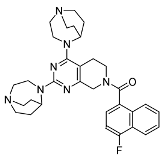
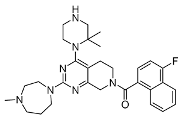
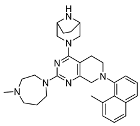
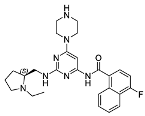
3.2. Chemistry
3.2.1. (2,4-Dichloro-5,8-dihydropyrido[3,4-d]pyrimidin-7(6H)-yl)(4-fluoronaphthalen-1-yl)methanone) (7)
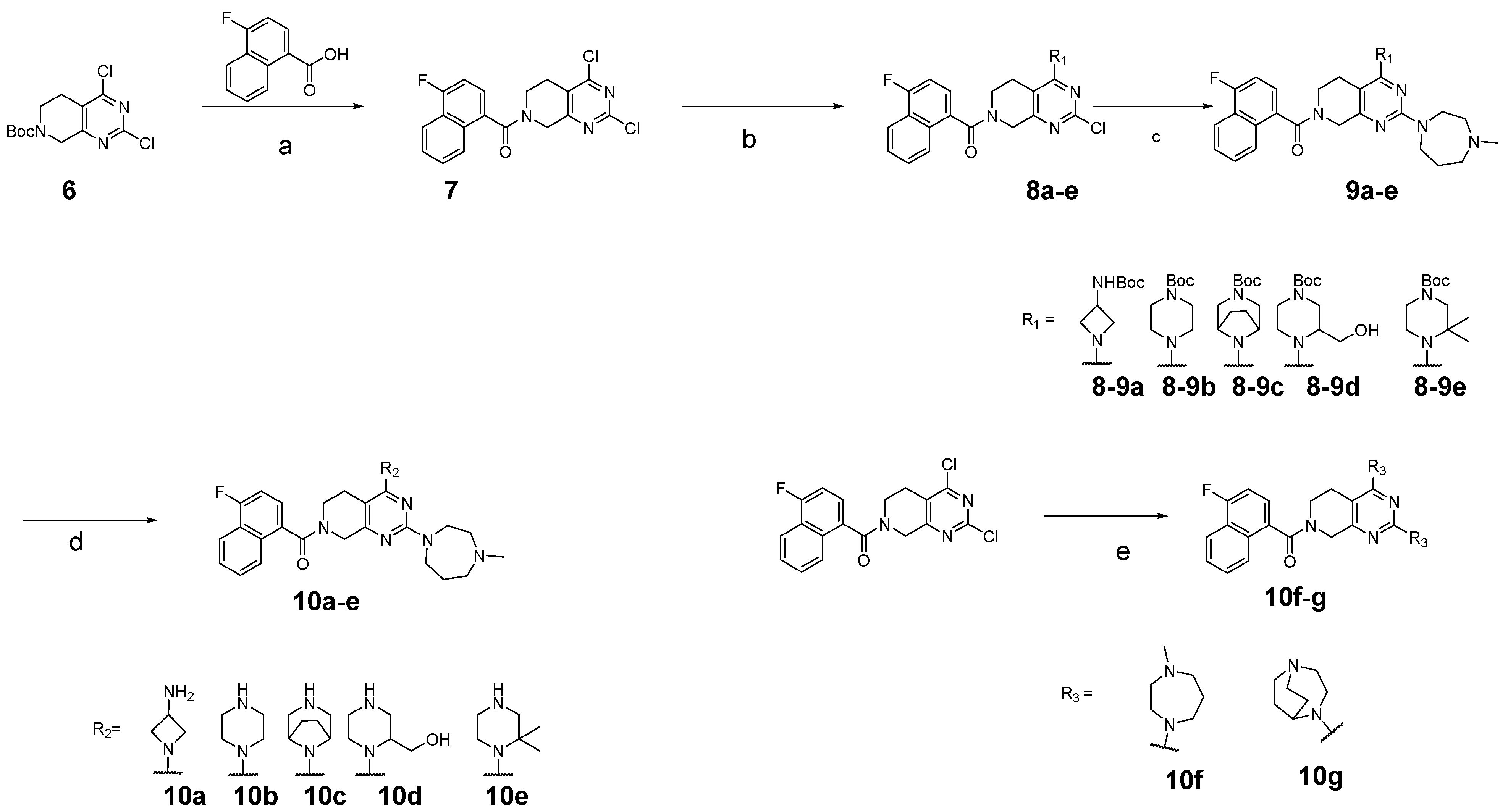
In a single-necked flask, under room-temperature conditions, tert-butyl-2,4-dichloro-5,8-dihydropyrido[3,4-d]pyrimidine (1.00 g, 3.29 mmol) was suspended in dichloromethane (10 mL). As stirring was ongoing, trifluoroacetic acid (5 mL) was introduced, and the mixture was stirred continuously at this temperature for approximately 2 h. The dichloromethane and excess trifluoroacetic acid were removed using a rotary evaporator, yielding the trifluoroacetate salt of 2,4-dichloro-5,6,7,8-tetrahydropyrido[3,4-d]pyrimidine. In another single-necked flask, 4-fluoro-1-naphthoic acid (0.63 g, 3.29 mmol) was suspended in DMF (N,N-dimethylformamide, 10 mL) and cooled to around 0 °C using an ice-salt bath. HATU (O-(7-azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate, 1.50 g, 3.95 mmol) and DIEA (N,N-Diisopropylethylamine, 1.28 g, 9.87 mmol) were added sequentially and slowly. The reaction mixture was stirred continuously at this temperature for about 10 min, after which a DMF (5 mL) solution of the aforementioned trifluoroacetate salt was introduced drop by drop. Following this addition, the reaction-derived mixture was allowed to reach the temperature of the surroundings naturally and was stirred at room temperature for an additional 16 h. The completion of the reaction was monitored by means of TLC (thin-layer chromatography). The reaction mixture was then slowly poured into water (50 mL) to form a slurry, which was filtered. Water (20 mL × 3) was used to wash the filter cake, yielding 0.96 g of 7 (white solid, yield: 78.05%).
3.2.2. Tert-butyl 4-(2-chloro-7-(4-fluoro-1-naphthoyl)-5,6,7,8-tetrahydropyrido[3,4-d]pyrimidin-4-yl)piperazine-1-carboxylate (8b)
In a single-necked flask, 7 (100 mg, 0.27 mmol) was dissolved in DMF (5 mL). N-Boc-piperazine (50 mg, 0.27 mmol) and DIEA (70 mg, 0.54 mmol) were added sequentially. After the addition was complete, the mixture of the reaction was agitated at a temperature of 50 °C for approximately 5 h. The completion of the reaction was monitored by means of TLC. Subsequently, ethyl acetate (50 mL) and water (20 mL) were used to extract the reaction mixture. The organic layer was rinsed three times with 20 mL portions of saturated saline solution. Subsequently, it was dehydrated using anhydrous sodium sulfate and then reduced in volume via rotary evaporation. Silica gel column chromatography was utilized to purify the crude product (petroleum ether:ethyl acetate = 5:1 to 3:1), yielding 105 mg of 8b (white solid, yield: 74.47%). 1H NMR (600 MHz, Chloroform-d) δ 8.23–8.10 (m, 1H), 7.84 (m, 1H), 7.65–7.53 (m, 2H), 7.47–7.36 (m, 1H), 7.24–7.14 (m, 1H), 5.22–4.68 (m, 1H), 4.38–4.22 (m, 1H), 4.06 (m, 1H), 3.62–3.32 (m, 9H), 2.90–2.74 (m, 1H), 2.49 (m, 1H), 1.48 (m, 9H). 13C NMR (100 MHz, Chloroform-d) δ 169.3, 165.9, 162.7, 161.0, 158.0, 154.8, 128.5, 127.1, 125.2, 125.1, 124.8, 124.4, 124.1, 123.94, 121.4, 113.1, 109.2, 109.0, 80.5, 51.5, 57.6, 47.6, 46.5, 44.2, 39.4, 28.5, 26.4.
3.2.3. Tert-butyl 4-(7-(4-fluoro-1-naphthoyl)-2-(4-methyl-1,4-diazepan-1-yl)-5,6,7,8-tetrahydropyrido[3,4-d]pyrimidin-4-yl)piperazine-1-carboxylate (9b)
In a single-necked flask, 8b (50 mg, 0.10 mmol), Cs2CO3 (65 mg, 0.20 mmol), and N-methyl-homopiperazine (23 mg, 0.20 mmol) were mixed in DMF (5 mL). The mixture underwent stirring at 120 °C over a period of 16 h, and the reaction progress was monitored by means of TLC. After the starting materials were mostly consumed, the reactant blend was subjected to extraction using ethyl acetate (50 mL) and water (20 mL). The organic layer was then rinsed thrice with 10-milliliter aliquots of saturated saltwater. After that, it was dehydrated with anhydrous sodium sulfate and finally had its volume reduced. The crude product was purified via thin-layer chromatography using a solvent system of dichloromethane/methanol = 10:1, yielding 38 mg of 8b (white solid, yield: 63.33%). 1H NMR (400 MHz, Chloroform-d) δ 8.21–8.09 (m, 1H), 7.91–7.78 (m, 1H), 7.65–7.51 (m, 2H), 7.45–7.35 (m, 1H), 7.17 (q, J = 9.6 Hz, 1H), 4.80 (m, 1H), 4.14 (d, J = 9.2 Hz, 1H), 4.00 (m, 2H), 3.85–3.75 (m, 2H), 3.66–3.58 (m, 1H), 3.56–3.50 (m, 2H), 3.45 (t, J = 4.8 Hz, 2H), 3.33 (t, J = 4.8 Hz, 3H), 3.24 (d, J = 5.8 Hz, 2H), 2.88–2.80 (m, 1H), 2.78–2.71 (m, 2H), 2.70–2.58 (m, 2H), 2.49 (s, 1H), 2.39 (d, J = 5.8 Hz, 3H), 2.16–1.93 (m, 2H), 1.47 (d, J = 11.6 Hz, 9H). 13C NMR (101 MHz, Chloroform-d) δ 169.2, 165.8, 161.7, 161.0, 159.4, 154.9, 131.3, 130.1, 128.2, 126.9, 124.8, 124.1, 120.6, 109.0, 103.9, 103.4, 80.1, 58.3, 57.1, 51.6, 51.4, 47.7, 47.5, 46.3, 45.4, 45.0, 44.6, 40.4, 29.8, 28.5, 26.8, 25.6.
