
3-(3-Azabicyclo[2, 2, 1]heptan-2-yl)-1,2,4-oxadiazoles as Novel Potent DPP-4 Inhibitors to Treat T2DM

 ,
,  , , and
, , and
Abstract
1. Introduction
2. Results
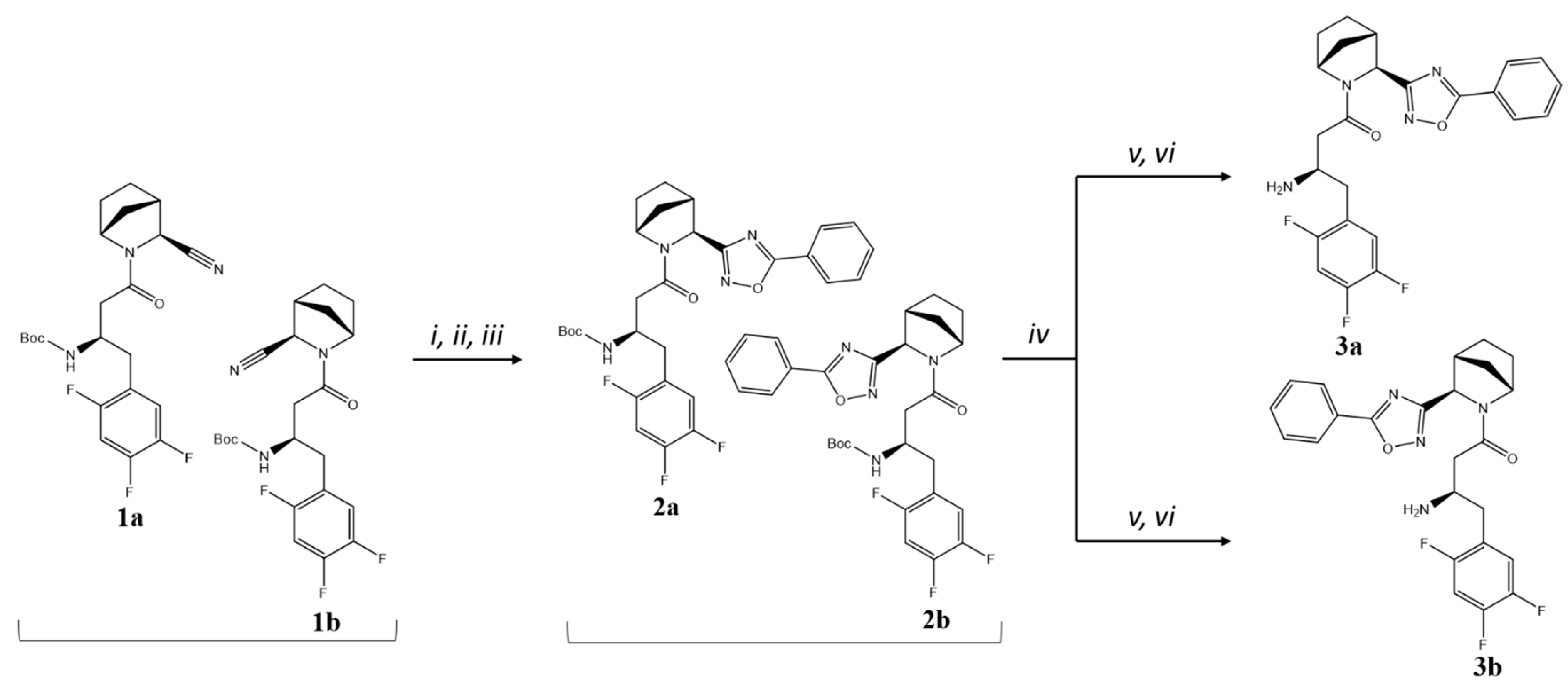
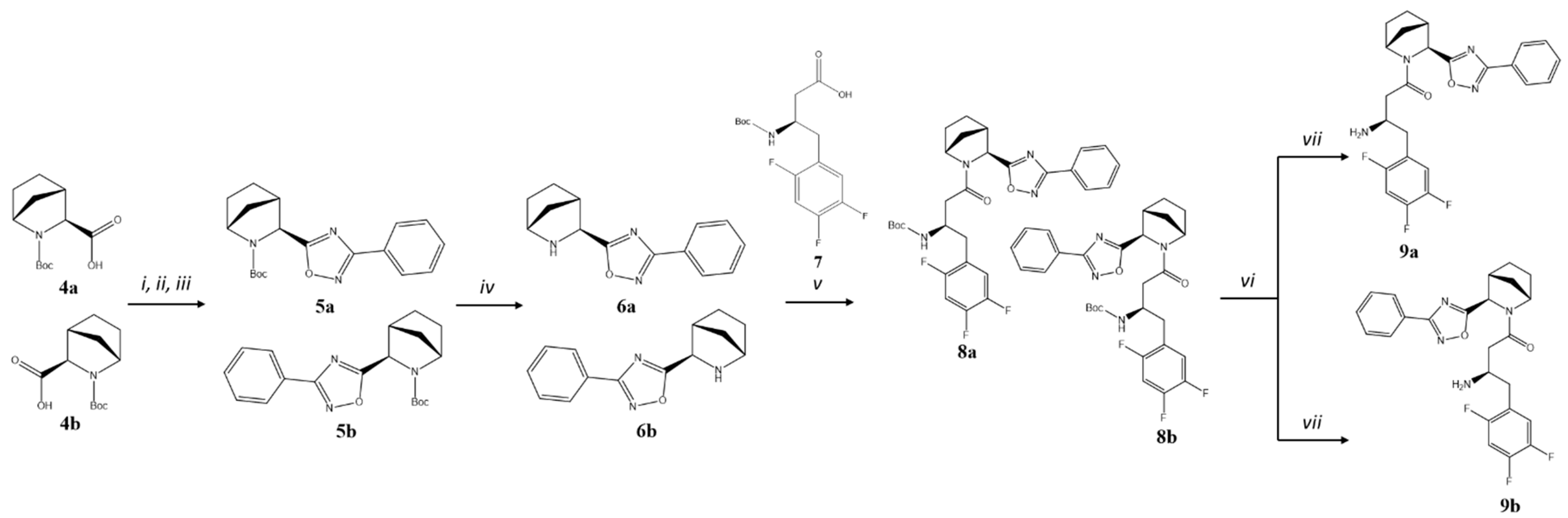
2.1. Chemical Synthesis
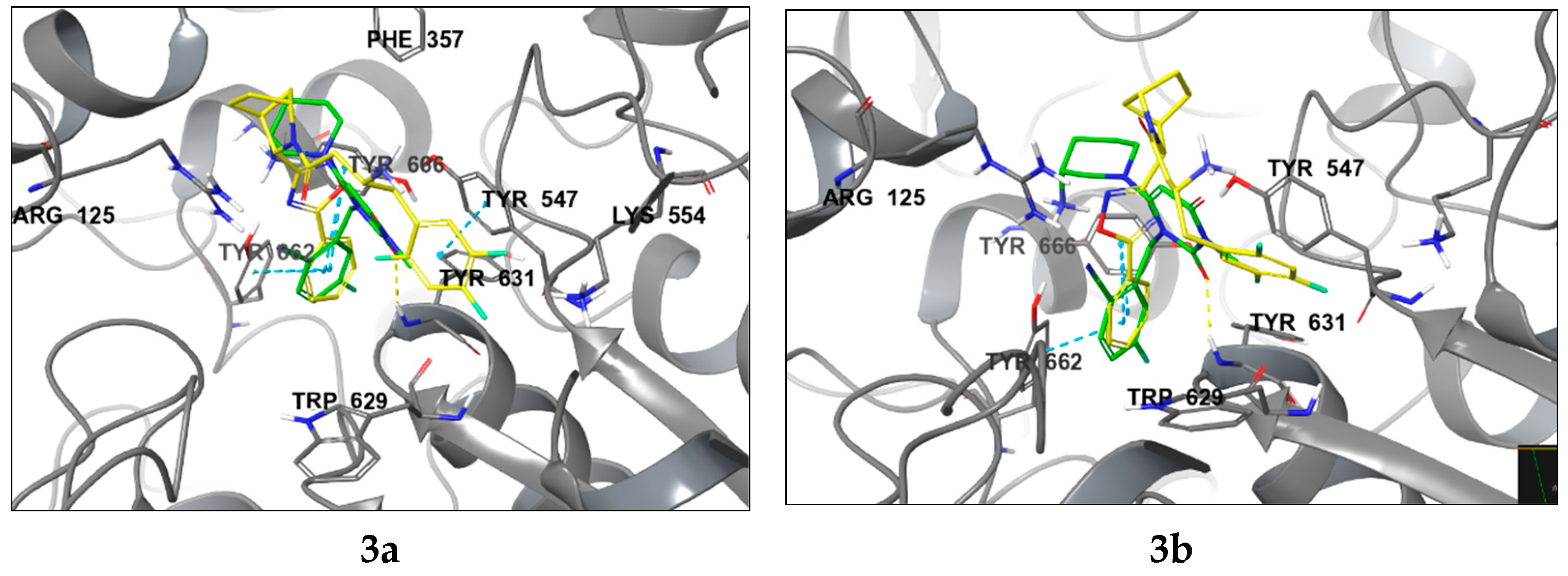
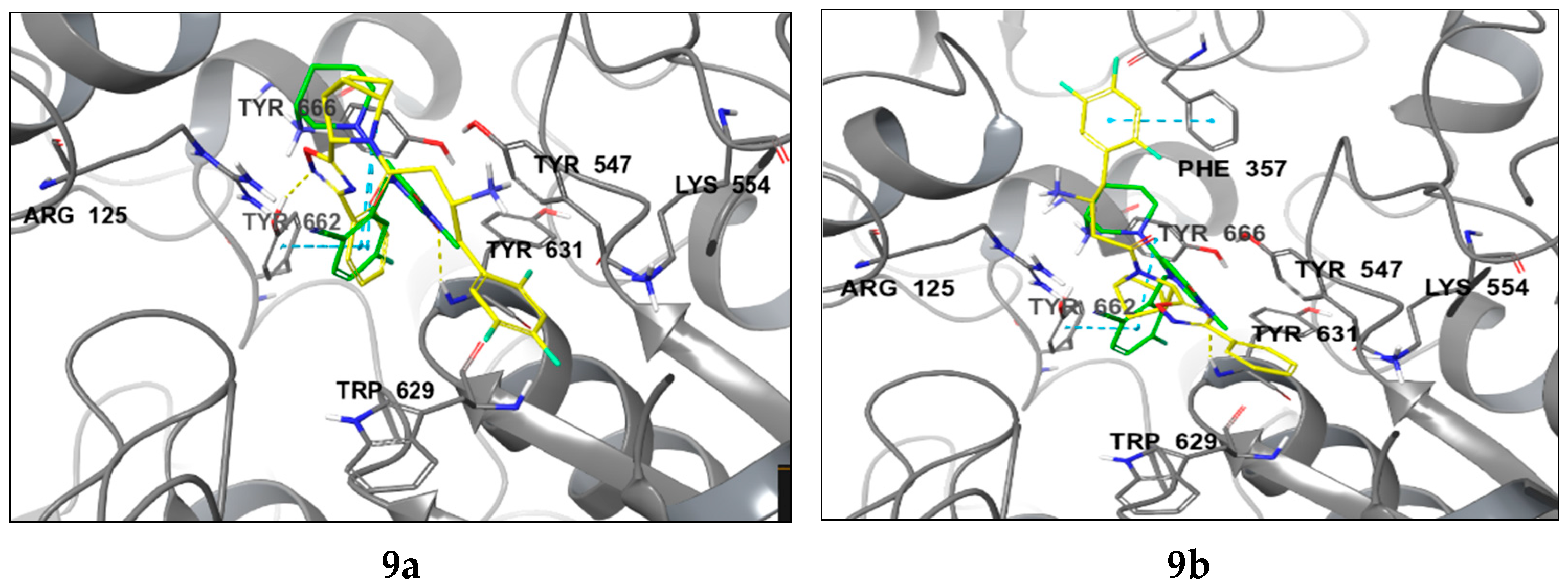
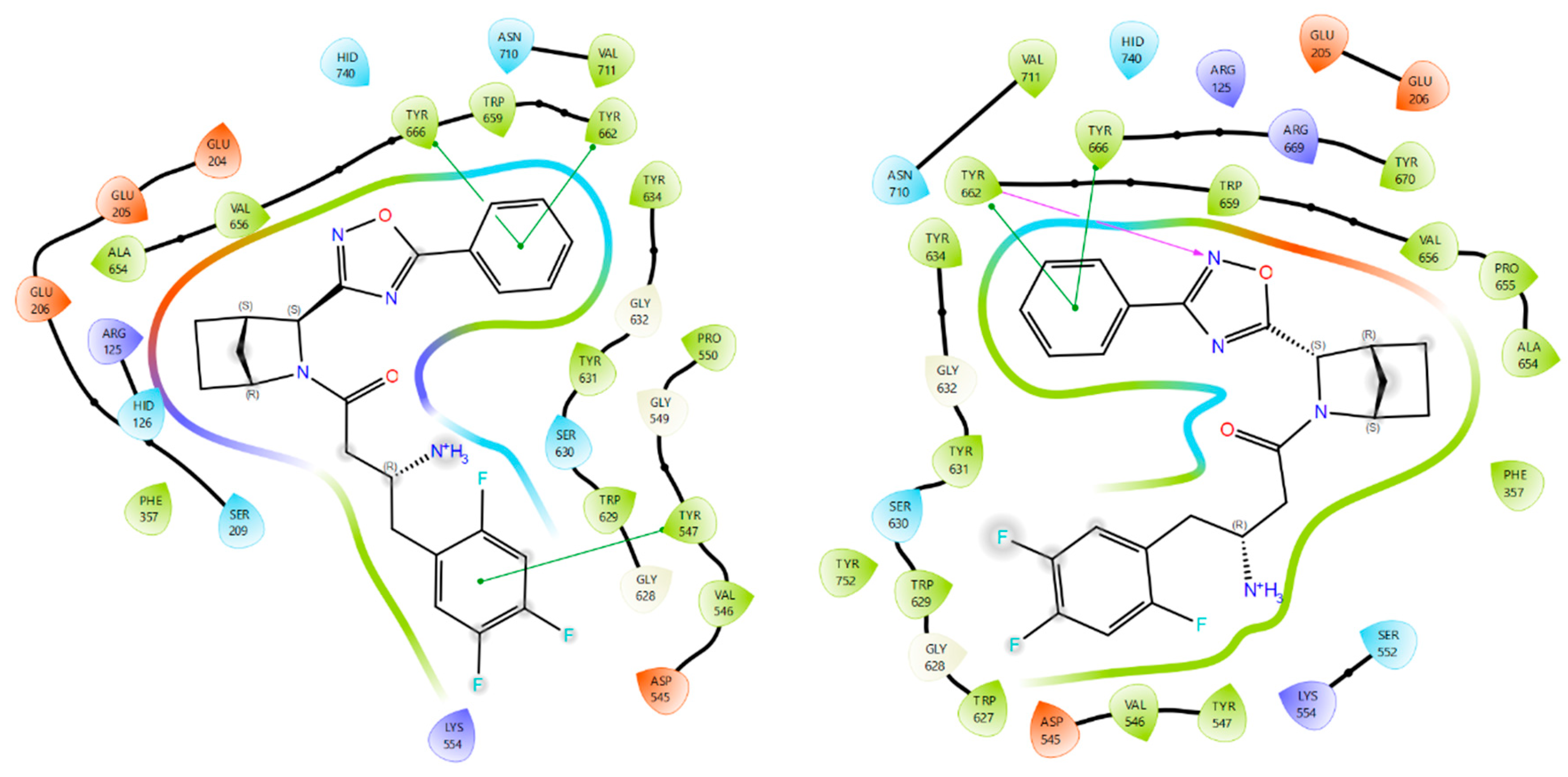
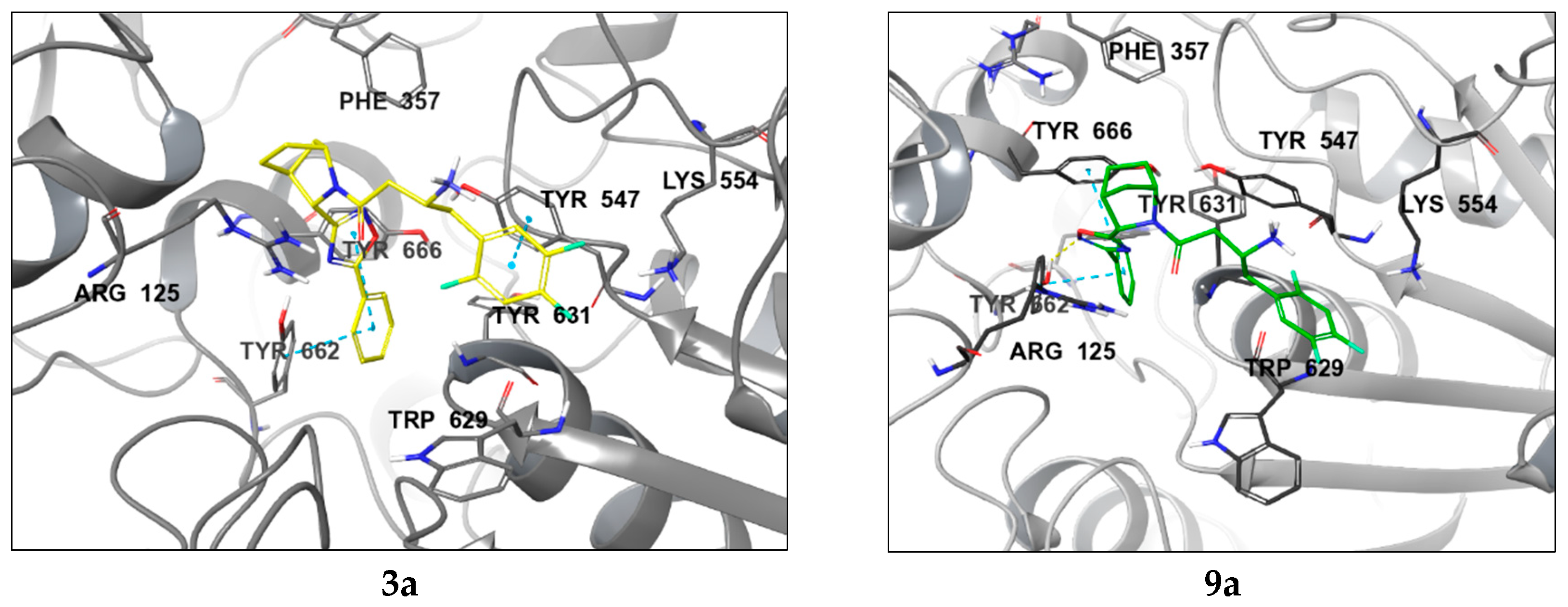
2.2. Molecular Modelling
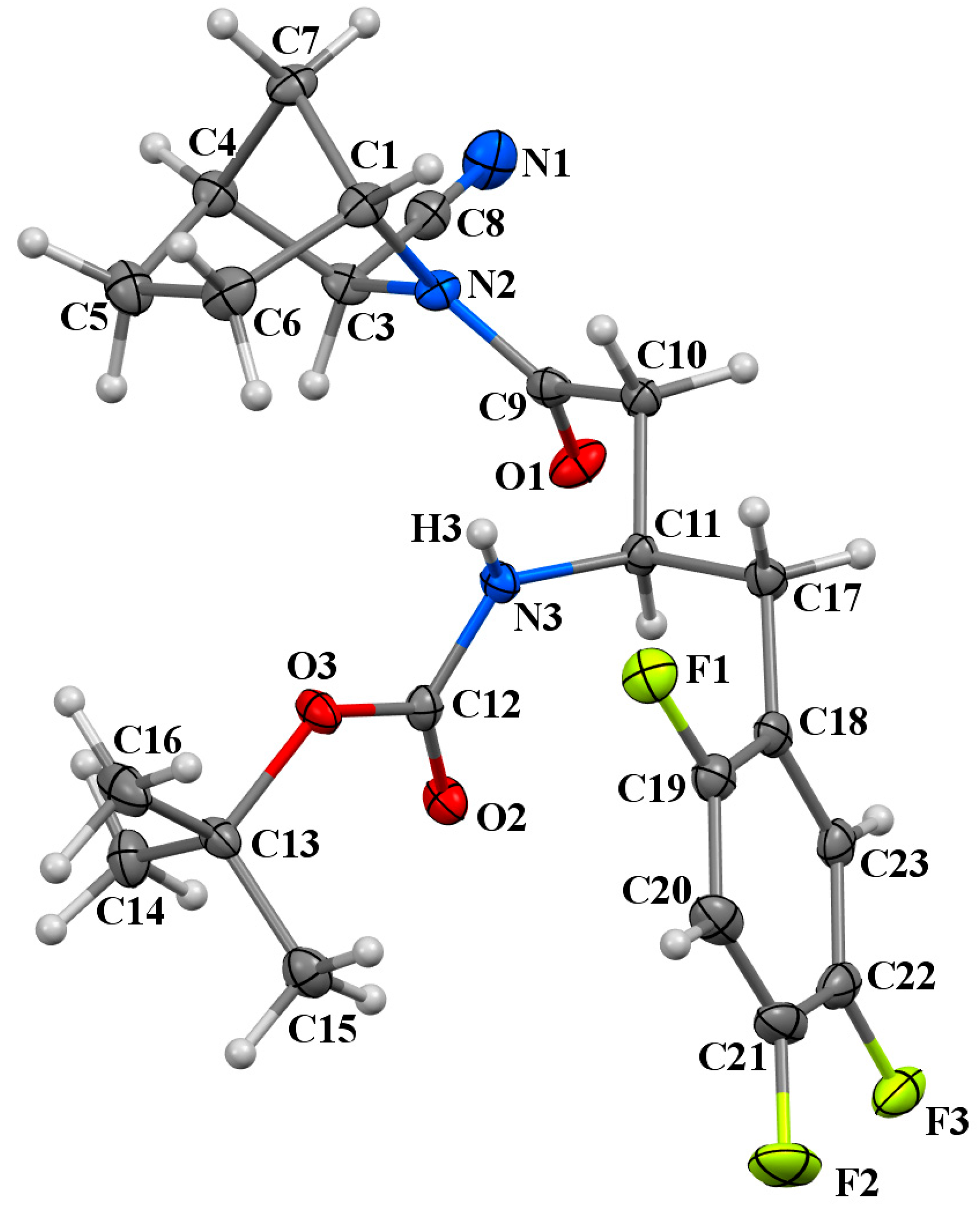
2.3. X-Ray Diffraction Study
2.4. Inhibitory Activity Assays
3. Discussion
4. Materials and Methods
4.1. Chemical Synthesis
4.2. Molecular Modelling
4.3. Structure and Purity Confirmation
4.4. X-Ray Diffraction Study
4.5. Inhibitory Activity Evaluation
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kahn, S.E.; Hull, R.L.; Utzschneider, K.M. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature 2006, 444, 840–846. [Google Scholar] [CrossRef] [PubMed]
- Halban, P.A.; Polonsky, K.S.; Bowden, D.W.; Hawkins, M.A.; Ling, C.; Mather, K.J.; Powers, A.C.; Rhodes, C.J.; Sussel, L.; Weir, G.C. β-Cell failure in type 2 diabetes: Postulated mechanisms and prospects for prevention and treatment. Diabetes Care 2014, 37, 1751–1758. [Google Scholar] [CrossRef] [PubMed]
