

Harnessing the Therapeutic Potential of Pomegranate Peel-Derived Bioactive Compounds in Pancreatic Cancer: A Computational Approach



Abstract
1. Introduction
2. Results
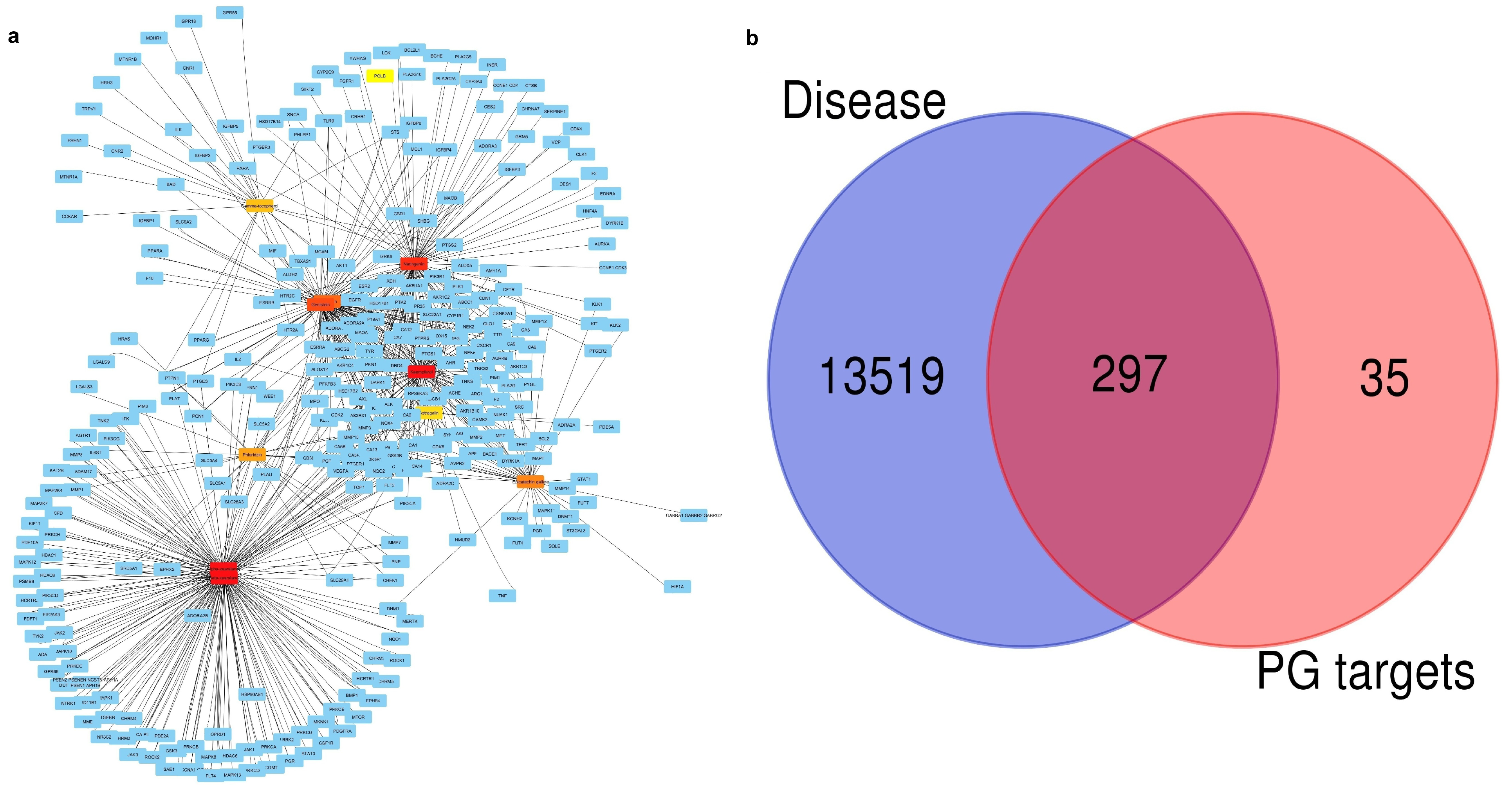
2.1. Finding Active Compounds and Common Target Proteins
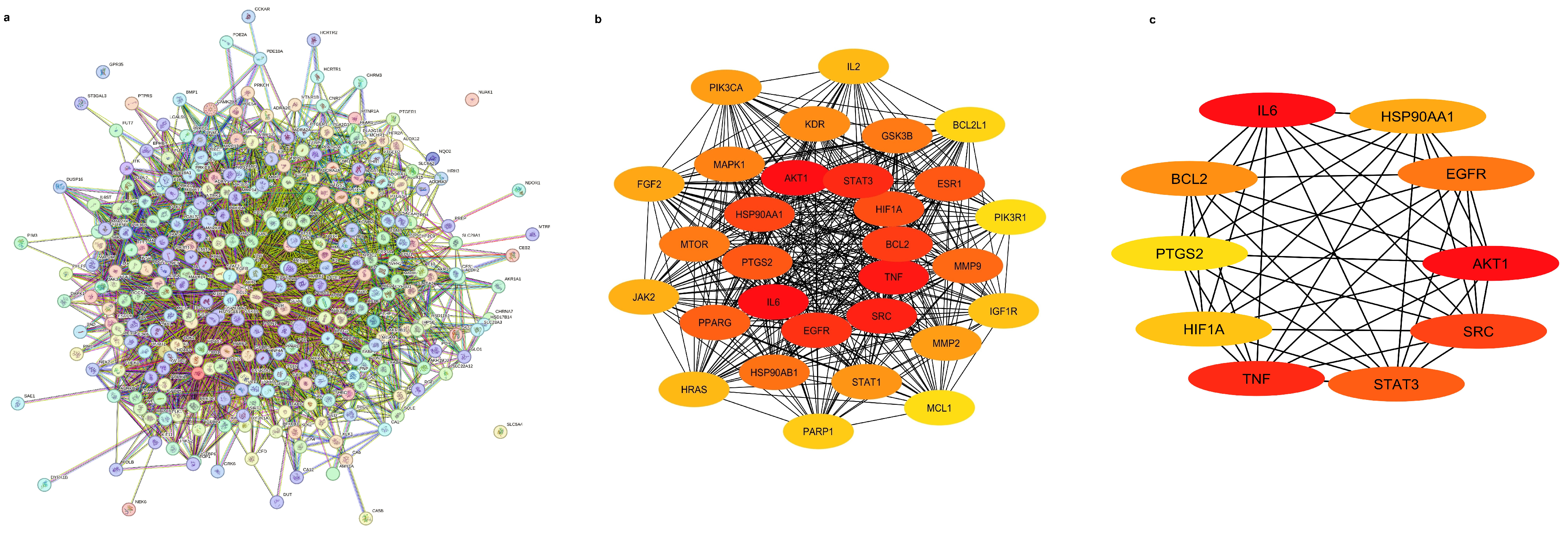
2.2. Investigation of Protein–Protein Network Interactions
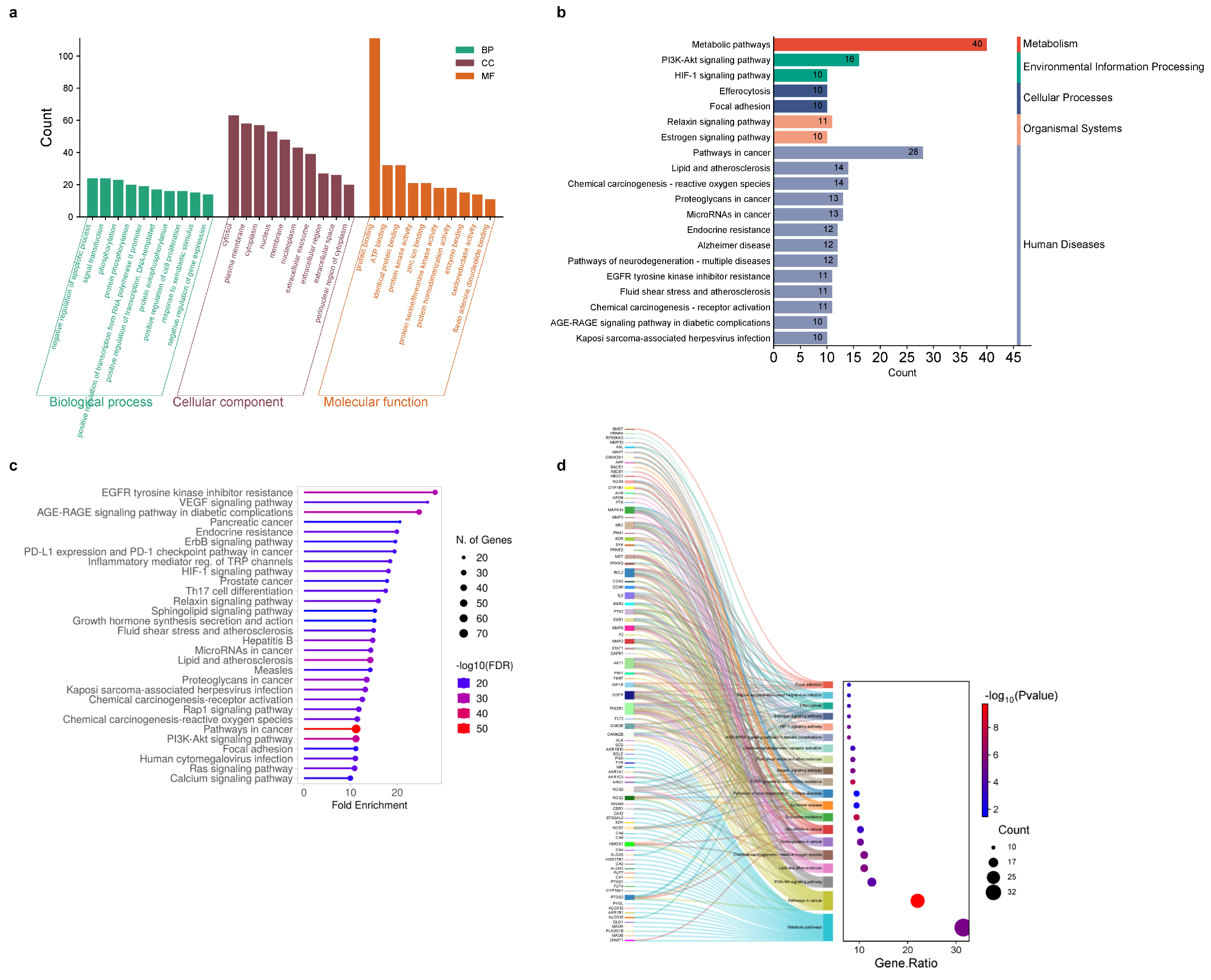
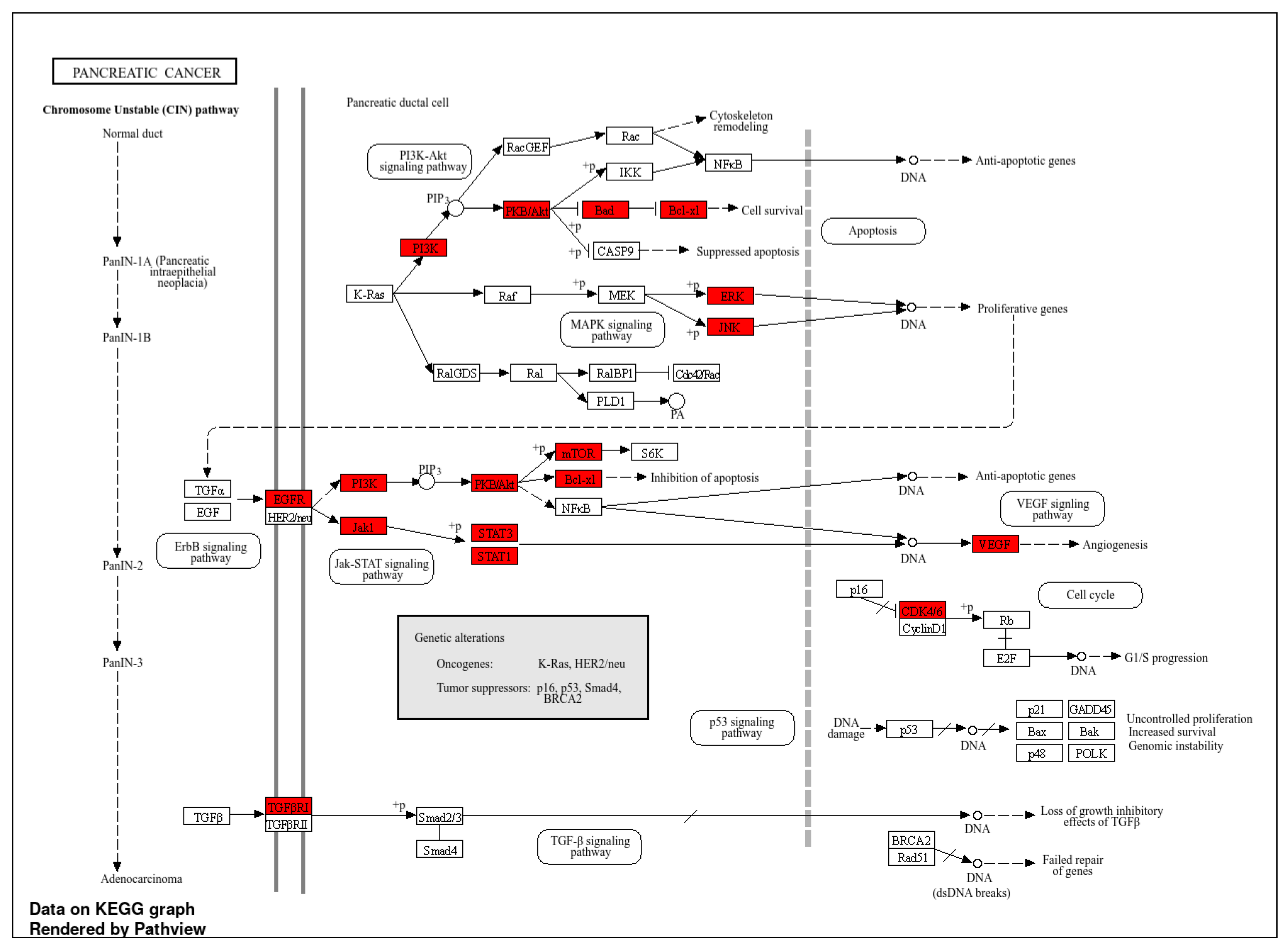
2.3. Go and KEGG Terms Analysis
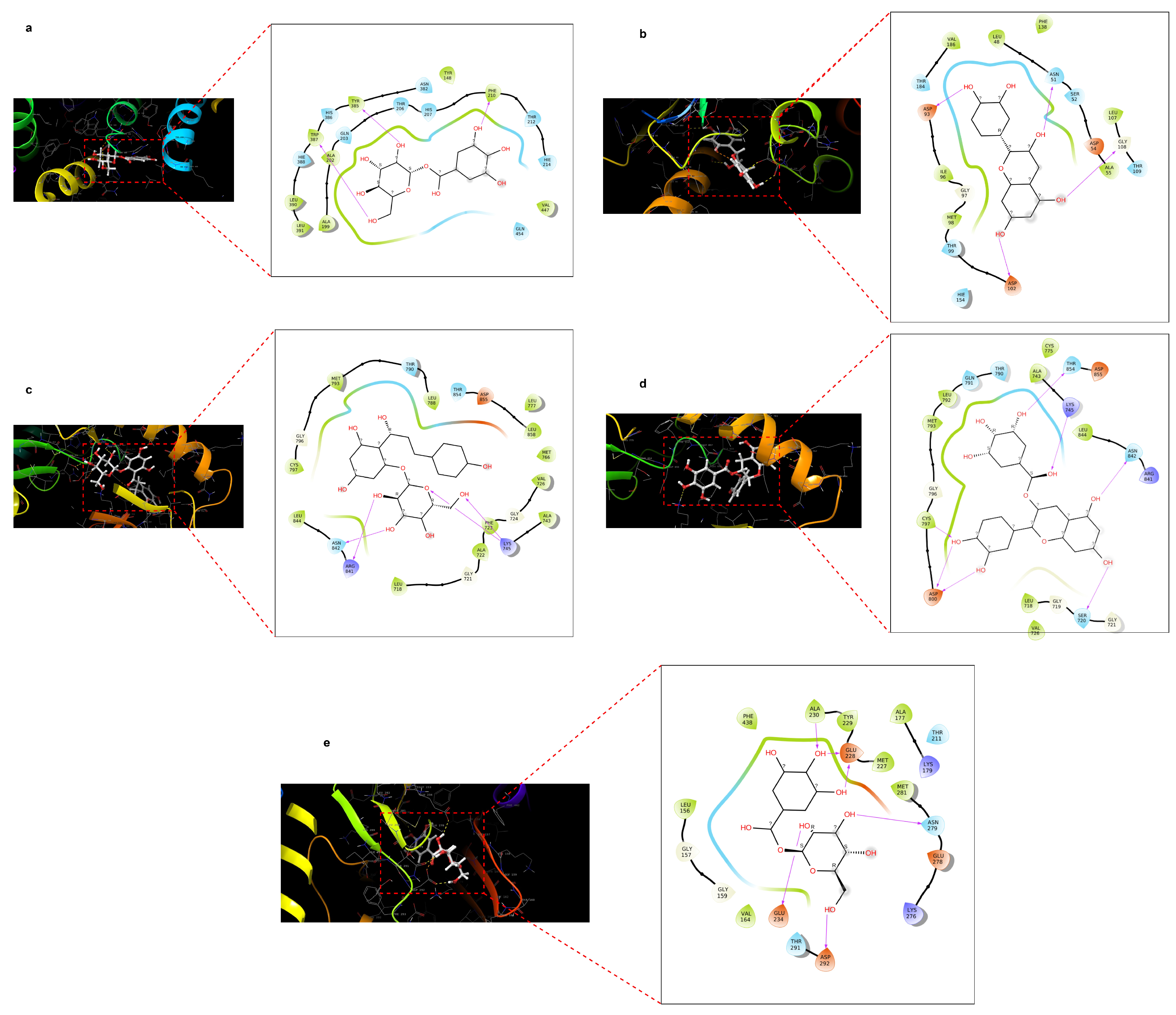
2.4. Docking Score of Core Targets with Compounds
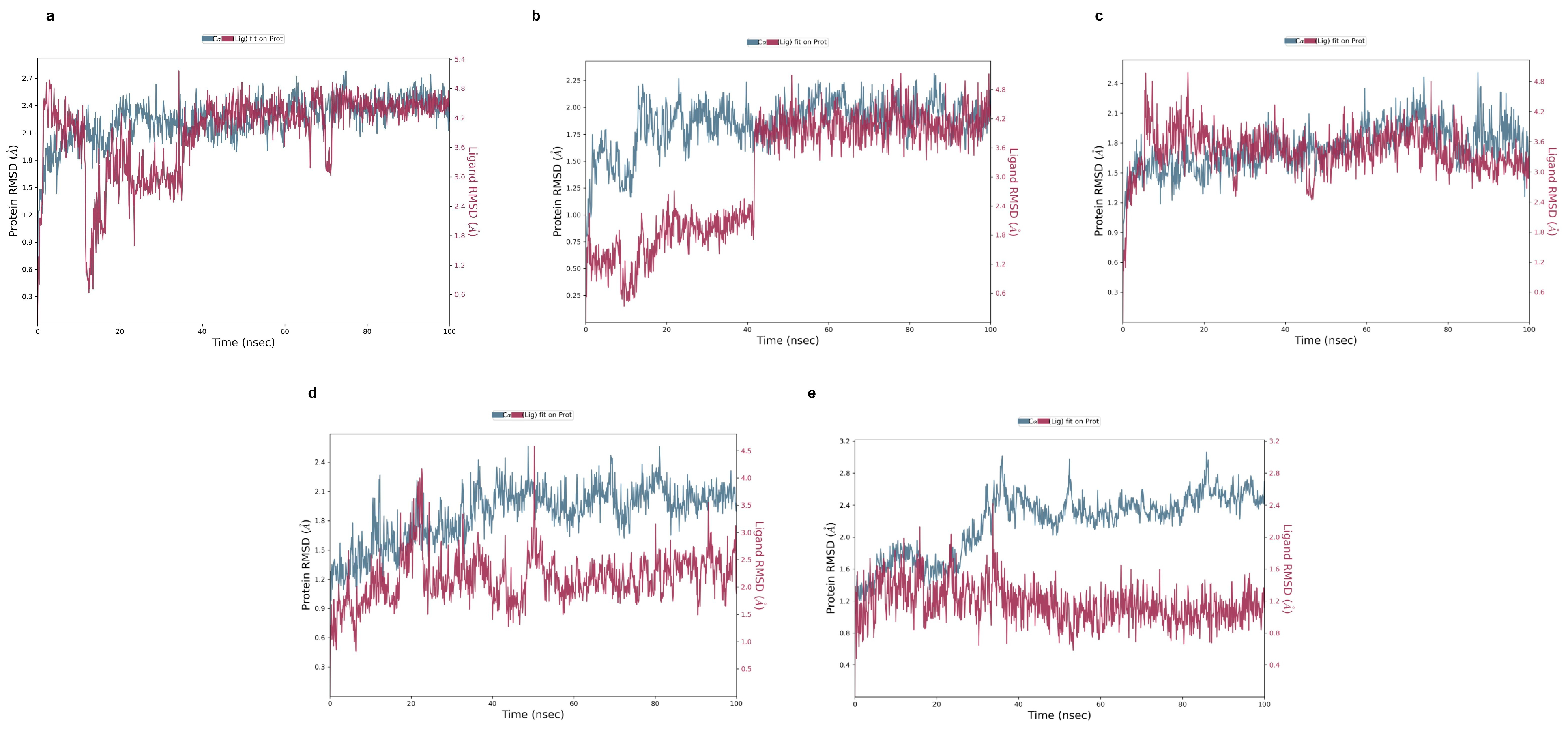
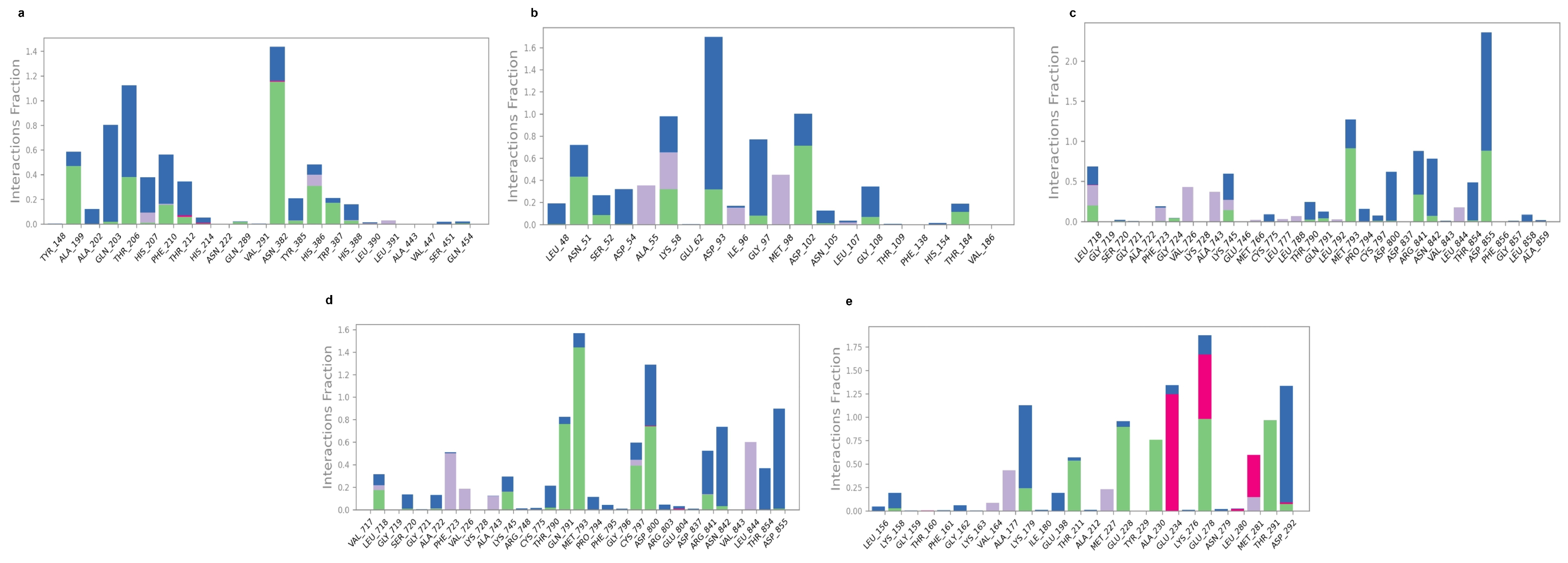
2.5. Molecular Dynamic Simulation
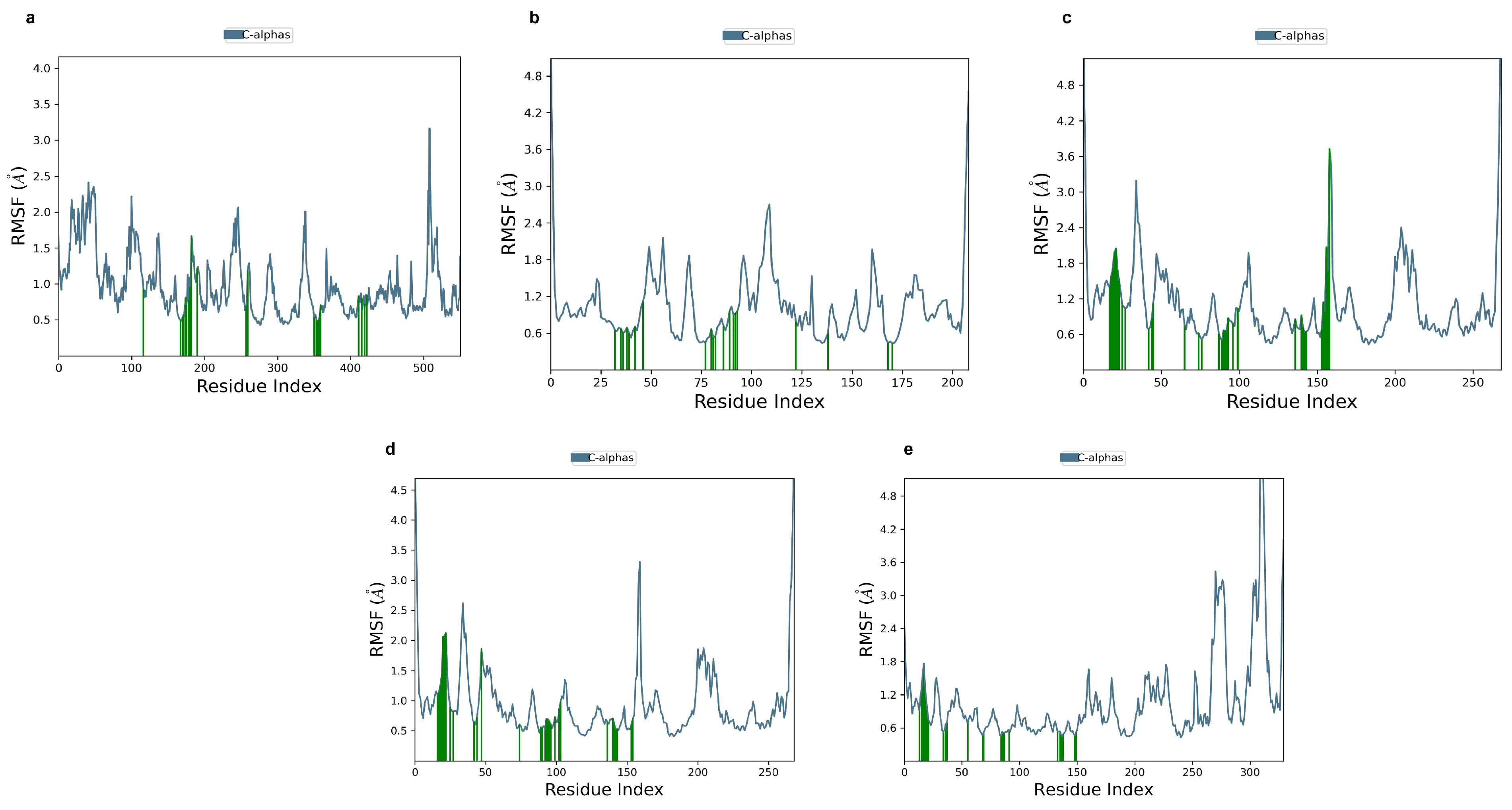
2.6. Ligand-Free Protein RMSF
2.7. In Silico ADME, Toxicity, and Lipinski and Pfizer’s Rule
3. Discussion
4. Materials and Methods
4.1. Screening of Bioactive Compounds
4.2. Identification of Pancreatic Cancer and the Compound’s Target Genes
4.3. Exploration of Overlapping Genes
4.4. Hub Gene Analysis Through Protein–Protein Network
4.5. Bioinformatics Technique for GO and KEGG Analysis
4.6. Molecular Docking of Hub Genes
4.6.1. Ligand Preparation
4.6.2. Protein Preparation and Docking Setup
4.7. Validation of Molecular Docking
4.8. Assessment of Protein–Ligand Complex Stability
4.9. In Silico Pharmacokinetics (ADME) and Toxicity Assessment
4.10. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| PC | Pancreatic cancer |
| PG | Pomegranate |
| PP | Pomegranate peel |
| GO | Gene ontology |
| KEGG | Kyoto Encyclopedia of genes and genomes |
| DL | Drug likeness |
| BO | Bio-availability |
| PPI | Protein–protein interaction |
| STRING | Search tool for the retrieval of interacting genes/proteins |
| DC | Degree of connectivity |
| CC | Closeness centrality |
| BP | Biological processes |
| MF | Molecular function |
| DAVID | Database for annotation, visualization, and integrated discovery |
| RCSB | Research collaboratory for structural bioinformatics |
| PDB | Protein data bank |
| RMSD | Root mean square deviation |
| RMSF | Root mean square fluctuation |
| AKT1 | RAC-alpha serine/threonine-protein kinase |
| IL6 | Interleukin-6 |
| TNF | Tumor necrosis factor |
| STAT3 | Signal transducer and activator of transcription 3 |
| EGFR | Epidermal growth factor receptor |
| BCL2 | Apoptosis regulator Bcl2 |
| HSP90AA1 | Heat shock protein HSP90-alpha |
