
Multidrug Resistance: Are We Still Afraid of the Big Bad Wolf



Abstract
1. Introduction
2. Methodology
3. Mechanisms Contributing to MDR
3.1. Transporters
3.1.1. ABCB1/MDR1/P-gp Transporter
3.1.2. ABCC1/MRP1 Transporter
3.1.3. ABCC10/MRP7 Transporter
3.1.4. ABCG2/BCRP/MXR Transporter
3.2. DNA Damage Response
3.2.1. Resistance to PARP Inhibitors
3.2.2. Resistance to Polθ Inhibitors
3.2.3. Resistance to ATR Inhibitors
3.2.4. WRN Inhibitors
3.2.5. Resistance to Topoisomerase Inhibitors
3.3. The Role of Apoptosis in MDR
3.4. Glutathione S-Transferases
4. Resistance to Molecularly Targeted Therapy
5. Strategies to Overcome Multidrug Resistance in Cancer
5.1. Nanotechnology-Based Drug Delivery Systems
5.2. RNA Interference and CRISPR/Cas9 Technology
5.3. Natural Modulators
5.4. Physical Approaches
5.5. Immunotherapy
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gillet, J.; Gottesman, M. Mechanisms of Multidrug Resistance in Cancer BT-Multi-Drug Resistance in Cancer; Zhou, J., Ed.; Humana Press: Totowa, NJ, USA, 2010. [Google Scholar]
- Szakács, G.; Paterson, J.K.; Ludwig, J.A.; Booth-Genthe, C.; Gottesman, M.M. Targeting multidrug resistance in cancer. Nat. Rev. Drug Discov. 2006, 5, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, M.N.; Slotnick, I.J.; Journey, L. In vitro studies with HeLa cell lines sensitive and resistant to actinomycin D. Ann. N. Y. Acad. Sci. 1960, 89, 474–483. [Google Scholar] [CrossRef] [PubMed]
- Emran, T.B.; Shahriar, A.; Mahmud, A.R.; Rahman, T.; Abir, M.H.; Siddiquee, M.F.; Ahmed, H.; Rahman, N.; Nainu, F.; Wahyudin, E.; et al. Multidrug Resistance in Cancer: Understanding Molecular Mechanisms, Immunoprevention and Therapeutic Approaches. Front. Oncol. 2022, 12, 891652. [Google Scholar] [CrossRef]
- Biedler, J.L.; Riehm, H. Cellular resistance to actinomycin D in Chinese hamster cells in vitro: Cross-resistance, radioautographic, and cytogenetic studies. Cancer Res. 1970, 30, 1174–1184. [Google Scholar]
- Juliano, R.L.; Ling, V. A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim. Biophys. Acta (BBA)-Biomembr. 1976, 455, 152–162. [Google Scholar] [CrossRef]
- Roninson, I.B.; Chin, J.E.; Choi, K.G.; Gros, P.; Housman, D.E.; Fojo, A.; Shen, D.-W.; Gottesman, M.M.; Pastan, I. Isolation of human mdr DNA sequences amplified in multidrug-resistant KB carcinoma cells. Proc. Natl. Acad. Sci. USA 1986, 83, 4538–4542. [Google Scholar] [CrossRef] [PubMed]
- Cole, S.; Bhardwaj, G.; Gerlach, J.; Mackie, J.; Grant, C.; Almquist, K.; Stewart, A.; Kurz, E.; Duncan, A.; Deeley, R.G. Overexpression of a transporter gene in a multidrug-resistant human lung cancer cell line. Science 1992, 258, 1650–1654. [Google Scholar] [CrossRef] [PubMed]
- Mirski, S.E.; Gerlach, J.H.; Cole, S.P. Multidrug resistance in a human small cell lung cancer cell line selected in adriamycin. Cancer Res. 1987, 47, 2594–2598. [Google Scholar] [PubMed]
- Doyle, L.A.; Yang, W.; Abruzzo, L.V.; Krogmann, T.; Gao, Y.; Rishi, A.K.; Ross, D.D. A multidrug resistance transporter from human MCF-7 breast cancer cells. Proc. Natl. Acad. Sci. USA 1998, 95, 15665–15670. [Google Scholar] [CrossRef]
- Miyake, K.; Mickley, L.; Litman, T.; Zhan, Z.; Robey, R.; Cristensen, B.; Brangi, M.; Greenberger, L.; Dean, M.; Fojo, T. Molecular cloning of cDNAs which are highly overexpressed in mitoxantrone-resistant cells: Demonstration of homology to ABC transport genes. Cancer Res. 1999, 59, 8–13. [Google Scholar]
- Sevick, E.M.; Jain, R.K. Geometric resistance to blood flow in solid tumors perfused ex vivo: Effects of tumor size and perfusion pressure. Cancer Res. 1989, 49, 3506–3512. [Google Scholar]
- Min, H.Y.; Lee, H.Y. Molecular targeted therapy for anticancer treatment. Exp. Mol. Med. 2022, 54, 1670–1694. [Google Scholar] [CrossRef]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef]
- Sottoriva, A.; Kang, H.; Ma, Z.; Graham, T.A.; Salomon, M.P.; Zhao, J.; Marjoram, P.; Siegmund, K.; Press, M.F.; Shibata, D.; et al. A Big Bang model of human colorectal tumor growth. Nat. Genet. 2015, 47, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Amir el, A.D.; Davis, K.L.; Tadmor, M.D.; Simonds, E.F.; Levine, J.H.; Bendall, S.C.; Shenfeld, D.K.; Krishnaswamy, S.; Nolan, G.P.; Pe’er, D. viSNE enables visualization of high dimensional single-cell data and reveals phenotypic heterogeneity of leukemia. Nat. Biotechnol. 2013, 31, 545–552. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, L.J.; Galski, H.; Fojo, A.; Willingham, M.; Lai, S.-L.; Gazdar, A.; Pirker, R.; Green, A.; Crist, W.; Brodeur, G.M. Expression of multidrug resistance gene in human cancers. JNCI J. Natl. Cancer Inst. 1989, 81, 116–124. [Google Scholar] [CrossRef]
- W Robey, R.; R Massey, P.; Amiri-Kordestani, L.; E Bates, S. ABC transporters: Unvalidated therapeutic targets in cancer and the CNS. Anti-Cancer Agents Med. Chem. (Former. Curr. Med. Chem.—Anti-Cancer Agents) 2010, 10, 625–633. [Google Scholar] [CrossRef]
- Leonard, G.D.; Fojo, T.; Bates, S.E. The role of ABC transporters in clinical practice. Oncol. 2003, 8, 411–424. [Google Scholar] [CrossRef] [PubMed]
- Binkhathlan, Z.; Lavasanifar, A. P-glycoprotein inhibition as a therapeutic approach for overcoming multidrug resistance in cancer: Current status and future perspectives. Curr. Cancer Drug Targets 2013, 13, 326–346. [Google Scholar] [CrossRef]
- Fletcher, J.I.; Williams, R.T.; Henderson, M.J.; Norris, M.D.; Haber, M. ABC transporters as mediators of drug resistance and contributors to cancer cell biology. Drug Resist. Updat. 2016, 26, 1–9. [Google Scholar] [CrossRef]
- Li, W.; Zhang, H.; Assaraf, Y.G.; Zhao, K.; Xu, X.; Xie, J.; Yang, D.H.; Chen, Z.S. Overcoming ABC transporter-mediated multidrug resistance: Molecular mechanisms and novel therapeutic drug strategies. Drug Resist. Updat. 2016, 27, 14–29. [Google Scholar] [CrossRef] [PubMed]
- Borst, P.; Evers, R.; Kool, M.; Wijnholds, J. A family of drug transporters: The multidrug resistance-associated proteins. J. Natl. Cancer Inst. 2000, 92, 1295–1302. [Google Scholar] [CrossRef] [PubMed]
- Khunweeraphong, N.; Kuchler, K. Multidrug Resistance in Mammals and Fungi-From MDR to PDR: A Rocky Road from Atomic Structures to Transport Mechanisms. Int. J. Mol. Sci. 2021, 22, 4806. [Google Scholar] [CrossRef]
- Maimaitijiang, A.; He, D.; Li, D.; Li, W.; Su, Z.; Fan, Z.; Li, J. Progress in Research of Nanotherapeutics for Overcoming Multidrug Resistance in Cancer. Int. J. Mol. Sci. 2024, 25, 9973. [Google Scholar] [CrossRef]
- Ueda, K.; Cornwell, M.M.; Gottesman, M.M.; Pastan, I.; Roninson, I.B.; Ling, V.; Riordan, J.R. The mdrl gene, responsible for multidrug-resistance, codes for P-glycoprotein. Biochem. Biophys. Res. Commun. 1986, 141, 956–962. [Google Scholar] [CrossRef]
- Callaghan, R.; Luk, F.; Bebawy, M. Inhibition of the multidrug resistance P-glycoprotein: Time for a change of strategy? Drug Metab. Dispos. 2014, 42, 623–631. [Google Scholar] [CrossRef] [PubMed]
- Assaraf, Y.G.; Borgnia, M.J. Differential reversal of lipophilic antifolate resistance in mammalian cells with modulators of the multidrug resistance phenotype. Anti-Cancer Drugs 1993, 4, 395–406. [Google Scholar] [CrossRef]
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug resistance in cancer: Role of ATP–dependent transporters. Nat. Rev. Cancer 2002, 2, 48–58. [Google Scholar] [CrossRef]
- Sarkadi, B.; Homolya, L.; Szakács, G.; Váradi, A. Human multidrug resistance ABCB and ABCG transporters: Participation in a chemoimmunity defense system. Physiol. Rev. 2006, 86, 1179–1236. [Google Scholar] [CrossRef]
- Schinkel, A.H.; Mayer, U.; Wagenaar, E.; Mol, C.A.; Van Deemter, L.; Smit, J.J.; Van Der Valk, M.A.; Voordouw, A.C.; Spits, H.; Van Tellingen, O. Normal viability and altered pharmacokinetics in mice lacking mdr1-type (drug-transporting) P-glycoproteins. Proc. Natl. Acad. Sci. USA 1997, 94, 4028–4033. [Google Scholar] [CrossRef]
- Alvarez, M.; Paull, K.; Monks, A.; Hose, C.; Lee, J.-S.; Weinstein, J.; Grever, M.; Bates, S.; Fojo, T. Generation of a drug resistance profile by quantitation of mdr-1/P-glycoprotein in the cell lines of the National Cancer Institute Anticancer Drug Screen. J. Clin. Investig. 1995, 95, 2205–2214. [Google Scholar] [CrossRef] [PubMed]
- Schöndorf, T.; Kurbacher, C.M.; Göhring, U.-J.; Benz, C.; Becker, M.; Sartorius, J.; Kolhagen, H.; Mallman, P.; Neumann, R. Induction of MDR1-gene expression by antineoplastic agents in ovarian cancer cell lines. Anticancer. Res. 2002, 22, 2199–2203. [Google Scholar] [PubMed]
- Xu, X.; Leo, C.; Jang, Y.; Chan, E.; Padilla, D.; Huang, B.C.; Lin, T.; Gururaja, T.; Hitoshi, Y.; Lorens, J.B. Dominant effector genetics in mammalian cells. Nat. Genet. 2001, 27, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Mickley, L.A.; Spengler, B.A.; Knutsen, T.A.; Biedler, J.L.; Fojo, T. Gene rearrangement: A novel mechanism for MDR-1 gene activation. J. Clin. Investig. 1997, 99, 1947–1957. [Google Scholar] [CrossRef] [PubMed]
- Bradshaw, D.M.; Arceci, R.J. Clinical relevance of transmembrane drug efflux as a mechanism of multidrug resistance. J. Clin. Oncol. 1998, 16, 3674–3690. [Google Scholar] [CrossRef]
- Merino, V.; Jimenez-Torres, N.V.; Merino-Sanjuan, M. Relevance of multidrug resistance proteins on the clinical efficacy of cancer therapy. Curr. Drug Deliv. 2004, 1, 203–212. [Google Scholar] [CrossRef]
- Nanayakkara, A.K.; Follit, C.A.; Chen, G.; Williams, N.S.; Vogel, P.D.; Wise, J.G. Targeted inhibitors of P-glycoprotein increase chemotherapeutic-induced mortality of multidrug resistant tumor cells. Sci. Rep. 2018, 8, 967. [Google Scholar] [CrossRef]
- Belpomme, D.; Gauthier, S.; Pujade-Lauraine, E.; Facchini, T.; Goudier, M.J.; Krakowski, I.; Netter-Pinon, G.; Frenay, M.; Gousset, C.; Marie, F.N.; et al. Verapamil increases the survival of patients with anthracycline-resistant metastatic breast carcinoma. Ann. Oncol. 2000, 11, 1471–1476. [Google Scholar] [CrossRef]
- Waghray, D.; Zhang, Q. Inhibit or Evade Multidrug Resistance P-Glycoprotein in Cancer Treatment. J. Med. Chem. 2018, 61, 5108–5121. [Google Scholar] [CrossRef]
- Dong, J.; Yuan, L.; Hu, C.; Cheng, X.; Qin, J.J. Strategies to overcome cancer multidrug resistance (MDR) through targeting P-glycoprotein (ABCB1): An updated review. Pharmacol. Ther. 2023, 249, 108488. [Google Scholar] [CrossRef]