- Classification and diagnosis of diabetes: Standards of Medical Care in Diabetes—2020. Diabetes Care 2020, 43, S14–S31. [CrossRef]
- Davies, M.J.; D’Alessio, D.A.; Fradkin, J.; Kernan, W.N.; Mathieu, C.; Mingrone, G.; Rossing, P.; Tsapas, A.; Wexler, D.J.; Buse, J.B. Management of hyperglycemia in type 2 diabetes, 2018. A consensus report by the American Diabetes Association (ADA) and the european association for the study of diabetes (EASD). Diabetes Care 2018, 41, 2669–2701. [Google Scholar] [CrossRef]
- Dahlén, A.D.; Dashi, G.; Maslov, I.; Attwood, M.M.; Jonsson, J.; Trukhan, V.; Schiöth, H.B. Trends in Antidiabetic Drug Discovery: FDA Approved Drugs, New Drugs in Clinical Trials and Global Sales. Front. Pharmacol. 2022, 12, 807548. [Google Scholar] [CrossRef]
- Drucker, D.J.; Nauck, M.A. The incretin system: Glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet 2006, 368, 1696–1705. [Google Scholar] [CrossRef]
- Gallwitz, B. Clinical use of DPP-4 inhibitors. Front. Endocrinol. 2019, 10, 389. [Google Scholar] [CrossRef]
