
Anti-Leukemic Profiling of Oxazole-Linked Oxadiazole Derivatives: A Computational and Kinetic Approach

 , , ,
, , , 
Abstract
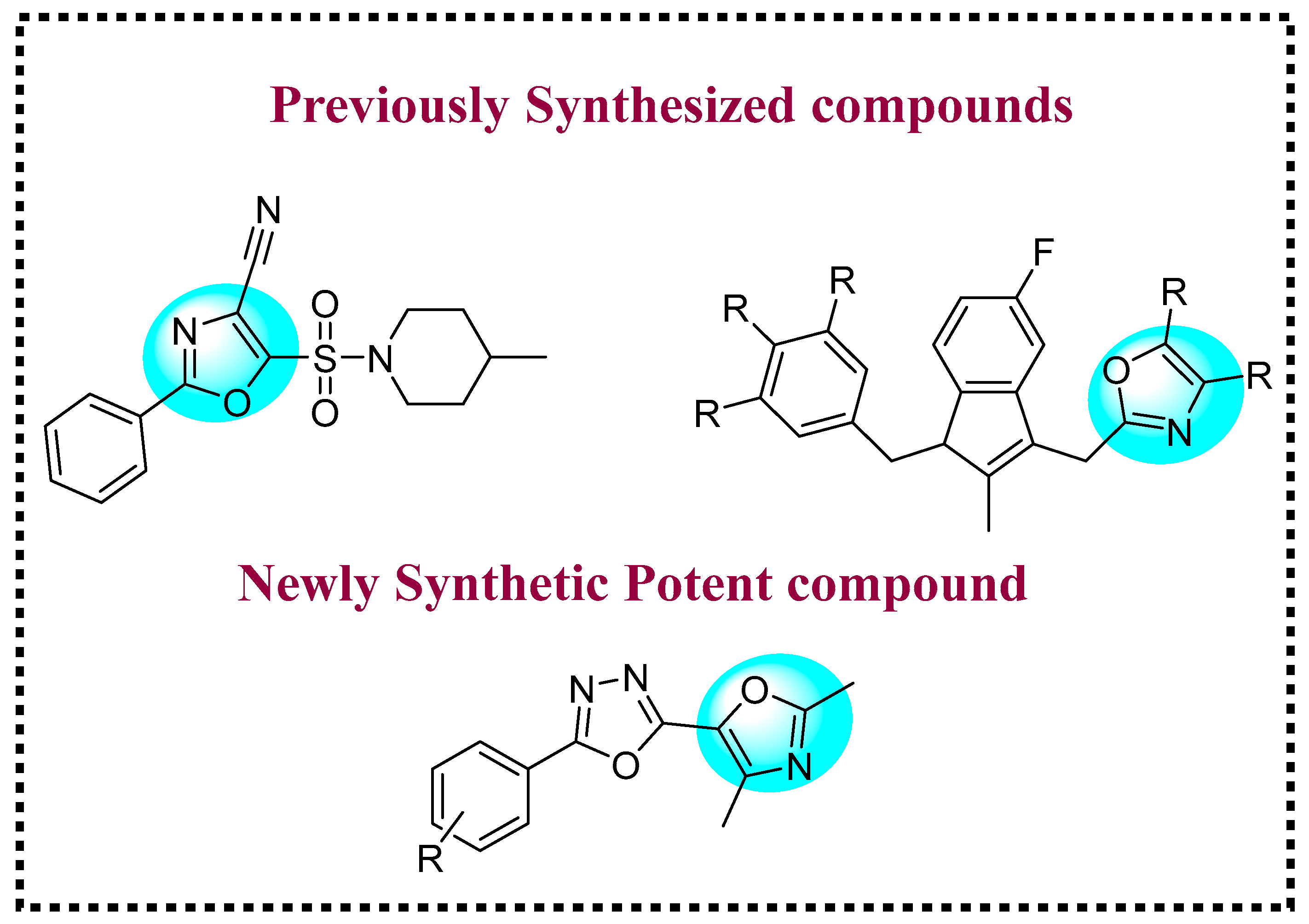
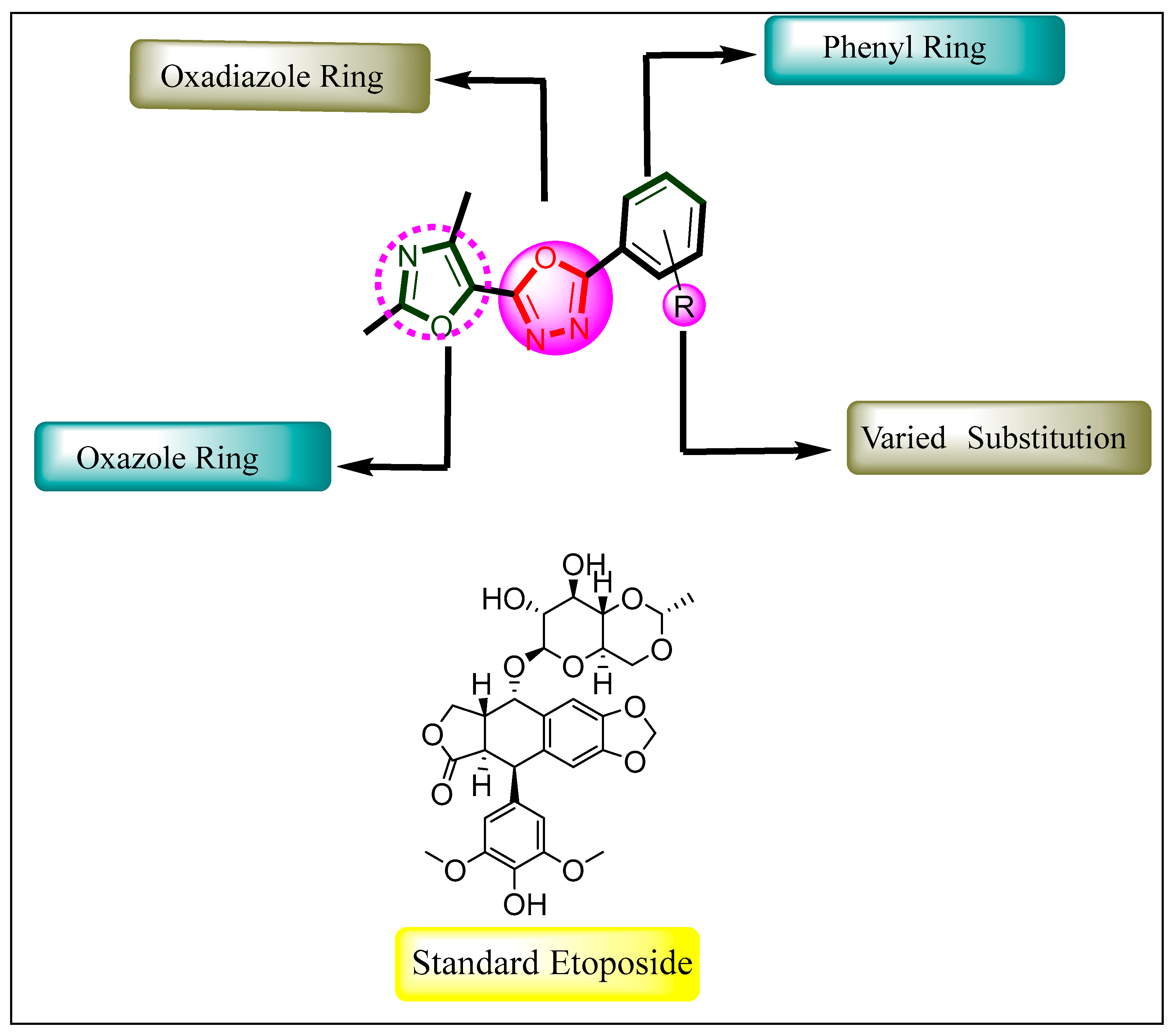
1. Introduction
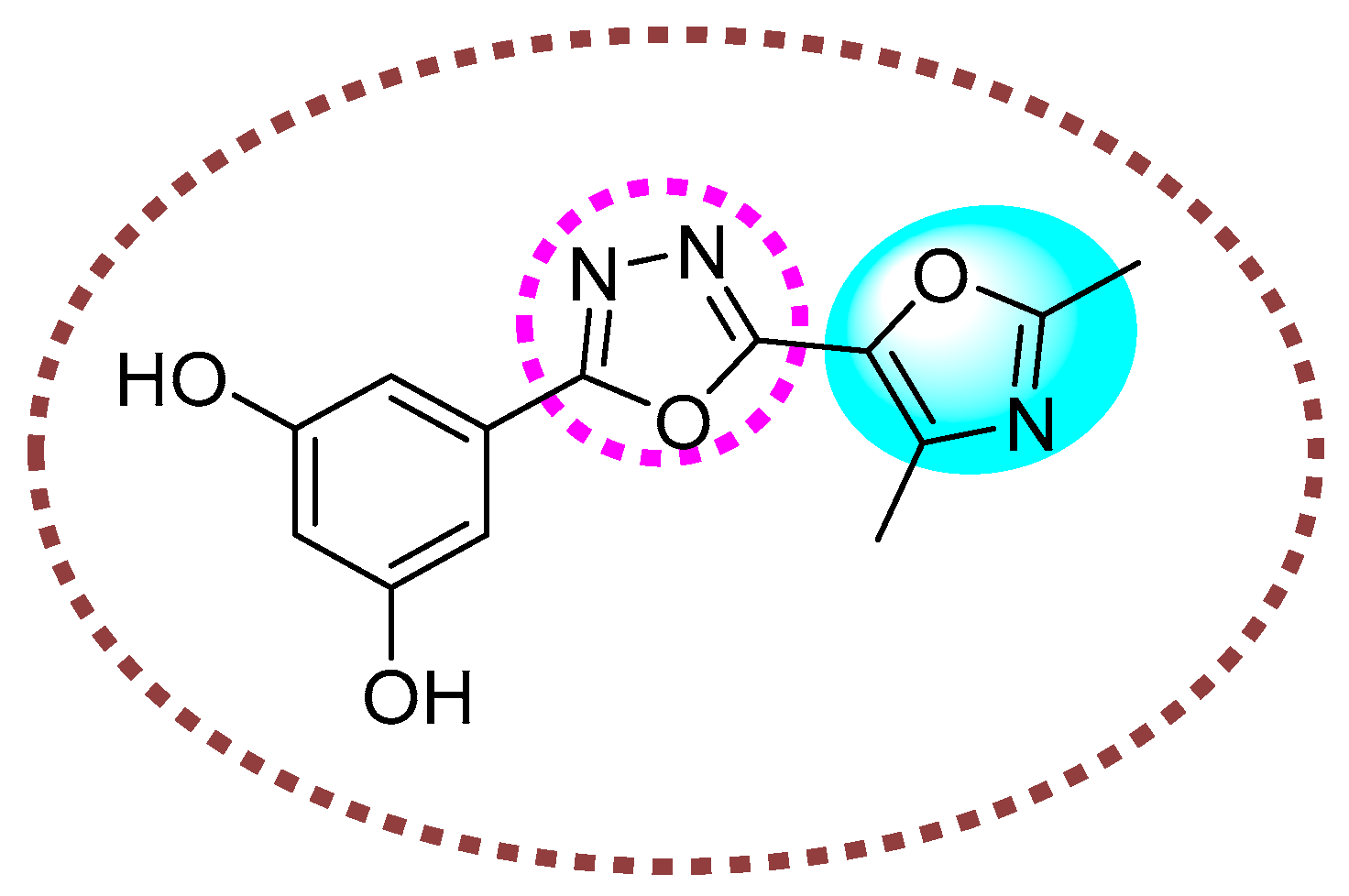
2. Results and Discussion
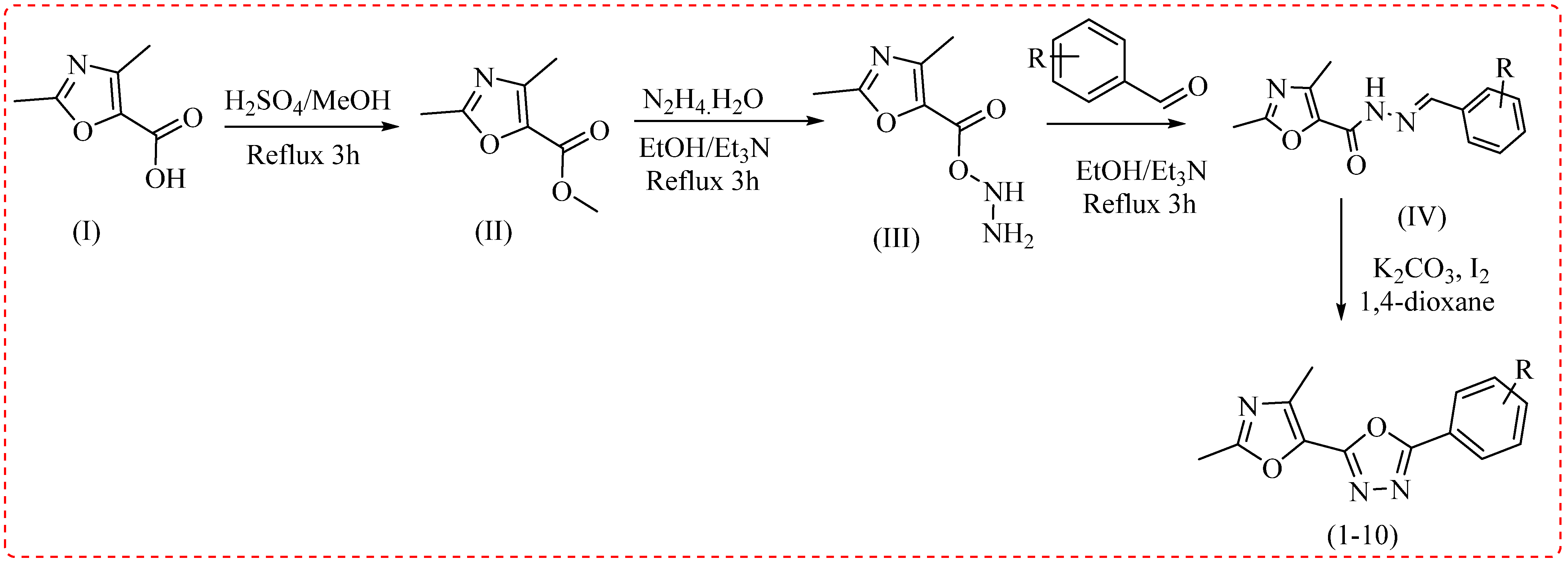
2.1. Chemistry
2.2. Spectral Study
2.3. Biological Activity
In Vitro Anti-Leukemia Profile
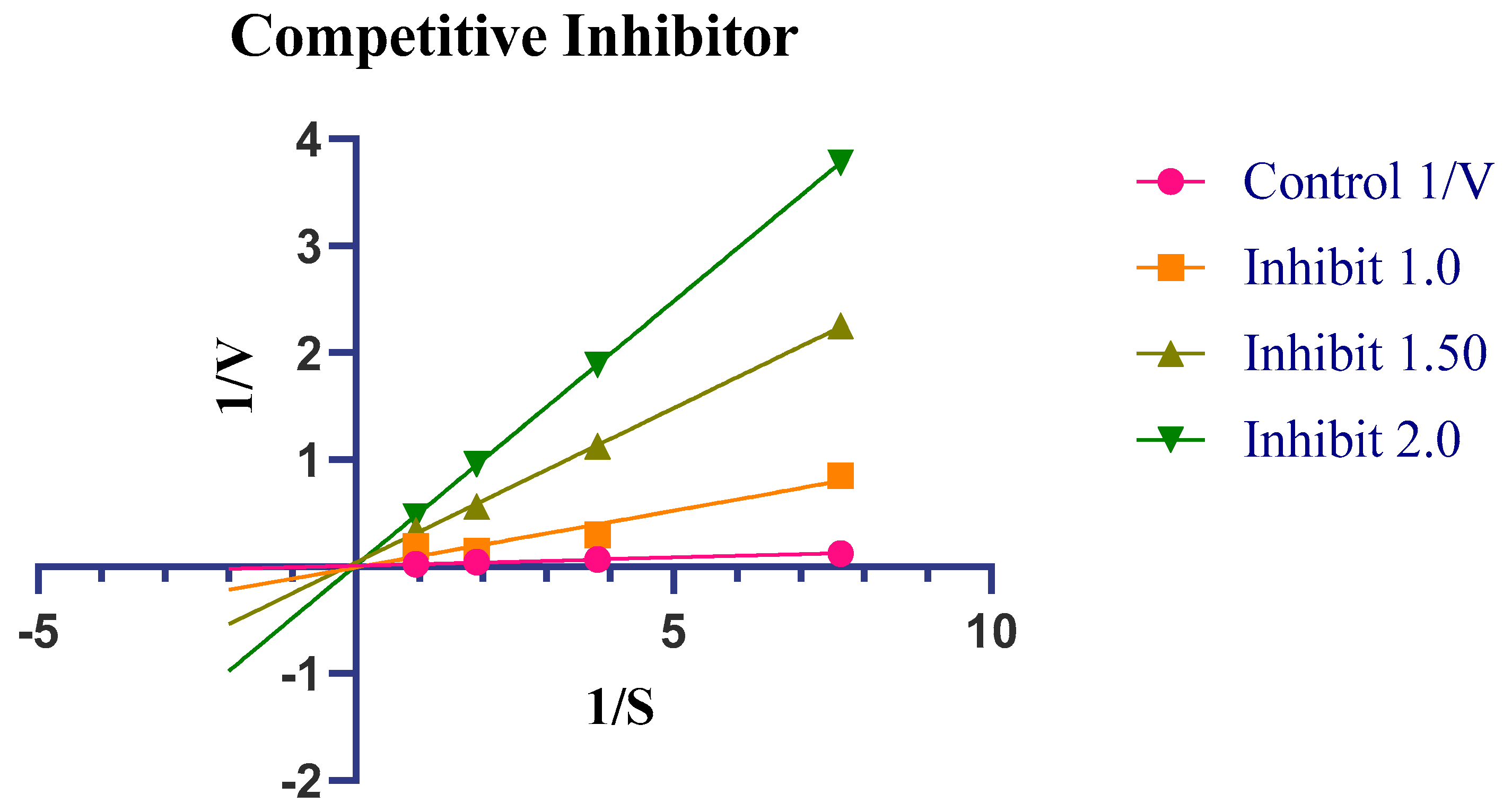
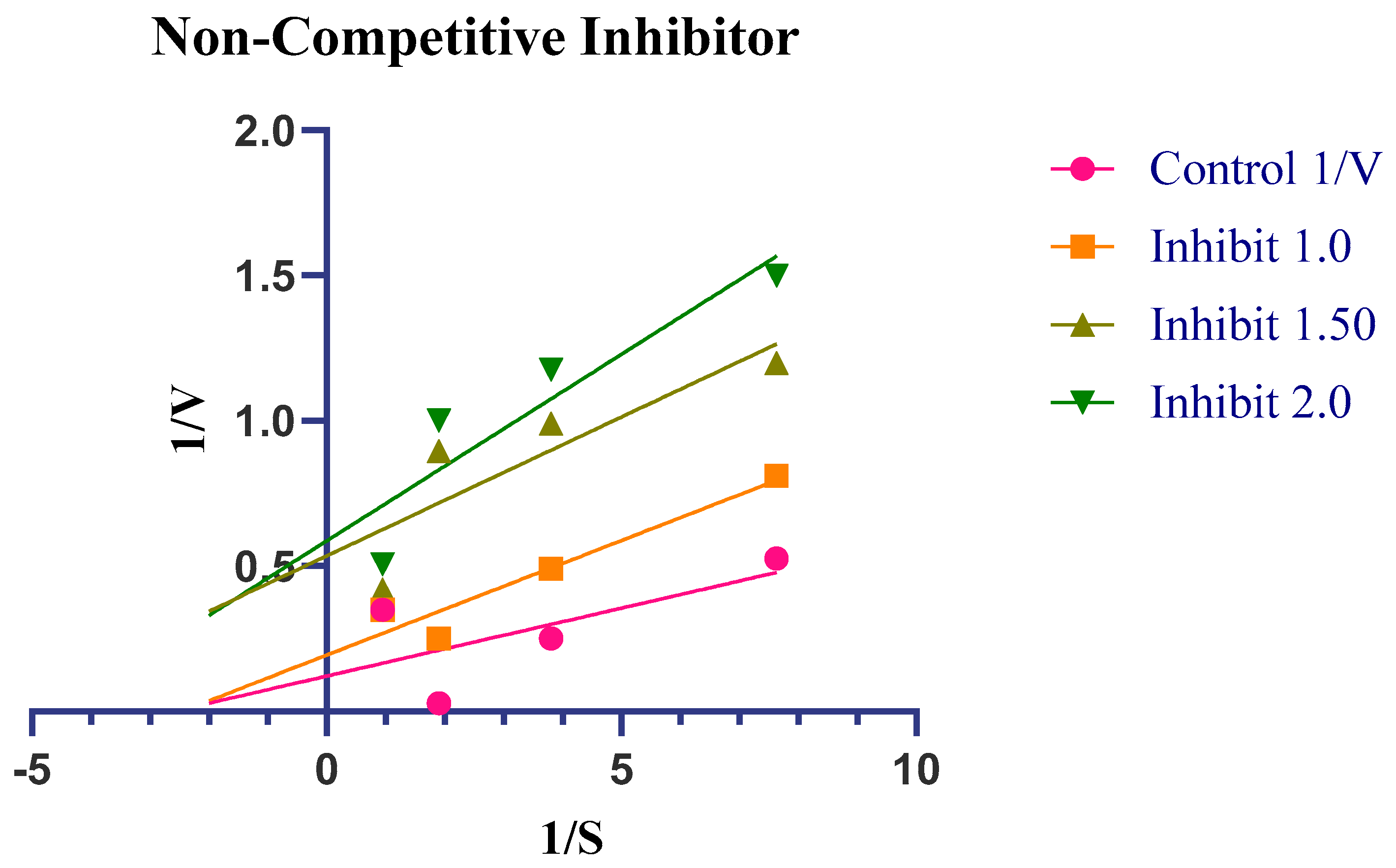
2.4. Enzyme Kinetics
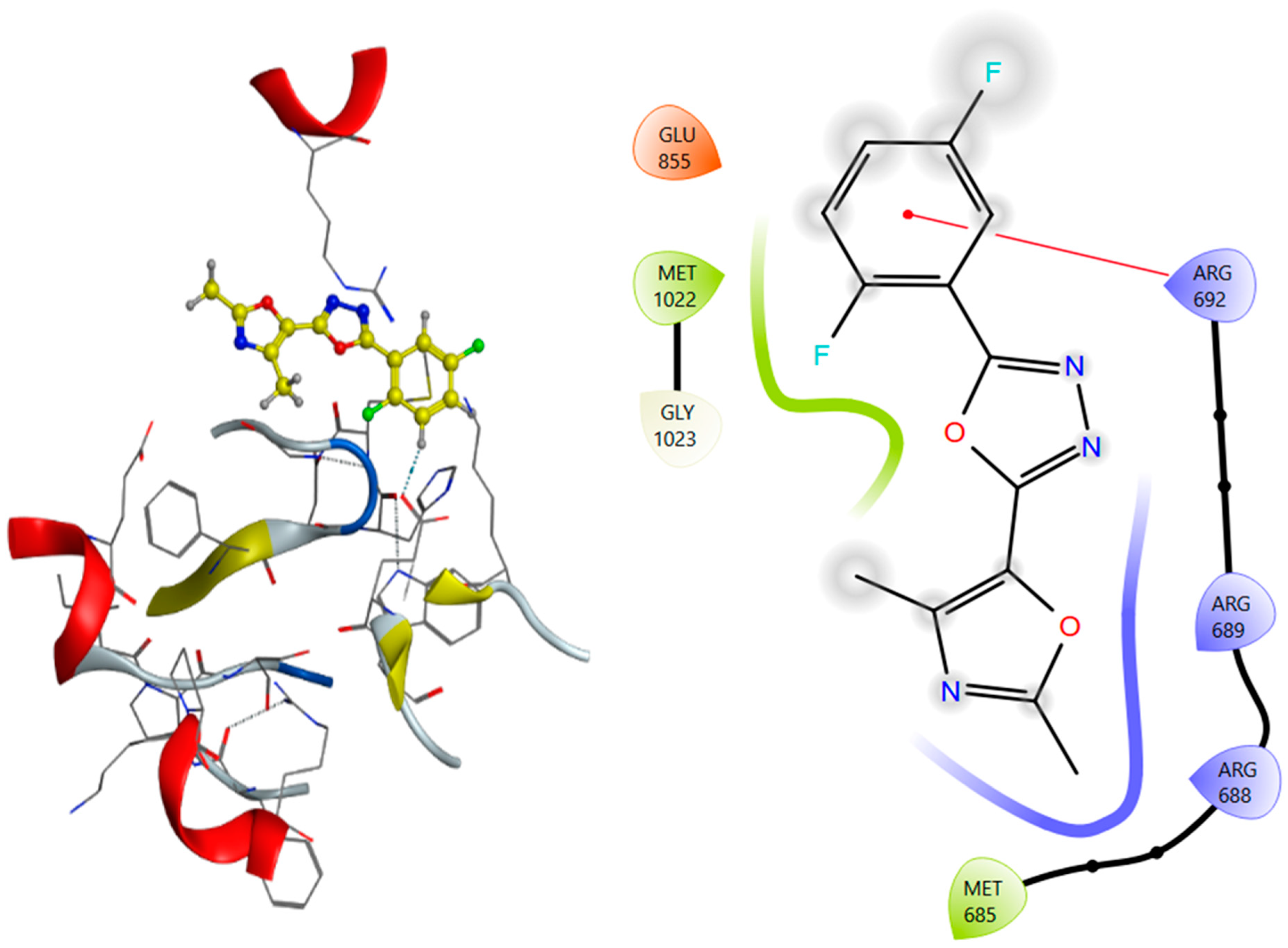
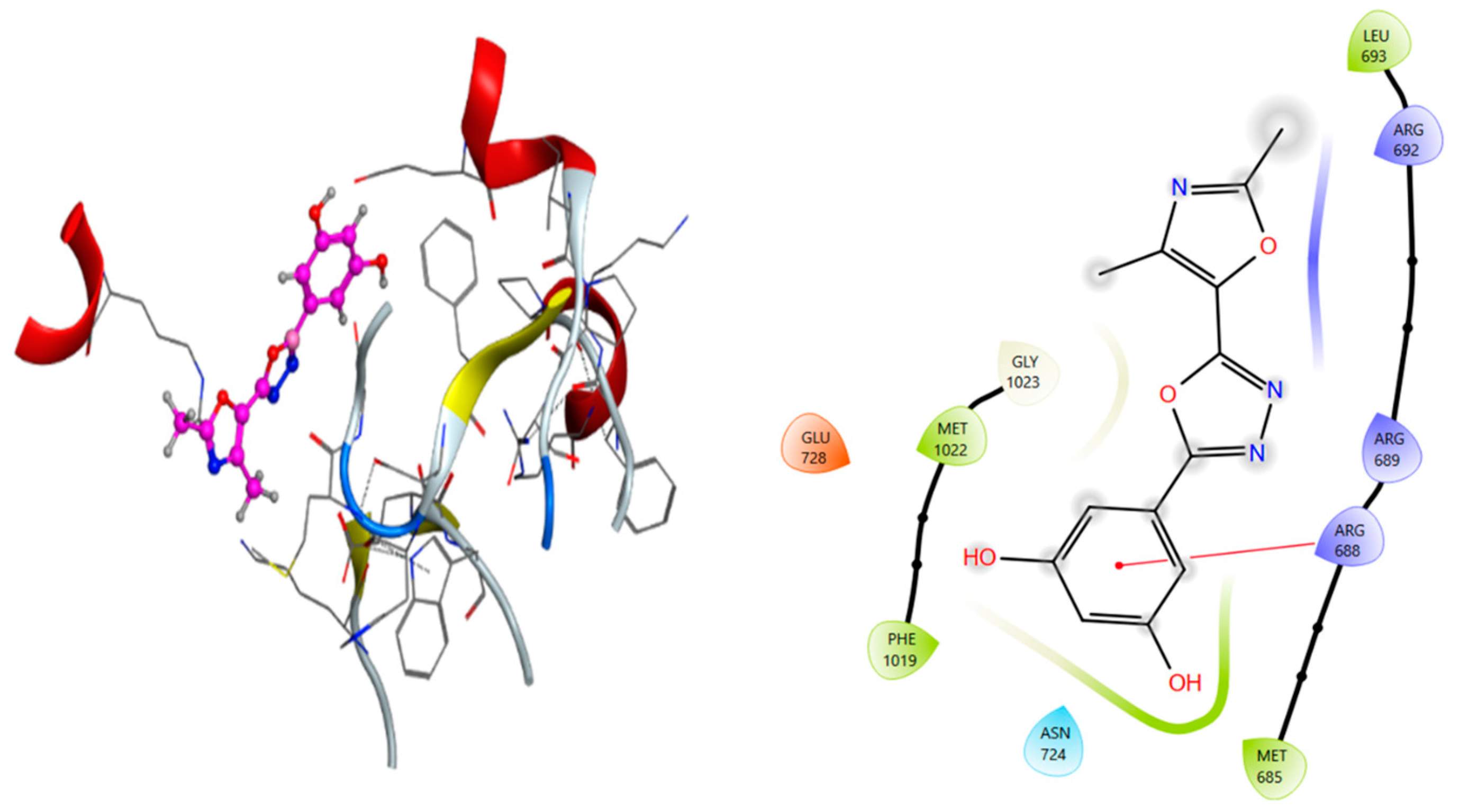
2.5. Molecular Docking Analysis
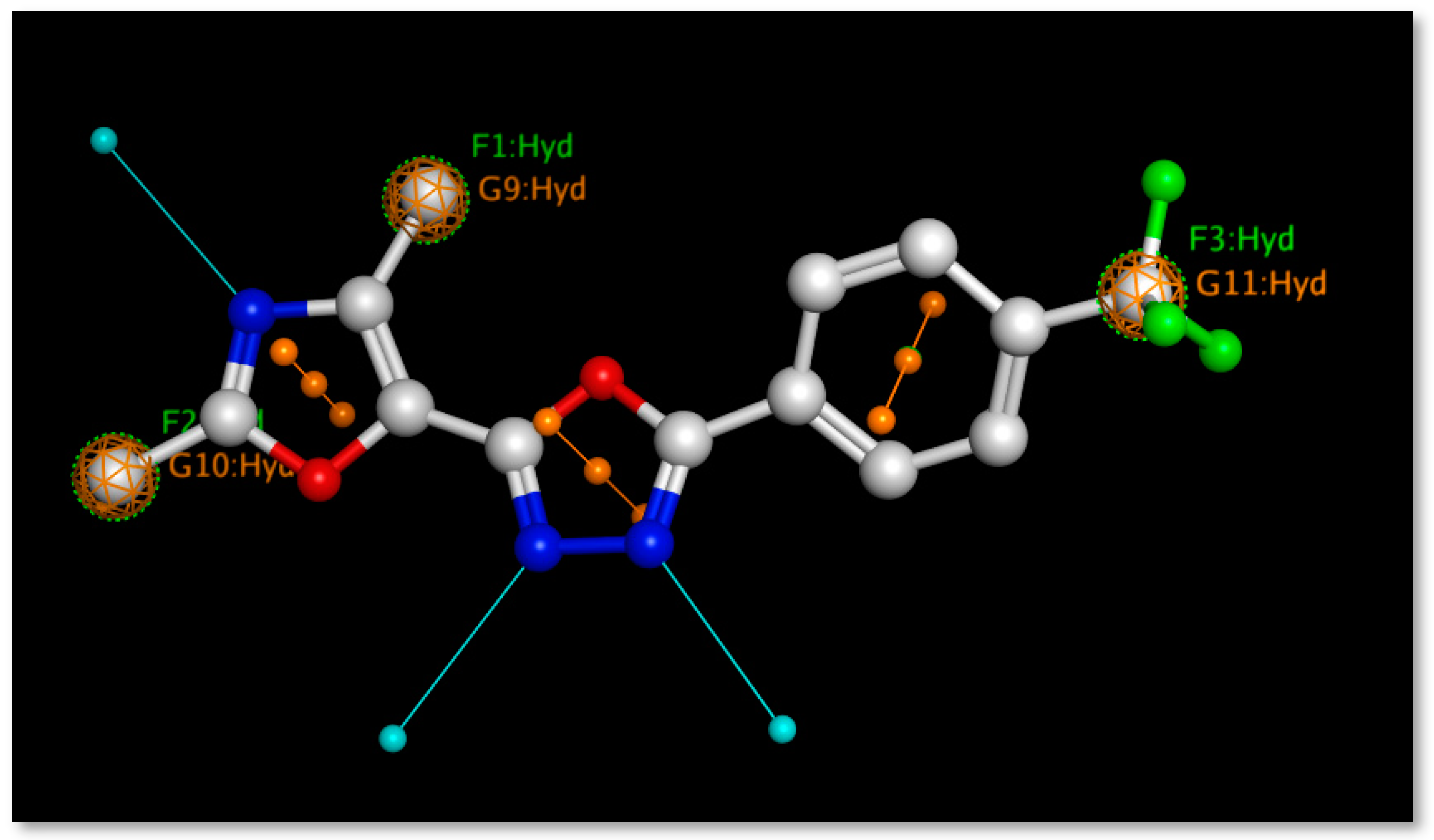
2.6. Pharmacophore Modeling
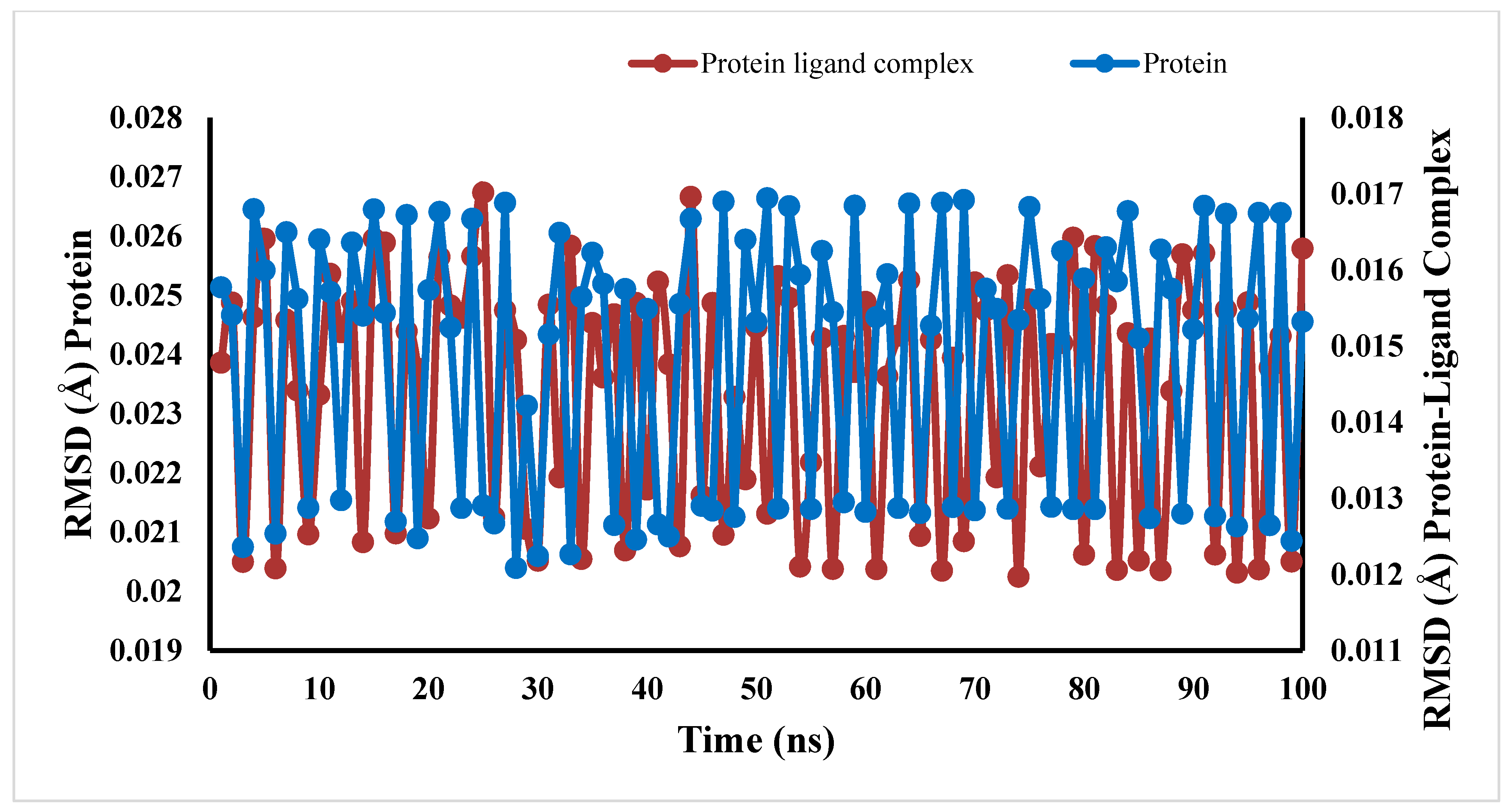
2.7. Molecular Dynamic Simulations
2.8. ADMET Analysis
2.9. DFT
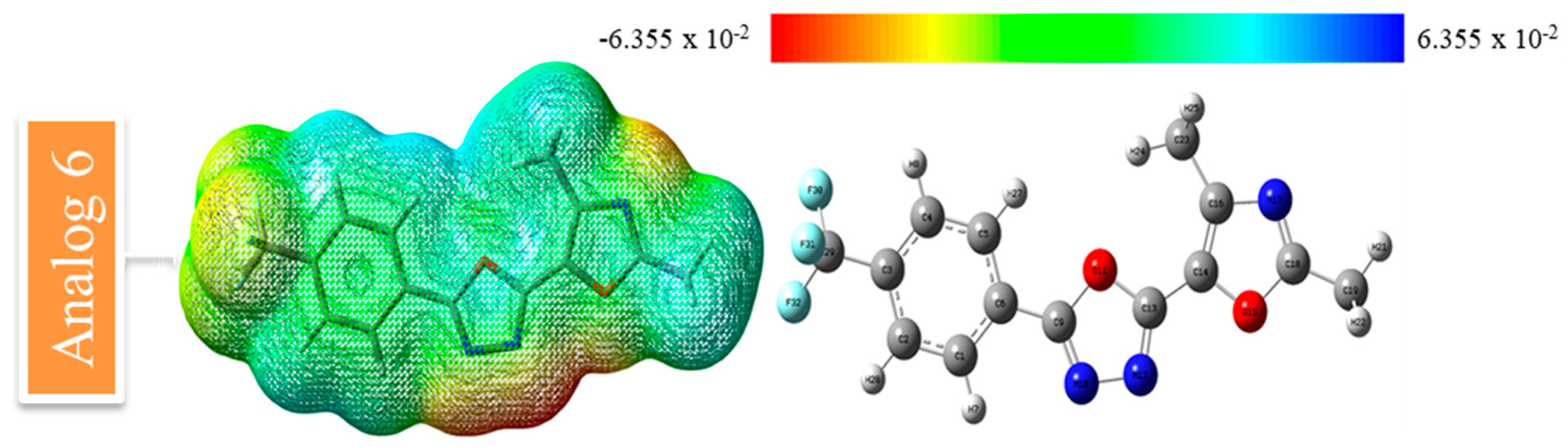
2.9.1. MESP Analysis
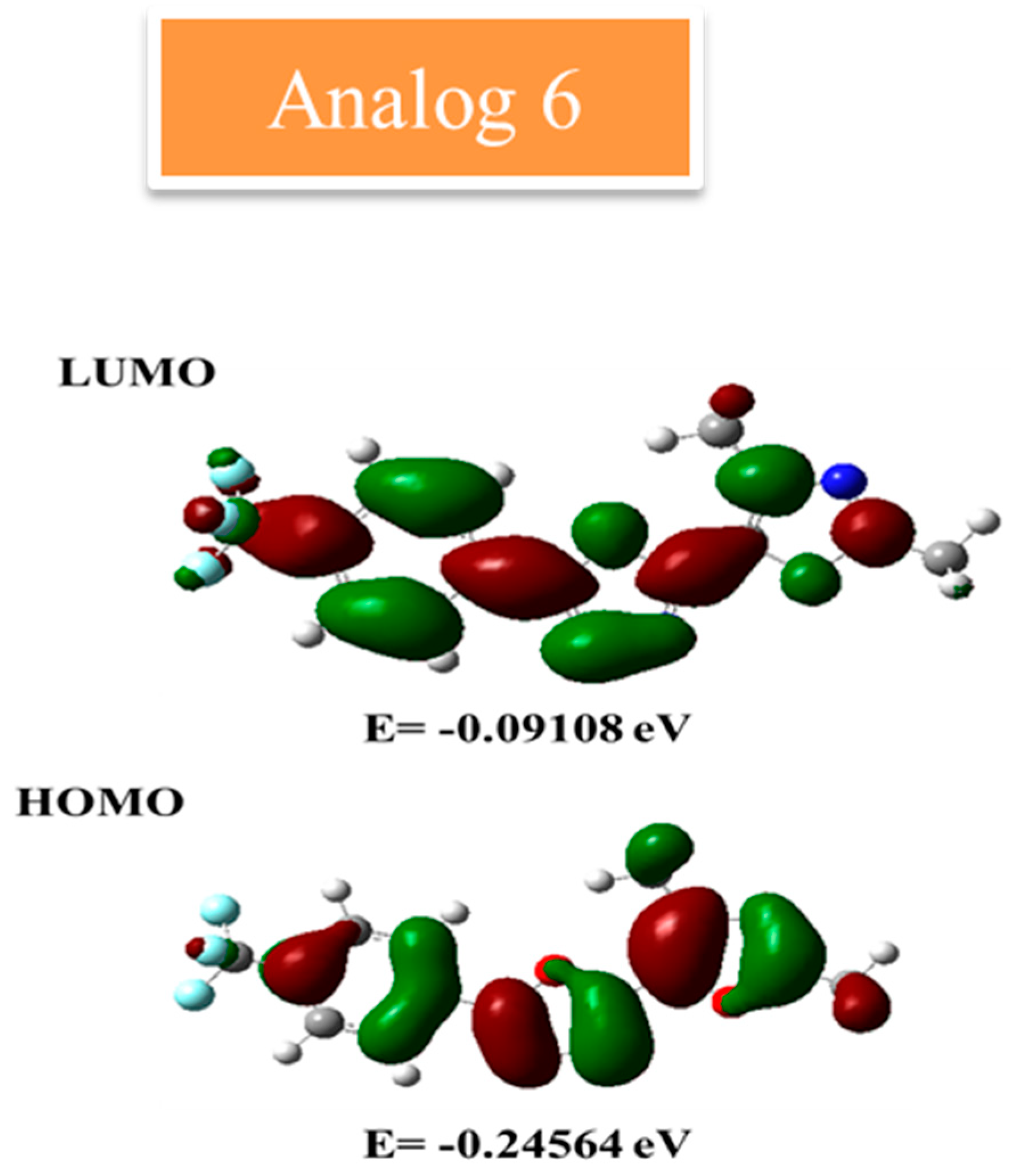
2.9.2. FMO Analysis
3. Materials and Methods
3.1. Materials
3.2. General Procedure to Obtain Derivatives (1–10)
3.3. Spectral Analysis
3.4. Protocol of Docking
3.5. Protocol of DFT
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Eid, M.M.A.; Rashed, A.N.Z.; Bulbul, A.A.M.; Podder, E. Mono-rectangular core photonic crystal fiber (MRC-PCF) for skin and blood cancer detection. Plasmonics 2021, 16, 717–727. