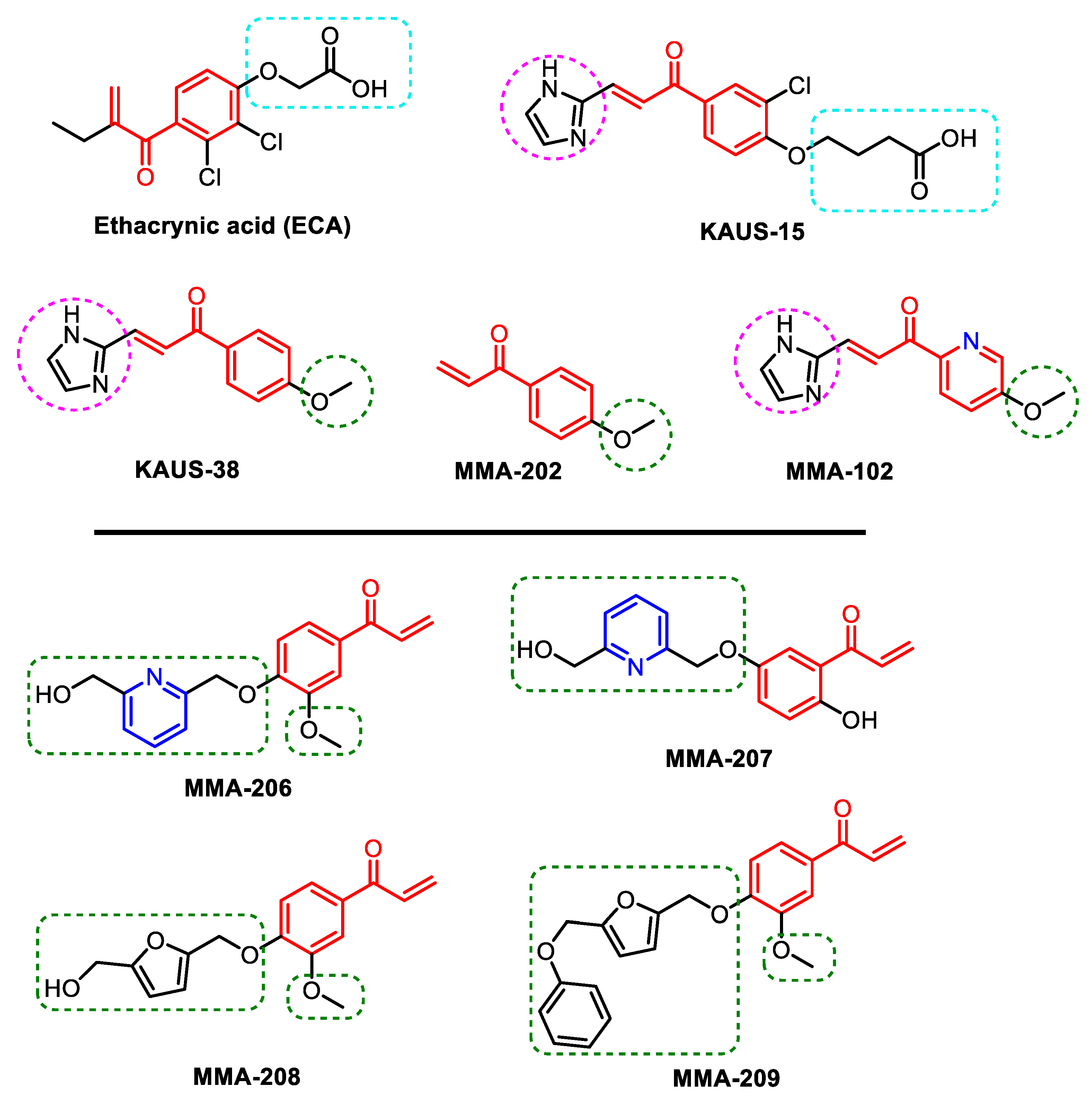
4.2. Compound Synthesis
Unless otherwise noted, reagents and solvents were used as purchased from commercial suppliers. Solvent removal was accomplished usually using a rotary evaporator at ~15 mm Hg pressure unless otherwise specified. Thin-layer chromatography (TLC) was performed using silica-gel 60 plates F254 (Merck KGaA, Darmstadt, Germany) and visualized by UV light (254 nm). Column chromatography and flash were carried out using silica gel (60–203 mesh and 40–60 mesh, respectively), unless otherwise specified. Proton nuclear magnetic resonance (1H NMR) spectra were recorded on a Bruker Avance II 300 MHz spectrometer (Bruker BioSpin, Billerica, MA, USA) at ambient temperature. Chemical shifts were calibrated to residual solvent peaks or internal tetramethylsilane (TMS). All target compounds and key intermediates were confirmed by 1H NMR (and 13C NMR where applicable) in appropriate deuterated solvents (e.g., DMSO-d_6 or CDCl_3), verifying their structures. Sample purity was assessed by ultra-performance liquid chromatography–mass spectrometry (UPLC–MS) using a Waters Acquity H-Class system equipped with a single-quadrupole QDa mass detector (Waters Corporation, Milford, MA, USA). UPLC separations employed a Phenomenex Kinetex EVO C_18 column (50 × 3 mm, 2.6 µm) (Phenomenex Inc, Torrance, CA, USA) with UV detection at 220 nm (and 254 nm). A short linear gradient (2–5 min runtime) of water and acetonitrile was used, under either acidic conditions (0.1% trifluoroacetic acid) or neutral buffered conditions (20 mM ammonium formate, pH 7.4), selected according to compound polarity. The UPLC–UV chromatograms typically showed a single major peak for each compound, and the QDa MS spectra displayed the expected molecular ion ([M+H]+ or Na+ adduct) for the product, confirming each compound’s identity and homogeneity. All final Michael acceptor analogues (MMA-206 through MMA-209) showed high purity by UPLC (generally >95% area under the curve) and gave NMR spectra consistent with their proposed structures.
