
Identification of 3-[(4-Acetylphenyl)(4-Phenylthiazol-2-Yl)Amino]Propanoic Acid Derivatives as Promising Scaffolds for the Development of Novel Anticancer Candidates Targeting SIRT2 and EGFR

 ,
,  , , and
, , and
Abstract

1. Introduction
2. Results
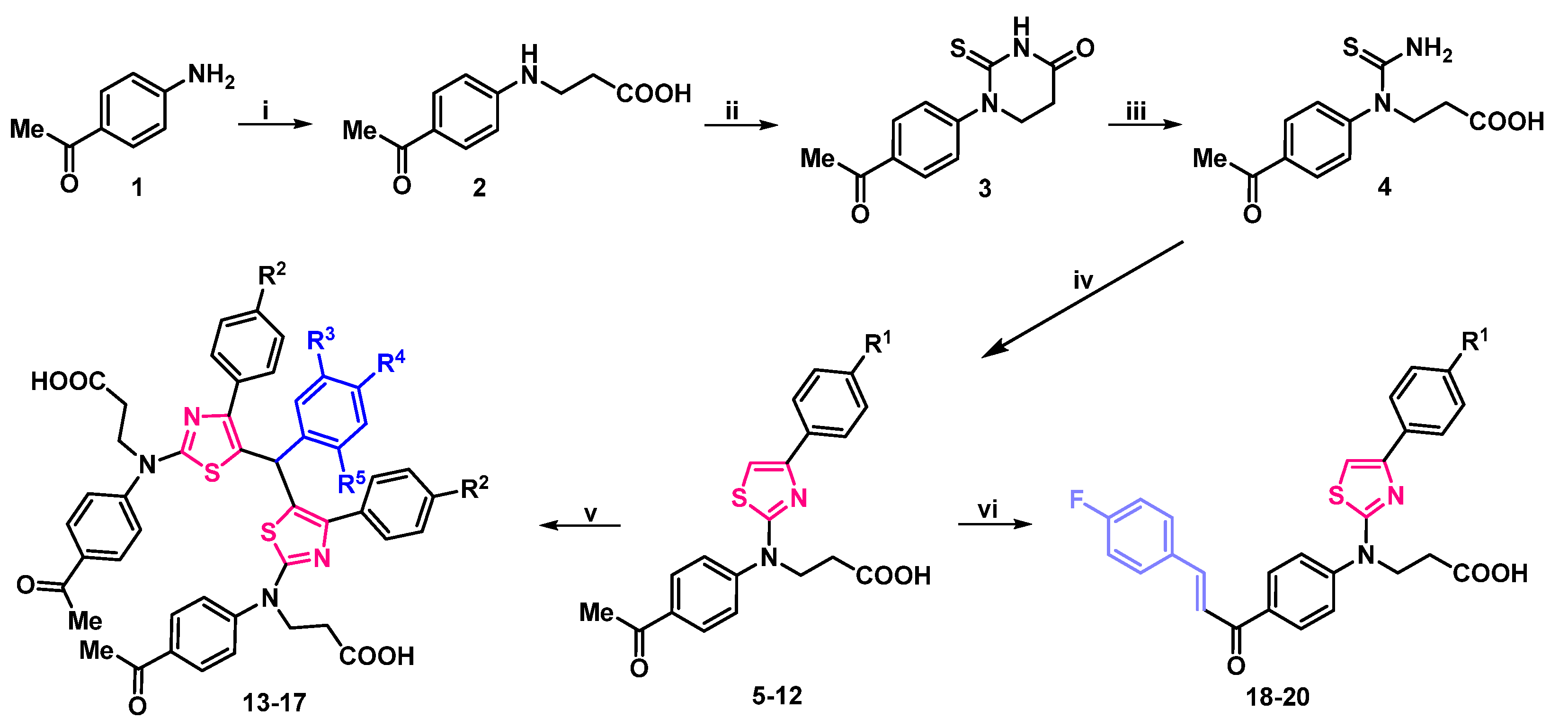
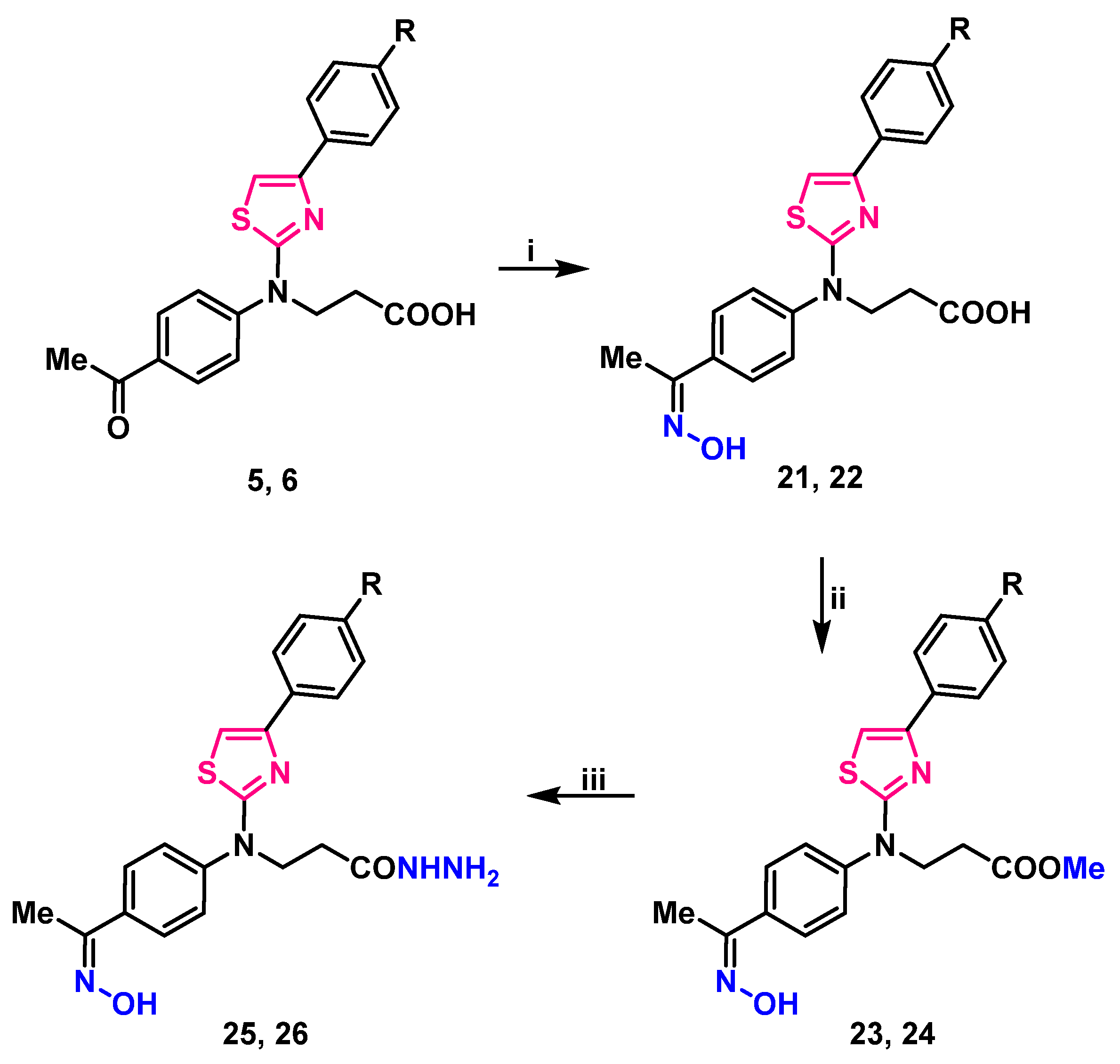
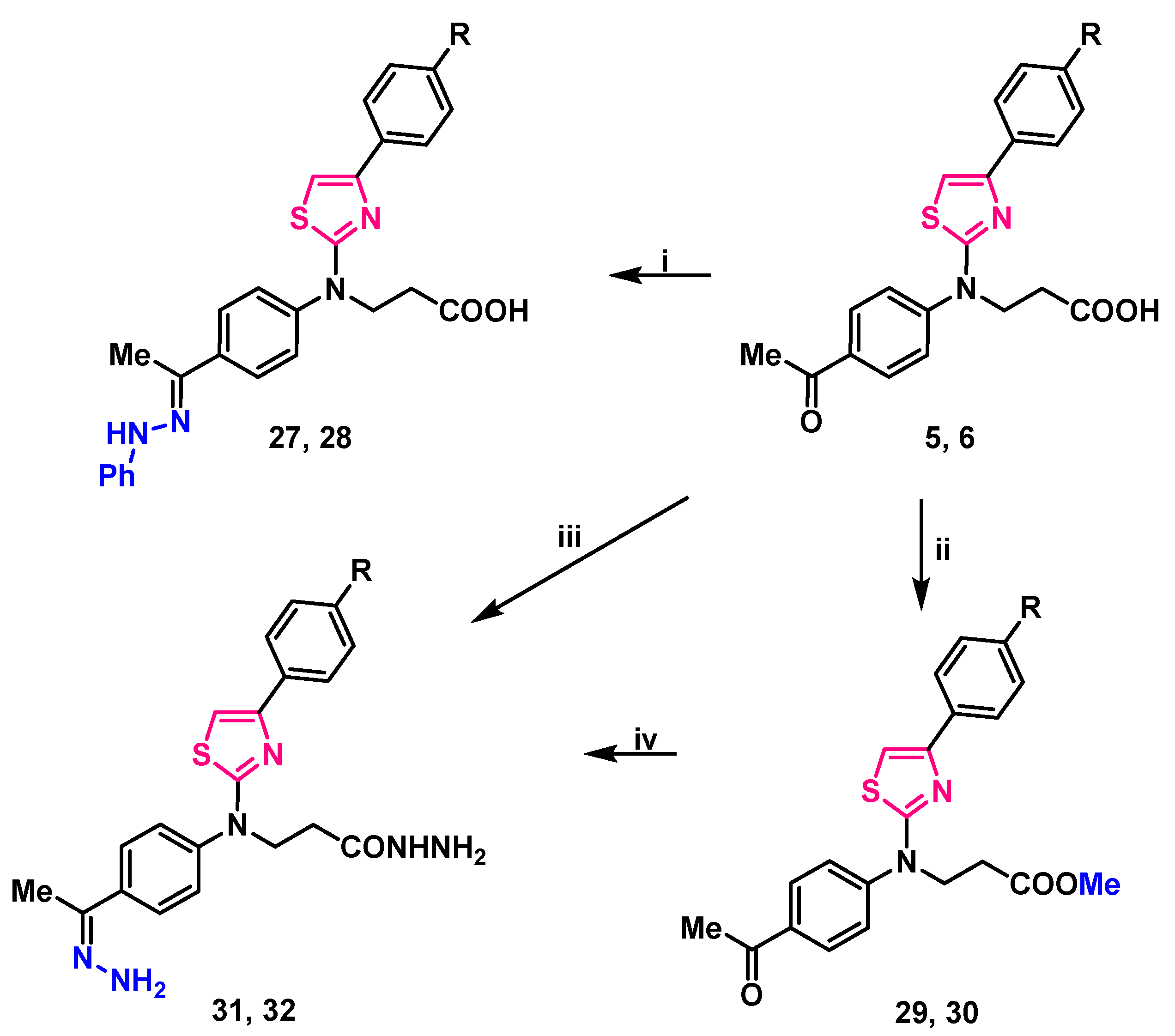
2.1. Synthesis
2.2. Structure-Dependent Antiproliferative Activity of 3-[(4-Acetylphenyl)(4-Phenylthiazol-2-Yl)Amino]Propanoic Acid Derivatives
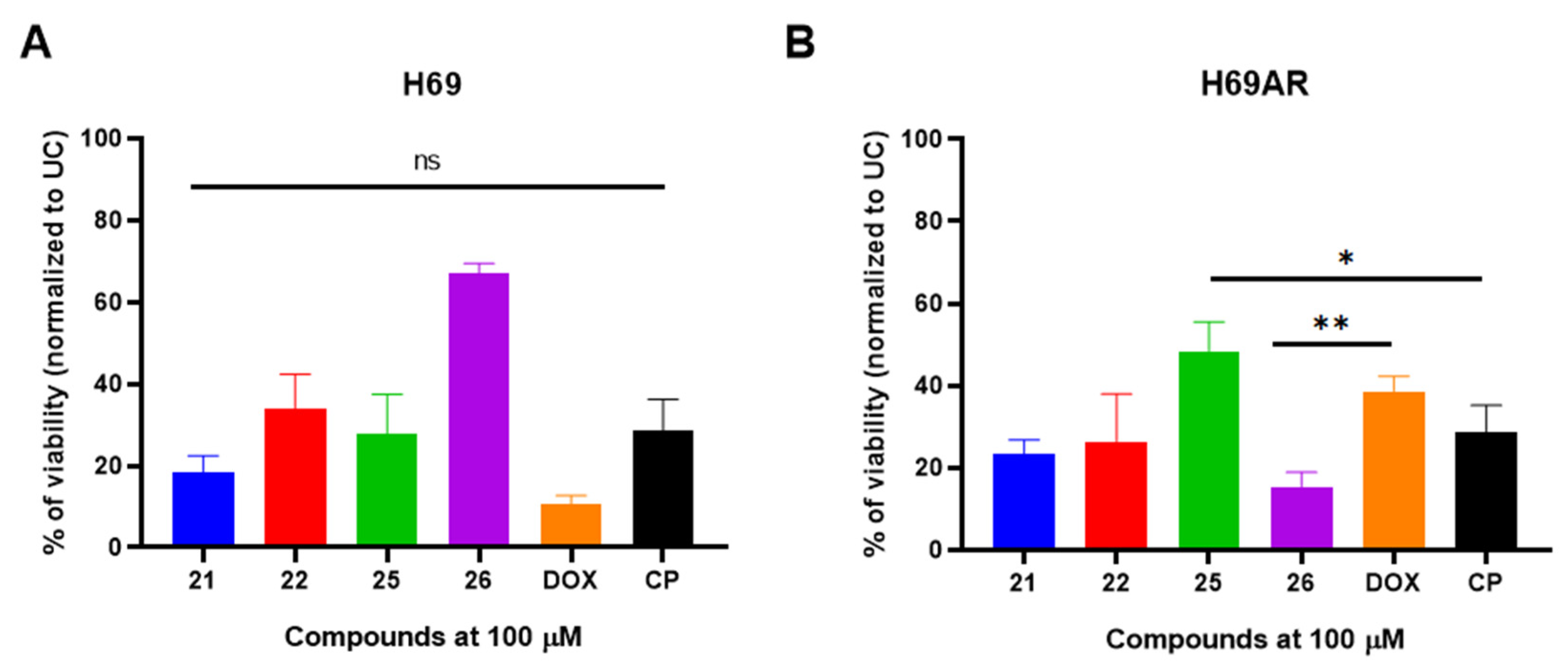
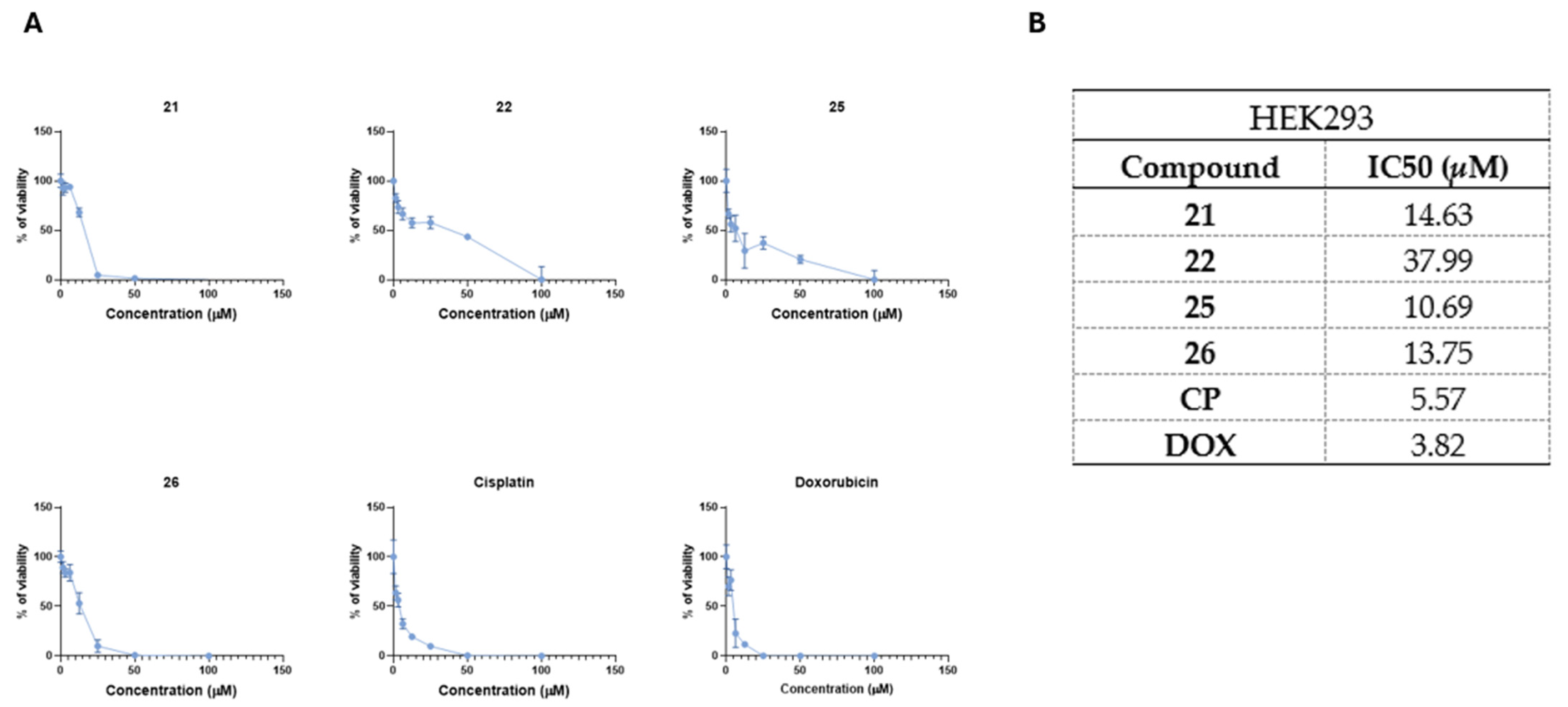
2.3. Most Promising 3-[(4-Acetylphenyl)(4-Phenylthiazol-2-Yl)Amino]Propanoic Acid Derivatives Exhibits Antiproliferative Activity Against Drug Sensitive H69 and Resistant H69AR Cells
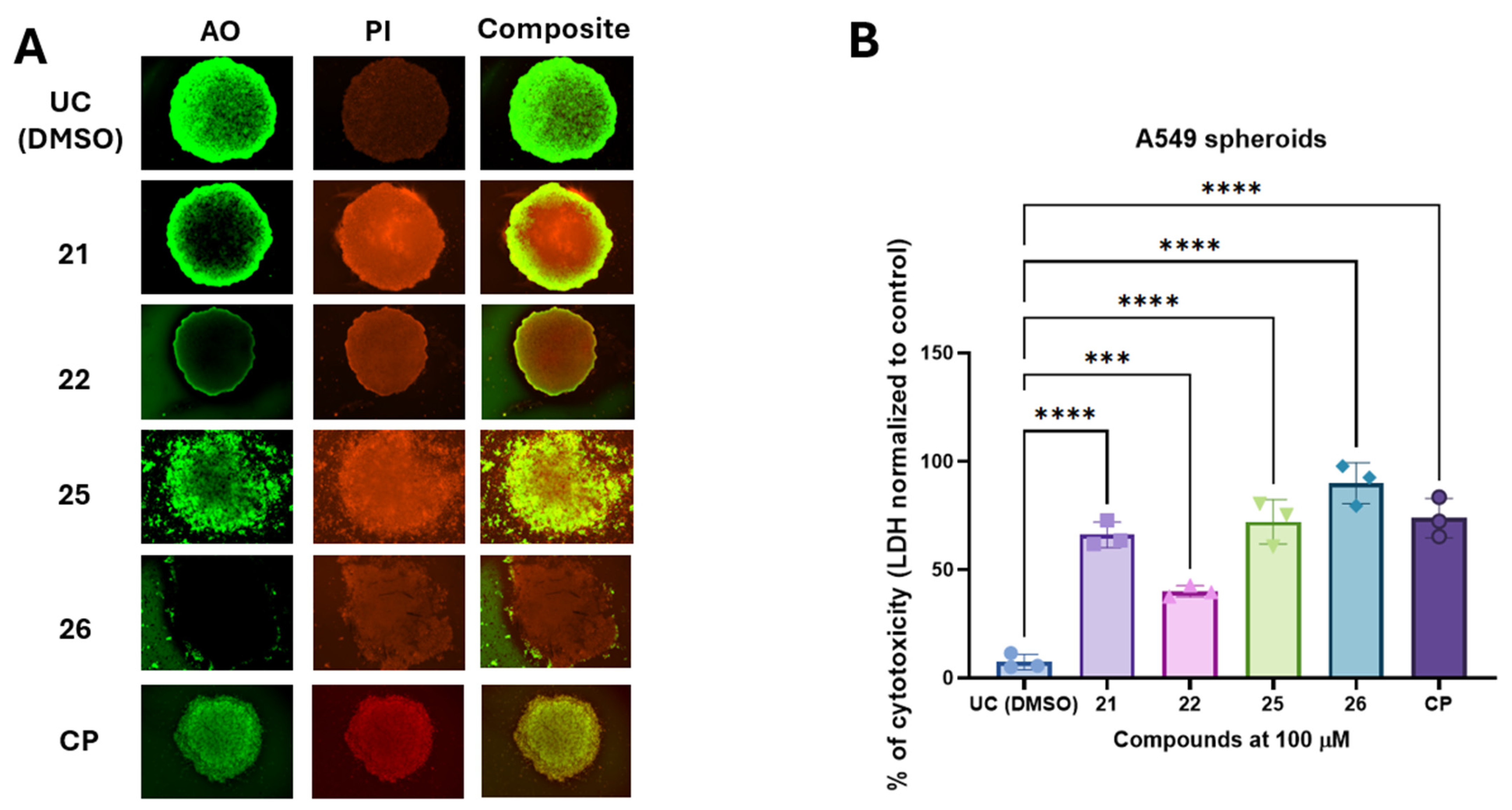
2.4. The 3-[(4-Acetylphenyl)(4-Phenylthiazol-2-Yl)Amino]propanoic Acid Derivatives 21, 22, 25, and 26 with Hydroxyimino Moiety Induces the Cytotoxic Activity in A549 Derived Cancer Spheroids
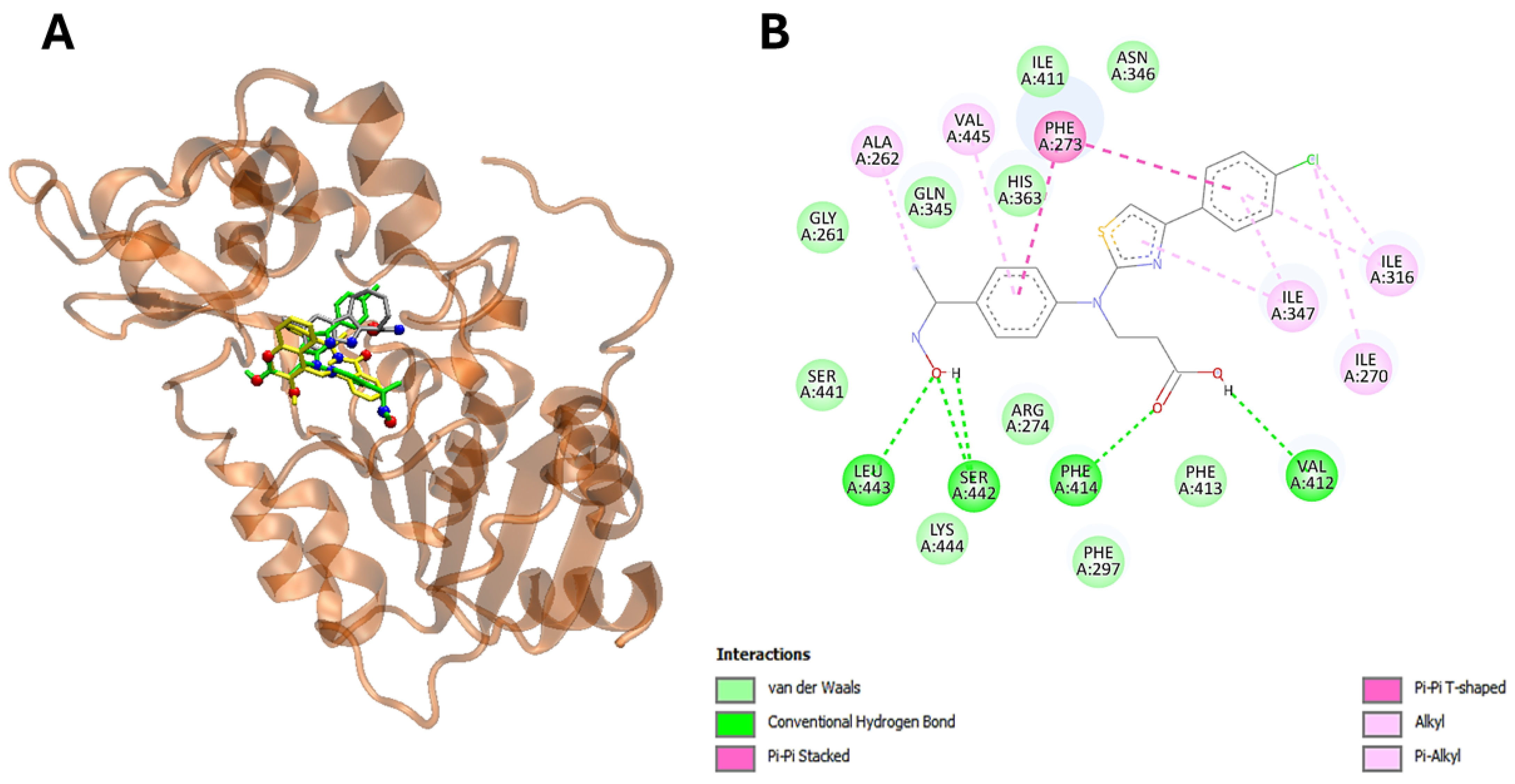
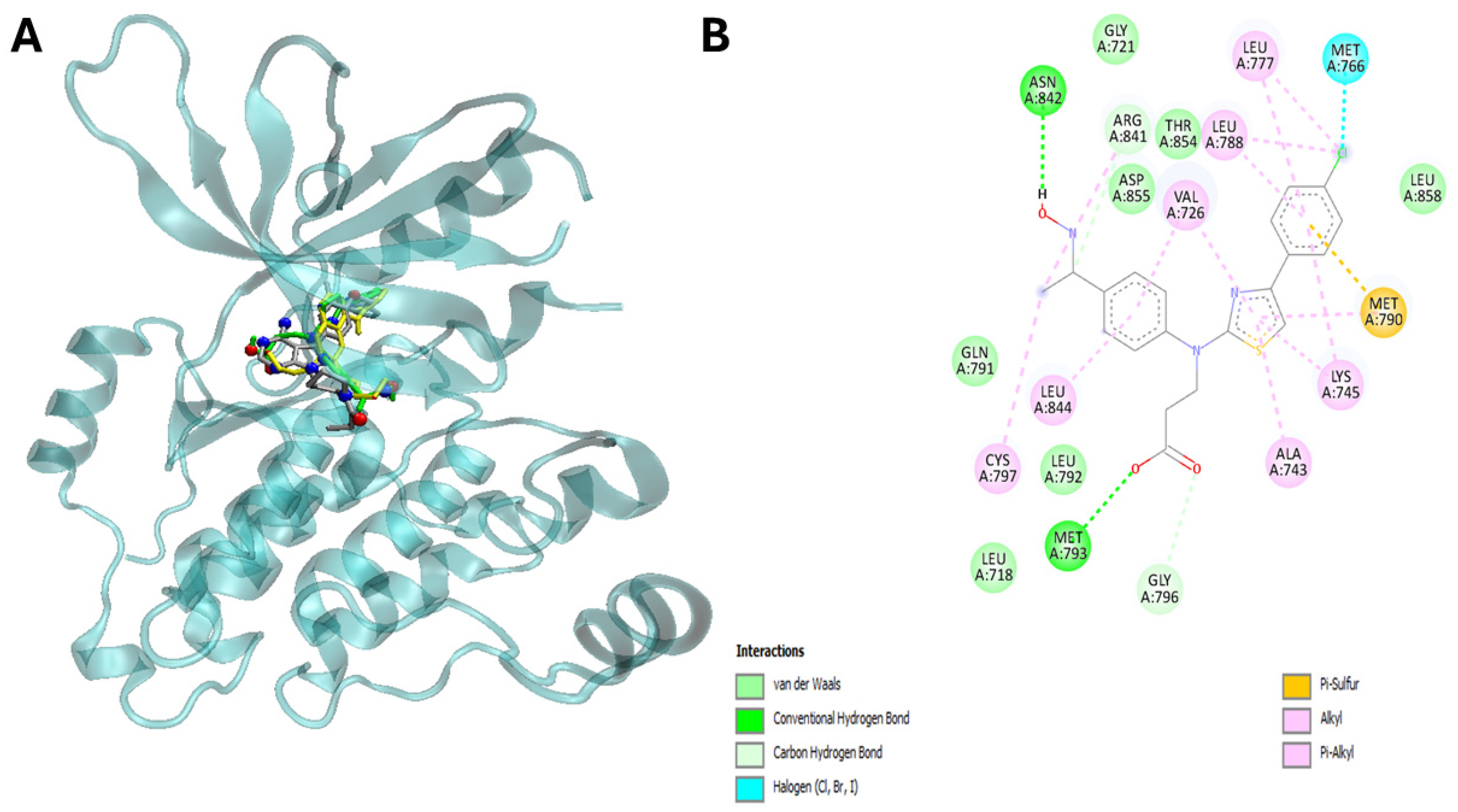
2.5. Molecular Docking Studies
3. Discussion
4. Materials and Methods
4.1. Synthesis
4.2. General Procedure for the Synthesis of 3
4.3. General Procedure for the Synthesis of 4
4.4. General Procedure for the Synthesis of 5–12
4.5. General Procedure for the Synthesis of 13–17
4.6. General Procedure for the Synthesis of 18–20
4.7. General Procedure for the Synthesis of Oximes 21, 22
4.8. General Procedure for the Synthesis of Esters 23, 24
4.9. General Procedure for the Preparation of Hydrazides 25, 26
4.10. General Procedure for the Preparation of Compounds 27, 28
4.11. General Procedure for the Preparation of Compounds 29, 30
4.12. General Procedures for the Preparation of Compounds 31, 32
4.13. Preparation of the Test Compounds and Screening Library
4.14. Cell Lines and Culture Conditions
4.15. MTT Based Cell Viability Assay
4.16. Generation of A549 3D Spheroids
4.17. Acridine Orange/Propidium Iodide (AO/PI) Staining of A549 Spheroids
4.18. Compound-Induced Cytotoxicity Evaluation in A549 Spheroids Using LDH Assay
4.19. IC50 Determination
4.20. The Protein Target Preparation
4.21. Ligand Preparation
4.22. Docking of Ligand–Protein Interaction
4.23. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Filho, A.M.; Laversanne, M.; Ferlay, J.; Colombet, M.; Piñeros, M.; Znaor, A.; Parkin, D.M.; Soerjomataram, I.; Bray, F. The GLOBOCAN 2022 Cancer Estimates: Data Sources, Methods, and a Snapshot of the Cancer Burden Worldwide. Int. J. Cancer 2025, 156, 1336–1346. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.J.; Lei, K.F.; Han, F. Tumor microenvironment: Recent advances in various cancer treatments. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 3855–3864. [Google Scholar]
