1. Introduction
Glycogen synthase kinase-3 beta (GSK3β) is a serine/threonine kinase that plays a pivotal role in various cellular signaling pathways, influencing metabolism, cell proliferation, apoptosis, and neurodevelopment. Unlike most kinases, GSK3β remains constitutively active under basal conditions and is negatively regulated by phosphorylation at serine 9 (S9) by kinases such as AKT and protein kinase A (PKA) [
1]. The functional diversity of GSK3Β is reflected in its involvement in numerous physiological processes, ranging from neuronal differentiation and synapse formation to neuroinflammation and neurodegenerative disorders. Dysregulation of GSK3Β activity has been directly linked to neurological diseases, including Alzheimer’s disease (AD), Parkinson’s disease (PD), bipolar disorder, schizophrenia, and ischemic stroke, making it a potential therapeutic target for drug intervention [
2]. Given its extensive role in neurophysiology and neuropathology, targeting GSK3Β through selective inhibitors has gained significant interest in recent years. However, the therapeutic implications of GSK3β inhibition remain complex, particularly due to concerns regarding off-target effects, systemic toxicity, and the paradoxical role of GSK3β as both a tumor suppressor and tumor promoter in different cellular contexts. Consequently, identifying potent, selective, and pharmacologically viable inhibitors of GSK3β remains a significant challenge in drug discovery [
3].
Recent advancements in computational drug discovery and machine learning-based screening approaches have opened new avenues for identifying novel GSK3β inhibitors with high selectivity and efficacy [
4]. Traditional high-throughput screening (HTS) approaches, although effective, are often labor-intensive, time-consuming, and limited by the diversity of screened chemical libraries. In contrast, machine learning (ML)-driven virtual screening techniques offer a more efficient and systematic approach to identifying potential drug candidates [
5]. These computational methods integrate quantitative structure–activity relationship (QSAR) models, molecular docking, and molecular dynamics (MD) simulations to evaluate the binding affinity, stability, and pharmacokinetic properties of candidate inhibitors [
6]. Additionally, the application of deep learning models in molecular property prediction has significantly improved hit-to-lead optimization, allowing for the rational design of novel GSK3β inhibitors with enhanced specificity and bioavailability [
7].
Several small-molecule inhibitors, such as Tideglusib [
8] and CHIR99021 [
9], have been identified using computational approaches, demonstrating promising preclinical efficacy in reducing tau hyperphosphorylation, mitigating synaptic dysfunction, and enhancing neuronal survival. However, despite these advancements, the full therapeutic potential of GSK3β inhibition has yet to be realized, necessitating further exploration into its structural dynamics, binding interactions, and downstream effects in neurological disorders. The pathophysiological significance of GSK3β in neurodegeneration is largely attributed to its role in tau phosphorylation and amyloid-beta (Aβ) regulation, both of which are hallmark features of Alzheimer’s disease. GSK3β-mediated phosphorylation of microtubule-associated tau protein leads to its aggregation into neurofibrillary tangles (NFTs), contributing to synaptic dysfunction and neuronal apoptosis [
10]. Furthermore, GSK3β has been shown to modulate Aβ production by regulating β-secretase (BACE1) activity, further exacerbating neurodegeneration [
11]. Beyond AD, GSK3β hyperactivation has been implicated in PD, where it facilitates dopaminergic neuronal loss, oxidative stress, and α-synuclein aggregation. Emerging evidence also suggests that GSK3β plays a regulatory role in psychiatric disorders, including bipolar disorder and schizophrenia, where its dysregulation disrupts synaptic plasticity, neurotransmitter balance, and neuroinflammatory responses. Given these diverse yet interrelated roles, targeting GSK3β through selective inhibitors is a potential neuroprotective intervention strategy across multiple CNS disorders [
12].
Despite significant advancements in GSK3β-targeted drug discovery, challenges remain in developing highly selective inhibitors that effectively cross the blood–brain barrier (BBB) while minimizing adverse effects. The pharmacokinetic limitations and toxicity concerns associated with broad-spectrum GSK3β inhibition necessitate a rational drug design approach that incorporates machine learning-driven compound screening, molecular docking, and in silico pharmacological profiling [
13]. In this study, we employ an integrated computational approach to construct a library of potential GSK3β inhibitors, leveraging machine learning algorithms for ligand-based screening, structure-based docking simulations, and molecular dynamics evaluations. By systematically evaluating binding affinities, ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) properties, and target selectivity, this study aims to identify novel GSK3β inhibitors with optimal drug-like properties. To further refine drug discovery, a natural compound library has been developed and systematically screened to identify selective GSK3β inhibitors using machine learning algorithms. The model construction process involved feature selection techniques, followed by the application of machine learning algorithms such as Support Vector Machine (SVM), Random Forest (RF), K-Nearest Neighbors (KNN), and deep learning-based predictive models, enabling the identification of potential inhibitors with high specificity and favorable pharmacokinetic properties. This integrated ML-driven strategy enhances the accuracy of virtual screening, optimizing compound selection for experimental validation. The findings from this research may contribute to the development of next-generation therapeutics for neurodegenerative and neuropsychiatric disorders, advancing our understanding of GSK3β modulation as a viable drug target.
