Abstract
Alkaloids are nitrogen-containing compounds naturally occurring in plants, microorganisms, and marine organisms. Potent biological activities have been reported to date, ranging from neuroprotective to antioxidant and anticancer effects. Alkaloids have recently gained attention as potential antifungal agents for crop protection due to their broad spectrum of activity, eco-friendly nature, and ability to overcome some of the issues associated with synthetic fungicides, such as resistance development and environmental contamination. Several efforts have been made to obtain natural and nature-derived alkaloids endowed with significant activity against numerous pathogenic fungal strains. In this review, we collect synthetic strategies developed over the past decade to produce alkaloid fungicides for crop protection. Special emphasis is given to recent advancements in obtaining pure natural compounds and more potent analogs endowed with tailored and optimized properties.
1. Introduction
Globally, 2.4 billion people struggle daily with food insecurity, facing either moderate or severe shortages that impact their lives and safety. The global population is expected to grow by approximately 30% by 2050, increasing food insecurity and limiting the possibility of achieving the zero-hunger goal established by the United Nations in 2015 [1]. Agriculture plays a pivotal role in food waste, recording losses both in pre-and post-harvest handling, mainly because of the heavy impact of plant diseases. Of these, pathogenic fungi infections alone are responsible for 70–80% of the world’s major crop devastations and are considered a serious threat to global food security [2].
Fungicides are one of the most valuable means to combat this emergency. However, their efficiency in dealing with the insurgence of infections in intensive cultivation is slowed by the development of resistant strains, improper crop rotations, the presence of spores in the soil, and monocultures [3,4].
Additionally, the use of synthetic agrochemicals is raising strong concerns among consumers and researchers due to their hazards to the environment, mainly because of their slow turnover and their long-lasting toxic remains. For example, lipophilic residues can be detected in both water and soil, damaging the non-target animals and plants, as they act on basic biological processes not specific to fungi (e.g., energy production) [3]. The health hazards to humans, related to the use of pesticides, are also worth mentioning, as they are considered responsible for an increased risk of developing cancers and cardiovascular, neurological, and endocrine-related diseases [5]. Consequently, in recent years, there has been increased attention paid to the replacement of synthetic agrochemicals with safer molecules, such as natural and nature-derived compounds.
Natural products (NPs) represent between 30% and 35% of the drugs approved by the FDA over the last 35 years. The high structural diversity, specificity, and novel mode of action of natural bioactive compounds make nature an endless source of suitable drug candidates. These are usually secondary metabolites produced in low amounts in response to biotic or abiotic stress conditions, and their composition can vary due to the environment or the sourcing season [6]. Moreover, since these compounds could be bio-transformed by plants and microorganisms into safe metabolites, they usually display a more rapid turnover and a subsequent mild impact on the environment.
Alkaloids are a heterogeneous and wide class of compounds which are characterized by the presence of at least one atom of nitrogen. They are primarily derived from plants, fungi, and some marine organisms [7]. Their pharmacological properties have been deeply studied over the last centuries, starting with the well-known example of morphine [8]. Neuroprotective effects, as well as antioxidant, antimicrobial, and anticancer properties, are only some of the bioactivities ascribed to these compounds [9,10,11]. In particular, antiviral, antibacterial, and antifungal activities have been reported; moreover, in recent years, their potential as a treatment for phytopathogenic fungi has raised interest among the scientific community [12,13].
In the framework of crop protection, alkaloids exhibit a broad spectrum of activities against different fungal species, including those responsible for plant diseases, such as Fusarium, Botrytis, and Alternaria. Additionally, with the rising concerns about the environmental impact of synthetic fungicides, alkaloids offer a more eco-friendly alternative. Many of them degrade naturally in the environment, as they are taken up and accumulated by acceptor plants, thus reducing the risk of soil and water contamination [14]. Finally, alkaloids may reduce the likelihood of resistance development in fungal populations, thanks to their diverse chemical structures and modes of action (e.g., inhibition of cell wall synthesis, disruption of membrane integrity, inhibition of enzymes necessary for fungal metabolism, and DNA damage).
The interest in developing synthetic approaches to attain bioactive natural alkaloids is widely observed in the literature. This is mainly due to the difficulty of extracting them from their natural matrices, driving up the cost and effort needed to gather the compounds of interest in reasonable yields and purities. As a matter of fact, alkaloid availability is often constrained by factors like specific plant cultivation conditions, slow growth rates, and difficulties in large-scale harvesting. Under these circumstances, chemical preparation offers a more controlled, scalable, and cost-effective method of production, enabling the obtainment of alkaloids in the necessary quantities to carry out bioactivity studies and potentially, effective crop protection. Moreover, synthetic approaches open the door to explore structure–activity relationships (SAR), enabling structural modifications of selected alkaloids to improve their effectiveness against fungal pathogens while minimizing negative impacts on non-target organisms. In this way, alkaloid analogs and derivatives can be tailored to produce desirable properties, such as increased solubility, enhanced stability against environmental stressors (e.g., heat and light), and more efficient delivery to target fungi.
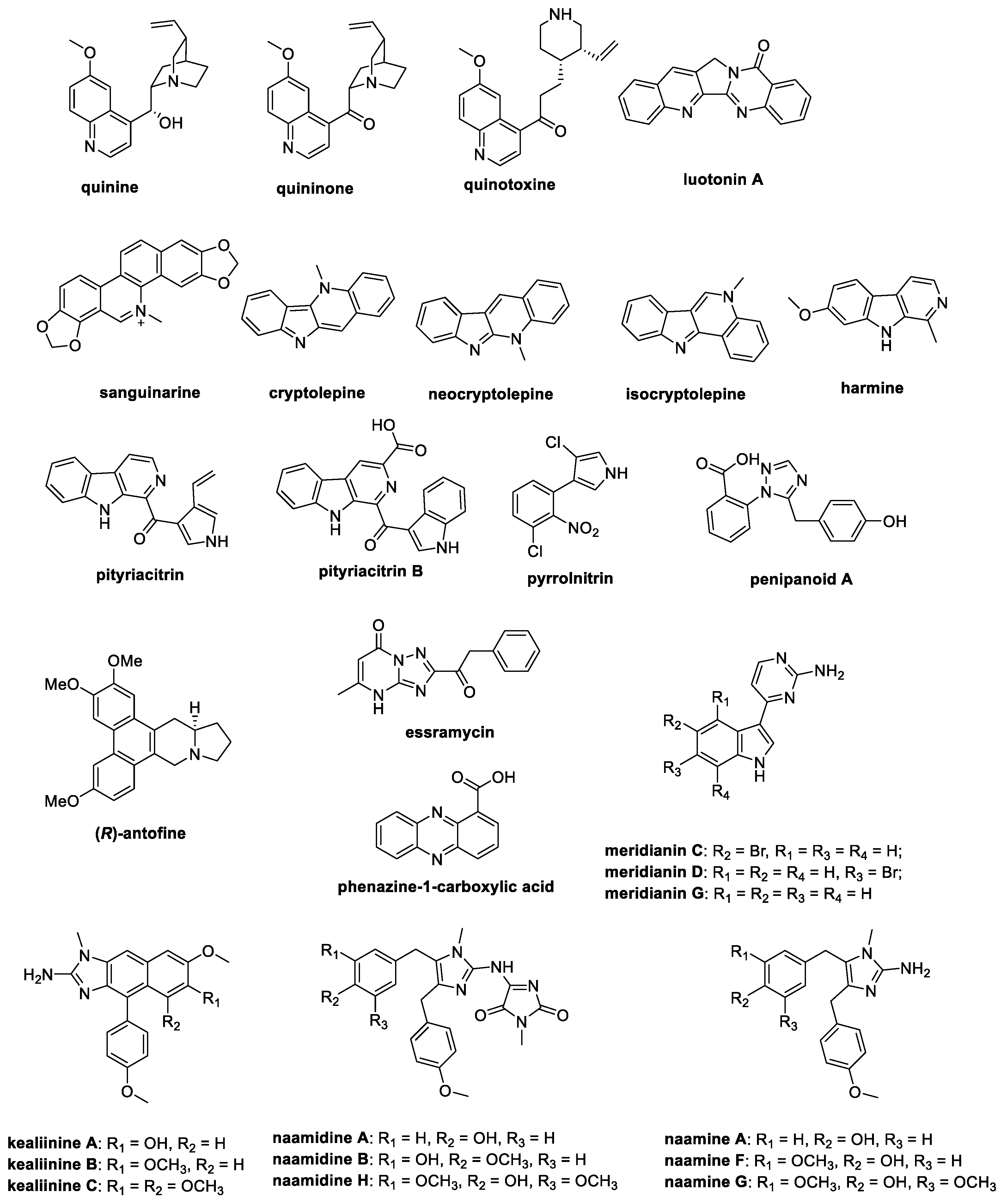
This review aims to perfuse the literature from the last ten years to obtain information regarding the total synthesis of natural alkaloids with antifungal activity in the field of crop protection, exploiting the SciFinder®, PubMed, and Scopus databases. Derivatives and analogs have been wittingly excluded from this work to highlight the variety and potential of natural parent compounds. For this reason, the attention will focus on the most investigated classes of alkaloids as antifungal agents, i.e., quinoline-, isoquinoline-, and indole-containing alkaloids, together with substituted indoles, imidazoles, and pyrroles, with the major purpose of supporting research groups working on the synthesis and investigation of such chemotypes as crop protectors (Figure 1).
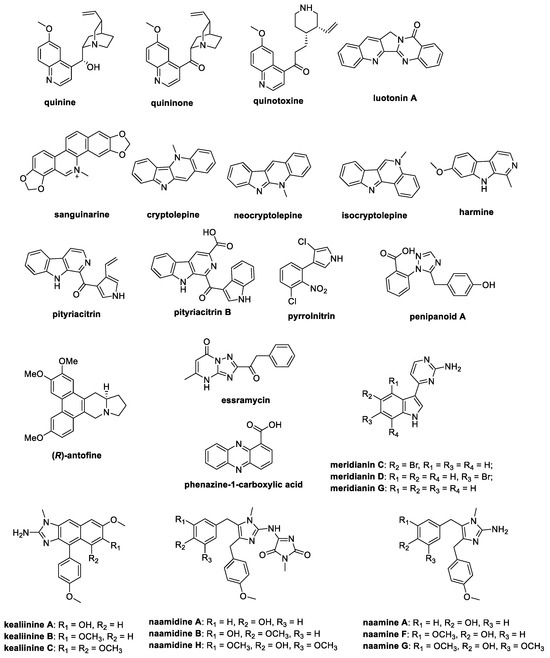
Figure 1.
Chemical structures of the natural antifungal alkaloids whose synthesis is reported in this paper.
2. Quinoline- and Isoquinoline-Containing Alkaloids
2.1. Quinine and Quinine Analogs (from Plants)
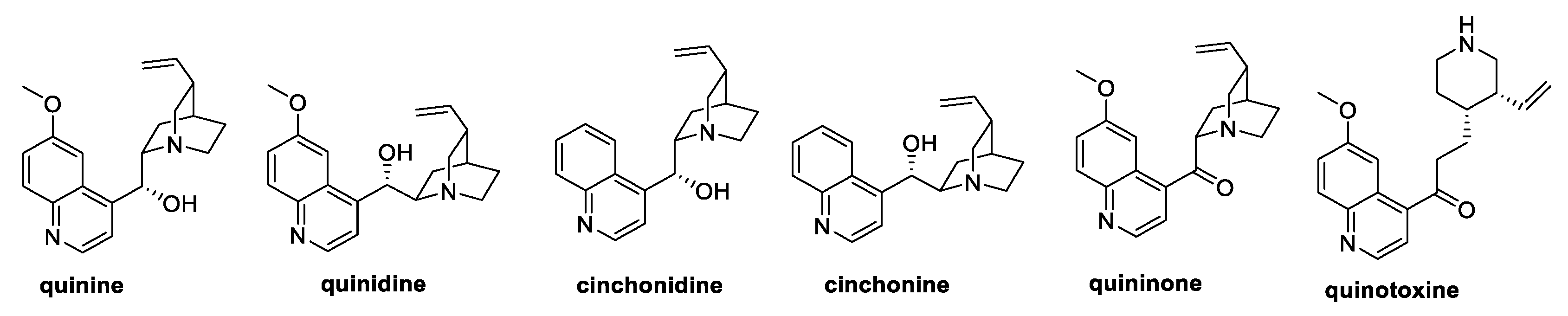
Among the natural quinoline alkaloids, quinine (Figure 2) is undeniably one of the most studied. Isolated from the bark of several species of Cinchona and Remijia trees, quinine shows several biological activities, ranging from antimalarial, anti-inflammatory, and anticancer to antifungal types [15,16]. Specifically, quinine was reported to inhibit mycelia growth of F. graminearum by 46%, whereas a 30% inhibition was observed against P. zeae, R. solani, M. melonis, and M. oryzae [15].
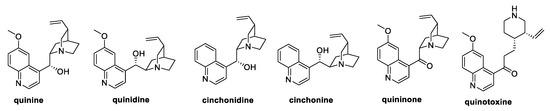
Figure 2.
Structure of quinine and its natural analogs.
Several natural quinine analogs, like quinidine, cinchonidine, cinchonine, quininone, and quinotoxine (Figure 1), were also evaluated as antifungal agents against pathogenic fungi, showing weak antifungal activities at 100 µg/mL concentration [15].
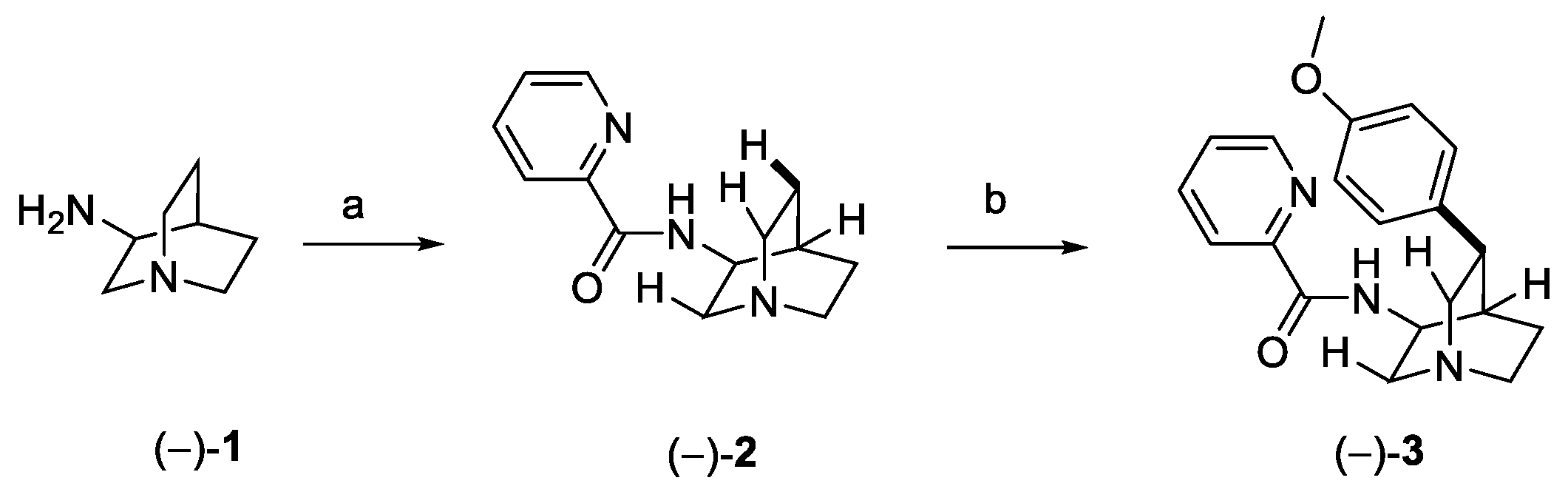
The stereoselective total synthesis of quinine was reported for the first time in 2001 in 15 steps, starting from enantiomerically pure (S)-4-vinyl butyrolactone [17], while several new synthetic routes have been developed to obtain this relevant compound over the last decades [18,19]. In 2018, O’Donovan et al. reported a novel approach based on two stereoselective key steps: (1) a C–H activation followed by (2) an aldol reaction, in order to connect the two heterocyclic moieties of quinine [20]. The C(sp3)-H activation (Scheme 1) was accomplished by treating (−)-3-aminoquinuclidine (1) with 2-picolinic acid to obtain compound (−)-2, with the amide fragment acting as the directing group towards the selective activation of the H at C(5). The Pd-catalyzed formation of a new C(sp3)–C(sp2) bond with the anisole fragment allowed the obtainment of enantiomerically pure (−)-3 with complete regio- and stereoselectivity.
