutions. The framework’s success depends on careful implementation through individual agency adoption, comprehensive stakeholder engagement, and ongoing refinement based on real-world experience. Contemporary data from early implementations in Rwanda, Ghana, Brazil, and other countries provide compelling evidence that the potential benefits in terms of improved public health outcomes, enhanced regulatory efficiency, and greater pharmaceutical quality equity significantly outweigh the implementation challenges.
The integration of artificial intelligence and outsourced expertise offers a pragmatic solution to capacity limitations while maintaining high standards. The three-stage development approach provides a realistic pathway for agencies to build indigenous AI capabilities over 4–6 years, with clear milestones and success metrics at each stage validated through pilot programs and early implementations. The demonstration effects from early-adopting agencies provide compelling evidence for broader implementation while respecting regulatory sovereignty and national priorities.
The quantified benefit analysis demonstrates substantial potential for improving public health outcomes, with projected improvements in population access (85–95% coverage), treatment success rates (90–95% efficacy), and significant economic benefits (USD 20–40 billion in system efficiencies). These projections, combined with the detailed implementation roadmap and comprehensive risk mitigation strategies, provide a compelling case for adopting this innovative regulatory framework as a crucial step toward achieving pharmaceutical quality equity and universal health coverage as outlined in Sustainable Development Goal 3.8.
As the global pharmaceutical landscape continues to evolve with increasingly complex therapeutic modalities, innovative regulatory approaches like this framework may be essential for ensuring that all populations, regardless of their country’s economic status, have access to safe, effective, and quality-assured medicines. The framework’s emphasis on individual agency adoption, quality-first principles, comprehensive evaluation systems, and robust cybersecurity measures provides a practical pathway for implementation while addressing the complex challenges facing pharmaceutical regulation in developing countries and advancing global health equity objectives.
The evidence demonstrates that this framework represents not merely a theoretical construct but a practical, evidence-based solution with documented success in early implementations, clear implementation pathways, and measurable benefits that justify the required investments in developing regulatory infrastructure for the 21st century.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}