
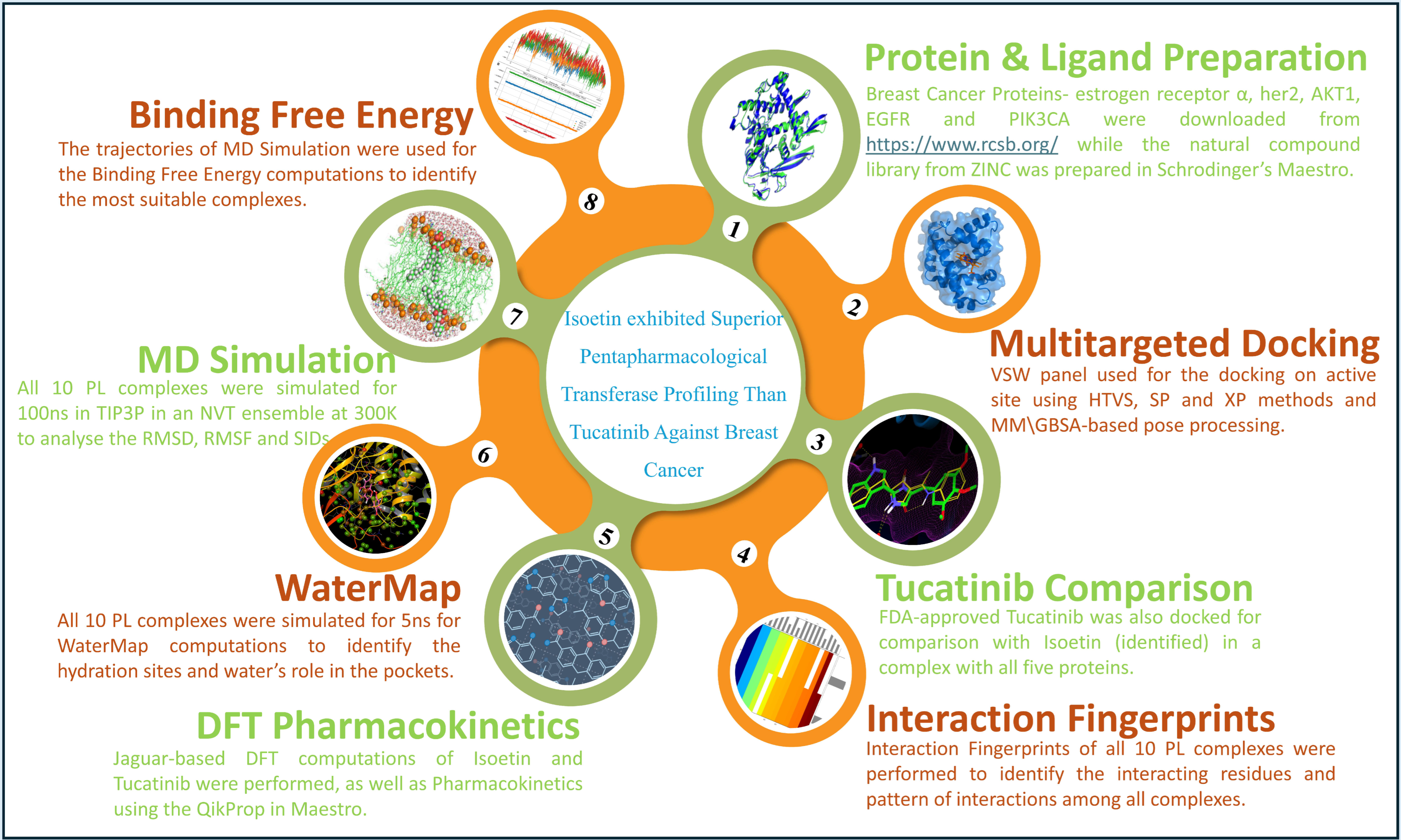
Isoetin from Isoetaceae Exhibits Superior Pentatransferase Inhibition in Breast Cancer: Comparative Computational Profiling with FDA-Approved Tucatinib

 , , , , , , , , and
, , , , , , , , and
Abstract
1. Introduction
2. Results
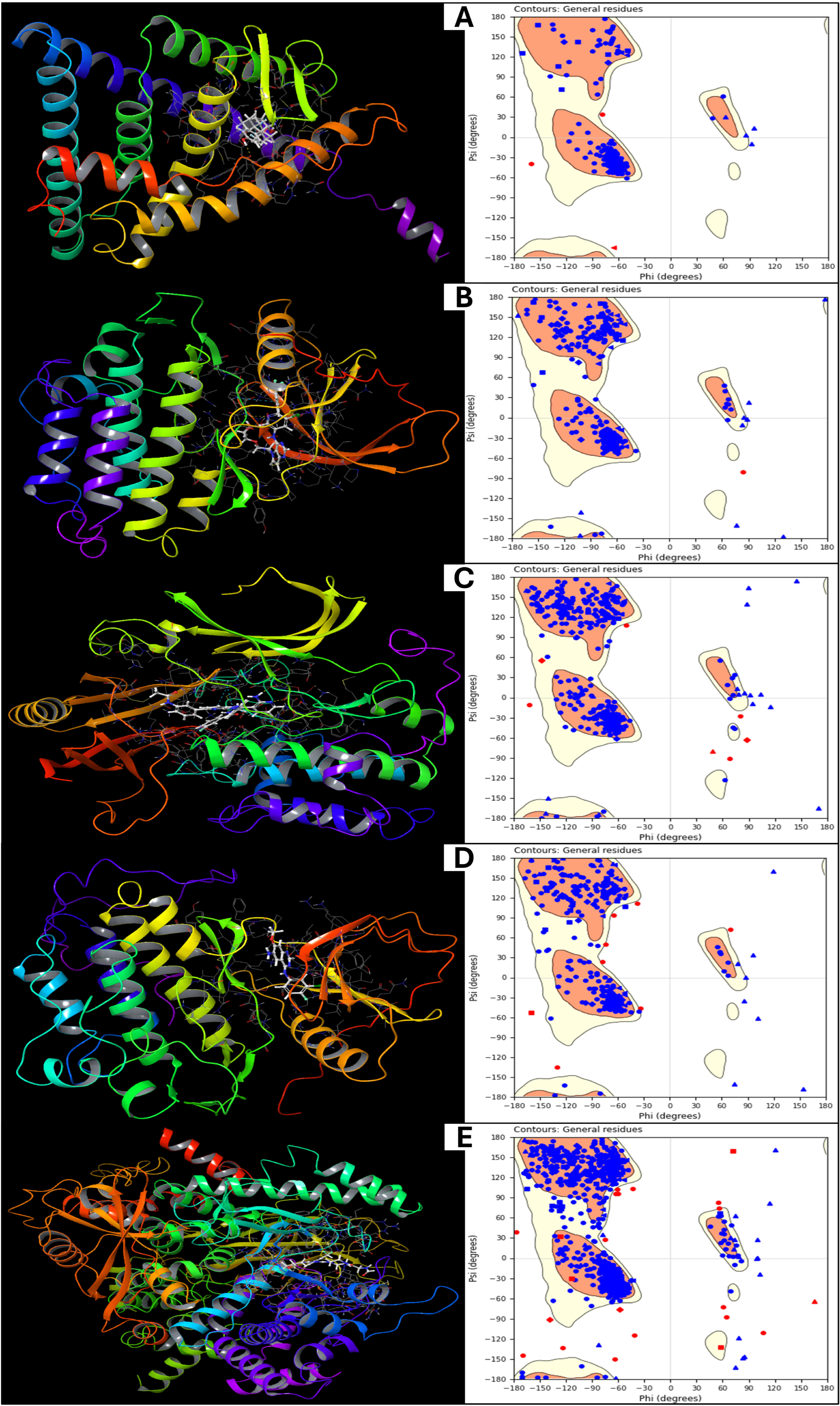
