

Doxorubicin-Induced Cardiotoxicity and the Emerging Role of SGLT2 Inhibitors: From Glycemic Control to Cardio-Oncology

 ,
,  ,
,  ,
,  , and
, and 
Abstract
1. Introduction
2. Methodology and Study Selection
3. Doxorubicin-Induced Cardiovascular Toxicity: Overview of Mechanisms and Challenges
4. Risk Assessment and Surveillance of Anthracycline Cardiotoxicity
4.1. Risk Stratification Before Anthracycline Treatment
4.2. Cancer Therapy–Related Cardiac Dysfunction Definition
4.3. Ongoing Surveillance During Anthracycline Treatment
5. Doxorubicin-Induced Cardiovascular Toxicity: Current and Future Treatments
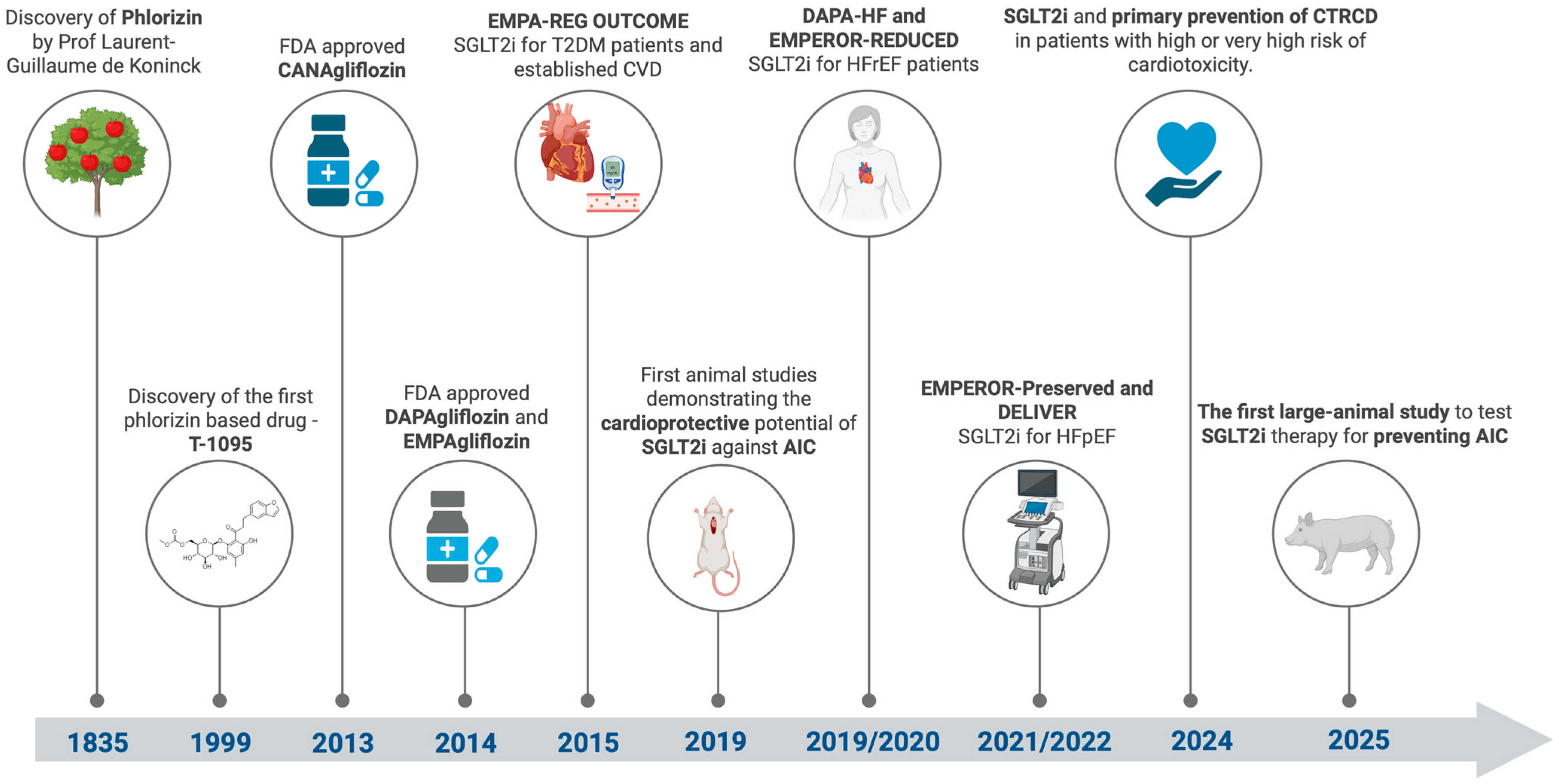
6. SGLT2 Inhibitors: A Journey from Discovery to Cardio-Oncology
7. Preventing Anthracycline Cardiotoxicity Using SGLT2 Inhibitors in Preclinical Models
8. Preventing Anthracycline Cardiotoxicity Using SGLT2 Inhibitors in Clinical Models
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
References
- Anand, U.; Dey, A.; Chandel, A.K.S.; Sanyal, R.; Mishra, A.; Pandey, D.K.; De Falco, V.; Upadhyay, A.; Kandimalla, R.; Chaudhary, A.; et al. Cancer Chemotherapy and beyond: Current Status, Drug Candidates, Associated Risks and Progress in Targeted Therapeutics. Genes Dis. 2023, 10, 1367–1401. [Google Scholar] [CrossRef] [PubMed]
- Delou, J.M.A.; Souza, A.S.O.; Souza, L.C.M.; Borges, H.L. Highlights in Resistance Mechanism Pathways for Combination Therapy. Cells 2019, 8, 1013. [Google Scholar] [CrossRef] [PubMed]
- Christakis, P. The Birth of Chemotherapy at Yale. Bicentennial Lecture Series: Surgery Grand Round. Yale J. Biol. Med. 2011, 84, 169–172. [Google Scholar] [PubMed]
- Sritharan, S.; Sivalingam, N. A Comprehensive Review on Time-Tested Anticancer Drug Doxorubicin. Life Sci. 2021, 278, 119527. [Google Scholar] [CrossRef]
- Linschoten, M.; Kamphuis, J.A.M.; Van Rhenen, A.; Bosman, L.P.; Cramer, M.J.; Doevendans, P.A.; Teske, A.J.; Asselbergs, F.W. Cardiovascular Adverse Events in Patients with Non-Hodgkin Lymphoma Treated with First-Line Cyclophosphamide, Doxorubicin, Vincristine, and Prednisone (CHOP) or CHOP with Rituximab (R-CHOP): A Systematic Review and Meta-Analysis. Lancet Haematol. 2020, 7, e295–e308. [Google Scholar] [CrossRef]
- Schmittlutz, K.; Marks, R. Current Treatment Options for Aggressive Non-Hodgkin Lymphoma in Elderly and Frail Patients: Practical Considerations for the Hematologist. Ther. Adv. Hematol. 2021, 12, 2040620721996484. [Google Scholar] [CrossRef]
- Chatterjee, K.; Zhang, J.; Honbo, N.; Karliner, J.S. Doxorubicin Cardiomyopathy. Cardiology 2010, 115, 155–162. [Google Scholar] [CrossRef]
- Cardinale, D.; Iacopo, F.; Cipolla, C.M. Cardiotoxicity of Anthracyclines. Front. Cardiovasc. Med. 2020, 7, 26. [Google Scholar] [CrossRef]
- Cardinale, D.; Biasillo, G.; Cipolla, C.M. Curing Cancer, Saving the Heart: A Challenge That Cardioncology Should Not Miss. Curr. Cardiol. Rep. 2016, 18, 51. [Google Scholar] [CrossRef]
- Lyon, A.R.; López-Fernández, T.; Couch, L.S.; Asteggiano, R.; Aznar, M.C.; Bergler-Klein, J.; Boriani, G.; Cardinale, D.; Cordoba, R.; Cosyns, B.; et al. 2022 ESC Guidelines on Cardio-Oncology Developed in Collaboration with the European Hematology Association (EHA), the European Society for Therapeutic Radiology and Oncology (ESTRO) and the International Cardio-Oncology Society (IC-OS). Eur. Heart J. 2022, 43, 4229–4361. [Google Scholar] [CrossRef]
- Albini, A.; Pennesi, G.; Donatelli, F.; Cammarota, R.; De Flora, S.; Noonan, D.M. Cardiotoxicity of Anticancer Drugs: The Need for Cardio-Oncology and Cardio-Oncological Prevention. JNCI J. Natl. Cancer Inst. 2010, 102, 14–25. [Google Scholar] [CrossRef]
- Vafa, R.G.; Sabahizadeh, A.; Mofarrah, R. Guarding the Heart: How SGLT-2 Inhibitors Protect against Chemotherapy-Induced Cardiotoxicity. Curr. Probl. Cardiol. 2024, 49, 102350. [Google Scholar] [CrossRef] [PubMed]
- Vallon, V.; Thomson, S.C. Targeting Renal Glucose Reabsorption to Treat Hyperglycaemia: The Pleiotropic Effects of SGLT2 Inhibition. Diabetologia 2017, 60, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Osataphan, N.; Abdel-Qadir, H.; Zebrowska, A.M.; Borowiec, A. Sodium-Glucose Cotransporter 2 Inhibitors During Cancer Therapy: Benefits, Risks, and Ongoing Clinical Trials. Curr. Oncol. Rep. 2024, 26, 1188–1196. [Google Scholar] [CrossRef] [PubMed]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. 2023 Focused Update of the 2021 ESC Guidelines for the Diagnosis and Treatment of Acute and Chronic Heart Failure. Eur. Heart J. 2023, 44, 3627–3639. [Google Scholar] [CrossRef]
- McMurray, J.J.V.; Solomon, S.D.; Inzucchi, S.E.; Køber, L.; Kosiborod, M.N.; Martinez, F.A.; Ponikowski, P.; Sabatine, M.S.; Anand, I.S.; Bělohlávek, J.; et al. Dapagliflozin in Patients with Heart Failure and Reduced Ejection Fraction. N. Engl. J. Med. 2019, 381, 1995–2008. [Google Scholar] [CrossRef]
- Packer, M.; Anker, S.D.; Butler, J.; Filippatos, G.; Pocock, S.J.; Carson, P.; Januzzi, J.; Verma, S.; Tsutsui, H.; Brueckmann, M.; et al. Cardiovascular and Renal Outcomes with Empagliflozin in Heart Failure. N. Engl. J. Med. 2020, 383, 1413–1424. [Google Scholar] [CrossRef]
- Pabel, S.; Hamdani, N.; Luedde, M.; Sossalla, S. SGLT2 Inhibitors and Their Mode of Action in Heart Failure—Has the Mystery Been Unravelled? Curr. Heart Fail. Rep. 2021, 18, 315–328. [Google Scholar] [CrossRef]
- Oh, C.-M.; Cho, S.; Jang, J.-Y.; Kim, H.; Chun, S.; Choi, M.; Park, S.; Ko, Y.-G. Cardioprotective Potential of an SGLT2 Inhibitor Against Doxorubicin-Induced Heart Failure. Korean Circ. J. 2019, 49, 1183. [Google Scholar] [CrossRef]
- Gongora, C.A.; Drobni, Z.D.; Quinaglia Araujo Costa Silva, T.; Zafar, A.; Gong, J.; Zlotoff, D.A.; Gilman, H.K.; Hartmann, S.E.; Sama, S.; Nikolaidou, S.; et al. Sodium-Glucose Co-Transporter-2 Inhibitors and Cardiac Outcomes Among Patients Treated With Anthracyclines. JACC Heart Fail. 2022, 10, 559–567. [Google Scholar] [CrossRef]
- Quagliariello, V.; De Laurentiis, M.; Rea, D.; Barbieri, A.; Monti, M.G.; Carbone, A.; Paccone, A.; Altucci, L.; Conte, M.; Canale, M.L.; et al. The SGLT-2 Inhibitor Empagliflozin Improves Myocardial Strain, Reduces Cardiac Fibrosis and pro-Inflammatory Cytokines in Non-Diabetic Mice Treated with Doxorubicin. Cardiovasc. Diabetol. 2021, 20, 150. [Google Scholar] [CrossRef] [PubMed]
- Hendryx, M.; Dong, Y.; Ndeke, J.M.; Luo, J. Sodium-Glucose Cotransporter 2 (SGLT2) Inhibitor Initiation and Hepatocellular Carcinoma Prognosis. PLoS ONE 2022, 17, e0274519. [Google Scholar] [CrossRef] [PubMed]
