
New Challenging Systemic Therapies for Juvenile Scleroderma: A Comprehensive Review

 ,
,  and
and 
Abstract
1. Introduction
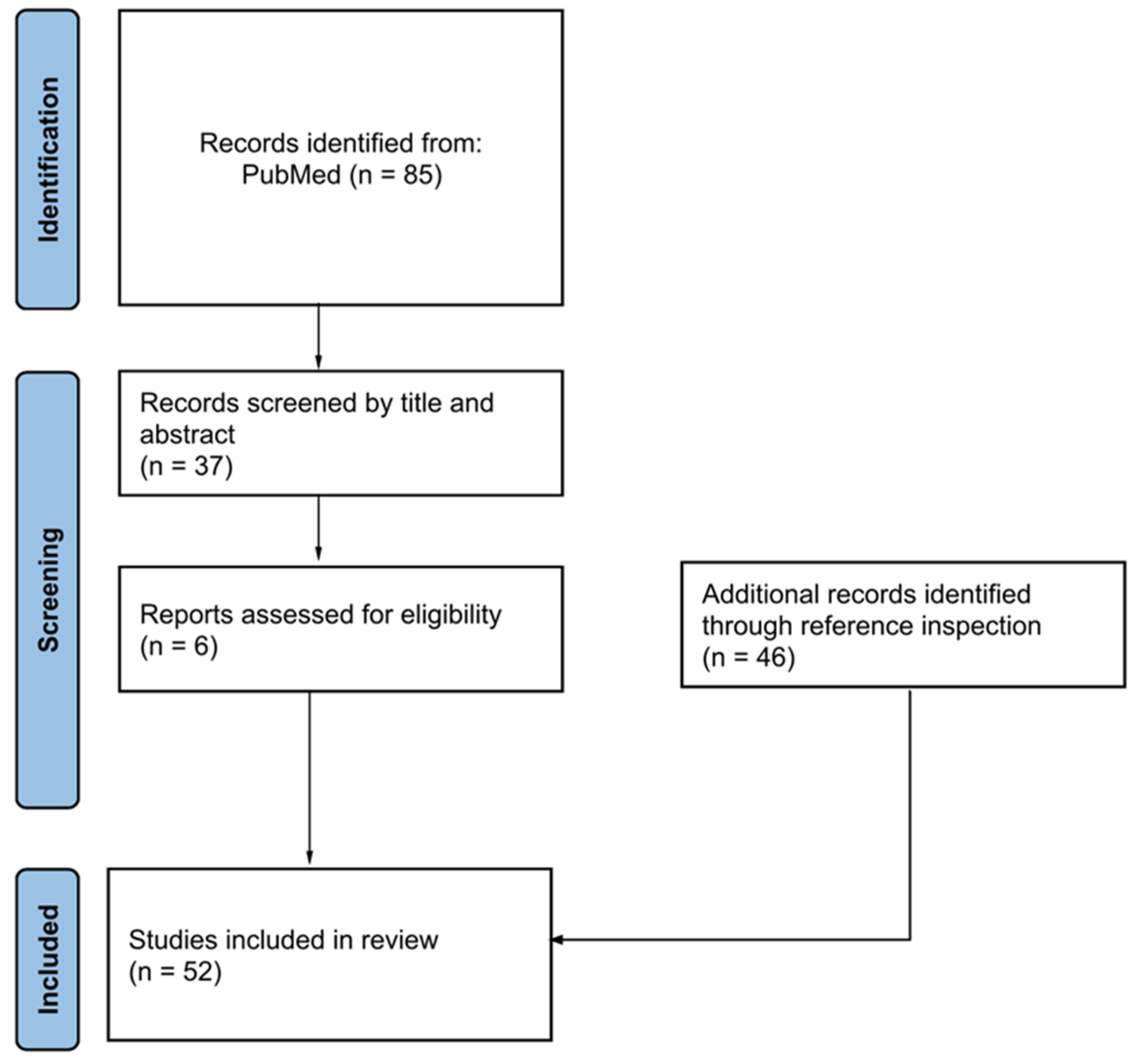
2. Methods
3. Results
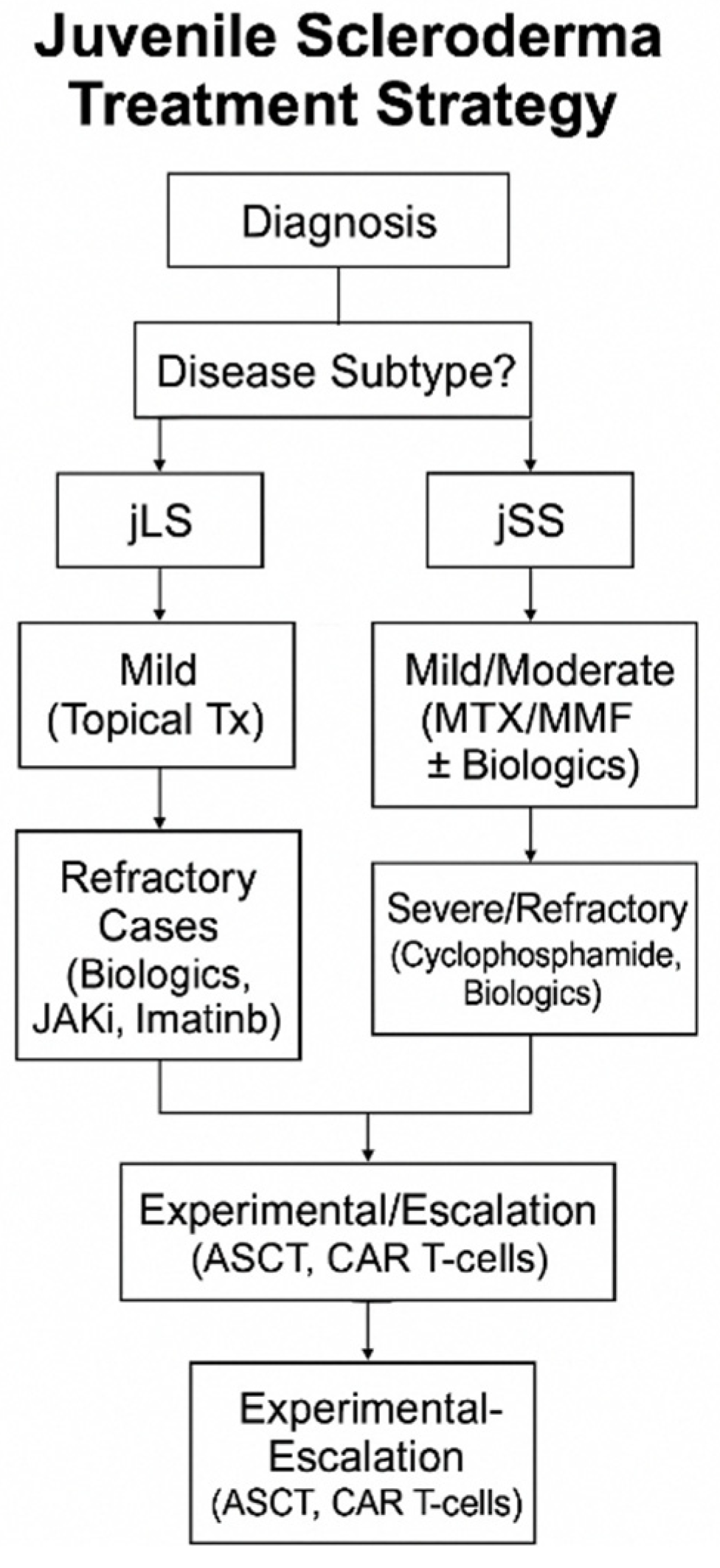
3.1. Juvenile Localized Scleroderma (jLS)
3.1.1. Tocilizumab and Sarilumab
3.1.2. Abatacept
3.1.3. Imatinib
3.1.4. Janus Kinase Inhibitors (JAK)
3.1.5. Autologous Stem Cell Transplantation (ASCT)
3.2. Juvenile Systemic Sclerosis (jSS)
3.2.1. Rituximab
3.2.2. Tocilizumab
3.2.3. Janus Kinase (JAK) Inhibitors
3.2.4. Abatacept
3.2.5. Imatinib and Nintedanib
3.2.6. Pamrevlumab
3.2.7. Autologous Stem Cell Transplantation (ASCT)
3.2.8. Chimeric Antigen Receptor (CAR-T) Cell Therapy
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bhat, A. Juvenile scleroderma. Indian J. Pediatr. 2024, 91, 1049–1055. [Google Scholar] [CrossRef] [PubMed]
- Martini, A. Juvenile systemic scleroderma. Curr. Rheumatol. Rep. 2001, 3, 387–390. [Google Scholar] [CrossRef] [PubMed]
- Li, S.C. Scleroderma in children and adolescents: Localized scleroderma and systemic sclerosis. Pediatr. Clin. North Am. 2018, 65, 757–781. [Google Scholar] [CrossRef]
- Laxer, R.M.; Zulian, F. Localized scleroderma. Curr. Opin. Rheumatol. 2006, 18, 606–613. [Google Scholar] [CrossRef]
- Foeldvari, I.; Wulffraat, N. Recognition and management of scleroderma in children. Paediatr. Drugs 2001, 3, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Zulian, F.; Woo, P.; Athreya, B.H.; Laxer, R.M.; Medsger, T.A., Jr.; Lehman, T.J.; Cerinic, M.M.; Martini, G.; Ravelli, A.; Russo, R.; et al. The Pediatric Rheumatology European Society/American College of Rheumatology/European League against Rheumatism provisional classification criteria for juvenile systemic sclerosis. Arthritis Care Res. 2007, 57, 203–212. [Google Scholar] [CrossRef]
- Jacobe, H.; Ahn, C.; Arnett, F.C.; Reveille, J.D. Major histocompatibility complex class I and class II alleles may confer susceptibility to or protection against morphea: Findings from the Morphea in Adults and Children cohort. Arthritis Rheumatol. 2014, 66, 3170–3177. [Google Scholar] [CrossRef]
- Stern, E.P.; Denton, C.P. The pathogenesis of systemic sclerosis. Rheum. Dis. Clin. North Am. 2015, 41, 367–382. [Google Scholar] [CrossRef]
- Torok, K.S.; Li, S.C.; Jacobe, H.M.; Taber, S.F.; Stevens, A.M.; Zulian, F.; Lu, T.T. Immunopathogenesis of pediatric localized scleroderma. Front. Immunol. 2019, 10, 908. [Google Scholar] [CrossRef]