2.1. Validation and Analysis of Protein Structures
2.2. Analysis of Multitargeted Molecular Docking Results and Control Comparison
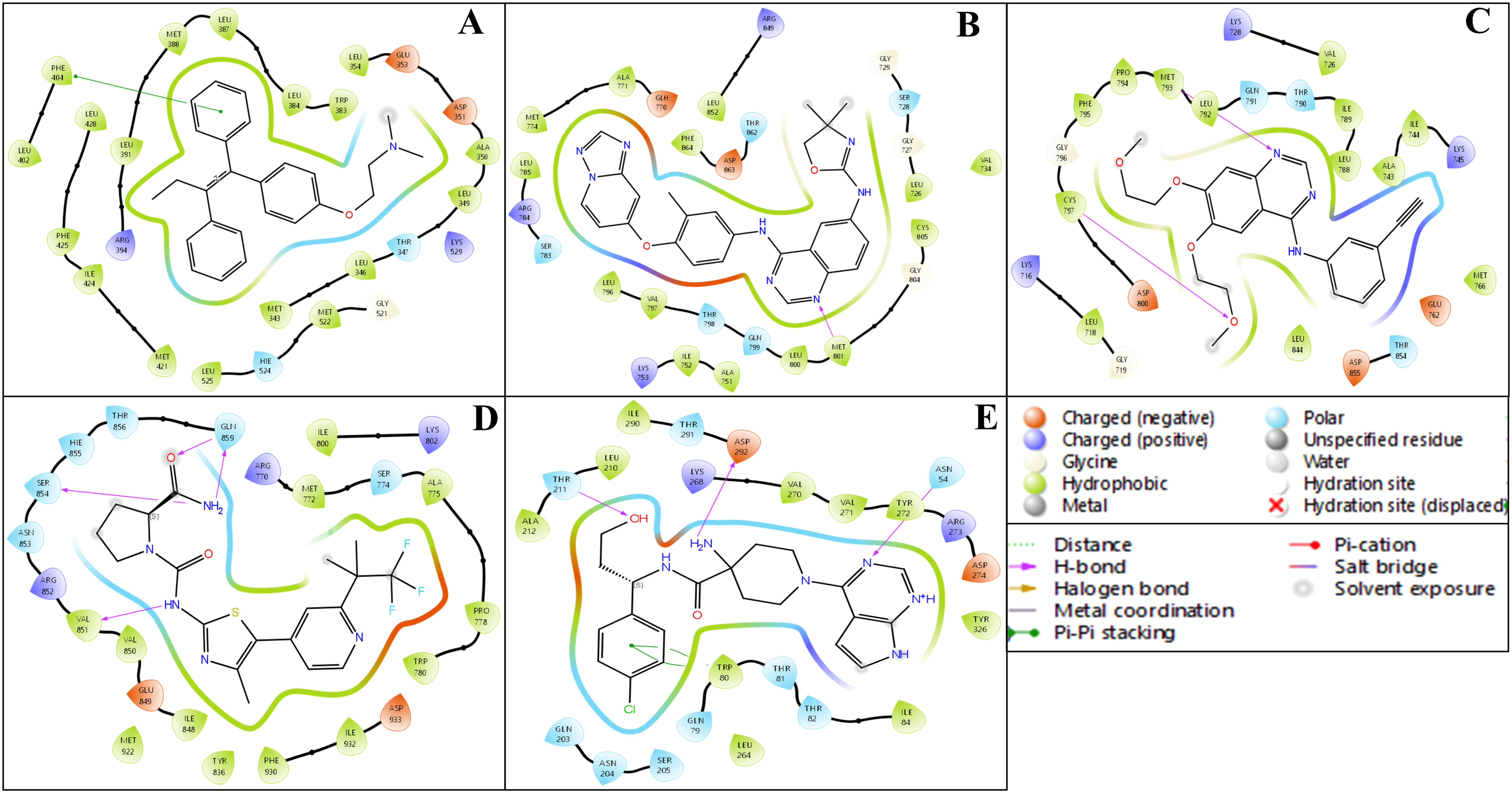
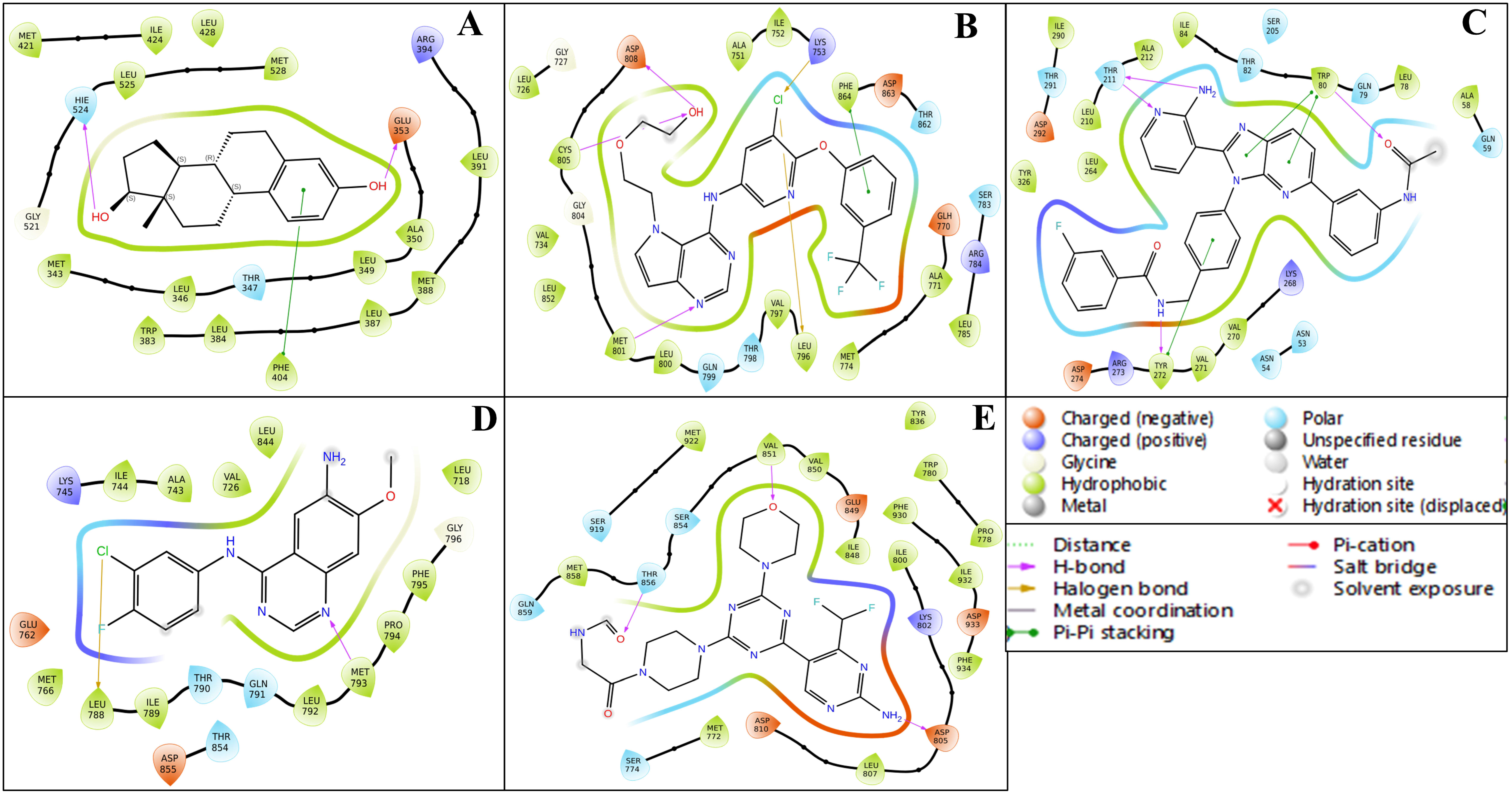
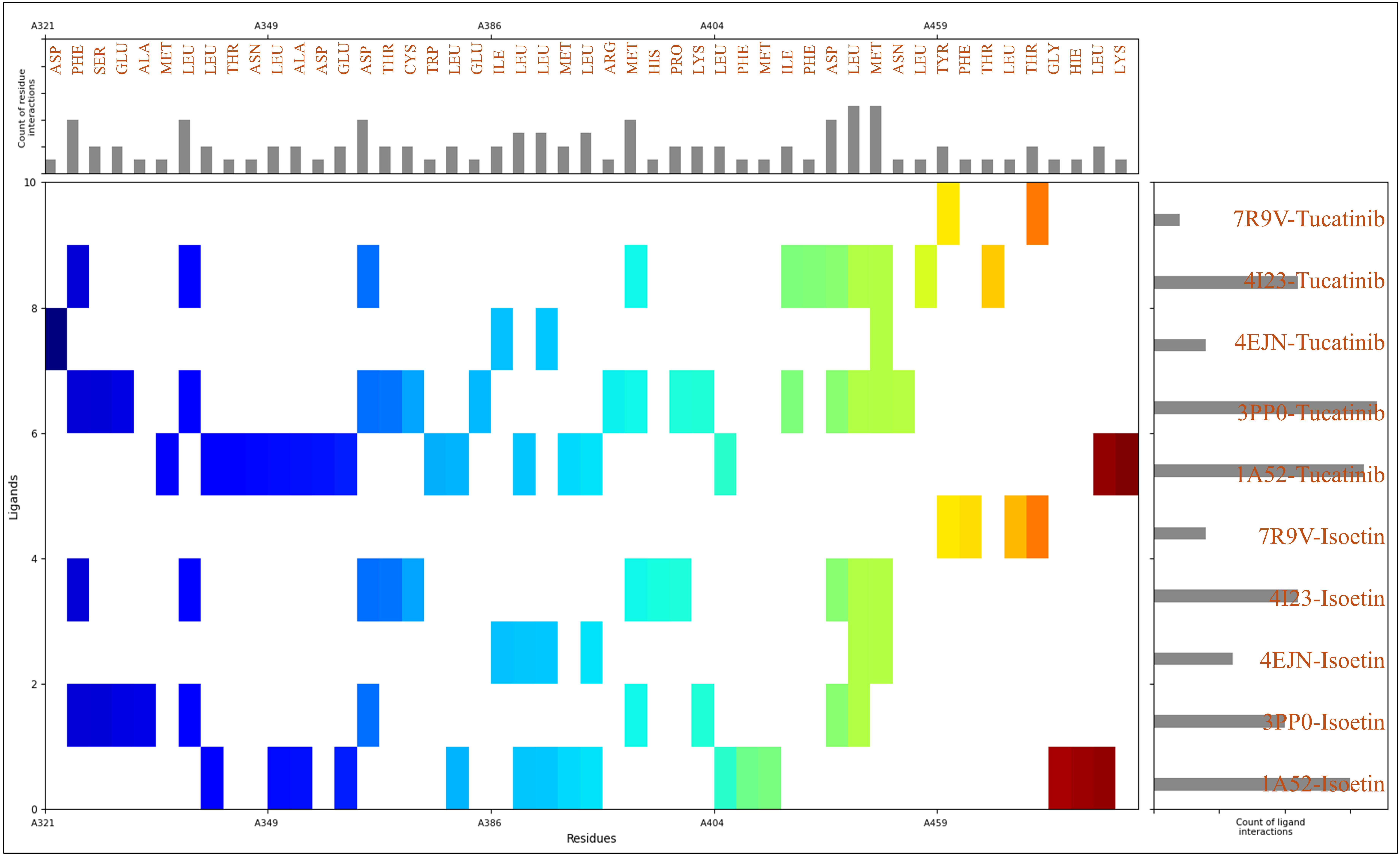
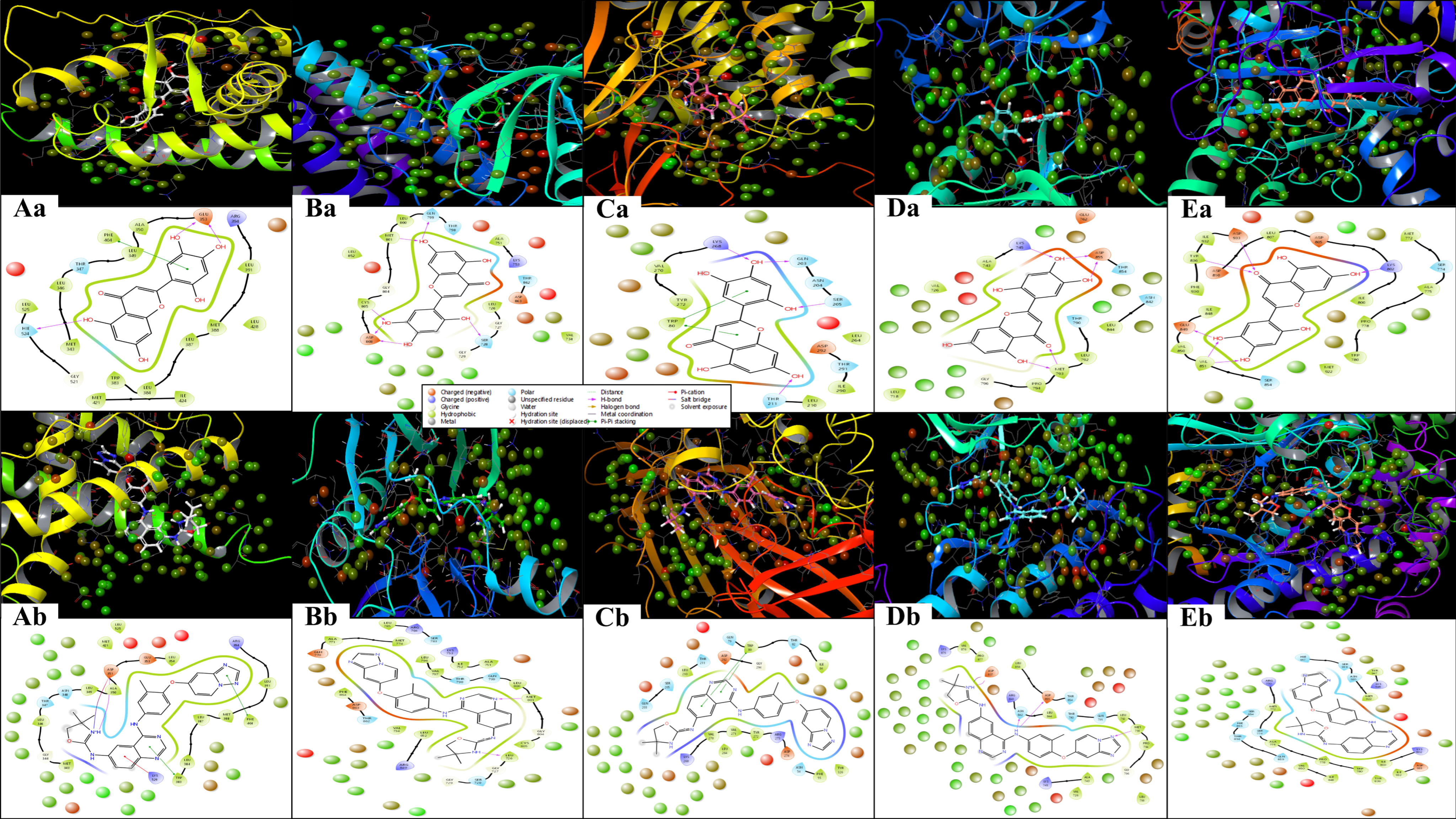
2.3. Analysis of Interactions Patterns