2. Results
2.1. Overview of QSAR Model Development
QSAR modeling was performed to predict the biological activity of compounds targeting GSK3β using key molecular descriptors. A dataset of 3092 compounds was curated, with 2164 used for training and 928 for testing. Feature selection identified essential descriptors, including molecular weight, ALogP, hydrogen bond properties, rotatable bonds, and topological indices, ensuring optimal model performance. Three machine learning models including SVM, RF, and KNN were applied in the present study. SVM classified compounds using hyperplane optimization, RF improved prediction accuracy through ensemble learning, and KNN classified compounds based on molecular similarity. These models were assessed for predictive accuracy, ensuring reliable identification of potential GSK3β inhibitors.
2.2. Feature Selection and Data Pre-Processing
The chosen descriptors were essential for capturing the physicochemical, geometric, electronic, and molecular characteristics relevant to GSK3β inhibition. The dataset comprised 3092 compounds, of which 2164 were used for training and 928 for testing. Data pre-processing included normalization techniques to ensure model robustness. The selected features were categorized into five main groups based on their relevance to molecular interactions and drug-like properties, as summarized in
Table 1 and illustrated in
Figure 1. To ensure consistency and eliminate bias, the dataset was normalized before training using Min–Max Scaling to rescale feature values between 0 and 1, preventing dominance by features with larger ranges, and Standardization (Z-score normalization) where necessary to maintain uniform variance across features.
As no experimentally validated GSK3β inhibitor labels were included, active/inactive compound definitions were derived through property-driven heuristics based on pharmacophoric alignment with known GSK3β binding site features. This approach enables unbiased screening of natural product libraries and is supported by previous ML-based screening studies in kinase and neurotherapeutic drug discovery.
Feature selection was conducted as a critical step prior to model development to ensure that only the most informative, non-redundant, and biologically relevant molecular descriptors were used during training. Initially, a comprehensive descriptor matrix comprising 797 features was generated using the PaDEL-Descriptor toolkit, including 663 one-dimensional (1D) and two-dimensional (2D) descriptors and 134 three-dimensional (3D) descriptors. These captured diverse chemical information across physicochemical, topological, geometrical, and electronic domains. To reduce dimensionality and avoid multicollinearity, descriptors with low variance (threshold < 0.01) were eliminated, followed by correlation filtering (Pearson r > 0.9) to remove redundant variables.