| HIF1A | Hypoxia-inducible factor 1-alpha |
| PTGS2 | Prostaglandin G/H synthase 2 |
References
- An, J.; An, S.; Choi, M.; Jung, J.H.; Kim, B. Natural Products for Esophageal Cancer Therapy: From Traditional Medicine to Modern Drug Discovery. Int. J. Mol. Sci. 2022, 23, 13558. [Google Scholar] [CrossRef] [PubMed]
- Hameed, B.S.; Krishnan, U.M. Artificial Intelligence-Driven Diagnosis of Pancreatic Cancer. Cancers 2022, 14, 5382. [Google Scholar] [CrossRef] [PubMed]
- Klatte, D.C.F.; Wallace, M.B.; Löhr, M.; Bruno, M.J.; Van Leerdam, M.E. Hereditary pancreatic cancer. Best Pract. Res. Clin. Gastroenterol. 2022, 58–59, 101783. [Google Scholar] [CrossRef]
- Xia, H.; Wang, N.; Zhang, Y.; Huang, X.; Wang, Y. The therapeutic potential of natural products for treating pancreatic cancer. Front. Pharmacol. 2022, 13, 1051952. [Google Scholar]
- Irena, I.; Ilic, M. International patterns in incidence and mortality trends of pancreatic cancer in the last three decades: A joinpoint regression analysis. World J. Gastroenterol. 2022, 28, 4698. [Google Scholar]
- Asiamah, I.; Obiri, S.A.; Tamekloe, W.; Armah, F.F.; Borquaye, L.S. Applications of molecular docking in natural products-based drug discovery. Sci. Afr. 2023, 20, e01593. [Google Scholar] [CrossRef]
- Xiang, L. Natural products as inhibitors against pancreatic cancer cell proliferation and invasion: Possible mechanisms. Am. J. Cancer Res. 2024, 14, 2695–2713. [Google Scholar]
- Xu, C.; Pascual-Sabater, S.; Fillat, C.; Goel, A. The LAMB3-EGFR signaling pathway mediates synergistic Anti-Cancer effects of berberine and emodin in Pancreatic cancer. Biochem. Pharmacol. 2024, 228, 116509. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Li, J.; Xiong, R.-G.; Wu, S.-X.; Huang, S.-Y.; Zhou, D.-D.; Saimaiti, A.; Shang, A.; Feng, Y.; Gan, R.-Y.; et al. Bioactive compounds and health benefits of pomegranate: An updated narrative review. Food Biosci. 2023, 53, 102629. [Google Scholar] [CrossRef]
- El Hosry, L.; Bou-Mitri, C.; Dargham, M.B.; Jaoudeh, M.A.; Farhat, A.; Hayek, J.E.; Mosleh, J.M.B.; Bou-Maroun, E. Phytochemical composition, biological activities and antioxidant potential of pomegranate fruit, juice and molasses: A review. Food Biosci. 2023, 55, 103034. [Google Scholar] [CrossRef]
- Peršurić, Ž.; Martinović, L.S.; Malenica, M.; Gobin, I.; Pedisić, S.; Dragović-Uzelac, V.; Pavelić, S.K. Assessment of the Biological Activity and Phenolic Composition of Ethanol Extracts of Pomegranate (Punica granatum L.) Peels. Mol. 2020, 25, 5916. [Google Scholar] [CrossRef] [PubMed]