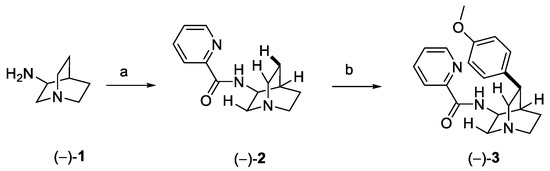
Scheme 1.
C(sp3)–H activation of 3-aminoquinuclidine and synthesis of intermediate 3: (a) picolinic acid, CDI, DMF; (b) 4-iodoanisole, Pd(OAc)2, Ag2CO3, DMF.
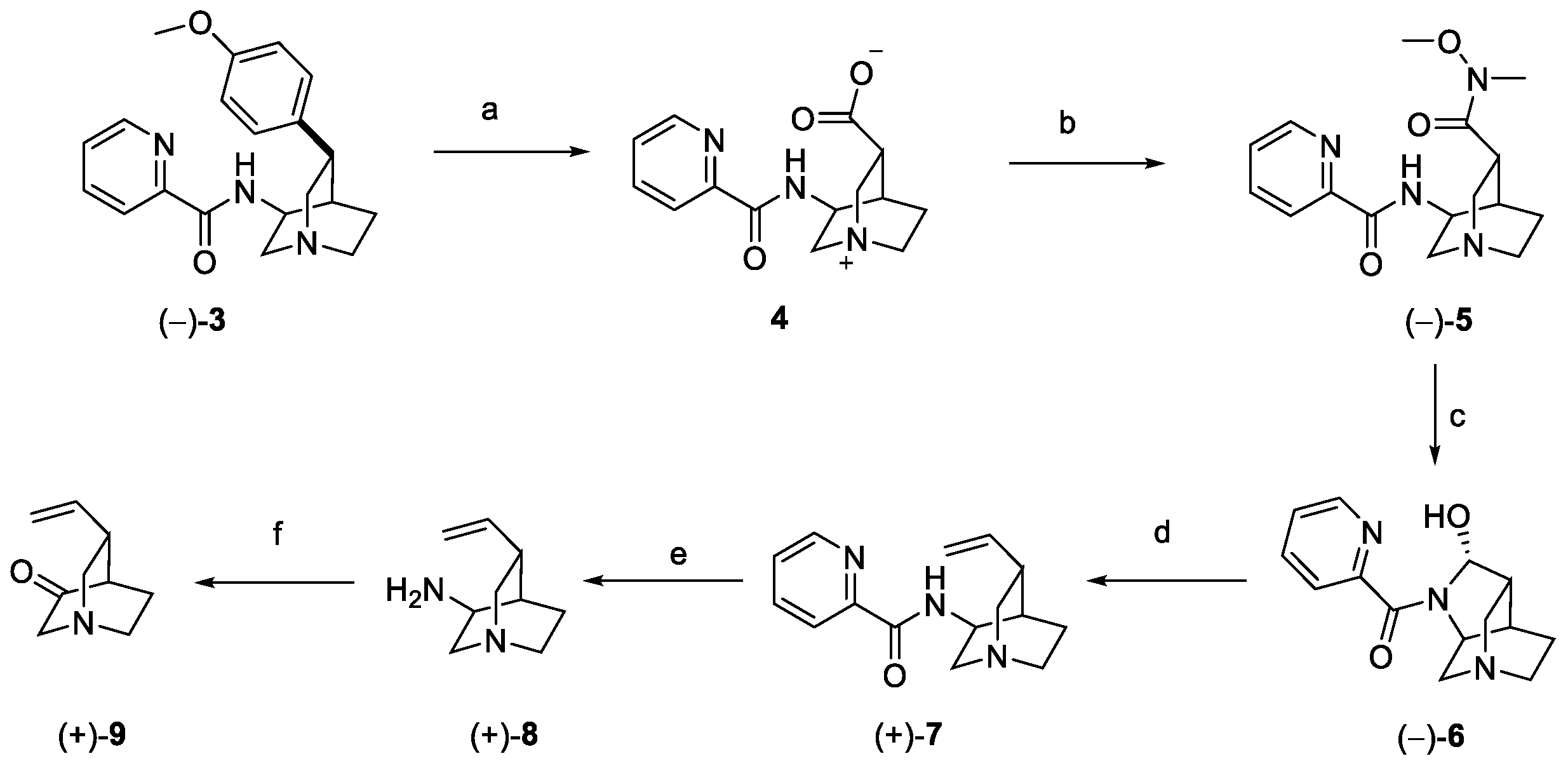
Intermediate 3 underwent oxidative degradation of the benzyl moiety under ruthenium catalysis, leading to zwitterion carboxylic acid 4 (Scheme 2). Generation of the Weinreb amide 5, followed by DIBAL-H mediated reduction, yielded optically pure hemiaminal 6. Wittig olefination with methylenetriphenylphosphorane afforded alkenyl-quinuclidine 7, further converted to the free amine 8 under reducing conditions. Final oxidation in the presence of IBX generated optically pure 9, as the key building block for the synthesis of quinine [20].
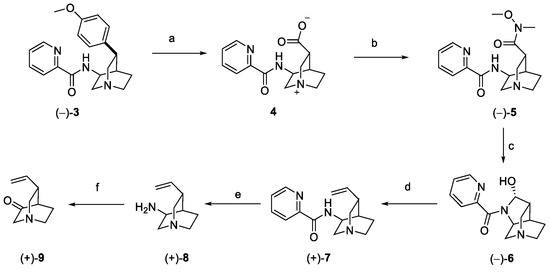
Scheme 2.
Synthesis of building block 9. (a) RuCl3, NaIO4, H2O, EtOAc, CH3CN; (b) HATU, MeNHOMe·HCl, Et3N, DMF; (c) DIBAL-H, DCM; (d) Ph3PMeBr, LiHMDS, DMSO, THF; (e) Zn, HCl, Zn(OTf)2, H2O; (f) IBX, p-TsOH, CH3CN.
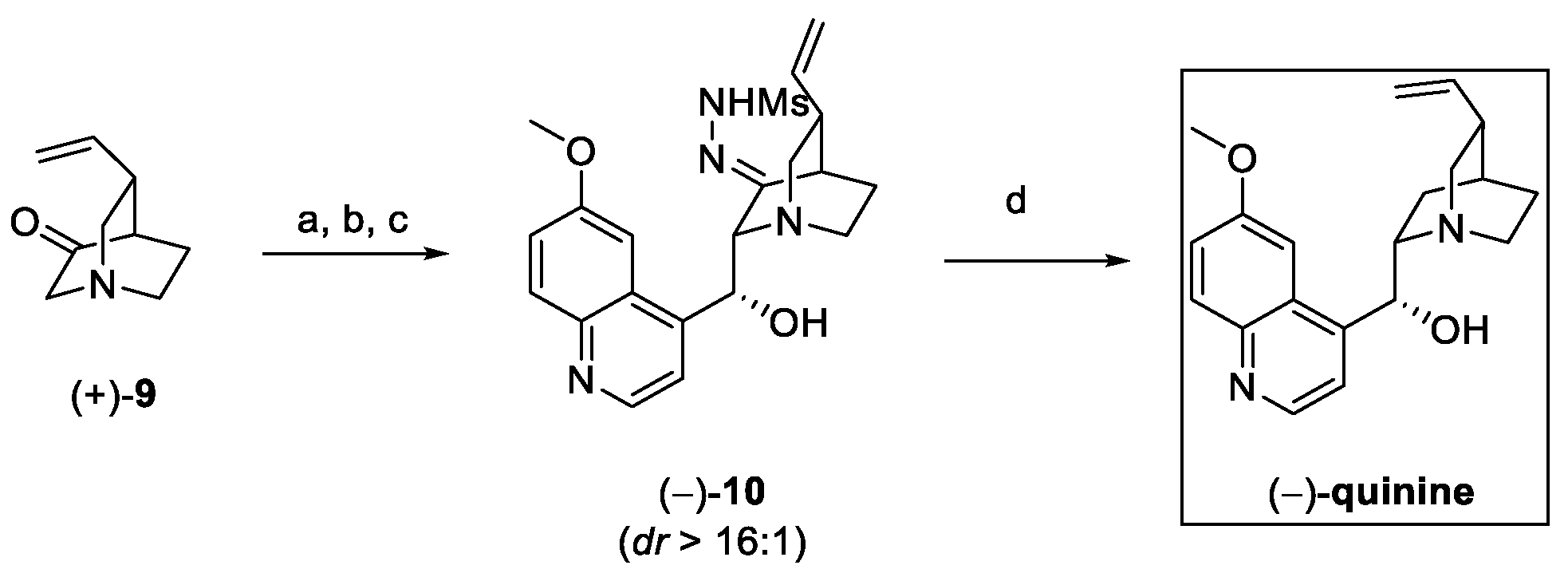
Aldol reaction between 9 and the suitable quinoline carbaldehyde was followed by the in situ conversion of the ketone to sulfonylhydrazone (10). Under these conditions, epimerization-free reduction in presence of LiAlH4 takes place, affording (−)-quinine in 10 steps, with a 5% overall yield (Scheme 3).
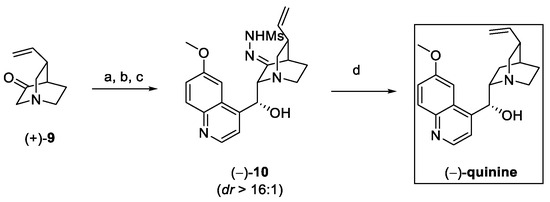
Scheme 3.
Conversion of (+)-9 to (−)-quinine. (a) LiHMDS, THF; (b) 6-methoxyquinoline-4-carbaldehyde; (c) Ti(O-iPr)3Cl, MsNHNH2; (d) LiAlH4, MeOH, THF.
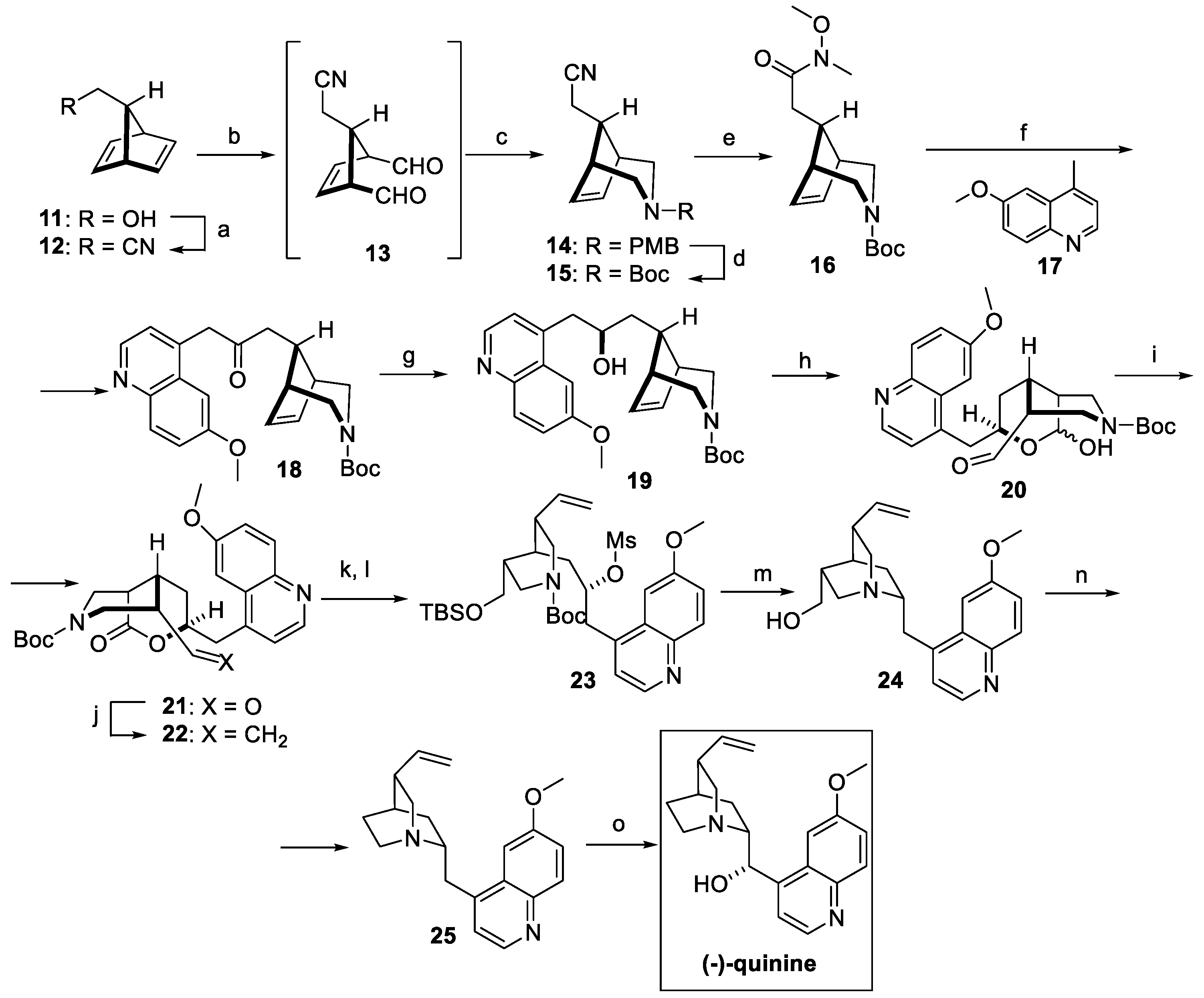
A “local desymmetrization” approach was exploited by Lee and Chen for the synthesis of pure quinine and quinidine, both obtained starting from the same racemic intermediate (Scheme 4) [21]. Commercially available primary alcohol 11 was converted to nitrile 12 under Mitsunobu conditions. The resulting diene was dihydroxylated and underwent oxidative cleavage to afford dialdehyde 13. Ring closing by reductive amination yielded the protected piperidine fused bicyclic 14, which was converted to carbamate 15 and then to Weinreb amide 16. The quinoline fragment was introduced by reaction with 6-methoxylepidine 17 in the presence of LDA (compound 18), which was further reduced to racemic alcohol 19. Functionalization of the endocyclic double bond resulted in a mixture of hemiacetals 20, further oxidated with DMP as the key desymmetrization step to obtain lactone 21 as a single stereoisomer, then converted to the vinyl substituent under modified Julia conditions (compound 22). Ketone reduction and conversion to mesylate 23 allowed for, upon Boc deprotection, the nucleophilic substitution and formation of the characteristic quinuclidine moiety (compound 24). Swern oxidation of the primary alcohol resulted in the corresponding aldehyde, which underwent Rh-catalyzed deformylation to dehydroxyquinine 25, which was selectively hydroxylated to natural quinine, obtained with an overall yield of 17% [21].
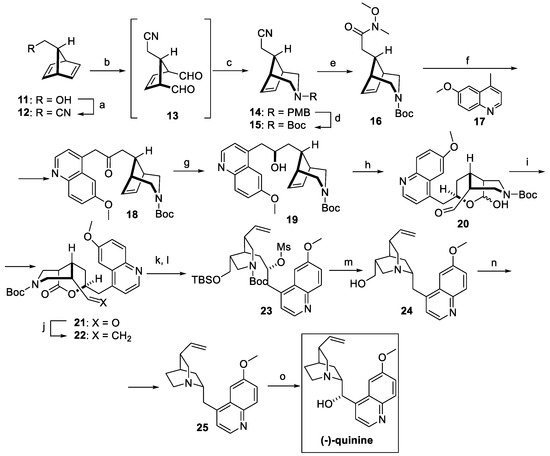
Scheme 4.