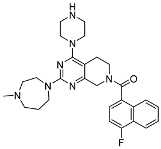
3.2.4. (4-Fluoronaphthalen-1-yl)(2-(4-methyl-1,4-diazepan-1-yl)-4-(piperazin-1-yl)-5,8-dihydropyrido[3,4-d]pyrimidin-7(6H)-yl)methanone (10b)
In a single-necked flask, 9b (25 mg, 0.04 mmol) was solubilized in 5 mL of DCM. As the solution was being stirred, 2 mL of trifluoroacetic acid was gradually added drop by drop. Once the addition of trifluoroacetic acid was finished, the resulting reaction mixture was continuously agitated at an ambient temperature for 2 h. The disappearance of the starting material was confirmed by TLC. The solvent was then removed using a rotary evaporator. The remaining substance was neutralized using a saturated sodium bicarbonate solution. Subsequently, extraction was carried out with a combination of ethyl acetate (30 mL) and water (10 mL). The organic layer was then rinsed three times, each time with 10 mL of saturated saline solution. After that, it was dried using anhydrous sodium sulfate and then concentrated. The unrefined product obtained was then purified through the use of silica gel column chromatography (dichloromethane/methanol = 10:1 to 5:1), yielding 15 mg of 10b (white solid, yield: 71.43%). Mp 128.5–131.2 °C. 1H NMR (400 MHz, Chloroform-d) δ 8.20–8.11 (m, 1H), 7.90–7.81 (m, 1H), 7.63–7.52 (m, 2H), 7.45–7.36 (m, 1H), 7.22–7.12 (m, 1H), 4.82 (d, J = 8.6 Hz, 1H), 4.13 (d, J = 9.2 Hz, 1H), 4.01 (t, J = 5.4 Hz, 1H), 3.92 (t, J = 4.9 Hz, 1H), 3.85–3.72 (m, 2H), 3.67–3.59 (m, 1H), 3.40–3.32 (m, 3H), 3.29–3.20 (m, 2H), 2.98 (t, J = 4.9 Hz, 2H), 2.94–2.86 (m, 2H), 2.74 (t, J = 5.4 Hz, 2H), 2.65–2.56 (m, 2H), 2.55–2.49 (m, 1H), 2.45–2.30 (m, 4H), 2.04–1.85 (m, 2H), 1.41–1.33 (m, 1H). 13C NMR (101 MHz, Chloroform-d) δ 169.2, 165.7, 161.4, 160.7, 159.1, 158.1, 131.4, 130.3, 128.3, 126.9, 124.9, 124.1, 121.2, 109.2, 103.6, 64.3, 58.6, 57.4, 52.0, 49.0, 47.6, 46.7, 46.0, 45.7, 45.3, 40.5, 29.8, 27.5. ESI-HRMS m/z calculated for C28H35FN7O+ 504.2882 [M + H]+, found 504.2886 [M + H]+.
3.2.5. Tert-butyl 4-(2-chloro-7-(4-fluoro-1-naphthoyl)-5,6,7,8-tetrahydropyrido[3,4-d]pyrimidin-4-yl)-3-(hydroxymethyl)piperazine-1-carboxylate (8d)
The synthesis of compound 8d was carried out following a procedure analogous to that of 8b. The starting material, 1-Boc-3-hydroxymethylpiperazine, was used as the reactant. The purification process involved column chromatography with a gradient elution system of petroleum ether/ethyl acetate (3:1 to 1:1). The target compound 8d was obtained as a white solid with a yield of 62.92%. 1H NMR (400 MHz, Chloroform-d) δ 8.21–8.09 (m, 1H), 7.89–7.76 (m, 1H), 7.65–7.53 (m, 2H), 7.44–7.36 (m, 1H), 7.21–7.11 (m, 1H), 5.15–4.76 (m, 1H), 4.27 (s, 2H), 4.21–3.83 (m, 4H), 3.83–3.68 (m, 2H), 3.67–3.47 (m, 1H), 3.47–3.21 (m, 2H), 3.19–2.96 (m, 2H), 2.86–2.80 (m, 1H), 2.64–2.36 (m, 1H), 1.46 (d, J = 12.0 Hz, 9H).
3.2.6. Tert-butyl 4-(7-(4-fluoro-1-naphthoyl)-2-(4-methyl-1,4-diazepan-1-yl)-5,6,7,8-tetrahydropyrido[3,4-d]pyrimidin-4-yl)-3-(hydroxymethyl)piperazine-1-carboxylate (9d)
Compound 9d was synthesized using a similar procedure to that employed for 9b, with 8d serving as the starting material. The target compound 9d was obtained as a white solid with a yield of 64.38%. 1H NMR (400 MHz, Methanol-d4) δ 8.28–8.08 (m, 1H), 7.87–7.70 (m, 1H), 7.66–7.56 (m, 2H), 7.55–7.38 (m, 1H), 7.36–7.17 (m, 1H), 4.80–4.72 (m, 1H), 4.14–3.92 (m, 4H), 3.92–3.84 (m, 2H), 3.79 (t, J = 6.3 Hz, 1H), 3.74–3.67 (m, 2H), 3.67–3.52 (m, 3H), 3.29–3.25 (m, 2H), 3.24–3.07 (m, 2H), 2.94–2.83 (m, 2H), 2.75–2.57 (m, 4H), 2.40 (m, 3H), 2.05–1.82 (m, 2H), 1.44 (d, J = 10.3 Hz, 9H). 13C NMR (101 MHz, Methanol-d4) δ 161.8, 161.4, 152.5, 151.1, 150.9, 147.4, 123.2, 121.9, 120.1, 118.9, 116.4, 116.2, 115.6, 111.9, 100.7, 95.5, 72.0, 50.6, 49.6, 48.5, 47.5, 43.3, 36.9, 36.8, 36.0, 33.8, 31.6, 19.1, 18.3, 16.3.
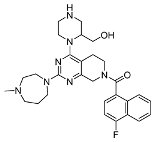
3.2.7. (4-Fluoronaphthalen-1-yl)(4-(2-(hydroxymethyl)piperazin-1-yl)-2-(4-methyl-1,4-diazepan-1-yl)-5,8-dihydropyrido[3,4-d]pyrimidin-7(6H)-yl)methanone (10d)
Referring to the process for 10b, 10d was obtained as a white solid with a yield of 79.32%. Mp 183.2–184.7 °C. 1H NMR (600 MHz, Methanol-d4) δ 8.24–8.15 (m, 1H), 7.86 (d, J = 25.0 Hz, 1H), 7.75–7.60 (m, 2H), 7.52 (d, J = 25.0 Hz, 1H), 7.30 (t, J = 9.1 Hz, 1H), 4.24–4.06 (m, 2H), 4.04–3.84 (m, 5H), 3.81–3.60 (m, 4H), 3.23–3.07 (m, 3H), 3.06–2.95 (m, 3H), 2.93–2.72 (m, 5H), 2.51 (m, 4H), 2.11–1.91 (m, 3H), 1.60 (s, 1H). 13C NMR (151 MHz, Methanol-d4) δ 170.0, 165.7, 160.9, 159.3, 158.4, 131.0, 130.0, 129.5, 128.2, 127.0, 124.5, 124.3, 123.6, 120.5, 108.7, 104.0, 72.5, 63.1, 59.9, 57.6, 56.5, 54.2, 51.4, 44.8, 44.1, 42.6, 40.1, 29.4, 26.0. ESI-HRMS m/z calculated for C29H37FN7O2+ 534.2987 [M + H]+, found 534.2995 [M + H]+.
3.2.8. Tert-butyl 4-(2-chloro-7-(4-fluoro-1-naphthoyl)-5,6,7,8-tetrahydropyrido[3,4-d]pyrimidin-4-yl)-3,3-dimethylpiperazine-1-carboxylate (8e)
8e was synthesized and processed using a method similar to that of 8b. The starting material, 1-Boc-3,3-dimethylpiperazine, was used as the reactant. The desired product was isolated as a white solid in 64.28% yield.