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Daltveit, D.S.; Morgan, E.; Colombet, M.; Steliarova-Foucher, E.; Bendahhou, K.; Marcos-Gragera, R.; Rongshou, Z.; Smith, A.; Wei, H.; Soerjomataram, I. Global patterns of leukemia by subtype, age, and sex in 185 countries in 2022. Leukemia 2025, 39, 412–419. [Google Scholar] [CrossRef]
- Watts, J.; Nimer, S. Recent advances in the understanding and treatment of acute myeloid leukemia. F1000Research 2018, 7, 1196. [Google Scholar] [CrossRef]
- Liu, X.; Ye, F.; Wu, J.; How, B.; Li, W.; Zhang, D.Y. Signaling proteins and pathways affected by flavonoids in leukemia cells. Nutr. Cancer 2015, 67, 238–249. [Google Scholar] [CrossRef]
- Lee, K.; Chung, K.; Seo, J.; Yim, H.; Park, J.; Choi, K. Sulfuretin from heartwood of Rhus verniciflua triggers apoptosis through activation of Fas, Caspase-8, and the mitochondrial death pathway in HL-60 human leukemia cells. J. Cell. Biochem. 2012, 113, 2835–2844. [Google Scholar] [CrossRef]
- Howlader, N.; Noone, A.M.; Krapcho, M.; Garshell, J.; Neyman, N.; Altekruse, S.F.; Kosary, C.L.; Yu, M.; Ruhl, J.; Tatalovich, Z. SEER cancer statistics review, 1975–2010, Bethesda, MD. Natl. Cancer Inst. 2013, 21, 12. [Google Scholar]
- Goutam, D.; Sailaja, S. Classification of acute myelogenous leukemia in blood microscopic images using supervised classifier. In Proceedings of the 2015 IEEE International Conference on Engineering and Technology (ICETECH), Coimbatore, India, 20 March 2015; pp. 1–5. [Google Scholar]