- Li, S.C.; Torok, K.S.; Pope, E.; Dedeoglu, F.; Hong, S.; Jacobe, H.T.; Rabinovich, C.E.; Laxer, R.M.; Higgins, G.C.; Ferguson, P.J.; et al. Development of consensus treatment plans for juvenile localized scleroderma: A roadmap toward comparative effectiveness studies in juvenile localized scleroderma. Arthritis Care Res. 2012, 64, 1175–1185. [Google Scholar] [CrossRef]
- Kreuter, A.; Krieg, T.; Worm, M.; Wenzel, J.; Moinzadeh, P.; Kuhn, A.; Aberer, E.; Scharffetter-Kochanek, K.; Horneff, G.; Reil, E.; et al. German guidelines for the diagnosis and therapy of localized scleroderma. J. Dtsch. Dermatol. Ges. 2016, 14, 199–216. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, T.; Okiyama, N.; Takamuki, R.; Inoue, S.; Saito, A.; Nakamura, Y.; Ishitsuka, Y.; Watanabe, R.; Fujisawa, Y.; Fujimoto, M. Juvenile case of multiple morphea profunda resulting in joint contracture that was successfully treated with cyclosporin A: A case report and review of the published works. J. Dermatol. 2019, 46, 354–357. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.B.; Blixt, E.K.; Drage, L.A.; El-Azhary, R.A.; Wetter, D.A. Treatment of morphea with hydroxychloroquine: A retrospective review of 84 patients at Mayo Clinic, 1996–2013. J. Am. Acad. Dermatol. 2019, 80, 1658–1663. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Castro, L.; de las Heras, E.; Moreno, C.; Fleta-Asín, B.; Muñoz-Zato, E.; Carrillo, R.; Jaén, P. Eosinophilic fasciitis/generalized morphea overlap successfully treated with azathioprine. Int. J. Dermatol. 2014, 53, 1386–1388. [Google Scholar] [CrossRef]
- Thomas, R.M.; Worswick, S.; Aleshin, M. Retinoic acid for treatment of systemic sclerosis and morphea: A literature review. Dermatol. Ther. 2017, 30, e12455. [Google Scholar] [CrossRef]
- Kromer, C.; Mitterlechner, L.; Langer, N.; Schön, M.P.; Mössner, R. Response of recalcitrant generalized morphea to intravenous immunoglobulins (IVIg): Three cases and a review of the literature. Eur. J. Dermatol. 2021, 31, 822–829. [Google Scholar] [CrossRef]
- Chimenti, M.S.; Teoli, M.; Di Stefani, A.; Giunta, A.; Esposito, M.; Perricone, R. Resolution with rituximab of localized scleroderma occurring during etanercept treatment in a patient with rheumatoid arthritis. Eur. J. Dermatol. 2013, 23, 273–274. [Google Scholar] [CrossRef]