- Hopper, E.; Belinsky, M.G.; Zeng, H.; Tosolini, A.; Testa, J.R.; Kruh, G.D. Analysis of the structure and expression pattern of MRP7 (ABCC10), a new member of the MRP subfamily. Cancer Lett. 2001, 162, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Deeley, R.G.; Westlake, C.; Cole, S.P. Transmembrane transport of endo-and xenobiotics by mammalian ATP-binding cassette multidrug resistance proteins. Physiol. Rev. 2006, 86, 849–899. [Google Scholar] [CrossRef]
- Poku, V.O.; Iram, S.H. A critical review on modulators of Multidrug Resistance Protein 1 in cancer cells. PeerJ 2022, 10, e12594. [Google Scholar] [CrossRef] [PubMed]
- Sodani, K.; Patel, A.; Kathawala, R.J.; Chen, Z.-S. Multidrug resistance associated proteins in multidrug resistance. Chin. J. Cancer 2012, 31, 58. [Google Scholar] [CrossRef]
- Brevig, T.; Krühne, U.; Kahn, R.A.; Ahl, T.; Beyer, M.; Pedersen, L.H. Hydrodynamic guiding for addressing subsets of immobilized cells and molecules in microfluidic systems. BMC Biotechnol. 2003, 3, 10. [Google Scholar] [CrossRef]
- Bakos, É.; Evers, R.; Sinkó, E.; Váradi, A.; Borst, P.; Sarkadi, B. Interactions of the human multidrug resistance proteins MRP1 and MRP2 with organic anions. Mol. Pharmacol. 2000, 57, 760–768. [Google Scholar] [CrossRef]
- Zhou, S.-F.; Wang, L.-L.; Di, Y.M.; Xue, C.C.; Duan, W.; Li, C.G.; Li, Y. Substrates and inhibitors of human multidrug resistance associated proteins and the implications in drug development. Curr. Med. Chem. 2008, 15, 1981–2039. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-S.; Min, Y.-D.; Choi, C.-H. Double-edged sword of chemosensitizer: Increase of multidrug resistance protein (MRP) in leukemic cells by an MRP inhibitor probenecid. Biochem. Biophys. Res. Commun. 2001, 283, 64–71. [Google Scholar] [CrossRef]
- O’Connor, R.; O’Leary, M.; Ballot, J.; Collins, C.; Kinsella, P.; Mager, D.; Arnold, R.; O’Driscoll, L.; Larkin, A.; Kennedy, S. A phase I clinical and pharmacokinetic study of the multi-drug resistance protein-1 (MRP-1) inhibitor sulindac, in combination with epirubicin in patients with advanced cancer. Cancer Chemother. Pharmacol. 2007, 59, 79–87. [Google Scholar] [CrossRef]
- Tivnan, A.; Zakaria, Z.; O’Leary, C.; Kögel, D.; Pokorny, J.L.; Sarkaria, J.N.; Prehn, J.H. Inhibition of multidrug resistance protein 1 (MRP1) improves chemotherapy drug response in primary and recurrent glioblastoma multiforme. Front. Neurosci. 2015, 9, 218. [Google Scholar] [CrossRef]
- Kathawala, R.J.; Wang, Y.-J.; Ashby Jr, C.R.; Chen, Z.-S. Recent advances regarding the role of ABC subfamily C member 10 (ABCC10) in the efflux of antitumor drugs. Chin. J. Cancer 2014, 33, 223. [Google Scholar] [CrossRef] [PubMed]
- Hopper-Borge, E.; Xu, X.; Shen, T.; Shi, Z.; Chen, Z.-S.; Kruh, G.D. Human Multidrug Resistance Protein 7 (ABCC10) Is a Resistance Factor for Nucleoside Analogues and Epothilone B. Cancer Res. 2008, 69, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.S.; Tiwari, A.K. Multidrug resistance proteins (MRPs/ABCCs) in cancer chemotherapy and genetic diseases. FEBS J. 2011, 278, 3226–3245. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Huang, Y.; Shi, J.; Dai, Y.; Wu, L.; Zhou, H. ABCC10 plays a significant role in the transport of gefitinib and contributes to acquired resistance to gefitinib in NSCLC. Front. Pharmacol. 2018, 9, 1312. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.-Q.; Xie, Y.; Cao, L.-Q.; Fleishman, J.S.; Chen, Y.; Wu, T.; Yang, D.-H. The role of ABCC10/MRP7 in anti-cancer drug resistance and beyond. Drug Resist. Updates 2024, 73, 101062. [Google Scholar] [CrossRef] [PubMed]
- Mao, Q.; Unadkat, J.D. Role of the breast cancer resistance protein (BCRP/ABCG2) in drug transport—An update. AAPS J. 2015, 17, 65–82. [Google Scholar] [CrossRef]
- Zattoni, I.F.; Delabio, L.C.; de Paula Dutra, J.; Kita, D.H.; Scheiffer, G.; Hembecker, M.; da Silva Pereira, G.; Moure, V.R.; Valdameri, G. Targeting breast cancer resistance protein (BCRP/ABCG2): Functional inhibitors and expression modulators. Eur. J. Med. Chem. 2022, 237, 114346. [Google Scholar] [CrossRef] [PubMed]
- Hira, D.; Terada, T. BCRP/ABCG2 and high-alert medications: Biochemical, pharmacokinetic, pharmacogenetic, and clinical implications. Biochem. Pharmacol. 2018, 147, 201–210. [Google Scholar] [CrossRef]
- Nakanishi, T.; Doyle, L.A.; Hassel, B.; Wei, Y.; Bauer, K.S.; Wu, S.; Pumplin, D.W.; Fang, H.-B.; Ross, D.D. Functional characterization of human breast cancer resistance protein (BCRP, ABCG2) expressed in the oocytes of Xenopus laevis. Mol. Pharmacol. 2003, 64, 1452–1462. [Google Scholar] [CrossRef]
- Volk, E.L.; Farley, K.M.; Wu, Y.; Li, F.; Robey, R.W.; Schneider, E. Overexpression of wild-type breast cancer resistance protein mediates methotrexate resistance. Cancer Res. 2002, 62, 5035–5040. [Google Scholar]
- Robey, R.W.; Honjo, Y.; van de Laar, A.; Miyake, K.; Regis, J.T.; Litman, T.; Bates, S.E. A functional assay for detection of the mitoxantrone resistance protein, MXR (ABCG2). Biochim. Biophys. Acta (BBA)-Biomembr. 2001, 1512, 171–182. [Google Scholar] [CrossRef]
- Ross, D.D.; Yang, W.; Abruzzo, L.V.; Dalton, W.S.; Schneider, E.; Lage, H.; Dietel, M.; Greenberger, L.; Cole, S.P.; Doyle, L.A. Atypical multidrug resistance: Breast cancer resistance protein messenger RNA expression in mitoxantrone-selected cell lines. J. Natl. Cancer Inst. 1999, 91, 429–433. [Google Scholar] [CrossRef] [PubMed]
- Maliepaard, M.; van Gastelen, M.A.; de Jong, L.A.; Pluim, D.; van Waardenburg, R.C.; Ruevekamp-Helmers, M.C.; Floot, B.G.; Schellens, J.H. Overexpression of the BCRP/MXR/ABCP gene in a topotecan-selected ovarian tumor cell line. Cancer Res. 1999, 59, 4559–4563. [Google Scholar] [PubMed]
- Wu, C.P.; Hsiao, S.H.; Huang, Y.H.; Hung, L.C.; Yu, Y.J.; Chang, Y.T.; Hung, T.H.; Wu, Y.S. Sitravatinib Sensitizes ABCB1- and ABCG2-Overexpressing Multidrug-Resistant Cancer Cells to Chemotherapeutic Drugs. Cancers 2020, 12, 195. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Qi, J.; Dong, Z.; Zhang, J.-T. Dynamic vs static ABCG2 inhibitors to sensitize drug resistant cancer cells. PLoS ONE 2010, 5, e15276. [Google Scholar] [CrossRef] [PubMed]
- Ding, R.; Shi, J.; Pabon, K.; Scotto, K.W. Xanthines down-regulate the drug transporter ABCG2 and reverse multidrug resistance. Mol. Pharmacol. 2012, 81, 328–337. [Google Scholar] [CrossRef]
- Ee, P.R.; He, X.; Ross, D.D.; Beck, W.T. Modulation of breast cancer resistance protein (BCRP/ABCG2) gene expression using RNA interference. Mol. Cancer Ther. 2004, 3, 1577–1584. [Google Scholar] [CrossRef]
- Jia, P.; Wu, S.-B.; Xu, Q.; Wu, M.-F.; Gao, Q.-L.; Liao, G.-N.; Lu, Y.-P.; Ma, D. Antisense oligonucleotide reverses topotecan-resistant ovarian cancer cells. Ai Zheng = Aizheng = Chin. J. Cancer 2003, 22, 1296–1300. [Google Scholar]
- Kowalski, P.; Stein, U.; Scheffer, G.L.; Lage, H. Modulation of the atypical multidrug-resistant phenotype by a hammerhead ribozyme directed against the ABC transporter BCRP/MXR/ABCG2. Cancer Gene Ther. 2002, 9, 579–586. [Google Scholar] [CrossRef]
- Huynh, T.; Norris, M.D.; Haber, M.; Henderson, M.J. ABCC4/MRP4: A MYCN-regulated transporter and potential therapeutic target in neuroblastoma. Front. Oncol. 2012, 2, 178. [Google Scholar] [CrossRef]
- Balça-Silva, J.; Matias, D.; Carmo, A.D.; Sarmento-Ribeiro, A.B.; Lopes, M.C.; Moura-Neto, V. Cellular and molecular mechanisms of glioblastoma malignancy: Implications in resistance and therapeutic strategies. Semin. Cancer Biol. 2019, 58, 130–141. [Google Scholar] [CrossRef] [PubMed]
- de Faria, G.P.; de Oliveira, J.A.; de Oliveira, J.G.; Romano Sde, O.; Neto, V.M.; Maia, R.C. Differences in the expression pattern of P-glycoprotein and MRP1 in low-grade and high-grade gliomas. Cancer Investig. 2008, 26, 883–889. [Google Scholar] [CrossRef] [PubMed]
- Mohri, M.; Nitta, H.; Yamashita, J. Expression of multidrug resistance-associated protein (MRP) in human gliomas. J. Neurooncol 2000, 49, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Calatozzolo, C.; Gelati, M.; Ciusani, E.; Sciacca, F.L.; Pollo, B.; Cajola, L.; Marras, C.; Silvani, A.; Vitellaro-Zuccarello, L.; Croci, D.; et al. Expression of drug resistance proteins Pgp, MRP1, MRP3, MRP5 and GST-pi in human glioma. J. Neurooncol 2005, 74, 113–121. [Google Scholar] [CrossRef]
- Ota, I.; Sakurai, A.; Toyoda, Y.; Morita, S.; Sasaki, T.; Chishima, T.; Yamakado, M.; Kawai, Y.; Ishidao, T.; Lezhava, A.; et al. Association between breast cancer risk and the wild-type allele of human ABC transporter ABCC11. Anticancer. Res. 2010, 30, 5189–5194. [Google Scholar]
- Dvorak, P.; Pesta, M.; Soucek, P. ABC gene expression profiles have clinical importance and possibly form a new hallmark of cancer. Tumour Biol. 2017, 39, 1010428317699800. [Google Scholar] [CrossRef] [PubMed]
- Hlaváč, V.; Brynychová, V.; Václavíková, R.; Ehrlichová, M.; Vrána, D.; Pecha, V.; Koževnikovová, R.; Trnková, M.; Gatěk, J.; Kopperová, D.; et al. The expression profile of ATP-binding cassette transporter genes in breast carcinoma. Pharmacogenomics 2013, 14, 515–529. [Google Scholar] [CrossRef]
- Lang, T.; Justenhoven, C.; Winter, S.; Baisch, C.; Hamann, U.; Harth, V.; Ko, Y.D.; Rabstein, S.; Spickenheuer, A.; Pesch, B.; et al. The earwax-associated SNP c.538G>A (G180R) in ABCC11 is not associated with breast cancer risk in Europeans. Breast Cancer Res. Treat. 2011, 129, 993–999. [Google Scholar] [CrossRef] [PubMed]
- Yamada, A.; Ishikawa, T.; Ota, I.; Kimura, M.; Shimizu, D.; Tanabe, M.; Chishima, T.; Sasaki, T.; Ichikawa, Y.; Morita, S.; et al. High expression of ATP-binding cassette transporter ABCC11 in breast tumors is associated with aggressive subtypes and low disease-free survival. Breast Cancer Res. Treat. 2013, 137, 773–782. [Google Scholar] [CrossRef]
- Yoshiura, K.; Kinoshita, A.; Ishida, T.; Ninokata, A.; Ishikawa, T.; Kaname, T.; Bannai, M.; Tokunaga, K.; Sonoda, S.; Komaki, R.; et al. A SNP in the ABCC11 gene is the determinant of human earwax type. Nat. Genet. 2006, 38, 324–330. [Google Scholar] [CrossRef]
- Bhangal, G.; Halford, S.; Wang, J.; Roylance, R.; Shah, R.; Waxman, J. Expression of the multidrug resistance gene in human prostate cancer. Urol. Oncol. 2000, 5, 118–121. [Google Scholar] [CrossRef] [PubMed]
- Demidenko, R.; Razanauskas, D.; Daniunaite, K.; Lazutka, J.R.; Jankevicius, F.; Jarmalaite, S. Frequent down-regulation of ABC transporter genes in prostate cancer. BMC Cancer 2015, 15, 683. [Google Scholar] [CrossRef] [PubMed]