- Kaur, R.; Bhardwaj, A.; Gupta, S. Cancer treatment therapies: Traditional to modern approaches to combat cancers. Mol. Biol. Rep. 2023, 50, 9663–9676. [Google Scholar] [CrossRef]
- Papież, M.A.; Krzyściak, W. Biological Therapies in the Treatment of Cancer-Update and New Directions. Int. J. Mol. Sci. 2021, 22, 11694. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. Small molecule inhibitors targeting the EGFR/ErbB family of protein-tyrosine kinases in human cancers. Pharmacol. Res. 2019, 139, 395–411. [Google Scholar] [CrossRef] [PubMed]
- Voldborg, B.R.; Damstrup, L.; Spang-Thomsen, M.; Poulsen, H.S. Epidermal growth factor receptor (EGFR) and EGFR mutations, function and possible role in clinical trials. Ann. Oncol. 1997, 8, 1197–1206. [Google Scholar] [CrossRef]
- Singh, D.; Attri, B.K.; Gill, R.K.; Bariwal, J. Review on EGFR Inhibitors: Critical Updates. Mini Rev. Med. Chem. 2016, 16, 1134–1166. [Google Scholar] [CrossRef]
- Chen, G.; Huang, P.; Hu, C. The role of SIRT2 in cancer: A novel therapeutic target. Int. J. Cancer 2020, 147, 3297–3304. [Google Scholar] [CrossRef]
- Zheng, M.; Hu, C.; Wu, M.; Chin, Y.E. Emerging role of SIRT2 in non-small cell lung cancer. Oncol. Lett. 2021, 22, 731. [Google Scholar] [CrossRef]
- Pun, R.; Kumari, N.; Monieb, R.H.; Wagh, S.; North, B.J. BubR1 and SIRT2: Insights into aneuploidy, aging, and cancer. Semin. Cancer Biol. 2024, 106–107, 201–216. [Google Scholar] [CrossRef]
- Dahiya, R.; Dahiya, S.; Fuloria, N.K.; Kumar, S.; Mourya, R.; Chennupati, S.V.; Jankie, S.; Gautam, H.; Singh, S.; Karan, S.K.; et al. Natural Bioactive Thiazole-Based Peptides from Marine Resources: Structural and Pharmacological Aspects. Mar. Drugs 2020, 18, 329. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.C.; Sun, J.; Wartmann, M.; Lerner, R.A. Synthesis of Epothilone Analogues by Antibody-Catalyzed Resolution of Thiazole Aldol Synthons on a Multigram Scale. Biological Consequences of C-13 Alkylation of Epothilones. ChemBioChem 2001, 2, 656–665. [Google Scholar] [CrossRef] [PubMed]
- Davyt, D.; Serra, G. Thiazole and Oxazole Alkaloids: Isolation and Synthesis. Mar. Drugs 2010, 8, 2755–2780. [Google Scholar] [CrossRef]
- Paerl, R.W.; Bertrand, E.M.; Rowland, E.; Schatt, P.; Mehiri, M.; Niehaus, T.D.; Hanson, A.D.; Riemann, L.; Bouget, F. Carboxythiazole is a Key Microbial Nutrient Currency and Critical Component of Thiamin Biosynthesis. Sci. Rep. 2018, 8, 5940. [Google Scholar] [CrossRef] [PubMed]