- Yu, Z.; Liu, L.; Shu, Q.; Li, D.; Wang, R. Leukemia stem cells promote chemoresistance by inducing downregulation of lumican in mesenchymal stem cells. Oncol. Lett. 2019, 18, 4317–4327. [Google Scholar] [CrossRef]
- Zhang, J.; Gu, Y.; Chen, B. Mechanisms of drug resistance in acute myeloid leukemia. OncoTargets Ther. 2019, 12, 1937–1945. [Google Scholar] [CrossRef]
- Crossnohere, N.L.; Richardson, D.R.; Reinhart, C.; Donoghue, B.O.; Love, S.M.; Smith, B.D.; Bridges, J.F.P. Side effects from acute myeloid leukemia treatment: Results from a national survey. Curr. Med. Res. Opin. 2019, 35, 1965–1970. [Google Scholar] [CrossRef]
- Naimi, A.; Entezari, A.; Hagh, M.F.; Hassanzadeh, A.; Saraei, R.; Solali, S. Quercetin sensitizes human myeloid leukemia KG-1 cells against TRAIL-induced apoptosis. J. Cell. Physiol. 2019, 234, 13233–13241. [Google Scholar] [CrossRef] [PubMed]
- Lichtman, M.A. Obesity and the risk for a hematological malignancy: Leukemia, lymphoma, or myeloma. Oncologist 2010, 15, 1083–1101. [Google Scholar] [CrossRef] [PubMed]
- Diller, L. Adult primary care after childhood acute lymphoblastic leukemia. N. Engl. J. Med. 2011, 365, 1417–1424. [Google Scholar] [CrossRef]