Model-based feature prioritization was then performed using Recursive Feature Elimination (RFE) in conjunction with Random Forest (RF) classification. This process identified the top-ranked predictors based on their importance to classification accuracy. The five most influential descriptors contributing to GSK3β inhibitory potency in the present study were: pIC50, Molecular Weight (MW), ALogP (lipophilicity), Hydrogen Bond Acceptors (HBA), and McGowan Volume. These features not only had the highest Gini importance scores in the RF model but also align with established pharmacological principles relevant to kinase inhibition. Specifically, pIC50 encodes the potency of known ligands, anchoring the model to real bioactivity trends. ALogP and HBA describe lipophilicity and hydrogen bonding potential, which are critical for forming stable interactions with key GSK3β residues such as ASP200, CYS199, and LEU188. MW and McGowan Volume relate to the steric and volumetric compatibility of ligands with the narrow ATP-binding pocket of GSK3β.
2.3. Performance of Machine Learning Models
Machine learning models were employed to predict the biological activity of natural compounds against GSK3β, focusing on classification accuracy, sensitivity, specificity, precision, and AUC-ROC. The three models were evaluated using a dataset of 3092 compounds, with 2164 compounds for training and 928 for testing. Performance metrics were assessed using cross-validation to ensure robustness.
Table 2 summarizes the classification performance across models, while
Table 3 presents additional evaluation metrics, including RMSE and R
2 scores.
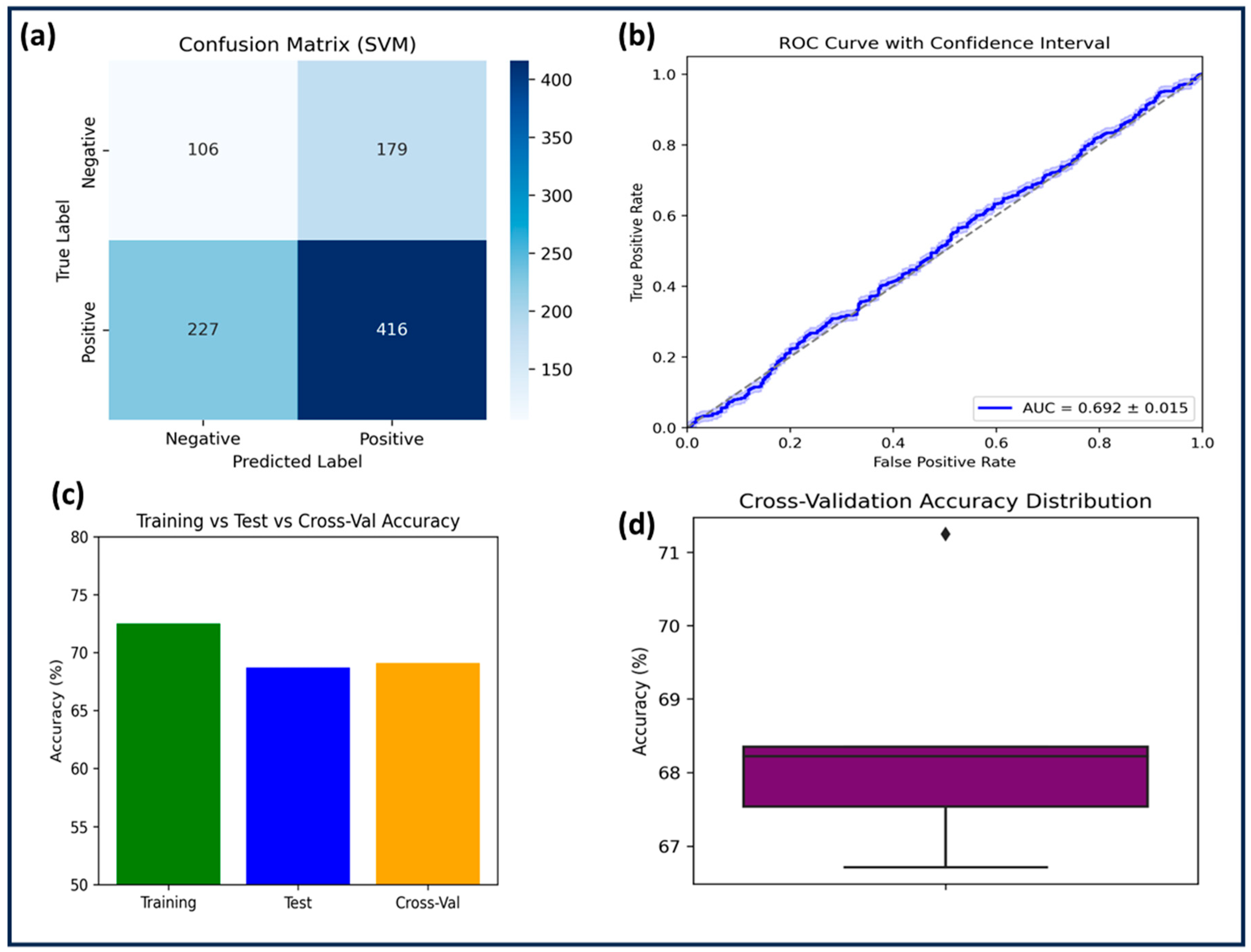
2.3.1. Support Vector Machine (SVM)
The SVM model was trained on 2164 compounds and tested on 928 compounds using an RBF kernel with C = 1.0 and γ = 1.0. Feature scaling was applied, but no data augmentation was performed. SVM achieved a training accuracy of 72.5% and a test accuracy of 68.7%, with a cross-validation accuracy of 69.1% ± 1.4. Despite moderate specificity (0.71), the model suffered from low sensitivity (0.64) and precision (0.62), resulting in an F1-score of 0.63. A negative R
2 score (−0.0013) and a high RMSE (0.4822 on the test set) indicated poor predictive power and possible overfitting, as confirmed by the performance gap between training and test accuracy (
Table 2 and
Table 3). The negative R
2 score suggests that the regression model performed worse than a simple horizontal mean predictor, meaning the predicted values deviated more from the actual values than the mean of the target variable would. This behavior often reflects that the model failed to capture underlying nonlinear patterns in the data, or that the feature–target relationship was not adequately learned given the kernel or hyperparameter configuration. In this case, it is likely that the model’s decision boundary was too rigid or misaligned with the complex, multi-dimensional feature space relevant to GSK3β inhibition. The AUC-ROC (0.692 ± 0.015,
Figure 2) suggested that SVM performed only slightly above random classification. Statistical comparison with RF (
p = 0.0021) confirmed significant performance differences. Given its inability to generalize well, SVM was deemed the least effective among the three models.
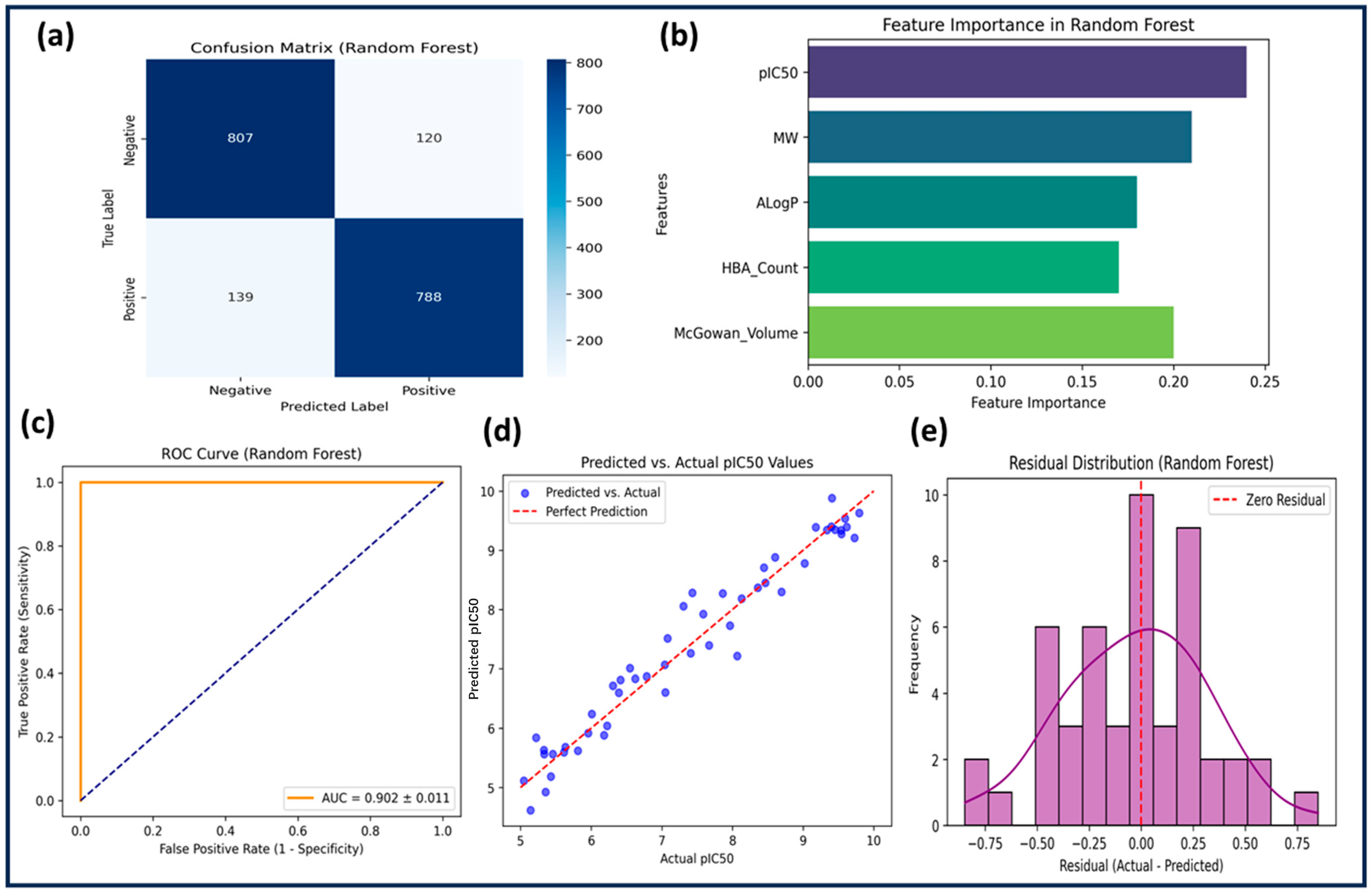
2.3.2. Random Forest (RF)