3.2.9. Tert-butyl 4-(7-(4-fluoro-1-naphthoyl)-2-(4-methyl-1,4-diazepan-1-yl)-5,6,7,8-tetrahydropyrido[3,4-d]pyrimidin-4-yl)-3,3-dimethylpiperazine-1-carboxylate (9e)
The preparation of compound 9e, including its synthesis and post-treatment, was conducted using a method similar to the one employed for intermediate 9b. Ultimately, the desired compound 9e was isolated as a white solid, achieving a yield of 44.18%.
3.2.10. (4-(2,2-Dimethylpiperazin-1-yl)-2-(4-methyl-1,4-diazepan-1-yl)-5,8-dihydropyrido[3,4-d]pyrimidin-7(6H)-yl)(4-fluoronaphthalen-1-yl)methanone (10e)
Compound 10e was synthesized and post-treated using a method similar to that for intermediate 10b, yielding a white solid at 78.65%. Mp 151.3–133.8 °C. ESI-HRMS m/z calculated for C29H37FN7O2+ 532.3195 [M + H]+, found 532.3172 [M + H]+.
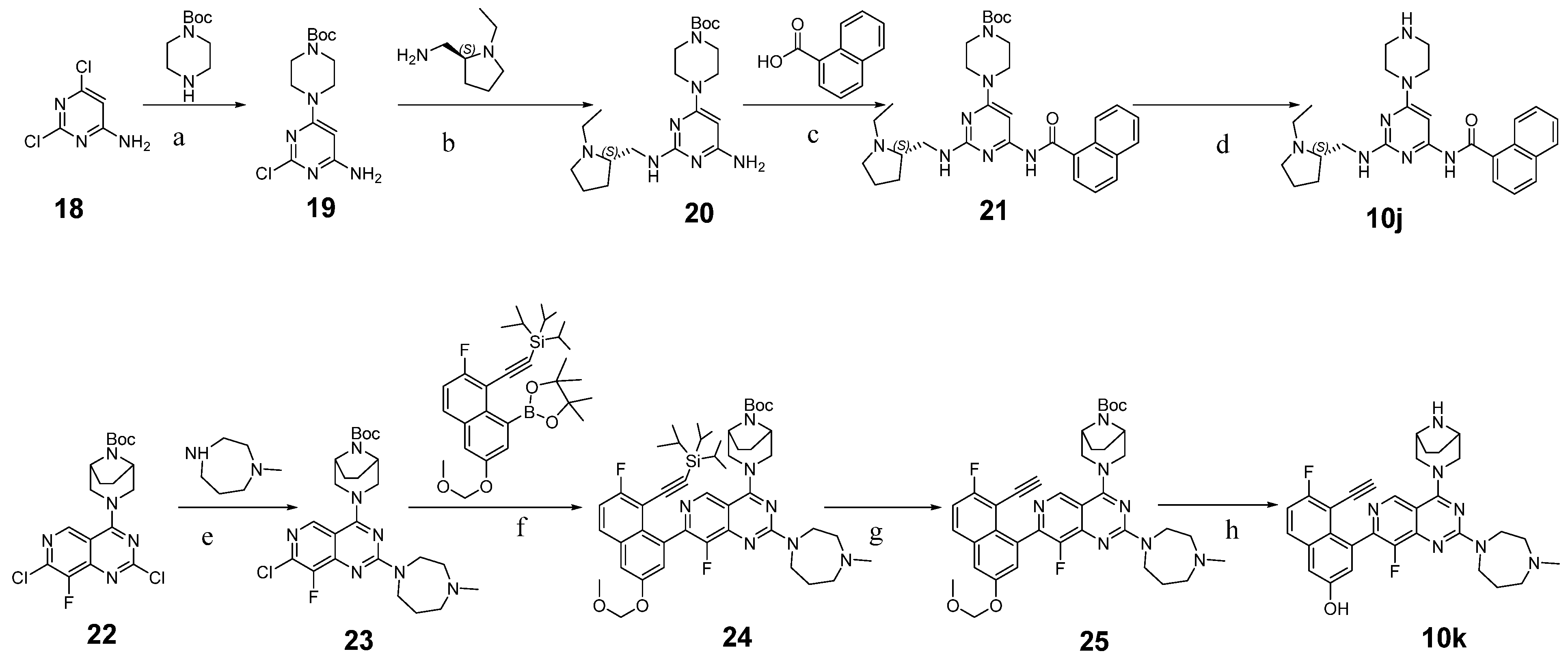
3.2.11. Tert-butyl 4-(6-amino-2-chloropyrimidin-4-yl)piperazine-1-carboxylate (19)
19 was synthesized and processed post-synthetically using a method akin to that for intermediate 8b. The starting material, 4-amino-2,6-dichloropyrimidine, was used as the reactant. The target compound 19 was obtained as a white solid with a yield of 79.28%. 1H NMR (400 MHz, DMSO-d6) δ 6.79 (s, 2H), 5.75 (s, 1H), 3.69–3.50 (m, 4H), 3.33 (d, J = 4.2 Hz, 4H), 1.41 (s, 9H). 13C NMR (101 MHz, DMSO-d6) δ 165.0, 160.8, 158.1, 153.9, 92.4, 79.0, 43.0, 28.1.
3.2.12. Tert-butyl (S)-4-(6-amino-2-(((1-ethylpyrrolidin-2-yl)methyl)amino)pyrimidin-4-yl)piperazine-1-carboxylate (20)
Compound 20 was synthesized similarly to 9b, using (S)-2-aminomethyl-1-ethylpyrrolidine as the starting material. The product was isolated as a white solid with 58.92% yield. 1H NMR (400 MHz, Chloroform-d) δ 5.15 (s, 1H), 4.95 (s, 1H), 4.29 (s, 2H), 3.68 (m, 4H), 3.42 (m, 5H), 3.28 (d, J = 9.3 Hz, 2H), 2.97–2.71 (m, 2H), 2.40–2.18 (m, 2H), 2.01–1.64 (m, 4H), 1.47 (s, 9H), 1.14 (t, J = 7.2 Hz, 3H). 13C NMR (101 MHz, Chloroform-d) δ 164.4, 164.0, 161.9, 155.1, 79.8, 74.7, 63.8, 53.7, 48.9, 43.7, 42.8, 28.5, 22.9, 13.4.
3.2.13. Tert-butyl (S)-4-(6-(1-naphthamido)-2-(((1-ethylpyrrolidin-2-yl)methyl)amino)pyrimidin-4-yl)piperazine-1-carboxylate (21)
In a single-necked flask under nitrogen protection, 100 mg (0.53 mmol) of 4-fluoro-1-naphthoic acid was dispersed in 5 mL of DMF. An ice-salt bath was employed to bring the temperature of the resulting mixture down to around 0 °C. HATU (240 mg, 0.63 mmol) and DIEA (164 mg, 1.26 mmol) were added slowly and sequentially. Once the addition process concluded, the blend was continuously agitated at the same temperature for 10 min. Subsequently, a solution composed of 20 (244 mg, 0.80 mmol) in 2 mL of DMF was slowly introduced drop by drop. After that, the reaction mixture was left to reach room temperature on its own and then transferred to a 50 °C oil bath, where it was stirred for 16 h. The mixture from the reaction was subjected to extraction using a combination of ethyl acetate (50 mL) and water (20 mL). The organic layer obtained was then rinsed with a saturated sodium chloride solution (20 mL × 3). Subsequently, it was dried using anhydrous sodium sulfate and then the solvent was removed to concentrate the mixture. The resulting unpurified product was then refined through the application of silica gel plate chromatography (dichloromethane/methanol = 10:1), yielding 21 (140 mg, white solid, yield: 45.90%). 1H NMR (600 MHz, Chloroform-d) δ 9.10 (s, 1H), 8.13 (dd, J = 8.4, 1.4 Hz, 1H), 8.04 (m, 1H), 7.92 (d, J = 8.4 Hz, 1H), 7.59–7.53 (m, 2H), 7.49 (s, 1H), 7.43 (dd, J = 7.8, 5.2 Hz, 1H), 7.16 (dd, J = 9.9, 7.8 Hz, 1H), 3.84–3.78 (m, 1H), 3.76 (t, J = 5.4 Hz, 4H), 3.63 (m, 1H), 3.44 (t, J = 5.2 Hz, 5H), 3.35 (s, 1H), 3.11 (dd, J = 13.0, 7.0 Hz, 1H), 2.84–2.73 (m, 2H), 2.04 (s, 1H), 1.86 (s, 2H), 1.66 (s, 1H), 1.46 (s, 9H), 1.16 (s, 3H). 13C NMR (151 MHz, Chloroform-d) δ 164.7, 164.2, 162.8, 160.1, 158.9, 154.9, 136.0, 131.2, 128.6, 127.4, 125.1, 124.9, 124.2, 121.1, 109.6, 91.8, 80.3, 66.5, 53.7, 50.1, 43.6, 42.1, 36.6, 31.6, 29.8, 28.5, 22.6, 11.3.