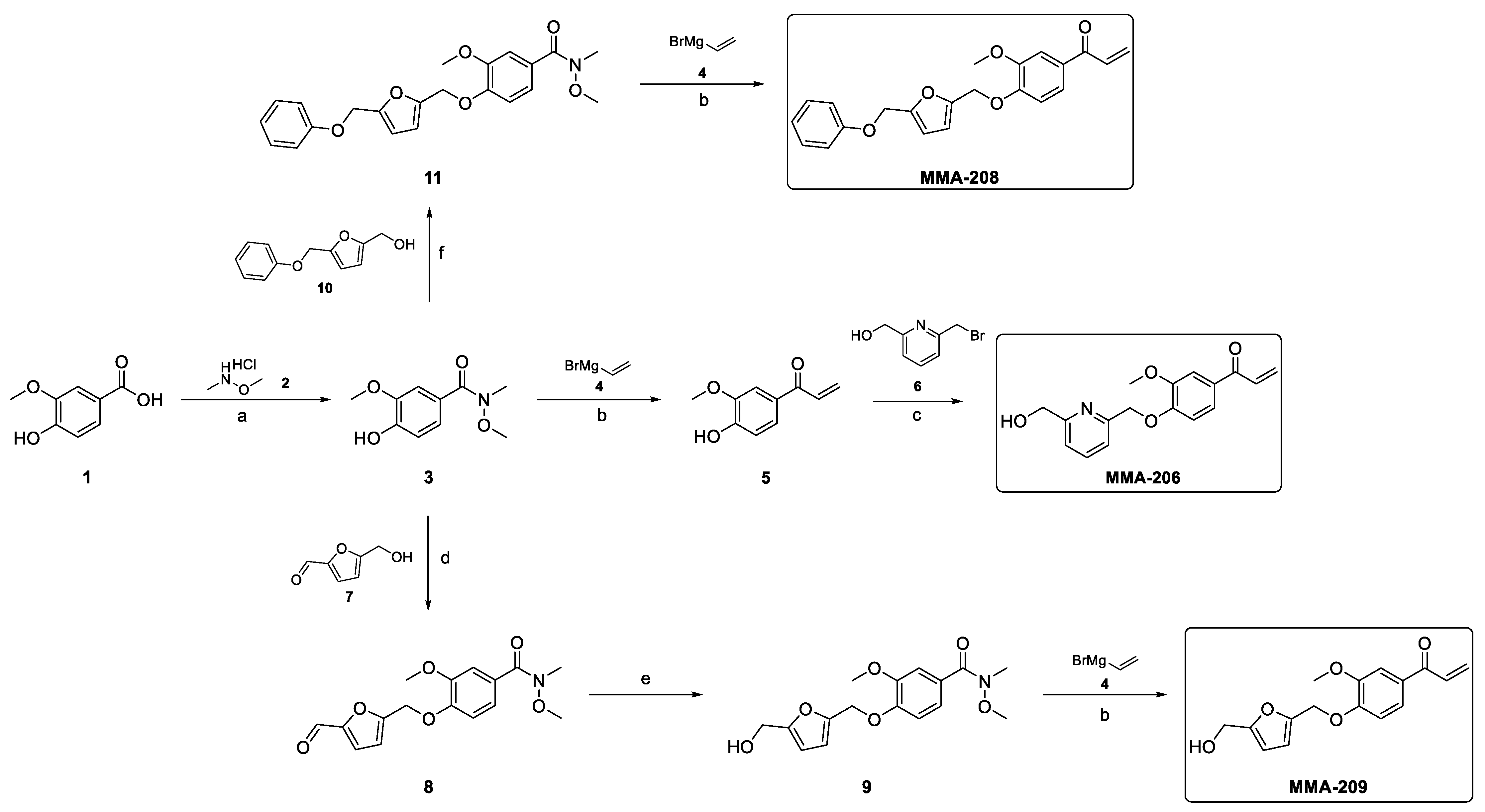
Preparation of 4-Hydroxy-N,3-dimethoxy-N-methylbenzamide (3)
Vanillic acid (1, 5.0 g, 29.7 mmol, 1.0 equiv) was dissolved in dry DMF (37.5 mL), and N,O-Dimethylhydroxylamine HCl (2, 3.57 g, 36.6 mmol, 1.23 equiv) was added. The mixture was cooled to 0 °C, and HOBt (602 mg, 4.46 mmol, 0.15 equiv), N-Methylmorpholine (NMM, 6.86 mL, 62.4 mmol, 2.1 equiv), and 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDCI, 6.84 g, 35.7 mmol, 1.2 equiv) were added. The resultant mixture was stirred at 0 °C for 75 min and at rt overnight. The progress of the reaction was monitored by TLC (dichloromethane-MeOH = 95:5) and LCMS analyses. After completion, the mixture was diluted with dichloromethane and extracted with citric acid (10%, twice). The organic phase was dried over Na2SO4, filtered and concentrated. The crude product was purified by column chromatography (eluent: 0–5% MeOH in dichloromethane) and recrystallized from Diisopropyl ether (DIPE) to afford the intermediate compound 4-Hydroxy-N,3-dimethoxy-N-methylbenzamide (3, 4.71 g, 75% yield) as a white crystal. 1H-NMR (300 MHz, DMSO-d6) δ 9.57 (s, 1H), 7.20 (d, J = 1.7 Hz, 1H), 7.15 (dd, J = 8.2, 1.8 Hz, 1H), 6.81 (d, J = 8.2 Hz, 1H), 3.79 (s, 3H), 3.57 (s, 3H), 3.22 (s, 3H). MS m/z 212 [M+H]+; UPLC-MS using (220 nm) 84% (AUC). Melting point: 101–102 °C.