- Kurzinski, K.; Torok, K.S. Cytokine profiles in localized scleroderma and relationship to clinical features. Cytokine 2011, 55, 157–164. [Google Scholar] [CrossRef]
- Foeldvari, I.; Anton, J.; Friswell, M.; Bica, B.; de Inocencio, J.; Aquilani, A.; Helmus, N. Tocilizumab is a promising treatment option for therapy resistant juvenile localized scleroderma patients. J. Scleroderma Relat. Disord. 2017, 2, 203–207. [Google Scholar] [CrossRef]
- Kelsey, C.E.; Torok, K.S. The localized scleroderma cutaneous assessment tool: Responsiveness to change in a pediatric clinical population. J. Am. Acad. Dermatol. 2013, 69, 214–220. [Google Scholar] [CrossRef]
- Khanna, D.; Denton, C.P.; Jahreis, A.; van Laar, J.M.; Frech, T.M.; Anderson, M.E.; Baron, M.; Chung, L.; Fierlbeck, G.; Lakshminarayanan, S.; et al. Safety and efficacy of subcutaneous tocilizumab in adults with systemic sclerosis (faSScinate): A phase 2, randomised, controlled trial. Lancet 2016, 387, 2630–2640. [Google Scholar] [CrossRef] [PubMed]
- Lythgoe, H.; Baildam, E.; Beresford, M.W.; Cleary, G.; McCann, L.J.; Pain, C.E. Tocilizumab as a potential therapeutic option for children with severe, refractory juvenile localized scleroderma. Rheumatology 2018, 57, 398–401. [Google Scholar] [CrossRef] [PubMed]
- Martini, G.; Campus, S.; Raffeiner, B.; Boscarol, G.; Meneghel, A.; Zulian, F. Tocilizumab in two children with pansclerotic morphoea: A hopeful therapy for refractory cases? Clin. Exp. Rheumatol. 2017, 35 (Suppl. S106), 211–213. [Google Scholar]
- Zhang, A.; Nocton, J.; Chiu, Y. A case of pansclerotic morphea treated with tocilizumab. JAMA Dermatol. 2019, 155, 388–389. [Google Scholar] [CrossRef]
- Ventéjou, S.; Schwieger-Briel, A.; Nicolai, R.; Christen-Zaech, S.; Schnider, C.; Hofer, M.; Bogiatzi, S.; Hohl, D.; De Benedetti, F.; Morren, M.-A. Case report: Pansclerotic morphea-clinical features, differential diagnoses and modern treatment concepts. Front. Immunol. 2021, 12, 656407. [Google Scholar] [CrossRef]
- Lamb, Y.N.; Deeks, E.D. Sarilumab: A review in moderate to severe rheumatoid arthritis. Drugs 2018, 78, 929–940. [Google Scholar] [CrossRef] [PubMed]
- Papara, C.; De Luca, D.A.; Bieber, K.; Vorobyev, A.; Ludwig, R.J. Morphea: The 2023 update. Front. Med. 2023, 10, 1108623. [Google Scholar] [CrossRef]