Total synthesis of (−)-quinine starting from intermediate 11: (a) acetone cyanohydrin, PPh3, DEAD, Et2O; (b) OsO4, NMO, acetone/H2O, then Pb(OAc)4, DCM; (c) NaBH3CN, PMBNH2, AcOH, MeOH; (d) triphosgene, Et3N, DCM, then NaHCO3, Boc2O; (e) NaOH/MeOH, then EDC, HOBt, Et3N, MeNH(OMe)•HCl, DCM; (f) 17, LDA, THF; (g) NaBH4, MeOH; (h) OsO4, NaIO4, 2,6-lutidine, dioxane/H2O; (i) DMP, DCM; (j) MeSO2PT, KHMDS, THF; (k) LiAlH4, THF; (l) TBSOTf, 2,6-lutidine, DCM, then MsCl, Et3N, DCM; (m) DCM/TFA, then NaHCO3, MeCN; (n) (COCl)2, DMSO, Et3N, DCM, then [Rh(COD)Cl]2, dppp, diglyme; (o) NaH/DMSO, then O2.
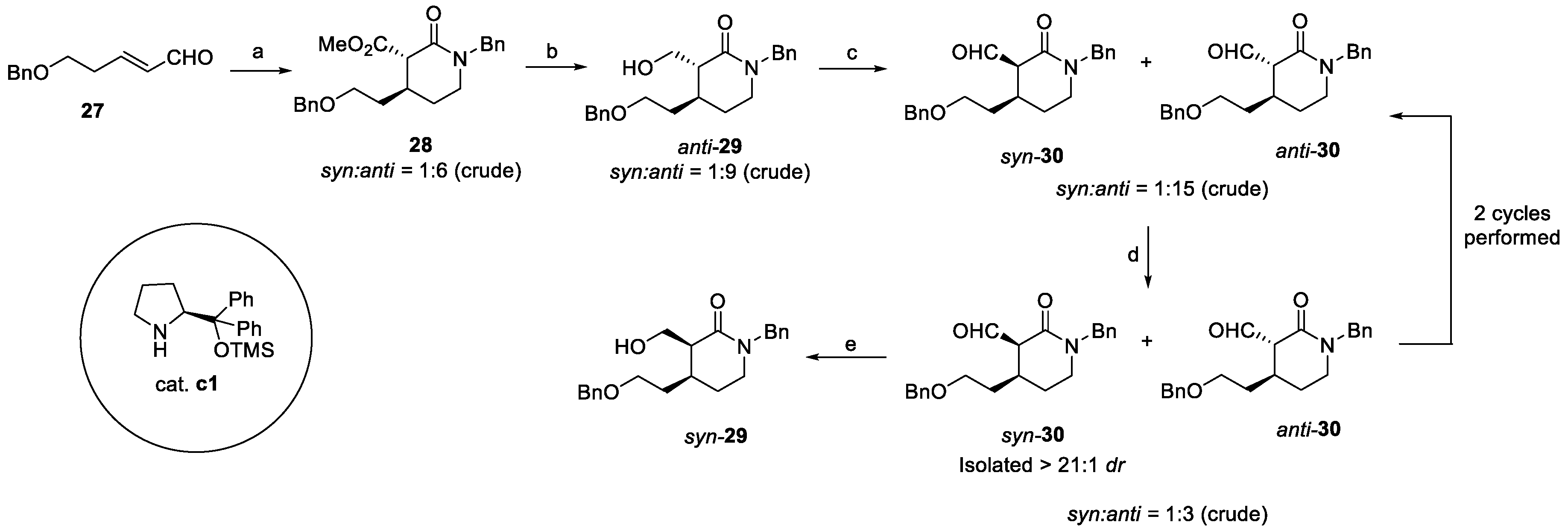
Lastly, Jiang et al. synthetized quinine via catalytic enantioselective cascade transformations [22]. In this case, starting from α,β-unsaturated aldehyde 27, a one-pot cascade in the presence of dimethyl malonate led to the α-substituted lactam anti-28, with 6:1 dr and 97% ee (Scheme 5). Reduction to primary alcohol afforded anti-29, which was further oxidized to aldehyde 30. Mild piperidine-catalyzed epimerization allowed for the isolation of syn-30 with >21:1 dr, which was ultimately reduced to the desired syn-29 [22].
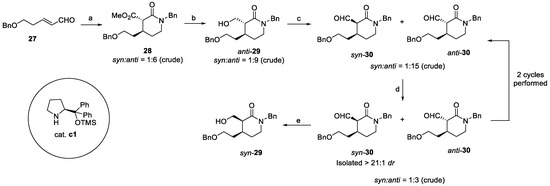
Scheme 5.
Synthesis of key intermediate syn-29: (a) dimethyl malonate, 10 mol% c1, EtOH, then BnNH2, NaBH(OAc)3, DCM, 97% ee, 85%; (b) LiAlH4, THF, >21:1 dr (isolated), 78%; (c) (COCl)2, DMSO, DCM, then TEA, 9:1 dr (isolated), 82%; (d) pyrrolidine, AcOH, DCM; (e) NaBH4, MeOH.
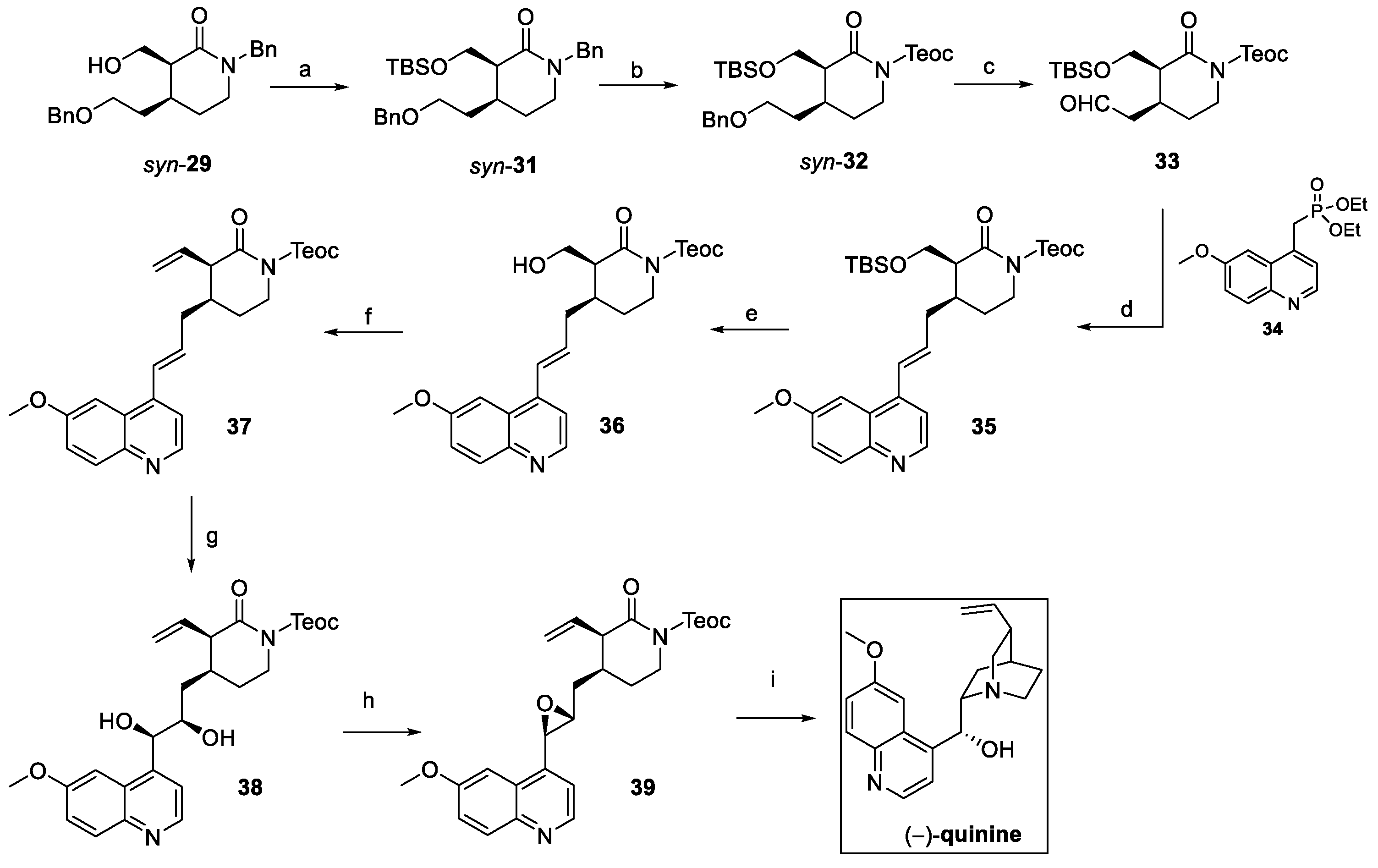
Such an approach was crucial to guarantee the required stereochemical control so that the debenzylation and further oxidation of suitably protected syn-31 could afford aldehyde 33 (Scheme 6). The Horner–Emmons reaction with the phosphonated quinoline 34 yielded olefin 35, treated to obtain the vinyl key intermediate 37. Catalytic chemo- and enantioselective dihydroxylation of the internal double bond using AD-mix-β gave diol 38, as a precursor of epoxide 39. Final conversion of the lactam ring to piperidine and the deprotection of the TEOC resulted in the stereoselective intramolecular cyclization on the epoxide, affording (−)-quinine in 20% overall yield from syn-29 [22].
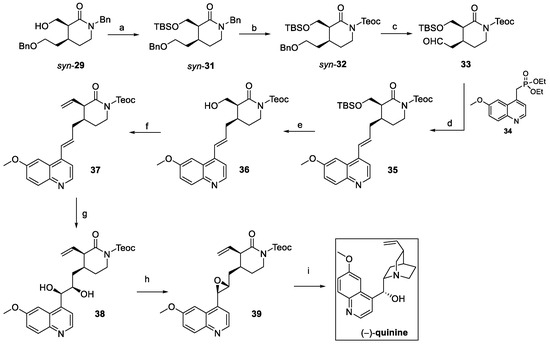
Scheme 6.
Total synthesis of (–)-quinine starting from intermediate isolated stereoisomer syn-29: (a) TBSCl, imidazole, dry DMF; (b) TMSCH2CH2OH, triphosgene, K2CO3, toluene; (c) i. Pd/C, H2, MeOH, ii. (COCl)2, DMSO, DCM, then TEA; (d) 34, NaH, THF; (e) HCl/MeOH; (f) i. (COCl)2, DMSO, DCM, then TEA; ii. Ph3P+CH3Br–, KOt-Bu, THF; (g) ADmix-β, CH3SO2NH2, t-BuOH, H2O, >21:1 dr, 82%; (h) i. trimethylorthoacetate, PPTS, DCM; ii. TMSCl, DCM; iii. K2CO3, MeOH; (i) CsF, t-BuOH, DMF.
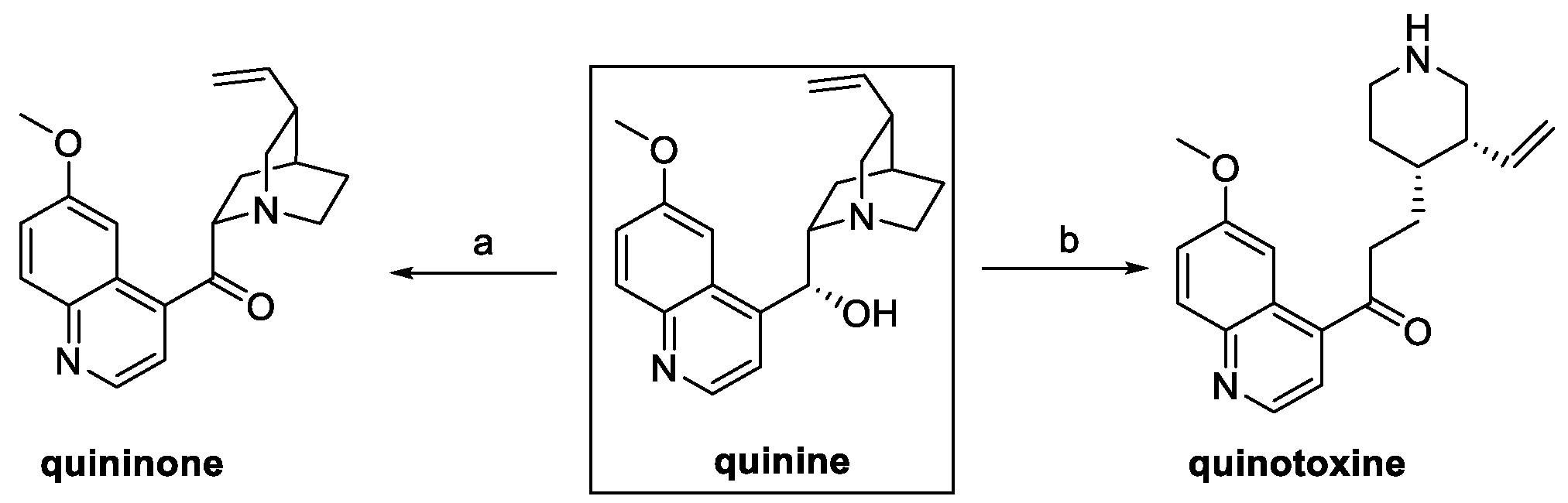
Among the natural quinine analogs, quininone and quinotoxine can be considered as derivatives of quinine itself. In fact, oxidation of the secondary alcohol in the presence of potassium tert-butoxide and benzophenone yielded quininone, whereas the boiling aqueous acetic acid treatment led to the quinuclidine bicyclic opening and hydroxyl group oxidation to quinotoxine (Scheme 7) [15].
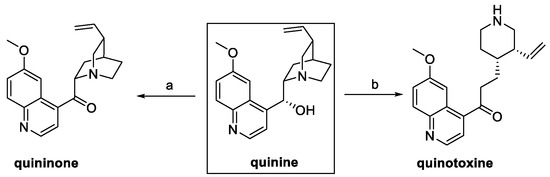
Scheme 7.
Synthesis of quininone and quinotoxine starting from quinine. (a) t-BuOK, benzophenone, toluene; (b) H2O/AcOH.