- Mishra, D.K.; Chen, Z.; Wu, Y.; Sarkissyan, M.; Koeffler, H.P.; Vadgama, J.V. Global methylation pattern of genes in androgen-sensitive and androgen-independent prostate cancer cells. Mol. Cancer Ther. 2010, 9, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Shannon, W.D.; Duncan, J.; Scheffer, G.L.; Scheper, R.J.; McLeod, H.L. Expression of drug pathway proteins is independent of tumour type. J. Pathol. 2006, 209, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Hedditch, E.L.; Gao, B.; Russell, A.J.; Lu, Y.; Emmanuel, C.; Beesley, J.; Johnatty, S.E.; Chen, X.; Harnett, P.; George, J.; et al. ABCA transporter gene expression and poor outcome in epithelial ovarian cancer. J. Natl. Cancer Inst. 2014, 106, dju149. [Google Scholar] [CrossRef]
- Chien, J.; Fan, J.B.; Bell, D.A.; April, C.; Klotzle, B.; Ota, T.; Lingle, W.L.; Gonzalez Bosquet, J.; Shridhar, V.; Hartmann, L.C. Analysis of gene expression in stage I serous tumors identifies critical pathways altered in ovarian cancer. Gynecol. Oncol. 2009, 114, 3–11. [Google Scholar] [CrossRef]
- Vesel, M.; Rapp, J.; Feller, D.; Kiss, E.; Jaromi, L.; Meggyes, M.; Miskei, G.; Duga, B.; Smuk, G.; Laszlo, T.; et al. ABCB1 and ABCG2 drug transporters are differentially expressed in non-small cell lung cancers (NSCLC) and expression is modified by cisplatin treatment via altered Wnt signaling. Respir. Res. 2017, 18, 52. [Google Scholar] [CrossRef]
- Ciccia, A.; Elledge, S.J. The DNA Damage Response: Making It Safe to Play with Knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef]
- O’Connor, M.J. Targeting the DNA Damage Response in Cancer. Mol. Cell 2015, 60, 547–560. [Google Scholar] [CrossRef]
- Morales, J.; Li, L.; Fattah, F.J.; Dong, Y.; Bey, E.A.; Patel, M.; Gao, J.; Boothman, D.A. Review of Poly (ADP-ribose) Polymerase (PARP) Mechanisms of Action and Rationale for Targeting in Cancer and Other Diseases. Crit. Rev. Eukaryot. Gene Expr. 2014, 24, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Bhamidipati, D.; Haro-Silerio, J.I.; Yap, T.A.; Ngoi, N. PARP inhibitors: Enhancing efficacy through rational combinations. Br. J. Cancer 2023, 129, 904–916. [Google Scholar] [CrossRef] [PubMed]
- Chen, A. PARP inhibitors: Its role in treatment of cancer. Chin. J. Cancer 2011, 30, 463–471. [Google Scholar] [CrossRef]
- Kotsopoulos, J.; Chen, Z.; Vallis, K.A.; Poll, A.; Ainsworth, P.; Narod, S.A. DNA repair capacity as a possible biomarker of breast cancer risk in female BRCA1 mutation carriers. Br. J. Cancer 2007, 96, 118–125. [Google Scholar] [CrossRef]
- Martinez-Useros, J.; Garcia-Foncillas, J. The Role of BRCA2 Mutation Status as Diagnostic, Predictive, and Prognosis Biomarker for Pancreatic Cancer. Biomed. Res. Int. 2016, 2016, 1869304. [Google Scholar] [CrossRef]
- Phan, Z.; Ford, C.E.; Caldon, C.E. DNA repair biomarkers to guide usage of combined PARP inhibitors and chemotherapy: A meta-analysis and systematic review. Pharmacol. Res. 2023, 196, 106927. [Google Scholar] [CrossRef]
- Xie, Y.; Xiao, D.; Li, D.; Peng, M.; Peng, W.; Duan, H.; Yang, X. Combined strategies with PARP inhibitors for the treatment of BRCA wide type cancer. Front. Oncol. 2024, 14, 1441222. [Google Scholar] [CrossRef]
- Aftimos, P.G.; Oliveira, M.; Punie, K.; Boni, V.; Hamilton, E.P.; Gucalp, A.; Shah, P.D.; de Miguel, M.J.; Sharma, P.; Bauman, L.; et al. A phase 1b/2 study of the BET inhibitor ZEN-3694 in combination with talazoparib for treatment of patients with TNBC without gBRCA1/2 mutations. J. Clin. Oncol. 2022, 40, 1023. [Google Scholar] [CrossRef]
- Alvarez Secord, A.; O’Malley, D.M.; Sood, A.K.; Westin, S.N.; Liu, J.F. Rationale for combination PARP inhibitor and antiangiogenic treatment in advanced epithelial ovarian cancer: A review. Gynecol. Oncol. 2021, 162, 482–495. [Google Scholar] [CrossRef]
- Soung, Y.-H.; Chung, J. Combination Treatment Strategies to Overcome PARP Inhibitor Resistance. Biomolecules 2023, 13, 1480. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Waggoner, S.; Vidal, G.A.; Mita, M.; Moroney, J.W.; Holloway, R.; Van Le, L.; Sachdev, J.C.; Chapman-Davis, E.; Colon-Otero, G.; et al. Single-Arm Phases 1 and 2 Trial of Niraparib in Combination With Pembrolizumab in Patients With Recurrent Platinum-Resistant Ovarian Carcinoma. JAMA Oncol. 2019, 5, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Nedeljkovic, M.; Damjanovic, A. Mechanisms of Chemotherapy Resistance in Triple-Negative Breast Cancer-How We Can Rise to the Challenge. Cells 2019, 8, 957. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.M.; Gupta, G.P. Polymerase θ inhibition steps on the cGAS pedal. J. Clin. Investig. 2023, 133, e170660. [Google Scholar] [CrossRef]
- Baxter, J.S.; Zatreanu, D.; Pettitt, S.J.; Lord, C.J. Resistance to DNA repair inhibitors in cancer. Mol. Oncol. 2022, 16, 3811–3827. [Google Scholar] [CrossRef]
- Kumar, R.J.; Chao, H.X.; Simpson, D.A.; Feng, W.; Cho, M.-G.; Roberts, V.R.; Sullivan, A.R.; Shah, S.J.; Wozny, A.-S.; Fagan-Solis, K.; et al. Dual inhibition of DNA-PK and DNA polymerase theta overcomes radiation resistance induced by p53 deficiency. NAR Cancer 2020, 2, zcaa038. [Google Scholar] [CrossRef]
- Yano, K.; Shiotani, B. Emerging strategies for cancer therapy by ATR inhibitors. Cancer Sci. 2023, 114, 2709–2721. [Google Scholar] [CrossRef]
- Cimprich, K.A.; Cortez, D. ATR: An essential regulator of genome integrity. Nat. Rev. Mol. Cell Biol. 2008, 9, 616–627. [Google Scholar] [CrossRef] [PubMed]
- Ngoi, N.Y.L.; Pilié, P.G.; McGrail, D.J.; Zimmermann, M.; Schlacher, K.; Yap, T.A. Targeting ATR in patients with cancer. Nat. Rev. Clin. Oncol. 2024, 21, 278–293. [Google Scholar] [CrossRef]
- O’Leary, P.C.; Chen, H.; Doruk, Y.U.; Williamson, T.; Polacco, B.; McNeal, A.S.; Shenoy, T.; Kale, N.; Carnevale, J.; Stevenson, E.; et al. Resistance to ATR Inhibitors Is Mediated by Loss of the Nonsense-Mediated Decay Factor UPF2. Cancer Res. 2022, 82, 3950–3961. [Google Scholar] [CrossRef]
- Kim, H.; Xu, H.; George, E.; Hallberg, D.; Kumar, S.; Jagannathan, V.; Medvedev, S.; Kinose, Y.; Devins, K.; Verma, P.; et al. Combining PARP with ATR inhibition overcomes PARP inhibitor and platinum resistance in ovarian cancer models. Nat. Commun. 2020, 11, 3726. [Google Scholar] [CrossRef]
- Yap, T.A.; Tan, D.S.P.; Terbuch, A.; Caldwell, R.; Guo, C.; Goh, B.C.; Heong, V.; Haris, N.R.M.; Bashir, S.; Drew, Y.; et al. First-in-Human Trial of the Oral Ataxia Telangiectasia and RAD3-Related (ATR) Inhibitor BAY 1895344 in Patients with Advanced Solid Tumors. Cancer Discov. 2021, 11, 80–91. [Google Scholar] [CrossRef]
- Lu, H.; Davis, A.J. Human RecQ Helicases in DNA Double-Strand Break Repair. Front. Cell Dev. Biol. 2021, 9, 640755. [Google Scholar] [CrossRef] [PubMed]
- Luo, J. WRN protein and Werner syndrome. N. Am. J. Med. Sci. 2010, 3, 205–207. [Google Scholar] [CrossRef] [PubMed]
- Parker, M.J.; Lee, H.; Yao, S.; Irwin, S.; Hwang, S.; Belanger, K.; de Mare, S.W.; Surgenor, R.; Yan, L.; Gee, P.; et al. Identification of 2-Sulfonyl/Sulfonamide Pyrimidines as Covalent Inhibitors of WRN Using a Multiplexed High-Throughput Screening Assay. Biochemistry 2023, 62, 2147–2160. [Google Scholar] [CrossRef] [PubMed]
- Heuser, A.; Abdul Rahman, W.; Bechter, E.; Blank, J.; Buhr, S.; Erdmann, D.; Fontana, P.; Mermet-Meillon, F.; Meyerhofer, M.; Strang, R.; et al. Challenges for the Discovery of Non-Covalent WRN Helicase Inhibitors. ChemMedChem 2024, 19, e202300613. [Google Scholar] [CrossRef]
- Xie, H.; Zhang, J. Targeting werner helicase with synthetic lethality in microsatellite instability cancers: Promising therapeutic approaches. MedComm–Oncol. 2024, 3, e82. [Google Scholar] [CrossRef]
- Bjornsti, M.A.; Kaufmann, S.H. Topoisomerases and cancer chemotherapy: Recent advances and unanswered questions. F1000Res 2019, 8, F1000 Faculty Rev-1704. [Google Scholar] [CrossRef]
- Postow, L.; Crisona, N.J.; Peter, B.J.; Hardy, C.D.; Cozzarelli, N.R. Topological challenges to DNA replication: Conformations at the fork. Proc. Natl. Acad. Sci. USA 2001, 98, 8219–8226. [Google Scholar] [CrossRef]
- Racko, D.; Benedetti, F.; Goundaroulis, D.; Stasiak, A. Chromatin Loop Extrusion and Chromatin Unknotting. Polymers 2018, 10, 1126. [Google Scholar] [CrossRef] [PubMed]
- McKinnon, P.J. Topoisomerases and the regulation of neural function. Nat. Rev. Neurosci. 2016, 17, 673–679. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y.; Nussenzweig, A.; Takeda, S.; Austin, C. Human topoisomerases and their roles in genome stability and organization. Nat. Rev. Mol. Cell Biol. 2022, 23, 407–427. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Saha, L.K.; Saha, S.; Jo, U.; Pommier, Y. Debulking of topoisomerase DNA-protein crosslinks (TOP-DPC) by the proteasome, non-proteasomal and non-proteolytic pathways. DNA Repair 2020, 94, 102926. [Google Scholar] [CrossRef] [PubMed]
- Yakkala, P.A.; Penumallu, N.R.; Shafi, S.; Kamal, A. Prospects of Topoisomerase Inhibitors as Promising Anti-Cancer Agents. Pharmaceuticals 2023, 16, 1456. [Google Scholar] [CrossRef]
- Lohri, A.; van Hille, B.; Bacchi, M.; Fopp, M.; Joncourt, F.; Reuter, J.; Cerny, T.; Fey, M.F.; Herrmann, R. Five putative drug resistance parameters (MDR1/P-glycoprotein, MDR-associated protein, glutathione-S-transferase, bcl-2 and topoisomerase IIalpha) in 57 newly diagnosed acute myeloid leukaemias. Swiss Group for Clinical Cancer Research (SAKK). Eur. J. Haematol. 1997, 59, 206–215. [Google Scholar] [CrossRef] [PubMed]
- Bondarev, A.D.; Jonsson, J.; Chubarev, V.N.; Tarasov, V.V.; Lagunas-Rangel, F.A.; Schioth, H.B. Recent developments of topoisomerase inhibitors: Clinical trials, emerging indications, novel molecules and global sales. Pharmacol. Res. 2024, 209, 107431. [Google Scholar] [CrossRef]
- Delgado, J.L.; Hsieh, C.M.; Chan, N.L.; Hiasa, H. Topoisomerases as anticancer targets. Biochem. J. 2018, 475, 373–398. [Google Scholar] [CrossRef]
- Sharma, N.K.; Bahot, A.; Sekar, G.; Bansode, M.; Khunteta, K.; Sonar, P.V.; Hebale, A.; Salokhe, V.; Sinha, B.K. Understanding Cancer’s Defense against Topoisomerase-Active Drugs: A Comprehensive Review. Cancers 2024, 16, 680. [Google Scholar] [CrossRef]
- Sordet, O.; Khan, Q.A.; Kohn, K.W.; Pommier, Y. Apoptosis induced by topoisomerase inhibitors. Curr. Med. Chem. Anticancer. Agents 2003, 3, 271–290. [Google Scholar] [CrossRef]