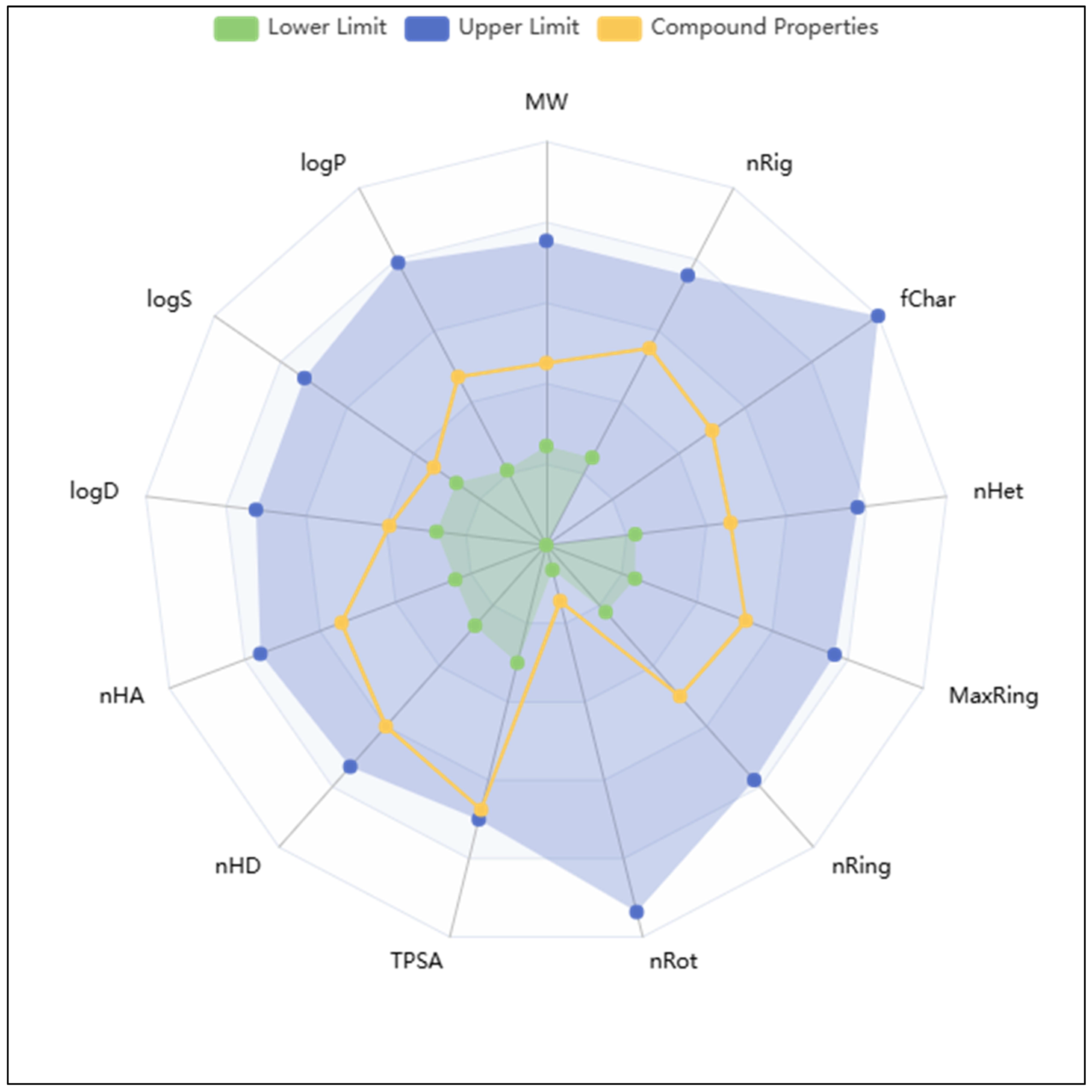
2.4. Analysis of Pharmacokinetics, DFT Computations, and Control Comparison
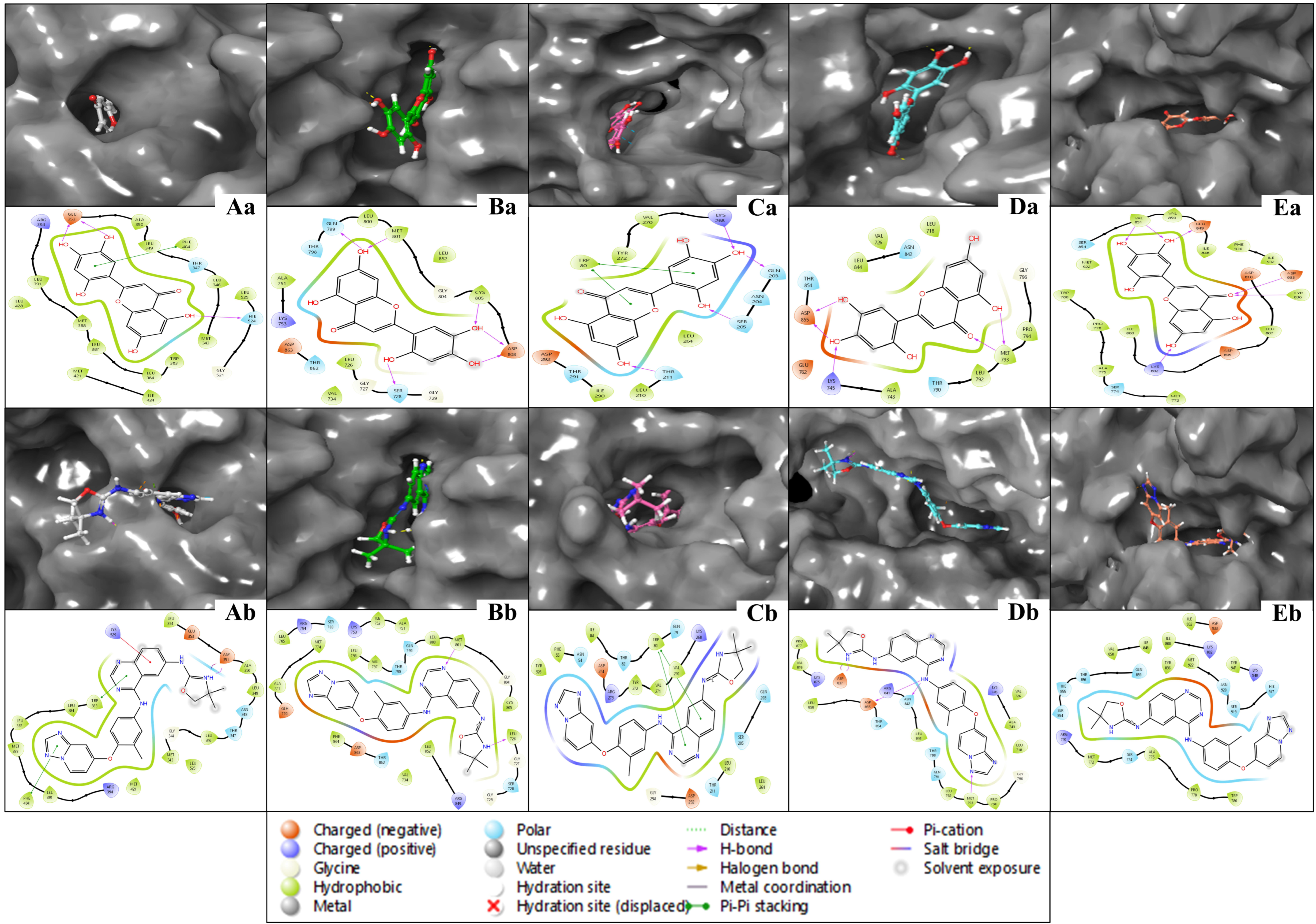
2.5. Analysis of WaterMap Results and Control Comparison
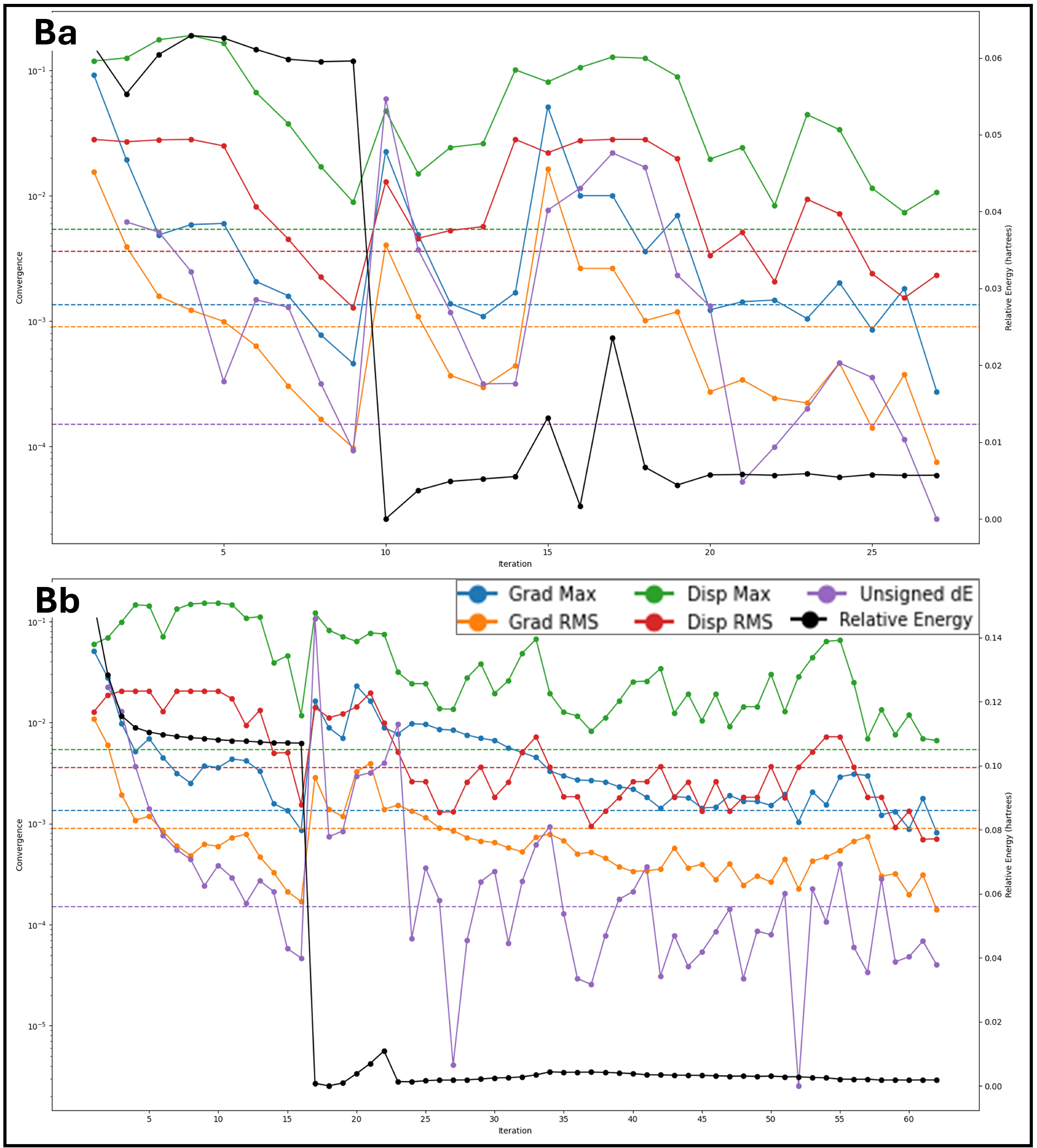
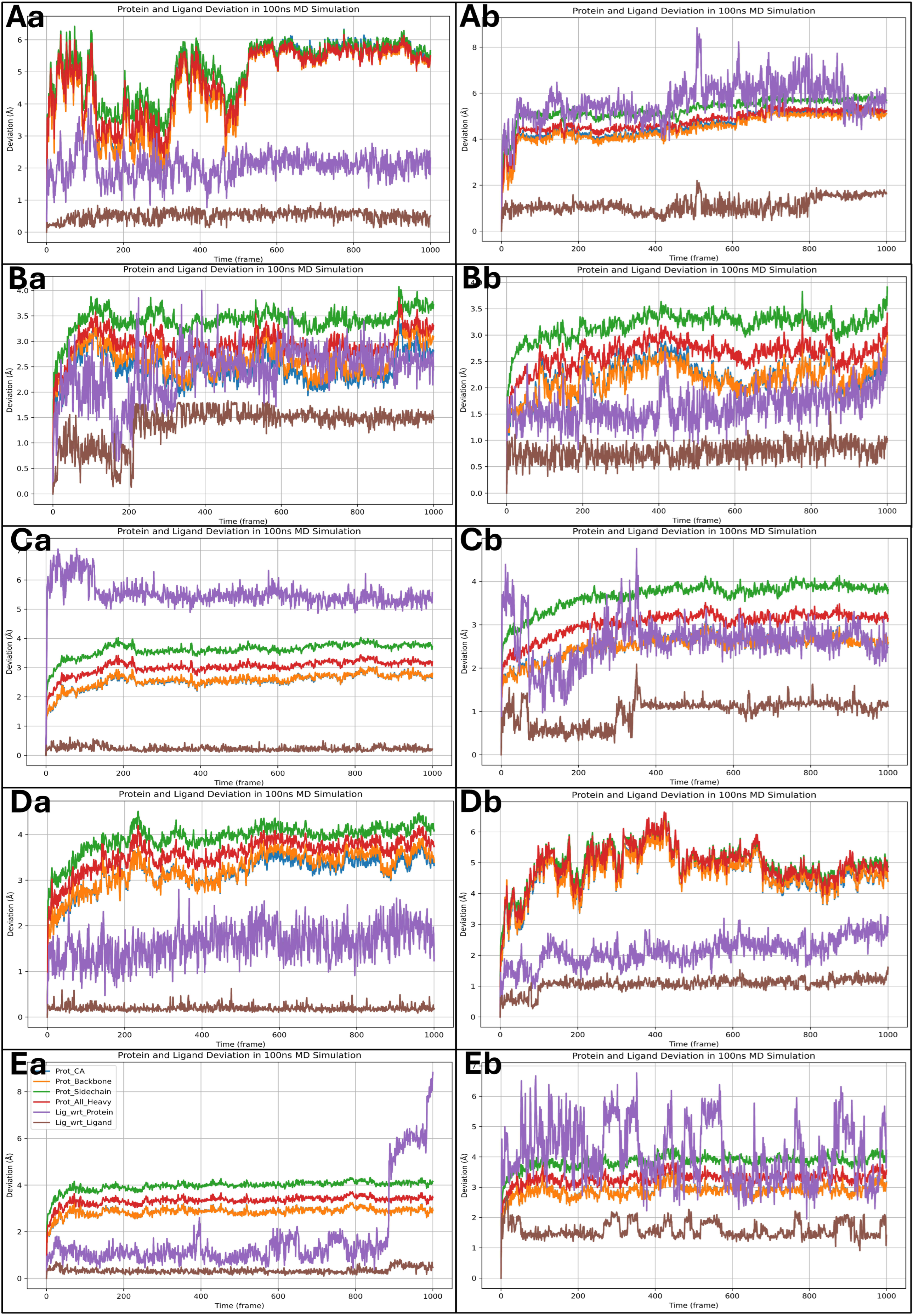
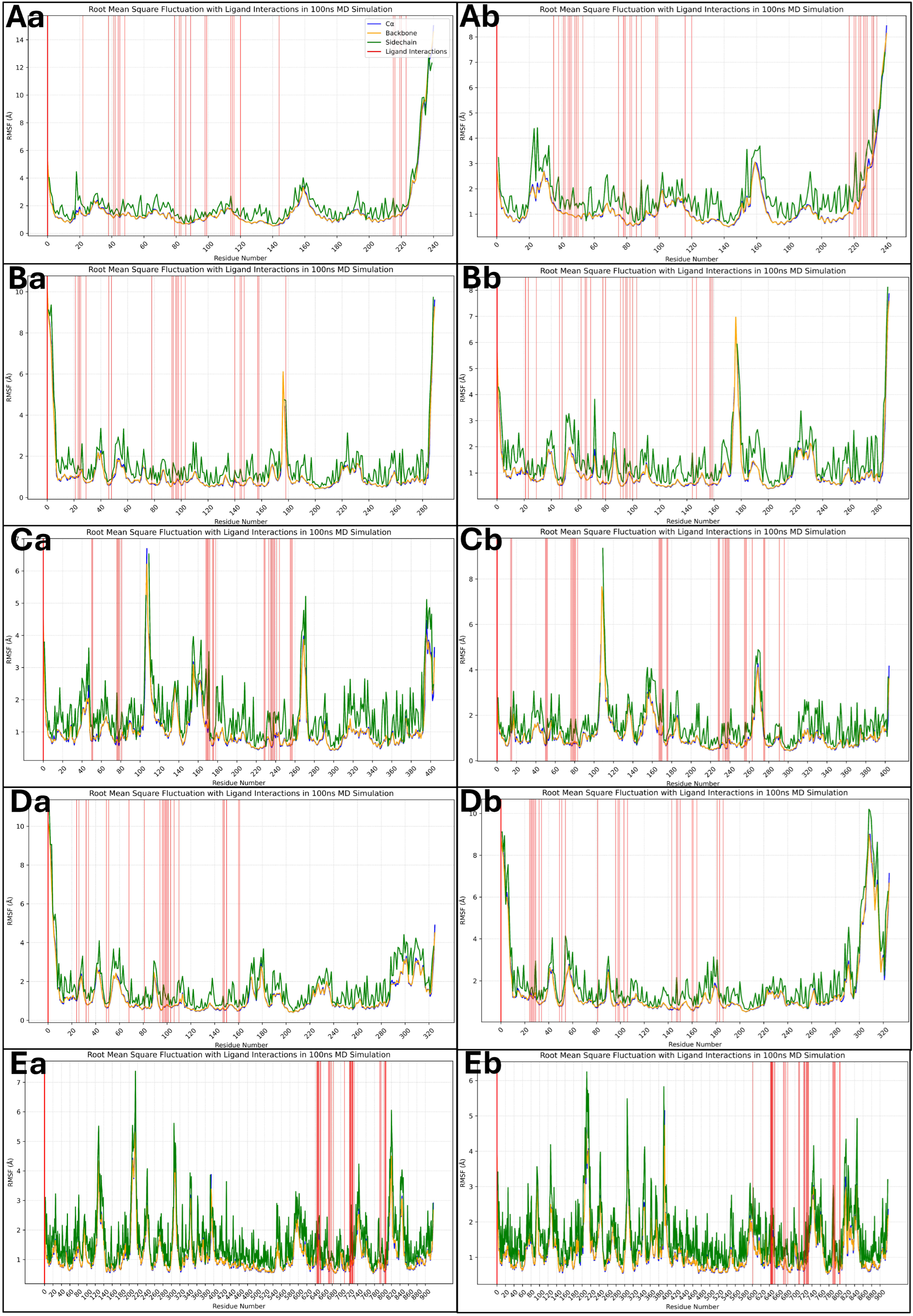