- Biernacki, K.; Daśko, M.; Ciupak, O.; Kubiński, K.; Rachon, J.; Demkowicz, S. Novel 1, 2, 4-oxadiazole derivatives in drug discovery. Pharmaceuticals 2020, 13, 111. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, S.; Asati, V.; Singh, J.; Roy, P.P. 1, 3, 4-Oxadiazoles: An emerging scaffold to target growth factors, enzymes and kinases as anticancer agents. Eur. J. Med. Chem. 2015, 97, 124–141. [Google Scholar] [CrossRef]
- Glomb, T.; Szymankiewicz, K.; Świątek, P. Anti-cancer activity of derivatives of 1, 3, 4-oxadiazole. Molecules 2018, 23, 3361. [Google Scholar] [CrossRef]
- Zhu, W.; Bao, X.; Ren, H.; Liao, P.; Zhu, W.; Yan, Y.; Wang, L.; Chen, Z. Design, synthesis, and pharmacological evaluation of 5-oxo-1, 2, 4-oxadiazole derivatives as AT1 antagonists with antihypertension activities. Clin. Exp. Hypertens. 2016, 38, 435–442. [Google Scholar] [CrossRef]
- Ibrahim, M.T.; Uzairu, A.; Shallangwa, G.A.; Ibrahim, A. In-silico studies of some oxadiazoles derivatives as anti-diabetic compounds. J. King Saud Univ. 2020, 32, 423–432. [Google Scholar] [CrossRef]
- Caputo, F.; Corbetta, S.; Piccolo, O.; Vigo, D. Seeking for selectivity and efficiency: New approaches in the synthesis of raltegravir. Org. Process Res. Dev. 2020, 24, 1149–1156. [Google Scholar] [CrossRef]
- Tassinari, M.; Lena, A.; Butovskaya, E.; Pirota, V.; Nadai, M.; Freccero, M.; Doria, F.; Richter, S.N. A fragment-based approach for the development of G-quadruplex ligands: Role of the amidoxime moiety. Molecules 2018, 23, 1874. [Google Scholar] [CrossRef]