- Fage, S.W.; Arvesen, K.B.; Olesen, A.B. Abatacept improves skin-score and reduces lesions in patients with localized scleroderma: A case series. Acta Derm.-Venereol. 2018, 98, 465–466. [Google Scholar] [CrossRef]
- Kalampokis, I.; Yi, B.Y.; Smidt, A.C. Abatacept in the treatment of localized scleroderma: A pediatric case series and systematic literature review. Semin. Arthritis Rheum. 2020, 50, 645–656. [Google Scholar] [CrossRef]
- Distler, J.H.; Jüngel, A.; Huber, L.C.; Schulze-Horsel, U.; Zwerina, J.; Gay, R.E.; Michel, B.A.; Hauser, T.; Schett, G.; Gay, S.; et al. Imatinib mesylate reduces production of extracellular matrix and prevents development of experimental dermal fibrosis. Arthritis Rheum. 2007, 56, 311–322. [Google Scholar] [CrossRef]
- Coelho-Macias, V.; Mendes-Bastos, P.; Assis-Pacheco, F.; Cardoso, J. Imatinib: A novel treatment approach for generalized morphea. Int. J. Dermatol. 2014, 53, 1299–1302. [Google Scholar] [CrossRef] [PubMed]
- Alcántara-Reifs, C.M.; Garnacho-Saucedo, G.M.; Salido-Vallejo, R.; de la Corte-Sánchez, S.; García-Nieto, A.V. Imatinib treatment of therapy resistant generalized deep morphea. Dermatol. Ther. 2015, 28, 271–273. [Google Scholar] [CrossRef] [PubMed]
- Moinzadeh, P.; Krieg, T.; Hunzelmann, N. Imatinib treatment of generalized localized scleroderma (morphea). J. Am. Acad. Dermatol. 2010, 63, e102–e104. [Google Scholar] [CrossRef]
- Inamo, Y.; Ochiai, T. Successful combination treatment of a patient with progressive juvenile localized scleroderma (morphea) using imatinib, corticosteroids, and methotrexate. Pediatr. Dermatol. 2012, 30, e191–e193. [Google Scholar] [CrossRef]
- Damsky, W.; Patel, D.; Garelli, C.J.; Garg, M.; Wang, A.; Dresser, K.; Deng, A.; Harris, J.E.; Richmond, J.; King, B. Jak inhibition prevents bleomycin-induced fibrosis in mice and is effective in patients with morphea. J. Investig. Dermatol. 2020, 140, 1446–1449.e4. [Google Scholar] [CrossRef]
- Tang, J.-C.; Zheng, W.-Y.; Han, G.-M.; Liu, S.-F.; Yang, B. Successful treatment of paediatric morphea with tofacitinib. Acta Derm.-Venereol. 2023, 103, adv4805. [Google Scholar] [CrossRef] [PubMed]
- Soh, H.J.; Samuel, C.; Heaton, V.; Renton, W.D.; Cox, A.; Munro, J. Challenges in the diagnosis and treatment of disabling pansclerotic morphea of childhood: Case-based review. Rheumatol. Int. 2019, 39, 933–941. [Google Scholar] [CrossRef]