- Petrou, A.; Fesatidou, M.; Geronikaki, A. Thiazole Ring—A Biologically Active Scaffold. Molecules 2021, 26, 3166. [Google Scholar] [CrossRef]
- Makam, P.; Thakur, P.K.; Kannan, T. In Vitro and in Silico Antimalarial Activity of 2-(2-Hydrazinyl)Thiazole Derivatives. Eur. J. Pharm. Sci. 2013, 52, 138–145. [Google Scholar] [CrossRef]
- Borcea, A.; Ionuț, I.; Crișan, O.; Oniga, O. An Overview of the Synthesis and Antimicrobial, Antiprotozoal, and Antitumor Activity of Thiazole and Bisthiazole Derivatives. Molecules 2021, 26, 624. [Google Scholar] [CrossRef]
- Pattan, S.R.; Dighe, N.S.; Nirmal, S.A.; Merekar, A.N.; Laware, R.B.; Shinde, H.V.; Musmade, D.S. Synthesis and Biological Evaluation of some Substituted Amino Thiazole Derivatives. Asian J. Res. Chem. 2009, 2, 196. [Google Scholar]
- Karaburun, A.Ç.; Acar Çevik, U.; Osmaniye, D.; Sağlık, B.N.; Kaya Çavuşoğlu, B.; Levent, S.; Özkay, Y.; Koparal, A.S.; Behçet, M.; Kaplancıklı, Z.A. Synthesis and Evaluation of New 1,3,4-Thiadiazole Derivatives as Potent Antifungal Agents. Molecules 2018, 23, 3129. [Google Scholar] [CrossRef]
- Villegas, C.; González-Chavarría, I.; Burgos, V.; Iturra-Beiza, H.; Ulrich, H.; Paz, C. Epothilones as Natural Compounds for Novel Anticancer Drugs Development. Int. J. Mol. Sci. 2023, 24, 6063. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Xie, Z.; Ruan, W.; Tang, Q.; Qiao, D.; Zhu, W. Thiazole-Based Analogues as Potential Antibacterial Agents Against Methicillin-Resistant Staphylococcus Aureus (MRSA) and their SAR Elucidation. Eur. J. Med. Chem. 2023, 259, 115689. [Google Scholar] [CrossRef] [PubMed]
- Thakur, S.; Sharma, R.; Yadav, R.; Sardana, S. The Potential of Thiazole Derivatives as Antimicrobial Agents. Chem. Proc. 2022, 12, 36. [Google Scholar]
- Al-Salmi, F.A.; Alrohaimi, A.H.; Behery, M.E.; Megahed, W.; Abu Ali, O.A.; Elsaid, F.G.; Fayad, E.; Mohammed, F.Z.; Keshta, A.T. Anticancer Studies of Newly Synthesized Thiazole Derivatives: Synthesis, Characterization, Biological Activity, and Molecular Docking. Crystals 2023, 13, 1546. [Google Scholar] [CrossRef]
- Mohanty, P.; Behera, S.; Behura, R.; Shubhadarshinee, L.; Mohapatra, P.; Kumar Barick, A.; Ranjan Jali, B. Antibacterial Activity of Thiazole and its Derivatives: A Review. Biointerface Res. Appl. Chem. 2022, 12, 2171–2195. [Google Scholar]
- Niu, Z.; Wang, Y.; Zhang, S.; Li, Y.; Chen, X.; Wang, S.; Liu, H. Application and Synthesis of Thiazole Ring in Clinically Approved Drugs. Eur. J. Med. Chem. 2023, 250, 115172. [Google Scholar] [CrossRef]