- Boot, A.; Liu, M.; Stantial, N.; Shah, V.; Yu, W.; Nitiss, K.C.; Nitiss, J.L.; Jinks-Robertson, S.; Rozen, S.G. Recurrent mutations in topoisomerase IIalpha cause a previously undescribed mutator phenotype in human cancers. Proc. Natl. Acad. Sci. USA 2022, 119, e2114024119. [Google Scholar] [CrossRef] [PubMed]
- Ganapathi, R.N.; Ganapathi, M.K. Mechanisms regulating resistance to inhibitors of topoisomerase II. Front. Pharmacol. 2013, 4, 89. [Google Scholar] [CrossRef] [PubMed]
- Gongora, C.; Vezzio-Vie, N.; Tuduri, S.; Denis, V.; Causse, A.; Auzanneau, C.; Collod-Beroud, G.; Coquelle, A.; Pasero, P.; Pourquier, P.; et al. New Topoisomerase I mutations are associated with resistance to camptothecin. Mol. Cancer 2011, 10, 64. [Google Scholar] [CrossRef] [PubMed]
- El-Readi, M.Z.; Al-Abd, A.M.; Althubiti, M.A.; Almaimani, R.A.; Al-Amoodi, H.S.; Ashour, M.L.; Wink, M.; Eid, S.Y. Multiple Molecular Mechanisms to Overcome Multidrug Resistance in Cancer by Natural Secondary Metabolites. Front. Pharmacol. 2021, 12, 658513. [Google Scholar] [CrossRef] [PubMed]
- Madkour, M.M.; Ramadan, W.S.; Saleh, E.; El-Awady, R. Epigenetic modulations in cancer: Predictive biomarkers and potential targets for overcoming the resistance to topoisomerase I inhibitors. Ann. Med. 2023, 55, 2203946. [Google Scholar] [CrossRef] [PubMed]
- Curtin, N.J. DNA repair dysregulation from cancer driver to therapeutic target. Nat. Rev. Cancer 2012, 12, 801–817. [Google Scholar] [CrossRef]
- Pommier, Y.; Huang, S.Y.; Gao, R.; Das, B.B.; Murai, J.; Marchand, C. Tyrosyl-DNA-phosphodiesterases (TDP1 and TDP2). DNA Repair 2014, 19, 114–129. [Google Scholar] [CrossRef]
- Pommier, Y.; Sun, Y.; Huang, S.N.; Nitiss, J.L. Roles of eukaryotic topoisomerases in transcription, replication and genomic stability. Nat. Rev. Mol. Cell Biol. 2016, 17, 703–721. [Google Scholar] [CrossRef]
- Ceccaldi, R.; Rondinelli, B.; D’Andrea, A.D. Repair Pathway Choices and Consequences at the Double-Strand Break. Trends Cell Biol. 2016, 26, 52–64. [Google Scholar] [CrossRef]
- Stingele, J.; Habermann, B.; Jentsch, S. DNA-protein crosslink repair: Proteases as DNA repair enzymes. Trends Biochem. Sci. 2015, 40, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Antony, S.; Agama, K.K.; Miao, Z.H.; Takagi, K.; Wright, M.H.; Robles, A.I.; Varticovski, L.; Nagarajan, M.; Morrell, A.; Cushman, M.; et al. Novel indenoisoquinolines NSC 725776 and NSC 724998 produce persistent topoisomerase I cleavage complexes and overcome multidrug resistance. Cancer Res. 2007, 67, 10397–10405. [Google Scholar] [CrossRef]
- Kummar, S.; Chen, A.; Gutierrez, M.; Pfister, T.D.; Wang, L.; Redon, C.; Bonner, W.M.; Yutzy, W.; Zhang, Y.; Kinders, R.J.; et al. Clinical and pharmacologic evaluation of two dosing schedules of indotecan (LMP400), a novel indenoisoquinoline, in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2016, 78, 73–81. [Google Scholar] [CrossRef]
- Drummond, D.C.; Noble, C.O.; Guo, Z.; Hong, K.; Park, J.W.; Kirpotin, D.B. Development of a highly active nanoliposomal irinotecan using a novel intraliposomal stabilization strategy. Cancer Res. 2006, 66, 3271–3277. [Google Scholar] [CrossRef] [PubMed]
- Frampton, J.E. Liposomal Irinotecan: A Review in Metastatic Pancreatic Adenocarcinoma. Drugs 2020, 80, 1007–1018. [Google Scholar] [CrossRef] [PubMed]
- Kalra, A.V.; Kim, J.; Klinz, S.G.; Paz, N.; Cain, J.; Drummond, D.C.; Nielsen, U.B.; Fitzgerald, J.B. Preclinical activity of nanoliposomal irinotecan is governed by tumor deposition and intratumor prodrug conversion. Cancer Res. 2014, 74, 7003–7013. [Google Scholar] [CrossRef]
- Song, S.; Sun, D.; Wang, H.; Wang, J.; Yan, H.; Zhao, X.; Fawcett, J.P.; Xu, X.; Cai, D.; Gu, J. Full-profile pharmacokinetics, anticancer activity and toxicity of an extended release trivalent PEGylated irinotecan prodrug. Acta Pharm. Sin. B 2023, 13, 3444–3453. [Google Scholar] [CrossRef]
- Modi, S.; Saura, C.; Yamashita, T.; Park, Y.H.; Kim, S.B.; Tamura, K.; Andre, F.; Iwata, H.; Ito, Y.; Tsurutani, J.; et al. Trastuzumab Deruxtecan in Previously Treated HER2-Positive Breast Cancer. N. Engl. J. Med. 2020, 382, 610–621. [Google Scholar] [CrossRef]
- Gu, Y.; Yang, R.; Zhang, Y.; Guo, M.; Takehiro, K.; Zhan, M.; Yang, L.; Wang, H. Molecular mechanisms and therapeutic strategies in overcoming chemotherapy resistance in cancer. Mol. Biomed. 2025, 6, 2. [Google Scholar] [CrossRef]
- Xu, Y.; Her, C. Inhibition of Topoisomerase (DNA) I (TOP1): DNA Damage Repair and Anticancer Therapy. Biomolecules 2015, 5, 1652–1670. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Boise, L.H.; Shanmugam, M. Cancer Metabolism and the Evasion of Apoptotic Cell Death. Cancers 2019, 11, 1144. [Google Scholar] [CrossRef]
- Jan, R.; Chaudhry, G.E. Understanding Apoptosis and Apoptotic Pathways Targeted Cancer Therapeutics. Adv. Pharm. Bull. 2019, 9, 205–218. [Google Scholar] [CrossRef]
- Qian, S.; Wei, Z.; Yang, W.; Huang, J.; Yang, Y.; Wang, J. The role of BCL-2 family proteins in regulating apoptosis and cancer therapy. Front. Oncol. 2022, 12, 985363. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, K.; Van Bockstaele, D.R.; Berneman, Z.N. Apoptosis: Mechanisms and relevance in cancer. Ann. Hematol. 2005, 84, 627–639. [Google Scholar] [CrossRef]
- Kumar, R.; Herbert, P.E.; Warrens, A.N. An introduction to death receptors in apoptosis. Int. J. Surg. 2005, 3, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Pistritto, G.; Trisciuoglio, D.; Ceci, C.; Garufi, A.; D’Orazi, G. Apoptosis as anticancer mechanism: Function and dysfunction of its modulators and targeted therapeutic strategies. Aging 2016, 8, 603–619. [Google Scholar] [CrossRef] [PubMed]
- Plati, J.; Bucur, O.; Khosravi-Far, R. Dysregulation of apoptotic signaling in cancer: Molecular mechanisms and therapeutic opportunities. J. Cell. Biochem. 2008, 104, 1124–1149. [Google Scholar] [CrossRef]
- Roberts, A.W. Therapeutic development and current uses of BCL-2 inhibition. Hematol. Am. Soc. Hematol. Educ. Program. 2020, 2020, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Yue, X.; Chen, Q.; He, J. Combination strategies to overcome resistance to the BCL2 inhibitor venetoclax in hematologic malignancies. Cancer Cell Int. 2020, 20, 524. [Google Scholar] [CrossRef]
- Chen, Y.; Yuan, L.; Lu, X.; Wang, X.; Zhang, Q.; Wang, X.; Zhao, X. Efficacy of venetoclax and rituximab in the treatment of concurrent acute myeloid leukemia and untreated chronic lymphocytic leukemia: A case report and literature review. Oncol. Lett. 2024, 28, 393. [Google Scholar] [CrossRef]
- Chien, K.S.; Rodriguez-Sevilla, J.J.; Alvarado, Y.; Montalban-Bravo, G.; Hammond, D.E.; Swaminathan, M.; Bazinet, A.; Kimberley, J.; Bodden, K.; Schneider, H.; et al. A phase I study of the myeloid cell leukemia 1 (MCL1) inhibitor tapotoclax (AMG 176) in patients with myelodysplastic syndromes after hypomethylating agent failure. Leuk. Res. 2024, 147, 107602. [Google Scholar] [CrossRef]
- Tao, Z.F.; Hasvold, L.; Wang, L.; Wang, X.; Petros, A.M.; Park, C.H.; Boghaert, E.R.; Catron, N.D.; Chen, J.; Colman, P.M.; et al. Discovery of a Potent and Selective BCL-XL Inhibitor with in Vivo Activity. ACS Med. Chem. Lett. 2014, 5, 1088–1093. [Google Scholar] [CrossRef]
- Wu, G.; Zhang, C.; Xu, L.; Chen, H.; Fan, X.; Sun, B.; Tang, Q.; Zhan, Y.; Chen, T.; Wang, X. BAK plays a key role in A-1331852-induced apoptosis in senescent chondrocytes. Biochem. Biophys. Res. Commun. 2022, 609, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.; Smith, D.C.; Wang, S. Small-molecule SMAC mimetics as new cancer therapeutics. Pharmacol. Ther. 2014, 144, 82–95. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, R.M.; Muqbil, I.; Lowe, L.; Yedjou, C.; Hsu, H.Y.; Lin, L.T.; Siegelin, M.D.; Fimognari, C.; Kumar, N.B.; Dou, Q.P.; et al. Broad targeting of resistance to apoptosis in cancer. Semin. Cancer Biol. 2015, 35 (Suppl. S0), S78–S103. [Google Scholar] [CrossRef] [PubMed]
- Algarin, E.M.; Diaz-Tejedor, A.; Mogollon, P.; Hernandez-Garcia, S.; Corchete, L.A.; San-Segundo, L.; Martin-Sanchez, M.; Gonzalez-Mendez, L.; Schoumacher, M.; Banquet, S.; et al. Preclinical evaluation of the simultaneous inhibition of MCL-1 and BCL-2 with the combination of S63845 and venetoclax in multiple myeloma. Haematologica 2020, 105, e116–e120. [Google Scholar] [CrossRef]
- Grundy, M.; Balakrishnan, S.; Fox, M.; Seedhouse, C.H.; Russell, N.H. Genetic biomarkers predict response to dual BCL-2 and MCL-1 targeting in acute myeloid leukaemia cells. Oncotarget 2018, 9, 37777–37789. [Google Scholar] [CrossRef] [PubMed]
- Montero, J.; Haq, R. Adapted to Survive: Targeting Cancer Cells with BH3 Mimetics. Cancer Discov. 2022, 12, 1217–1232. [Google Scholar] [CrossRef]
- Dogan, E.; Kara, H.G.; Kosova, B.; Cetintas, V.B. Targeting Apoptosis to Overcome Chemotherapy Resistance. In Metastasis; Sergi, C.M., Ed.; Exon Publications: Brisbane, Australia, 2022. [Google Scholar]
- Hersey, P.; Zhang, X.D. Treatment combinations targeting apoptosis to improve immunotherapy of melanoma. Cancer Immunol. Immunother. 2009, 58, 1749–1759. [Google Scholar] [CrossRef]
- Adjei, I.M.; Blanka, S. Modulation of the tumor microenvironment for cancer treatment: A biomaterials approach. J. Funct. Biomater. 2015, 6, 81–103. [Google Scholar] [CrossRef]
- Kundu, M.; Butti, R.; Panda, V.K.; Malhotra, D.; Das, S.; Mitra, T.; Kapse, P.; Gosavi, S.W.; Kundu, G.C. Modulation of the tumor microenvironment and mechanism of immunotherapy-based drug resistance in breast cancer. Mol. Cancer 2024, 23, 92. [Google Scholar] [CrossRef]
- Li, Y.; Liu, J.; Gao, L.; Liu, Y.; Meng, F.; Li, X.; Qin, F.X. Targeting the tumor microenvironment to overcome immune checkpoint blockade therapy resistance. Immunol. Lett. 2020, 220, 88–96. [Google Scholar] [CrossRef]
- Zhao, Z.; Li, T.; Sun, L.; Yuan, Y.; Zhu, Y. Potential mechanisms of cancer-associated fibroblasts in therapeutic resistance. Biomed. Pharmacother. 2023, 166, 115425. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; Abrams, S.L.; Steelman, L.S.; Cocco, L.; Ratti, S.; Martelli, A.M.; Lombardi, P.; Gizak, A.; Duda, P. APR-246-The Mutant TP53 Reactivator-Increases the Effectiveness of Berberine and Modified Berberines to Inhibit the Proliferation of Pancreatic Cancer Cells. Biomolecules 2022, 12, 276. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.J.; Chee, C.E.; Huang, S.; Sinicrope, F.A. The role of autophagy in cancer: Therapeutic implications. Mol. Cancer Ther. 2011, 10, 1533–1541. [Google Scholar] [CrossRef]