Synthesis of MMA-206
Preparation of 1-(4-Hydroxy-3-methoxyphenyl)prop-2-en-1-one (5)
The Weinreb amide 3 (1.5 g, 7.1 mmol, 1.0 equiv) was dissolved in anhydrous THF (35 mL) under argon atmosphere, and the resulting solution was cooled to −78 °C. Then, a solution of vinyl magnesium bromide (4, 28.4 mL, 28.4 mmol, 4.0 equiv, 1M in THF) was added dropwise. After complete addition, the cooling bath was removed, and the mixture was allowed to warm to room temperature. The progress of the reaction was monitored by TLC (dichloromethane-MeOH = 95:5) and LCMS analyses. After completion, the mixture was poured into a vigorously stirred solution of 1M HCl (100 mL) and stirred for 15 min. The resulting mixture was extracted with dichloromethane twice, and the combined organic layer was dried over MgSO4, filtered and concentrated. The crude product was purified by column chromatography (eluent: 0–20% EtOAc in n-heptane) to afford the intermediate compound 1-(4-Hydroxy-3-methoxyphenyl)prop-2-en-1-one (5, 0.98 g, 78% yield) as a light brown oil. 1H-NMR (300 MHz, DMSO-d6) δ 10.11 (s, 1H), 7.60 (d, J = 8.4 Hz, 1H), 7.52 (s, 1H), 7.43 (dd, J = 16.9, 10.4 Hz, 1H), 6.89 (d, J = 8.3 Hz, 1H), 6.29 (d, J = 16.9 Hz, 1H), 5.88 (dd, J = 10.3, 1.7 Hz, 1H), 3.84 (s, 3H). MS m/z 179 [M+H]+; UPLC-MS (220 nm) 99% (AUC).
Preparation of 1-(4-((6-(Hydroxymethyl)pyridin-2-yl)methoxy)-3-methoxyphenyl)prop-2-en-1-one (MMA-206)
The enone intermediate 5 (0.3 g, 1.68 mmol, 1.0 equiv) was combined with (6-(bromomethyl)pyridin-2-yl)methanol (6) (0.544 g, 2.69 mmol, 1.6 equiv) and K2CO3 (0.7 g, 5.05 mmol, 3.0 equiv) in MeCN (5 mL) and stirred at 60 °C for 5 h. The progress of the reaction was monitored by TLC (cyclohexane-EtOAc = 1:2) and LCMS analyses. After completion, the mixture was filtered off, and the inorganic salt was washed with dichloromethane. The filtrate was concentrated under reduced pressure, and the crude product was purified by column chromatography (eluent: 0–50% EtOAc in cyclohexane) to afford the desired product (MMA-206, 130 mg, 26% yield) as an off-white crystal. 1H-NMR (300 MHz, CDCl3) δ 7.73 (t, J = 7.7 Hz, 1H), 7.62 (d, J = 1.6 Hz, 1H), 7.54 (dd, J = 8.3, 1.7 Hz, 1H), 7.45 (d, J = 7.5 Hz, 1H), 7.25–7.07 (m, 2H), 6.92 (d, J = 8.4 Hz, 1H), 6.44 (dd, J = 17.0, 1.6 Hz, 1H), 5.89 (dd, J = 10.5, 1.5 Hz, 1H), 5.38 (s, 2H), 4.80 (s, 2H), 4.00 (s, 3H), 3.72 (s, 1H). 13C-NMR (300 MHz, CDCl3) δ 189.18, 158.64, 155.52, 152.13, 149.69, 137.68, 131.93, 131.01, 129.42, 123.24, 119.83, 119.57, 112.09, 111.32, 71.25, 63.97, 56.15. MS m/z 300 [M+H]+; UPLC-MS (220 nm) 95% (AUC). Melting point: 81–82 °C.