- Basak, D.; Gamez, D.; Deb, S. SGLT2 Inhibitors as Potential Anticancer Agents. Biomedicines 2023, 11, 1867. [Google Scholar] [CrossRef]
- Songbo, M.; Lang, H.; Xinyong, C.; Bin, X.; Ping, Z.; Liang, S. Oxidative Stress Injury in Doxorubicin-Induced Cardiotoxicity. Toxicol. Lett. 2019, 307, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Schirone, L.; D’Ambrosio, L.; Forte, M.; Genovese, R.; Schiavon, S.; Spinosa, G.; Iacovone, G.; Valenti, V.; Frati, G.; Sciarretta, S. Mitochondria and Doxorubicin-Induced Cardiomyopathy: A Complex Interplay. Cells 2022, 11, 2000. [Google Scholar] [CrossRef]
- Lyu, Y.L.; Kerrigan, J.E.; Lin, C.-P.; Azarova, A.M.; Tsai, Y.-C.; Ban, Y.; Liu, L.F. Topoisomerase IIβ–Mediated DNA Double-Strand Breaks: Implications in Doxorubicin Cardiotoxicity and Prevention by Dexrazoxane. Cancer Res. 2007, 67, 8839–8846. [Google Scholar] [CrossRef]
- Shinlapawittayatorn, K.; Chattipakorn, S.C.; Chattipakorn, N. The Effects of Doxorubicin on Cardiac Calcium Homeostasis and Contractile Function. J. Cardiol. 2022, 80, 125–132. [Google Scholar] [CrossRef]
- Sun, M.; Zhang, X.; Tan, B.; Zhang, Q.; Zhao, X.; Dong, D. Potential Role of Endoplasmic Reticulum Stress in Doxorubicin-Induced Cardiotoxicity-an Update. Front. Pharmacol. 2024, 15, 1415108. [Google Scholar] [CrossRef]
- Christidi, E.; Brunham, L.R. Regulated Cell Death Pathways in Doxorubicin-Induced Cardiotoxicity. Cell Death Dis. 2021, 12, 339. [Google Scholar] [CrossRef]
- Tadokoro, T.; Ikeda, M.; Ide, T.; Deguchi, H.; Ikeda, S.; Okabe, K.; Ishikita, A.; Matsushima, S.; Koumura, T.; Yamada, K.; et al. Mitochondria-Dependent Ferroptosis Plays a Pivotal Role in Doxorubicin Cardiotoxicity. JCI Insight. 2023, 8, e169756. [Google Scholar] [CrossRef]
- Maayah, Z.H.; Takahara, S.; Dyck, J.R.B. The Beneficial Effects of Reducing NLRP3 Inflammasome Activation in the Cardiotoxicity and the Anti-Cancer Effects of Doxorubicin. Arch. Toxicol. 2021, 95, 1–9. [Google Scholar] [CrossRef]
- Hutchins, E.; Yang, E.H.; Stein-Merlob, A.F. Inflammation in Chemotherapy-Induced Cardiotoxicity. Curr. Cardiol. Rep. 2024, 26, 1329–1340. [Google Scholar] [CrossRef] [PubMed]
- Koleini, N.; Kardami, E. Autophagy and Mitophagy in the Context of Doxorubicin-Induced Cardiotoxicity. Oncotarget 2017, 8, 46663–46680. [Google Scholar] [CrossRef] [PubMed]
- Kong, C.-Y.; Guo, Z.; Song, P.; Zhang, X.; Yuan, Y.-P.; Teng, T.; Yan, L.; Tang, Q.-Z. Underlying the Mechanisms of Doxorubicin-Induced Acute Cardiotoxicity: Oxidative Stress and Cell Death. Int. J. Biol. Sci. 2022, 18, 760–770. [Google Scholar] [CrossRef]
- Cappetta, D.; De Angelis, A.; Sapio, L.; Prezioso, L.; Illiano, M.; Quaini, F.; Rossi, F.; Berrino, L.; Naviglio, S.; Urbanek, K. Oxidative Stress and Cellular Response to Doxorubicin: A Common Factor in the Complex Milieu of Anthracycline Cardiotoxicity. Oxidative Med. Cell. Longev. 2017, 2017, 1521020. [Google Scholar] [CrossRef]
- Štěrba, M.; Popelová, O.; Vávrová, A.; Jirkovský, E.; Kovaříková, P.; Geršl, V.; Šimůnek, T. Oxidative Stress, Redox Signaling, and Metal Chelation in Anthracycline Cardiotoxicity and Pharmacological Cardioprotection. Antioxid. Redox Signal. 2013, 18, 899–929. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Fang, L.; Li, H.; Li, Z.; Lyu, L.; Wang, H.; Xiao, J. Astragaloside IV Alleviates Doxorubicin Induced Cardiomyopathy by Inhibiting NADPH Oxidase Derived Oxidative Stress. Eur. J. Pharmacol. 2019, 859, 172490. [Google Scholar] [CrossRef]
- Vitale, R.; Marzocco, S.; Popolo, A. Role of Oxidative Stress and Inflammation in Doxorubicin-Induced Cardiotoxicity: A Brief Account. Int. J. Mol. Sci. 2024, 25, 7477. [Google Scholar] [CrossRef]
- Avagimyan, A.; Pogosova, N.; Kakturskiy, L.; Sheibani, M.; Challa, A.; Kogan, E.; Fogacci, F.; Mikhaleva, L.; Vandysheva, R.; Yakubovskaya, M.; et al. Doxorubicin-Related Cardiotoxicity: Review of Fundamental Pathways of Cardiovascular System Injury. Cardiovasc. Pathol. 2024, 73, 107683. [Google Scholar] [CrossRef]
- Pohjoismäki, J.L.; Goffart, S. The Role of Mitochondria in Cardiac Development and Protection. Free. Radic. Biol. Med. 2017, 106, 345–354. [Google Scholar] [CrossRef]
- Robichaux, D.J.; Harata, M.; Murphy, E.; Karch, J. Mitochondrial Permeability Transition Pore-Dependent Necrosis. J. Mol. Cell. Cardiol. 2023, 174, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, P.; Gerle, C.; Halestrap, A.P.; Jonas, E.A.; Karch, J.; Mnatsakanyan, N.; Pavlov, E.; Sheu, S.-S.; Soukas, A.A. Identity, Structure, and Function of the Mitochondrial Permeability Transition Pore: Controversies, Consensus, Recent Advances, and Future Directions. Cell Death Differ. 2023, 30, 1869–1885. [Google Scholar] [CrossRef]
- Kwong, J.Q.; Molkentin, J.D. Physiological and Pathological Roles of the Mitochondrial Permeability Transition Pore in the Heart. Cell Metab. 2015, 21, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Nishi, M.; Wang, P.; Hwang, P.M. Cardiotoxicity of Cancer Treatments: Focus on Anthracycline Cardiomyopathy. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 2648–2660. [Google Scholar] [CrossRef]
- Shandilya, M.; Sharma, S.; Prasad Das, P.; Charak, S. Molecular-Level Understanding of the Anticancer Action Mechanism of Anthracyclines. In Advances in Precision Medicine Oncology; Arnouk, H., Abdul Rasool Hassan, B., Eds.; IntechOpen: London, UK, 2021. [Google Scholar] [CrossRef]
- Goffart, S.; Hangas, A.; Pohjoismäki, J.L.O. Twist and Turn—Topoisomerase Functions in Mitochondrial DNA Maintenance. Int. J. Mol. Sci. 2019, 20, 2041. [Google Scholar] [CrossRef]
- Gyöngyösi, M.; Lukovic, D.; Zlabinger, K.; Spannbauer, A.; Gugerell, A.; Pavo, N.; Traxler, D.; Pils, D.; Maurer, G.; Jakab, A.; et al. Liposomal Doxorubicin Attenuates Cardiotoxicity via Induction of Interferon-Related DNA Damage Resistance. Cardiovasc. Res. 2019, 116, 970–982. [Google Scholar] [CrossRef] [PubMed]
- Frank, N.E.; Cusack, B.J.; Talley, T.T.; Walsh, G.M.; Olson, R.D. Comparative Effects of Doxorubicin and a Doxorubicin Analog, 13-Deoxy, 5-Iminodoxorubicin (GPX-150), on Human Topoisomerase IIβ Activity and Cardiac Function in a Chronic Rabbit Model. Investig. New Drugs 2016, 34, 693–700. [Google Scholar] [CrossRef]