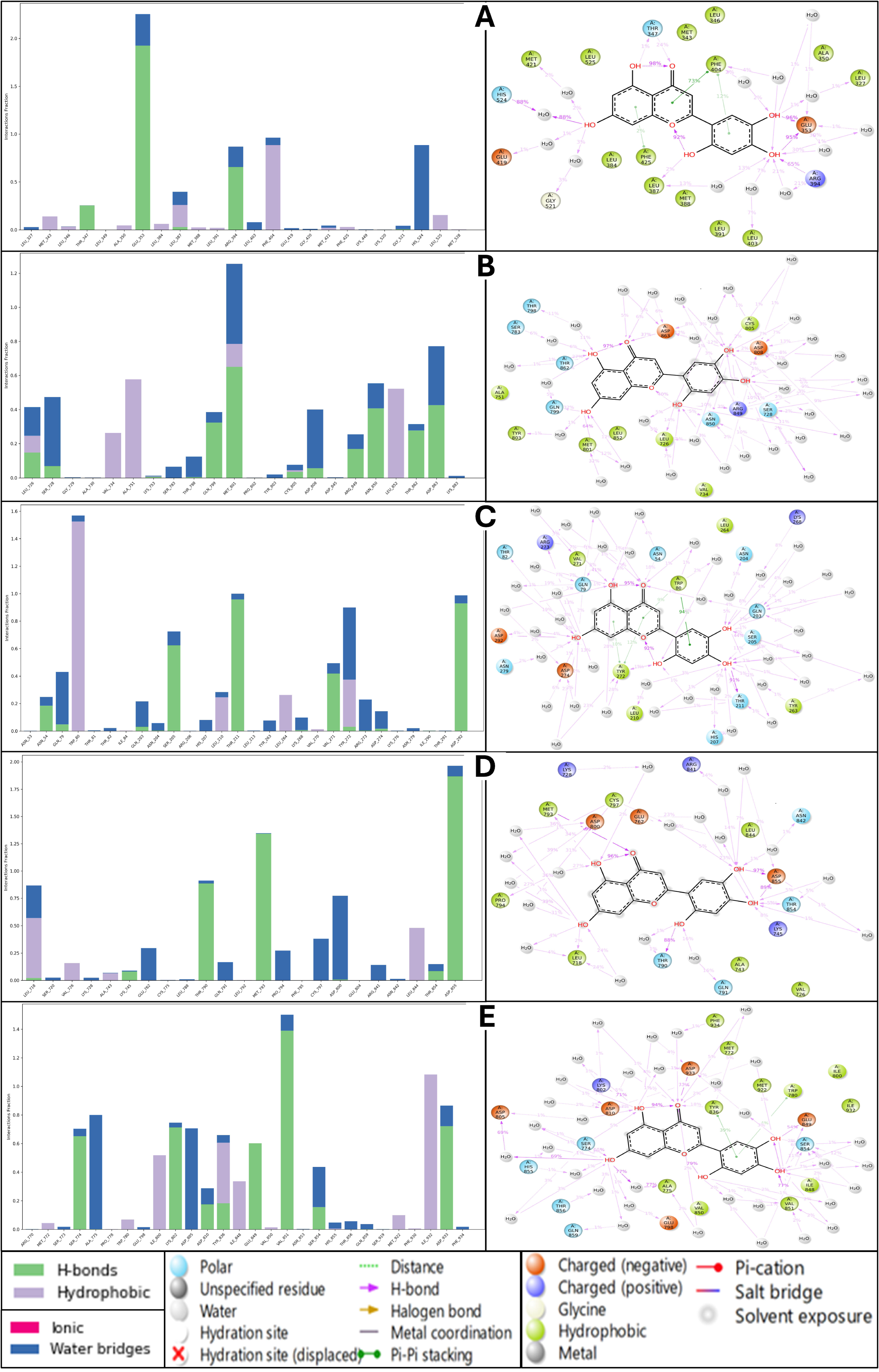
2.6. Analysis of Molecular Dynamics Simulation Trajectories and Control Comparison
2.6.1. Analysis of Root Mean Square Deviation
2.6.2. Analysis of Root Mean Square Fluctuations
2.6.3. Analysis of Simulation Interaction Diagram
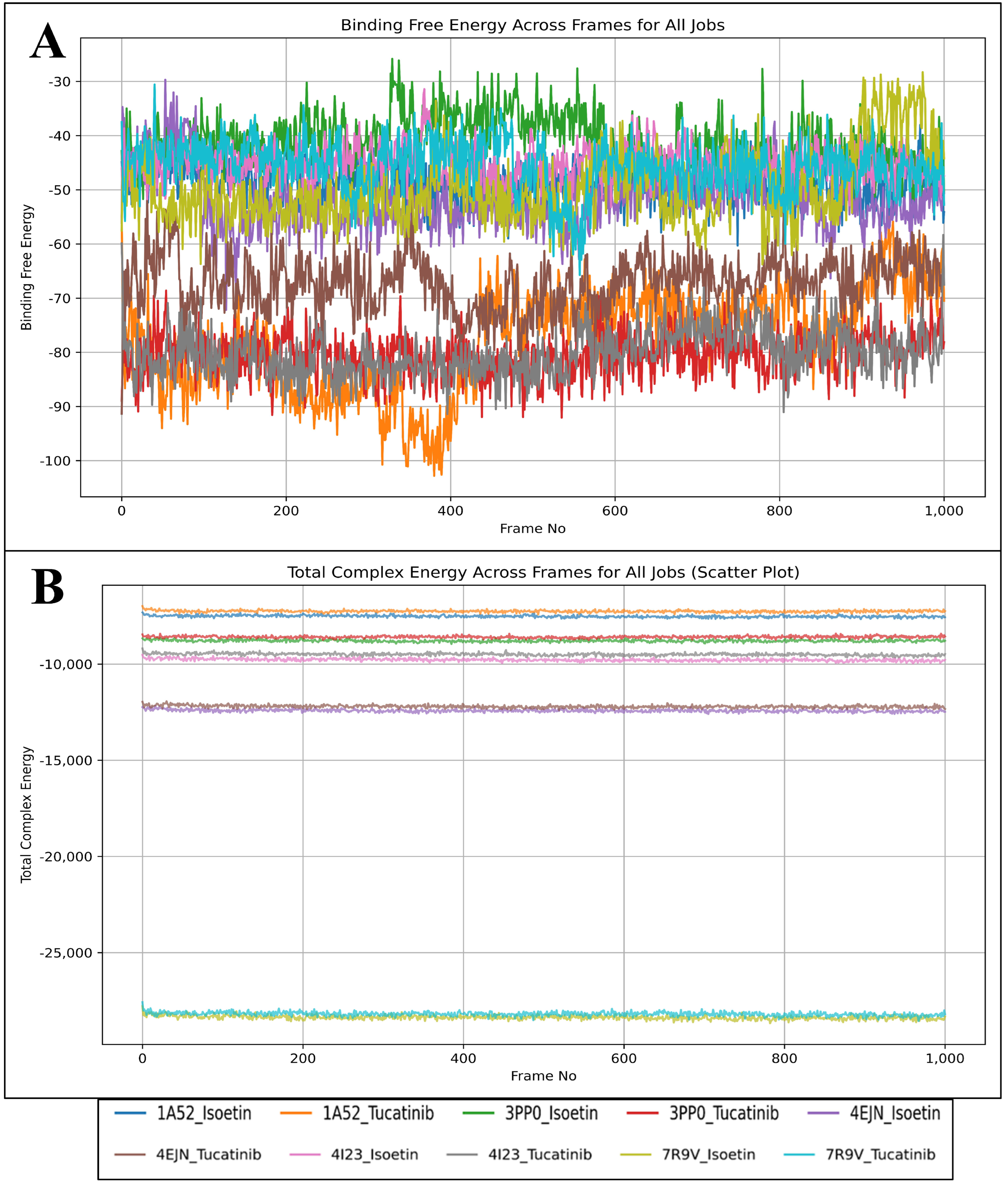