- Inzucchi, S.E.; Bergenstal, R.M.; Buse, J.B.; Diamant, M.; Ferrannini, E.; Nauck, M.; Peters, A.L.; Tsapas, A.; Wender, R.; Matthews, D.R. Management of Hyperglycemia in Type 2 Diabetes, 2015: A Patient-Centered Approach: Update to a position statement of the american diabetes association and the european association for the study of diabetes. Diabetes Care 2015, 38, 140–149. [Google Scholar] [CrossRef]
- Thornberry, N.; Weber, A. Discovery of JANUVIA™ (Sitagliptin), a Selective Dipeptidyl Peptidase IV Inhibitor for the Treatment of Type2 Diabetes. Curr. Top. Med. Chem. 2007, 7, 557–568. [Google Scholar] [CrossRef]
- Augeri, D.J.; Robl, J.A.; Betebenner, D.A.; Magnin, D.R.; Khanna, A.; Robertson, J.G.; Wang, A.; Simpkins, L.M.; Taunk, P.; Huang, Q.; et al. Discovery and preclinical profile of saxagliptin (BMS-477118): A highly potent, long-acting, orally active dipeptidyl peptidase IV inhibitor for the treatment of type 2 diabetes. J. Med. Chem. 2005, 48, 5025–5037. [Google Scholar] [CrossRef]
- Feng, J.; Zhang, Z.; Wallace, M.B.; Stafford, J.A.; Kaldor, S.W.; Kassel, D.B.; Navre, M.; Shi, L.; Skene, R.J.; Asakawa, T.; et al. Discovery of Alogliptin: A Potent, Selective, Bioavailable, and Efficacious Inhibitor of Dipeptidyl Peptidase IV. J. Med. Chem. 2008, 51, 4357. [Google Scholar] [CrossRef]