The RF model outperformed both SVM and KNN, achieving a training accuracy of 88.4% and a test accuracy of 83.6%. Feature selection was performed using feature importance scores, and hyperparameters were optimized with n_estimators = 200, max_depth = 10, min_samples_split = 4, and min_samples_leaf = 2. The cross-validation accuracy was 84.2% ± 0.9, confirming low variance. Sensitivity (0.85) and specificity (0.87) were high, with precision at 0.84 and an F1-score of 0.845. The model demonstrated superior predictive power, with an R
2 score of 0.6321 and the lowest RMSE (0.3214) among all models (
Table 2 and
Table 3).
Figure 3 presents the ROC curve (AUC = 0.902 ± 0.011), further confirming strong classification performance. Feature importance analysis identified pIC50, ALogP, MW, and HBA_Count as the most influential descriptors. The residual plot confirmed minimal systematic error. RF demonstrated high generalization capability and reliability, making it the best model for identifying potential GSK3β inhibitors.
2.3.3. K-Nearest Neighbors (KNN)
The KNN model, using neighbors = 7, a distance-weighted voting scheme, and the Euclidean metric, achieved a training accuracy of 80.1% and a test accuracy of 76.2%. The cross-validation accuracy was 77.4% ± 1.2, indicating stable performance. Sensitivity (0.76) and specificity (0.78) were moderate, with an F1-score of 0.755. The R
2 score (0.4756) was lower than RF but significantly better than SVM. The RMSE (0.4123) suggested an intermediate level of prediction error (
Table 2 and
Table 3).
Figure 4 presents the ROC curve (AUC = 0.837 ± 0.014), showing that KNN outperformed SVM but was less predictive than RF. Accuracy and RMSE trends across different
k-values indicated that the optimal value (
k = 7) balanced overfitting and underfitting. Despite its reasonable performance, KNN struggled with large datasets due to computational inefficiency and limited ability to distinguish active from inactive compounds compared to RF.
2.4. Model Comparison
The comparative evaluation of the three machine learning models: SVM, KNN, and RF, was conducted in order to increase predictive performance to facilitate systematic interpretation. SVM, utilizing a radial basis function (RBF) kernel with hyperparameters C = 1.0 and γ = 1.0, performed the weakest. Despite appropriate feature scaling, it yielded a test accuracy of 68.7%, negative R2 (−0.0013), and the highest RMSE (0.4822), indicating that the model was not able to effectively generalize beyond the training data. The rigid decision boundary imposed by its hyperparameters likely contributed to this poor performance. The AUC-ROC of 0.692 ± 0.015 supports this limited classification ability.
Next, the KNN model, configured with k = 7 and a distance-weighted Euclidean metric, demonstrated improved generalization with a test accuracy of 76.2%, an R2 of 0.4756, and RMSE of 0.4123. These results suggest that the chosen k-value effectively balanced overfitting and underfitting, although performance remained lower than that of RF. The AUC-ROC of 0.837 ± 0.014 confirmed moderate discriminative power.