2.7. Analysis of Binding Free Energy Computation and Control Comparison
3. Discussion
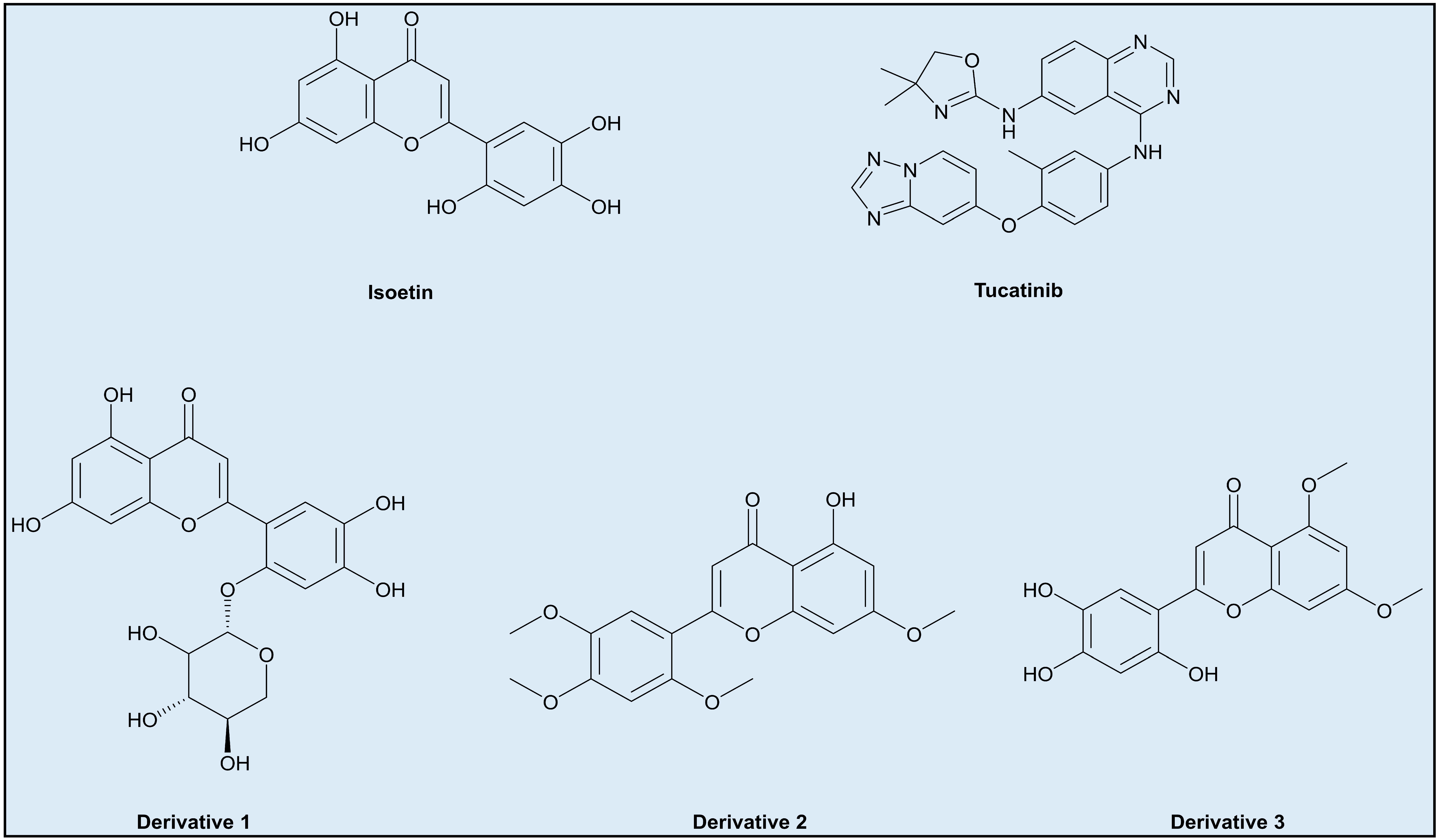
- Incorporating additional aromatic or larger groups in the structure at positions where the interfering groups in Isoetin are hydrophobic interactions, but near the residues involved in hydrogen bonding, such as derivative 1 (Figure 14).