- Petrie, M.; Borlaug, B.; Buchholtz, K.; Ducharme, A.; Hvelplund, A.; Ping, C.L.S.; Mendieta, G.; Hardt-Lindberg, S.Ø.; Voors, A.A.; Ridker, P.M. HERMES: Effects Of Ziltivekimab Versus Placebo On Morbidity And Mortality In Patients With Heart Failure With Mildly Reduced Or Preserved Ejection Fraction And Systemic Inflammation. J. Card. Fail. 2024, 30, 126. [Google Scholar] [CrossRef]
- Sun, Z.; Fang, C.; Xu, S.; Wang, B.; Li, D.; Liu, X.; Mi, Y.; Guo, H.; Jiang, J. SIRT3 Attenuates Doxorubicin-Induced Cardiotoxicity by Inhibiting NLRP3 Inflammasome via Autophagy. Biochem. Pharmacol. 2023, 207, 115354. [Google Scholar] [CrossRef]
- Bloom, M.W.; Vo, J.B.; Rodgers, J.E.; Ferrari, A.M.; Nohria, A.; Deswal, A.; Cheng, R.K.; Kittleson, M.M.; Upshaw, J.N.; Palaskas, N.; et al. Cardio-Oncology and Heart Failure: A Scientific Statement from the Heart Failure Society of America. J. Card. Fail. 2024, 31, 415–455. [Google Scholar] [CrossRef]
- Xie, S.; Sun, Y.; Zhao, X.; Xiao, Y.; Zhou, F.; Lin, L.; Wang, W.; Lin, B.; Wang, Z.; Fang, Z.; et al. An Update of the Molecular Mechanisms Underlying Anthracycline Induced Cardiotoxicity. Front. Pharmacol. 2024, 15, 1406247. [Google Scholar] [CrossRef] [PubMed]
- Hershman, D.L.; Eisenberger, A.; Wang, J.; Jacobson, J.; Grann, V.; McBride, R.; Tsai, W.; Neugut, A. Doxorubicin, Cardiac Risk Factors and Cardiac Toxicity in Elderly Patients with Diffuse b-Cell Non-Hodgkin’s Lymphoma. J. Clin. Oncol. 2007, 25 (Suppl. S18). [Google Scholar] [CrossRef]
- Qin, A.; Thompson, C.L.; Silverman, P. Predictors of Late-Onset Heart Failure in Breast Cancer Patients Treated with Doxorubicin. J. Cancer Surviv. 2015, 9, 252–259. [Google Scholar] [CrossRef]
- Salz, T.; Zabor, E.C.; De Nully Brown, P.; Dalton, S.O.; Raghunathan, N.J.; Matasar, M.J.; Steingart, R.; Vickers, A.J.; Svenssen Munksgaard, P.; Oeffinger, K.C.; et al. Preexisting Cardiovascular Risk and Subsequent Heart Failure Among Non-Hodgkin Lymphoma Survivors. J. Clin. Oncol. 2017, 35, 3837–3843. [Google Scholar] [CrossRef] [PubMed]
- Swain, S.M.; Whaley, F.S.; Ewer, M.S. Congestive Heart Failure in Patients Treated with Doxorubicin: A Retrospective Analysis of Three Trials. Cancer 2003, 97, 2869–2879. [Google Scholar] [CrossRef]
- Von Hoff, D.D.; Layard, M.W.; Basa, P.; Davis, H.L.; Von Hoff, A.L.; Rozencweig, M.; Muggia, F.M. Risk Factors for Doxorubicin-Lnduced Congestive Heart Failure. Ann. Intern. Med. 1979, 91, 710–717. [Google Scholar] [CrossRef]
- Russell, S.D.; Blackwell, K.L.; Lawrence, J.; Pippen, J.E.; Roe, M.T.; Wood, F.; Paton, V.; Holmgren, E.; Mahaffey, K.W. Independent Adjudication of Symptomatic Heart Failure With the Use of Doxorubicin and Cyclophosphamide Followed by Trastuzumab Adjuvant Therapy: A Combined Review of Cardiac Data from the National Surgical Adjuvant Breast and Bowel Project B-31 and the North Central Cancer Treatment Group N9831 Clinical Trials. J. Clin. Oncol. 2010, 28, 3416–3421. [Google Scholar] [CrossRef]
- Myrehaug, S.; Pintilie, M.; Yun, L.; Crump, M.; Tsang, R.W.; Meyer, R.M.; Sussman, J.; Yu, E.; Hodgson, D.C. A Population-Based Study of Cardiac Morbidity among Hodgkin Lymphoma Patients with Preexisting Heart Disease. Blood 2010, 116, 2237–2240. [Google Scholar] [CrossRef]
- Montalvo, R.N.; Doerr, V.; Nguyen, B.L.; Kelley, R.C.; Smuder, A.J. Consideration of Sex as a Biological Variable in the Development of Doxorubicin Myotoxicity and the Efficacy of Exercise as a Therapeutic Intervention. Antioxidants 2021, 10, 343. [Google Scholar] [CrossRef]
- Podyacheva, E.Y.; Kushnareva, E.A.; Karpov, A.A.; Toropova, Y.G. Analysis of Models of Doxorubicin-Induced Cardiomyopathy in Rats and Mice. A Modern View from the Perspective of the Pathophysiologist and the Clinician. Front. Pharmacol. 2021, 12, 670479. [Google Scholar] [CrossRef]
- Moulin, M.; Piquereau, J.; Mateo, P.; Fortin, D.; Rucker-Martin, C.; Gressette, M.; Lefebvre, F.; Gresikova, M.; Solgadi, A.; Veksler, V.; et al. Sexual Dimorphism of Doxorubicin-Mediated Cardiotoxicity: Potential Role of Energy Metabolism Remodeling. Circ. Heart Fail. 2015, 8, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Lipshultz, S.E.; Scully, R.E.; Lipsitz, S.R.; Sallan, S.E.; Silverman, L.B.; Miller, T.L.; Barry, E.V.; Asselin, B.L.; Athale, U.; Clavell, L.A.; et al. Assessment of Dexrazoxane as a Cardioprotectant in Doxorubicin-Treated Children with High-Risk Acute Lymphoblastic Leukaemia: Long-Term Follow-up of a Prospective, Randomised, Multicentre Trial. Lancet Oncol. 2010, 11, 950–961. [Google Scholar] [CrossRef]
- Armstrong, G.T.; Chen, Y.; Yasui, Y.; Leisenring, W.; Gibson, T.M.; Mertens, A.C.; Stovall, M.; Oeffinger, K.C.; Bhatia, S.; Krull, K.R.; et al. Reduction in Late Mortality among 5-Year Survivors of Childhood Cancer. N. Engl. J. Med. 2016, 374, 833–842. [Google Scholar] [CrossRef]
- Biro, F.M.; Pinney, S.M.; Huang, B.; Baker, E.R.; Walt Chandler, D.; Dorn, L.D. Hormone Changes in Peripubertal Girls. J. Clin. Endocrinol. Metab. 2014, 99, 3829–3835. [Google Scholar] [CrossRef] [PubMed]
- Magdy, T.; Burridge, P.W. Use of Hipsc to Explicate Genomic Predisposition to AnthrAcycline-Induced Cardiotoxicity. Pharmacogenomics 2021, 22, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Fonoudi, H.; Jouni, M.; Cejas, R.B.; Magdy, T.; Blancard, M.; Ge, N.; Shah, D.A.; Lyra-Leite, D.M.; Neupane, A.; Gharib, M.; et al. Functional Validation of Doxorubicin-Induced Cardiotoxicity-Related Genes. JACC CardioOncol. 2024, 6, 38–50. [Google Scholar] [CrossRef]
- Curigliano, G.; Lenihan, D.; Fradley, M.; Ganatra, S.; Barac, A.; Blaes, A.; Herrmann, J.; Porter, C.; Lyon, A.R.; Lancellotti, P.; et al. Management of Cardiac Disease in Cancer Patients throughout Oncological Treatment: ESMO Consensus Recommendations. Ann. Oncol. 2020, 31, 171–190. [Google Scholar] [CrossRef]
- Lyon, A.R.; Dent, S.; Stanway, S.; Earl, H.; Brezden-Masley, C.; Cohen-Solal, A.; Tocchetti, C.G.; Moslehi, J.J.; Groarke, J.D.; Bergler-Klein, J.; et al. Baseline Cardiovascular Risk Assessment in Cancer Patients Scheduled to Receive Cardiotoxic Cancer Therapies: A Position Statement and New Risk Assessment Tools from the Cardio-Oncology Study Group of the Heart Failure Association of the European Society of Cardiology in Collaboration with the International Cardio-Oncology Society. Eur. J. Heart Fail. 2020, 22, 1945–1960. [Google Scholar] [CrossRef]
- Pareek, N.; Cevallos, J.; Moliner, P.; Shah, M.; Tan, L.L.; Chambers, V.; Baksi, A.J.; Khattar, R.S.; Sharma, R.; Rosen, S.D.; et al. Activity and Outcomes of a Cardio-oncology Service in the United Kingdom—A Five-year Experience. Eur. J. Heart Fail. 2018, 20, 1721–1731. [Google Scholar] [CrossRef]