3.2.14. (S)-N-(2-(((1-Ethylpyrrolidin-2-yl)methyl)amino)-6-(piperazin-1-yl)pyrimidin-4-yl)-1-naphthamide (10j)
Compound 10j was prepared using a method similar to 10b, yielding 75.56% of the product as a white solid. Mp 133.7–136.3 °C. 1H NMR (400 MHz, Chloroform-d) δ 8.17–8.11 (m, 1H), 7.99 (d, J = 8.0 Hz, 1H), 7.62–7.51 (m, 2H), 7.44 (dd, J = 8.0, 5.3 Hz, 1H), 7.16 (t, J = 10.2, 8.0 Hz, 1H), 3.83 (t, J = 5.3 Hz, 4H), 3.71–3.61 (m, 1H), 3.62–3.45 (m, 2H), 3.20–3.05 (m, 1H), 2.91 (t, J = 5.3 Hz, 4H), 2.83–2.75 (m, 1H), 2.66–2.49 (m, 1H), 2.42–2.32 (m, 1H), 2.08–1.91 (m, 3H), 1.74–1.61 (m, 1H), 1.43–1.39 (m, 1H), 0.89–0.85 (m, 3H). ESI-HRMS m/z calculated for C26H33FN7O+ 478.2725 [M + H]+, found 478.2775 [M + H]+.
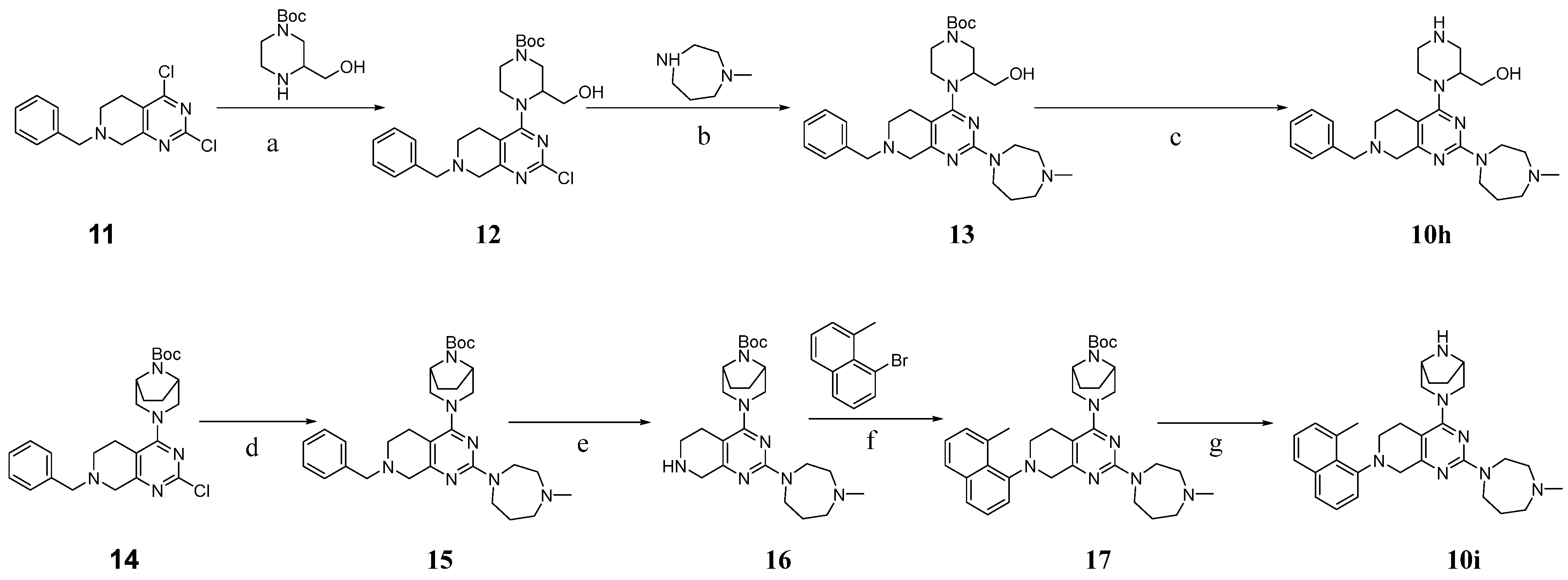
3.2.15. Tert-butyl 4-(7-benzyl-2-chloro-5,6,7,8-tetrahydropyrido[3,4-d]pyrimidin-4-yl)-3-(hydroxymethyl)piperazine-1-carboxylate (12)
In a single-necked flask, 2,4-dichloro-5,6,7,8-tetrahydro-7-(phenylmethyl)-pyrido[3,4-d]pyrimidine (2.00 g, 6.80 mmol) was dissolved in DMF (20 mL). 3-Hydroxymethyl-1-Boc-piperazine (1.47 g, 6.80 mmol) and DIEA (1.77 g, 13.60 mmol) were added. The reaction mixture was stirred at 60 °C under nitrogen protection overnight, and the progress was monitored by TLC. The reaction mixture underwent liquid–liquid extraction with ethyl acetate (200 mL) and water (100 mL). After phase separation, the organic phase was washed with brine (saturated NaCl solution, 50 mL × 3) and subsequently dried over anhydrous sodium sulfate. Following solvent evaporation to concentrate the crude material, purification was achieved by means of silica gel thin-layer chromatography (petroleum ether:ethyl acetate = 3:1 to 0:1), yielding 12 (2.66 g, white solid, yield: 82.74%). 1H NMR (400 MHz, Chloroform-d) δ 7.36–7.32 (m, 3H), 7.32–7.27 (m, 2H), 4.26–4.19 (m, 1H), 4.18–3.96 (m, 1H), 3.93–3.74 (m, 2H), 3.73–3.67 (m, 3H), 3.67–3.53 (m, 2H), 3.29 (t, J = 13.0 Hz, 1H), 3.21–2.90 (m, 2H), 2.87–2.73 (m, 1H), 2.73–2.59 (m, 3H), 2.42–2.05 (m, 1H), 1.47 (s, 9H). 13C NMR (101 MHz, Chloroform-d) δ 171.2, 166.0, 165.8, 162.6, 157.0, 137.1, 129.2, 128.5, 127.6, 113.8, 80.7, 62.2, 60.4, 57.9, 55.7, 49.5, 42.6, 28.4, 27.3, 21.1, 14.2.
3.2.16. Tert-butyl 4-(7-benzyl-2-(4-methyl-1,4-diazepan-1-yl)-5,6,7,8-tetrahydropyrido[3,4-d]pyrimidin-4-yl)-3-(hydroxymethyl)piperazine-1-carboxylate (13)
In a single-necked flask, 12 (1.50 g, 3.17 mmol) was dissolved in DMF (15 mL). N-methylhomopiperazine (722 mg, 6.34 mmol) and Cs2CO3 (2.07 g, 6.34 mmol) were added. The mixture was placed in a 120 °C oil bath and stirred for 16 h. The crude reaction mixture was partitioned between ethyl acetate and water. The ethyl acetate layer was then sequentially rinsed with saturated NaCl solution (3 × 50 mL), dried with anhydrous Na2SO4, and evaporated to dryness. Final purification was accomplished through flash column chromatography on silica gel (dichloromethane/methanol = 8:1 to 5:1), yielding the product (0.92 g, white solid, yield: 52.67%). 1H NMR (400 MHz, DMSO-d6) δ 7.40–7.18 (m, 5H), 4.78–3.94 (m, 1H), 3.94–3.66 (m, 3H), 3.66–3.52 (m, 6H), 3.52–3.37 (m, 3H), 3.30–3.04 (m, 4H), 3.04–2.75 (m, 3H), 2.63–2.51 (m, 2H), 2.45–2.25 (m, 4H), 2.17 (s, 3H), 1.75 (p, J = 6.0 Hz, 2H), 1.36 (s, 9H). 13C NMR (151 MHz, DMSO-d6) δ 165.1, 163.4, 159.1, 154.9, 138.7, 129.4, 128.8, 127.6, 103.6, 79.4, 62.1, 58.9, 58.6, 58.2, 57.2, 55.5, 50.9, 46.7, 45.9, 45.8, 42.6, 28.5, 27.7, 26.3.
3.2.17. (1-(7-Benzyl-2-(4-methyl-1,4-diazepan-1-yl)-5,6,7,8-tetrahydropyrido [3,4-d]pyrimidin-4-yl)piperazin-2-yl)methanol (10h)
Compound 10h was synthesized according to a process similar to that for 10b. The desired compound 10h was acquired as a white solid, achieving a yield of 75.92%. 1H NMR (600 MHz, Methanol-d4) δ 7.48 (d, J = 7.4 Hz, 2H), 7.39 (t, J = 7.4 Hz, 2H), 7.37–7.33 (m, 1H), 4.45 (s, 2H), 4.04 (s, 2H), 3.96 (s, 2H), 3.91–3.76 (m, 2H), 3.67–3.52 (m, 3H), 3.47–3.38 (m, 2H), 3.30–3.25 (m, 2H), 3.24–3.15 (m, 2H), 3.11–3.05 (m, 1H), 3.03–2.98 (m, 2H), 2.94 (d, J = 12.7 Hz, 1H), 2.90 (s, 3H), 2.80–2.68 (m, 2H), 2.24–2.14 (m, 2H). 13C NMR (151 MHz, Methanol-d4) δ 168.0, 162.8, 160.4, 135.9, 131.3, 129.8, 129.4, 103.2, 66.7, 62.5, 57.7, 57.0, 52.9, 50.9, 46.2, 45.9, 45.1, 44.1, 43.3, 43.2, 25.7, 21.3. Mp 113.7–116.2 °C. ESI-HRMS m/z calculated for C25H38N7O+ 452.3132 [M + H]+, found 452.3129 [M + H]+.