- Changing the axial position of the aromatic groups in Isoetin to better align them for π–π stacking against aromatic residues like phenylalanine (Phe) or tryptophan (Trp), key to stabilising interactions in Tucatinib. Substituents that enhance the planarity or electron density of these aromatic rings of Isoetin would favour π–π interactions, such as derivative 2 (Figure 14).
- Designing derivatives of Isoetin involves the substitution of specific positions on the benzene rings to improve π–π stacking; electron-donating groups like methoxy group may also contribute to the same by arranging the aromatic rings into favourable positions for stacking, present in derivative 3 (Figure 14).
4. Methods
4.1. Ligand Library and Protein Structural Data Collection and Preparation
4.2. Receptor Grid Generation and Multitargeted Molecular Docking Studies and Control Comparison
4.3. Molecular Interaction Fingerprints
4.4. Density Functional Theory and Pharmacokinetics
4.5. WaterMap Studies
4.6. Molecular Dynamics Simulation Studies and Binding Free Energy Calculations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Obeagu, E.I.; Obeagu, G.U. Breast cancer: A review of risk factors and diagnosis. Medicine 2024, 103, e36905. [Google Scholar] [CrossRef] [PubMed]
- Smolarz, B.; Nowak, A.Z.; Romanowicz, H. Breast cancer—Epidemiology, classification, pathogenesis and treatment (review of literature). Cancers 2022, 14, 2569. [Google Scholar] [CrossRef]
- Arnold, M.; Morgan, E.; Rumgay, H.; Mafra, A.; Singh, D.; Laversanne, M.; Vignat, J.; Gralow, J.R.; Cardoso, F.; Siesling, S. Current and future burden of breast cancer: Global statistics for 2020 and 2040. Breast 2022, 66, 15–23. [Google Scholar] [CrossRef]
- Contiero, P.; Boffi, R.; Borgini, A.; Fabiano, S.; Tittarelli, A.; Mian, M.; Vittadello, F.; Epifani, S.; Ardizzone, A.; Cirilli, C. Causes of death in women with breast cancer: A risks and rates study on a population-based cohort. Front. Oncol. 2023, 13, 1270877. [Google Scholar] [CrossRef] [PubMed]
- Venkitaraman, A.R. How do mutations affecting the breast cancer genes BRCA1 and BRCA2 cause cancer susceptibility? DNA Repair 2019, 81, 102668. [Google Scholar] [CrossRef]
- Iranmakani, S.; Mortezazadeh, T.; Sajadian, F.; Ghaziani, M.F.; Ghafari, A.; Khezerloo, D.; Musa, A.E. A review of various modalities in breast imaging: Technical aspects and clinical outcomes. Egypt. J. Radiol. Nucl. Med. 2020, 51, 57. [Google Scholar] [CrossRef]
- Tung, N.; Battelli, C.; Allen, B.; Kaldate, R.; Bhatnagar, S.; Bowles, K.; Timms, K.; Garber, J.E.; Herold, C.; Ellisen, L. Frequency of mutations in individuals with breast cancer referred for BRCA 1 and BRCA 2 testing using next-generation sequencing with a 25-gene panel. Cancer 2015, 121, 25–33. [Google Scholar] [CrossRef]
- Tang, Y.; Wang, Y.; Kiani, M.F.; Wang, B. Classification, treatment strategy, and associated drug resistance in breast cancer. Clin. Breast Cancer 2016, 16, 335–343. [Google Scholar] [CrossRef]
- Kandasamy, T.; Sen, P.; Ghosh, S.S. Multi-targeted drug repurposing approach for breast cancer via integrated functional network analysis. Mol. Inform. 2022, 41, 2100300. [Google Scholar] [CrossRef]
- Hu, W.; Tan, C.; He, Y.; Zhang, G.; Xu, Y.; Tang, J. Functional miRNAs in breast cancer drug resistance. Oncotargets Ther. 2018, 11, 1529–1541. [Google Scholar] [CrossRef]
- Natarajan, K.; Xie, Y.; Baer, M.R.; Ross, D.D. Role of breast cancer resistance protein (BCRP/ABCG2) in cancer drug resistance. Biochem. Pharmacol. 2012, 83, 1084–1103. [Google Scholar] [CrossRef]
- Doostmohammadi, A.; Jooya, H.; Ghorbanian, K.; Gohari, S.; Dadashpour, M. Potentials and future perspectives of multi-target drugs in cancer treatment: The next generation anti-cancer agents. Cell Commun. Signal. 2024, 22, 228. [Google Scholar] [CrossRef]
- Ahmad, S.; Raza, K. Identification of 5-nitroindazole as a multitargeted inhibitor for CDK and transferase kinase in lung cancer: A multisampling algorithm-based structural study. Mol. Divers. 2024, 28, 1189–1202. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Singh, V.; Gautam, H.K.; Raza, K. Multisampling-based docking reveals Imidazolidinyl urea as a multitargeted inhibitor for lung cancer: An optimisation followed multi-simulation and in-vitro study. J. Biomol. Struct. Dyn. 2024, 42, 2494–2511. [Google Scholar] [CrossRef]
- Olawale, F.; Iwaloye, O.; Elekofehinti, O.O.; Kikiowo, B.; Oluwarotimi, E.A.; Ilesanmi, K.M.; Akinropo, I.D.; Akinlosotu, O.B.; Adegboyega, A.E.; Ologuntere, T.E. A multi-target approach for the discovery of anti breast cancer agents from plants secondary metabolites. Lett. Drug Des. Discov. 2021, 18, 1009–1023. [Google Scholar] [CrossRef]
- Tanenbaum, D.M.; Wang, Y.; Williams, S.P.; Sigler, P.B. Crystallographic comparison of the estrogen and progesterone receptor’s ligand binding domains. Proc. Natl. Acad. Sci. USA 1998, 95, 5998–6003. [Google Scholar] [CrossRef]
- Aertgeerts, K.; Skene, R.; Yano, J.; Sang, B.-C.; Zou, H.; Snell, G.; Jennings, A.; Iwamoto, K.; Habuka, N.; Hirokawa, A. Structural analysis of the mechanism of inhibition and allosteric activation of the kinase domain of HER2 protein. J. Biol. Chem. 2011, 286, 18756–18765. [Google Scholar] [CrossRef]
- Ashwell, M.A.; Lapierre, J.-M.; Brassard, C.; Bresciano, K.; Bull, C.; Cornell-Kennon, S.; Eathiraj, S.; France, D.S.; Hall, T.; Hill, J. Discovery and optimization of a series of 3-(3-Phenyl-3 H-imidazo [4,5-b] pyridin-2-yl) pyridin-2-amines: Orally bioavailable, selective, and potent ATP-independent Akt inhibitors. J. Med. Chem. 2012, 55, 5291–5310. [Google Scholar] [CrossRef]
- Gajiwala, K.S.; Feng, J.; Ferre, R.; Ryan, K.; Brodsky, O.; Weinrich, S.; Kath, J.C.; Stewart, A. Insights into the aberrant activity of mutant EGFR kinase domain and drug recognition. Structure 2013, 21, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Borsari, C.; Keles, E.; McPhail, J.A.; Schaefer, A.; Sriramaratnam, R.; Goch, W.; Schaefer, T.; De Pascale, M.; Bal, W.; Gstaiger, M. Covalent proximity scanning of a distal cysteine to target PI3Kα. J. Am. Chem. Soc. 2022, 144, 6326–6342. [Google Scholar] [CrossRef]
- Sahu, A.; Ahmad, S.; Imtiyaz, K.; Kizhakkeppurath Kumaran, A.; Islam, M.; Raza, K.; Easwaran, M.; Kurukkan Kunnath, A.; Rizvi, M.A.; Verma, S. In-silico and in-vitro study reveals Ziprasidone as a potential aromatase inhibitor against breast carcinoma. Sci. Rep. 2023, 13, 16545. [Google Scholar]
- Jha, V.; Devkar, S.; Gharat, K.; Kasbe, S.; Matharoo, D.K.; Pendse, S.; Bhosale, A.; Bhargava, A. Screening of phytochemicals as potential inhibitors of breast cancer using structure based multitargeted molecular docking analysis. Phytomed. Plus 2022, 2, 100227. [Google Scholar] [CrossRef]
- Aloqbi, A.A.; Alahdal, H.; Alqosaibi, A.I.; Alnamshan, M.M.; Al-Dhuayan, I.S.; Al-Eidan, A.A.; Alzahrani, H.A.; ALaqeel, N.K.; Alsharif, F.H.; Al Tuwaijri, A. Lucidin from Rubia cordifolia Outperforms FDA-Approved Lapatinib as a Potential Multitargeted Candidate for Breast Cancer Signalling Proteins. Pharmaceuticals 2025, 18, 68. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, N.A.; Huang, H.K.; McDermott, M.S.; Madrid, A.M.; Luo, T.; Ayala, R.; Issakhanian, S.; Gong, K.W.; Lu, M.; Zhang, J. Tucatinib has selective activity in HER2-positive cancers and significant combined activity with approved and novel breast cancer–targeted therapies. Mol. Cancer Ther. 2022, 21, 751–761. [Google Scholar] [CrossRef]