- Selahvarzi, A.; Ramezan, Y.; Sanjabi, M.R.; Namdar, B.; Akbarmivehie, M.; Mirsaeedghazi, H.; Azarikia, F. Optimization of ultrasonic-assisted extraction of phenolic compounds from pomegranate and orange peels and their antioxidant activity in a functional drink. Food Biosci. 2022, 49, 101918. [Google Scholar] [CrossRef]
- Zhang, M.; Zhang, Y.; Guo, X.; Chen, Y.; Li, H.; Zhou, H.; Sun, S.; Ren, Q.; Simal-Gandara, J.; Sun, J.; et al. Extraction, purification and anticancer activity studies on triterpenes from pomegranate peel. Food Funct. 2024, 15, 6914–6928. [Google Scholar] [CrossRef]
- Mohlamonyane, M.J.; Adeyemi, J.O.; Fawole, O.A. Pomegranate fruit peel: A sustainable bioresource in food preservation. Food Biosci. 2024, 62, 105532. [Google Scholar] [CrossRef]
- Siddiqui, S.A.; Singh, S.; Nayik, G.A. Bioactive compounds from pomegranate peels—Biological properties, structure–function relationships, health benefits and food applications—A comprehensive review. J. Funct. Foods 2024, 116, 106132. [Google Scholar] [CrossRef]
- Xiang, Q.; Li, M.; Wen, J.; Ren, F.; Yang, Z.; Jiang, X.; Chen, Y. The bioactivity and applications of pomegranate peel extract: A review. J. Food Biochem. 2022, 46, e14105. [Google Scholar] [CrossRef]
- Luís, C.; Sousa, A.P.; Costa, R.; Maduro, A.T.; Pais, P.J.; Sá, S.; Gestoso, Á.; Fernandes, F.; Jerónimo, E.; Soares, R.; et al. Exploring the Anti-Cancer Properties of Pomegranate Peel Aqueous Extract. Appl. Sci. 2023, 13, 11773. [Google Scholar] [CrossRef]
- Wang, K.; Miao, X.; Kong, F.; Huang, S.; Mo, J.; Jin, C.; Zheng, Y. Integrating Network Pharmacology and Experimental Verification to Explore the Mechanism of Effect of Zuojin Pills in Pancreatic Cancer Treatment. Drug Des. Devel. Ther. 2021, 15, 3749–3764. [Google Scholar] [CrossRef]
- Pokhrel, A.; Chong, K.T.; Tayara, H. Therapeutic potential of curcuminoids in type 2 diabetes mellitus (T2DM): Insights from network pharmacology, molecular docking, and dynamics simulations. Food Biosci. 2025, 68, 106406. [Google Scholar] [CrossRef]
- Araújo, P.H.F.; Ramos, R.S.; Cruz, J.N.D.; Silva, S.G.; Ferreira, E.F.B.; Lima, L.R.D.; Macêdo, W.J.C.; Espejo-Román, J.M.; Campos, J.M.; Santos, C.B.R. Identification of Potential COX-2 Inhibitors for the Treatment of Inflammatory Diseases Using Molecular Modeling Approaches. Molecules 2020, 25, 4183. [Google Scholar] [CrossRef]