The Random Forest model exhibited the highest performance, benefiting from ensemble-based learning and robust hyperparameter optimization. With n_estimators = 200, max_depth = 10, min_samples_split = 4, and min_samples_leaf = 2, RF achieved a test accuracy of 83.6%, the highest R2 score (0.6321), and the lowest RMSE (0.3214) among all models. Its AUC-ROC of 0.902 ± 0.011 underscores its superior classification ability. Feature importance analysis confirmed that descriptors such as pIC50, ALogP, molecular weight, and HBA were key contributors.
To evaluate whether the observed differences in predictive performance between models were statistically significant, a paired t-test was conducted using the cross-validation accuracy scores obtained across ten independent folds. Specifically, the test compared fold-wise classification accuracy of the SVM model against the RF model, which showed the highest overall performance. The resulting p-value (p = 0.0021) indicated a statistically significant improvement in accuracy by RF over SVM at the 95% confidence level. This test supports the conclusion that RF’s superior classification results are not due to random variation but are instead consistently observed across multiple data splits.
2.5. Virtual Screening
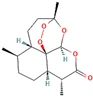
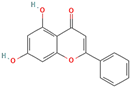
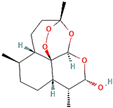
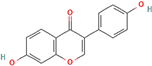
Following model training and evaluation, predictions from all three ML were applied to the full dataset of 3092 natural compounds obtained from the Selleckchem natural product library. To increase prediction confidence, only those compounds classified as “active” by all three models (i.e., overlapping inliers) were retained. This consensus approach yielded a final set of 181 compounds, each consistently predicted to exhibit potential GSK3β inhibitory activity. The virtual screening of 181 compounds yielded binding affinities ranging from −3.2 kcal/mol to −8.6 kcal/mol. The top-scoring compounds with binding affinities better than −8.0 kcal/mol were analyzed for molecular interactions (
Table 4). The most relevant hydrogen bond interaction observed was with ASP200, while the remaining interactions were predominantly hydrophobic in nature. The most frequent hydrophobic interactions involved VAL70, CYS199, LYS85, ALA83, and LEU188. These residues played a crucial role in stabilizing ligand binding within the GSK3Β active site. The 68827-GSK3Β complex (artemisinin) exhibited the best binding affinity of −8.6 kcal/mol, followed closely by chrysin (−8.5 kcal/mol), dihydroartemisinin (−8.3 kcal/mol), costunolide (−8.2 kcal/mol), and daidzein (−8.1 kcal/mol). The visualization of these interactions within the binding pocket is presented in
Figure 5, while the corresponding 2D interaction maps are provided in
Figure 6.
2.6. Molecular Dynamics Simulations
The MD simulation of the top compound 68827 in complex with GSK3Β provides crucial insights into its stability, flexibility, and overall interaction behavior within the binding pocket of the protein.
2.6.1. RMSD Analysis
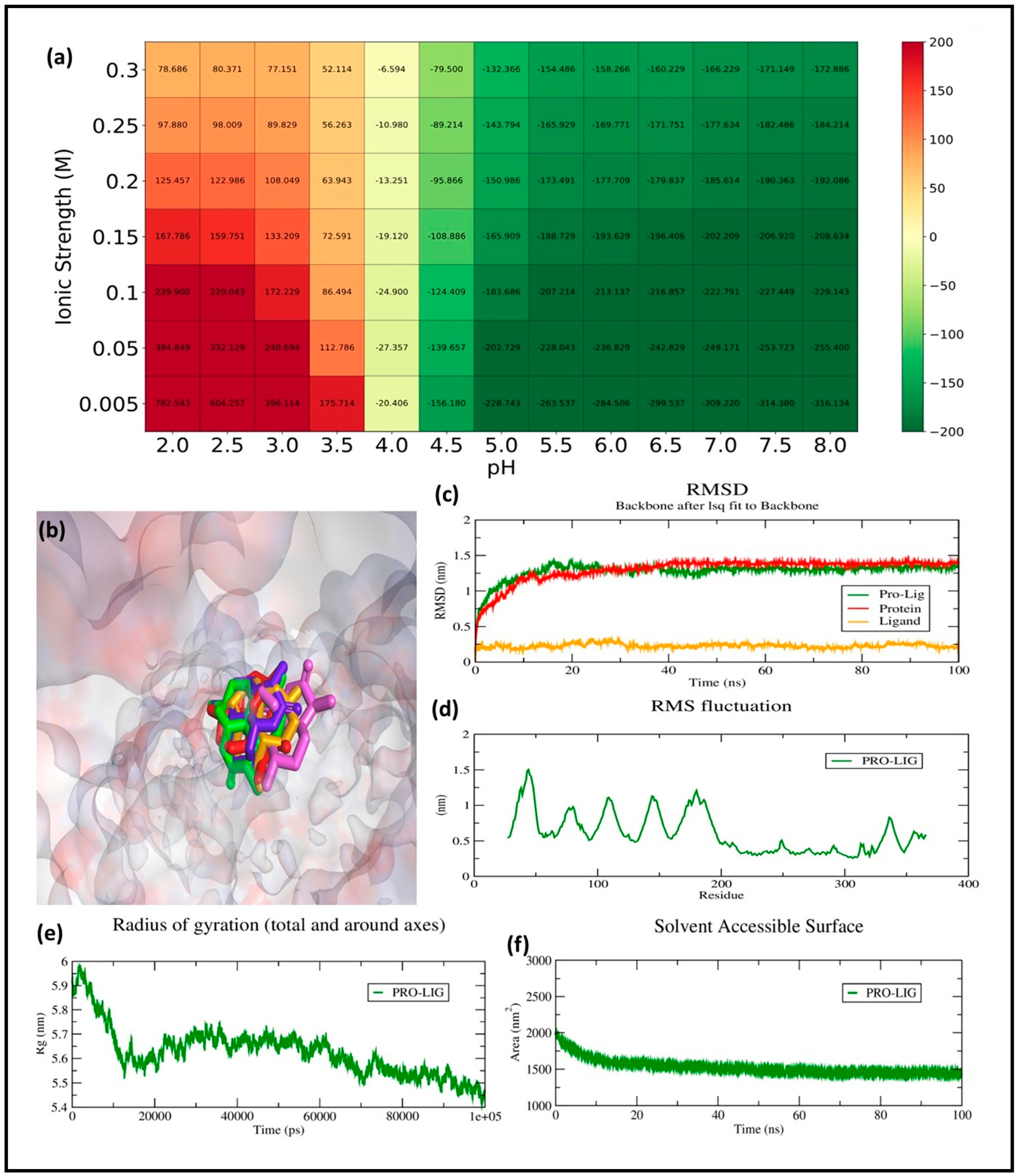
The RMSD analysis was carried out to evaluate the conformational stability and binding dynamics of the GSK3β–ligand complex over a 100 ns molecular dynamics (MD) simulation. The RMSD trajectory of the protein backbone (red line) shows an initial rise during the equilibration phase, reaching ~1.25 nm within the first 10 ns, after which it stabilizes and fluctuates around a mean of 1.38 ± 0.07 nm. This indicates that the protein undergoes expected relaxation and structural adjustment early in the simulation but maintains conformational stability for the remaining duration.
The ligand RMSD (orange line), computed after aligning the complex to the protein backbone, remained consistently low throughout the simulation, averaging 0.26 ± 0.03 nm. This minimal fluctuation confirms that the ligand remains stably bound within the active site, without significant drift or conformational rearrangement. Importantly, this behavior implies retention of the docking pose, suggesting a well-accommodated fit between the ligand and the GSK3β binding pocket.