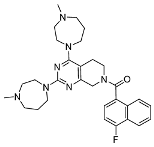
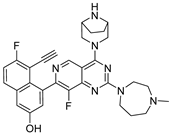
3.2.18. (2,4-Bis(4-methyl-1,4-diazepan-1-yl)-5,8-dihydropyrido[3,4-d]pyrimidin-7(6H)-yl)(4-fluoronaphthalen-1-yl)methanone (10f)
In a single-necked flask, 7 (40 mg, 0.11 mmol) was suspended in DMF (5 mL). Cs2CO3 (108 mg, 0.33 mmol) and N-methylhomopiperazine (25 mg, 0.22 mmol) were added. The mixture was stirred at 120 °C for 16 h and the reaction progress was monitored by TLC. After the reaction was nearly complete, the mixture underwent ethyl acetate/water extraction. The organic extract underwent three brine washes (5 mL each), was dried over sodium sulfate, and evaporated. Final isolation employed silica gel column chromatography (dichloromethane/methanol = 8:1 to 5:1), yielding 10f (45 mg, white solid, yield: 77.23%). Mp 115.5–118.2 °C. 1H NMR (600 MHz, Chloroform-d) δ 8.15 (t, J = 8.1 Hz, 1H), 7.91–7.82 (m, 1H), 7.61–7.53 (m, 2H), 7.44–7.35 (m, 1H), 7.22–7.09 (m, 1H), 4.86–4.06 (m, 2H), 4.04–3.85 (m, 2H), 3.81–3.69 (m, 3H), 3.67–3.53 (m, 4H), 3.35–3.23 (m, 1H), 2.90–2.65 (m, 4H), 2.63–2.54 (m, 4H), 2.52–2.43 (m, 2H), 2.42–2.26 (m, 6H), 2.02–1.95 (m, 2H), 1.95–1.82 (m, 2H). Mp 115.2–118.2 °C. ESI-HRMS m/z calculated for C30H39FN7O+ 532.3195 [M + H]+, found 532.3196 [M + H]+.
3.2.19. (2,4-Di(1,4-diazabicyclo[3.2.2]nonan-4-yl)-5,8-dihydropyrido[3,4-d]pyrimidin-7(6H)-yl)(4-fluoronaphthalen-1-yl)methanone (10g)
Compound 10g was prepared from 1,4-diazabicyclo[3.2.2]nonane following a method analogous to that used for 10f, affording a white solid with 69.38% yield. Mp 123.6–126.2 °C. ESI-HRMS m/z calculated for C32H39FN7O+ 556.3195 [M + H]+, found 556.3193 [M + H]+.
3.2.20. Tert-butyl (1-(2-chloro-7-(4-fluoro-1-naphthoyl)-5,6,7,8-tetrahydropyrido[3,4-d]pyrimidin-4-yl)azetidin-3-yl)carbamate (8a)
Compound 8a was obtained using a method similar to that of 8b, starting from 3-(Boc-amino)azetidine. The product was isolated as a white solid with 77.18% yield.
3.2.21. Tert-butyl (1-(7-(4-fluoro-1-naphthoyl)-2-(4-methyl-1,4-diazepan-1-yl)-5,6,7,8-tetrahydropyrido[3,4-d]pyrimidin-4-yl)azetidin-3-yl)carbamate (9a)
Compound 9a was synthesized from 8a using a procedure similar to that for 9b, yielding a white solid with 64.37% yield.
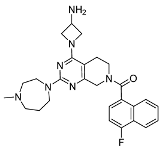
3.2.22. (4-(3-Aminoazetidin-1-yl)-2-(4-methyl-1,4-diazepan-1-yl)-5,8-dihydropyrido[3,4-d]pyrimidin-7(6H)-yl)(4-fluoronaphthalen-1-yl)methanone (10a)
The synthesis of compound 10a was carried out following a procedure analogous to that for 10b. The starting material, 9a, was used as the reactant. The target compound 10a was obtained as a white solid with a yield of 81.53%. Mp 119.5–121.4 °C. 1H NMR (600 MHz, Methanol-d4) δ 8.22–8.15 (m, 1H), 7.87–7.78 (m, 1H), 7.68–7.62 (m, 2H), 7.55–7.45 (m, 1H), 7.35–7.25 (m, 1H), 4.75 (d, J = 10.7 Hz, 1H), 4.53–4.44 (m, 1H), 4.37 (t, J = 7.9 Hz, 1H), 4.23–4.08 (m, 1H), 4.04–4.00 (m, 2H), 3.95–3.91 (m, 1H), 3.88 (t, J = 6.4 Hz, 1H), 3.82 (s, 1H), 3.69–3.63 (m, 1H), 3.63–3.50 (m, 1H), 3.41 (dt, J = 11.3, 5.9 Hz, 1H), 3.25 (t, J = 5.3 Hz, 1H), 3.17–3.13 (m, 1H), 3.06 (s, 1H), 3.03–2.99 (m, 1H), 2.87–2.80 (m, 1H), 2.78–2.76 (m, 2H), 2.66 (s, 1H), 2.57–2.40 (m, 2H), 2.20–2.12 (m, 1H), 2.03–1.97 (m, 1H). ESI-HRMS m/z calculated for C27H33FN7O+ 490.2725 [M + H]+, found 490.2730 [M + H]+.
3.2.23. Tert-butyl (1R,5S)-8-(2-chloro-7-(4-fluoro-1-naphthoyl)-5,6,7,8-tetrahydropyrido[3,4-d]pyrimidin-4-yl)-3,8-diazabicyclo[3.2.1]octane-3-carboxylate (8c)
8c was synthesized by adhering to the method used for compound 8b. Commencing with tert-butyl 3,8-diazabicyclo[3.2.1]octane-3-carboxylate as the starting material, compound 8c was obtained as a white solid with a yield of 71.78%. 1H NMR (600 MHz, Chloroform-d) δ 8.28–8.10 (m, 1H), 7.83 (m, 1H), 7.65–7.51 (m, 2H), 7.46–7.33 (m, 1H), 7.21–7.08 (m, 1H), 5.11–4.74 (m, 1H), 4.69–4.43 (m, 2H), 4.28–4.00 (m, 2H), 3.90 (dd, J = 37.2, 13.1 Hz, 1H), 3.75 (dd, J = 28.3, 13.1 Hz, 1H), 3.51–3.30 (m, 1H), 3.28–3.14 (m, 1H), 3.12–2.93 (m, 1H), 2.88–2.73 (m, 1H), 2.64–2.31 (m, 1H), 2.09–1.72 (m, 4H), 1.45 (d, J = 13.8 Hz, 9H).13C NMR (151 MHz, Chloroform-d) δ 169.2, 162.5, 162.1, 158.1, 155.9, 131.2, 129.3, 128.4, 127.0, 125.1, 124.8, 124.4, 124.1, 121.4, 111.6, 109.1, 109.0, 80.4, 55.7, 55.4, 51.5, 49.8, 48.9, 46.9, 44.1, 39.3, 28.5, 26.7, 26.1.
3.2.24. Tert-butyl (1R,5S)-8-(7-(4-fluoro-1-naphthoyl)-2-(4-methyl-1,4-diazepan-1-yl)-5,6,7,8-tetrahydropyrido[3,4-d]pyrimidin-4-yl)-3,8-diazabicyclo[3.2.1]octane-3-carboxylate (9c)
Compound 9c was prepared from 8c using the same method as 9b, affording a white solid with 59.77% yield.
3.2.25. (4-((1R,5S)-3,8-Diazabicyclo[3.2.1]octan-8-yl)-2-(4-methyl-1,4-diazepan-1-yl)-5,8-dihydropyrido[3,4-d]pyrimidin-7(6H)-yl)(4-fluoronaphthalen-1-yl)methanone (10c)
The synthesis of compound 10c followed the procedure for 10b, using 9c as the starting material, yielding 10c (white solid, 80.55% yield). Mp 147.4–150.1 °C. 1H NMR (600 MHz, Chloroform-d) δ 8.21–8.11 (m, 1H), 7.91–7.82 (m, 1H), 7.62–7.49 (m, 2H), 7.47–7.34 (m, 1H), 7.23–7.05 (m, 1H), 4.82 (q, J = 18.5 Hz, 1H), 4.38–4.32 (m, 1H), 4.26–4.17 (m, 1H), 4.18–3.96 (m, 2H), 3.88 (s, 1H), 3.75 (d, J = 33.5 Hz, 2H), 3.60 (s, 1H), 3.44–3.21 (m, 1H), 3.15 (d, J = 12.1 Hz, 1H), 3.07 (dd, J = 25.4, 12.1 Hz, 1H), 2.81–2.61 (m, 4H), 2.59–2.51 (m, 2H), 2.49–2.44 (m, 1H), 2.34 (d, J = 47.4 Hz, 3H), 2.06–1.79 (m, 7H), 1.46–1.36 (m, 1H). ESI-HRMS m/z calculated for C30H37FN7O+ 530.3027 [M + H]+, found 530.3043 [M + H]+.