- Zhou, J.; Gao, M.; Zhang, S.; Lu, X.; Lei, Z.; Cheng, T.; Liu, Y.; Tianshu, C. Retrospective comparative study of the efficacy of JAK inhibitor (tofacitinib) in the treatment of systemic sclerosis-associated interstitial lung disease. Clin. Rheumatol. 2023, 42, 2823–2832. [Google Scholar] [CrossRef]
- Moll, M.; Holzer, U.; Zimmer, C.; Rieber, N.; Kuemmerle-Deschner, J.B. Autologous stem cell transplantation in two children with disabling pansclerotic morphea. Pediatr. Rheumatol. Online J. 2011, 9 (Suppl. S1), 77. [Google Scholar] [CrossRef]
- Zulian, F.; Dal Pozzolo, R.; Meneghel, A.; Castaldi, B.; Marcolongo, R.; Caforio, A.L.P.; Martini, G. Rituximab for rapidly progressive juvenile systemic sclerosis. Rheumatology 2020, 59, 3793–3797. [Google Scholar] [CrossRef]
- Kaegi, C.; Wuest, B.; Schreiner, J.; Steiner, U.C.; Vultaggio, A.; Matucci, A.; Crowley, C.; Boyman, O. Systematic Review of safety and efficacy of rituximab in treating immune-mediated disorders. Front. Immunol. 2019, 10, 1990. [Google Scholar] [CrossRef]
- Adrovic, A.; Yildiz, M.; Haslak, F.; Koker, O.; Aliyeva, A.; Sahin, S.; Barut, K.; Kasapcopur, O. Tocilizumab therapy in juvenile systemic sclerosis: A retrospective single centre pilot study. Rheumatol. Int. 2020, 41, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Funauchi, M.; Kinoshita, K. Problems to consider before determining the regimen of the treatment for juvenile systemic sclerosis treatment: A case report where tocilizumab monotherapy succeeded efficiently and safely. Clin. Med. Insights Arthritis Musculoskelet. Disord. 2022, 15, 11795441211066307. [Google Scholar] [CrossRef] [PubMed]
- Pin, A.; Tesser, A.; Pastore, S.; Moressa, V.; Valencic, E.; Arbo, A.; Maestro, A.; Tommasini, A.; Taddio, A. Biological and clinical changes in a pediatric series treated with off-label JAK inhibitors. Int. J. Mol. Sci. 2020, 21, 7767. [Google Scholar] [CrossRef]
- Kalogerou, A.; Gelou, E.; Mountantonakis, S.; Settas, L.; Zafiriou, E.; Sakkas, L. Early T cell activation in the skin from patients with systemic sclerosis. Ann. Rheum. Dis. 2005, 64, 1233–1235. [Google Scholar] [CrossRef] [PubMed]
- Ponsoye, M.; Frantz, C.; Ruzehaji, N.; Nicco, C.; Elhai, M.; Ruiz, B.; Cauvet, A.; Pezet, S.; Brandely, M.L.; Batteux, F.; et al. Treatment with abatacept prevents experimental dermal fibrosis and induces regression of established inflammation-driven fibrosis. Ann. Rheum. Dis. 2016, 75, 2142–2149. [Google Scholar] [CrossRef]