- Shah, M.; Bibi, S.; Kamal, Z.; Al-Sabahi, J.N.; Alam, T.; Ullah, O.; Murad, W.; Rehman, N.U.; Al-Harrasi, A. Bridging the Chemical Profile and Biomedical Effects of Scutellaria edelbergii Essential Oils. Antioxidants 2022, 11, 1723. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.; Singh, J.P.; Kaur, A.; Singh, N. Phenolic compounds as beneficial phytochemicals in pomegranate (Punica Granatum L.) Peel: A Review. Food Chem. 2018, 261, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Mo, Y.; Ma, J.; Gao, W.; Zhang, L.; Li, J.; Li, J.; Zang, J. Pomegranate Peel as a Source of Bioactive Compounds: A Mini Review on Their Physiological Functions. Front. Nutr. 2022, 9, 887113. [Google Scholar] [CrossRef] [PubMed]
- Jia, C.-Y.; Li, J.-Y.; Hao, G.-F.; Yang, G.-F. A drug-likeness toolbox facilitates ADMET study in drug discovery. Drug Discov. Today 2020, 25, 248–258. [Google Scholar] [CrossRef]
- Tuo, Y.; Lu, X.; Tao, F.; Tukhvatshin, M.; Xiang, F.; Wang, X.; Shi, Y.; Lin, J.; Hu, Y. The Potential Mechanisms of Catechins in Tea for Anti-Hypertension: An Integration of Network Pharmacology, Molecular Docking, and Molecular Dynamics Simulation. Foods 2024, 13, 2685. [Google Scholar] [CrossRef]
- Rehan, M.; Ahmed, F.; Khan, M.I.; Ansari, H.R.; Shakil, S.; El-Araby, M.E.; Hosawi, S.; Saleem, M. Computational insights into the stereo-selectivity of catechins for the inhibition of the cancer therapeutic target EGFR kinase. Front. Pharmacol. 2024, 14, 1231671. [Google Scholar] [CrossRef]
- Al-Karmalawy, A.A.; Dahab, M.A.; Metwaly, A.M.; Elhady, S.S.; Elkaeed, E.B.; Eissa, I.H.; Darwish, K.M. Molecular Docking and Dynamics Simulation Revealed the Potential Inhibitory Activity of ACEIs Against SARS-CoV-2 Targeting the hACE2 Receptor. Front. Chem. 2021, 9, 661230. [Google Scholar] [CrossRef]
- Stanciu, S.; Ionita-Radu, F.; Stefani, C.; Miricescu, D.; Stanescu-Spinu, I.-I.; Greabu, M.; Totan, A.R.; Jinga, M. Targeting PI3K/AKT/mTOR Signaling Pathway in Pancreatic Cancer: From Molecular to Clinical Aspects. Int. J. Mol. Sci. 2022, 23, 10132. [Google Scholar] [CrossRef]
- Chen, C.; Jiang, Y.-P.; You, I.; Gray, N.S.; Lin, R.Z. Down-Regulation of AKT Proteins Slows the Growth of Mutant-KRAS Pancreatic Tumors. Cells 2024, 13, 1061. [Google Scholar] [CrossRef]