- Raisi-Estabragh, Z.; Murphy, A.C.; Ramalingam, S.; Scherrer-Crosbie, M.; Lopez-Fernandez, T.; Reynolds, K.L.; Aznar, M.; Lin, A.E.; Libby, P.; Cordoba, R.; et al. Cardiovascular Considerations Before Cancer Therapy. JACC CardioOncol. 2024, 6, 631–654. [Google Scholar] [CrossRef]
- Porta-Sánchez, A.; Gilbert, C.; Spears, D.; Amir, E.; Chan, J.; Nanthakumar, K.; Thavendiranathan, P. Incidence, Diagnosis, and Management of QT Prolongation Induced by Cancer Therapies: A Systematic Review. J. Am. Heart Assoc. 2017, 6, e007724. [Google Scholar] [CrossRef] [PubMed]
- Mazur, M.; Wang, F.; Hodge, D.O.; Siontis, B.L.; Beinborn, D.S.; Villarraga, H.R.; Lerman, A.; Friedman, P.A.; Herrmann, J. Burden of Cardiac Arrhythmias in Patients with Anthracycline-Related Cardiomyopathy. JACC Clin. Electrophysiol. 2017, 3, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Michel, L.; Mincu, R.I.; Mahabadi, A.A.; Settelmeier, S.; Al-Rashid, F.; Rassaf, T.; Totzeck, M. Troponins and Brain Natriuretic Peptides for the Prediction of Cardiotoxicity in Cancer Patients: A Meta-analysis. Eur. J. Heart Fail. 2020, 22, 350–361. [Google Scholar] [CrossRef]
- Pudil, R.; Mueller, C.; Čelutkienė, J.; Henriksen, P.A.; Lenihan, D.; Dent, S.; Barac, A.; Stanway, S.; Moslehi, J.; Suter, T.M.; et al. Role of Serum Biomarkers in Cancer Patients Receiving Cardiotoxic Cancer Therapies: A Position Statement from the Cardio-Oncology Study Group of the Heart Failure Association and the Cardio-Oncology Council of the European Society of Cardiology. Eur. J. Heart Fail. 2020, 22, 1966–1983. [Google Scholar] [CrossRef]
- De Michieli, L.; Jaffe, A.S. Cancer Therapy–Related Cardiac Dysfunction. JACC CardioOncol. 2024, 6, 96–98. [Google Scholar] [CrossRef]
- Camilli, M.; Cipolla, C.M.; Dent, S.; Minotti, G.; Cardinale, D.M. Anthracycline Cardiotoxicity in Adult Cancer Patients. JACC CardioOncol. 2024, 6, 655–677. [Google Scholar] [CrossRef]
- Shafi, A.; Siddiqui, N.; Imtiaz, S.; Din Sajid, M.U. Left Ventricular Systolic Dysfunction Predicted By Early Troponin I Release After Anthracycline Based Chemotherapy In Breast Cancer Patients. J. Ayub Med. Coll. Abbottabad 2017, 29, 266–269. [Google Scholar]
- Bisoc, A.; Ciurescu, D.; Rădoi, M.; Tântu, M.M.; Rogozea, L.; Sweidan, A.J.; Bota, D.A. Elevations in High-Sensitive Cardiac Troponin T and N-Terminal Prohormone Brain Natriuretic Peptide Levels in the Serum Can Predict the Development of Anthracycline-Induced Cardiomyopathy. Am. J. Ther. 2020, 27, e142–e150. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Machino-Ohtsuka, T.; Nakazawa, Y.; Iida, N.; Sasamura, R.; Bando, H.; Chiba, S.; Tasaka, N.; Ishizu, T.; Murakoshi, N.; et al. Early Detection and Prediction of Anthracycline-Induced Cardiotoxicity―A Prospective Cohort Study. Circ. J. 2024, 88, 751–759. [Google Scholar] [CrossRef]
- Cardinale, D.; Sandri, M.T.; Martinoni, A.; Tricca LabTech, A.; Civelli, M.; Lamantia, G.; Cinieri, S.; Martinelli, G.; Cipolla, C.M.; Fiorentini, C. Left Ventricular Dysfunction Predicted by Early Troponin I Release after High-Dose Chemotherapy. J. Am. Coll. Cardiol. 2000, 36, 517–522. [Google Scholar] [CrossRef]
- Pongprot, Y.; Sittiwangkul, R.; Charoenkwan, P.; Silvilairat, S. Use of Cardiac Markers for Monitoring of Doxorubixin-Induced Cardiotoxicity in Children with Cancer. J. Pediatr. Hematol./Oncol. 2012, 34, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Balážová, K.; Kubincová, D. Current Possibilities of Early Detection of Cardiotoxicity of Cytostatic Treatment. Klin. Onkol. 2020, 33, 208–213. [Google Scholar] [PubMed]
- Muckiene, G.; Vaitiekus, D.; Zaliaduonyte, D.; Zabiela, V.; Verseckaite-Costa, R.; Vaiciuliene, D.; Juozaityte, E.; Jurkevicius, R. Prognostic Impact of Global Longitudinal Strain and NT-proBNP on Early Development of Cardiotoxicity in Breast Cancer Patients Treated with Anthracycline-Based Chemotherapy. Medicina 2023, 59, 953. [Google Scholar] [CrossRef]
- Prasad, K.; Yadav, B.S.; Zohmangaihi, D.; Ballari, N.; Mehrotra, S. Evaluation of Anthracycline-Induced Cardiotoxicity Using Cardiac Biomarkers: A Prospective Study. Indian J. Cancer 2024, 10-4103. [Google Scholar] [CrossRef] [PubMed]
- Seropian, I.M.; Fontana Estevez, F.S.; Villaverde, A.; Cacciagiú, L.; Bustos, R.; Touceda, V.; Penas, F.; Selser, C.; Morales, C.; Miksztowicz, V.; et al. Galectin-3 Contributes to Acute Cardiac Dysfunction and Toxicity by Increasing Oxidative Stress and Fibrosis in Doxorubicin-Treated Mice. Int. J. Cardiol. 2023, 393, 131386. [Google Scholar] [CrossRef]
- Ky, B.; Putt, M.; Sawaya, H.; French, B.; Januzzi, J.L.; Sebag, I.A.; Plana, J.C.; Cohen, V.; Banchs, J.; Carver, J.R.; et al. Early Increases in Multiple Biomarkers Predict Subsequent Cardiotoxicity in Patients With Breast Cancer Treated with Doxorubicin, Taxanes, and Trastuzumab. J. Am. Coll. Cardiol. 2014, 63, 809–816. [Google Scholar] [CrossRef]
- Dean, M.; Kim, M.J.; Dimauro, S.; Tannenbaum, S.; Graham, G.; Liang, B.T.; Kim, A.S. Cardiac and Noncardiac Biomarkers in Patients Undergoing Anthracycline Chemotherapy—A Prospective Analysis. Cardio-Oncology 2023, 9, 23. [Google Scholar] [CrossRef]
- Cheung, Y.; Li, V.W.; Lai, C.T.; Shin, V.Y.; Keung, W.; Cheuk, D.K.; Kwong, A.; Li, R.A.; Chan, G.C. Circulating High-Sensitivity Troponin T and microRNAs as Markers of Myocardial Damage during Childhood Leukaemia Treatment. Pediatr. Res. 2021, 89, 1245–1252. [Google Scholar] [CrossRef]
- Nguyen, N.; Souza, T.; Kleinjans, J.; Jennen, D. Transcriptome Analysis of Long Noncoding RNAs Reveals Their Potential Roles in Anthracycline-Induced Cardiotoxicity. Non-Coding RNA Res. 2022, 7, 106–113. [Google Scholar] [CrossRef]
- Desai, V.G.; Vijay, V.; Lee, T.; Han, T.; Moland, C.L.; Phanavanh, B.; Herman, E.H.; Stine, K.; Fuscoe, J.C. MicroRNA-34a-5p as a Promising Early Circulating Preclinical Biomarker of Doxorubicin-induced Chronic Cardiotoxicity. J. Appl. Toxicol. 2022, 42, 1477–1490. [Google Scholar] [CrossRef]
- Murtagh, G.; Januzzi, J.L.; Scherrer-Crosbie, M.; Neilan, T.G.; Dent, S.; Ho, J.E.; Appadurai, V.; McDermott, R.; Akhter, N. Circulating Cardiovascular Biomarkers in Cancer Therapeutics-Related Cardiotoxicity: Review of Critical Challenges, Solutions, and Future Directions. J. Am. Heart Assoc. 2023, 12, e029574. [Google Scholar] [CrossRef]