- Sirhan, Z.; Thyagarajan, A.; Sahu, R.P. The efficacy of tucatinib-based therapeutic approaches for HER2-positive breast cancer. Mil. Med. Res. 2022, 9, 39. [Google Scholar] [CrossRef]
- Hoyek, C.; Zheng-Lin, B.; Jones, J.; Bekaii-Saab, T. Tucatinib in the treatment of HER2-overexpressing gastrointestinal cancers: Current insights and future prospects. Expert Opin. Investig. Drugs 2025, 34, 161–168. [Google Scholar] [CrossRef]
- Zhou, F.; Ding, K. Tucatinib (Tukysa): An Oral, Selective HER2 Inhibitor for the Treatment of HER2-Positive Solid Tumors. In Chemistry and Pharmacology of Drug Discovery; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2024; pp. 187–198. [Google Scholar]
- Sankarapandian, V.; Rajendran, R.L.; Miruka, C.O.; Sivamani, P.; Maran, B.A.V.; Krishnamoorthy, R.; Gangadaran, P.; Ahn, B.-C. A review on tyrosine kinase inhibitors for targeted breast cancer therapy. Pathol.-Res. Pract. 2024, 263, 155607. [Google Scholar]
- Liu, H.-N.; Zhu, Y.; Chi, Y.; Zhang, Y.; Li, X.; Wen, W.; Shan, L.-S.; Wang, Y.-T.; Dai, B. Synthetic routes and clinical application of Small-Molecule HER2 inhibitors for cancer therapy. Bioorg. Chem. 2024, 151, 107653. [Google Scholar] [CrossRef]
- Ahmad, S.; Bano, N.; Khanna, K.; Gupta, D.; Raza, K. Reporting multitargeted potency of Tiaprofenic acid against lung cancer: Molecular fingerprinting, MD simulation, and MTT-based cell viability assay studies. Int. J. Biol. Macromol. 2024, 276, 133872. [Google Scholar]
- Ahmad, S.; Bano, N.; Raza, K. Evaluating the polypharmacological potency of FEDPN from ChEMBL BioAssays against lung cancer EGFR, ALK, TrkA and KRAS proteins. Int. J. Biol. Macromol. 2025, 306, 141703. [Google Scholar] [CrossRef]
- Ahmad, S.; Raza, K. An extensive review on lung cancer therapeutics using machine learning techniques: State-of-the-art and perspectives. J. Drug Target. 2024, 32, 635–646. [Google Scholar] [CrossRef]
- Ahmad, S.; Singh, A.P.; Bano, N.; Raza, K.; Singh, J.; Medigeshi, G.R.; Pandey, R.; Gautam, H.K. Integrative analysis discovers Imidurea as dual multitargeted inhibitor of CD69, CD40, SHP2, lysozyme, GATA3, cCBL, and S-cysteinase from SARS-CoV-2 and M. tuberculosis. Int. J. Biol. Macromol. 2024, 270, 132332. [Google Scholar] [CrossRef] [PubMed]
- Karwasra, R.; Khanna, K.; Singh, S.; Ahmad, S.; Verma, S. The Incipient Role of Computational Intelligence in Oncology: Drug Designing, Discovery, and Development. In Computational Intelligence in Oncology; Springer: Singapore, 2022; Volume 1016, pp. 369–384. [Google Scholar]
- Tripathi, M.K.; Ahmad, S.; Tyagi, R.; Dahiya, V.; Yadav, M.K. Fundamentals of molecular modeling in drug design. In Computer Aided Drug Design (CADD): From Ligand-Based Methods to Structure-Based Approaches; Elsevier: Amsterdam, The Netherlands, 2022; pp. 125–155. [Google Scholar]
- Famuyiwa, S.O.; Ahmad, S.; Fakola, E.G.; Olusola, A.J.; Adesida, S.A.; Obagunle, F.O.; Raza, K.; Ugwo, J.P.; Oyelekan, E.I.; Faloye, K.O. Comprehensive computational studies of naturally occurring kuguacins as antidiabetic agents by targeting visfatin. Chem. Afr. 2023, 6, 1415–1427. [Google Scholar] [CrossRef]
- Maestro, S. Maestro; Schrödinger, LLC.: New York, NY, USA, 2024. [Google Scholar]
- Release, S. LigPrep; Schrödinger, LLC.: New York, NY, USA, 2024. [Google Scholar]
- Jorgensen, W.L.; Tirado-Rives, J. The OPLS [optimized potentials for liquid simulations] potential functions for proteins, energy minimizations for crystals of cyclic peptides and crambin. J. Am. Chem. Soc. 1988, 110, 1657–1666. [Google Scholar] [CrossRef] [PubMed]
- Shelley, J.C.; Cholleti, A.; Frye, L.L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: A software program for pK a prediction and protonation state generation for drug-like molecules. J. Comput.-Aided Mol. Des. 2007, 21, 681–691. [Google Scholar] [CrossRef]
- Ahmad, S.; Dahiya, V.; Vibhuti, A.; Pandey, R.P.; Tripathi, M.K.; Yadav, M.K. Therapeutic protein-based vaccines. In Protein-Based Therapeutics; Springer Nature: Singapore, 2023; pp. 355–384. [Google Scholar]
- Ahmad, S.; Kishan, A.; Chitkara, P.; Asiri, S.A.; Eswaran, M.; Mehta, S.; Alam, M. Natural product-based drug designing for treatment of human parasitic diseases. In Natural Product Based Drug Discovery Against Human Parasites: Opportunities and Challenges; Springer Nature: Singapore, 2023; pp. 37–59. [Google Scholar]
- Ahmad, S.; Pasha KM, M.; Raza, K.; Rafeeq, M.M.; Habib, A.H.; Eswaran, M.; Yadav, M.K. Reporting dinaciclib and theodrenaline as a multitargeted inhibitor against SARS-CoV-2: An in-silico study. J. Biomol. Struct. Dyn. 2023, 41, 4013–4023. [Google Scholar] [CrossRef]
- Ahmad, S.; Bano, N.; Raza, K. RCSB Protein Data Bank: Revolutionising drug discovery and design for over five decades. Med. Data Min. 2025, 8, 7–11. [Google Scholar] [CrossRef]
- Release, S. Schrödinger Suite 2024 Protein Preparation Wizard; Epik, Schrödinger, LLC.: New York, NY, USA, 2024. [Google Scholar]
- Olsson, M.H.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent treatment of internal and surface residues in empirical p K a predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef]
- Release, S. Receptor Grid Generation; Schrödinger, LLC.: New York, NY, USA, 2024. [Google Scholar]
- Release, S. Glide; Schrödinger, LLC.: New York, NY, USA, 2024. [Google Scholar]
- QikProp, S. Schrödinger Release 2024; Maestro LLC.: New York, NY, USA, 2024. [Google Scholar]
- Acharya, R.; Chacko, S.; Bose, P.; Lapenna, A.; Pattanayak, S.P. Structure based multitargeted molecular docking analysis of selected furanocoumarins against breast cancer. Sci. Rep. 2019, 9, 15743. [Google Scholar] [CrossRef]
- Rana, M.; Ahmedi, S.; Fatima, A.; Ahmad, S.; Siddiqui, N.; Raza, K.; Manzoor, N.; Javed, S. Synthesis, single crystal, TD-DFT, molecular dynamics simulation and DNA binding studies of carbothioamide analog. J. Mol. Struct. 2023, 1287, 135701. [Google Scholar] [CrossRef]
- Bochevarov, A.D.; Harder, E.; Hughes, T.F.; Greenwood, J.R.; Braden, D.A.; Philipp, D.M.; Rinaldo, D.; Halls, M.D.; Zhang, J.; Friesner, R.A. Jaguar: A high-performance quantum chemistry software program with strengths in life and materials sciences. Int. J. Quantum Chem. 2013, 113, 2110–2142. [Google Scholar] [CrossRef]
- Release, S. Jaguar; Schrödinger, LLC.: New York, NY, USA, 2024. [Google Scholar]
- Witte, J.; Mardirossian, N.; Neaton, J.B.; Head-Gordon, M. Assessing DFT-D3 damping functions across widely used density functionals: Can we do better? J. Chem. Theory Comput. 2017, 13, 2043–2052. [Google Scholar] [CrossRef] [PubMed]
- Carbó, R.; Riera, J.M. A General SCF Theory; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2012; Volume 5. [Google Scholar]
- Hamilton, T.P.; Pulay, P. Direct inversion in the iterative subspace (DIIS) optimization of open-shell, excited-state, and small multiconfiguration SCF wave functions. J. Chem. Phys. 1986, 84, 5728–5734. [Google Scholar] [CrossRef]
- Schlegel, H.B. Geometry optimization. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 1, 790–809. [Google Scholar] [CrossRef]
- Banerjee, P.; Kemmler, E.; Dunkel, M.; Preissner, R. ProTox 3.0: A webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2024, 52, W513–W520. [Google Scholar] [CrossRef]