- Boleto, G.; Guignabert, C.; Pezet, S.; Cauvet, A.; Sadoine, J.; Tu, L.; Nicco, C.; Gobeaux, C.; Batteux, F.; Allanore, Y.; et al. T-cell costimulation blockade is effective in experimental digestive and lung tissue fibrosis. Arthritis Res. Ther. 2018, 20, 197. [Google Scholar] [CrossRef]
- Khanna, D.; Spino, C.; Johnson, S.; Chung, L.; Whitfield, M.L.; Denton, C.P.; Berrocal, V.; Franks, J.; Mehta, B.; Molitor, J.; et al. Abatacept in early diffuse cutaneous systemic sclerosis: Results of a phase II investigator-initiated, multicenter, double-blind, randomized, placebo-controlled trial. Arthritis Rheumatol. 2019, 72, 125–136. [Google Scholar] [CrossRef]
- Liakouli, V.; Ciaffi, J.; Ursini, F.; Ruscitti, P.; Meliconi, R.; Ciccia, F.; Cipriani, P.; Giacomelli, R. Efficacy and safety of imatinib mesylate in systemic sclerosis. A systematic review and meta-analysis. Expert Rev. Clin. Immunol. 2020, 16, 931–942. [Google Scholar] [CrossRef]
- Boutel, M.; Boutou, A.; Pitsiou, G.; Garyfallos, A.; Dimitroulas, T. Efficacy and safety of nintedanib in patients with connective tissue disease-interstitial lung disease (CTD-ILD): A real-world single center experience. Diagnostics 2023, 13, 1221. [Google Scholar] [CrossRef]
- Lafyatis, R. Transforming growth factor β—At the centre of systemic sclerosis. Nat. Rev. Rheumatol. 2014, 10, 706–719. [Google Scholar] [CrossRef]
- Makino, K.; Makino, T.; Stawski, L.; Lipson, K.E.; Leask, A.; Trojanowska, M. Anti-connective tissue growth factor (CTGF/CCN2) monoclonal antibody attenuates skin fibrosis in mice models of systemic sclerosis. Arthritis Res. Ther. 2017, 19, 134. [Google Scholar] [CrossRef] [PubMed]
- Martini, A.; Maccario, R.; Ravelli, A.; Montagna, D.; De Benedetti, F.; Bonetti, F.; Viola, S.; Zecca, M.; Perotti, C.; Locatelli, F. Marked and sustained improvement two years after autologous stem cell transplantation in a girl with systemic sclerosis. Arthritis Rheum. 1999, 42, 807–811. [Google Scholar] [CrossRef]
- Dabas, P.; Danda, A. Revolutionizing cancer treatment: A comprehensive review of CAR-T cell therapy. Med. Oncol. 2023, 40, 275. [Google Scholar] [CrossRef] [PubMed]
- Dörner, T.; Furie, R. Novel paradigms in systemic lupus erythematosus. Lancet 2019, 393, 2344–2358. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, I.E.; Fujimoto, M.; Vencovsky, J.; Aggarwal, R.; Holmqvist, M.; Christopher-Stine, L.; Mammen, A.L.; Miller, F.W. Idiopathic inflammatory myopathies. Nat. Rev. Dis. Prim. 2021, 7, 86. [Google Scholar] [CrossRef]