- Hayes, J.D.; Flanagan, J.U.; Jowsey, I.R. Glutathione transferases. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 51–88. [Google Scholar] [CrossRef]
- Townsend, D.M.; Tew, K.D. The role of glutathione-S-transferase in anti-cancer drug resistance. Oncogene 2003, 22, 7369–7375. [Google Scholar] [CrossRef] [PubMed]
- Allocati, N.; Masulli, M.; Di Ilio, C.; Federici, L. Glutathione transferases: Substrates, inihibitors and pro-drugs in cancer and neurodegenerative diseases. Oncogenesis 2018, 7, 8. [Google Scholar] [CrossRef]
- Dirven, H.A.; van Ommen, B.; van Bladeren, P.J. Involvement of human glutathione S-transferase isoenzymes in the conjugation of cyclophosphamide metabolites with glutathione. Cancer Res. 1994, 54, 6215–6220. [Google Scholar] [PubMed]
- Karpusas, M.; Axarli, I.; Chiniadis, L.; Papakyriakou, A.; Bethanis, K.; Scopelitou, K.; Clonis, Y.D.; Labrou, N.E. The interaction of the chemotherapeutic drug chlorambucil with human glutathione transferase A1-1: Kinetic and structural analysis. PLoS ONE 2013, 8, e56337. [Google Scholar] [CrossRef]
- Morrow, C.S.; Smitherman, P.K.; Diah, S.K.; Schneider, E.; Townsend, A.J. Coordinated action of glutathione S-transferases (GSTs) and multidrug resistance protein 1 (MRP1) in antineoplastic drug detoxification. Mechanism of GST A1-1- and MRP1-associated resistance to chlorambucil in MCF7 breast carcinoma cells. J. Biol. Chem. 1998, 273, 20114–20120. [Google Scholar] [CrossRef]
- Leslie, E.M.; Deeley, R.G.; Cole, S.P. Multidrug resistance proteins: Role of P-glycoprotein, MRP1, MRP2, and BCRP (ABCG2) in tissue defense. Toxicol. Appl. Pharmacol. 2005, 204, 216–237. [Google Scholar] [CrossRef]
- Anwar, S.; Alrumaihi, F.; Sarwar, T.; Babiker, A.Y.; Khan, A.A.; Prabhu, S.V.; Rahmani, A.H. Exploring Therapeutic Potential of Catalase: Strategies in Disease Prevention and Management. Biomolecules 2024, 14, 697. [Google Scholar] [CrossRef] [PubMed]
- Laborde, E. Glutathione transferases as mediators of signaling pathways involved in cell proliferation and cell death. Cell Death Differ. 2010, 17, 1373–1380. [Google Scholar] [CrossRef] [PubMed]
- Sau, A.; Pellizzari Tregno, F.; Valentino, F.; Federici, G.; Caccuri, A.M. Glutathione transferases and development of new principles to overcome drug resistance. Arch. Biochem. Biophys. 2010, 500, 116–122. [Google Scholar] [CrossRef]
- Lv, N.; Huang, C.; Huang, H.; Dong, Z.; Chen, X.; Lu, C.; Zhang, Y. Overexpression of Glutathione S-Transferases in Human Diseases: Drug Targets and Therapeutic Implications. Antioxidants 2023, 12, 1970. [Google Scholar] [CrossRef] [PubMed]
- Oakley, A.J. Glutathione transferases: New functions. Curr. Opin. Struct. Biol. 2005, 15, 716–723. [Google Scholar] [CrossRef]
- Tabassum, D.P.; Polyak, K. Tumorigenesis: It takes a village. Nat. Rev. Cancer 2015, 15, 473–483. [Google Scholar] [CrossRef]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef]
- Bousoik, E.; Nabiee, R.; Amirrad, F.; Nichols, A.; Witt, R.; Mahdipoor, P.; Montazeri Aliabadi, H. Heterogeneity and Plasticity of Human Breast Cancer Cells in Response to Molecularly-Targeted Drugs. Front. Oncol. 2019, 9, 1070. [Google Scholar] [CrossRef] [PubMed]
- Holzel, M.; Bovier, A.; Tuting, T. Plasticity of tumour and immune cells: A source of heterogeneity and a cause for therapy resistance? Nat. Rev. Cancer 2013, 13, 365–376. [Google Scholar] [CrossRef]
- Capp, J.P. Cancer Stem Cells: From Historical Roots to a New Perspective. J. Oncol. 2019, 2019, 5189232. [Google Scholar] [CrossRef]
- Tu, S.M.; Guo, C.C.; Chow, D.S.; Zacharias, N.M. Stem Cell Theory of Cancer: Implications for Drug Resistance and Chemosensitivity in Cancer Care. Cancers 2022, 14, 1548. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.M.; Zhang, J.G.; Zhang, X.; Li, Q. Targeting cancer stem cells for reversing therapy resistance: Mechanism, signaling, and prospective agents. Signal Transduct. Target. Ther. 2021, 6, 62. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.C.; Lerou, P.H.; Lahav, G. Stem cells: Balancing resistance and sensitivity to DNA damage. Trends Cell Biol. 2014, 24, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.H.; Zhang, M.; Rajapakshe, K.; Coarfa, C.; Edwards, D.; Huang, S.; Rosen, J.M. Mammary Stem Cells and Tumor-Initiating Cells Are More Resistant to Apoptosis and Exhibit Increased DNA Repair Activity in Response to DNA Damage. Stem Cell Rep. 2015, 5, 378–391. [Google Scholar] [CrossRef]
- Moitra, K.; Lou, H.; Dean, M. Multidrug efflux pumps and cancer stem cells: Insights into multidrug resistance and therapeutic development. Clin. Pharmacol. Ther. 2011, 89, 491–502. [Google Scholar] [CrossRef]
- Qin, X.; Tape, C.J. Functional analysis of cell plasticity using single-cell technologies. Trends Cell Biol. 2024, 34, 854–864. [Google Scholar] [CrossRef]
- Fatma, H.; Siddique, H.R. Cancer cell plasticity, stem cell factors, and therapy resistance: How are they linked? Cancer Metastasis Rev. 2024, 43, 423–440. [Google Scholar] [CrossRef]
- Torborg, S.R.; Li, Z.; Chan, J.E.; Tammela, T. Cellular and molecular mechanisms of plasticity in cancer. Trends Cancer 2022, 8, 735–746. [Google Scholar] [CrossRef] [PubMed]
- Kroon, J.; Kooijman, S.; Cho, N.J.; Storm, G.; van der Pluijm, G. Improving Taxane-Based Chemotherapy in Castration-Resistant Prostate Cancer. Trends Pharmacol. Sci. 2016, 37, 451–462. [Google Scholar] [CrossRef]
- Shah, P.P.; Dupre, T.V.; Siskind, L.J.; Beverly, L.J. Common cytotoxic chemotherapeutics induce epithelial-mesenchymal transition (EMT) downstream of ER stress. Oncotarget 2017, 8, 22625–22639. [Google Scholar] [CrossRef]
- Terry, S.; Savagner, P.; Ortiz-Cuaran, S.; Mahjoubi, L.; Saintigny, P.; Thiery, J.P.; Chouaib, S. New insights into the role of EMT in tumor immune escape. Mol. Oncol. 2017, 11, 824–846. [Google Scholar] [CrossRef] [PubMed]
- Starska-Kowarska, K. Role of Mesenchymal Stem/Stromal Cells in Head and Neck Cancer-Regulatory Mechanisms of Tumorigenic and Immune Activity, Chemotherapy Resistance, and Therapeutic Benefits of Stromal Cell-Based Pharmacological Strategies. Cells 2024, 13, 1270. [Google Scholar] [CrossRef] [PubMed]
- Desbats, M.A.; Giacomini, I.; Prayer-Galetti, T.; Montopoli, M. Metabolic Plasticity in Chemotherapy Resistance. Front. Oncol. 2020, 10, 281. [Google Scholar] [CrossRef] [PubMed]
- Chae, Y.K.; Chang, S.; Ko, T.; Anker, J.; Agte, S.; Iams, W.; Choi, W.M.; Lee, K.; Cruz, M. Epithelial-mesenchymal transition (EMT) signature is inversely associated with T-cell infiltration in non-small cell lung cancer (NSCLC). Sci. Rep. 2018, 8, 2918. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.K.; Diamond, M.S.; Yuan, S.; Kemp, S.B.; Kahn, B.M.; Li, Q.; Lin, J.H.; Li, J.; Norgard, R.J.; Thomas, S.K.; et al. Plasticity-induced repression of Irf6 underlies acquired resistance to cancer immunotherapy in pancreatic ductal adenocarcinoma. Nat. Commun. 2024, 15, 1532. [Google Scholar] [CrossRef]
- Shien, K.; Papadimitrakopoulou, V.A.; Ruder, D.; Behrens, C.; Shen, L.; Kalhor, N.; Song, J.; Lee, J.J.; Wang, J.; Tang, X.; et al. JAK1/STAT3 Activation through a Proinflammatory Cytokine Pathway Leads to Resistance to Molecularly Targeted Therapy in Non-Small Cell Lung Cancer. Mol. Cancer Ther. 2017, 16, 2234–2245. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Y.; Chen, Y.P.; Li, Y.Q.; Liu, N.; Ma, J. Chemotherapeutic and targeted agents can modulate the tumor microenvironment and increase the efficacy of immune checkpoint blockades. Mol. Cancer 2021, 20, 27. [Google Scholar] [CrossRef] [PubMed]
- Kersh, A.E.; Ng, S.; Chang, Y.M.; Sasaki, M.; Thomas, S.N.; Kissick, H.T.; Lesinski, G.B.; Kudchadkar, R.R.; Waller, E.K.; Pollack, B.P. Targeted Therapies: Immunologic Effects and Potential Applications Outside of Cancer. J. Clin. Pharmacol. 2018, 58, 7–24. [Google Scholar] [CrossRef]
- Hsu, Y.F.; Ajona, D.; Corrales, L.; Lopez-Picazo, J.M.; Gurpide, A.; Montuenga, L.M.; Pio, R. Complement activation mediates cetuximab inhibition of non-small cell lung cancer tumor growth in vivo. Mol. Cancer 2010, 9, 139. [Google Scholar] [CrossRef]
- Joly, E.; Hudrisier, D. What is trogocytosis and what is its purpose? Nat. Immunol. 2003, 4, 815. [Google Scholar] [CrossRef]
- Antonarelli, G.; Taurelli Salimbeni, B.; Marra, A.; Esposito, A.; Locatelli, M.A.; Trapani, D.; Pescia, C.; Fusco, N.; Curigliano, G.; Criscitiello, C. The CDK4/6 inhibitors biomarker landscape: The most relevant biomarkers of response or resistance for further research and potential clinical utility. Crit. Rev. Oncol. Hematol. 2023, 192, 104148. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Wang, H.; Yan, Q.; Cui, J.; Chen, Y.; Ruan, S.; Yang, J.; Wu, Z.; Han, M.; Huang, S.; et al. Genome-wide CRISPR/Cas9 screening for drug resistance in tumors. Front. Pharmacol. 2023, 14, 1284610. [Google Scholar] [CrossRef]
- Xiang, L.; Rao, J.; Yuan, J.; Xie, T.; Yan, H. Single-Cell RNA-Sequencing: Opening New Horizons for Breast Cancer Research. Int. J. Mol. Sci. 2024, 25, 9482. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Song, Y.; Wang, L. Single-cell RNA sequencing reveals the landscape of the cellular ecosystem of primary hepatocellular carcinoma. Cancer Cell Int. 2024, 24, 379. [Google Scholar] [CrossRef]
- Khosroabadi, Z.; Azaryar, S.; Dianat-Moghadam, H.; Amoozgar, Z.; Sharifi, M. Single cell RNA sequencing improves the next generation of approaches to AML treatment: Challenges and perspectives. Mol. Med. 2025, 31, 33. [Google Scholar] [CrossRef]
- Saleh, R.O.; Hjazi, A.; Rab, S.O.; Uthirapathy, S.; Ganesan, S.; Shankhyan, A.; Ravi Kumar, M.; Sharma, G.C.; Kariem, M.; Ahmed, J.K. Single-cell RNA Sequencing Contributes to the Treatment of Acute Myeloid Leukaemia With Hematopoietic Stem Cell Transplantation, Chemotherapy, and Immunotherapy. J. Biochem. Mol. Toxicol. 2025, 39, e70218. [Google Scholar] [CrossRef] [PubMed]
- An, Y.; Zhou, L.; Huang, Z.; Nice, E.C.; Zhang, H.; Huang, C. Molecular insights into cancer drug resistance from a proteomics perspective. Expert. Rev. Proteom. 2019, 16, 413–429. [Google Scholar] [CrossRef] [PubMed]
- Della Corte, C.M.; Viscardi, G.; Di Liello, R.; Fasano, M.; Martinelli, E.; Troiani, T.; Ciardiello, F.; Morgillo, F. Role and targeting of anaplastic lymphoma kinase in cancer. Mol. Cancer 2018, 17, 30. [Google Scholar] [CrossRef] [PubMed]