- Fu, L.; Shi, S.; Yi, J.; Wang, N.; He, Y.; Wu, Z.; Peng, J.; Deng, Y.; Wang, W.; Wu, C. ADMETlab 3.0: An updated comprehensive online ADMET prediction platform enhanced with broader coverage, improved performance, API functionality and decision support. Nucleic Acids Res. 2024, 52, W422–W431. [Google Scholar] [CrossRef]
- Kaczor, A.A.; Zięba, A.; Matosiuk, D. The application of WaterMap-guided structure-based virtual screening in novel drug discovery. Expert Opin. Drug Discov. 2024, 19, 73–83. [Google Scholar] [CrossRef]
- Release, S. Desmond Molecular Dynamics System, DE Shaw Research, New York, NY, USA, 2024. Maestro-Desmond Interoperability Tools; Schrödinger: New York, NY, USA, 2024. [Google Scholar]
- Mark, P.; Nilsson, L. Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- McDonald, I. NpT-ensemble Monte Carlo calculations for binary liquid mixtures. Mol. Phys. 1972, 23, 41–58. [Google Scholar] [CrossRef]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.; Hou, T. End-point binding free energy calculation with MM/PBSA and MM/GBSA: Strategies and applications in drug design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDB ID | Case | Ligand | Resolution | Gridbox Xcent | Gridbox Xrange | Gridbox Ycent | Gridbox Yrange | Gridbox Zcent | Gridbox Zrange | |
|---|---|---|---|---|---|---|---|---|---|---|
| 1A52 | All | Native | 2.800 | 106.693 | 23.845 | 14.700 | 23.845 | 96.263 | 23.845 | |
| 3PP0 | 2.250 | 16.642 | 31.913 | 15.920 | 31.913 | 27.037 | 31.913 | |||
| 4EJN | 2.190 | 35.298 | 27.473 | 43.719 | 27.473 | 18.723 | 27.473 | |||
| 4I23 | 2.800 | −0.219 | 25.144 | −52.639 | 25.144 | −22.501 | 25.144 | |||
| 7R9V | 2.690 | −19.281 | 37.238 | 10.026 | 37.238 | 28.391 | 37.238 | |||
| PDB ID | Case | Ligand | Docking Score | RMSD | MM/GBSA ΔG_Bind | lig efficiency | lig efficiency | Prime H-bond | Prime Bond Covalent | Prime Coulomb |
| 1A52 | Native | Respective Native Ligands | −7.244 | 0.040 | −33.720 | −8.775 | −1.944 | −143.120 | 118.605 | −8762.180 |
| 3PP0 | −6.935 | 0.750 | −31.890 | −8.208 | −1.832 | −138.970 | 113.491 | −8526.400 | ||
| 4EJN | −7.801 | 0.110 | −36.840 | −9.112 | −1.964 | −149.060 | 124.310 | −8955.710 | ||
| 4I23 | −6.618 | 0.040 | −29.450 | −7.836 | −1.798 | −132.450 | 109.904 | −8234.570 | ||
| 7R9V | −7.004 | 0.220 | −34.290 | −8.554 | −1.917 | −145.610 | 120.387 | −8678.960 | ||
| PDB ID | Case | Ligand | Docking Score | MM/GBSA ΔG_Bind | lig efficiency | lig efficiency | Prime H-bond | Prime Bond Covalent | Prime Coulomb | |
| 1A52 | Identified | Isoetin | −10.397 | −31.810 | −7.776 | −2.169 | −135.030 | 105.074 | −7878.840 | |
| 3PP0 | Isoetin | −10.399 | −37.040 | −9.054 | −2.525 | −156.790 | 135.938 | −9008.030 | ||
| 4EJN | Isoetin | −9.901 | −47.310 | −11.564 | −3.225 | −199.250 | 209.235 | −11,727.040 | ||
| 4I23 | Isoetin | −9.639 | −36.000 | −8.800 | −2.455 | −158.390 | 144.182 | −9760.740 | ||
| 7R9V | Isoetin | −13.903 | −44.380 | −10.847 | −3.026 | −466.300 | 329.598 | −27,846.060 | ||
| 1A52 | Control | Tucatinib | −4.875 | −29.680 | −6.475 | −1.237 | −133.420 | 109.923 | −7640.020 | |
| 3PP0 | Tucatinib | −10.948 | −68.730 | −14.995 | −2.864 | −154.040 | 137.508 | −8813.450 | ||
| 4EJN | Tucatinib | −7.933 | −50.770 | −11.076 | −2.115 | −199.250 | 209.235 | −11,727.040 | ||
| 4I23 | Tucatinib | −5.782 | −54.390 | −11.866 | −2.266 | −157.410 | 146.888 | −9506.870 | ||
| 7R9V | Tucatinib | −6.319 | −29.400 | −6.414 | −1.225 | −466.300 | 329.598 | −27,846.060 | ||
| 1A52 | Control (Respective FDA-Approved) | Tamoxifen | −7.354 | −0.263 | −1.698 | −132.863 | 107.921 | −7509.527 | −7640.020 | |
| 3PP0 | Tucatinib | −10.948 | −68.730 | −14.995 | −2.864 | −154.040 | 137.508 | −8813.450 | ||
| 4EJN | Erlotinib | −8.679 | −0.299 | −1.987 | −156.149 | 146.785 | −9456.162 | −11,727.040 | ||
| 4I23 | Alpelisib | −8.842 | −0.295 | −2.009 | −466.296 | 329.598 | −27,846.064 | −9506.870 | ||
| 7R9V | Capivasertib | −6.961 | −0.232 | −1.582 | −199.252 | 209.235 | −11,727.038 | −27,846.060 | ||
| Descriptors | Isoetin | Tucatinib | Descriptors | Isoetin | Tucatinib |
|---|---|---|---|---|---|
| #NandO | 7 | 10 | PSA | 144.553 | 103.512 |
| #acid | 0 | 0 | % HumanOralAbs | 50.196 | 100 |
| #amide | 0 | 0 | QPPCaco | 15.721 | 496.085 |
| #amidine | 0 | 0 | QPPMDCK | 5.558 | 231.89 |
| #amine | 0 | 0 | Blood–Brain Barrier permeability (QPlogBB) | −2.477 | −1.396 |
| #in34 | 0 | 0 | hERG liability (QPlogHERG) | −5.064 | −7.499 |
| #in56 | 16 | 30 | QPlogKhsa | −0.342 | 0.752 |
| #metab | 5 | 2 | QPlogKp | −5.692 | −1.974 |
| #nonHatm | 22 | 36 | QPlogPC16 | 10.752 | 16.722 |
| #noncon | 0 | 2 | QPlogPo/w | 0.314 | 4.635 |
| #ringatoms | 16 | 30 | QPlogPoct | 18.481 | 26.796 |
| #rotor | 5 | 6 | QPlogPw | 14.424 | 15.029 |
| #rtvFG | 0 | 0 | QPlogS | −2.904 | −7.373 |
| #stars | 0 | 2 | QPpolrz | 27.481 | 53.452 |
| CNS | −2 | −2 | SASA | 518.893 | 831.91 |
| HumanOralAbs | 2 | 1 | SAamideO | 0 | 0 |
| RuleOfFive | 0 | 0 | SAfluorine | 0 | 0 |
| RuleOfThree | 1 | 1 | WPSA | 0 | 0 |
| ACxDN^.5/SA | 0.0202354 | 0.0135997 | accptHB | 5.25 | 8 |
| CIQPlogS | −4.043 | −7.401 | dip^2/V | 0.0193286 | 0.1185603 |
| EA(eV) | 0.318 | 1.006 | dipole | 4.094 | 13.266 |
| FISA | 295.205 | 137.124 | donorHB | 4 | 2 |
| FOSA | 0 | 269.985 | glob | 0.8476223 | 0.7564299 |
| IP(eV) | 8.912 | 8.156 | mol MW | 302.24 | 480.528 |
| Jm | 0.001 | 0 | volume | 867.306 | 1484.293 |
| PISA | 223.688 | 424.801 | Type | small | small |
| CYP1A2 inhibitior (Yes/No) | Yes | Yes | CYP2D6 inhibitior (Yes/No) | Yes | Yes |
| CYP2C19 inhibitior (Yes/No) | Yes | Yes | CYP3A4 inhibitior (Yes/No) | No | Yes |
| CYP2C9 inhibitior (Yes/No) | Yes | Yes | Synthetic accessibility | 3.12 | 4.01 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al Khzem, A.H.; Alturki, M.S.; Almuzaini, O.K.; Wali, S.M.; Almaghrabi, M.; Aldawsari, M.F.; Abduljabbar, M.H.; Alnemari, R.M.; Almalki, A.H.; Rants’o, T.A. Isoetin from Isoetaceae Exhibits Superior Pentatransferase Inhibition in Breast Cancer: Comparative Computational Profiling with FDA-Approved Tucatinib. Pharmaceuticals 2025, 18, 662. https://doi.org/10.3390/ph18050662
Al Khzem AH, Alturki MS, Almuzaini OK, Wali SM, Almaghrabi M, Aldawsari MF, Abduljabbar MH, Alnemari RM, Almalki AH, Rants’o TA. Isoetin from Isoetaceae Exhibits Superior Pentatransferase Inhibition in Breast Cancer: Comparative Computational Profiling with FDA-Approved Tucatinib. Pharmaceuticals. 2025; 18(5):662. https://doi.org/10.3390/ph18050662
Chicago/Turabian StyleAl Khzem, Abdulaziz H., Mansour S. Alturki, Ohood K. Almuzaini, Saad M. Wali, Mohammed Almaghrabi, Mohammed F. Aldawsari, Maram H. Abduljabbar, Reem M. Alnemari, Atiah H. Almalki, and Thankhoe A. Rants’o. 2025. "Isoetin from Isoetaceae Exhibits Superior Pentatransferase Inhibition in Breast Cancer: Comparative Computational Profiling with FDA-Approved Tucatinib" Pharmaceuticals 18, no. 5: 662. https://doi.org/10.3390/ph18050662
APA StyleAl Khzem, A. H., Alturki, M. S., Almuzaini, O. K., Wali, S. M., Almaghrabi, M., Aldawsari, M. F., Abduljabbar, M. H., Alnemari, R. M., Almalki, A. H., & Rants’o, T. A. (2025). Isoetin from Isoetaceae Exhibits Superior Pentatransferase Inhibition in Breast Cancer: Comparative Computational Profiling with FDA-Approved Tucatinib. Pharmaceuticals, 18(5), 662. https://doi.org/10.3390/ph18050662