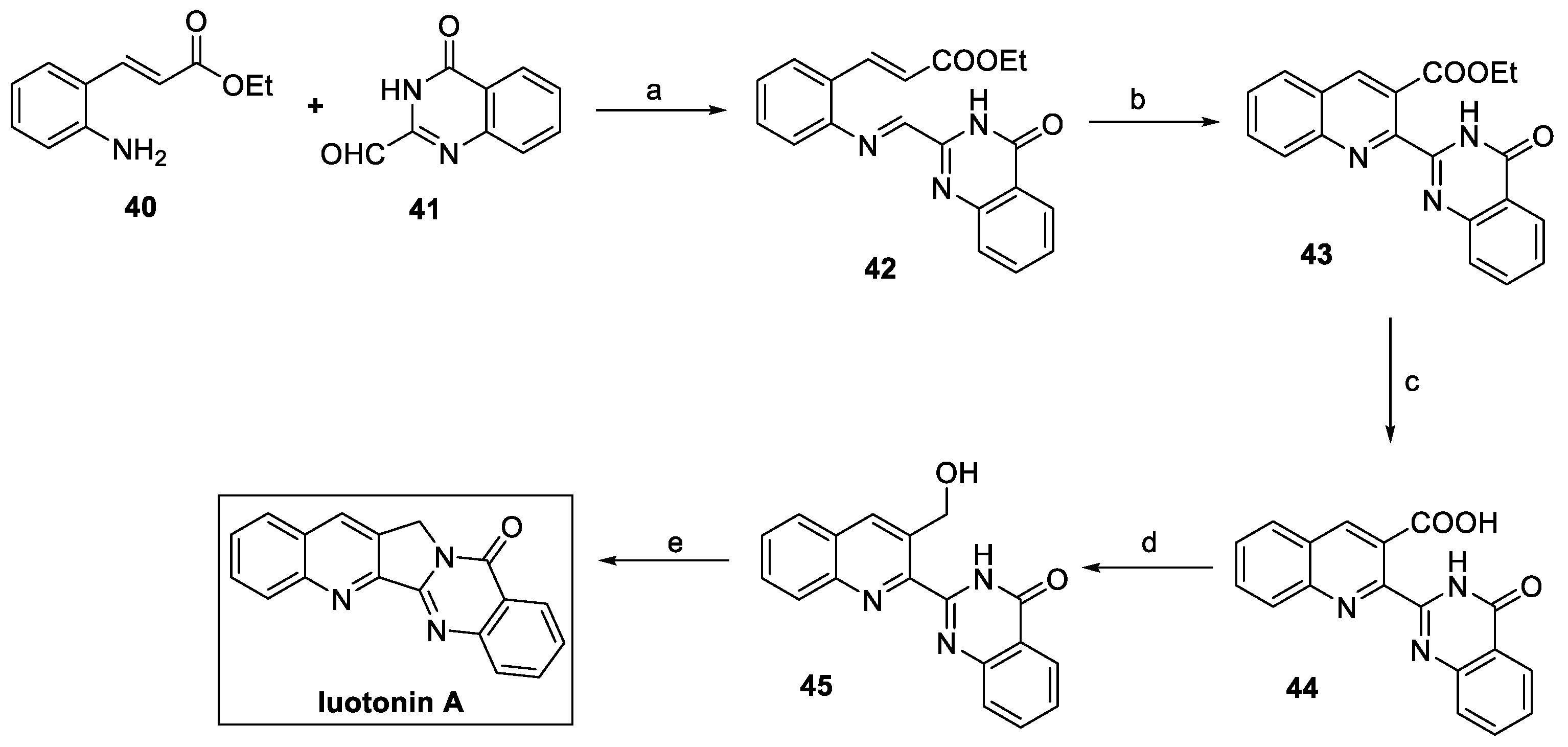
2.2. Luotonin A (from Plants)
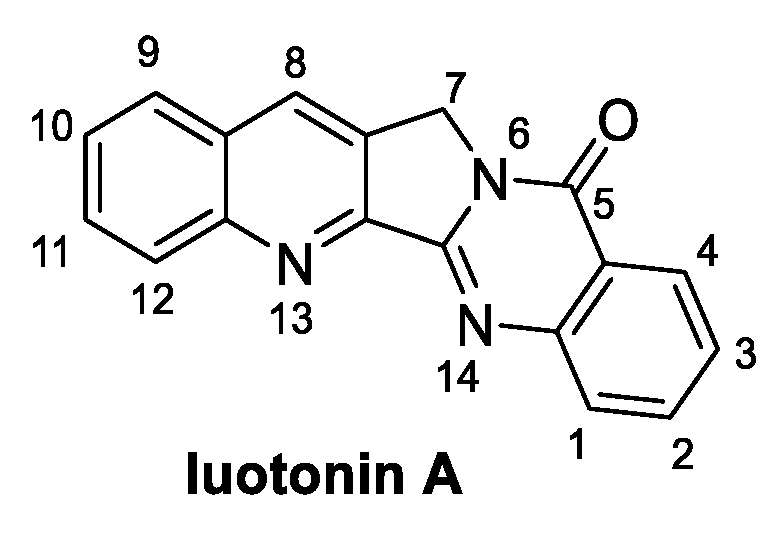
Luotonin A (Figure 3) is a quinoline alkaloid extracted from the Chinese medicinal plant Peganum nigellastrum. It specifically targets DNA topoisomerase I, resulting in a potential antitumoral natural compound [23]. Notably, evaluation of the antifungal activity of luotonin A resulted in an EC50 value of 0.50 mM against B. cinerea and M. oryzae [24]. Examples of its total synthesis have been reported at the end of the 20th century; however, over the last decade, several authors have proposed alternative synthetic routes, as discussed hereafter.
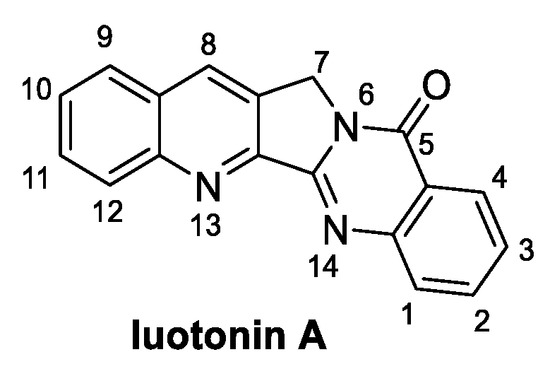
Figure 3.
Chemical structure of luotonin A.
In 2016, Kwon et al. described the total synthesis of luotonin A via the condensation between 2-amino cinnamate 40 and quinazolinone-2-carbaldehyde 41 (Scheme 8). The resulting aldimine 42, in this case, acts as a direct precursor of the fused pentacyclic moiety. Thermal electrocyclization under microwave irradiation at 160 °C generated the desired quinoline core (compound 43). Suitable functionalization of the ester moiety yielded alcohol 45, which finally underwent an intramolecular Mitsunobu reaction to the desired natural product luotonin A, with an overall yield of 32% in five steps [25].
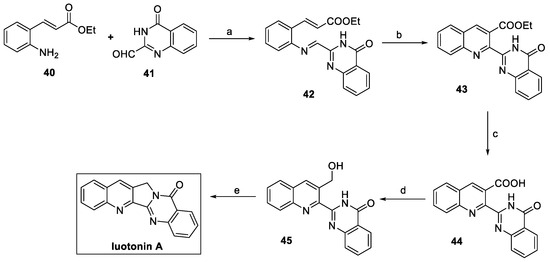
Scheme 8.
Synthesis of luotonin A. (a) MgSO4, Na2SO4, THF; (b) 1,2,4-trichlorobenzene, μW; (c) KOH, MeOH/H2O; (d) i. (COCl)2, DCM, DMSO; ii. NaBH4, THF; (e) PPh3, DIAD, THF.
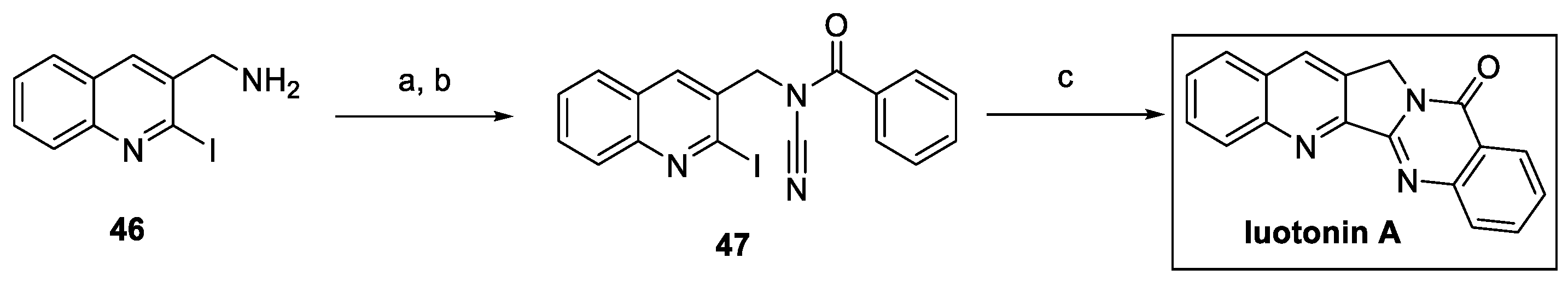
The synthesis of luotonin A was carried out by Baguia et al. in the framework of a copper-catalyzed photoinduced radical domino cyclization of ynamides and cyanamides [26]. In particular, aminomethylquinoline 46 was converted to N-benzoylcyanamide 47 by cyanation and benzoylation reactions (Scheme 9). The photocatalyzed cyclization of 47 led to the quinazoline core and the fused pentacyclic system of luotonin A by 5-exo-dig and 6-endo-trig consecutive cyclizations.
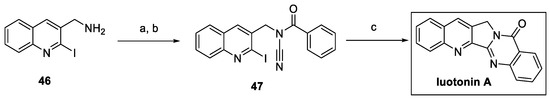
Scheme 9.
Synthesis of luotonin A by copper-catalyzed photoinduced radical domino cyclization reaction. (a) BrCN, Na2CO3, THF, then (b) BzCl, NaH, THF; (c) [(DPEphos)(bcp)Cu]PF6, Cy2NiBu, K2CO3, light (420 nm), MeCN.
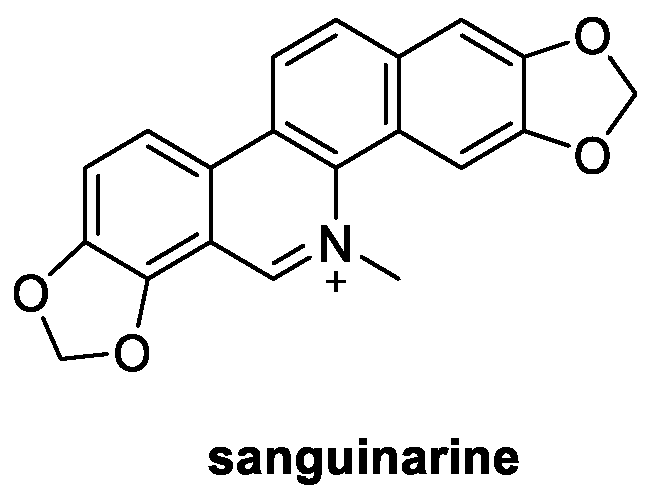
2.3. Sanguinarine (from Plants)
Extracted from the rhizomes of the plant species Sanguinaria canadensis, isoquinoline alkaloid sanguinarine (Figure 4) is present in nature both as a charged iminium (pH 2–6) or an uncharged alkanolamine (pH 6.5–9.0) [27]. From the biological point of view, sanguinarine is well known not only for its remarkable antitumoral effect but also for its antioxidant and antifungal activities [28,29]. In particular, the greatest inhibitory activity was observed against M. oryzae (EC50 = 6.96 μg/mL), which was even better than that of the positive control azoxystrobin (EC50 = 12.04 μg/mL). Indeed, treatment of M. oryzae mycelia with sanguinarine at 10 μg/mL induced morphology and cell membrane integrity damage. Furthermore, the reactive oxygen species production, mitochondrial membrane potential, and nuclear morphometry of the mycelia were changed, and the membrane function and cell proliferation of the mycelia were destroyed. Moreover, sanguinarine significantly suppressed the spore germination process in M. oryzae at the concentration of 50 μg/mL [29].
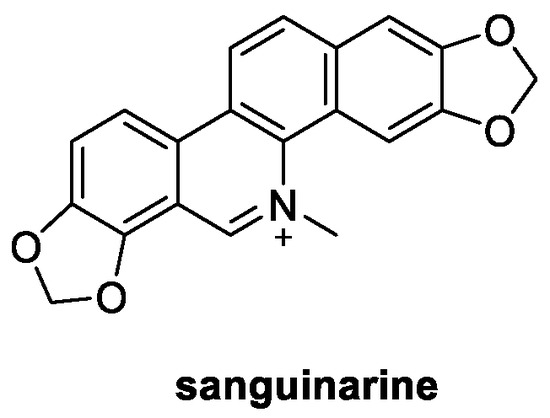
Figure 4.
Chemical structure of sanguinarine.
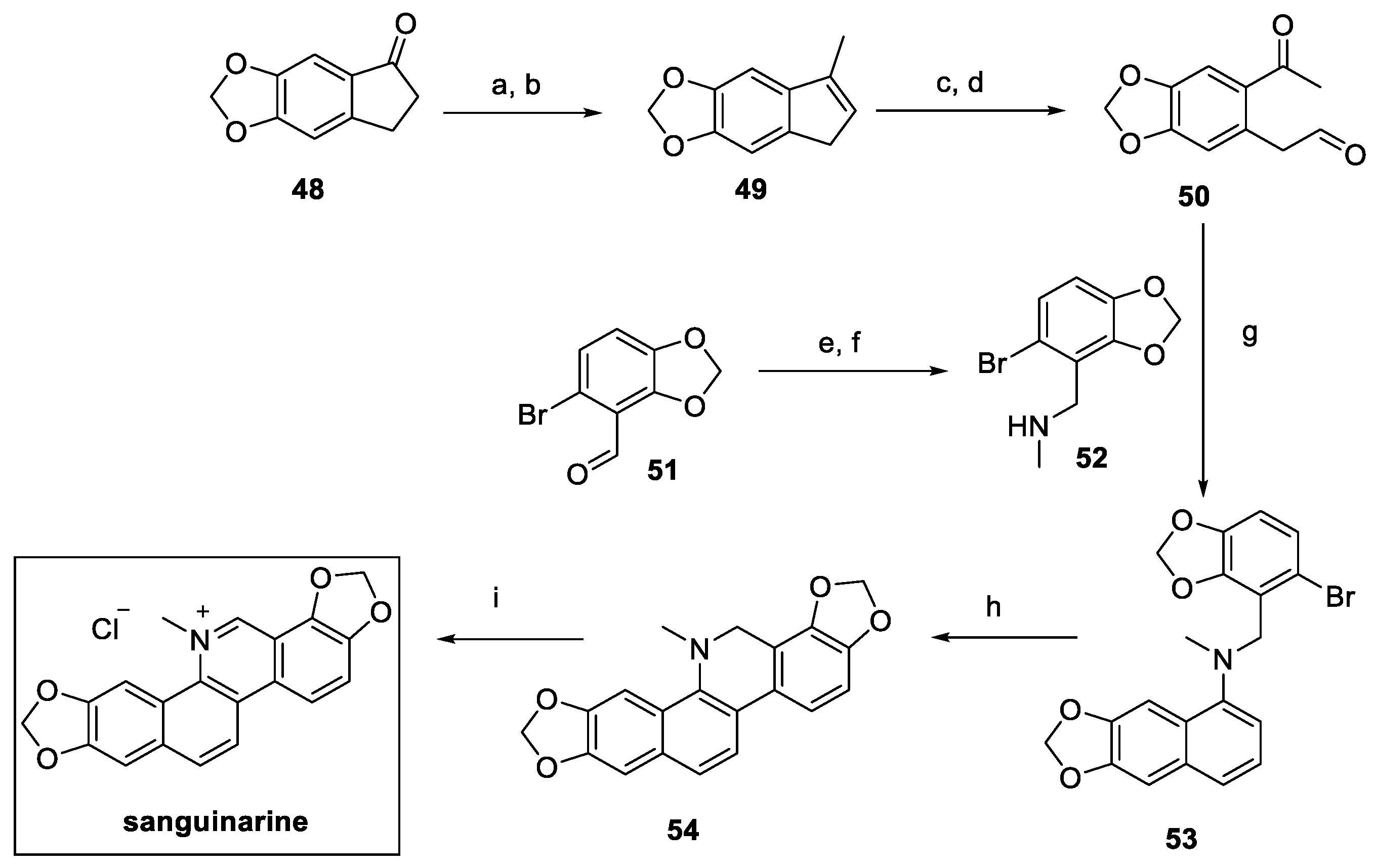
The total synthesis of sanguinarine was carried out by Tatton in 2014, starting from commercially available indanone 48. The introduction of the methyl group via methyllithium and further dehydration (compound 49, Scheme 10) was essential to guarantee the oxidative cleavage to 50, followed by the enamine-mediated cyclization to naphthylamine 53. Intramolecular substitution yielded the dihydro isoquinoline core in 54, oxidized by DDQ and treated with HCl to afford the desired natural product sanguinarine in the form of chloride salt [30].
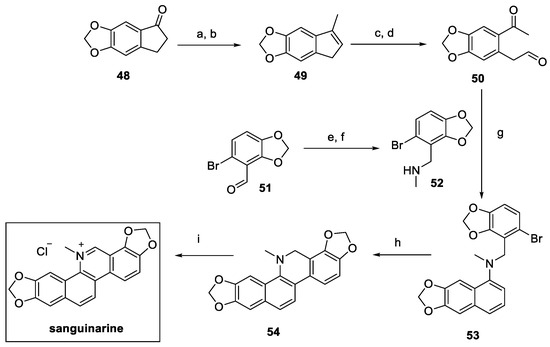
Scheme 10.