- Katayama, R.; Lovly, C.M.; Shaw, A.T. Therapeutic targeting of anaplastic lymphoma kinase in lung cancer: A paradigm for precision cancer medicine. Clin. Cancer Res. 2015, 21, 2227–2235. [Google Scholar] [CrossRef]
- Golding, B.; Luu, A.; Jones, R.; Viloria-Petit, A.M. The function and therapeutic targeting of anaplastic lymphoma kinase (ALK) in non-small cell lung cancer (NSCLC). Mol. Cancer 2018, 17, 52. [Google Scholar] [CrossRef]
- Hall, R.; Alasmari, A.; Mozaffari, S.; Mahdipoor, P.; Parang, K.; Montazeri Aliabadi, H. Peptide/Lipid-Associated Nucleic Acids (PLANAs) as a Multicomponent siRNA Delivery System. Mol. Pharm. 2021, 18, 986–1002. [Google Scholar] [CrossRef] [PubMed]
- Heuckmann, J.M.; Holzel, M.; Sos, M.L.; Heynck, S.; Balke-Want, H.; Koker, M.; Peifer, M.; Weiss, J.; Lovly, C.M.; Grutter, C.; et al. ALK mutations conferring differential resistance to structurally diverse ALK inhibitors. Clin. Cancer Res. 2011, 17, 7394–7401. [Google Scholar] [CrossRef] [PubMed]
- Laetsch, T.W.; Hong, D.S. Tropomyosin Receptor Kinase Inhibitors for the Treatment of TRK Fusion Cancer. Clin. Cancer Res. 2021, 27, 4974–4982. [Google Scholar] [CrossRef] [PubMed]
- Cocco, E.; Scaltriti, M.; Drilon, A. NTRK fusion-positive cancers and TRK inhibitor therapy. Nat. Rev. Clin. Oncol. 2018, 15, 731–747. [Google Scholar] [CrossRef]
- Hemming, M.L.; Nathenson, M.J.; Lin, J.R.; Mei, S.; Du, Z.; Malik, K.; Marino-Enriquez, A.; Jagannathan, J.P.; Sorger, P.K.; Bertagnolli, M.; et al. Response and mechanisms of resistance to larotrectinib and selitrectinib in metastatic undifferentiated sarcoma harboring oncogenic fusion of NTRK1. JCO Precis. Oncol. 2020, 4, 79–90. [Google Scholar] [CrossRef]
- MacFarland, S.P.; Naraparaju, K.; Iyer, R.; Guan, P.; Kolla, V.; Hu, Y.; Tan, K.; Brodeur, G.M. Mechanisms of Entrectinib Resistance in a Neuroblastoma Xenograft Model. Mol. Cancer Ther. 2020, 19, 920–926. [Google Scholar] [CrossRef] [PubMed]
- Scholl, S.; Fleischmann, M.; Schnetzke, U.; Heidel, F.H. Molecular Mechanisms of Resistance to FLT3 Inhibitors in Acute Myeloid Leukemia: Ongoing Challenges and Future Treatments. Cells 2020, 9, 2493. [Google Scholar] [CrossRef] [PubMed]
- Nitika; Wei, J.; Hui, A.M. Role of Biomarkers in FLT3 AML. Cancers 2022, 14, 1164. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol. Res. 2014, 79, 34–74. [Google Scholar] [CrossRef]
- Amelia, T.; Kartasasmita, R.E.; Ohwada, T.; Tjahjono, D.H. Structural Insight and Development of EGFR Tyrosine Kinase Inhibitors. Molecules 2022, 27, 819. [Google Scholar] [CrossRef]
- Guo, G.; Gong, K.; Wohlfeld, B.; Hatanpaa, K.J.; Zhao, D.; Habib, A.A. Ligand-Independent EGFR Signaling. Cancer Res. 2015, 75, 3436–3441. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, S. Lazertinib: First Approval. Drugs 2021, 81, 1107–1113. [Google Scholar] [CrossRef] [PubMed]
- Santarpia, M.; Liguori, A.; Karachaliou, N.; Gonzalez-Cao, M.; Daffina, M.G.; D’Aveni, A.; Marabello, G.; Altavilla, G.; Rosell, R. Osimertinib in the treatment of non-small-cell lung cancer: Design, development and place in therapy. Lung Cancer Targets Ther. 2017, 8, 109–125. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Tsui, S.T.; Liu, C.; Song, Y.; Liu, D. EGFR C797S mutation mediates resistance to third-generation inhibitors in T790M-positive non-small cell lung cancer. J. Hematol. Oncol. 2016, 9, 59. [Google Scholar] [CrossRef]
- Zhu, X.; Chen, L.; Liu, L.; Niu, X. EMT-Mediated Acquired EGFR-TKI Resistance in NSCLC: Mechanisms and Strategies. Front. Oncol. 2019, 9, 1044. [Google Scholar] [CrossRef]
- Miyamoto, K.; Ogino, H.; Kakimoto, T.; Matsumura, Y.; Haji, K.; Mitsuhashi, A.; Morita, Y.; Tsukazaki, Y.; Yabuki, Y.; Ozaki, R.; et al. Transformation of epidermal growth factor receptor mutated lung adenocarcinoma to small-cell carcinoma long after the cessation of tyrosine kinase inhibitor treatment: A case series and literature review. Respir. Med. Case Rep. 2024, 51, 102076. [Google Scholar] [CrossRef]
- Akbay, E.A.; Koyama, S.; Carretero, J.; Altabef, A.; Tchaicha, J.H.; Christensen, C.L.; Mikse, O.R.; Cherniack, A.D.; Beauchamp, E.M.; Pugh, T.J.; et al. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discov. 2013, 3, 1355–1363. [Google Scholar] [CrossRef]
- Odogwu, L.; Mathieu, L.; Goldberg, K.B.; Blumenthal, G.M.; Larkins, E.; Fiero, M.H.; Rodriguez, L.; Bijwaard, K.; Lee, E.Y.; Philip, R.; et al. FDA Benefit-Risk Assessment of Osimertinib for the Treatment of Metastatic Non-Small Cell Lung Cancer Harboring Epidermal Growth Factor Receptor T790M Mutation. Oncologist 2018, 23, 353–359. [Google Scholar] [CrossRef]
- Turke, A.B.; Zejnullahu, K.; Wu, Y.L.; Song, Y.; Dias-Santagata, D.; Lifshits, E.; Toschi, L.; Rogers, A.; Mok, T.; Sequist, L.; et al. Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell 2010, 17, 77–88. [Google Scholar] [CrossRef]
- Hong, L.; Zhang, J.; Heymach, J.V.; Le, X. Current and future treatment options for MET exon 14 skipping alterations in non-small cell lung cancer. Ther. Adv. Med. Oncol. 2021, 13, 1758835921992976. [Google Scholar] [CrossRef]
- Paik, P.K.; Felip, E.; Veillon, R.; Sakai, H.; Cortot, A.B.; Garassino, M.C.; Mazieres, J.; Viteri, S.; Senellart, H.; Van Meerbeeck, J.; et al. Tepotinib in Non-Small-Cell Lung Cancer with MET Exon 14 Skipping Mutations. N. Engl. J. Med. 2020, 383, 931–943. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, J.T.; Mollerup, J. Companion Diagnostics and Predictive Biomarkers for MET-Targeted Therapy in NSCLC. Cancers 2022, 14, 2150. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Li, Y.; Xiong, L.; Wang, W.; Wu, M.; Yuan, T.; Yang, W.; Tian, C.; Miao, Z.; Wang, T.; et al. Small molecules in targeted cancer therapy: Advances, challenges, and future perspectives. Signal Transduct. Target. Ther. 2021, 6, 201. [Google Scholar] [CrossRef] [PubMed]
- Wright, S.C.E.; Vasilevski, N.; Serra, V.; Rodon, J.; Eichhorn, P.J.A. Mechanisms of Resistance to PI3K Inhibitors in Cancer: Adaptive Responses, Drug Tolerance and Cellular Plasticity. Cancers 2021, 13, 1538. [Google Scholar] [CrossRef] [PubMed]
- Murali, I.; Kasar, S.; Naeem, A.; Tyekucheva, S.; Khalsa, J.K.; Thrash, E.M.; Itchaki, G.; Livitz, D.; Leshchiner, I.; Dong, S.; et al. Activation of the MAPK pathway mediates resistance to PI3K inhibitors in chronic lymphocytic leukemia. Blood 2021, 138, 44–56. [Google Scholar] [CrossRef]
- Meyer, L.A.; Slomovitz, B.M.; Djordjevic, B.; Westin, S.N.; Iglesias, D.A.; Munsell, M.F.; Jiang, Y.; Schmandt, R.; Broaddus, R.R.; Coleman, R.L.; et al. The search continues: Looking for predictive biomarkers for response to mammalian target of rapamycin inhibition in endometrial cancer. Int. J. Gynecol. Cancer 2014, 24, 713–717. [Google Scholar] [CrossRef] [PubMed]
- Prior, I.A.; Hood, F.E.; Hartley, J.L. The Frequency of Ras Mutations in Cancer. Cancer Res. 2020, 80, 2969–2974. [Google Scholar] [CrossRef]
- Yaeger, R.; Corcoran, R.B. Targeting Alterations in the RAF-MEK Pathway. Cancer Discov. 2019, 9, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Wellbrock, C.; Karasarides, M.; Marais, R. The RAF proteins take centre stage. Nat. Rev. Mol. Cell Biol. 2004, 5, 875–885. [Google Scholar] [CrossRef]
- Blair, H.A. Sotorasib: First Approval. Drugs 2021, 81, 1573–1579. [Google Scholar] [CrossRef]
- Koelblinger, P.; Thuerigen, O.; Dummer, R. Development of encorafenib for BRAF-mutated advanced melanoma. Curr. Opin. Oncol. 2018, 30, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Aplin, A.E.; Kaplan, F.M.; Shao, Y. Mechanisms of resistance to RAF inhibitors in melanoma. J. Investig. Dermatol. 2011, 131, 1817–1820. [Google Scholar] [CrossRef] [PubMed]
- Degirmenci, U.; Yap, J.; Sim, Y.R.M.; Qin, S.; Hu, J. Drug resistance in targeted cancer therapies with RAF inhibitors. Cancer Drug Resist. 2021, 4, 665–683. [Google Scholar] [CrossRef] [PubMed]
- Luebker, S.A.; Koepsell, S.A. Diverse Mechanisms of BRAF Inhibitor Resistance in Melanoma Identified in Clinical and Preclinical Studies. Front. Oncol. 2019, 9, 268. [Google Scholar] [CrossRef]
- Jing, J.; Greshock, J.; Holbrook, J.D.; Gilmartin, A.; Zhang, X.; McNeil, E.; Conway, T.; Moy, C.; Laquerre, S.; Bachman, K.; et al. Comprehensive predictive biomarker analysis for MEK inhibitor GSK1120212. Mol. Cancer Ther. 2012, 11, 720–729. [Google Scholar] [CrossRef]
- Olbryt, M. Potential Biomarkers of Skin Melanoma Resistance to Targeted Therapy-Present State and Perspectives. Cancers 2022, 14, 2315. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Kaldis, P. Cdks, cyclins and CKIs: Roles beyond cell cycle regulation. Development 2013, 140, 3079–3093. [Google Scholar] [CrossRef]
- Otto, T.; Sicinski, P. Cell cycle proteins as promising targets in cancer therapy. Nat. Rev. Cancer 2017, 17, 93–115. [Google Scholar] [CrossRef]
- Lu, J. Palbociclib: A first-in-class CDK4/CDK6 inhibitor for the treatment of hormone-receptor positive advanced breast cancer. J. Hematol. Oncol. 2015, 8, 98. [Google Scholar] [CrossRef]
- Huang, J.; Zheng, L.; Sun, Z.; Li, J. CDK4/6 inhibitor resistance mechanisms and treatment strategies (Review). Int. J. Mol. Med. 2022, 50, 128. [Google Scholar] [CrossRef]
- Papadimitriou, M.C.; Pazaiti, A.; Iliakopoulos, K.; Markouli, M.; Michalaki, V.; Papadimitriou, C.A. Resistance to CDK4/6 inhibition: Mechanisms and strategies to overcome a therapeutic problem in the treatment of hormone receptor-positive metastatic breast cancer. Biochim. Biophys. Acta Mol. Cell Res. 2022, 1869, 119346. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.Q.; Pan, X.H.; Wang, T.T.; Wang, J.; Yang, B.; He, Q.J.; Ding, L. Intrinsic and acquired resistance to CDK4/6 inhibitors and potential overcoming strategies. Acta Pharmacol. Sin. 2021, 42, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Bousoik, E.; Montazeri Aliabadi, H. “Do We Know Jack” About JAK? A Closer Look at JAK/STAT Signaling Pathway. Front. Oncol. 2018, 8, 287. [Google Scholar] [CrossRef] [PubMed]
- Seavey, M.M.; Dobrzanski, P. The many faces of Janus kinase. Biochem. Pharmacol. 2012, 83, 1136–1145. [Google Scholar] [CrossRef]