- Chawla, B.; Naaz, A.A. Exploring 1, 3, 4-oxadiazole scaffold for anti-inflammatory and analgesic activities: A review of literature from 2005–2016. Rev. Med. Chem. 2018, 18, 216–233. [Google Scholar] [CrossRef] [PubMed]
- Belaidi, S.; Mellaoui, M. Electronic Structure and Physical-Chemistry Property Relationship for Oxazole Derivatives by Ab Initio and DFT Methods. Org. Chem. Int. 2011, 2011, 254064. [Google Scholar] [CrossRef]
- Dabholkar, V.V.; Syed, S.A.S.A. Synthesis of novel oxazoles and their hydrazones. Rasayan J. Chem. 2010, 3, 761–765. [Google Scholar]
- Zahanich, I.; Kondratov, I.; Naumchyk, V.; Kheylik, Y.; Platonov, M.; Zozulya, S.; Krasavin, M. Phenoxymethyl 1, 3-oxazoles and 1, 2, 4-oxadiazoles as potent and selective agonists of free fatty acid receptor 1 (GPR40). Bioorg. Med. Chem. Lett. 2015, 25, 3105–3111. [Google Scholar] [CrossRef]
- Tandel, R.C.; Mammen, D. Synthesis and study of some compounds containing oxazolone ring, showing biological activity. Indian J. Chem.-Sect. B 2008, 47, 932–937. [Google Scholar] [CrossRef]
- Kachaeva, M.V.; Pilyo, S.G.; Zhirnov, B.V. Oxazole-5-sulfonamides as Novel Promising Anticancer lead Compounds. Int. J. Curr. Res. 2018, 10, 69410–69425. [Google Scholar]
- Khan, S.; Iqbal, T. A new synthetic approach for determination of thiadiazole clubbed sulfonamide as diabetic therapeutic with in silico modeling and kinetic study. J. Mol. Struct. 2025, 1338, 142261. [Google Scholar] [CrossRef]