Synthesis of sanguinarine chloride salt. (a) MeLi, CeCl3, THF, then (b) PPTS, DCM; (c) K2OsO2(OH)4, NMO, THF/H2O then (d) NaIO4·SiO2, Et2O; (e) MeNH2, EtOH, then (f) NaBH4, MeOH, quant; (g) ZnCl2, DCE; (h) phenanthroline, t-BuOK, benzene; (i) DDQ, NaOH, benzene then HCl.
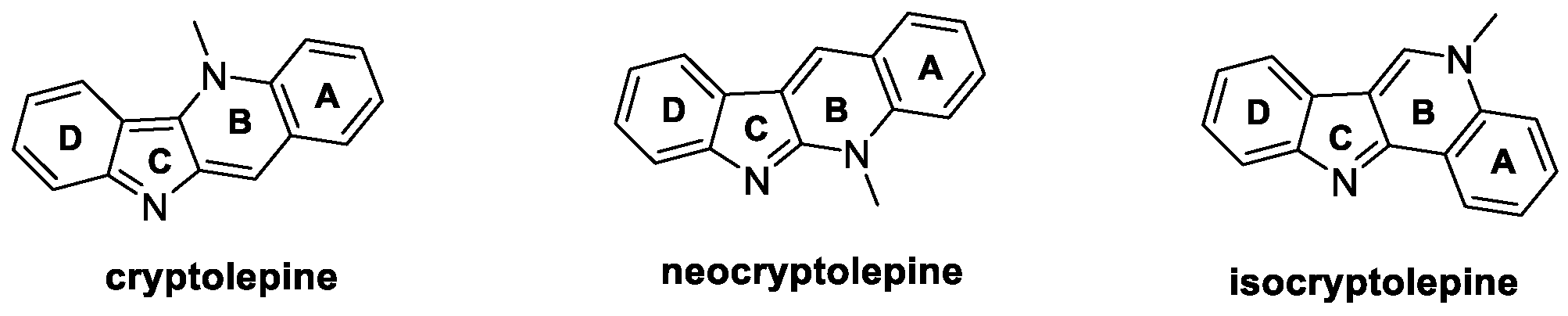
2.4. Cryptolepine, Neocryptolepine, and Isocryptolepine (from Plants)
Cryptolepine, neocryptolepine, and isocryptolepine (Figure 5) are isomeric indoloquinoline alkaloids isolated from the roots of Cryptolepis sanguinolenta (Periploaceae), a tropical shrub indigenous to West Africa, consisting of four fused rings (A–D, Figure 5) [31]. As for their biological potential, cryptolepine effectively inhibits S. sclerotiorum (EC50 = 5.507 μg/mL) and B. cinerea growth (EC50 = 0.050 μg/mL), whereas neocryptolepine is active against R. solani, both in vitro and in vivo. Shang et al. demonstrated that neocryptolepine can inhibit complex III by binding UQCRFS1 (cytochrome b-c1 complex subunit Rieske) protein, blocking the ion transfer, and affecting oxidative phosphorylation, ultimately leading to mycelium death [32]. In addition, neocryptolepine significantly inhibits sclerotia formation in R. solani [33]. Isocryptolepine shows moderate antifungal activity against F. graminearum and B. cinerea, with an inhibition of mycelia growth of 34% and 46%, respectively, at 50 μg/mL [34]. The total synthesis of the three natural alkaloids, along with the efforts aimed at their structural modifications over the last ten years, have been collected and are hereafter reported.
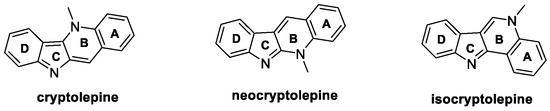
Figure 5.
Chemical structures of natural cryptolepine, neocryptolepine, and isocryptolepine.
2.4.1. Cryptolepine
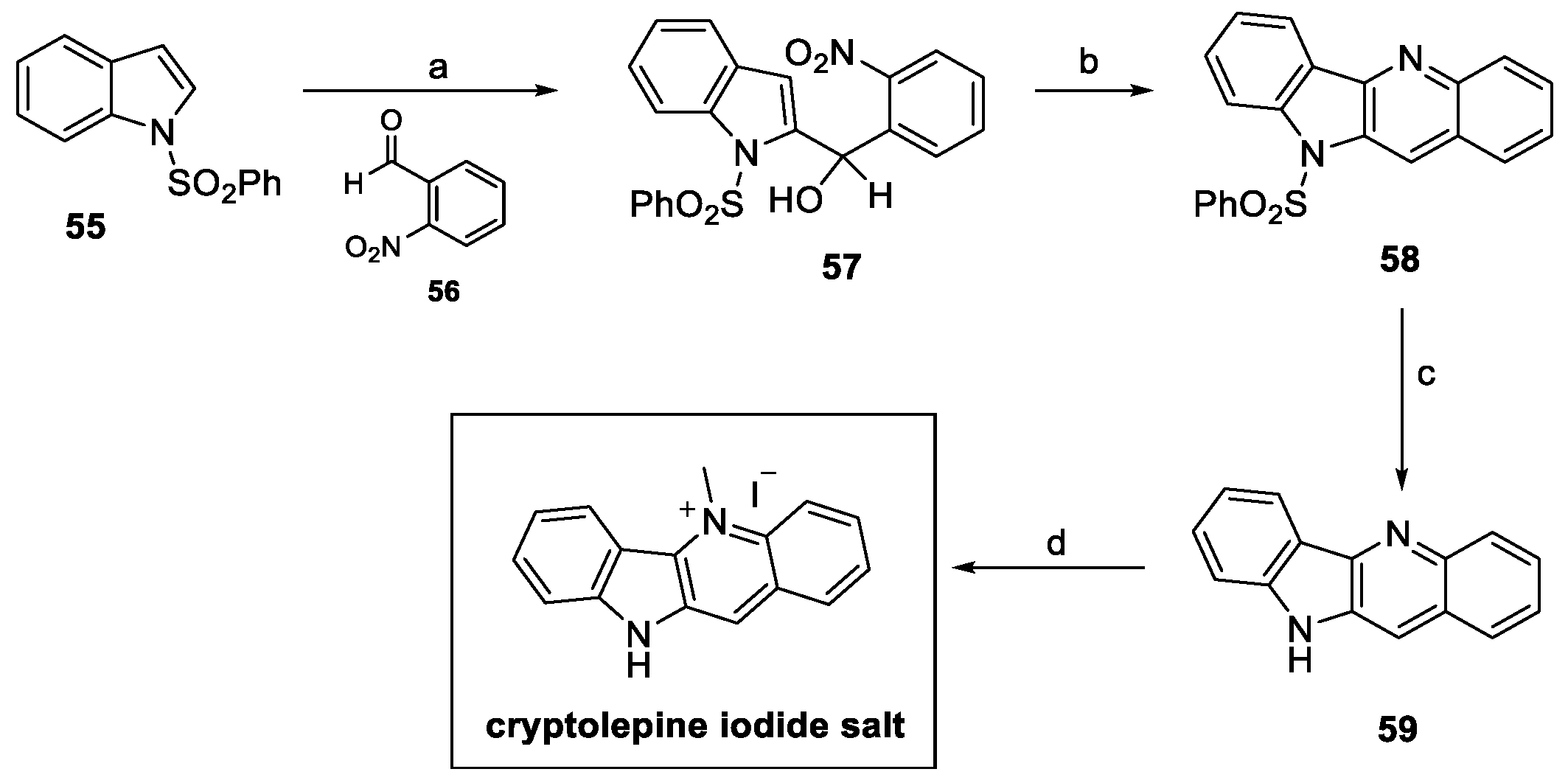
In 2016, Parvatkar and Parameswaran proposed a four-step pathway for the synthesis of cryptolepine salts, using a tandem reductive cyclization–dehydration protocol, as illustrated in Scheme 11 [31]. The C-2 lithiation of commercially available N-sulfonylindole 55 with n-BuLi allowed for functionalization with suitable benzaldehyde 56 to provide the corresponding 2-substituted indole derivative 57. Mo-catalyzed reductive cyclization and dehydration resulted in the formation of tetracyclic 58. Indole deprotection under alkaline conditions afforded compound 59, while chemoselective methylation at the pyridine nitrogen using methyl iodide in sulfolane provided the desired cryptolepine salt in a 17% yield over four steps [31].
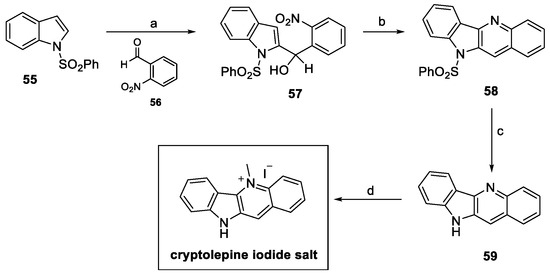
Scheme 11.
Synthesis of cryptolepine iodine salt. (a) n-BuLi, THF, then 56; (b) PPh3, MoO2Cl2(dmf)2, toluene; (c) aq. NaOH, MeOH; (d) CH3I, sulfolane.
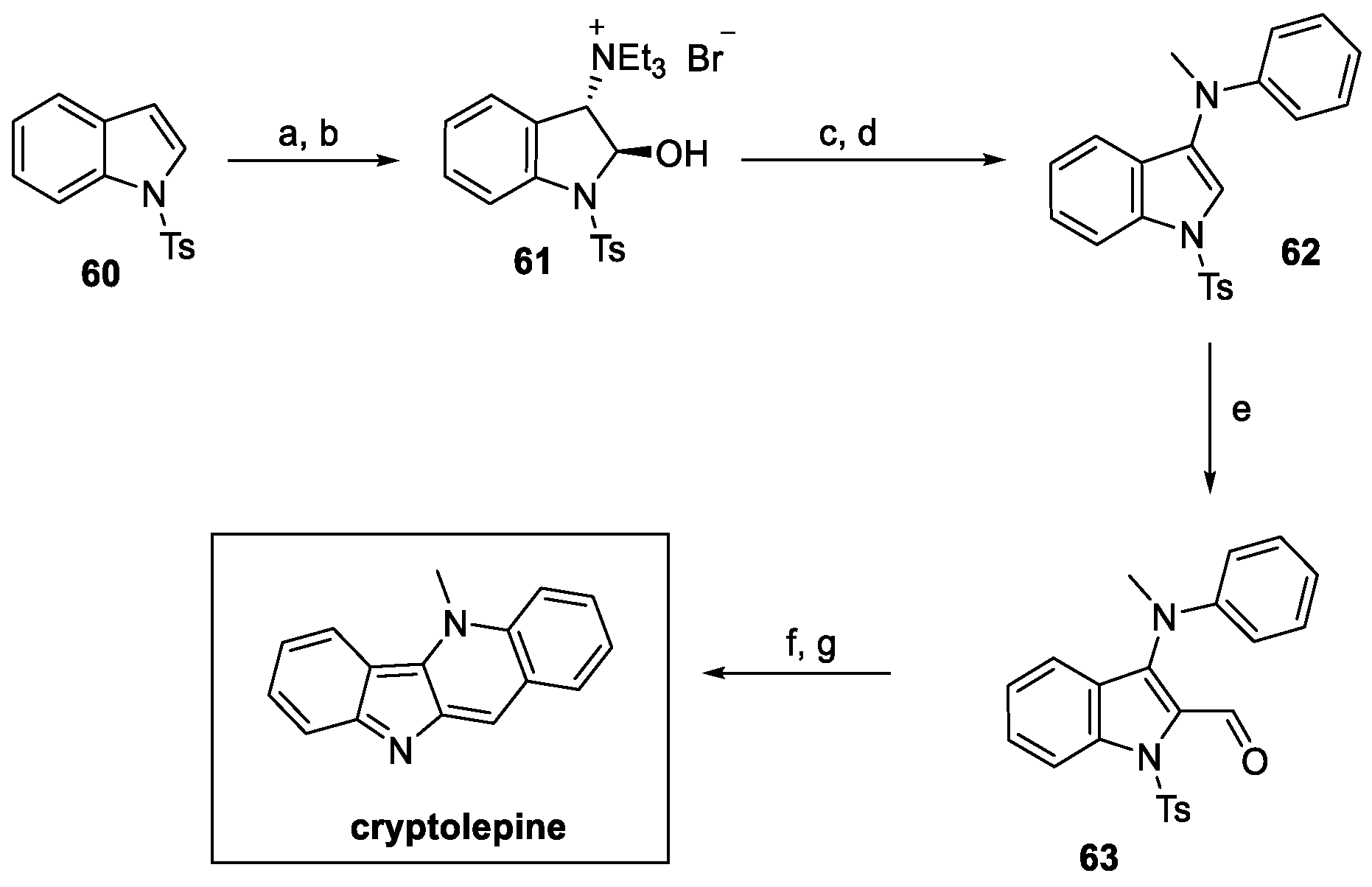
The following year, a novel methodology for the synthesis of cryptolepine was designed by Abe et al. (Scheme 12) [35]. One-pot functionalization of the double bond on tosyl indole 60 provided ammonium salt 61 in a very good yield. Substitution with N-methyl aniline and further dehydration resulted in the 3-substituted intermediate 62. Vilsmeier–Haack formylation at indole C(2) afforded 63, which underwent N-deprotection and ring closure to yield natural cryptolepine, with a 34% overall yield.
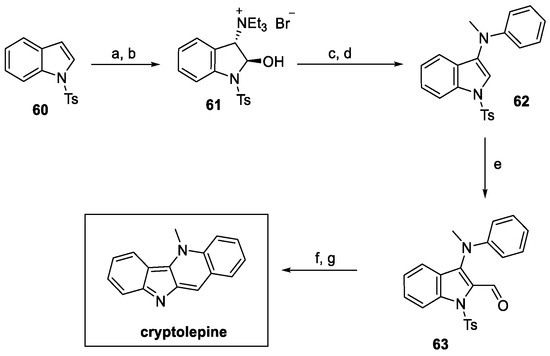
Scheme 12.
Synthesis of cryptolepine, starting from protected pyrrole 60. (a) NBS, H2O, acetone, then (b) Et3N; (c) N-methyl aniline, Et3N, EtOAc, then (d) BF3, EtOAc; (e) POCl3, DMF; (f) Me2NH·HCl, DMF, then (g) aq. Na2CO3.