- Sourav, C.; Chong, K.T.; Tayara, H. Exploring Nigella Sativa Anticancer. Prop. Using Netw. Pharmacol. Mol. Docking Mol. Dyn. Simul. Approach Non-Small Cell Lung Cancer. Food Biosci. 2025, 63, 105525. [Google Scholar] [CrossRef]
- Ma, X.; Liu, X.; Ou, K.; Zhang, M.; Gao, L.; Yang, L. Advanced pancreatic cancer with KRAS wild-type and EGFR-sensitive mutation respond favorably to furmonertinib: A case report. Front. Oncol. 2023, 13, 1151178. [Google Scholar] [CrossRef] [PubMed]
- Mucciolo, G.; Henríquez, J.A.; Jihad, M.; Teles, S.P.; Manansala, J.S.; Li, W.; Ashworth, S.; Lloyd, E.G.; Cheng, P.S.W.; Luo, W.; et al. EGFR-activated myofibroblasts promote metastasis of pancreatic cancer. Cancer Cell 2024, 42, 101–118.e11. [Google Scholar] [CrossRef]
- Jiao, J.; Cheng, C.; Xu, P.; Yang, P.; Zhang, K.; Jing, Y.; Chen, Z. Mechanisms of pancreatic tumor suppression mediated by Xiang-lian pill: An integrated in silico exploration and experimental validation. J. Ethnopharmacol. 2022, 298, 115586. [Google Scholar] [CrossRef] [PubMed]
- Shukla, V.; Chandra, V.; Sankhwar, P.; Popli, P.; Kaushal, J.B.; Sirohi, V.K.; Dwivedi, A. Phytoestrogen genistein inhibits EGFR/PI3K/NF-kB activation and induces apoptosis in human endometrial hyperplasial cells. RSC Adv. 2015, 5, 56075–56085. [Google Scholar] [CrossRef]
- Rahman, N.; Khan, H.; Zia, A.; Khan, A.; Fakhri, S.; Aschner, M.; Saso, K.G.L. Bcl-2 Modulation in p53 Signaling Pathway by Flavonoids: A Potential Strategy towards the Treatment of Cancer. Int. J. Mol. Sci. 2021, 22, 11315. [Google Scholar] [CrossRef]
- Ahmad, I.; Kuznetsov, A.E.; Pirzada, A.S.; Alsharif, K.F.; Daglia, M.; Khan, H. Computational pharmacology and computational chemistry of 4-hydroxyisoleucine: Physicochemical, pharmacokinetic, and DFT-based approaches. Front. Chem. 2023, 11, 1145974. [Google Scholar] [CrossRef]
- Niu, Z.; Xiao, X.; Wu, W.; Cai, Q.; Jiang, Y.; Jin, W.; Wang, M.; Yang, G.; Kong, L.; Jin, X.; et al. PharmaBench: Enhancing ADMET benchmarks with large language models. Sci. Data 2024, 11, 985. [Google Scholar] [CrossRef] [PubMed]
- Bitew, M.; Desalegn, T.; Demissie, T.B.; Belayneh, A.; Endale, M.; Eswaramoorthy, R. Pharmacokinetics and drug-likeness of antidiabetic flavonoids: Molecular docking and DFT study. PLoS ONE 2021, 16, e0260853. [Google Scholar] [CrossRef]
- Jeevan, K.; Palistha, S.; Tayara, H.; Chong, K.T. PUResNetV2.0: A deep learning model leveraging sparse representation for improved ligand binding site prediction. J. Cheminform. 2024, 16, 66. [Google Scholar] [CrossRef]