- Kyllo, T.; Singh, V.; Shim, H.; Latika, S.; Nguyen, H.M.; Chen, Y.; Terry, E.; Wulff, H.; Erickson, J.D. Riluzole and Novel Naphthalenyl Substituted Aminothiazole Derivatives Prevent Acute Neural Excitotoxic Injury in a Rat Model of Temporal Lobe Epilepsy. Neuropharmacology 2022, 224, 109349. [Google Scholar] [CrossRef]
- Donnelley, M.A.; Zhu, E.S.; Thompson, G.R. Isavuconazole in the Treatment of Invasive Aspergillosis and Mucormycosis Infections. Infect. Drug Resist. 2016, 9, 79–86. [Google Scholar]
- Miodragović, D.U.; Bogdanović, G.A.; Miodragović, Z.M.; Radulović, M.Đ.; Novaković, S.B.; Kaluđerović, G.N.; Kozłowski, H. Interesting Coordination Abilities of Antiulcer Drug Famotidine and Antimicrobial Activity of Drug and its Cobalt (III) Complex. J. Inorg. Biochem. 2006, 100, 1568–1574. [Google Scholar] [CrossRef]
- Wilby, M.J.; Hutchinson, P.J. The Pharmacology of Chlormethiazole: A Potential Neuroprotective Agent? CNS Drug Rev. 2004, 10, 281–294. [Google Scholar] [CrossRef]
- Sahil; Kaur, K.; Jaitak, V. Thiazole and Related Heterocyclic Systems as Anticancer Agents: A Review on Synthetic Strategies, Mechanisms of Action and SAR Studies. Curr. Med. Chem. 2022, 29, 4958–5009. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Liu, W.; Fan, M.; He, M.; Li, Y.; Peng, Z. Design, Synthesis and Biological Evaluation of Novel Thiazole-Naphthalene Derivatives as Potential Anticancer Agents and Tubulin Polymerisation Inhibitors. J. Enzyme Inhib. Med. Chem. 2021, 36, 1693–1701. [Google Scholar] [CrossRef] [PubMed]
- Shosha, M.I.; El-Ablack, F.; Saad, E.A. New Thiazole Derivative as a Potential Anticancer and Topoisomerase II Inhibitor. Sci. Rep. 2025, 15, 710. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishna, B.; Mondal, S.; Chakraborty, S. A New Stability Indicating Method Development and Validation Report for the Assay of Nivolumab by RP-UPLC. J. Pharm. Negat. Results 2022, 13, 1020–1032. [Google Scholar]
- Grybaitė, B.; Vaickelionienė, R.; Stasevych, M.; Komarovska-porokhnyavets, O.; Kantminienė, K.; Novikov, V.; Mickevičius, V. Synthesis and Antimicrobial Activity of Novel Thiazoles with Reactive Functional Groups. ChemistrySelect 2019, 4, 6965–6970. [Google Scholar] [CrossRef]
- Malūkaitė, D.; Grybaitė, B.; Vaickelionienė, R.; Vaickelionis, G.; Sapijanskaitė-Banevič, B.; Kavaliauskas, P.; Mickevičius, V. Synthesis of Novel Thiazole Derivatives Bearing β-Amino Acid and Aromatic Moieties as Promising Scaffolds for the Development of New Antibacterial and Antifungal Candidates Targeting Multidrug-Resistant Pathogens. Molecules 2022, 27, 74. [Google Scholar] [CrossRef]