- Denton, C.P.; Khanna, D. Systemic sclerosis. Lancet 2017, 390, 1685–1699. [Google Scholar] [CrossRef]
- Kansal, R.; Richardson, N.; Neeli, I.; Khawaja, S.; Chamberlain, D.; Ghani, M.; Ghani, Q.-U.; Balazs, L.; Beranova-Giorgianni, S.; Giorgianni, F.; et al. Sustained B cell depletion by CD19-targeted CAR T cells is a highly effective treatment for murine lupus. Sci. Transl. Med. 2019, 11, eaav1648. [Google Scholar] [CrossRef]
- Müller, F.; Taubmann, J.; Bucci, L.; Wilhelm, A.; Bergmann, C.; Völkl, S.; Aigner, M.; Rothe, T.; Minopoulou, I.; Tur, C.; et al. CD19 CAR T-cell therapy in autoimmune disease—A case series with follow-up. N. Engl. J. Med. 2024, 390, 687–700. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Major Criterion (Required) | Proximal Sclerosis/Induration of the Skin | |
|---|---|---|
| Minor criteria (involving specific districts of the body) | Skin | Sclerodactyly |
| Vascular | Raynaud’s phenomenon | |
| Nailfold videocapillaroscopy abnormalities | ||
| Digital tip ulcers | ||
| Gastrointestinal | Dysphagia | |
| Gastroesophageal reflux | ||
| Renal | Renal crisis | |
| New-onset arterial hypertension | ||
| Cardiac | Arrhythmias | |
| Heart failure | ||
| Respiratory | Pulmonary fibrosis (seen on HCRT or radiography of chest) | |
| Abnormal diffusing capacity of the lungs for carbon monoxide | ||
| Pulmonary hypertension | ||
| Musculoskeletal | Tendon friction rubs | |
| Arthritis | ||
| Myositis | ||
| Neurologic | Neuropathy | |
| Carpal tunnel syndrome | ||
| Serology | Anti-nuclear antibodies | |
| Selective autoantibodies for systemic sclerosis (anti-centromere, anti-topoisomerase I, anti-fibrillarin, anti-PM/Scl, anti-fibrillin, or anti-RNA polymerase I/III antibodies) | ||
| Drug | Number of Patients | Article (Year) |
|---|---|---|
| Tocilizumab | ||
| 8 mg/kg every 3–4 weeks SC | 3 | #19 (2017) |
| 8 mg/kg (>30 kg) SC 12 mg/kg (<30 kg) SC weeks 0, 2, and 4, and then every 4 weeks | 5 | #22 (2018) |
| 8 mg/kg every 4 weeks SC | 2 | #23 (2017) |
| 300 mg IV every 4 weeks | 1 | #24 (2019) |
| Abatacept | ||
| 10 mg/kg SC at days 0, 14 and 28, then once monthly | 8 | #29 (2020) |
| Imatinib | ||
| 235 mg/m2/day per os | 1 | #34 (2013) |
| JAK inhibitors | ||