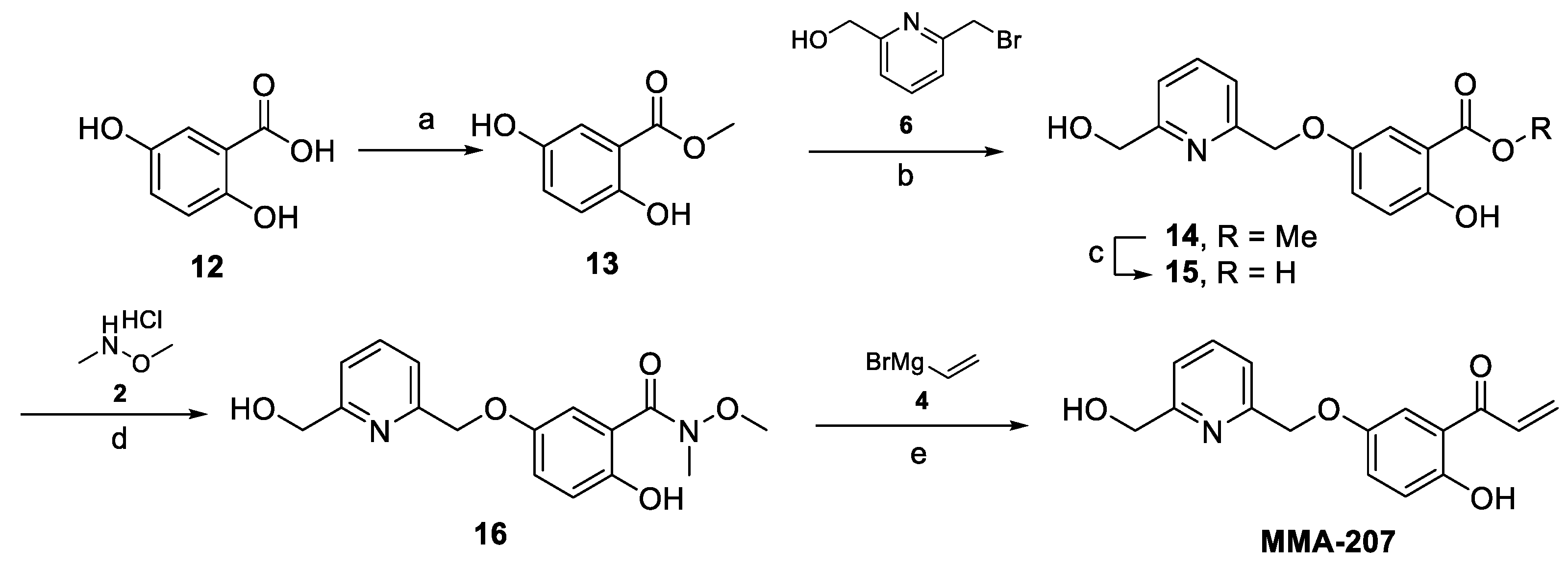
Synthesis of MMA-207
Preparation of Methyl 2,5-dihydroxybenzoate (13)
2,5-Dihydroxybenzoic acid (12, 2.0 g, 12.98 mmol, 1.0 equiv) was dissolved in MeOH (30 mL), SOCl2 (1.42 mL, 19.5 mmol, 1.5 equiv) was added dropwise at 0 °C, and the reaction mixture was refluxed overnight. The progress of the reaction was monitored by TLC (dichloromethane-MeOH = 9:1 + NH4OH) and LCMS analyses. After completion, the mixture was evaporated, and the crude product was co-evaporated with toluene to give the intermediate compound Methyl 2,5-dihydroxybenzoate (13, 2.18 g, 99.9% yield) as a white crystal, which was used in the next step without further purification. 1H-NMR (300 MHz, DMSO-d6) δ 9.92 (s, 1H), 9.21 (s, 1H), 7.14 (d, J = 3.0 Hz, 1H), 6.98 (dd, J = 8.9, 3.0 Hz, 1H), 6.82 (d, J = 8.8 Hz, 1H), 3.87 (s, 3H). MS m/z not detected (220 nm) 99% (AUC). Melting point: 92–93 °C.
Preparation of Methyl 2-hydroxy-5-((6-(hydroxymethyl)pyridin-2-yl)methoxy)benzoate (14)
Starting compound (13, 1.69 g, mmol, 1.0 equiv) was combined with (6-(bromomethyl)pyridin-2-yl)methanol (6, 2.03 g, 10.0 mmol, 1.0 equiv) and K2CO3 (2.5 g, 18.1 mmol, 1.8 equiv) in dry acetone (40 mL) and stirred at 50 °C for 48 h. The progress of the reaction was monitored by TLC (dichloromethane:MeOH-95:5) and LCMS analyses. After completion, the mixture was filtered off, and the inorganic salt was washed with dichloromethane. The filtrate was evaporated under reduced pressure, and the concentrate was dissolved in dichloromethane, washed with water (twice), dried over anhydrous MgSO4, and evaporated. The crude product was purified by column chromatography (eluent: 0–5% MeOH in dichloromethane) to afford the intermediate compound Methyl 2-hydroxy-5-((6-(hydroxymethyl)pyridin-2-yl)methoxy)benzoate (14, 1.32 g, 45% yield) as a yellow solid. 1H-NMR (300 MHz, DMSO-d6) δ 10.09 (s, 1H), 7.83 (t, J = 7.7 Hz, 1H), 7.53–7.32 (m, 3H), 7.26 (dd, J = 9.0, 3.1 Hz, 1H), 6.94 (d, J = 9.0 Hz, 1H), 5.44 (t, J = 5.8 Hz, 1H), 5.10 (s, 2H), 4.57 (d, J = 5.5 Hz, 2H), 3.89 (s, 3H). MS m/z 290 [M+H]+; UPLC-MS (220 nm) 75% (AUC). Melting point: 85–86 °C.