- Minickaitė, R.; Grybaitė, B.; Vaickelionienė, R.; Kavaliauskas, P.; Petraitis, V.; Petraitienė, R.; Tumosienė, I.; Jonuškienė, I.; Mickevičius, V. Synthesis of Novel Aminothiazole Derivatives as Promising Antiviral, Antioxidant and Antibacterial Candidates. Int. J. Mol. Sci. 2022, 23, 7688. [Google Scholar] [CrossRef]
- Kavaliauskas, P.; Acevedo, W.; Garcia, A.; Naing, E.; Grybaite, B.; Sapijanskaite-Banevic, B.; Grigaleviciute, R.; Petraitiene, R.; Mickevicius, V.; Petraitis, V. Exploring the Potential of Bis(Thiazol-5-yl)Phenylmethane Derivatives as Novel Candidates Against Genetically Defined Multidrug-Resistant Staphylococcus Aureus. PLoS ONE 2024, 19, e0300380. [Google Scholar] [CrossRef]
- Osorio-Lozada, A.; Tovar-Miranda, R.; Olivo, H.F. Biotransformation of N-Piperidinylacetophenone with Beauveria Bassiana ATCC-7159. J. Mol. Catal. B Enzym. 2008, 55, 30–36. [Google Scholar] [CrossRef]
- Batista, J.E.S.; Rodrigues, M.B.; Bristot, I.J.; Silva, V.; Bernardy, S.; Rodrigues, O.E.D.; Dornelles, L.; Carvalho, F.B.; Sousa, F.J.F.; Fernandes, M.C.; et al. Systematic screening of synthetic organochalcogen compounds with anticancer activity using human lung adenocarcinoma spheroids. Chem. Biol. Interact. 2024, 396, 111047. [Google Scholar] [CrossRef]
- Zakaria, N.H.; Saad, N.; Che Abdullah, C.A.; Mohd. Esa, N. The Antiproliferative Effect of Chloroform Fraction of Eleutherine Bulbosa (Mill.) Urb. on 2D- and 3D-Human Lung Cancer Cells (A549) Model. Pharmaceuticals 2023, 16, 936. [Google Scholar] [CrossRef] [PubMed]
- Mabela, C.M.; Gouws, C.; Pheiffer, W. Overcoming Obstacles in Three-Dimensional Cell Culture Model Establishment: Approaches for Growing A549 Non-Small Cell Lung Cancer Spheroids using a Clinostat System. J. Pharmacol. Toxicol. Methods 2024, 130, 107564. [Google Scholar] [CrossRef] [PubMed]
- Kavaliauskas, P.; Opazo, F.S.; Acevedo, W.; Petraitiene, R.; Grybaitė, B.; Anusevičius, K.; Mickevičius, V.; Belyakov, S.; Petraitis, V. Synthesis, Biological Activity, and Molecular Modelling Studies of Naphthoquinone Derivatives as Promising Anticancer Candidates Targeting COX-2. J. Pharm. 2022, 15, 541. [Google Scholar] [CrossRef]
- Kumar, P.; Nagarajan, A.; Uchil, P.D. Analysis of Cell Viability by the MTT Assay. Cold Spring Harb. Protoc. 2018. [Google Scholar] [CrossRef]
- Petzer, M.; Fobian, S.; Gulumian, M.; Steenkamp, V.; Cordier, W. A549 Alveolar Carcinoma Spheroids as a Cytotoxicity Platform for Carboxyl- and Amine-Polyethylene Glycol Gold Nanoparticles. Pharmacol. Res. Perspect. 2025, 13, e70051. [Google Scholar] [CrossRef]