- Mouli, H.M.C.; Harini, D.; Shaikh, N.; Khemchandani, R.; Shreya, S.; Jana, A.; Samanthula, G. In silico characterization of indole-substituted densely functionalized pyrrole against breast cancer: Integrating DFT, molecular docking, MD simulations, and ADME analysis. J. Mol. Struct. 2025, 1328, 141375. [Google Scholar] [CrossRef]
- Lu, C.; Wu, C.; Ghoreishi, D.; Chen, W.; Wang, L.; Damm, W.; Ross, G.A.; Dahlgren, M.K.; Russell, E.; Bargen, C.D.V.; et al. OPLS4: Improving Force Field Accuracy on Challenging Regimes of Chemical Space. J. Chem. Theory Comput. 2021, 17, 4291–4300. [Google Scholar] [CrossRef] [PubMed]
- Nassar, H.; Sippl, W.; Dahab, R.A.; Taha, M. Molecular docking, molecular dynamics simulations and in vitro screening reveal cefixime and ceftriaxone as GSK3b covalent inhibitors. RSC Adv. 2023, 13, 11278–11290. [Google Scholar] [CrossRef] [PubMed]
- Sabarees, G.; Velmurugan, V.; Solomon, V.R. Molecular docking and molecular dynamics simulations discover curcumin analogs as potential wound healing agents. Chem. Phys. Impact 2024, 8, 100441. [Google Scholar] [CrossRef]
- Bhaumik, S.; Sarkar, A.; Debnath, S.; Debnath, B.; Ghosh, R.; Zaki, M.E.A.; Al-Hussain, S.A. Alpha-Glucosidase inhibitory potential of Oroxylum indicum using molecular docking, molecular dynamics, and in vitro evaluation. Saudi Pharm. J. 2024, 32, 102095. [Google Scholar] [CrossRef]
- Tripathi, S.K.; Muttineni, R.; Singh, S.K. Extra precision docking, free energy calculation and molecular dynamics simulation studies of CDK2 inhibitors. J. Theor. Biol. 2013, 334, 87–100. [Google Scholar] [CrossRef]
- Gheidari, D.; Mehrdad, M.; Bayat, M. Synthesis, docking, MD simulation, ADMET, drug likeness, and DFT studies of novel furo[2,3-b]indol-3a-ol as promising Cyclin-dependent kinase 2 inhibitors. Sci. Rep. 2024, 14, 3084. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | CID No. | Molecular Formula | Molecular Weight (gm/mol) | Drug Likeness (DL) ≥ 0.18 | Oral Bioavailabilty (OB) ≥ 0.3 |
|---|---|---|---|---|---|
| Catechin | 9064 | C15H14O6 | 290.27 | 0.64 | 0.55 |
| Epicatechin | 72276 | C15H14O6 | 290.27 | 0.64 | 0.55 |
| Naringenin | 439246 | C15H12O5 | 272.25 | 0.82 | 0.55 |
| Phloridzin | 6072 | C21H24O10 | 436.41 | 0.66 | 0.55 |
| Genistein | 5280961 | C15H10O5 | 270.24 | 0.44 | 0.55 |
| Gamma-Tocopherol | 92729 | C28H48O2 | 416.68 | 0.48 | 0.55 |