Preparation of 2-Hydroxy-5-((6-(hydroxymethyl)pyridin-2-yl)methoxy)benzoic acid (15)
Starting ester (14, 1.27 g, 4.4 mmol, 1.0 equiv) was dissolved in MeOH (14 mL), 1M LiOH solution (15.5 mL, 15.4 mmol, 3.5 equiv) was added, and the mixture was stirred at 45 °C for 6 h. The progress of the reaction was monitored by TLC (dichloromethane-MeOH = 95:5 and 95:5 + AcOH) and LCMS analyses. After completion, the mixture was poured into water, its pH was adjusted to 2–3 by the addition of 1M aq. HCl, and the precipitate was filtered off, washed with water, and dried to obtain the intermediate compound 2-Hydroxy-5-((6-(hydroxymethyl)pyridin-2-yl)methoxy)benzoic acid (15, 1.07 g, 88% yield) as a white crystal. 1H-NMR (300 MHz, DMSO-d6) δ 10.99 (s, 1H), 7.83 (t, J = 7.7 Hz, 1H), 7.49–7.29 (m, 3H), 7.24 (dd, J = 9.0, 3.1 Hz, 1H), 6.91 (d, J = 9.0 Hz, 1H), 5.42 (s, 1H), 5.10 (s, 2H), 4.57 (s, 2H). MS m/z 276 [M+H]+; UPLC-MS (220 nm) 95% (AUC). Melting point: 242–243 °C.
Preparation of 2-Hydroxy-5-((6-(hydroxymethyl)pyridin-2-yl)methoxy)-N-methoxy-N-methylbenzamide (16)
This Weinreb amide was prepared according to the procedure described for the preparation of 3. The crude product was purified by column chromatography (eluent: 0–5% MeOH in dichloromethane) to afford the intermediate compound 2-Hydroxy-5-((6-(hydroxymethyl)pyridin-2-yl)methoxy)-N-methoxy-N-methylbenzamide (16, 0.81 g, 66% yield) as a white crystal. MS m/z 319 [M+H]+; UPLC-MS (220 nm) 95% (AUC), which was used for the next reaction without further purification.
Preparation of 1-(2-Hydroxy-5-((6-(hydroxymethyl)pyridin-2-yl)methoxy)phenyl)prop-2-en-1-one (MMA-207)
The intermediate 16 (0.81 g, mmol, 1.0 equiv) was dissolved in anhydrous THF (12.5 mL) under argon atmosphere, and the resulting solution was cooled to −78 °C. Then, a solution of vinyl magnesium bromide (4, 10.2 mL, 10.2 mmol, 4.0 equiv, 1M in THF) was added dropwise. After complete addition, the mixture was stirred at −78 °C for 2.5 h; then, it was allowed to warm to 0 °C while stirring. The progress of the reaction was monitored by LCMS analysis. After completion, the mixture was poured into a vigorously stirred cold solution of 1 M HCl (50 mL) and stirred for 15 min. The resulting mixture was extracted with dichloromethane twice and the combined organic layers dried over MgSO4 and concentrated. The crude product was purified by preparative HPLC to afford the final compound 1-(2-Hydroxy-5-((6-(hydroxymethyl)pyridin-2-yl)methoxy)phenyl)prop-2-en-1-one (MMA-207, 0.12 g, 16.5% yield) as a yellowish crystal. 1H-NMR (300 MHz, CDCl3) δ 12.13 (s, 1H), 7.76 (t, J = 7.7 Hz, 1H), 7.46 (d, J = 7.6 Hz, 1H), 7.38 (d, J = 3.0 Hz, 1H), 7.32–7.16 (m, 3H), 6.99 (d, J = 9.1 Hz, 1H), 6.56 (dd, J = 16.9, 1.6 Hz, 1H), 5.99 (dd, J = 10.6, 1.5 Hz, 1H), 5.21 (s, 2H), 4.81 (s, 2H), 3.65 (s, 1H). 13C-NMR (300 MHz, CDCl3) δ 194.08, 170.08, 158.62, 158.34, 155.99, 150.47, 137.64, 131.02, 130.67, 125.34, 120.08, 119.58, 118.96, 114.19, 71.48, 64.02. MS m/z 286 [M+Na]+; UPLC-MS using (220 nm) 95% (AUC). Melting point: 75–78 °C.
Synthesis of MMA-208
Preparation of 5-(Phenoxymethyl)furan-2-carbaldehyde (10)
Starting aldehyde 5-hydroxymethylfurfural (5-HMF, 7, 2.0 g, 15.86 mmol, 1.0 equiv) was dissolved in THF (104 mL); then, phenol (2.24 g, 1.5 equiv), TPP (2.24 g, 23.8 mmol, 1.5 equiv), and DIAD (4.67 mL, 23.8 mmol, 1.5 equiv) were added. The resultant mixture was stirred at rt overnight. After completion, the solvent was evaporated under reduced pressure, and the residue was partitioned between dichloromethane and 10% aq. NaOH solution. After separation, the organic phase was dried over MgSO4, filtered, and evaporated. The crude product was purified by column chromatography (eluent: 0–30% EtOAc in cyclohexane) to afford the intermediate product 5-(Phenoxymethyl)furan-2-carbaldehyde (10, PMFC). 1H-NMR (CDCl3, 300 MHz): δ 9.60 (s, 1H), 7.54 (d, J = 3.5 Hz, 1H), 7.32 (td, J = 8.0, 1.9 Hz, 2H), 7.04 (d, J = 7.9 Hz, 2H), 6.98 (t, J = 7.3 Hz, 1H), 6.87 (d, J = 3.5 Hz, 1H), 5.20 (s, 1H). MS m/z 203 [M+H]+; UPLC-MS (220 nm) 98% (AUC). Mp: 95–96 °C.