- Luan, Q.; Becker, J.H.; Macaraniag, C.; Massad, M.G.; Zhou, J.; Shimamura, T.; Papautsky, I. Non-Small Cell Lung Carcinoma Spheroid Models in Agarose Microwells for Drug Response Studies. Lab Chip 2022, 22, 2364. [Google Scholar] [CrossRef] [PubMed]
- Moraes, G.D.S.; Wink, M.R.; Klamt, F.; Silva, A.O.; Da Cruz Fernandes, M. Simplified Low-Cost Methodology to Establish, Histologically Process and Analyze Three-Dimensional Cancer Cell Spheroid Arrays. Eur. J. Cell Biol. 2020, 99, 151095. [Google Scholar] [CrossRef]
- Berman, H.; Westbrook, M.; Feng, J.; Gilliland, Z.; Bhat, G.; Weissig, V.H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Bethune, G.; Bethune, D.; Ridgway, N.; Xu, Z. Epidermal growth factor receptor (EGFR) in lung cancer: An overview and update. J. Thorac. Dis. 2010, 2, 48–51. [Google Scholar]
- Bodén, E.; Sveréus, F.; Olm, F.; Lindstedt, S. A Systematic Review of Mesenchymal Epithelial Transition Factor (MET) and Its Impact in the Development and Treatment of Non-Small-Cell Lung Cancer. Cancers 2023, 15, 3827. [Google Scholar] [CrossRef]
- Chen, Y.; Nowak, I.; Huang, J.; Keng, P.C.; Sun, H.; Xu, H.; Wei, G.; Lee, S.O. Erk/MAP kinase signaling pathway and neuroendocrine differentiation of non-small-cell lung cancer. J. Thorac. Oncol. 2014, 9, 50–58. [Google Scholar] [CrossRef]
- Dassault Systèmes BIOVIA. Discovery Studio Visualizer, version 20.1.0; Dassault Systèmes: San Diego, CA, USA, 2019; Volume 19295. [Google Scholar]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2014, 4, 17. [Google Scholar] [CrossRef]
- Houlbrook, S.; Harris, A.L.; Carmichael, J.; Stratford, I.J. Relationship between topoisomerase II levels and resistance to topoisomerase II inhibitors in lung cancer cell lines. Anticancer Res. 1996, 16, 1603–1610. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Inoue, Y.; Nikolic, A.; Farnsworth, D.; Shi, R.; Johnson, F.D.; Liu, A.; Ladanyi, M.; Somwar, R.; Gallo, M.; Lockwood, W.W. Extracellular signal-regulated kinase mediates chromatin rewiring and lineage transformation in lung cancer. ELife 2021, 10, e66524. [Google Scholar] [CrossRef]
- Jung, K.H.; Noh, J.H.; Kim, J.K.; Eun, J.W.; Bae, H.J.; Xie, H.J.; Chang, Y.G.; Kim, M.G.; Park, H.; Lee, J.Y.; et al. HDAC2 overexpression confers oncogenic potential to human lung cancer cells by deregulating expression of apoptosis and cell cycle proteins. J. Cell. Biochem. 2012, 113, 2167–2177. [Google Scholar] [CrossRef]