| Daidzein | 5281708 | C15H10O4 | 254.24 | 0.29 | 0.55 |
| Quinic acid | 6508 | C7H12O6 | 192.17 | 0.19 | 0.56 |
| 1-O-Galloyl-beta-D-glucose | 124021 | C13H16O10 | 332.26 | 0.81 | 0.55 |
| Palmitelaidic acid | 5282745 | C16H30O2 | 254.41 | 0.87 | 0.56 |
| Epicatechin gallate | 107905 | C22H18O10 | 442.37 | 0.93 | 0.55 |
| Alpha-Zearalanol | 2999413 | C18H26O5 | 322.4 | 0.5 | 0.55 |
| Beta-Zearalanol | 65434 | C18H26O5 | 322.4 | 0.5 | 0.55 |
| Astragalin | 5282102 | C21H22O11 | 448.4 | 0.67 | 14.03 |
| Kaempferol | 5280863 | C15H10O6 | 286.24 | 0.5 | 0.55 |
| Target Gene | UniProt ID | Protein Name | Degree | BC | CC |
|---|---|---|---|---|---|
| AKT1 | P31749 | RAC-alpha serine/threonine-protein kinase | 158 | 0.067 | 0.678 |
| IL6 | P05231 | Interleukin-6 | 157 | 0.066 | 0.678 |
| TNF | P01375 | Tumor Necrosis Factor | 152 | 0.058 | 0.669 |
| SRC | P12931 | Proto-oncogene tyrosine-protein kinase Src | 134 | 0.081 | 0.633 |
| STAT3 | P40763 | Signal transducer and activator of transcription 3 | 130 | 0.032 | 0.633 |
| EGFR | P00533 | Epidermal growth factor receptor | 129 | 0.037 | 0.630 |
| BCL2 | P10415 | Apoptosis Regulator Bcl2 | 121 | 0.020 | 0.617 |
| HSP90AA1 | P07900 | Heat shock protein HSP90-alpha | 119 | 0.030 | 0.612 |
| HIF1A | Q16665 | Hypoxia inducible factor 1-alpha | 117 | 0.026 | 0.608 |
| PTGS2 | P35354 | Prostaglandin G/H synthase 2 | 114 | 0.040 | 0.612 |
| Compounds | AKT1 | EGFR | BCL2 | HSP90AA1 | PTGS2 |
|---|---|---|---|---|---|
| Catechin | −6.66 | −6.45 | −5.61 | −8.18 | −7.28 |
| Epicatechin | −6.53 | −6.63 | −5.82 | −7.93 | −7.06 |
| Naringenin | −6.33 | −6.91 | −6.00 | −7.85 | −7.44 |
| Phloridzin | −6.92 | −6.44 | −6.02 | −7.08 | −6.73 |
| Genistein | −6.25 | −8.10 | −5.40 | −5.61 | −7.34 |
| Daidzein | −5.73 | −7.56 | −5.11 | −5.84 | −7.34 |
| Quinic acid | −4.92 | −6.54 | −5.04 | −5.77 | −6.47 |
| 1-O-Galloyl-beta-D-glucose | −6.37 | −7.11 | −4.92 | −7.65 | −6.35 |
| Epicatechin gallate | −5.63 | −6.39 | −6.32 | −7.92 | −7.92 |
| Astragalin | −4.42 | −5.81 | −6.09 | −7.92 | −9.00 |
| Kaempferol | −6.25 | −6.99 | −6.67 | −7.11 | −6.96 |
| Compounds | Absorption | Distribution | Metabolism (CYP Inhibitor) | Excretion | Log S | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CaCO2 | P-gp | HIA | BBB | VDss | 1A2 | 2C19 | 2C9 | 2D6 | 3A4 | CL | T1/2 | ||
| Catechin | −6.04 | – | — | — | 1.15 | — | — | — | — | — | 14.9 | 2.14 | −2.28 |
| Epicatechin | −6.04 | – | — | — | 1.15 | — | — | — | — | — | 14.9 | 2.14 | −2.28 |