- Khan, S.; Iqbal, T.; Hussain, R.; Khan, Y.; Fiaz, Z.; Rahim, F.; Darwish, H.W. Synthesis, Characterizations, Anti-Diabetic and Molecular Modeling Approaches of Hybrid Indole-Oxadiazole Linked Thiazolidinone Derivatives. Pharmaceuticals 2024, 17, 1428. [Google Scholar] [CrossRef]
- Iqbal, T.; Khan, S.; Rahim, F.; Tayyab, M.; Hussain, R.; Khan, Y.; Fiaz, Z.; Bibi, S.; Ullah, H.; Alzahrani, A.Y.; et al. Insights into the role of potent thiadiazole based Schiff base derivatives in targeting type-II diabetes: A combine in-vitro and in-silico approaches. J. Mole Struct. 2025, 1321, 140000. [Google Scholar] [CrossRef]
- Khan, S.; Hussain, R.; Khan, Y.; Iqbal, T.; Shoaib, K.; Jehangir, U.; Khowdiary, M.M.; Felemban, S. Developing Correlation Between In silico Molecular Modeling and In vitro α-Amylase, Anti-Cancer and SARS-CoV-2 Studies of Bis-Indole Derived Triazine-Based Thiazolidinone Hybrid Derivatives. Chem. Select. 2024, 9, 202400686. [Google Scholar] [CrossRef]
- Ghasemi, M.; Mahdavi, M.; Dehghan, M.; Eftekharian, S.; Mojtabavi, M.A.; Faramarzi, A.; Iraji, A. Substituted piperazine conjugated to quinoline-thiosemicarbazide as potent α-glucosidase inhibitors to target hyperglycemia. Sci. Rep. 2025, 15, 1871. [Google Scholar]