- Talpaz, M.; Kiladjian, J.J. Fedratinib, a newly approved treatment for patients with myeloproliferative neoplasm-associated myelofibrosis. Leukemia 2021, 35, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Daver, N.; Kantarjian, H.M.; Verstovsek, S.; Ravandi, F. The role of JAK pathway dysregulation in the pathogenesis and treatment of acute myeloid leukemia. Clin. Cancer Res. 2013, 19, 327–335. [Google Scholar] [CrossRef]
- Bhagwat, N.; Levine, R.L.; Koppikar, P. Sensitivity and resistance of JAK2 inhibitors to myeloproliferative neoplasms. Int. J. Hematol. 2013, 97, 695–702. [Google Scholar] [CrossRef]
- Greuber, E.K.; Smith-Pearson, P.; Wang, J.; Pendergast, A.M. Role of ABL family kinases in cancer: From leukaemia to solid tumours. Nat. Rev. Cancer 2013, 13, 559–571. [Google Scholar] [CrossRef]
- Rossari, F.; Minutolo, F.; Orciuolo, E. Past, present, and future of Bcr-Abl inhibitors: From chemical development to clinical efficacy. J. Hematol. Oncol. 2018, 11, 84. [Google Scholar] [CrossRef]
- Tan, F.H.; Putoczki, T.L.; Stylli, S.S.; Luwor, R.B. Ponatinib: A novel multi-tyrosine kinase inhibitor against human malignancies. Onco Targets Ther. 2019, 12, 635–645. [Google Scholar] [CrossRef]
- Weisberg, E.; Manley, P.; Mestan, J.; Cowan-Jacob, S.; Ray, A.; Griffin, J.D. AMN107 (nilotinib): A novel and selective inhibitor of BCR-ABL. Br. J. Cancer 2006, 94, 1765–1769. [Google Scholar] [CrossRef] [PubMed]
- Braun, T.P.; Eide, C.A.; Druker, B.J. Response and Resistance to BCR-ABL1-Targeted Therapies. Cancer Cell 2020, 37, 530–542. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Kok, C.H.; Saunders, V.A.; Wang, J.; McLean, J.A.; Hughes, T.P.; White, D.L. Modelling ponatinib resistance in tyrosine kinase inhibitor-naive and dasatinib resistant BCR-ABL1+ cell lines. Oncotarget 2018, 9, 34735–34747. [Google Scholar] [CrossRef]
- Lee, C.H.; Hsu, K.W.; Hsieh, Y.Y.; Li, W.T.; Long, Y.; Lin, C.Y.; Chen, S.H. Unveiling IL6R and MYC as Targeting Biomarkers in Imatinib-Resistant Chronic Myeloid Leukemia through Advanced Non-Invasive Apoptosis Detection Sensor Version 2 Detection. Cells 2024, 13, 616. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, S.S.; Muiwo, P.; Ahmad, H.M.; Pandey, R.M.; Singh, S.; Bakhshi, S.; Kumar, L.; Bhattacharya, A.; Gupta, Y.K. miR-505-5p and miR-193b-3p: Potential biomarkers of imatinib response in patients with chronic myeloid leukemia. Leuk. Lymphoma 2017, 58, 1981–1984. [Google Scholar] [CrossRef]
- Martellucci, S.; Clementi, L.; Sabetta, S.; Mattei, V.; Botta, L.; Angelucci, A. Src Family Kinases as Therapeutic Targets in Advanced Solid Tumors: What We Have Learned so Far. Cancers 2020, 12, 1448. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Ahler, E.; Simon, J.J.; Fang, L.; Potter, Z.E.; Sitko, K.A.; Stephany, J.J.; Guttman, M.; Fowler, D.M.; Maly, D.J. Profiling of drug resistance in Src kinase at scale uncovers a regulatory network coupling autoinhibition and catalytic domain dynamics. Cell Chem. Biol. 2024, 31, 207–220 e211. [Google Scholar] [CrossRef] [PubMed]
- Nagaraj, N.S.; Washington, M.K.; Merchant, N.B. Combined blockade of Src kinase and epidermal growth factor receptor with gemcitabine overcomes STAT3-mediated resistance of inhibition of pancreatic tumor growth. Clin. Cancer Res. 2011, 17, 483–493. [Google Scholar] [CrossRef]
- Crook, T.; Patil, D.; Nagarkar, R.; Gaya, A.; Plowman, N.; Limaye, S.; Srivastava, N.; Akolkar, D.; Ranade, A.; Bhatt, A.; et al. Angiogenesis Inhibitors in Personalized Combination Regimens for the Treatment of Advanced Refractory Cancers. Front. Mol. Med. 2021, 1, 749283. [Google Scholar] [CrossRef]
- Andrae, J.; Gallini, R.; Betsholtz, C. Role of platelet-derived growth factors in physiology and medicine. Genes. Dev. 2008, 22, 1276–1312. [Google Scholar] [CrossRef]
- Beenken, A.; Mohammadi, M. The FGF family: Biology, pathophysiology and therapy. Nat. Rev. Drug Discov. 2009, 8, 235–253. [Google Scholar] [CrossRef] [PubMed]
- Salvatore, D.; Santoro, M.; Schlumberger, M. The importance of the RET gene in thyroid cancer and therapeutic implications. Nat. Rev. Endocrinol. 2021, 17, 296–306. [Google Scholar] [CrossRef]
- Bergers, G.; Hanahan, D. Modes of resistance to anti-angiogenic therapy. Nat. Rev. Cancer 2008, 8, 592–603. [Google Scholar] [CrossRef] [PubMed]
- Pircher, A.; Johrer, K.; Kocher, F.; Steiner, N.; Graziadei, I.; Heidegger, I.; Pichler, R.; Leonhartsberger, N.; Kremser, C.; Kern, J.; et al. Biomarkers of evasive resistance predict disease progression in cancer patients treated with antiangiogenic therapies. Oncotarget 2016, 7, 20109–20123. [Google Scholar] [CrossRef] [PubMed]
- Couvreur, P.; Vauthier, C. Nanotechnology: Intelligent design to treat complex disease. Pharm. Res. 2006, 23, 1417–1450. [Google Scholar] [CrossRef] [PubMed]
- Talens-Visconti, R.; Diez-Sales, O.; de Julian-Ortiz, J.V.; Nacher, A. Nanoliposomes in Cancer Therapy: Marketed Products and Current Clinical Trials. Int. J. Mol. Sci. 2022, 23, 4249. [Google Scholar] [CrossRef]
- Chaurasia, M.; Singh, R.; Sur, S.; Flora, S.J.S. A review of FDA approved drugs and their formulations for the treatment of breast cancer. Front. Pharmacol. 2023, 14, 1184472. [Google Scholar] [CrossRef]
- Sadat Tabatabaei Mirakabad, F.; Nejati-Koshki, K.; Akbarzadeh, A.; Yamchi, M.R.; Milani, M.; Zarghami, N.; Zeighamian, V.; Rahimzadeh, A.; Alimohammadi, S.; Hanifehpour, Y.; et al. PLGA-based nanoparticles as cancer drug delivery systems. Asian Pac. J. Cancer Prev. 2014, 15, 517–535. [Google Scholar] [CrossRef]
- Russo, A.; Pellosi, D.S.; Pagliara, V.; Milone, M.R.; Pucci, B.; Caetano, W.; Hioka, N.; Budillon, A.; Ungaro, F.; Russo, G.; et al. Biotin-targeted Pluronic((R)) P123/F127 mixed micelles delivering niclosamide: A repositioning strategy to treat drug-resistant lung cancer cells. Int. J. Pharm. 2016, 511, 127–139. [Google Scholar] [CrossRef]
- Lu, H.L.; Syu, W.J.; Nishiyama, N.; Kataoka, K.; Lai, P.S. Dendrimer phthalocyanine-encapsulated polymeric micelle-mediated photochemical internalization extends the efficacy of photodynamic therapy and overcomes drug-resistance in vivo. J. Control. Release 2011, 155, 458–464. [Google Scholar] [CrossRef]
- Pan, L.; Liu, J.; He, Q.; Wang, L.; Shi, J. Overcoming multidrug resistance of cancer cells by direct intranuclear drug delivery using TAT-conjugated mesoporous silica nanoparticles. Biomaterials 2013, 34, 2719–2730. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Ling, G.; Pan, X.; Sun, J.; Zhang, T.; Pu, X.; Yin, S.; He, Z. Novel nanostructured lipid-dextran sulfate hybrid carriers overcome tumor multidrug resistance of mitoxantrone hydrochloride. Nanomedicine 2012, 8, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Goh, B.; Lu, W.; Zhang, Q.; Chang, A.; Liu, X.Y.; Tan, T.M.; Lee, H. In vitro cytotoxicity of Stealth liposomes co-encapsulating doxorubicin and verapamil on doxorubicin-resistant tumor cells. Biol. Pharm. Bull. 2005, 28, 822–828. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Zhang, D.; Zhang, Q.; Chen, Y.; Zheng, D.; Hao, L.; Duan, C.; Jia, L.; Liu, G.; Liu, Y. Synergistic effect of folate-mediated targeting and verapamil-mediated P-gp inhibition with paclitaxel-polymer micelles to overcome multi-drug resistance. Biomaterials 2011, 32, 9444–9456. [Google Scholar] [CrossRef]
- Jia, L.; Li, Z.; Shen, J.; Zheng, D.; Tian, X.; Guo, H.; Chang, P. Multifunctional mesoporous silica nanoparticles mediated co-delivery of paclitaxel and tetrandrine for overcoming multidrug resistance. Int. J. Pharm. 2015, 489, 318–330. [Google Scholar] [CrossRef] [PubMed]
- Sousa, C.; Videira, M. Dual Approaches in Oncology: The Promise of siRNA and Chemotherapy Combinations in Cancer Therapies. Onco 2025, 5, 2. [Google Scholar] [CrossRef]
- Pallathadka, H.; Jabir, M.; Rasool, K.H.; Hanumanthaiah, M.; Sharma, N.; Pramanik, A.; Rab, S.O.; Jawad, S.F.; Oghenemaro, E.F.; Mustafa, Y.F. siRNA-based therapy for overcoming drug resistance in human solid tumours; molecular and immunological approaches. Hum. Immunol. 2025, 86, 111221. [Google Scholar] [CrossRef]
- Yang, Y.; Qiu, J.-G.; Li, Y.; Di, J.-M.; Zhang, W.-J.; Jiang, Q.-W.; Zheng, D.-W.; Chen, Y.; Wei, M.-N.; Huang, J.-R. Targeting ABCB1-mediated tumor multidrug resistance by CRISPR/Cas9-based genome editing. Am. J. Transl. Res. 2016, 8, 3986. [Google Scholar]
- Ha, J.S.; Byun, J.; Ahn, D.R. Overcoming doxorubicin resistance of cancer cells by Cas9-mediated gene disruption. Sci. Rep. 2016, 6, 22847. [Google Scholar] [CrossRef]
- Ryu, J.Y.; Choi, Y.J.; Won, E.J.; Hui, E.; Kim, H.S.; Cho, Y.S.; Yoon, T.J. Gene editing particle system as a therapeutic approach for drug-resistant colorectal cancer. Nano Res. 2020, 13, 1576–1585. [Google Scholar] [CrossRef]
- Mintz, R.L.; Lao, Y.H.; Chi, C.W.; He, S.; Li, M.; Quek, C.H.; Shao, D.; Chen, B.; Han, J.; Wang, S. CRISPR/Cas9-mediated mutagenesis to validate the synergy between PARP1 inhibition and chemotherapy in BRCA1-mutated breast cancer cells. Bioeng. Transl. Med. 2020, 5, e10152. [Google Scholar] [CrossRef] [PubMed]
- Messersmith, W.A.; Ahnen, D.J. Targeting EGFR in colorectal cancer. N. Engl. J. Med. 2008, 359, 1834–1836. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.; Min, L.; Seebacher, N.A.; Li, X.; Zhou, Y.; Hornicek, F.J.; Wei, Y.; Tu, C.; Duan, Z. Targeting mutant TP53 as a potential therapeutic strategy for the treatment of osteosarcoma. J. Orthop. Res. 2019, 37, 789–798. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.; Wan, J.; Nur, A.A.; Dou, P.; Mankin, H.; Liu, T.; Ouyang, Z. Targeting CD44 by CRISPR-Cas9 in Multi-Drug Resistant Osteosarcoma Cells. Cell Physiol. Biochem. 2018, 51, 1879–1893. [Google Scholar] [CrossRef] [PubMed]
- Coleman, N.; Zhang, B.; Byers, L.A.; Yap, T.A. The role of Schlafen 11 (SLFN11) as a predictive biomarker for targeting the DNA damage response. Br. J. Cancer 2021, 124, 857–859. [Google Scholar] [CrossRef] [PubMed]
- Lok, B.H.; Gardner, E.E.; Schneeberger, V.E.; Ni, A.; Desmeules, P.; Rekhtman, N.; de Stanchina, E.; Teicher, B.A.; Riaz, N.; Powell, S.N.; et al. PARP Inhibitor Activity Correlates with SLFN11 Expression and Demonstrates Synergy with Temozolomide in Small Cell Lung Cancer. Clin. Cancer Res. 2017, 23, 523–535. [Google Scholar] [CrossRef]
- Murai, J.; Feng, Y.; Yu, G.K.; Ru, Y.; Tang, S.W.; Shen, Y.; Pommier, Y. Resistance to PARP inhibitors by SLFN11 inactivation can be overcome by ATR inhibition. Oncotarget 2016, 7, 76534–76550. [Google Scholar] [CrossRef]