The protein–ligand complex RMSD (green line), which accounts for the full interaction system, follows a trajectory similar to the protein backbone, reaching an average of ~1.45 ± 0.06 nm during the stable phase. This trajectory reflects the combined influence of minor backbone movements and ligand–protein interaction dynamics. The absence of sharp deviations after the 20 ns mark further suggests that the complex has equilibrated and maintained its integrity throughout the simulation (
Figure 7c). The RMSD profiles demonstrate that the binding of the ligand contributes to the stabilization of the GSK3β backbone, reinforcing the structural rigidity of the receptor’s active site. The low ligand RMSD combined with the stable protein dynamics implies a strong and persistent interaction, likely involving key residues within the ATP-binding pocket. These results align well with the docking outcomes and validate the compound’s potential as a stable GSK3β inhibitor candidate under physiological conditions.
2.6.2. RMSF Analysis
RMSF measures the flexibility of individual residues in a biomolecular system during a simulation, where higher values indicate greater flexibility and lower values suggest structural rigidity. In the given RMSF graph, fluctuations range from 0.2 nm to approximately 1.6 nm, with the highest peaks occurring around residues 50, 150, and 250, reaching about 1.5 nm. These peaks suggest highly flexible regions, likely corresponding to loop or terminal regions. Most residues exhibit fluctuations between 0.3 and 0.8 nm, indicating moderate flexibility. The lowest fluctuations, around 0.2 nm, occur in structured regions, likely α-helices or β-sheets, demonstrating stability (
Figure 7d).
To evaluate the environmental sensitivity of the protein–ligand binding interaction, a systematic analysis of binding free energy was conducted across varying pH values (2.0–8.0) and ionic strengths (0.005–1.0 M). The resulting heatmap (
Figure 7a) revealed a distinct trend where binding affinity improved with both increasing pH and ionic strength. At acidic conditions (pH 2.0–4.0), the binding energies were predominantly positive or weakly negative, indicating reduced or destabilized complex formation under such proton-rich environments. However, as the pH transitioned towards neutral and basic ranges, particularly between pH 6.0 and 8.0, a substantial enhancement in binding strength was observed. The most favorable interaction occurred at pH 8.0 and an ionic strength of 1.0 M, where the binding free energy reached a minimum of −316.13 kJ/mol. This suggests that electrostatic stabilization plays a pivotal role in driving the binding event, likely through ionic screening effects that reduce repulsive forces and stabilize hydrogen bonding networks. Moreover, across all tested pH conditions, higher ionic strength consistently correlated with more negative free energy values, reinforcing the conclusion that ionic composition of the solvent environment critically influences the thermodynamics of ligand binding.
2.6.3. Rg Analysis
Rg is a crucial parameter in molecular dynamics that reflects the compactness and overall structural stability of a biomolecule. A higher Rg indicates an expanded conformation, while a lower Rg suggests a more compact structure. The initial Rg value is around 5.9 nm, indicating a relatively extended structure. A gradual decrease in Rg is observed over time, stabilizing between 5.5 and 5.6 nm after approximately 20 ns. This suggests that the system undergoes a compaction phase, likely due to stabilization in the presence of a ligand or solvent effects. The fluctuations in Rg after stabilization are minimal, indicating a stable and folded structure over time. The observed decrease and stabilization in Rg imply that the protein–ligand complex adopts a more compact and stable conformation, suggesting structural convergence (
Figure 7e).