- Eckhardt, M.; Langkopf, E.; Mark, M.; Tadayyon, M.; Thomas, L.; Nar, H.; Pfrengle, W.; Guth, B.; Lotz, R.; Sieger, P.; et al. 8-(3-(R)-aminopiperidin-1-yl)-7-but-2-ynyl-3-methyl-1-(4-methyl-quinazolin- 2-ylmethyl)-3,7-dihydropurine-2,6-dione (BI 1356), a highly potent, selective, long-acting, and orally bioavailable DPP-4 inhibitor for the treatment of type 2 diabetes. J. Med. Chem. 2007, 50, 6450–6453. [Google Scholar] [CrossRef] [PubMed]
- Maslov, I.O.; Zinevich, T.V.; Kirichenko, O.G.; Trukhan, M.V.; Shorshnev, S.V.; Tuaeva, N.O.; Gureev, M.A.; Dahlén, A.D.; Porozov, Y.B.; Schiöth, H.B.; et al. Design, Synthesis and Biological Evaluation of Neogliptin, a Novel 2-Azabicyclo[2.2.1]heptane-Based Inhibitor of Dipeptidyl Peptidase-4 (DPP-4). Pharmaceuticals 2022, 15, 273. [Google Scholar] [CrossRef] [PubMed]
- Nordhoff, S.; Bulat, S.; Cerezo-Gálvez, S.; Hill, O.; Hoffmann-Enger, B.; López-Canet, M.; Rosenbaum, C.; Rummey, C.; Thiemann, M.; Matassa, V.G.; et al. The design of potent and selective inhibitors of DPP-4: Optimization of ADME properties by amide replacements. Bioorg. Med. Chem. Lett. 2009, 19, 6340–6345. [Google Scholar] [CrossRef]