- Čelutkienė, J.; Pudil, R.; López-Fernández, T.; Grapsa, J.; Nihoyannopoulos, P.; Bergler-Klein, J.; Cohen-Solal, A.; Farmakis, D.; Tocchetti, C.G.; Von Haehling, S.; et al. Role of Cardiovascular Imaging in Cancer Patients Receiving Cardiotoxic Therapies: A Position Statement on Behalf of the Heart Failure Association (HFA), the European Association of Cardiovascular Imaging (EACVI) and the Cardio-Oncology Council of the European Society of Cardiology (ESC). Eur. J. Heart Fail. 2020, 22, 1504–1524. [Google Scholar] [CrossRef]
- Tan, T.C.; Scherrer-Crosbie, M. Assessing the Cardiac Toxicity of Chemotherapeutic Agents: Role of Echocardiography. Curr. Cardiovasc. Imaging Rep. 2012, 5, 403–409. [Google Scholar] [CrossRef]
- Araujo-Gutierrez, R.; Chitturi, K.R.; Xu, J.; Wang, Y.; Kinder, E.; Senapati, A.; Chebrolu, L.B.; Kassi, M.; Trachtenberg, B.H. Baseline Global Longitudinal Strain Predictive of Anthracycline-Induced Cardiotoxicity. Cardio-Oncology 2021, 7, 4. [Google Scholar] [CrossRef]
- Upshaw, J.N.; Finkelman, B.; Hubbard, R.A.; Smith, A.M.; Narayan, H.K.; Arndt, L.; Domchek, S.; DeMichele, A.; Fox, K.; Shah, P.; et al. Comprehensive Assessment of Changes in Left Ventricular Diastolic Function with Contemporary Breast Cancer Therapy. JACC Cardiovasc. Imaging 2020, 13, 198–210. [Google Scholar] [CrossRef]
- Saunderson, C.E.D.; Plein, S.; Manisty, C.H. Role of Cardiovascular Magnetic Resonance Imaging in Cardio-Oncology. Eur. Heart J. Cardiovasc. Imaging 2021, 22, 383–396. [Google Scholar] [CrossRef]
- Baldassarre, L.A.; Ganatra, S.; Lopez-Mattei, J.; Yang, E.H.; Zaha, V.G.; Wong, T.C.; Ayoub, C.; DeCara, J.M.; Dent, S.; Deswal, A.; et al. Advances in Multimodality Imaging in Cardio-Oncology. J. Am. Coll. Cardiol. 2022, 80, 1560–1578. [Google Scholar] [CrossRef]
- Mecinaj, A.; Gulati, G.; Ree, A.H.; Gravdehaug, B.; Røsjø, H.; Steine, K.; Wisløff, T.; Geisler, J.; Omland, T.; Heck, S.L. Impact of the ESC Cardio-Oncology Guidelines Biomarker Criteria on Incidence of Cancer Therapy–Related Cardiac Dysfunction. JACC CardioOncol. 2024, 6, 83–95. [Google Scholar] [CrossRef]
- Mabudian, L.; Jordan, J.H.; Bottinor, W.; Hundley, W.G. Cardiac MRI Assessment of Anthracycline-Induced Cardiotoxicity. Front. Cardiovasc. Med. 2022, 9, 903719. [Google Scholar] [CrossRef] [PubMed]
- Burrage, M.K.; Ferreira, V.M. The Use of Cardiovascular Magnetic Resonance as an Early Non-Invasive Biomarker for Cardiotoxicity in Cardio-Oncology. Cardiovasc. Diagn. Ther. 2020, 10, 610–624. [Google Scholar] [CrossRef] [PubMed]
- Macedo, A.V.S.; Hajjar, L.A.; Lyon, A.R.; Nascimento, B.R.; Putzu, A.; Rossi, L.; Costa, R.B.; Landoni, G.; Nogueira-Rodrigues, A.; Ribeiro, A.L.P. Efficacy of Dexrazoxane in Preventing Anthracycline Cardiotoxicity in Breast Cancer. JACC CardioOncol. 2019, 1, 68–79. [Google Scholar] [CrossRef]
- De Baat, E.C.; Mulder, R.L.; Armenian, S.; Feijen, E.A.; Grotenhuis, H.; Hudson, M.M.; Mavinkurve-Groothuis, A.M.; Kremer, L.C.; Van Dalen, E.C. Dexrazoxane for Preventing or Reducing Cardiotoxicity in Adults and Children with Cancer Receiving Anthracyclines. Cochrane Database Syst. Rev. 2022, 9, CD014638. [Google Scholar] [CrossRef]
- Varghese, S.S.; Eekhoudt, C.R.; Jassal, D.S. Mechanisms of Anthracycline-Mediated Cardiotoxicity and Preventative Strategies in Women with Breast Cancer. Mol. Cell. Biochem. 2021, 476, 3099–3109. [Google Scholar] [CrossRef]
- Zheng, H.; Kobrinsky, B.; Katz, S.; Speyer, J.L. Cardiac Effects of Cancer Therapy. In Abeloff’s Clinical Oncology; Elsevier: Amsterdam, The Netherlands, 2014; pp. 858–873.e4. [Google Scholar] [CrossRef]
- Chow, E.J.; Aggarwal, S.; Doody, D.R.; Aplenc, R.; Armenian, S.H.; Baker, K.S.; Bhatia, S.; Blythe, N.; Colan, S.D.; Constine, L.S. Dexrazoxane and Long-Term Heart Function in Survivors of Childhood Cancer. J. Clin. Oncol. 2023, 41, 2248–2257. [Google Scholar] [CrossRef]
- Krone, R.J.; Merchant, A.; Mitchell, J.D. Cardioprotection Using Doxorubicin: The Role of Dexrazoxane. In Pharmaceutical Science; Rauf, A., Ed.; IntechOpen: London, UK, 2024; Volume 5. [Google Scholar] [CrossRef]
- Tebbi, C.K.; London, W.B.; Friedman, D.; Villaluna, D.; De Alarcon, P.A.; Constine, L.S.; Mendenhall, N.P.; Sposto, R.; Chauvenet, A.; Schwartz, C.L. Dexrazoxane-Associated Risk for Acute Myeloid Leukemia/Myelodysplastic Syndrome and Other Secondary Malignancies in Pediatric Hodgkin’s Disease. J. Clin. Oncol. 2007, 25, 493–500. [Google Scholar] [CrossRef]
- Asselin, B.L.; Devidas, M.; Chen, L.; Franco, V.I.; Pullen, J.; Borowitz, M.J.; Hutchison, R.E.; Ravindranath, Y.; Armenian, S.H.; Camitta, B.M.; et al. Cardioprotection and Safety of Dexrazoxane in Patients Treated for Newly Diagnosed T-Cell Acute Lymphoblastic Leukemia or Advanced-Stage Lymphoblastic Non-Hodgkin Lymphoma: A Report of the Children’s Oncology Group Randomized Trial Pediatric Oncology Group 9404. J. Clin. Oncol. 2016, 34, 854–862. [Google Scholar] [CrossRef]
- Abdel-Qadir, H.; Bobrowski, D.; Zhou, L.; Austin, P.C.; Calvillo-Argüelles, O.; Amir, E.; Lee, D.S.; Thavendiranathan, P. Statin Exposure and Risk of Heart Failure After Anthracycline- or Trastuzumab-Based Chemotherapy for Early Breast Cancer: A Propensity Score-Matched Cohort Study. J. Am. Heart Assoc. 2021, 10, e018393. [Google Scholar] [CrossRef]
- Oh, J.; Lee, B.S.; Lim, G.; Lim, H.; Lee, C.J.; Park, S.; Lee, S.-H.; Chung, J.H.; Kang, S.-M. Atorvastatin Protects Cardiomyocyte from Doxorubicin Toxicity by Modulating Survivin Expression through FOXO1 Inhibition. J. Mol. Cell. Cardiol. 2020, 138, 244–255. [Google Scholar] [CrossRef]
- Kuşçu, G.C.; Gürel, Ç.; Buhur, A.; Karabay Yavaşoğlu, N.Ü.; Köse, T.; Yavaşoğlu, A.; Oltulu, F. Fluvastatin Alleviates Doxorubicin-Induced Cardiac and Renal Toxicity in Rats via Regulation of Oxidative Stress, Inflammation, and Apoptosis Associated Genes Expressions. Drug Chem. Toxicol. 2023, 46, 400–411. [Google Scholar] [CrossRef]
- Hundley, W.G.; D’Agostino, R., Jr.; Crotts, T.; Craver, K.; Hackney, M.H.; Jordan, J.H.; Ky, B.; Wagner, L.I.; Herrington, D.M.; Yeboah, J.; et al. Statins and left ventricular ejection fraction following doxorubicin treatment. NEJM Evid. 2022, 1, EVIDoa2200097. [Google Scholar] [CrossRef] [PubMed]
- Neilan, T.G.; Quinaglia, T.; Onoue, T.; Mahmood, S.S.; Drobni, Z.D.; Gilman, H.K.; Smith, A.; Heemelaar, J.C.; Brahmbhatt, P.; Ho, J.S.; et al. Atorvastatin for Anthracycline-Associated Cardiac Dysfunction: The STOP-CA Randomized Clinical Trial. JAMA 2023, 330, 528. [Google Scholar] [CrossRef]