3.2.26. Tert-butyl (1R,5S)-3-(7-benzyl-2-(4-methyl-1,4-diazepan-1-yl)-5,6,7,8-tetrahydropyrido[3,4-d]pyrimidin-4-yl)-3,8-diazabicyclo[3.2.1]octane-8-carboxylate (15)
15 was synthesized following the procedure for 9b, using tert-butyl 3-(3-[2-chloro-5,6,7,8-tetrahydro-7-(benzyl)pyrido[3,4-d]pyridin-4-yl])diazabicyclo[3.2.1]octane-8-carboxylate as the starting material, affording compound 23 as a white solid with 71.57% yield. 1H NMR (600 MHz, DMSO-d6) δ 7.76–7.27 (m, 4H), 4.71–4.24 (m, 2H), 4.16 (s, 2H), 4.03–3.39 (m, 9H), 3.39–3.21 (m, 6H), 3.11–2.99 (m, 2H), 2.95–2.82 (m, 1H), 2.81–2.68 (m, 3H), 2.66–2.54 (m, 1H), 2.46–1.96 (m, 2H), 1.91–1.66 (m, 3H), 1.40 (s, 9H).
3.2.27. Tert-butyl (1R,5S)-3-(2-(4-methyl-1,4-diazepan-1-yl)-5,6,7,8-tetrahydropyrido[3,4-d]pyrimidin-4-yl)-3,8-diazabicyclo[3.2.1]octane-8-carboxylate (16)
In a single-necked flask, 15 (130 mg, 0.24 mmol) was dissolved in methanol (10 mL). To the solution was added palladium on carbon (10%, 20 mg). The flask was purged with hydrogen gas three times to remove air and stirred at room temperature under a hydrogen atmosphere for 16 h. The reaction progress was monitored by TLC, indicating completion. The mixture was filtered through a pad of Celite, and the filter cake was washed with methanol (3 × 5 mL). The filtrate was concentrated under reduced pressure to afford crude 16 (white solid), which was used directly in the next step without further purification. 1H NMR (600 MHz, DMSO-d6) δ 4.15 (s, 2H), 3.87–3.60 (m, 6H), 3.60–3.15 (m, 5H), 3.07–2.90 (m, 2H), 2.83 (t, J = 5.5 Hz, 1H), 2.63 (d, J = 18.2 Hz, 1H), 2.60–2.53 (m, 2H), 2.49–2.46 (m, 1H), 2.46–2.38 (m, 2H), 2.30–2.19 (m, 2H), 2.00–1.85 (m, 1H), 1.84–1.80 (m, 2H), 1.79 (s, 3H), 1.41 (s, 9H).
3.2.28. Tert-butyl (1R,5S)-3-(2-(4-methyl-1,4-diazepan-1-yl)-7-(8-methylnaphthalen-1-yl)-5,6,7,8-tetrahydropyrido[3,4-d]pyrimidin-4-yl)-3,8-diazabicyclo[3.2.1]octane-8-carboxylate (17)
In a single-necked flask, 16 (100 mg, 0.22 mmol), 1-bromo-8-methylnaphthalene (73 mg, 0.33 mmol), tris(dibenzylideneacetone)dipalladium(0) (18 mg, 0.02 mmol), 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene (XPhos) (12 mg, 0.02 mmol), cesium carbonate (144 mg, 0.44 mmol) and 1,4-dioxane (5 mL) were added sequentially. The air in the flask was replaced with nitrogen, and the reaction proceeded at 100 °C for 16 h before being filtered. The collected solids were rinsed with ethyl acetate (20 mL × 3), while the combined filtrates were concentrated and subjected to silica gel column chromatography (gradient:DCM:MeOH = 15:1 to 8:1) to afford 17 (33 mg, brown solid, 25.19% yield). 1H NMR (600 MHz, Methanol-d4) δ 8.34–7.82 (m, 1H), 7.73 (m, 1H), 7.61–7.43 (m, 2H), 7.43–7.34 (m, 1H), 7.30–7.17 (m, 1H), 4.30–4.18 (m, 2H), 4.10–3.92 (m, 4H), 3.88–3.70 (m, 3H), 3.68–3.41 (m, 4H), 3.38–3.32 (m, 1H), 3.24 (t, J = 5.2 Hz, 1H), 3.11–2.96 (m, 2H), 2.88 (s, 2H), 2.86–2.78 (m, 3H), 2.75–2.69 (m, 1H), 2.62–2.57 (m, 1H), 2.21–2.13 (m, 2H), 2.06–1.70 (m, 4H), 1.48–1.44 (m, 9H), 1.27–1.23 (m, 2H).
3.2.29. 4-((1R,5S)-3,8-Diazabicyclo[3.2.1]octan-3-yl)-2-(4-methyl-1,4-diazepan-1-yl)-7-(8-methylnaphthalen-1-yl)-5,6,7,8-tetrahydropyrido[3,4-d]pyrimidine (10i)
The synthesis of compound 10i followed the procedure for 10b, yielding 10i (white solid, 77.23% yield). 1H NMR (600 MHz, Methanol-d4) δ 8.23 (m, 1H), 7.98 (dd, J = 35.3, 7.6 Hz, 1H), 7.89–7.65 (m, 1H), 7.62 (td, J = 7.6, 3.6 Hz, 1H), 7.59–7.37 (m, 1H), 7.36–7.12 (m, 1H), 5.12 (s, 1H), 4.52–4.10 (m, 5H), 4.03–3.35 (m, 10H), 3.35–3.30 (m, 4H), 3.27–3.14 (m, 1H), 2.99–2.90 (m, 3H), 2.85–2.57 (m, 1H), 2.57–1.85 (m, 7H). Mp 125.1–128.2 °C. ESI-HRMS m/z calculated for C30H40N7+ 498.3340 [M + H]+, found 498.3345 [M + H]+.
3.2.30. Tert-butyl (1R,5S)-3-(7-chloro-8-fluoro-2-(4-methyl-1,4-diazepan-1-yl)pyrido[4,3-d]pyrimidin-4-yl)-3,8-diazabicyclo[3.2.1]octane-8-carboxylate (23)
In a single-necked flask, tert-butyl 3-(2,7-dichloro-8-fluoropyrido[4,3-d]pyrimidin-4-yl)-3,8-diazabicyclo[3.2.1]octane-8-carboxylate (0.50 g, 1.17 mmol) was suspended in DMF (10 mL). N-methylhomopiperazine (267 mg, 2.34 mmol) and DIEA (456 mg, 3.51 mmol) were introduced into the reaction vessel, followed by stirring at 80 °C for 16 h. After completion, the crude product was partitioned between ethyl acetate (100 mL) and water (30 mL). The organic layer underwent three brine washes (30 mL each), was dried over Na2SO4 and was concentrated in vacuo. Purification via silica gel column chromatography yielded the target compound (gradient: DCM:MeOH = 8:1 to 5:1) to afford compound 23 (440 mg, white solid, 74.58% yield). 1H NMR (600 MHz, Chloroform-d) δ 8.51 (s, 1H), 4.45–4.21 (m, 4H), 4.17–3.78 (m, 4H), 3.65–3.41 (m, 2H), 2.93–2.61 (m, 4H), 2.50–2.38 (m, 3H), 2.18–2.06 (m, 2H), 1.99–1.89 (m, 2H), 1.78–1.69 (m, 2H), 1.50 (s, 9H). 13C NMR (151 MHz, Chloroform-d) δ 163.9, 163.8, 159.6, 153.3, 150.5, 150.5, 148.9, 147.1, 143.6, 143.5, 137.2, 137.1, 137.1, 109.1, 80.2, 58.2, 58.0, 57.2, 57.0, 54.0, 53.3, 46.6, 46.3, 46.2, 28.4, 27.2, 26.5.
3.2.31. Tert-butyl (1R,5S)-3-(8-fluoro-7-(7-fluoro-3-(methoxymethoxy)-8-((triisopropylsilyl)ethynyl)naphthalen-1-yl)-2-(4-methyl-1,4-diazepan-1-yl)pyrido[4,3-d]pyrimidin-4-yl)-3,8-diazabicyclo[3.2.1]octane-8-carboxylate (24)
In a single-necked flask, 23 (400 mg, 0.79 mmol), ((2-fluoro-6-(methoxymethoxy)-8-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)naphthalen-1-yl)ethynyl)triisopropylsilane (609 mg, 1.19 mmol), K3PO4 (502 mg, 2.37 mmol), Ad2nBuP-Pd-G3 (58 mg, 0.08 mmol) and 1,4-dioxane (5 mL) were combined. Under a nitrogen atmosphere, the reaction mixture was maintained at 80 °C with continuous stirring for 5 h. The reaction was diluted with ethyl acetate (30 mL), and the solid was filtered under reduced pressure. The collected solids were rinsed with ethyl acetate (3 × 10 mL), and the combined washes were concentrated. The residue was purified via silica gel column chromatography (gradient: DCM:MeOH = 10:1 to 7:1) to afford 24 (430 mg, white solid, 63.70% yield). 1H NMR (600 MHz, Chloroform-d) δ 8.89 (s, 1H), 7.77 (dd, J = 9.0, 5.6 Hz, 1H), 7.48 (d, J = 2.6 Hz, 1H), 7.32–7.29 (m, 1H), 7.29–7.28 (m, 1H), 7.28–7.27 (m, 1H), 7.24 (s, 1H), 5.33–5.24 (m, 3H), 4.69–3.93 (m, 10H), 2.73–2.52 (m, 4H), 2.02–2.01 (m, 2H), 2.00 (s, 3H), 1.76–1.72 (m, 4H), 1.51 (s, 9H), 1.25–1.24 (m, 3H), 0.87 (dd, J = 15.8, 7.4 Hz, 18H).