- Caneschi, W.; Enes, K.B.; de Mendonça, C.C.; Fernandes, F.d.S.; Miguel, F.B.; Martins, J.d.S.; Le Hyaric, M.; Pinho, R.R.; Duarte, L.M.; de Oliveira, M.A.L.; et al. Synthesis and anticancer evaluation of new lipophilic 1,2,4 and 1,3,4-oxadiazoles. Eur. J. Med. Chem. 2019, 165, 18–30. [Google Scholar] [CrossRef]
- Da Wu, K.; Chen, G.S.; Liu, J.R.; Hsieh, C.E.; Chern, J.W. Acrylamide Functional Group Incorporation Improves Drug-like Properties: An Example with EGFR Inhibitors. ACS Med. Chem. Lett. 2019, 10, 22–26. [Google Scholar] [CrossRef]
- Glomb, T.; Świątek, P. Antimicrobial activity of 1,3,4-oxadiazole derivatives. Int. J. Mol. Sci. 2021, 22, 6979. [Google Scholar] [CrossRef]
- Zhou, Y.; Varadharaj, S.; Zhao, X.; Parinandi, N.; Flavahan, N.A.; Zweier, J.L. Acetylcholine causes endothelium-dependent contraction of mouse arteries. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, 1027–1033. [Google Scholar] [CrossRef]