PMFC (1.69 g, 8.36 mmol, 1.0 equiv) was dissolved in a mixture of MeOH (15 mL) and dichloromethane (15 mL) under N2 atmosphere and cooled to 0 °C. NaBH4 (0.38 g, 10.0 mmol, 1.2 equiv) was added portion-wise, and the mixture was stirred at 0 °C for 15 min. Then, the cooling bath was removed, and the mixture was allowed to warm to room temperature and stirred for 1 h. The progress of the reaction was monitored by TLC (n-heptane-EtOAc = 3:2) and LCMS analyses. After completion, the reaction was quenched with water. The volatile solvent was removed under reduced pressure, and the residual aqueous solution was extracted with EtOAc. The organic layer was dried over MgSO4, filtered, and evaporated. The crude product was purified by column chromatography (eluent: 0–5% MeOH in dichloromethane) to afford the intermediate product 5-(Phenoxymethyl)furan-2-carbaldehyde (10, 1.56 g, 91% yield) as a yellowish solid. 1H-NMR (300 MHz, DMSO-d6) δ 7.30 (t, J = 8.0 Hz, 2H), 7.02 (d, = 8.0 Hz, 2H), 6.95 (t, J = 7.3 Hz, 1H), 6.50 (d, J = 3.1 Hz, 1H), 6.27 (d, J = 3.0 Hz, 1H), 5.24 (t, J = 5.3 Hz, 1H), 5.01 (s, 2H), 4.38 (d, J = 5.1 Hz, 2H). MS m/z 187 [M+H-H2O]+; UPLC-MS (220 nm) 97% (AUC). Melting point: 37–39 °C.
Preparation of N,3-Dimethoxy-N-methyl-4-((5-(phenoxymethyl)furan-2-yl)methoxy)benzamide (11)
The phenolic intermediate 3 (1.0 g, 4.74 mmol, 1.0 equiv) was dissolved in THF (30 mL); then, the alcohol reagent 10 (1.16 g, 5.68 mmol, 1.2 equiv), Triphenylphosphine (TPP, 1.86 g, 7.1 mmol, 1.5 equiv), and Diisopropyl azodicarboxylate (DEAD, 1.11 mL, 7.1 mmol, 1.5 equiv) were added. The resultant mixture was stirred at rt overnight. The progress of the reaction was monitored by TLC (dichloromethane-MeOH = 95:5) and LCMS analyses. After completion, the solvent was evaporated under reduced pressure, and the residue was partitioned between EtOAc and water. After separation, the organic phase was dried over MgSO4, filtered, and evaporated. The crude product was purified by column chromatography (eluent: 0–50% EtOAc in cyclohexane) to afford the intermediate compound N,3-Dimethoxy-N-methyl-4-((5-(phenoxymethyl)furan-2-yl)methoxy)benzamide (11, 1.81 g impure, 96% yield, contaminated with some triphenylphosphine oxide) as a yellowish oil. 1H NMR (300 MHz, DMSO-d6) δ 7.29 (dd, J = 16.2, 8.5 Hz, 3H), 7.22 (s, 1H), 7.16 (d, J = 8.2 Hz, 1H), 7.03 (d, J = 7.9 Hz, 2H), 6.96 (t, J = 7.2 Hz, 1H), 6.61 (s, 2H), 5.08 (d, J = 11.7 Hz, 4H), 3.78 (s, 3H), 3.57 (s, 3H), 3.23 (s, 3H). MS m/z 398 [M+H]+; (220 nm) 55% (AUC).