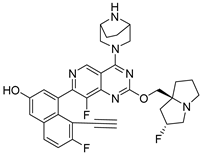
3.2.32. 4-(4-((1R,5S)-3,8-Diazabicyclo[3.2.1]octan-3-yl)-8-fluoro-2-(4-methyl-1,4-diazepan-1-yl)pyrido[4,3-d]pyrimidin-7-yl)-5-ethynyl-6-fluoronaphthalen-2-ol (10k)
In a single-necked flask, 24 (100 mg, 0.12 mmol) was suspended in DMF (5 mL). Following the addition of CsF (89 mg, 0.58 mmol), the reaction was maintained at 50 °C in an oil bath for 2 h, monitored by TLC until complete conversion to the desilylated intermediate occurred. After triturating the reaction mixture in water (20 mL), the resulting solid was collected by filtration and subjected to three distilled water washes (5 mL each), affording 25. 25 was redissolved in dichloromethane (10 mL), and the solution was cooled to 0 °C in an ice bath. A solution of HCl in 1,4-dioxane (4 mol/L, 1.2 mL) was introduced dropwise. After full addition, the reaction temperature was gradually raised to an ambient temperature and maintained with stirring for 5 h. The mixture was concentrated under reduced pressure, adjusted to pH 8–9 with saturated sodium bicarbonate and processed with ethyl acetate (50 mL)-water (10 mL) biphasic extraction. After brine (10 mL × 3) washing (saturated) and drying (Na2SO4), the organic extract was concentrated, with the resulting residue chromatographed on silica gel (gradient: DCM:MeOH = 10:1 to 7:1) to afford 10k (31 mg, pale yellow solid, 47.23% yield). 1H NMR (600 MHz, Methanol-d4) δ 8.75 (s, 1H), 7.87–7.81 (m, 1H), 7.35–7.27 (m, 2H), 7.18 (t, J = 2.7 Hz, 1H), 4.50 (d, J = 12.9 Hz, 1H), 4.44–4.34 (m, 1H), 4.04 (d, J = 14.1 Hz, 2H), 3.97 (s, 2H), 3.69 (s, 2H), 3.61 (dd, J = 25.7, 12.9 Hz, 3H), 2.93–2.80 (m, 2H), 2.75–2.63 (m, 2H), 2.44 (s, 3H), 2.11–2.03 (m, 2H), 1.96–1.82 (m, 5H). Mp 198.2–201.3 °C. ESI-HRMS m/z calculated for C31H32F2N7O+ 556.2631 [M + H]+, found 556.2628 [M + H]+.
3.2.33. Tert-butyl 4-(2-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)ethoxy)ethyl)-1,4-diazepane-1-carboxylate (28)
In a single-necked flask, 1,4-diazepane-1-carboxylic acid tert-butyl ester (1.00 g, 5.00 mmol), 27 (2.58 g, 5.00 mmol), DMSO (10 mL) and DIEA (1.3 g, 10.00 mmol) were added sequentially. Under a nitrogen purge, the reaction was maintained at 70 °C with continuous stirring for 4 h, with reaction progress monitored by TLC until completion. The reaction mixture was extracted with ethyl acetate (100 mL)/water (50 mL). The organic extract underwent brine (50 mL × 3) washing, Na2SO4 drying and vacuum concentration. Purification by column chromatography afforded product 28 (2.12 g, yellow solid, 78.08% yield). 1H NMR (400 MHz, Chloroform-d) δ 7.48 (dd, J = 8.5, 7.1 Hz, 1H), 7.09 (d, J = 7.1 Hz, 1H), 6.89 (d, J = 8.5 Hz, 1H), 6.53–6.43 (m, 1H), 4.96–4.83 (m, 1H), 3.66 (t, J = 5.5 Hz, 2H), 3.59 (t, J = 5.5 Hz, 2H), 3.50 (t, J = 5.5 Hz, 2H), 3.46–3.43 (m, 4H), 3.39 (t, J = 6.3 Hz, 2H), 3.00–2.95 (m, 1H), 2.93–2.90 (m, 1H), 2.87–2.81 (m, 1H), 2.77–2.75 (m, 2H), 2.70–2.66 (m, 2H), 2.15–2.08 (m, 1H), 1.93–1.83 (m, 2H), 1.44 (s, 9H).
3.2.34. 4-((2-(2-(1,4-Diazepan-1-yl)ethoxy)ethyl)amino)-2-(2,6-dioxopiperidin-3-yl)isoindoline-1,3-dione (29)
In a single-necked flask, 28 (2.0 g, 3.68 mmol) was dissolved in DCM (10 mL). We added TFA (5 mL) dropwise to the stirring solution and continued stirring at an ambient temperature for 1 h. Reaction completion was confirmed by TLC monitoring. The DCM and trifluoroacetic acid were evaporated under reduced pressure. The residue was adjusted to alkaline pH with saturated sodium bicarbonate solution and extracted with dichloromethane (100 mL)/water (50 mL). The organic layer underwent brine (50 mL × 3) washing, dehydration with anhydrous Na2SO4 and solvent evaporation to afford the crude product 29 (2.40 g), which was directly used in the next step.
3.2.35. (2S,4R)-1-((S)-2-(6-Bromohexanamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)pyrrolidine-2-carboxamide (31)
In a single-necked flask, 6-bromohexanoic acid (300 mg, 1.44 mmol) was dissolved in DMF. The mixture was cooled to 0 °C using an ice-salt bath. While stirring, HATU (657 mg, 1.73 mmol) and DIEA (281 mg, 2.16 mmol) were slowly added sequentially. After complete addition, the reaction was maintained at this temperature for 5 min. Following gradual addition of the VHL ligand (700 mg, 1.44 mmol), the reaction was warmed to an ambient temperature and stirred for 12 h. The crude product was then partitioned between ethyl acetate (50 mL) and water (30 mL). The organic extract underwent brine washing (20 mL × 3), Na2SO4 drying and vacuum concentration. Purification by column chromatography afforded product 31 (680 mg, off-white solid, 76.23% yield). 1H NMR (600 MHz, Chloroform-d) δ 8.67 (s, 1H), 7.43 (d, J = 7.9 Hz, 1H), 7.38 (d, J = 8.1 Hz, 2H), 7.35 (d, J = 8.1 Hz, 2H), 6.28 (d, J = 8.8 Hz, 1H), 5.06 (t, J = 7.2 Hz, 1H), 4.67 (t, J = 7.8 Hz, 1H), 4.56 (d, J = 8.8 Hz, 1H), 4.49 (qd, J = 4.6, 3.2, 2.4 Hz, 1H), 4.01 (d, J = 11.2 Hz, 1H), 3.61 (dd, J = 11.2, 4.0 Hz, 1H), 3.41–3.30 (m, 2H), 2.50 (d, J = 1.0 Hz, 3H), 2.44 (ddd, J = 12.8, 7.5, 4.9 Hz, 1H), 2.17 (t, J = 7.5 Hz, 2H), 2.09–1.98 (m, 1H), 1.82 (p, J = 7.1 Hz, 2H), 1.60 (q, J = 7.6 Hz, 2H), 1.46 (d, J = 7.0 Hz, 3H), 1.42 (dt, J = 12.3, 4.4 Hz, 2H), 1.02 (s, 9H). 13C NMR (151 MHz, Chloroform-d) δ 173.1, 171.9, 169.7, 162.5, 150.3, 148.3, 143.1, 131.5, 130.7, 129.4, 126.4, 69.8, 58.5, 57.4, 56.5, 48.7, 36.4, 36.0, 35.5, 35.1, 33.4, 32.2, 31.3, 27.6, 26.4, 24.5, 22.1, 15.9.
3.2.36. Tert-butyl 4-(6-(((S)-1-((2S,4R)-4-hydroxy-2-(((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-6-oxohexyl)-1,4-diazepane-1-carboxylate (32)
Compound 32 was obtained in a 71.24% yield as an off-white solid, using 31 as the starting material and employing the same procedure as that for compound 28. 1H NMR (600 MHz, Chloroform-d) δ 8.65 (s, 1H), 7.44 (dd, J = 25.3, 7.8 Hz, 1H), 7.36 (q, J = 7.8 Hz, 4H), 6.28–6.19 (m, 1H), 5.27 (s, 1H), 5.06 (m, 1H), 4.68 (t, J = 8.0 Hz, 1H), 4.57 (d, J = 8.4 Hz, 1H), 3.99 (d, J = 10.5 Hz, 1H), 3.60 (d, J = 10.5 Hz, 1H), 3.51–3.45 (m, 2H), 3.43–3.35 (m, 4H), 2.89–2.82 (m, 2H), 2.60–2.55 (m, 2H), 2.50 (s, 3H), 2.47–2.43 (m, 1H), 2.23–2.10 (m, 2H), 2.07–2.00 (m, 1H), 1.87–1.72 (m, 4H), 1.61–1.57 (m, 2H), 1.47–1.45 (m, 2H), 1.44 (s, 9H), 1.40 (s, 3H), 1.02 (s, 9H). 13C NMR (151 MHz, Chloroform-d) δ 173.5, 172.2, 169.9, 169.8, 155.6, 150.4, 148.6, 143.3, 131.6, 131.0, 129.6, 126.5, 79.4, 69.9, 58.7, 57.5, 56.8, 55.9, 54.6, 48.9, 45.9, 45.1, 36.4, 35.7, 35.3, 28.6, 27.1, 27.0, 26.6, 22.7, 22.3, 16.1, 14.2.