- Sepehrmansourie, H.; Azimi, M.; Ebadi, A.; Chehardoli, G.; Zolfigol, M.A.; Amanlou, M.; Montazer, M.N.; Mahdavi, M.; Najafi, Z. Novel coumarin-based acetohydrazide-1, 2, 3-triazole derivatives as urease enzyme inhibitors: Synthesis, in vitro evaluation, and molecular dynamics simulation studies. Heliyon 2025, 11, e41321. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M. Gaussian 09, Revision d. 01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Khan, A.U.; Muhammad, S.; Khera, R.A.; Shehzad, R.A.; Ayub, K.; Iqbal, J. DFT study of superhalogen (AlF4) doped boron nitride for tuning their nonlinear optical properties. Optik 2021, 231, 166464. [Google Scholar] [CrossRef]
- Esrafili, M.D.; Saeidi, N. DFT calculations on the catalytic oxidation of CO over Si-doped (6,0) boron nitride nanotubes. Struct. Chem. 2015, 27, 595–604. [Google Scholar] [CrossRef]
- Liu, H.B.; Gao, W.W.; Tangadanchu, V.K.R.; Zhou, C.H.; Geng, R.X. Novel aminopyrimidinyl benzimidazoles as potentially antimicrobial agents: Design, synthesis and biological evaluation. Eur. J. Med. Chem. 2018, 143, 66–84. [Google Scholar] [CrossRef]
- Marinho, E.S.; Marinho, M.M. A DFT study of synthetic drug topiroxostat: MEP, HOMO, LUMO. Int. J. Sci. Eng. Res. 2016, 7, 1264. [Google Scholar]
- Molecular Operating Environment (MOE), version 2016.08; Chemical Computing Group ULC: Montreal, QC, Canada, 2016.
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Molecular Operating Environment (MOE), 2015.10; Chemical Computing Group ULC: Montreal, QC, Canada, 2015.