- Nordhoff, S.; Cerezo-Gálvez, S.; Deppe, H.; Hill, O.; López-Canet, M.; Rummey, C.; Thiemann, M.; Matassa, V.G.; Edwards, P.J.; Feurer, A. Discovery of β-homophenylalanine based pyrrolidin-2-ylmethyl amides and sulfonamides as highly potent and selective inhibitors of dipeptidyl peptidase IV. Bioorg. Med. Chem. Lett. 2009, 19, 4201–4203. [Google Scholar] [CrossRef]
- Ruan, B.; Zhang, Y.; Tadesse, S.; Preston, S.; Taki, A.C.; Jabbar, A.; Hofmann, A.; Jiao, Y.; Garcia-Bustos, J.; Harjani, J.; et al. Synthesis and structure-activity relationship study of pyrrolidine-oxadiazoles as anthelmintics against Haemonchus contortus. Eur. J. Med. Chem. 2020, 190, 112100. [Google Scholar] [CrossRef]
- De Vasconcelos, N.M.; Vliegen, G.; Gonçalves, A.; De Hert, E.; Martín-Pérez, R.; Van Opdenbosch, N.; Jallapally, A.; Geiss-Friedlander, R.; Lambeir, A.-M.; Augustyns, K.; et al. DPP8/DPP9 inhibition elicits canonical Nlrp1b in fl ammasome hallmarks in murine macrophages. Life Sci. Alliance 2019, 2, e201900313. [Google Scholar] [CrossRef] [PubMed]
- Repasky, M.P.; Shelley, M.; Friesner, R.A. Flexible Ligand Docking with Glide. Curr. Protoc. Bioinform. 2007, 18, 8.12.1–8.12.36. [Google Scholar] [CrossRef] [PubMed]