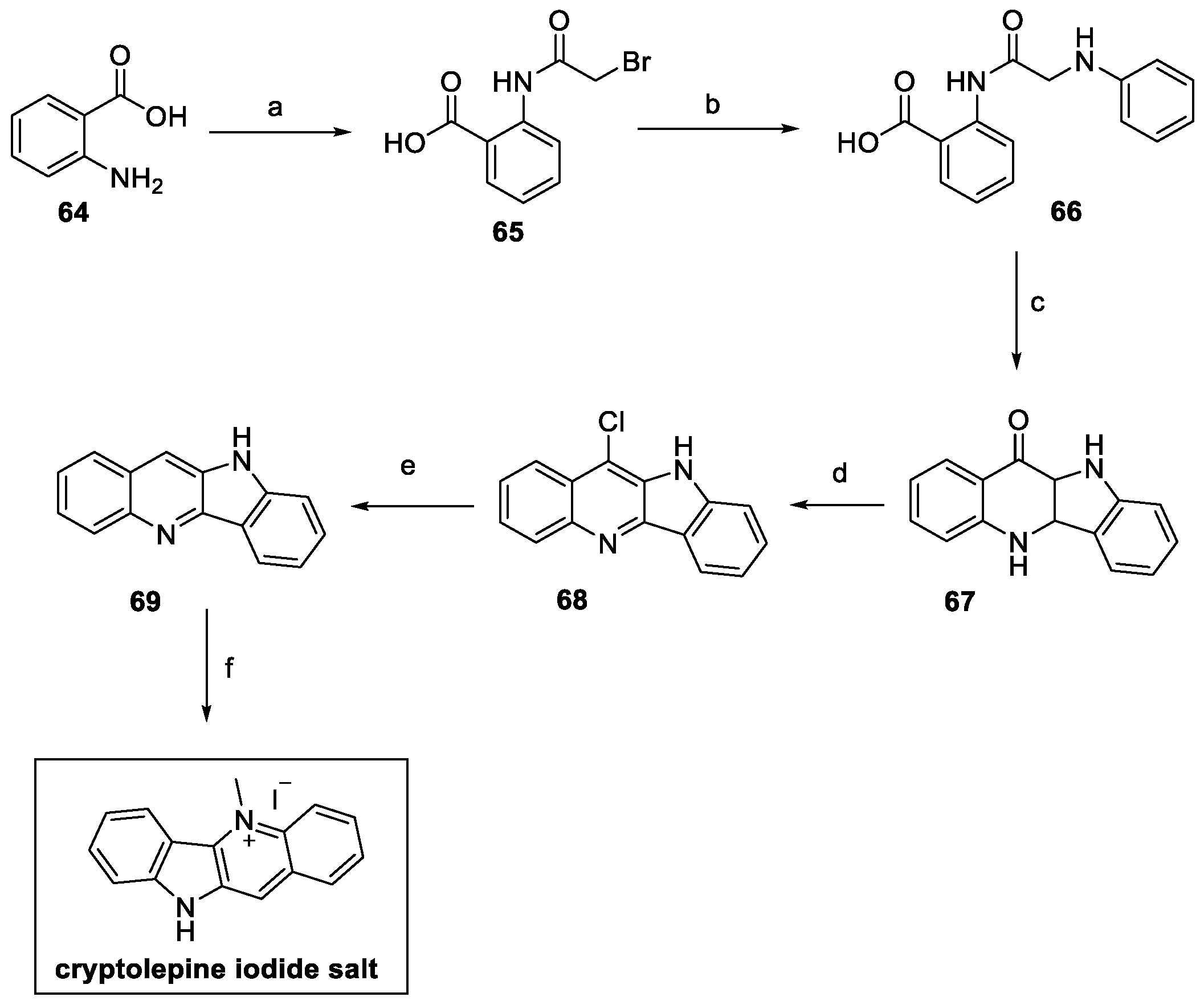
Following a different approach, cryptolepine was synthesized by Mudududdla et al., starting from commercially available anthranilic acid 64, as depicted in Scheme 13 [36]. Bromoacetylation and alkylation with aniline led to benzoic acid 66. Friedel–Crafts reaction and subsequent dehydration–cyclization catalyzed by polyphosphoric acid (PPA) provided access to indolo[3,2-b]quinolin-11-one 67. The POCl3-mediated chlorination yieldied the quinoline skeleton on 68, hydrogenated at high pressure, in the presence of a catalytic amount of Pd, to afford compound 69. Final N-methylation with methyl iodide yielded cryptolepine iodide salt, with an overall yield of 16%, starting from anthranilic acid.
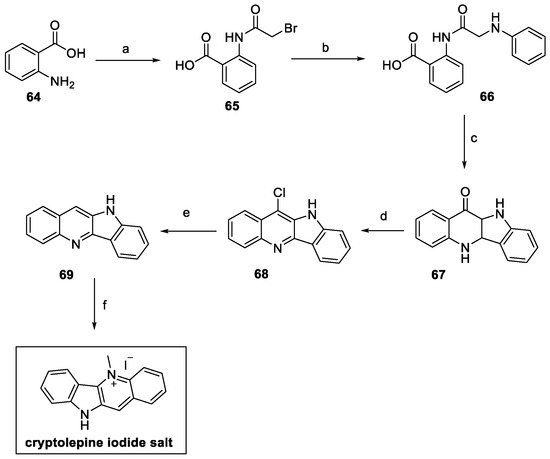
Scheme 13.
Synthesis of cryptolepine, starting from anthranilic acid 64: (a) bromoacetyl bromide, 1,4-dioxane/DMF 1:1; (b) aniline, DMF; (c) PPA; (d) POCl3; (e) H2, Pd/C, AcOH, NaOAc, 60 psi; (f) MeI, DMF.
2.4.2. Neocryptolepine
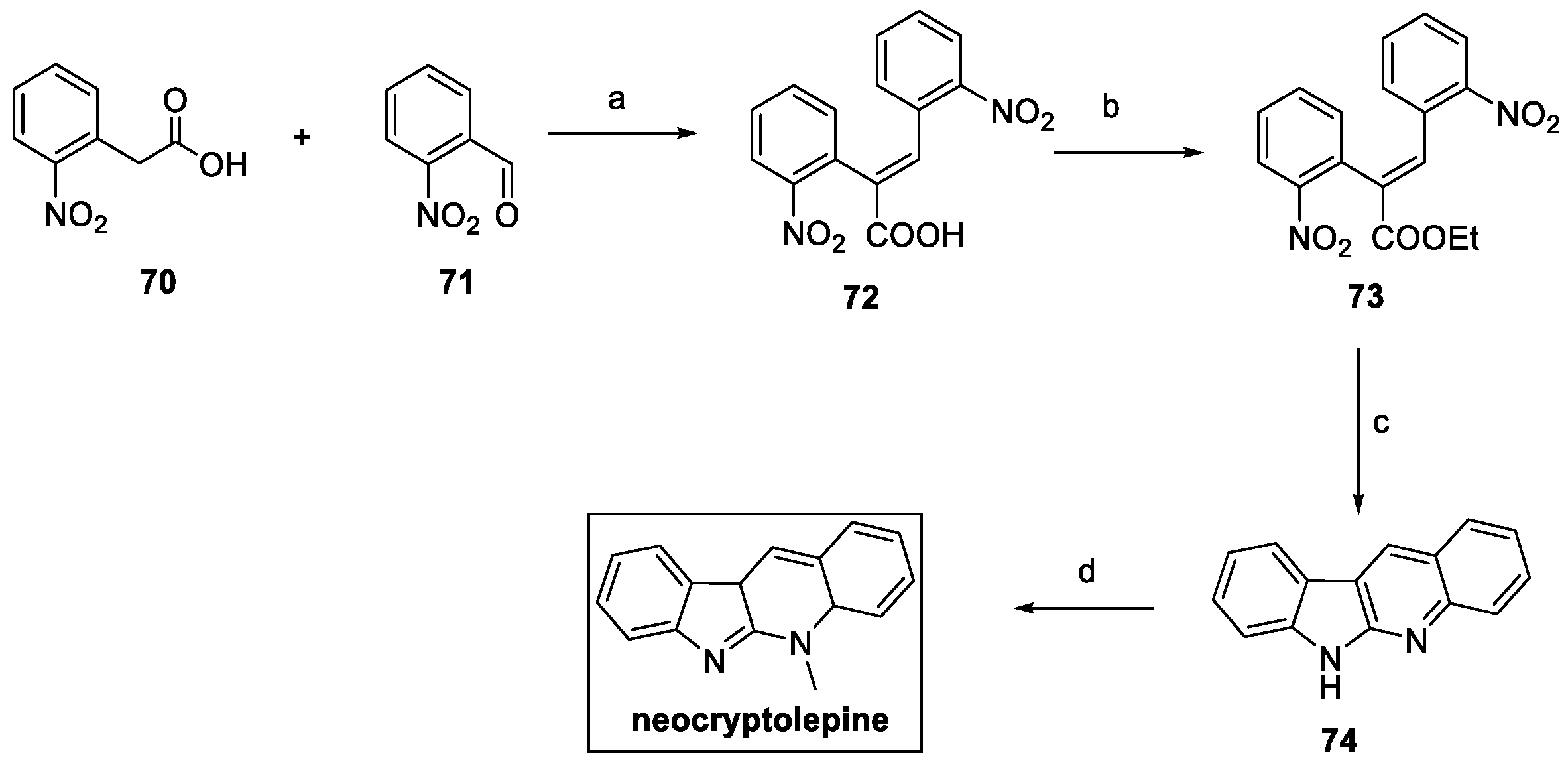
Neocryptolepine was synthesized following a domino strategy, starting from o-nitrophenylacetic acid 70. (Scheme 14) [31]. Perkin reaction with o-nitrobenzaldehyde 71 yielded the corresponding α,β-unsaturated acid 72, further converted to ethyl ester 73, in high yield. Iron powder in the presence of HCl allowed for the nitro reduction and ring closure via a domino reaction sequence, resulting in indoloquinoline 74, a naturally occurring alkaloid also known as norcryptotackieine. Methylation of the quinoline nitrogen using dimethyl sulfate afforded neocryptolepine in around a 40% overall yield in four steps.
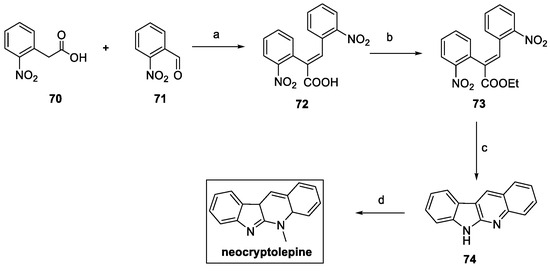
Scheme 14.
Synthesis of neocryptolepine. (a) Ac2O, Et3N, reflux; (b) EtOH, H2SO4, 24 h; (c) Fe/HCl, AcOH:EtOH:H2O; (d) Me2SO4, DMF, μW, or Me2SO4, MeCN.
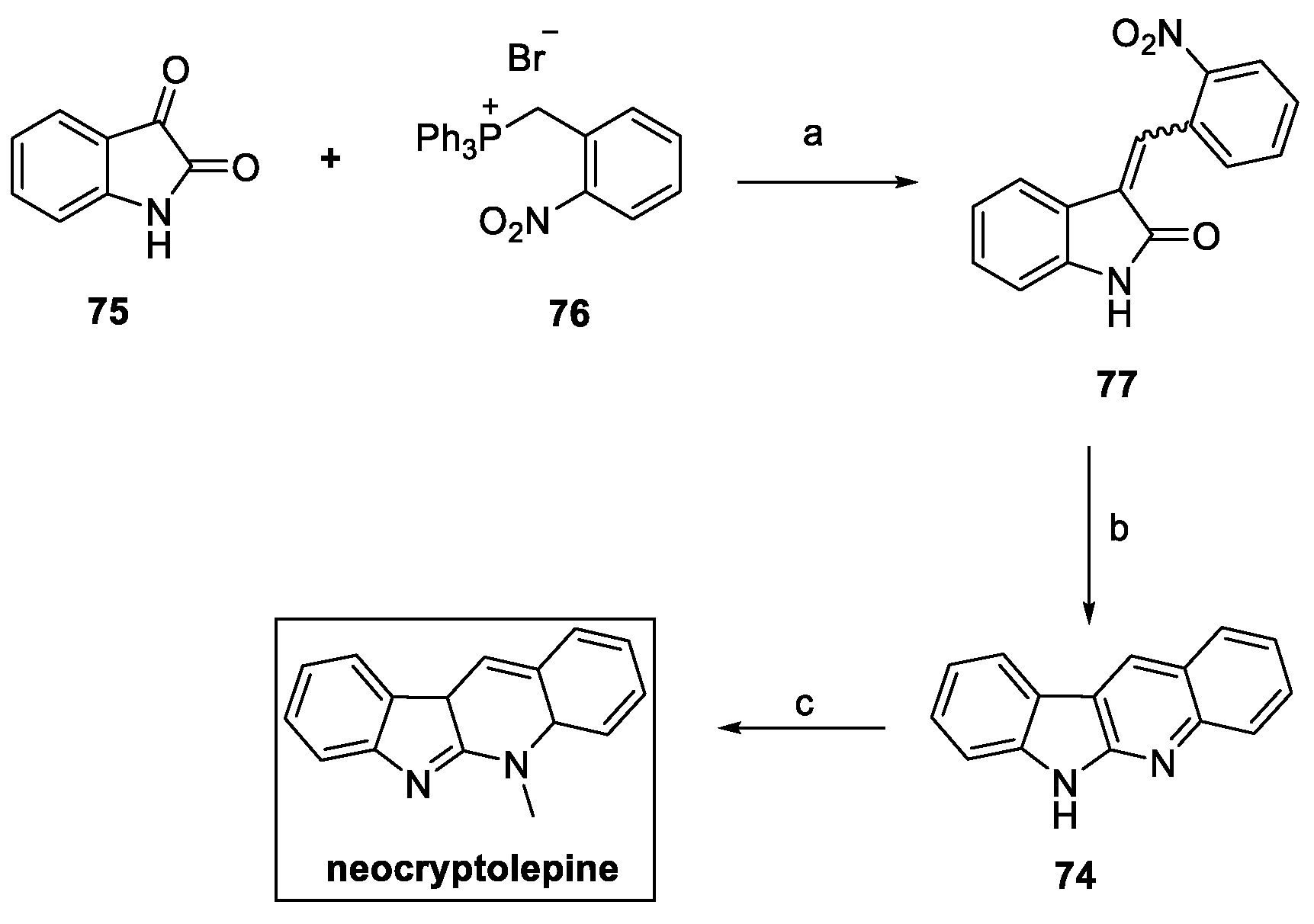
With the purpose of improving the overall yield of the pathway, an alternative method for the preparation of neocryptolepine was reported by the same authors (Scheme 15). In this case, Wittig olefination on isatin 75 with the phosphonium salt 76 yielded a 2:1 ratio of E/Z isomers (77). Reduction of the resulting olefin with iron powder in an acidic environment provided the fused tetracycle 74, which was finally methylated to afford neocryptolepine with a 68% overall yield in just three steps [31].
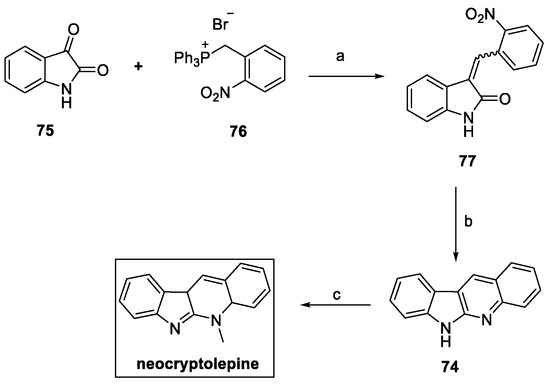
Scheme 15.
Optimized synthesis of neocryptolepine. (a) Et3N, CHCl3; (b) Fe/HCl, AcOH; (c) MeI, THF.
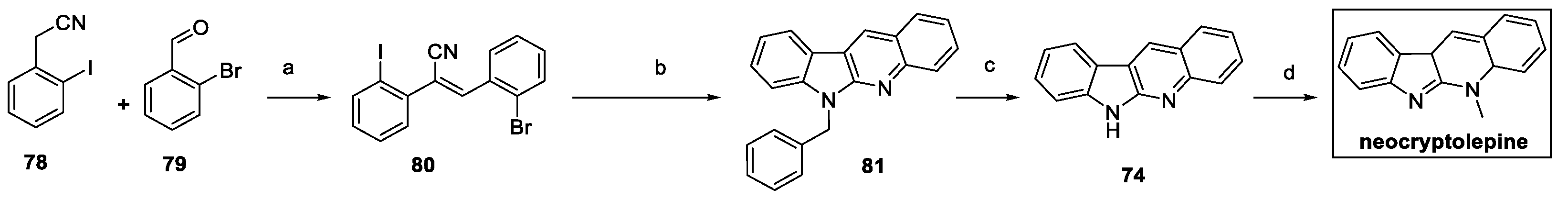
In 2023, neocryptolepine was synthesized by exploiting different substrates using a similar approach (Scheme 16) [37]. Condensation between commercially available 2-iodobenzyl cyanide 78 and 2-bromobenzaldehyde 79 delivered (E)-stilbene intermediate 80. Sequential Pd-catalyzed intermolecular and intramolecular double Buchwald−Hartwig C−N coupling with benzyl amine yielded the desired N6-benzylated norcryptotackieine 81. AlCl3-catalyzed debenzylation resulted in norcryptotackieine 74, which was finally methylated to neocryptolepine.
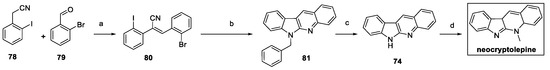
Scheme 16.