| Tofacitinib: 2.5 mg twice daily for two months, then once daily for 4 months then every other day, for 2 months per os | 1 | #38 (2023) |
| Tofacitinib: 5 mg, twice daily for 2 month, then 5 mg once daily per os | 1 | #38 (2023) |
| Ruxolitinib: 10 mg twice daily per os | 1 | #39 (2019) |
| Drug | Number of Patients | Article (Year) |
|---|---|---|
| Rituximab | ||
| 375 mg/m2 IV on day 0 and 14, at 3-month intervals | 4 | #40 (2020) |
| Tocilizumab | ||
| 8 mg/kg (children ≥ 30 kg) and 12 mg/kg (children < 30 kg) SC once every 4 weeks | 9 | #42 (2021) |
| 162 mg SC once every 2 weeks | 1 | #43 (2022) |
| JAK inhibitors | ||
| Tofacitinib: 5 mg twice per day | 1 | #44 (2020) |
| Abatacept | ||
| 125 mg SC | 88 | #48 (2020) |
| Imatinib and nintedanib | ||
| Imatinib: 200–400 mg/day orally used in adults in several early-phase studies; 235 mg/m2/day orally, adjusted to body surface area, in a pediatric case report | Unknown | #49 (2020) |
| Nintedanib: Approved adult dose: 150 mg orally twice daily (total daily dose: 300 mg); dose adjustments for children have not been formally defined due to lack of pediatric trials | Unknown | #50 (2023) |
| Pamrevlumab | ||
| Investigational dose in adults in clinical trials: 30 mg/kg intravenously every 3 weeks; no clinical data available for pediatric populations | Mice models | #52 (2017) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sassetti, C.; Borrelli, C.; Mazuy, M.; Guerriero, C.; Rigante, D.; Esposito, S. New Challenging Systemic Therapies for Juvenile Scleroderma: A Comprehensive Review. Pharmaceuticals 2025, 18, 643. https://doi.org/10.3390/ph18050643
Sassetti C, Borrelli C, Mazuy M, Guerriero C, Rigante D, Esposito S. New Challenging Systemic Therapies for Juvenile Scleroderma: A Comprehensive Review. Pharmaceuticals. 2025; 18(5):643. https://doi.org/10.3390/ph18050643
Chicago/Turabian StyleSassetti, Chiara, Claudia Borrelli, Martha Mazuy, Cristina Guerriero, Donato Rigante, and Susanna Esposito. 2025. "New Challenging Systemic Therapies for Juvenile Scleroderma: A Comprehensive Review" Pharmaceuticals 18, no. 5: 643. https://doi.org/10.3390/ph18050643
APA StyleSassetti, C., Borrelli, C., Mazuy, M., Guerriero, C., Rigante, D., & Esposito, S. (2025). New Challenging Systemic Therapies for Juvenile Scleroderma: A Comprehensive Review. Pharmaceuticals, 18(5), 643. https://doi.org/10.3390/ph18050643