- Grimshaw, C.E.; Jennings, A.; Kamran, R.; Ueno, H.; Nishigaki, N.; Kosaka, T.; Tani, A.; Sano, H.; Kinugawa, Y.; Koumura, E.; et al. Trelagliptin (syr-472, zafatek), novel once-weekly treatment for type 2 diabetes, inhibits dipeptidyl peptidase-4 (dpp-4) via a non-covalent mechanism. PLoS ONE 2016, 11, e0157509. [Google Scholar] [CrossRef] [PubMed]
- Suenaga, A.; Okimoto, N.; Hirano, Y.; Fukui, K. An efficient computational method for calculating ligand binding affinities. PLoS ONE 2012, 7, e42846. [Google Scholar] [CrossRef]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure | GScore (kcal/mol) | LE (kcal/atom) | Lipo (kcal/mol) | ΔG Lipo | ΔG | ΔG Strain |
| Trelagliptin | −8.33 | −0.32 | −1.27 | −2.20 | −30.19 | 0.01 |
| 3a | −6.54 | −0.20 | −2.04 | −8.16 | −29.17 | −4.96 |
| 3b | −5.85 | −0.18 | −1.35 | −2.68 | −32.96 | −1.19 |
| 9a | −6.37 | −0.19 | −2.78 | −8.04 | −31.11 | −4.26 |
| 9b | −6.01 | −0.18 | −1.33 | −2.96 | −29.72 | 2.32 |
| Structure | DPP-4 IC50, nM | DPP-8 IC50, nM | DPP-9 IC50, nM |
|---|---|---|---|
| Neogliptin [13] | 16.8 | >1000 | >1000 |
| 3a | 21.6 | >1000 | >1000 |
| 3b | >1000 | >1000 | >1000 |
| 9a | 4.3 | >1000 | >1000 |