- Qiu, Y.; Jiang, P.; Huang, Y. Anthracycline-Induced Cardiotoxicity: Mechanisms, Monitoring, and Prevention. Front. Cardiovasc. Med. 2023, 10, 1242596. [Google Scholar] [CrossRef]
- Heck, S.L.; Mecinaj, A.; Ree, A.H.; Hoffmann, P.; Schulz-Menger, J.; Fagerland, M.W.; Gravdehaug, B.; Røsjø, H.; Steine, K.; Geisler, J.; et al. Prevention of Cardiac Dysfunction During Adjuvant Breast Cancer Therapy (PRADA): Extended Follow-Up of a 2×2 Factorial, Randomized, Placebo-Controlled, Double-Blind Clinical Trial of Candesartan and Metoprolol. Circulation 2021, 143, 2431–2440. [Google Scholar] [CrossRef]
- Livi, L.; Barletta, G.; Martella, F.; Saieva, C.; Desideri, I.; Bacci, C.; Del Bene, M.R.; Airoldi, M.; Amoroso, D.; Coltelli, L.; et al. Cardioprotective Strategy for Patients With Nonmetastatic Breast Cancer Who Are Receiving an Anthracycline-Based Chemotherapy: A Randomized Clinical Trial. JAMA Oncol. 2021, 7, 1544. [Google Scholar] [CrossRef]
- Mecinaj, A.; Gulati, G.; Heck, S.; Holte, E.; Fagerland, M.; Larsen, A.; Blix, E.S.; Geisler, J.; Wethal, T.; Omland, T. Rationale and Design of the PRevention of cArdiac Dysfunction during Adjuvant Breast Cancer Therapy (PRADA II) Trial: A Randomized, Placebo-Controlled, Multicenter Trial. Cardio-Oncology 2021, 7, 33. [Google Scholar] [CrossRef]
- Jörgens, V. The Roots of SGLT Inhibition: Laurent-Guillaume de Koninck, Jean Servais Stas and Freiherr Josef von Mering. Acta Diabetol. 2019, 56, 29–31. [Google Scholar] [CrossRef]
- Bebernitz, G. Sodium-Glucose Cotransporters. In Comprehensive Medicinal Chemistry III; Elsevier: Amsterdam, The Netherlands, 2017; pp. 491–511. [Google Scholar] [CrossRef]
- Oku, A.; Ueta, K.; Arakawa, K.; Ishihara, T.; Nawano, M.; Kuronuma, Y.; Matsumoto, M.; Saito, A.; Tsujihara, K.; Anai, M.; et al. T-1095, an Inhibitor of Renal Na+-Glucose Cotransporters, May Provide a Novel Approach to Treating Diabetes. Diabetes 1999, 48, 1794–1800. [Google Scholar] [CrossRef]
- Tsimihodimos, V.; Filippas-Ntekouan, S.; Elisaf, M. SGLT1 Inhibition: Pros and Cons. Eur. J. Pharmacol. 2018, 838, 153–156. [Google Scholar] [CrossRef] [PubMed]
- Perkovic, V.; Jardine, M.J.; Neal, B.; Bompoint, S.; Heerspink, H.J.L.; Charytan, D.M.; Edwards, R.; Agarwal, R.; Bakris, G.; Bull, S.; et al. Canagliflozin and Renal Outcomes in Type 2 Diabetes and Nephropathy. N. Engl. J. Med. 2019, 380, 2295–2306. [Google Scholar] [CrossRef]
- Cannon, C.P.; Pratley, R.; Dagogo-Jack, S.; Mancuso, J.; Huyck, S.; Masiukiewicz, U.; Charbonnel, B.; Frederich, R.; Gallo, S.; Cosentino, F.; et al. Cardiovascular Outcomes with Ertugliflozin in Type 2 Diabetes. N. Engl. J. Med. 2020, 383, 1425–1435. [Google Scholar] [CrossRef]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef] [PubMed]
- Wiviott, S.D.; Raz, I.; Bonaca, M.P.; Mosenzon, O.; Kato, E.T.; Cahn, A.; Silverman, M.G.; Zelniker, T.A.; Kuder, J.F.; Murphy, S.A.; et al. Dapagliflozin and Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2019, 380, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Heidenreich, P.A.; Bozkurt, B.; Aguilar, D.; Allen, L.A.; Byun, J.J.; Colvin, M.M.; Deswal, A.; Drazner, M.H.; Dunlay, S.M.; Evers, L.R.; et al. 2022 AHA/ACC/HFSA Guideline for the Management of Heart Failure: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation 2022, 145, e263–e421. [Google Scholar] [CrossRef]
- Stevens, P.E.; Ahmed, S.B.; Carrero, J.J.; Foster, B.; Francis, A.; Hall, R.K.; Herrington, W.G.; Hill, G.; Inker, L.A.; Kazancıoğlu, R.; et al. KDIGO 2024 Clinical Practice Guideline for the Evaluation and Management of Chronic Kidney Disease. Kidney Int. 2024, 105, S117–S314. [Google Scholar] [CrossRef]
- Yang, C.-C.; Chen, Y.-T.; Wallace, C.G.; Chen, K.-H.; Cheng, B.-C.; Sung, P.-H.; Li, Y.-C.; Ko, S.-F.; Chang, H.-W.; Yip, H.-K. Early Administration of Empagliflozin Preserved Heart Function in Cardiorenal Syndrome in Rat. Biomed. Pharmacother. 2019, 109, 658–670. [Google Scholar] [CrossRef]
- Quagliariello, V.; Canale, M.L.; Bisceglia, I.; Iovine, M.; Paccone, A.; Maurea, C.; Scherillo, M.; Merola, A.; Giordano, V.; Palma, G.; et al. Sodium-Glucose Cotransporter 2 Inhibitor Dapagliflozin Prevents Ejection Fraction Reduction, Reduces Myocardial and Renal NF-κB Expression and Systemic pro-Inflammatory Biomarkers in Models of Short-Term Doxorubicin Cardiotoxicity. Front. Cardiovasc. Med. 2024, 11, 1289663. [Google Scholar] [CrossRef]
- Chang, W.-T.; Lin, Y.-W.; Ho, C.-H.; Chen, Z.-C.; Liu, P.-Y.; Shih, J.-Y. Dapagliflozin Suppresses ER Stress and Protects Doxorubicin-Induced Cardiotoxicity in Breast Cancer Patients. Arch. Toxicol. 2021, 95, 659–671. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, P.-L.; Chu, P.-M.; Cheng, H.-C.; Huang, Y.-T.; Chou, W.-C.; Tsai, K.-L.; Chan, S.-H. Dapagliflozin Mitigates Doxorubicin-Caused Myocardium Damage by Regulating AKT-Mediated Oxidative Stress, Cardiac Remodeling, and Inflammation. Int. J. Mol. Sci. 2022, 23, 10146. [Google Scholar] [CrossRef]
- Wang, C.-Y.; Chen, C.-C.; Lin, M.-H.; Su, H.-T.; Ho, M.-Y.; Yeh, J.-K.; Tsai, M.-L.; Hsieh, I.-C.; Wen, M.-S. TLR9 Binding to Beclin 1 and Mitochondrial SIRT3 by a Sodium-Glucose Co-Transporter 2 Inhibitor Protects the Heart from Doxorubicin Toxicity. Biology 2020, 9, 369. [Google Scholar] [CrossRef]
- Zhang, W.; Lu, J.; Wang, Y.; Sun, P.; Gao, T.; Xu, N.; Zhang, Y.; Xie, W. Canagliflozin Attenuates Lipotoxicity in Cardiomyocytes by Inhibiting Inflammation and Ferroptosis through Activating AMPK Pathway. Int. J. Mol. Sci. 2023, 24, 858. [Google Scholar] [CrossRef]
- Chen, W.; Zhang, Y.; Wang, Z.; Tan, M.; Lin, J.; Qian, X.; Li, H.; Jiang, T. Dapagliflozin Alleviates Myocardial Ischemia/Reperfusion Injury by Reducing Ferroptosis via MAPK Signaling Inhibition. Front. Pharmacol. 2023, 14, 1078205. [Google Scholar] [CrossRef]
- Wallenius, K.; Kroon, T.; Hagstedt, T.; Löfgren, L.; Sörhede-Winzell, M.; Boucher, J.; Lindén, D.; Oakes, N.D. The SGLT2 Inhibitor Dapagliflozin Promotes Systemic FFA Mobilization, Enhances Hepatic β-Oxidation, and Induces Ketosis. J. Lipid Res. 2022, 63, 100176. [Google Scholar] [CrossRef]
- Hundertmark, M.J.; Adler, A.; Antoniades, C.; Coleman, R.; Griffin, J.L.; Holman, R.R.; Lamlum, H.; Lee, J.; Massey, D.; Miller, J.J.J.J.; et al. Assessment of Cardiac Energy Metabolism, Function, and Physiology in Patients With Heart Failure Taking Empagliflozin: The Randomized, Controlled EMPA-VISION Trial. Circulation 2023, 147, 1654–1669. [Google Scholar] [CrossRef]