- Frisch, M.J. Gaussian 98, Revision A.9; Gaussian Inc.: Pittsburgh, PA, USA, 1998. [Google Scholar]
- El-Baz, A.F.; Sorour, N.M.; Shetaia, Y.M. Trichosporon jirovecii-mediated synthesis of cadmium sulfide nanoparticles. J. Basic Microbiol. 2016, 56, 520–530. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Flurry, R.L., Jr. Molecular Orbital Theory of Bonding in Organic Molecules; Marcel Dekker: New York, NY, USA, 1968. [Google Scholar]
- Seboletswe, P.; Kumar, G.; Gcabashe, N.; Olofinsan, K.; Idris, A.; Islam, S.; Singh, P. Benzylidenehydrazine Derivatives: Synthesis, Antidiabetic Evaluation, Antioxidation, Mode of Inhibition, DFT and Molecular Docking Studies. Chem. Biodivers. 2025, 22, e202401556. [Google Scholar]

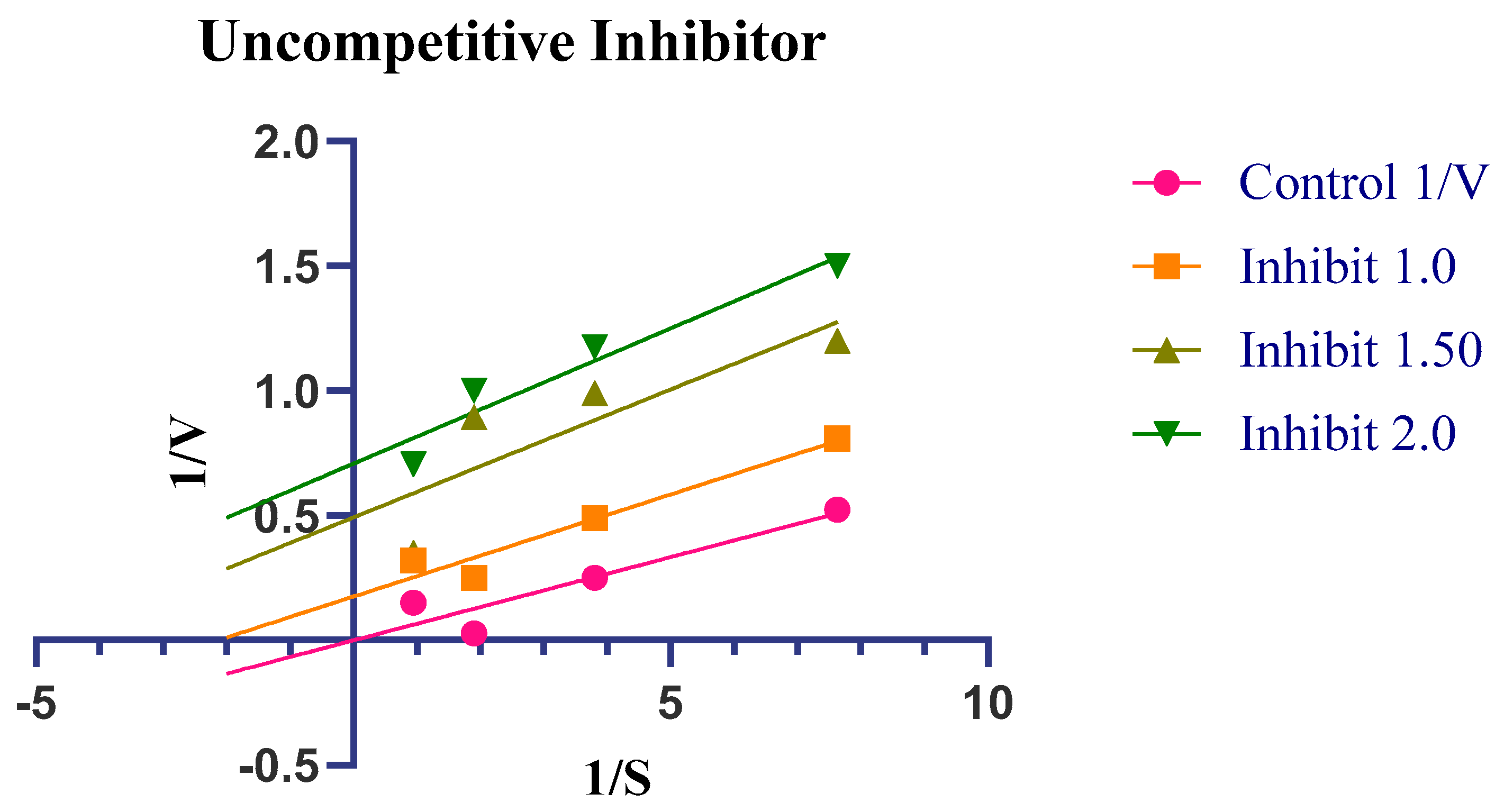


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S/No | R Groups | IC50 = HL-60 µM ± SEM | IC50 = PLB-985 µM ± SEM 1 |
|---|---|---|---|
| 1 | 4-NO2 | 16.20 ± 0.10 | 19.40 ± 0.30 |
| 2 | 4-Cl | 18.20 ± 0.30 | 23.30 ± 0.60 |
| 3 | 3-NO2-4-Br | 25.30 ± 0.10 | 31.30 ± 0.70 |
| 4 | 3-NO2-4-Br | 26.80 ± 0.40 | 33.20 ± 0.50 |
| 5 | 2,5-F | 9.10 ± 0.20 | 13.90 ± 0.50 |
| 6 | 4-CF3 | 8.50 ± 0.30 | 12.50 ± 0.20 |
| 7 | 2-F-4-OH | 15.20 ± 0.60 | 18.30 ± 0.80 |
| 8 | 3-Cl-4-F | 16.20 ± 0.20 | 21.10 ± 0.40 |
| 9 | 4-F | 13.30 ± 0.30 | 19.10 ± 0.60 |
| 10 | 3,5-OH | 9.80 ± 0.20 | 14.60 ± 0.20 |
| Standard drug Etoposide | 10.50 ± 0.20 | 15.20 ± 0.30 | |
| Compound | Mol.wt (g/mol) | Number of H-Bond Acceptors | Number of H-Bond Donors | Log S (ESOL) | Solubility | Log Po/w (ILOGP) | Log Kp (Skin Permeation) cm/s | Lipinski | Ghose | Veber | Egan | Muegge | Bioavailability Score | PAINS | Brenk | Leadlikeness | Synthetic Accessibility |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 5 | 277.23 | 7 | 0 | −3.61 | 6.76 × 10−2 mg/mL; 2.44 × 10−4 mol/L | 2.99 | −6.19 | Yes; 0 violation | Yes | Yes | Yes | Yes | 0.55 | 0 alert | 0 alert | Yes | 3.04 |
| 6 | 309.24 | 8 | 0 | −4.12 | 2.35 × 10−2 mg/mL; 7.59 × 10−5 mol/L | 3.03 | −5.91 | Yes; 0 violation | Yes | Yes | Yes | Yes | 0.55 | 0 alert | 0 alert | Yes | 3.12 |
| 10 | 273.24 | 7 | 2 | −3.01 | 2.64 × 10−1 mg/mL; 9.67 × 10−4 mol/L | 2.13 | −6.82 | Yes; 0 violation | Yes | Yes | Yes | Yes | 0.55 | 0 alert | 0 alert | Yes | 2.99 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khowdiary, M.M.; Khan, S.; Iqbal, T.; Rehman, W.; Hayat, A.; Hussain, R.; Shaaer, N.A.L.; Kashtoh, H. Anti-Leukemic Profiling of Oxazole-Linked Oxadiazole Derivatives: A Computational and Kinetic Approach. Pharmaceuticals 2025, 18, 625. https://doi.org/10.3390/ph18050625
Khowdiary MM, Khan S, Iqbal T, Rehman W, Hayat A, Hussain R, Shaaer NAL, Kashtoh H. Anti-Leukemic Profiling of Oxazole-Linked Oxadiazole Derivatives: A Computational and Kinetic Approach. Pharmaceuticals. 2025; 18(5):625. https://doi.org/10.3390/ph18050625
Chicago/Turabian StyleKhowdiary, Manal M., Shoaib Khan, Tayyiaba Iqbal, Wajid Rehman, Azam Hayat, Rafaqat Hussain, Nehad A. L. Shaaer, and Hamdy Kashtoh. 2025. "Anti-Leukemic Profiling of Oxazole-Linked Oxadiazole Derivatives: A Computational and Kinetic Approach" Pharmaceuticals 18, no. 5: 625. https://doi.org/10.3390/ph18050625
APA StyleKhowdiary, M. M., Khan, S., Iqbal, T., Rehman, W., Hayat, A., Hussain, R., Shaaer, N. A. L., & Kashtoh, H. (2025). Anti-Leukemic Profiling of Oxazole-Linked Oxadiazole Derivatives: A Computational and Kinetic Approach. Pharmaceuticals, 18(5), 625. https://doi.org/10.3390/ph18050625