- Krook, M.A.; Reeser, J.W.; Ernst, G.; Barker, H.; Wilberding, M.; Li, G.; Chen, H.Z.; Roychowdhury, S. Fibroblast growth factor receptors in cancer: Genetic alterations, diagnostics, therapeutic targets and mechanisms of resistance. Br. J. Cancer 2021, 124, 880–892. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, J.; Sun, W.; Cao, J.; Ma, Z. COX-2 in lung cancer: Mechanisms, development, and targeted therapies. Chronic. Dis. Transl. Med. 2024, 10, 281–292. [Google Scholar] [CrossRef]
- Miettinen, M.; Rikala, M.S.; Rys, J.; Lasota, J.; Wang, Z.F. Vascular Endothelial Growth Factor Receptor 2 as a Marker for Malignant Vascular Tumors and Mesothelioma. Am. J. Surg. Pathol. 2012, 36, 629–639. [Google Scholar] [CrossRef]
- Mollerup, S.; Jørgensen, K.; Berge, G.; Haugen, A. Expression of estrogen receptors α and β in human lung tissue and cell lines. Lung Cancer 2002, 37, 153–159. [Google Scholar] [CrossRef]
- Sakon, K.; Sasaki, M.; Tanaka, K.; Mizunaga, T.; Yano, K.; Kawamura, Y.; Okada, A.; Ikeda, T.; Tanabe, S.; Takamori, A.; et al. Intratumoral gene expression of dihydrofolate reductase and folylpoly-c-glutamate synthetase affects the sensitivity to 5-fluorouracil in non-small cell lung cancer. Discov. Oncol. 2021, 12, 19. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Yeung, F.; Hoberg, J.E.; Ramsey, C.S.; Keller, M.D.; Jones, D.R.; Frye, R.A.; Mayo, M.W. Modulation of NF-κB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 2004, 23, 2369–2380. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | R | IC50 (µM) |
|---|---|---|
| 21 | H | 5.42 |
| 22 | Cl | 2.47 |
| 25 | H | 8.05 |
| 26 | Cl | 25.40 |
| CP | - | 11.71 |
| DOX | - | 3.02 |
| Compounds | Target Proteins | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| COX-2 | FGFR-2 | EGFR | HER2 | c-MET | ERK2 | MEK1 | TPK | PD2 | SIRT1 | |
| 21 | −8.2 | −9.0 | −9.4 | −8.9 | −8.6 | −8.1 | −8.1 | −7.9 | −9.7 | −9.5 |
| 22 | −8.5 | −8.9 | −9.5 | −8.6 | −8.5 | −7.6 | −8.2 | −8.2 | −9.1 | −9.7 |
| 25 | −8.6 | −9.5 | −9.3 | −8.7 | −8.5 | −8.0 | −8.6 | −8.4 | −9.5 | −10.0 |
| 26 | −8.9 | −8.3 | −9.2 | −8.9 | −8.5 | −8.5 | −8.0 | −8.4 | −9.3 | −10.2 |
| P aver. | −8.55 | −8.93 | −9.35 | −8.78 | −8.53 | −8.05 | −8.23 | −8.23 | −9.40 | −9.85 |
| Compound | ΔGbin (kcal/mol) | H Bonds and Hydrophobic Contacts in the Binding Site |
|---|---|---|
| SIRT1 | ||
| 22 | −9.7 | Gly261, Ala262, Ile270, Phe273, Arg274, Phe297, Ile316, Gln345, Asn346, Ile347, His363, Ile411, Val412, Phe413, Phe414, Ser441, Ser442, Leu443, Lys444, Val445 |
| Ligand 1 [a] | −11.2 | Ala262, Ser265, Ile270, Pro271, Phe273, Ile279, Phe297, Ile316, Gln345, Asn346, Ile347, Asp348, His363, Ile411, Val412, Phe413 |