3.2.37. Tert-butyl 4-(6-(((S)-1-((2S,4R)-4-hydroxy-2-(((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-6-oxohexyl)-1,4-diazepane-1-carboxylate (33)
Compound 33 was synthesized following the procedure described for compound 29, using 32 as the starting material. The crude product 33 was obtained and directly used in the next step without further purification.1H NMR (600 MHz, Chloroform-d) δ 8.66 (d, J = 1.1 Hz, 1H), 7.43 (d, J = 7.8 Hz, 1H), 7.39–7.37 (m, 2H), 7.36–7.33 (m, 2H), 6.31 (d, J = 8.8 Hz, 1H), 5.07 (t, J = 7.2 Hz, 2H), 4.67 (t, J = 8.0 Hz, 1H), 4.59 (d, J = 8.8 Hz, 1H), 4.49 (s, 1H), 3.99 (d, J = 11.0 Hz, 1H), 3.60 (dd, J = 11.0, 3.7 Hz, 1H), 3.13–2.99 (m, 4H), 2.72 (q, J = 6.3, 5.8 Hz, 2H), 2.70–2.61 (m, 2H), 2.55–2.50 (m, 4H), 2.50–2.47 (m, 2H), 2.40 (m, 1H), 2.26–2.15 (m, 2H), 2.12–2.05 (m, 1H), 1.84 (t, J = 6.0 Hz, 2H), 1.61 (m, 2H), 1.49–1.45 (m, 4H), 1.44 (t, J = 7.0 Hz, 2H), 1.33 (dt, J = 14.3, 7.0 Hz, 2H), 1.02 (s, 9H). 13C NMR (151 MHz, Chloroform-d) δ 173.4, 171.7, 169.9, 150.2, 148.4, 143.2, 131.5, 130.8, 129.5, 126.4, 126.3, 69.7, 58.7, 57.5, 57.4, 57.0, 54.5, 54.1, 48.7, 46.8, 45.8, 35.8, 35.3, 28.0, 26.4, 24.5, 22.1, 16.0.
3.2.38. Tert-butyl (1R,5S)-3-(7-chloro-2-(4-(2-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)ethoxy)ethyl)-1,4-diazepan-1-yl)-8-fluoropyrido[4,3-d]pyrimidin-4-yl)-3,8-diazabicyclo[3.2.1]octane-8-carboxylate (23a)
Compound 23a was prepared from starting material 29 using the method applied for compound 23, yielding a yellow solid with a 52.78% yield. 1H NMR (600 MHz, Chloroform-d) δ 8.50 (s, 1H), 7.57–7.45 (m, 1H), 7.11 (d, J = 7.1 Hz, 1H), 6.90 (t, J = 9.1 Hz, 1H), 6.47 (d, J = 6.6 Hz, 1H), 4.90 (dd, J = 12.1, 5.4 Hz, 1H), 4.46–4.20 (m, 4H), 4.12–3.76 (m, 4H), 3.71–3.66 (m, 2H), 3.66–3.62 (m, 1H), 3.60–3.48 (m, 2H), 3.44 (t, J = 5.4 Hz, 2H), 3.09–2.59 (m, 8H), 2.16–1.98 (m, 2H), 1.95–1.89 (m, 2H), 1.78–1.71 (m, 2H), 1.69–1.58 (m, 3H), 1.50 (s, 9H). 13C NMR (151 MHz, Chloroform-d) δ 171.1, 168.4, 159.6, 153.5, 143.8, 136.2, 132.7, 116.9, 111.9, 110.4, 109.3, 80.5, 69.5, 69.4, 55.5, 54.9, 53.5, 49.0, 46.3, 42.4, 31.5, 28.6, 22.9.
3.2.39. Tert-butyl (1R,5S)-3-(2-(4-(2-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)ethoxy)ethyl)-1,4-diazepan-1-yl)-8-fluoro-7-(7-fluoro-3-(methoxymethoxy)-8-((triisopropylsilyl)ethynyl)naphthalen-1-yl)pyrido[4,3-d]pyrimidin-4-yl)-3,8-diazabicyclo[3.2.1]octane-8-carboxylate (24a)
Compound 24a was prepared from 23a as the starting material, using the method outlined for compound 24, resulting in a yellow solid with a yield of 42.38%. 1H NMR (600 MHz, DMSO-d6) δ 11.09 (s, 1H), 8.86 (s, 1H), 8.07 (dd, J = 9.1, 5.7 Hz, 1H), 7.76–7.67 (m, 1H), 7.54 (q, J = 10.4, 9.1 Hz, 2H), 7.29 (s, 1H), 7.13 (t, J = 8.4 Hz, 1H), 7.02 (t, J = 9.1 Hz, 1H), 6.60 (s, 1H), 5.35 (s, 2H), 5.11–4.97 (m, 1H), 4.63–4.52 (m, 1H), 4.27 (d, J = 26.5 Hz, 2H), 4.15–4.01 (m, 2H), 3.98–3.88 (m, 5H), 3.89–3.77 (m, 3H), 3.60–3.58 (m, 1H), 3.54 (t, J = 5.9 Hz, 2H), 3.49–3.44 (m, 2H), 3.31–3.29 (m, 1H), 3.28–3.24 (m, 1H), 3.19–3.15 (m, 3H), 2.91–2.83 (m, 1H), 2.82–2.73 (m, 1H), 2.72–2.59 (m, 4H), 2.02–1.97 (m, 1H), 1.91–1.87 (m, 2H), 1.72–1.60 (m, 2H), 1.47–1.43 (m, 9H), 1.24–1.21 (m, 3H), 1.07–1.06 (m, 18H).
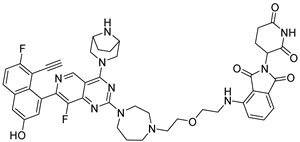
3.2.40. 4-((2-(2-(4-(4-((1R,5S)-3,8-Diazabicyclo[3.2.1]octan-3-yl)-7-(8-ethynyl-7-fluoro-3-hydroxynaphthalen-1-yl)-8-fluoropyrido[4,3-d]pyrimidin-2-yl)-1,4-diazepan-1-yl)ethoxy)ethyl)amino)-2-(2,6-dioxopiperidin-3-yl)isoindoline-1,3-dione (26a)
Compound 26a was synthesized following the procedure described for compound 10k, using 24a as the starting material. The target compound 26a was obtained as a yellow solid with a yield of 64.25%. 1H NMR (400 MHz, DMSO-d6) δ 11.09 (s, 1H), 7.61–7.54 (m, 1H), 7.14 (d, J = 8.5 Hz, 1H), 7.04 (d, J = 7.0 Hz, 1H), 6.60 (s, 1H), 5.05 (dd, J = 12.9, 5.3 Hz, 1H), 3.62 (t, J = 5.4 Hz, 2H), 3.59–3.55 (m, 2H), 3.55–3.52 (m, 2H), 3.52–3.44 (m, 8H), 3.42–3.36 (m, 2H), 2.99–2.79 (m, 1H), 2.65–2.51 (m, 2H), 2.12–1.94 (m, 1H). Mp 127.2–130.4 °C. ESI-HRMS m/z calculated for C47H47F2N10O6+ 885.3643 [M + H]+, found 885.3638 [M + H]+.
3.2.41. Tert-butyl (1R,5S)-3-(7-chloro-8-fluoro-2-(4-(6-(((S)-1-((2S,4R)-4-hydroxy-2-(((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-6-oxohexyl)-1,4-diazepan-1-yl)pyrido[4,3-d]pyrimidin-4-yl)-3,8-diazabicyclo[3.2.1]octane-8-carboxylate (23b)
Compound 23b was obtained using 33 as the starting material and the method outlined for compound 23, yielding a pale yellow solid at 27.20%.
3.2.42. Tert-butyl (1R,5S)-3-(8-fluoro-7-(7-fluoro-3-(methoxymethoxy)-8-((triisopropylsilyl)ethynyl)naphthalen-1-yl)-2-(4-(6-(((S)-1-((2S,4R)-4-hydroxy-2-(((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-6-oxohexyl)-1,4-diazepan-1-yl)pyrido[4,3-d]pyrimidin-4-yl)-3,8-diazabicyclo[3.2.1]octane-8-carboxylate (24b)
Compound 24b was prepared using the method outlined for compound 24a, with 23b as the starting material, yielding a white solid at 43.62%.
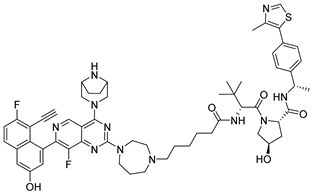
3.2.43. (2S,4R)-1-((S)-2-(6-(4-(4-((1R,5S)-3,8-Diazabicyclo[3.2.1]octan-3-yl)-7-(8-ethynyl-7-fluoro-3-hydroxynaphthalen-1-yl)-8-fluoropyrido[4,3-d]pyrimidin-2-yl)-1,4-diazepan-1-yl)hexanamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)pyrrolidine-2-carboxamide (26b)
Compound 26b was synthesized following the procedure described for compound 10k, using 24b as the starting material, yielding a white solid at 34.65%. Mp 116.3–118.7 °C. ESI-HRMS m/z calculated for C59H70F2N11O5S+ 1082.5245 [M + H]+, found 1082.5252 [M + H]+.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}