| 9b | >1000 | >1000 | >1000 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zinevich, T.V.; Maslov, I.O.; Kirichenko, O.G.; Shorshnev, S.V.; Gureev, M.A.; Dolgushin, F.M.; Porozov, Y.B.; Trukhan, V.M. 3-(3-Azabicyclo[2, 2, 1]heptan-2-yl)-1,2,4-oxadiazoles as Novel Potent DPP-4 Inhibitors to Treat T2DM. Pharmaceuticals 2025, 18, 642. https://doi.org/10.3390/ph18050642
Zinevich TV, Maslov IO, Kirichenko OG, Shorshnev SV, Gureev MA, Dolgushin FM, Porozov YB, Trukhan VM. 3-(3-Azabicyclo[2, 2, 1]heptan-2-yl)-1,2,4-oxadiazoles as Novel Potent DPP-4 Inhibitors to Treat T2DM. Pharmaceuticals. 2025; 18(5):642. https://doi.org/10.3390/ph18050642
Chicago/Turabian StyleZinevich, Tatiana V., Ivan O. Maslov, Olga G. Kirichenko, Sergey V. Shorshnev, Maxim A. Gureev, Fedor M. Dolgushin, Yuri B. Porozov, and Vladimir M. Trukhan. 2025. "3-(3-Azabicyclo[2, 2, 1]heptan-2-yl)-1,2,4-oxadiazoles as Novel Potent DPP-4 Inhibitors to Treat T2DM" Pharmaceuticals 18, no. 5: 642. https://doi.org/10.3390/ph18050642
APA StyleZinevich, T. V., Maslov, I. O., Kirichenko, O. G., Shorshnev, S. V., Gureev, M. A., Dolgushin, F. M., Porozov, Y. B., & Trukhan, V. M. (2025). 3-(3-Azabicyclo[2, 2, 1]heptan-2-yl)-1,2,4-oxadiazoles as Novel Potent DPP-4 Inhibitors to Treat T2DM. Pharmaceuticals, 18(5), 642. https://doi.org/10.3390/ph18050642