| Sirtinol [b] | −11.7 | Gly261, Ala262, Phe273, Arg274, Tyr280, Gln294, Phe297, Gln345, Asn346, Ile347, His363, Ile411, Val412, Phe413, Phe414, Val445 |
| EGFR | ||
| 22 | −9.5 | Leu718, Gly721, Val726, Ala743, Lys745, Met766, Leu777, Leu788, Met790, Gln791, Leu792, Met793, Gly796, Cys797, Arg841, Asn842, Leu844, Thr854, Asp855, Leu858 |
| Ligand 2 [a] | −6.2 | Leu718, Gly719, Ser720, Val726, Ala743, Ile744, Lys745, Cys775, Arg776, Leu777, Leu788, Ile789, Met790, Gln791, Leu792, Met793, Gly796, Cys797, Asp800, Arg841, Leu844, Thr854, Asp855, Phe856, Leu858 |
| Erlotinib [b] | −8.6 | Leu718, Gly721, Val726, Ala743, Lys745, Met766, Cys775, Arg776, Leu777, Leu788, Met790, Leu792, Met793, Gly796, Arg841, Asn842, Leu844, Thr854, Asp855, Phe856, Leu858 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Golcienė, B.; Kavaliauskas, P.; Acevedo, W.; Sapijanskaitė-Banevič, B.; Grybaitė, B.; Grigalevičiūtė, R.; Petraitienė, R.; Petraitis, V.; Mickevičius, V. Identification of 3-[(4-Acetylphenyl)(4-Phenylthiazol-2-Yl)Amino]Propanoic Acid Derivatives as Promising Scaffolds for the Development of Novel Anticancer Candidates Targeting SIRT2 and EGFR. Pharmaceuticals 2025, 18, 733. https://doi.org/10.3390/ph18050733
Golcienė B, Kavaliauskas P, Acevedo W, Sapijanskaitė-Banevič B, Grybaitė B, Grigalevičiūtė R, Petraitienė R, Petraitis V, Mickevičius V. Identification of 3-[(4-Acetylphenyl)(4-Phenylthiazol-2-Yl)Amino]Propanoic Acid Derivatives as Promising Scaffolds for the Development of Novel Anticancer Candidates Targeting SIRT2 and EGFR. Pharmaceuticals. 2025; 18(5):733. https://doi.org/10.3390/ph18050733
Chicago/Turabian StyleGolcienė, Božena, Povilas Kavaliauskas, Waldo Acevedo, Birutė Sapijanskaitė-Banevič, Birutė Grybaitė, Ramunė Grigalevičiūtė, Rūta Petraitienė, Vidmantas Petraitis, and Vytautas Mickevičius. 2025. "Identification of 3-[(4-Acetylphenyl)(4-Phenylthiazol-2-Yl)Amino]Propanoic Acid Derivatives as Promising Scaffolds for the Development of Novel Anticancer Candidates Targeting SIRT2 and EGFR" Pharmaceuticals 18, no. 5: 733. https://doi.org/10.3390/ph18050733
APA StyleGolcienė, B., Kavaliauskas, P., Acevedo, W., Sapijanskaitė-Banevič, B., Grybaitė, B., Grigalevičiūtė, R., Petraitienė, R., Petraitis, V., & Mickevičius, V. (2025). Identification of 3-[(4-Acetylphenyl)(4-Phenylthiazol-2-Yl)Amino]Propanoic Acid Derivatives as Promising Scaffolds for the Development of Novel Anticancer Candidates Targeting SIRT2 and EGFR. Pharmaceuticals, 18(5), 733. https://doi.org/10.3390/ph18050733