Synthesis of neocryptolepine, starting from cyano substrate 78. (a) K2CO3, MeOH; (b) benzyl amine, Pd2(dba)3, xantphos, t-BuONa, toluene; (c) anhydrous AlCl3, benzene; (d) MeI, aq. KOH, THF [37].
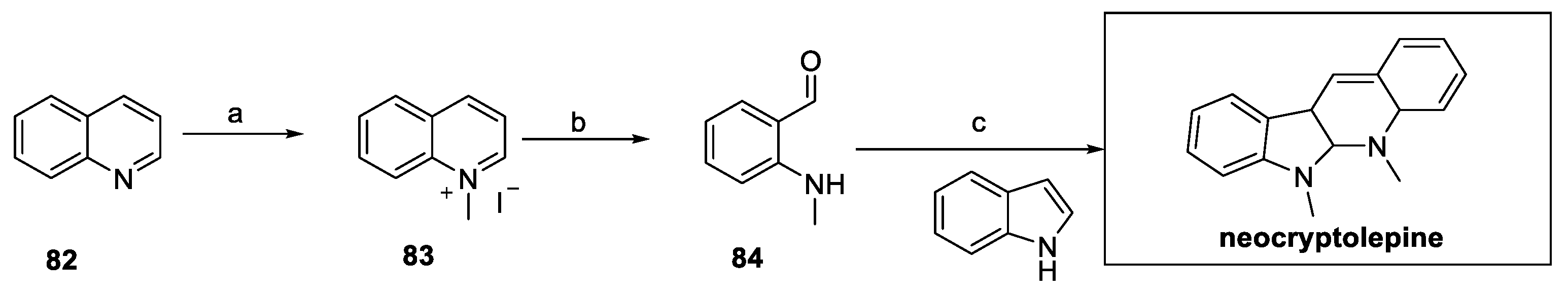
Neocryptolepine and its derivatives were synthetized by Zhu’s group and screened as fungicides against R. solani, B. cinerea, F. graminearum, M. melonis, S. sclerotiorum, and M. oryzae [38]. In particular, quinoline 82 was converted to the quinolonium salt 83 and underwent oxidative cleavage to access amino benzaldehyde 84 (Scheme 17). In this case, the tetrahydroquinoline moiety was obtained by treatment with indole in the presence of catalytic amounts of p-toluenesulfonic acid, thus affording the desired natural product.
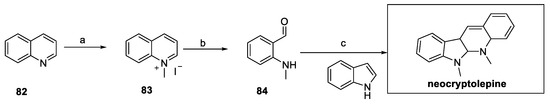
Scheme 17.
Synthesis of neocryptolepine, starting from substituted indole 82. (a) iPrOH, MeI; (b) KOH, H2O2; (c) indole, p-TSOH, EtOH.
2.4.3. Isocryptolepine
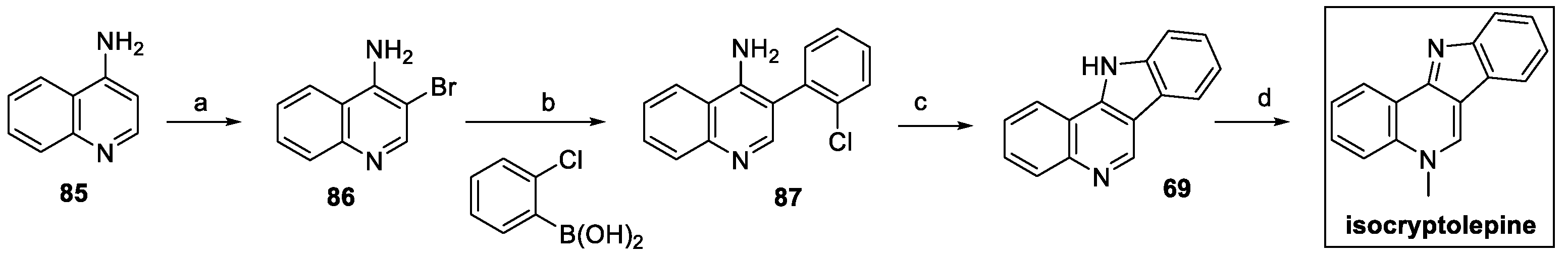
Isolation of isocryptolepine from the natural source (Cryptolepis sanguinolenta) and its chemical synthesis were thoroughly reported by Thobokholt et al. in their review in 2020 [39]. However, in 2021 a new, more efficient method was proposed using modifications of commercially available 4-aminoquinoline 85 (Scheme 18) [40]. Bromination at C(3) was followed by Suzuki–Miyaura coupling with 2-chlorophenylboronic acid to yield 87 in a very high yield. Intramolecular nucleophilic aromatic substitution provided indoloquinoline 69, which was finally methylated to isocryptolepine, with a 61% overall yield in four steps.
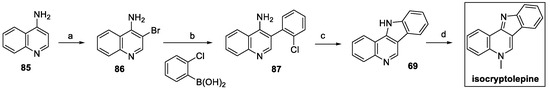
Scheme 18.
Synthesis of isocryptolepine, starting from 4-amino quinoline 85. (a) Br2, AcOH; (b) Pd(PPh3)4, K2CO3, toluene:EtOH:H2O 3:2:1; (c) t-BuOK, DMSO; (d) MeI, toluene.
3. Indole-Containing Alkaloids
3.1. β-Carbolines: Harmine, Norharmane, Harmaline, Harmol, Harmane, and Harmalol (from Plants)
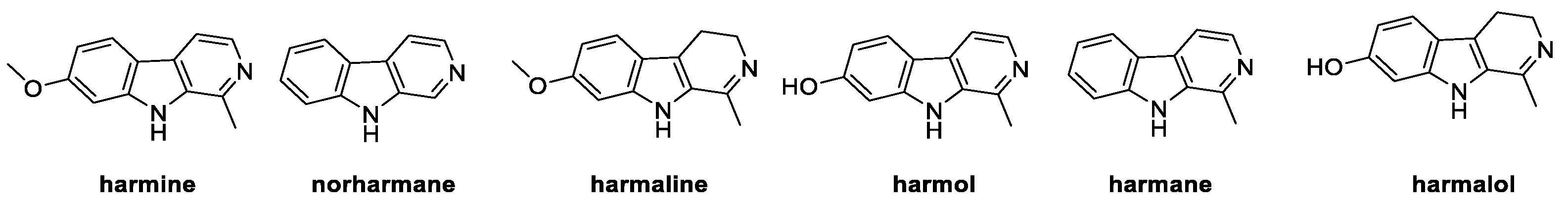
β-carbolines belong to a class of natural alkaloids widely distributed in plants and food, as well as in marine microorganisms, insects, and mammalians. Isolated from Peganum harmala for the first time in 1841, these heterocyclic compounds are characterized by a 9H-pyrido[3,4-b]indole core [41], endowed with antifungal activity. Six natural carbolines, namely harmine, norharmane, harmaline, harmol, harmane, and harmalol, were studied for their growth inhibition effect on strains of P. digitatum, F. oxysporum, and B. cinerea, which are mainly caused by cellular membrane damage and ROS accumulation (Figure 6) [41].
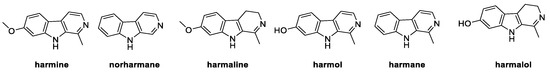
Figure 6.
Chemical structure of natural β-carbolines.
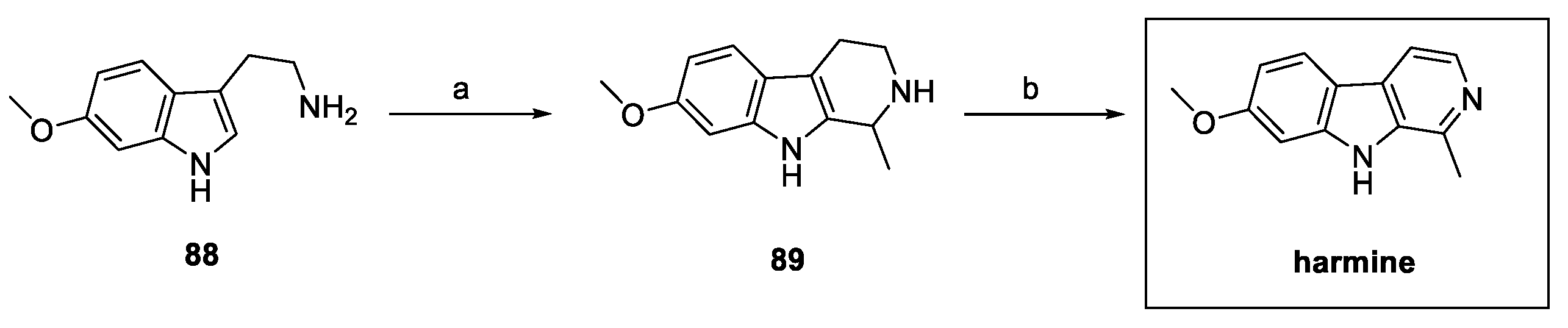
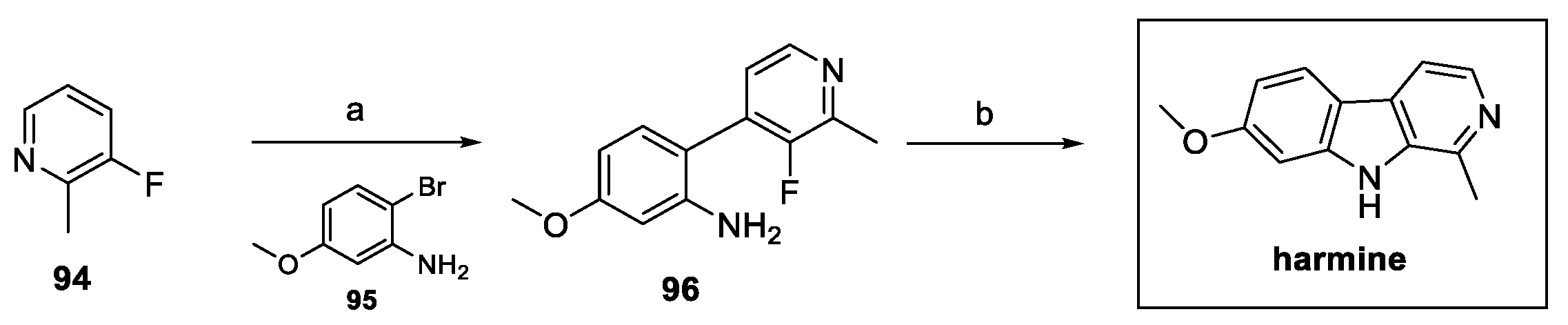
In recent years, different authors have reported the synthesis of harmine, commonly obtained by a Pictet–Spengler reaction between previously prepared 6-methoxy tryptamine 88 (obtained in four steps) [42] and acetaldehyde, followed by dehydrogenation on a Pd/C catalyst (Scheme 19) [43].
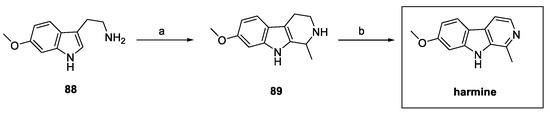
Scheme 19.
Classical synthesis of harmine via Pictet–Spengler reaction: (a) 0.1 N HCl, acetaldehyde; (b) 5% Pd/C, dry toluene.
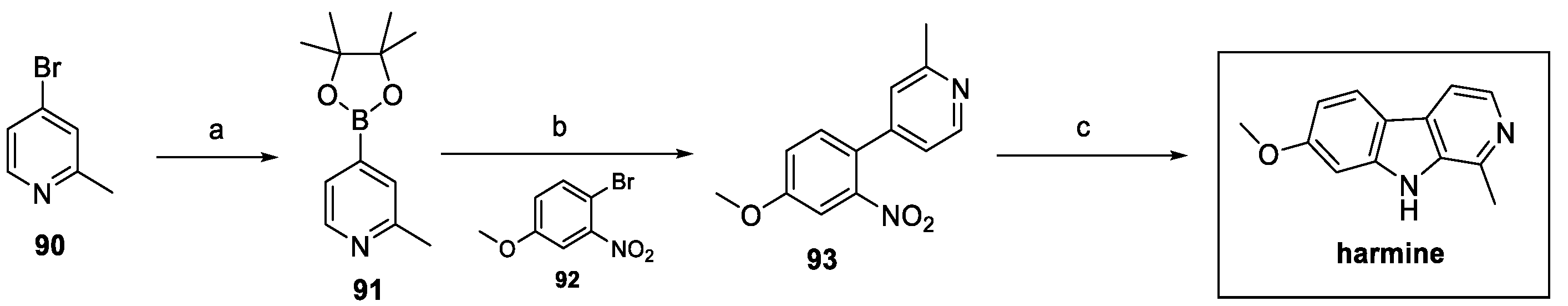
In order to design a shorter synthesis of harmine, W. Liu et al. proposed a new three-step route, as shown in Scheme 20. Starting from 4-bromo-2-methylpyridine 90, borylation and Suzuki coupling with the suitable bromide 92 afforded aryl pyridine 93, which then underwent Cadogan cyclization to obtain the desired carboline harmine [44].
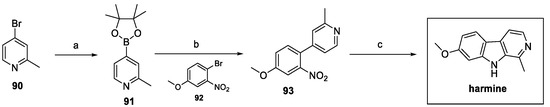
Scheme 20.
Synthesis of harmine via Suzuki reaction: (a) bis(pinacolato)diboron, Pd(dppf)Cl2, KOAc, dry 1,4-dioxane; (b) Pd(dppf)Cl2, Cs2CO3, 1,4-dioxane/H2O; (c) PPh3, o-DCB.
Several natural β-carbolines, including harmine, were synthesized in two steps, following the approach proposed by Sathiyalingama et al. (Scheme 21). Direct harmine precursor 96 was obtained via tandem reaction (lithiation, transmetalation, Negishi coupling), starting from fluoropyridine 94. Final intramolecular nucleophilic aromatic substitution resulted in the desired natural compound [45].
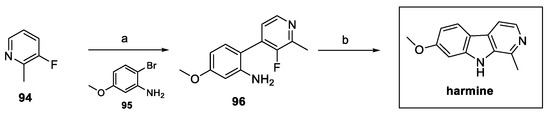
Scheme 21.
Synthesis of harmine via Negishi coupling: (a) i. LDA, THF; ii. ZnCl2; iii. Pd XPhos G3, THF; (b) NaHMDS, THF.
3.2. Pityriacitrin and Pityriacitrin B (from Bacteria and Marine Environment)
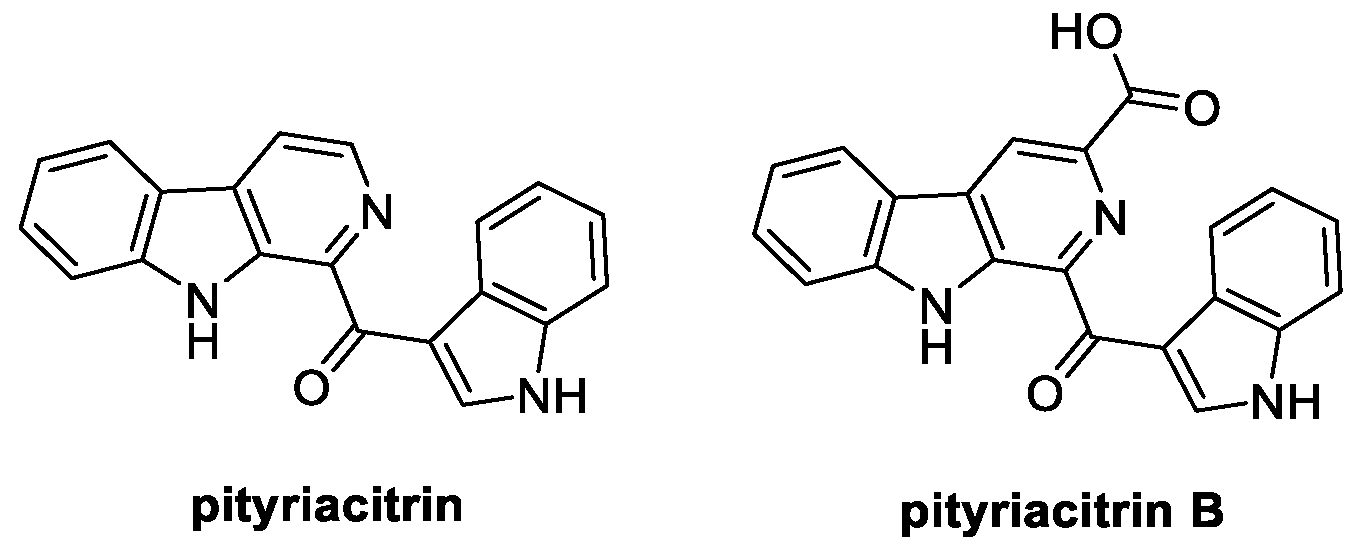
Characterized by a β-carboline core structure bearing an acyl portion at C(1), pityriacitrin was first extracted and isolated from the marine bacterium Paracoccus sp. in 1999 (Figure 7) [46]. Instead, its carboxylated analogue pityriacitrin B naturally occurs in the human pathogenic yeast Malassezia furfur; moreover, it is known as a secondary metabolite obtained from the marine fungus Dichotomomyces cejpii and from Chinese Burkholderia sp. NBF227.