- Packer, M. Critical Reanalysis of the Mechanisms Underlying the Cardiorenal Benefits of SGLT2 Inhibitors and Reaffirmation of the Nutrient Deprivation Signaling/Autophagy Hypothesis. Circulation 2022, 146, 1383–1405. [Google Scholar] [CrossRef]
- Piras, L.; Zuccanti, M.; Tini Melato, G.; Volpe, M.; Tocci, G.; Barbato, E.; Battistoni, A. Double Duty: SGLT2 Inhibitors as Cardioprotective and Anticancer Allies. Hearts 2024, 5, 529–546. [Google Scholar] [CrossRef]
- Naeimzadeh, Y.; Tajbakhsh, A.; Nemati, M.; Fallahi, J. Exploring the Anti-Cancer Potential of SGLT2 Inhibitors in Breast Cancer Treatment in Pre-Clinical and Clinical Studies. Eur. J. Pharmacol. 2024, 978, 176803. [Google Scholar] [CrossRef]
- Pandey, A.; Alcaraz, M.; Saggese, P.; Soto, A.; Gomez, E.; Jaldu, S.; Yanagawa, J.; Scafoglio, C. Exploring the Role of SGLT2 Inhibitors in Cancer: Mechanisms of Action and Therapeutic Opportunities. Cancers 2025, 17, 466. [Google Scholar] [CrossRef]
- Sabatino, J.; De Rosa, S.; Tammè, L.; Iaconetti, C.; Sorrentino, S.; Polimeni, A.; Mignogna, C.; Amorosi, A.; Spaccarotella, C.; Yasuda, M.; et al. Empagliflozin Prevents Doxorubicin-Induced Myocardial Dysfunction. Cardiovasc. Diabetol. 2020, 19, 66. [Google Scholar] [CrossRef]
- Chen, M. Empagliflozin Attenuates Doxorubicin-Induced Cardiotoxicity by Activating AMPK/SIRT-1/PGC-1α-Mediated Mitochondrial Biogenesis. Toxicol. Res. 2023, 12, 216–223. [Google Scholar] [CrossRef]
- Guo, Z.; Javaheri, A. Empagliflozin to Prevent Doxorubicin Cardiotoxicity. JACC CardioOncol. 2025, 7, 185–187. [Google Scholar] [CrossRef]
- Barış, V.Ö.; Dinçsoy, A.B.; Gedikli, E.; Zırh, S.; Müftüoğlu, S.; Erdem, A. Empagliflozin Significantly Prevents the Doxorubicin-Induced Acute Cardiotoxicity via Non-Antioxidant Pathways. Cardiovasc. Toxicol. 2021, 21, 747–758. [Google Scholar] [CrossRef] [PubMed]
- Medina-Hernández, D.; Cádiz, L.; Mastrangelo, A.; Moreno-Arciniegas, A.; Fernández Tocino, M.; Cueto Becerra, A.A.; Díaz-Guerra Priego, A.; Skoza, W.A.; Higuero-Verdejo, M.I.; López-Martín, G.J.; et al. SGLT2i Therapy Prevents Anthracycline-Induced Cardiotoxicity in a Large Animal Model by Preserving Myocardial Energetics. JACC CardioOncol. 2025, 7, 171–184. [Google Scholar] [CrossRef]
- Hu, J.; Xu, J.; Tan, X.; Li, D.; Yao, D.; Xu, B.; Lei, Y. Dapagliflozin Protects against Dilated Cardiomyopathy Progression by Targeting NLRP3 Inflammasome Activation. Naunyn-Schmiedebergs Arch. Pharmacol. 2023, 396, 1461–1470. [Google Scholar] [CrossRef] [PubMed]
- Ulusan, S. Dapagliflozin May Protect Against Doxorubicin-Induced Cardiotoxicity. Anatol. J. Cardiol. 2023, 27, 339–347. [Google Scholar] [CrossRef]
- Kim, D.; Jang, G.; Hwang, J.; Wei, X.; Kim, H.; Son, J.; Rhee, S.-J.; Yun, K.-H.; Oh, S.-K.; Oh, C.-M.; et al. Combined Therapy of Low-Dose Angiotensin Receptor–Neprilysin Inhibitor and Sodium-Glucose Cotransporter-2 Inhibitor Prevents Doxorubicin-Induced Cardiac Dysfunction in Rodent Model with Minimal Adverse Effects. Pharmaceutics 2022, 14, 2629. [Google Scholar] [CrossRef]
- Chang, W.-T.; Shih, J.-Y.; Lin, Y.-W.; Chen, Z.-C.; Kan, W.-C.; Lin, T.-H.; Hong, C.-S. Dapagliflozin Protects against Doxorubicin-Induced Cardiotoxicity by Restoring STAT3. Arch. Toxicol. 2022, 96, 2021–2032. [Google Scholar] [CrossRef] [PubMed]
- Avagimyan, A.; Sheibani, M.; Pogosova, N.; Mkrtchyan, L.; Yeranosyan, H.; Aznauryan, A.; Sahaakyan, K.; Fogacci, F.; Cicero, A.; Shafie, D.; et al. Possibilities of Dapagliflozin-Induced Cardioprotection on Doxorubicin + Cyclophosphamide Mode of Chemotherapy-Induced Cardiomyopathy. Int. J. Cardiol. 2023, 391, 131331. [Google Scholar] [CrossRef]
- Maurea, N.; Palma, G.; Luciano, A.; Maurea, F.; Bruzzese, F.; Paccone, A.; Izzo, F.; Barbato, M.; Arianna, R.; Giacobbe, I.; et al. Dapagliflozin Improves Ejection Fraction, Radial/Longitudinal Strain and Reduces Systemic H-FABP and pro-Inflammatory Cytokines in Models of Dororubicin/Trastuzumab Induced Cardiotoxicity. Eur. Heart J. Cardiovasc. Imaging 2025, 26 (Supp. S1), jeae333.184. [Google Scholar] [CrossRef]
- Maurea, N.; Quagliariello, V.; Coppola, C.; Rea, D.; Barbieri, A.; Arra, C.; Botti, G. Empaglifozin Has Cardioprotective and Anti-Inflammatory Effects during Doxorubicin Treatment: A Preclinical Study. J. Clin. Oncol. 2019, 37 (Suppl. S15), e23057. [Google Scholar] [CrossRef]
- Satyam, S.M.; Bairy, L.K.; Shetty, P.; Sainath, P.; Bharati, S.; Ahmed, A.Z.; Singh, V.K.; Ashwal, A.J. Metformin and Dapagliflozin Attenuate Doxorubicin-Induced Acute Cardiotoxicity in Wistar Rats: An Electrocardiographic, Biochemical, and Histopathological Approach. Cardiovasc. Toxicol. 2023, 23, 107–119. [Google Scholar] [CrossRef]
- Chang, H.-Y.; Hsu, H.-C.; Fang, Y.-H.; Liu, P.-Y.; Liu, Y.-W. Empagliflozin Attenuates Doxorubicin-Induced Cardiotoxicity by Inhibiting the JNK Signaling Pathway. Biomed. Pharmacother. 2024, 176, 116759. [Google Scholar] [CrossRef] [PubMed]
- Tabowei, G.; Dadzie, S.K.; Perswani, P.; Nawaz, S.; Kaur, M.; Moqattash, M.; Wei, C.R.; Hirani, S. Efficacy of Sodium-Glucose Cotransporter 2 Inhibitors in Preventing Heart Failure in Patients Receiving Anthracycline-Based Cancer Therapy: A Systematic Review and Meta-Analysis. Cureus 2024, 16, e60086. [Google Scholar] [CrossRef]
- Bhalraam, U.; Veerni, R.B.; Paddock, S.; Meng, J.; Piepoli, M.; López-Fernández, T.; Tsampasian, V.; Vassiliou, V.S. Impact of Sodium–Glucose Cotransporter-2 Inhibitors on Heart Failure Outcomes in Cancer Patients and Survivors: A Systematic Review and Meta-Analysis. Eur. J. Prev. Cardiol. 2025, zwaf026. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.-H.; Chiang, C.-H.; Chiang, C.-H.; Ma, K.S.-K.; Peng, C.-Y.; Hsia, Y.P.; Horng, C.-S.; Chen, C.-Y.; Chang, Y.-C.; See, X.Y.; et al. Impact of Sodium-Glucose Cotransporter-2 Inhibitors on Heart Failure and Mortality in Patients with Cancer. Heart 2023, 109, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Hwang, H.-J.; Kim, M.; Jun, J.E.; Yon, D.K. Sodium-Glucose Cotransporter-2 Inhibitors Improve Clinical Outcomes in Patients with Type 2 Diabetes Mellitus Undergoing Anthracycline-Containing Chemotherapy: An Emulated Target Trial Using Nationwide Cohort Data in South Korea. Sci. Rep. 2023, 13, 21756. [Google Scholar] [CrossRef]
- Abdel-Qadir, H.; Carrasco, R.; Austin, P.C.; Chen, Y.; Zhou, L.; Fang, J.; Su, H.M.H.; Lega, I.C.; Kaul, P.; Neilan, T.G.; et al. The Association of Sodium-Glucose Cotransporter 2 Inhibitors With Cardiovascular Outcomes in Anthracycline-Treated Patients With Cancer. JACC CardioOncol. 2023, 5, 318–328. [Google Scholar] [CrossRef]