2.6.4. SASA Analysis
SASA is an important parameter in molecular dynamics, representing the extent of a molecule’s surface exposed to the solvent. Changes in SASA can indicate protein folding, complex formation, or ligand binding. Initially, SASA is around 2000 nm
2, suggesting a relatively expanded protein conformation with a large solvent-exposed surface. Over the first 20 ns, SASA decreases steadily, indicating structural compaction and reduced solvent exposure. After approximately 20 ns, SASA stabilizes around 1500 nm
2, suggesting that the structure maintains a stable, compact form for the remainder of the simulation. The fluctuations in SASA remain small, further supporting that the system has reached equilibrium. The significant decrease in SASA over time suggests protein compaction and structural stabilization, possibly due to ligand-induced folding, complex formation, or a shift toward a more hydrophobic core, ultimately confirming that the protein remains in a stable structural state (
Figure 7f).
2.7. MMPBSA Calculations
The MM-PBSA binding free energy analysis of the GSK3β–artemisinin complex revealed that the interaction is predominantly stabilized by van der Waals and electrostatic forces. Specifically, the van der Waals contribution was −223.98 ± 15.53 kJ/mol, indicating strong hydrophobic packing between artemisinin and the residues within the GSK3β active site. The electrostatic interaction energy was also significantly favorable at −86.20 ± 14.16 kJ/mol, reflecting effective charge-based interactions such as potential hydrogen bonds and salt bridges. These attractive forces were partially counterbalanced by a positive polar solvation energy of +196.71 ± 11.79 kJ/mol, which represents the energetic cost of desolvating the ligand and binding site in a polar aqueous environment. In contrast, the non-polar solvation energy, estimated via SASA, contributed −21.60 ± 0.75 kJ/mol, further supporting the binding through hydrophobic effects.
3. Discussion
Natural compounds have historically played a crucial role in drug development, offering diverse chemical structures and bioactivities that have been refined through evolutionary processes [
14]. Their natural origins make them valuable as potential therapeutics, with many currently approved drugs being derived from plant, microbial, or marine sources. Some of the most well-known examples include paclitaxel (Taxol) from the Pacific yew tree, which is used in cancer treatment [
15], and artemisinin, derived from Artemisia annua, which serves as a highly effective antimalarial agent [
16]. One of the key advantages of natural compounds is their inherent biological activity, often fine-tuned to interact with specific biomolecules, making them excellent starting points for drug discovery. Additionally, they often exhibit structural complexity that is difficult to achieve through synthetic chemistry alone, providing unique scaffolds for the design of new drugs. However, despite these advantages, natural compounds also present significant challenges in drug development. Many of them are large and complex molecules, which can lead to issues related to bioavailability, solubility, and metabolic stability [
17]. Their large molecular size and high lipophilicity may hinder their ability to cross biological membranes, thereby limiting their effectiveness as drugs. Another major concern is toxicity; while natural compounds have evolved to interact with biological targets, some may also affect off-target pathways, leading to adverse effects [
18]. For example, digitoxin and ouabain, derived from Digitalis species, are potent cardiac glycosides that can cause fatal toxicity if not administered at precise doses [
19]. Furthermore, identifying truly drug-like compounds from the vast pool of natural molecules is challenging due to the sheer chemical diversity and the need to balance potency with safety. Thus, filtering out promising candidates for further development requires sophisticated screening and optimization techniques, which have been revolutionized by modern computational approaches.
With advances in computational techniques, the field of drug discovery has significantly benefited from in silico methods, which allow for efficient screening and prediction of bioactivity, thereby overcoming many limitations associated with traditional experimental approaches. One widely used computational strategy is network pharmacology, which maps interactions between bioactive compounds and biological networks to predict potential therapeutic effects. This holistic approach allows researchers to understand polypharmacological properties and identify multitarget drugs [
20]. Another powerful method is quantitative QSAR modeling, which uses mathematical algorithms to correlate molecular structures with biological activity, enabling the prediction of drug-like properties in novel compounds [
21]. Pharmacophore modeling is another essential computational approach that identifies key molecular features responsible for binding to a specific biological target, facilitating the design of optimized compounds with improved efficacy and selectivity. Virtual screening, which encompasses docking studies and molecular dynamics simulations, enables the rapid screening of vast chemical libraries against specific protein targets, reducing the time and cost required for experimental validation [
22]. Pharmacokinetic modeling helps predict absorption, distribution, metabolism, and excretion (ADME) properties, ensuring that candidate molecules possess desirable drug-like characteristics before advancing to in vitro and in vivo testing [
23]. Furthermore, artificial intelligence (AI) and ML have emerged as transformative tools in drug discovery, capable of analyzing vast datasets to identify patterns and optimize lead compounds. AI-driven drug design allows for rapid hypothesis generation, improving hit-to-lead optimization by predicting binding affinities, toxicity, and metabolic stability [
24]. The integration of these computational approaches has accelerated drug discovery, particularly in the field of natural product-based therapeutics, by enabling more precise identification of promising candidates while minimizing the risks associated with experimental trial-and-error approaches.