Preparation of 1-(3-Methoxy-4-((5-(phenoxymethyl)furan-2-yl)methoxy)phenyl)prop-2-en-1-one (MMA-208)
This final compound was prepared following the procedure described for the preparation of 5. Starting Weinreb amide 11 (1.0 g, 2.51 mmol, 1.0 equiv) was dissolved in anhydrous THF (12 mL) under argon atmosphere, and the resulting solution was cooled to −78 °C. Then, a solution of vinyl magnesium bromide (1M in THF, 10 mL, 10.1 mmol, 4.0 equiv) was added dropwise. After complete addition, the mixture was stirred at −78 °C for 2.5 h; then, it was allowed to warm to 0 °C and stirred for 30 min. The progress of the reaction was monitored by LCMS analysis. After completion, the mixture was poured into a vigorously stirred cold solution of 1 M HCl (50 mL) and stirred for 15 min. The resulting mixture was extracted with dichloromethane twice, and the combined organic layer was dried over MgSO4, filtered and concentrated. The crude product was purified by preparative HPLC to afford the final compound 1-(3-Methoxy-4-((5-(phenoxymethyl)furan-2-yl)methoxy)phenyl)prop-2-en-1-one (MMA-208, 120 mg, 13% yield) as a yellowish crystal. 1H-NMR (300 MHz, DMSO-d6) δ 7.71 (dd, J = 8.5, 1.7 Hz, 1H), 7.62–7.38 (m, 2H), 7.29 (dd, J = 14.3, 8.4 Hz, 3H), 7.03 (d, J = 7.9 Hz, 2H), 6.96 (t, J = 7.2 Hz, 1H), 6.62 (dd, J = 9.7, 3.1 Hz, 2H), 6.32 (dd, J = 16.9, 2.0 Hz, 1H), 5.92 (dd, J = 10.4, 1.9 Hz, 1H), 5.17 (s, 2H), 5.06 (s, 2H), 3.83 (s, 3H). 13C-NMR (300 MHz, DMSO-d6) δ 200.35, 164.78, 158.38, 152.32, 151.51, 150.56, 149.53, 132.63, 130.57, 130.01, 129.75, 123.66, 121.47, 115.21, 112.91, 112.57, 111.91, 111.40, 63.33, 62.75, 62.04, 56.00. MS m/z 387 [M+H+Na]+; UPLC-MS (220 nm) 98% (AUC). Melting point: 87–89 °C.
Synthesis of MMA-209
Preparation of 4-((5-Formylfuran-2-yl)methoxy)-N,3-dimethoxy-N-methylbenzamide (8)
The phenolic intermediate 3 (1.37 g, 6.5 mmol, 1.0 equiv) was dissolved in THF (41 mL); then, 5-(Hydroxymethyl)furfural (5-HMF, 7, 982 mg, 7.8 mmol, 1.2 equiv), TPP (2.55 g, 9.72 mmol, 1.5 equiv), and DIAD (1.91 mL, 9.72 mmol, 1.5 equiv) were added. The resultant mixture was stirred at rt overnight. The progress of the reaction was monitored by TLC (dichloromethane-MeOH = 95:5) and LCMS analyses. After completion, the solvent was evaporated under reduced pressure, and the residue was partitioned between EtOAc and water. After separation, the organic phase was dried over MgSO4, filtered, and evaporated. The crude product was purified by column chromatography (eluent: 0–60% EtOAc in cyclohexane) to afford the intermediate product 4-((5-Formylfuran-2-yl)methoxy)-N,3-dimethoxy-N-methylbenzamide (8, 1.39 g, 67% yield) as a yellowish-white solid. 1H-NMR (300 MHz, DMSO-d6) δ 9.62 (s, 1H), 7.55 (d, J = 3.5 Hz, 1H), 7.30–7.09 (m, 3H), 6.89 (d, J = 3.5 Hz, 1H), 5.24 (s, 2H), 3.79 (s, 3H), 3.57 (s, 3H), 3.24 (s, 3H). MS m/z 320 [M+H]+; UPLC-MS (220 nm) 96% (AUC). Melting point: 102–103 °C.
Preparation of 4-((5-(Hydroxymethyl)furan-2-yl)methoxy)-N,3-dimethoxy-N-methylbenzamide (9)
Starting aldehyde (8 1.365 g, 4.27 mmol, 1.0 equiv) was dissolved in MeOH (13.5 mL) under N2 atmosphere and cooled to 0 °C; then, NaBH4 (0.324 g, 8.55 mmol, 2.0 equiv) was added portion-wise, and it was stirred at 0 °C for 15 min. Then, the cooling bath was removed, and the mixture was allowed to warm to room temperature and stirred for 1 h. The progress of the reaction was monitored by TLC (dichloromethane-MeOH = 95:5) and LCMS analyses. After completion, the reaction was quenched with water. The volatile solvent was removed under reduced pressure, and the residual aqueous solution was extracted with EtOAc. The organic layer was dried over MgSO4, filtered, and evaporated. The crude product was purified by column chromatography (eluent: 0–5% MeOH in dichloromethane) to afford the intermediate product 4-((5-(Hydroxymethyl)furan-2-yl)methoxy)-N,3-dimethoxy-N-methylbenzamide (9, 763 mg, 55% yield) as an off-white solid. 1H-NMR (300 MHz, DMSO-d6) δ 7.29–7.09 (m, 3H), 6.53 (d, J = 3.1 Hz, 1H), 6.29 (d, J = 3.0 Hz, 1H), 5.27 (t, J = 5.8 Hz, 1H), 5.05 (s, 2H), 4.39 (d, J = 5.7 Hz, 2H), 3.77 (s, 3H), 3.58 (s, 3H), 3.24 (s, 3H). MS m/z 344 [M+Na]+; UPLC-MS using Waters method 5min_slow_A1B1_EVO (220 nm) 95% (AUC). Melting point: 104–105 °C.