- Avula, V.; Sharma, G.; Kosiborod, M.N.; Vaduganathan, M.; Neilan, T.G.; Lopez, T.; Dent, S.; Baldassarre, L.; Scherrer-Crosbie, M.; Barac, A.; et al. SGLT2 Inhibitor Use and Risk of Clinical Events in Patients With Cancer Therapy–Related Cardiac Dysfunction. JACC Heart Fail. 2024, 12, 67–78. [Google Scholar] [CrossRef]
- Henson, B.D.; Bale-Neary, C.A.; Mecaskey, R.; Gbujie, O.; Zhan, M.; Rao, K.; Carbone, S. Sodium–Glucose Cotransporter 2 Inhibitors, Malnutrition, Cachexia, and Survival in Patients with Heart Failure with a History of Anthracycline Treatment. J. Cardiovasc. Pharmacol. 2024, 84, 486–489. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Author/Year | Species | DOX Dosage and Administration | SGLT2 Inhibitor | Cardiovascular Assessment |
|---|---|---|---|---|
| Oh Et Al. (2019) [19] | Male C57BL/6J (B6J) | 15 mg/kg i.p. single dose | EMPA 300 mg/kg in NCD for 2 weeks | ↑ Fractional shortening; ↓ LV hypertrophy; ↓ Perivascular and interstitial fibrosis; |
| Maurea et al. (2019) [153] | Male C57BL6 mice | 2.25 mg/kg/day i.p. | EMPA 10 mg/kg/day | Prevented GLS reduction; |
| Sabatino et al. (2020) [142] | Male C57Bl/6 mice | 15 mg/kg i.p. single dose/4 mg/kg/week for 5 weeks | EMPA 10 mg/kg/day | Improved BP values and LV systolic function; ↓ Myocardial fibrosis; |
| Wang et al. (2020) [133] | Male C57BL/6 mice | 5 mg/kg/week for 4 weeks i.v. | EMPA 0.05 mg/kg/day for 5 weeks | ↑ Fractional shortening; ↑ Cardiac contractility; ↓ Myocardial fibrosis; ↓ BNP and cTnT; |
| Quagliariello Et Al. (2021) [21] | Female C57Bl/6 mice | 2.17 mg/kg/day for 7 days | EMPA 10 mg/kg/day for 10 days | Attenuated fractional shortening and LVEF decline; |
| Baris et al. (2021) [145] | Male Sprague Dawley rats | 18 mg/kg for 7 days i.p. cumulative dose | EMPA 10 mg/kg for 14 days | ↑ LVEF; ↓ QTc interval and myofibril loss; |
| Chang et al. (2022) [150] | Male Sprague Dawley rats | 5 mg/kg/week for 4 weeks i.p. | DAPA 10 mg/kg/day for 6 weeks | ↑ Fractional shortening; ↑ LVEF; ↓ Hemodynamic changes in echo parameters; ↓ Myocardial fibrosis; |
| Hsieh et al. (2022) [132] | Sprague Dawley rats | 3 mg/kg/week for 4 weeks i.p. | DAPA 0.1 mg/kg/day for 4 weeks | Attenuated LVEF decline and cardiac hypertrophy; |
| Satyam et al. (2023) [154] | Adult Wister rats | 20 mg/kg i.p. single dose | DAPA 0.9 mg/kg/day for 8 days | Attenuated CK-MB increase and pathological ECG changes (e.g., QTc prolongation); |
| Chen et al. (2023) [143] | Male Sprague Dawley rats | 2.5 mg/kg/twice a week for 4 weeks i.p. | EMPA 30 mg/kg/day for 4 weeks | ↑ LVEF and fractional shortening; ↓ NT-proBNP and cTnT; |
| Quagliariello Et Al. (2024) [130] | Female C57Bl/6 mice | 2.17 mg/kg/day i.p. | DAPA 10 mg/kg/day | Attenuated LVEF decline and cardiac strain impairment; |
| Chang et al. (2024) [155] | C57Bl/6 mice | 5 mg/kg/week for 4 weeks | EMPA 10 mg/kg/day for 5 weeks | Attenuated LVEF and fractional shortening decline; |
| Medina-Hernández et al. (2025) [146] | Female large white pigs | 6 triweekly i.v. injections (1.8 mg/kg each) | EMPA 10/20 mg/day | Improved LVEF and cardiac energetics in a dose-dependent manner; |
| Maurea et al. (2025) [152] | Female C57Bl/6 mice | 2.17 mg/kg/day i.p. for 10 days | DAPA 12 mg/kg for 10 days | ↑ LVEF and fractional shortening; |
| Author/Year | Population | Final Cohort | SGLT2 Inhibitor | Key Findings |
|---|---|---|---|---|
| Gongora et al. (2022) [20] | Cancer patients diagnosed with DM prior to anthracycline treatment | 32 SGLT2i recipients/96 SGLT2i non-recipients | EMPA 16 pts CANA 11 pts DAPA 5 pts | Patients on SGLT2 inhibitors: ↓ Cardiac events post anthracycline therapy; ↓ HF admissions; ↓ Rate of cardiac dysfunction; No new cases of AIC observed; |
| Chiang et al. (2023) [158] | Adult patients with T2DM diagnosed with cancer | 878 SGLT2i recipients/7556 SGLT2i non-recipients | EMPA CANA DAPA | Patients on SGLT2 inhibitors: ↓ HF admissions; ↑ Overall survival; |
| Hwang et al. (2023) [159] | Adult patients newly diagnosed with cancer undergoing anthracycline-based chemotherapy | 7800 non-DM/779 SGLT2i recipients/2337 SGLT2i non-recipients | EMPA CANA DAPA | Patients on SGLT2 inhibitors: ↓ Cardiovascular composite outcome (stroke, MI, HF admissions, death); |
| Abdel-Qadir et al. (2023) [160] | Patients over 65 years old with DM and no prior HF undergoing anthracycline treatment | 99 SGLT2i recipients/834 SGLT2i non-recipients | EMPA CANA DAPA | Patients on SGLT2 inhibitors: ↓ HF hospitalizations; |
| Avula et al. (2024) [161] | Patients aged ≥18 years with T2DM, cancer, and exposure to anthracyclines | 640 SGLT2i recipients/640 SGLT2i non-recipients | EMPA CANA DAPA | ↓ Acute HF exacerbations; ↓ All-cause mortality; Fewer hospitalizations and emergency department visits; ↓ Incidence of AF; ↓ Occurrence of AKI and need for renal replacement therapy; |
| Henson et al. (2024) [162] | Patients with HF previously treated with anthracyclines | 1323 SGLT2i recipients/1323 SGLT2i non-recipients | EMPA CANA DAPA | ↓ All-cause mortality; |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goje, I.-D.; Goje, G.-I.; Ordodi, V.L.; Ciobotaru, V.G.; Ivan, V.S.; Buzaș, R.; Tunea, O.; Bojin, F.; Lighezan, D.-F. Doxorubicin-Induced Cardiotoxicity and the Emerging Role of SGLT2 Inhibitors: From Glycemic Control to Cardio-Oncology. Pharmaceuticals 2025, 18, 681. https://doi.org/10.3390/ph18050681
Goje I-D, Goje G-I, Ordodi VL, Ciobotaru VG, Ivan VS, Buzaș R, Tunea O, Bojin F, Lighezan D-F. Doxorubicin-Induced Cardiotoxicity and the Emerging Role of SGLT2 Inhibitors: From Glycemic Control to Cardio-Oncology. Pharmaceuticals. 2025; 18(5):681. https://doi.org/10.3390/ph18050681
Chicago/Turabian StyleGoje, Iacob-Daniel, Greta-Ionela Goje, Valentin Laurențiu Ordodi, Valentina Gabriela Ciobotaru, Vlad Sabin Ivan, Roxana Buzaș, Oana Tunea, Florina Bojin, and Daniel-Florin Lighezan. 2025. "Doxorubicin-Induced Cardiotoxicity and the Emerging Role of SGLT2 Inhibitors: From Glycemic Control to Cardio-Oncology" Pharmaceuticals 18, no. 5: 681. https://doi.org/10.3390/ph18050681
APA StyleGoje, I.-D., Goje, G.-I., Ordodi, V. L., Ciobotaru, V. G., Ivan, V. S., Buzaș, R., Tunea, O., Bojin, F., & Lighezan, D.-F. (2025). Doxorubicin-Induced Cardiotoxicity and the Emerging Role of SGLT2 Inhibitors: From Glycemic Control to Cardio-Oncology. Pharmaceuticals, 18(5), 681. https://doi.org/10.3390/ph18050681