- Han, H.A.; Pang, J.K.S.; Soh, B.S. Mitigating off-target effects in CRISPR/Cas9-mediated in vivo gene editing. J. Mol. Med. 2020, 98, 615–632. [Google Scholar] [CrossRef]
- Lino, C.A.; Harper, J.C.; Carney, J.P.; Timlin, J.A. Delivering CRISPR: A review of the challenges and approaches. Drug Deliv. 2018, 25, 1234–1257. [Google Scholar] [CrossRef]
- Zhang, X.H.; Tee, L.Y.; Wang, X.G.; Huang, Q.S.; Yang, S.H. Off-target Effects in CRISPR/Cas9-mediated Genome Engineering. Mol. Ther. Nucleic Acids 2015, 4, e264. [Google Scholar] [CrossRef]
- Joshi, P.; Vishwakarma, R.A.; Bharate, S.B. Natural alkaloids as P-gp inhibitors for multidrug resistance reversal in cancer. Eur. J. Med. Chem. 2017, 138, 273–292. [Google Scholar] [CrossRef] [PubMed]
- Youns, M.; Abdel Halim Hegazy, W. The Natural Flavonoid Fisetin Inhibits Cellular Proliferation of Hepatic, Colorectal, and Pancreatic Cancer Cells through Modulation of Multiple Signaling Pathways. PLoS ONE 2017, 12, e0169335. [Google Scholar] [CrossRef]
- Wang, P.; Yang, H.L.; Yang, Y.J.; Wang, L.; Lee, S.C. Overcome Cancer Cell Drug Resistance Using Natural Products. Evid. Based Complement. Altern. Med. 2015, 2015, 767136. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Pan, S.; Zhang, J.; Xu, J.; Zhang, R.; Zhang, Y.; Fu, Z.; Wang, Y.; Hu, C.; Xu, Z. The role of hyperthermia in the treatment of tumor. Crit. Rev. Oncol. Hematol. 2024, 204, 104541. [Google Scholar] [CrossRef] [PubMed]
- Vilas-Boas, V.; Espiña, B.; Kolen’ko, Y.V.; Bañobre-López, M.; Brito, M.; Martins, V.; Duarte, J.A.; Petrovykh, D.Y.; Freitas, P.; Carvalho, F. Effectiveness and Safety of a Nontargeted Boost for a CXCR4-Targeted Magnetic Hyperthermia Treatment of Cancer Cells. Acs Omega 2019, 4, 1931–1940. [Google Scholar] [CrossRef]
- Franke, K.; Kettering, M.; Lange, K.; Kaiser, W.A.; Hilger, I. The exposure of cancer cells to hyperthermia, iron oxide nanoparticles, and mitomycin C influences membrane multidrug resistance protein expression levels. Int. J. Nanomed. 2013, 8, 351–363. [Google Scholar] [CrossRef]
- Deng, Z.; Yan, F.; Jin, Q.; Li, F.; Wu, J.; Liu, X.; Zheng, H. Reversal of multidrug resistance phenotype in human breast cancer cells using doxorubicin-liposome-microbubble complexes assisted by ultrasound. J. Control. Release 2014, 174, 109–116. [Google Scholar] [CrossRef]
- Yin, T.; Wang, P.; Li, J.; Wang, Y.; Zheng, B.; Zheng, R.; Cheng, D.; Shuai, X. Tumor-penetrating codelivery of siRNA and paclitaxel with ultrasound-responsive nanobubbles hetero-assembled from polymeric micelles and liposomes. Biomaterials 2014, 35, 5932–5943. [Google Scholar] [CrossRef]
- Du, B.; Shim, J.S. Targeting Epithelial-Mesenchymal Transition (EMT) to Overcome Drug Resistance in Cancer. Molecules 2016, 21, 965. [Google Scholar] [CrossRef]
- Xu, G.L.; Ni, C.F.; Liang, H.S.; Xu, Y.H.; Wang, W.S.; Shen, J.; Li, M.M.; Zhu, X.L. Upregulation of PD-L1 expression promotes epithelial-to-mesenchymal transition in sorafenib-resistant hepatocellular carcinoma cells. Gastroenterol. Rep. 2020, 8, 390–398. [Google Scholar] [CrossRef]
- Noman, M.Z.; Janji, B.; Abdou, A.; Hasmim, M.; Terry, S.; Tan, T.Z.; Mami-Chouaib, F.; Thiery, J.P.; Chouaib, S. The immune checkpoint ligand PD-L1 is upregulated in EMT-activated human breast cancer cells by a mechanism involving ZEB-1 and miR-200. Oncoimmunology 2017, 6, e1263412. [Google Scholar] [CrossRef] [PubMed]
- Curiel, T.J. Immunotherapy: A useful strategy to help combat multidrug resistance. Drug Resist. Updat. 2012, 15, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Su, L.; Ao, M.; Guo, X.; Cheng, C.; Luo, Y.; Xie, Z.; Wang, X.; Wang, J.; Liu, S.; et al. Amplified antitumor efficacy by a targeted drug retention and chemosensitization strategy-based “combo” nanoagent together with PD-L1 blockade in reversing multidrug resistance. J. Nanobiotechnol. 2021, 19, 200. [Google Scholar] [CrossRef] [PubMed]
- Aliabadi, H.M.; Mahdipoor, P.; Kucharsky, C.; Chan, N.; Uludag, H. Effect of siRNA pre-Exposure on Subsequent Response to siRNA Therapy. Pharm. Res. 2015, 32, 3813–3826. [Google Scholar] [CrossRef]
- Roell, K.R.; Havener, T.M.; Reif, D.M.; Jack, J.; McLeod, H.L.; Wiltshire, T.; Motsinger-Reif, A.A. Synergistic Chemotherapy Drug Response Is a Genetic Trait in Lymphoblastoid Cell Lines. Front. Genet. 2019, 10, 829. [Google Scholar] [CrossRef]
- Montazeri Aliabadi, H.; Manda, A.; Sidgal, R.; Chung, C. Targeting Breast Cancer: The Familiar, the Emerging, and the Uncharted Territories. Biomolecules 2023, 13, 1306. [Google Scholar] [CrossRef]
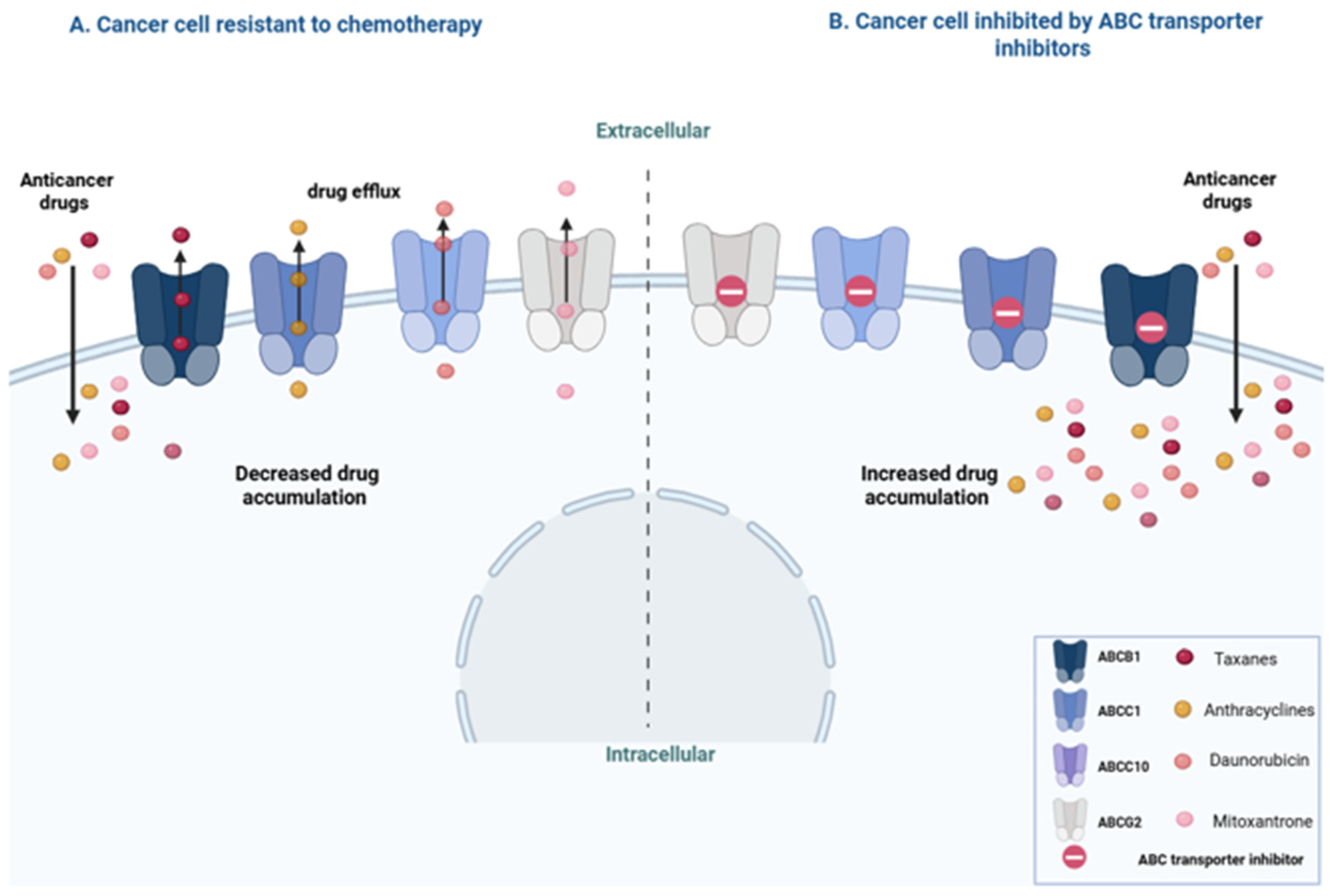
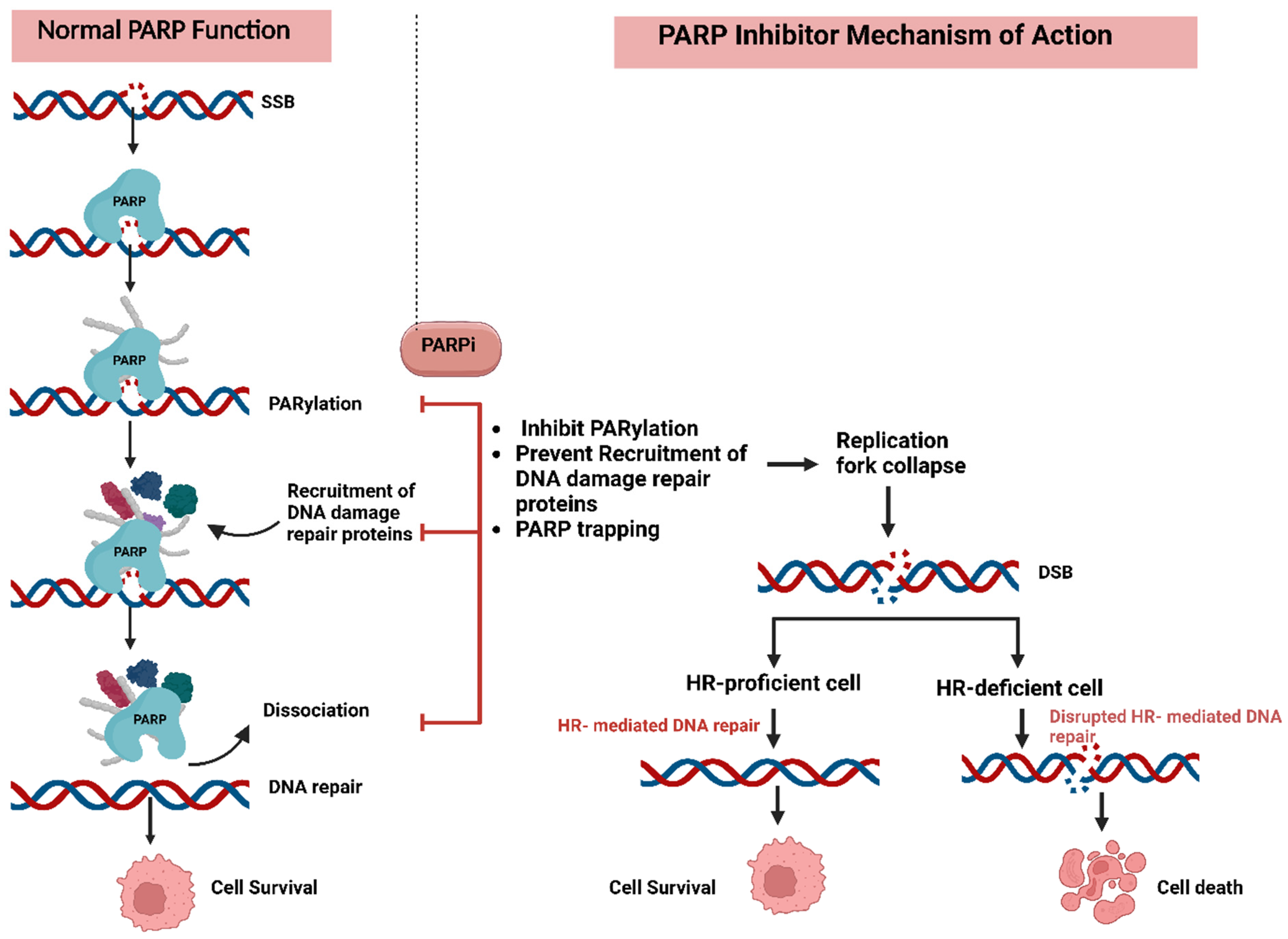





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer Type | ABC Transporter(s) | Expression (↑/↓) | Tumor Behavior/Outcome | Remarks |
|---|---|---|---|---|
| Neuroblastoma | ABCC4 | ↑ | Linked to MYCN amplification, advanced stage, lower event-free and overall survival | Independent prognostic marker after adjusting for age, stage, and MYCN [71]. |
| Glioblastoma | ABCB1, ABCC1 | ABCB1 ↓ in high grade ABCC1 ↑ in high grade | ABCC1 highly expressed in grade III–IV gliomas; associated with malignancy | ABCC1 may indicate undifferentiated glial phenotypes in glioblastoma [72,73,74,75]. |
| Breast Cancer | ABCC11, ABCC1, ABCC8, ABCF1 | ABCC11 ↑, ABCC1 ↑, ABCC8 ↓ | ABCC11 linked to aggressive subtypes (TNBC, HER2+); ABCC8 ↓ expression in higher grades | ABCC11 polymorphism (538G) associated with breast cancer in some populations [76,77,78,79,80,81]. |
| Prostate Cancer | ABCB1, ABCA1 | ABCB1 ↓ (also ↑ in some cases), ABCA1 ↓ | ABCB1 ↓ leads to androgen accumulation and tumor growth; ABCA1 ↓ leads to AKT pathway activation | ABCB1 expression varies by subtype; ABCA1 loss contributes to cholesterol-driven tumor progression [82,83,84,85]. |
| Ovarian Cancer | ABCA1, ABCA4 | ↑ | ABCA1 linked to reduced survival; ABCA4 ↑ in stage I epithelial ovarian cancer | ABCA1 inhibition reduces migration and proliferation in vitro [86,87]. |
| Non-Small Cell Lung Cancer (NSCLC) | ABCB1, ABCG2 | Differential: ↑ in adenocarcinoma (AC)/↓ in squamous cell carcinoma (SCC) | Higher expression in AC contributes to chemoresistance via increased efflux; cisplatin upregulates both in AC and SCC through beta-catenin activation | Wnt microenvironment dictates transporter expression: Wnt7b (canonical) ↑ ABC transporters in AC; Wnt5a (non-canonical) suppresses them in SCC. Cisplatin induces beta-catenin pathway, upregulating ABCB1 and ABCG2 even in SCC [88]. |
| Inhibitor Class | Targets | Key Agents | Cancer Types | Resistance Mechanisms | Biomarkers |
|---|---|---|---|---|---|
| ALK Inhibitors [220,221] | ALK | Crizotinib [221], Ceritinib, Alectinib [222], Brigatinib, Lorlatinib | NSCLC [223], neuroblastoma, lymphoma | CNS penetration limits (crizotinib), secondary ALK mutations | ALK mutations (e.g., G1202R) [224] |
| TRK/FLT3 Inhibitors [225,226] | TRKA/B/C, FLT3 | Larotrectinib, Entrectinib, Midostaurin | Sarcomas, breast cancer, AML | NTRK mutations (e.g., solvent-front) [227], PTEN loss [228], FLT3-ITD mutations [229] | FL3 mutations [230] |
| EGFR-Family Inhibitors [231,232,233] | EGFR, HER2, HER3, HER4 | Erlotinib, Gefitinib, Osimertinib, Mobocertinib, Trastuzumab | NSCLC, glioblastoma, breast cancer | T790M [234,235], C797S [236], bypass signaling EGFR exon 20 insertions, HER2 amplifications, downstream mutations (PIK3CA, AKT), EMT [237], SCLC transformation [238], BIM loss, PD-L1 overexpression [239] | EGFR T790M (for resistance to 1st/2nd gen TKIs → guides osimertinib use) [240] |
| MET Inhibitors [241] | MET | Tepotinib, Capmatinib [242] | NSCLC [242] | PI3K/AKT, RAS/RAF pathway overactivation [243] | MET dysregulation (e.g., exon 14 skipping, amplification) [244] |
| PI3K/mTOR Inhibitors [245] | PI3K isoforms, mTORC1/2 | Alpelisib, Copanlisib, Temsirolimus, Everolimus | Lymphoma, breast cancer | Feedback loops [246], ERK/MAPK crosstalk [247] | Positive pS6rp staining combined with KRAS mutation [248] |
| RAS/RAF/MEK Inhibitors | KRAS [249], BRAF [250], MEK [251] | Sotorasib [252], Dabrafenib, Trametinib [253] | Melanoma, colorectal, thyroid cancer | Pathway reactivation, compensatory signaling [254,255,256] | Lack of expression of DUSP6 [257], mutations in NRAS, RAC1, MAP2K1, MAP2K2, and NF1 [258] |
| CDK Inhibitors [259,260] | CDK4/6 | Abemaciclib, Ribociclib, Palbociclib [261] | Breast cancer | In-target mutations, off-target activation [262,263,264] | Biomarkers of inherent resistance (e.g., cyclin E1, Rb1) and acquired resistance (e.g., AURKA) [213] |
| JAK Inhibitors | JAK1/2/3, TYK2 [265,266] | Ruxolitinib, Fedratinib [267] | Myeloproliferative neoplasms [268] | Crosstalk with PI3K/AKT/MAPK, secondary mutations [269] | Secondary mutations on Jak2 [269] |
| BCR-ABL Inhibitors | BCR-ABL1 [270] | Imatinib, Nilotinib, Bosutinib [271], Ponatinib [272], Asciminib [273] | CML, ALL | T315I mutation [274], compound mutations [275], compensatory activation | IL6R, IL7R, and MYC expression [276], miRNAs [277] |
| SFK Inhibitors | Src-family kinases | Dasatinib (Broad Spectrum, Not Specific [278]) | Various | Lack of specific inhibitors, compensatory activation [279] | STAT3 overactivation [280] |
| Angiogenesis Inhibitors | VEGFR, PDGFR, FGFR [281,282,283], RET [284] | Sorafenib, Sunitinib, Lenvatinib, Pazopanib | Renal, thyroid, ovarian cancer | Common resistance as with other TKIs [285] | Circulating endothelial progenitor cells (CEP)/circulating endothelial cells (CEC) populations, proangiogenic cytokines and tumor endothelial markers (TEMs) [286] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alhazza, A.; Oyegbesan, A.; Bousoik, E.; Montazeri Aliabadi, H. Multidrug Resistance: Are We Still Afraid of the Big Bad Wolf. Pharmaceuticals 2025, 18, 895. https://doi.org/10.3390/ph18060895
Alhazza A, Oyegbesan A, Bousoik E, Montazeri Aliabadi H. Multidrug Resistance: Are We Still Afraid of the Big Bad Wolf. Pharmaceuticals. 2025; 18(6):895. https://doi.org/10.3390/ph18060895
Chicago/Turabian StyleAlhazza, Abdulelah, Adenike Oyegbesan, Emira Bousoik, and Hamidreza Montazeri Aliabadi. 2025. "Multidrug Resistance: Are We Still Afraid of the Big Bad Wolf" Pharmaceuticals 18, no. 6: 895. https://doi.org/10.3390/ph18060895
APA StyleAlhazza, A., Oyegbesan, A., Bousoik, E., & Montazeri Aliabadi, H. (2025). Multidrug Resistance: Are We Still Afraid of the Big Bad Wolf. Pharmaceuticals, 18(6), 895. https://doi.org/10.3390/ph18060895