Preparation of 1-(4-((5-(Hydroxymethyl)furan-2-yl)methoxy)-3-methoxyphenyl)prop-2-en-1-one (MMA-209)
Starting compound (16, 730 mg, 2.27 mmol, 1.0 equiv) was dissolved in anhydrous THF (11 mL) under argon atmosphere, and the resulting solution was cooled to −78 °C. Then, a solution of vinyl magnesium bromide (4, 9.1 mL, 9.1 mmol, 4.0 equiv, 1M in THF) was added dropwise. After complete addition, the mixture was stirred at −78 °C for 2.5 h; then, it was allowed to warm to 0 °C and stirred for 30 min. The progress of the reaction was monitored by LCMS analysis. After completion, the mixture was poured into a vigorously stirred cold solution of 1 M HCl (40 mL) and stirred for 15 min. The resulting mixture was extracted with dichloromethane twice, and the combined organic layer was dried over MgSO4, and concentrated. The crude product was purified by preparative HPLC and lyophilized to afford the final compound 1-(4-((5-(Hydroxymethyl)furan-2-yl)methoxy)-3-methoxyphenyl)prop-2-en-1-one (MMA-209, 32 mg, 5% yield) as a white crystal. 1H-NMR (300 MHz, DMSO-d6) δ 7.77–7.05 (m, 4H), 6.54 (d, J = 3.0 Hz, 1H), 6.29 (d, J = 2.8 Hz, 1H), 5.33–5.17 (m, 1H), 5.15–4.96 (m, 2H), 4.39 (d, J = 4.6 Hz, 2H), 3.88–3.70 (m, 3H). MS m/z 311 [M+Na]+; UPLC-MS (220 nm) 85% (AUC). Melting point: 89–91 °C.
Reactivity of MMA Compounds with Accessible Sulfhydryl Groups in Hb
The accessible sulfhydryl (SH) groups in Hb and their reactivity with the MMA compounds was quantified using a disulfide exchange reaction between the SH group of βCys93 and 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB, Ellman’s reagent) by measuring absorbance at 412 nm (ε = 14,150 M
−1 cm
−1) [
25]. An aqueous solution of Hb (50 μM in PBS) was mixed with MMA-206, MMA-207, MMA-208, or MMA-209, and the positive control ethacrynic acid (ECA) at 2mM final concentration in a final volume of 500 μL. The tubes were incubated at room temperature for 3h with shaking at 100rpm. The reaction mixture was then transferred to a microfiltration centrifugal tube (MWCO 10 kDa) and centrifuged at 7000 rpm for 30 min at 4 °C to separate Hb from excess reagents. Modified Hb was further washed with PBS and centrifuged again to a reaction volume of 100 μL. A total of 25 μL of each Hb solution was added to 475 µL of 100 mM potassium phosphate buffer, pH 8.0, and incubated at 25 °C for 1 h (non-DTNB control samples). Another 25 μL of each Hb solution was added to the phosphate buffer (465 μL) with 10 μL of DTNB (10 mM) and incubated at 25 °C for 1 h. Before centrifuging the non-DTNB control samples, the absorbance of each sample was noted at 576 nm to determine the concentration of Hb in each sample. Both sets of tubes were centrifuged using different centrifugal filters (7000 rpm, 20 min, 4 °C) to collect the yellow filtrate (2-nitro-5-thiobenzoate), which was quantified by measuring an absorbance at 412 nm.
In vitro Time-Dependent Hb Oxygen Equilibrium Curve Studies Using Normal Whole Blood
As previously reported [
25], normal whole blood samples (hematocrit 30%) in the presence of 2 mM concentration of the test compounds, MMA-206 and MMA-208, were incubated at 37 °C for 24 h with shaking (at 100 rpm). At 1.5, 3, 6, 12, and 24 h time intervals, aliquots of the mixture were removed and further incubated in TM8000 Thin film tonometer (Meon Medical Solutions, Graz, Austria) to equilibrate at oxygen tensions 6, 20, and 40 mmHg for approximately 10 min at 37 °C. The samples were then aspirated into an ABL 800 Automated Blood Gas Analyzer (Radiometer, Copenhagen, Denmark) to determine the partial pressure of oxygen (pO
2) and Hb oxygen saturation values (SO
2). The measured values of pO
2 (mmHg) and SO
2 at each oxygen tension values were then subjected to a non-linear regression analysis using the program Scientist (Micromath, Salt Lake City, UT, USA) to estimate P
50, as previously reported [
25]. The observed P
50 shifts values in %P
50 shifts were plotted as function of time (h).
In vitro Antisickling and Oxygen Equilibrium Curve Studies Using Human Homozygous Sickle Cell (SS) Blood
The compounds MMA-206 to -209 and the positive control MMA-202 were studied for their abilities to increase Hb affinity for oxygen and inhibit deoxygenated-induced RBC sickling (RBC morphology study), as previously published [
25]. Briefly, homozygous SS blood (hematocrit 20%) suspensions were incubated under air in the absence or presence of 0.5 mM, 1.0 mM, and 2 mM concentration of test compounds at 37 °C for 1 h. Following, the suspensions were incubated under hypoxic condition (2.5% O
2 gas/97.5% N
2 gas) at 37 °C for 2 h. Aliquot samples were fixed with 2% glutaraldehyde solution without exposure to air and then subjected to microscopic morphological analysis. The residual samples were washed in phosphate-buffered saline and hemolyzed in hypotonic lysis buffer for the Hb oxygen equilibrium curve (OEC) experiment. For the OEC study, approximately 100 μL aliquot samples from the lysate were added to 4 mL of 0.1 M potassium phosphate buffer, pH 7.0, in a cuvette and subjected to hemoximetry analysis, using Hemox™ Analyzer (TCS Scientific Corp., New Hope, PA, USA) to assess P
50 shifts.


 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}