In the present study, our objective was to screen a natural compound-based library specifically targeting GSK3β, a protein that plays a central role in neurodegenerative diseases such as Alzheimer’s disease AD and PD. GSK3β is involved in various cellular processes, including neuronal development, glucose metabolism, and inflammation. However, its hyperactivation has been linked to tau hyperphosphorylation, amyloid-beta aggregation, and neurodegeneration, making it a promising therapeutic target. Given the significance of GSK3β in neurodegenerative disorders, our study aimed to identify natural compounds capable of inhibiting this enzyme. To achieve this, we employed three machine-learning-based approaches including SVM, RF, and KNN to build predictive models for selecting promising GSK3β inhibitors from the natural compound library. These models were designed to enhance the accuracy of virtual screening by incorporating structure–activity relationships, molecular descriptors, and pharmacokinetic parameters. By integrating these computational techniques, we aimed to identify highly selective and potent natural inhibitors of GSK3β that could serve as potential candidates for neurodegenerative methods utilized in this study, i.e., SVM, RF, and KNN are widely used in drug discovery for predicting compound bioactivity and optimizing virtual screening processes. The SVM algorithm is a powerful supervised learning model that classifies compounds based on their molecular descriptors by constructing a hyperplane that separates active and inactive compounds. It is particularly effective in handling high-dimensional datasets and has been extensively used in QSAR modeling to identify potential drug candidates. RF, on the other hand, is an ensemble learning technique that builds multiple decision trees and aggregates their outputs to improve predictive accuracy. This method is highly robust against overfitting and can handle noisy datasets, making it ideal for identifying bioactive compounds with complex molecular interactions [
25]. The KNN algorithm is a distance-based classification method that assigns a compound’s activity based on the similarity of its molecular features to known active compounds [
26]. While computationally simple, KNN can be highly effective when combined with appropriate feature selection techniques. In our study, we trained these three models using a dataset of known GSK3β inhibitors, extracting key molecular descriptors and physicochemical properties to enhance model performance. After training and validation, the models were applied to screen the natural compound library, identifying lead candidates with high predicted binding affinity to GSK3β. Among the three methods, the Random Forest model demonstrated the highest accuracy and predictive performance, making it the most reliable approach for prioritizing compounds for further investigation.
Following virtual screening and machine learning-based selection, inlier compounds that were consistently identified as promising GSK3β inhibitors across all three models were extracted to construct the final compound library. These top hits were further evaluated through molecular docking and ADME predictions to ensure favorable pharmacokinetic and drug-likeness properties. One of the most significant findings in our study was the identification of Artemisinin as the top ranked GSK3β inhibitor. Artemisinin is a sesquiterpene lactone extracted from Artemisia annua, known primarily for its antimalarial properties. Discovered by Tu Youyou in 1972, it has been a cornerstone in malaria treatment due to its rapid action against
Plasmodium falciparum. Beyond malaria, artemisinin has demonstrated potential therapeutic effects in cancer, inflammation, and neurodegenerative diseases [
27]. Recent studies suggest that artemisinin can induce ferroptosis, a form of programmed cell death linked to iron metabolism, which may be beneficial in targeting cancer cells and neurodegeneration. Furthermore, artemisinin derivatives have shown activity against various pathogens, including viruses, protozoa, and fungi, broadening its potential therapeutic applications [
28].
Another key hit from our study was dihydroartemisinin, a derivative of artemisinin, which exhibited strong predicted GSK3β inhibition. Dihydroartemisinin has been reported to induce ferroptosis by promoting lysosomal degradation of ferritin, which may have implications for neurodegenerative disease treatment. Given that neurodegeneration is associated with oxidative stress and iron dysregulation, dihydroartemisinin’s ability to modulate ferroptosis pathways makes it a particularly intriguing candidate for further investigation [
29]. In the context of AD, recent studies have suggested that artemisinin and its derivatives can reduce Aβ deposition and tau hyperphosphorylation in transgenic mouse models. For instance, artemisinin treatment in APPswe/PS1dE9 transgenic mice significantly decreased amyloid plaque burden in the cortex and hippocampus, likely through the inhibition of β-secretase activity. Additionally, other artemisinin derivatives have been shown to reduce neuroinflammation by suppressing NF-κB signaling and decreasing pro-inflammatory cytokine levels, further supporting their potential neuroprotective effects [
30].
While current FDA-approved monoclonal antibody therapies for Alzheimer’s disease, such as aducanumab, lecanemab, and donanemab, target amyloid-beta plaques, their modest clinical benefits highlight the need for alternative therapeutic approaches. The ability of artemisinin and dihydroartemisinin to modulate multiple pathways, including GSK3β inhibition, ferroptosis regulation, and neuroinflammation reduction, presents a compelling case for their further development as neuroprotective agents. The top-performing compound identified in the screening process exhibited high binding affinity and selectivity towards the target implicated in neurodegenerative diseases. Molecular docking studies and molecular dynamics simulations confirmed its stability within the binding site, suggesting strong interactions with key residues responsible for disease progression. Future studies will focus on experimental validation of these computationally identified hits through in vitro and in vivo assays, as well as structural modifications to enhance their selectivity and pharmacokinetic profiles for potential clinical applications in neurodegenerative disease treatment.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}