| Phloridzin | −6.20 | - | — | — | 0.68 | — | — | — | — | ++ | 3.76 | 2.26 | −2.53 |
| Daidzein | −4.69 | - | — | — | 0.62 | +++ | +++ | +++ | +++ | ++ | 7.85 | 1.17 | −3.79 |
| Quinic acid | −6.31 | — | + | — | 0.37 | — | — | — | — | — | 1.57 | 3.35 | −0.10 |
| 1-O-Galloyl-beta-D-glucose | −6.39 | – | + | — | 0.37 | — | — | — | — | — | 3.68 | 2.37 | −1.29 |
| Epicatechin gallate | −6.51 | — | — | — | 0.44 | — | — | ++ | — | +++ | 9.65 | 2.08 | −3.70 |
| Kaempferol | −5.97 | – | — | — | 0.15 | +++ | – | ++ | — | +++ | 5.69 | 1.33 | −3.65 |
| Compounds | Carcinogenicity | Immunotoxicity | Mutagenicity | Cytotoxicity | Lipinski | Pfizer | PAINS |
|---|---|---|---|---|---|---|---|
| Catechin | Inactive | Inactive | Inactive | Inactive | Yes | Yes | 1 |
| Epicatechin | Inactive | Inactive | Inactive | Inactive | Yes | Yes | 1 |
| Phloridzin | Inactive | Inactive | Inactive | Inactive | Yes | Yes | 0 |
| Daidzein | Inactive | Inactive | Inactive | Inactive | Yes | Yes | 0 |
| Quinic acid | Inactive | Inactive | Inactive | Inactive | Yes | Yes | 0 |
| 1-O-Galloyl-beta-D-glucose | Inactive | Inactive | Inactive | Inactive | Yes | Yes | 1 |
| Epicatechin gallate | Inactive | Inactive | Inactive | Inactive | Yes | Yes | 1 |
| Kaempferol | Inactive | Inactive | Inactive | Inactive | Yes | Yes | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Majhi, R.; Kurmi, S.; Tayara, H.; Chong, K.T. Harnessing the Therapeutic Potential of Pomegranate Peel-Derived Bioactive Compounds in Pancreatic Cancer: A Computational Approach. Pharmaceuticals 2025, 18, 896. https://doi.org/10.3390/ph18060896
Majhi R, Kurmi S, Tayara H, Chong KT. Harnessing the Therapeutic Potential of Pomegranate Peel-Derived Bioactive Compounds in Pancreatic Cancer: A Computational Approach. Pharmaceuticals. 2025; 18(6):896. https://doi.org/10.3390/ph18060896
Chicago/Turabian StyleMajhi, Rita, Sagar Kurmi, Hilal Tayara, and Kil To Chong. 2025. "Harnessing the Therapeutic Potential of Pomegranate Peel-Derived Bioactive Compounds in Pancreatic Cancer: A Computational Approach" Pharmaceuticals 18, no. 6: 896. https://doi.org/10.3390/ph18060896
APA StyleMajhi, R., Kurmi, S., Tayara, H., & Chong, K. T. (2025). Harnessing the Therapeutic Potential of Pomegranate Peel-Derived Bioactive Compounds in Pancreatic Cancer: A Computational Approach. Pharmaceuticals, 18(6), 896. https://doi.org/10.3390/ph18060896