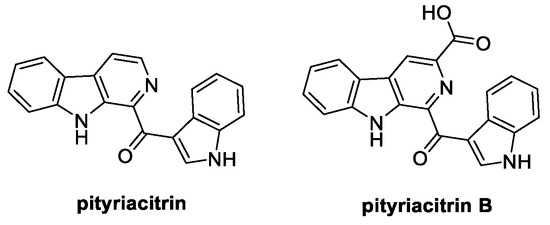
Figure 7.
Chemical structures of pityriacitrin and pityriacitrin B.
Antimalarial, antitumor, antioxidant, PLA2 regulation, and UV protection are among the biological activities reported for pityriacitrin, even though its application as a crop protector has only recently been described. Indeed, pityriacitrin significantly inhibits the growth rate of R. cerealis and S. sclerotiorum [47].
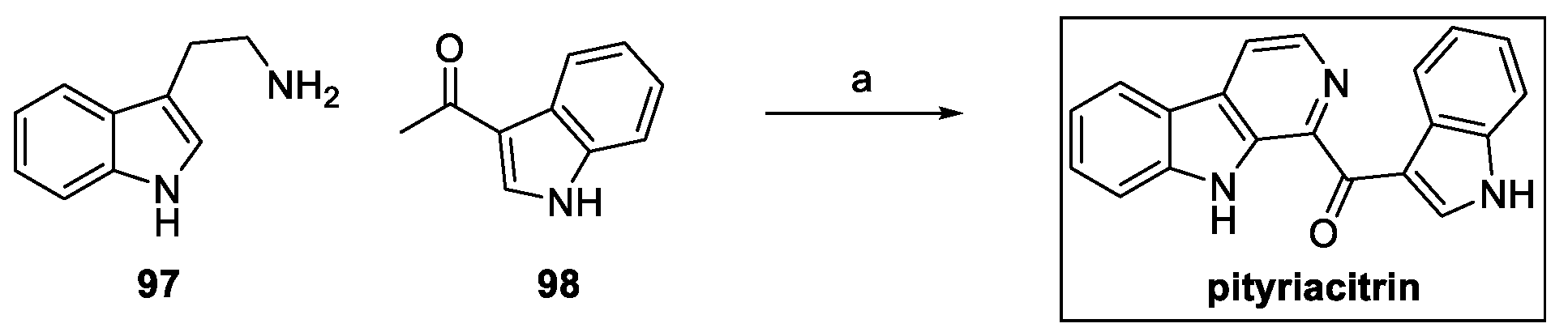
So far, five total syntheses have been reported in the literature, starting from the valuable work regarding the one-pot approach by Zhu et al. using tryptamine 97 and the carbonyl indole 98 under oxidative reaction conditions (Scheme 22) [48].
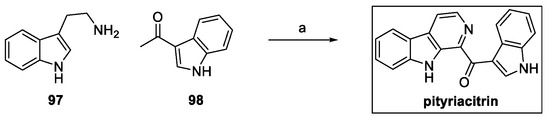
Scheme 22.
Synthesis of pityriacitrin: (a) I2, DMSO.
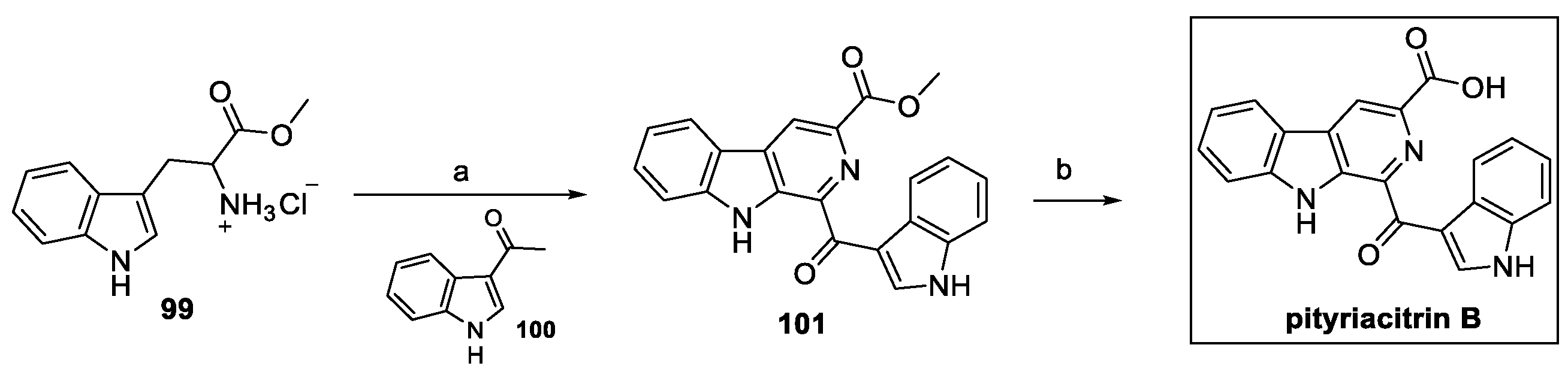
Starting from tryptophan methyl ester 99 allowed for the introduction of a carboxylic moiety so that heterocyclization with acetyl indole 100 and further hydrolysis could afford pityriacitrin B (Scheme 23) [49].
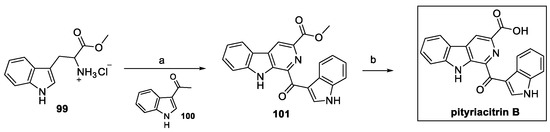
Scheme 23.
Synthesis of carboline pityriacitrin B. (a) I2, DMSO; (b) NaOH, MeOH, H2O.
3.3. Meridianin (from Marine Environment)
Meridianins are alkaloids isolated from marine organisms like the Antarctic tunicates Aplidium meridianum, falklandicum, and Synoicum sp. [50]. Starting in 1998, a total of eight meridianins have been collected and chemically characterized, with the core structure typically consisting of a brominated or hydroxylated indole, substituted at C(3) with a 2-aminopyrimidine moiety (Figure 8) [50].
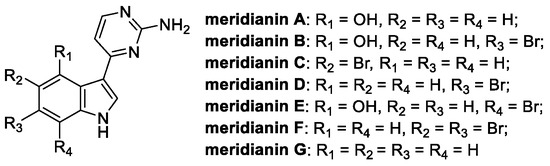
Figure 8.
Chemical structure of meridianins A-G [51].
Meridianins C, D, and G are endowed with a plethora of biological activities (anticancer, protein kinase inhibitory, antimalarial, antituberculosis, and anti-Alzheimer), including the fungicidal type [52]. Specifically, meridianin C has been reported as the most active, showing a broad-spectrum antifungal activity against several phytopathogenic fungal strains [51].
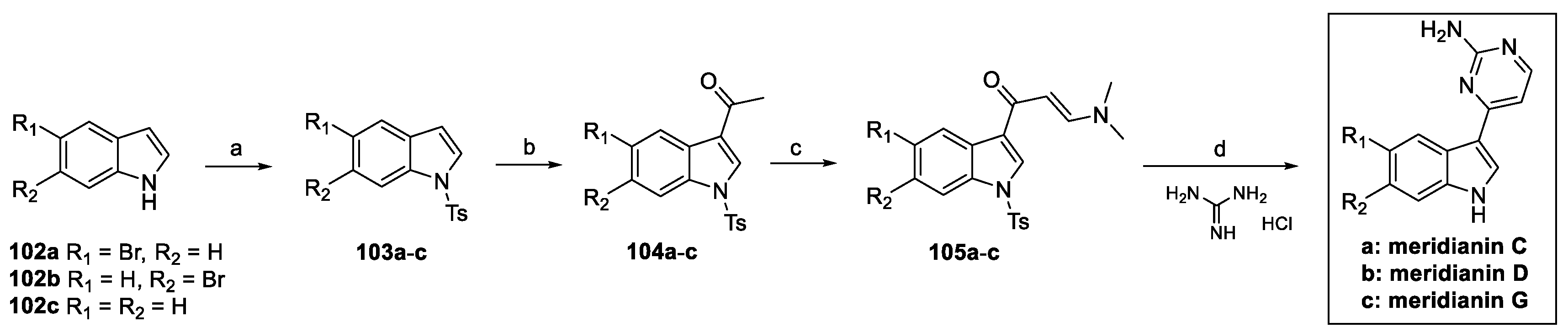
To date, several papers concerning the total synthesis of meridianins have been published [53,54,55]. Among them, the strategy reported by Molina, based on the Bredereck protocol, will hereafter be described [54]. Meridianin C, D, and G were synthetized, starting from the substituted indoles 102a–c (Scheme 24). Nitrogen tosylation and C(3)-acylation afforded 104a–c, which were further functionalized to obtain enaminones 105a–c. Bredereck cyclization and N-deprotection finally afforded the desired natural products [51].
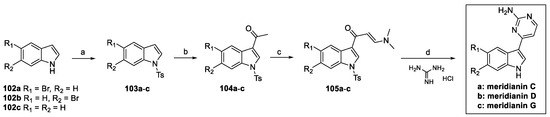
Scheme 24.
(a) TsCl, NaOH, H2O, n-Bu4NHSO4, toluene; (b) Ac2O, AlCl3, DCM; (c) DMF-DMA; (d) K2CO3, CH3OCH2CH2OH.
4. Miscellanea
4.1. Phenylpirrole: Pyrrolnitrin (from Bacteria)
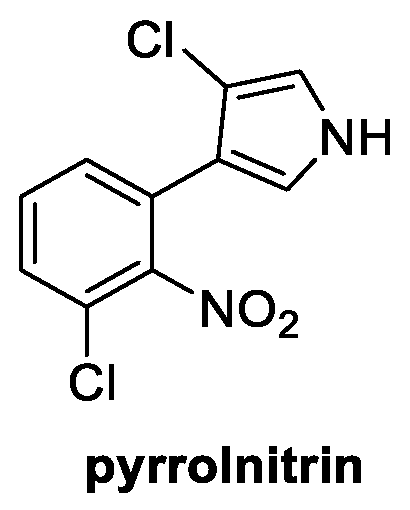
Pyrrolnitrin is an interesting natural compound well-known for its antifungal properties. Some of its derivatives are, in fact, already widely used in agriculture. It was firstly isolated in 1964 from Pseudomonas pyrrocinia and was later also identified in several other proteobacterial strains [56]. It is structurally characterized by a chlorinated pyrrole ring linked to a 2-nitro-3-cloro phenyl group (Figure 9). Pyrrolnitrin is known to reach the intracellular environment of the fungus via passive diffusion, targeting the mitochondrial electron transport chain. The increased oxidative stress and proliferation of oxygen free radicals are responsible for the damage occurring from the spore germination stage (inhibition of conidial germination at 0.4 µg/mL concentration) to the mycelia growth phase [57]. It is considered a broad-spectrum fungicide, with its strongest activity reported against Alternaria sp., B. cinerea, P. aphanidermatum, P. ultimun, R. solani, Rhizopus sp., A. niger, F. oxysporum, P. expansum, and S. rolfsii [58].
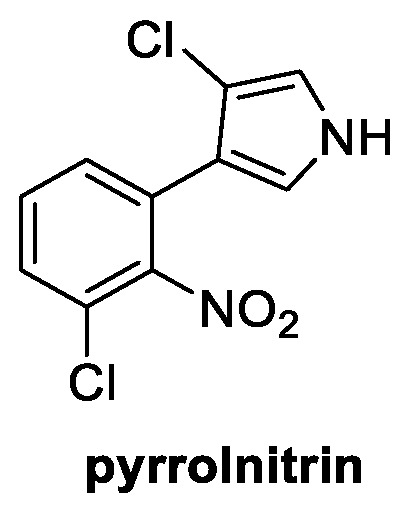
Figure 9.
Chemical structure of pyrrolnitrin.
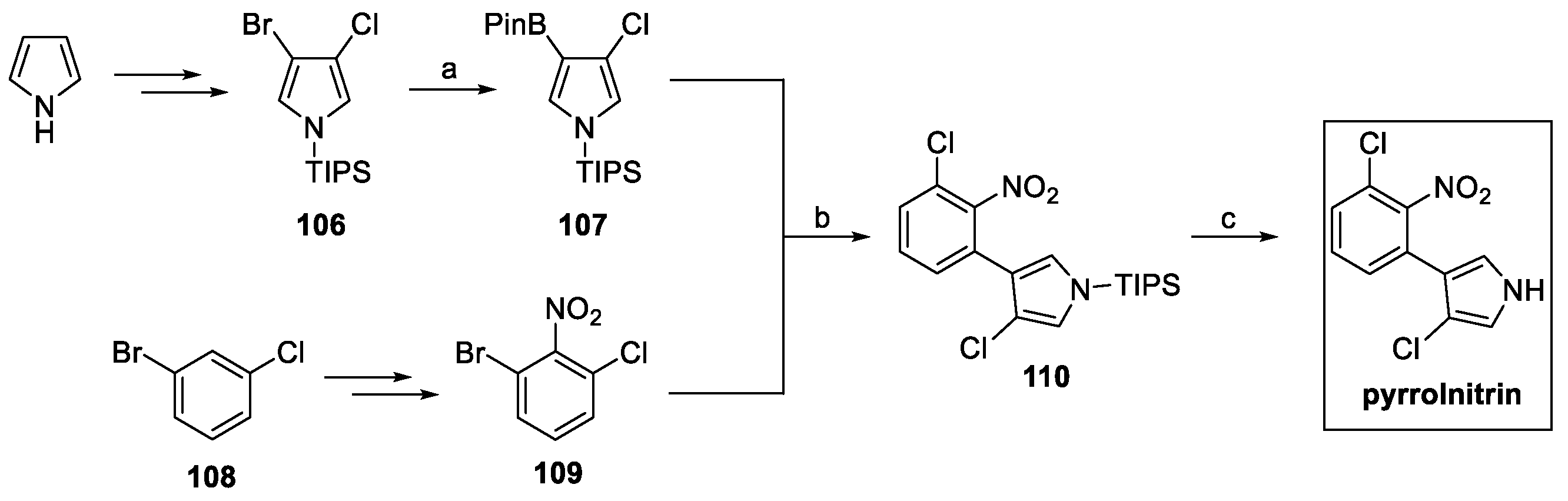
Pyrrolnitrin total synthesis was first approached by Nakano et al. in 1966 [59]. However, newer and more versatile approaches have been developed over the years, including the overexpression of its gene cluster in E. coli [60]. Morrison et al. reported a convenient synthetic pathway, starting from protected pyrrole (Scheme 25) [60].
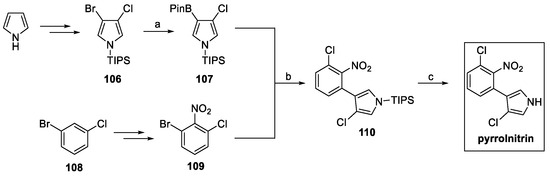
Scheme 25.
Synthesis of pyrrolnitrin. (a) HBPin, Et3N; (b) Pd(OAc)2:SPhos (1:2), K3PO4, n-BuOH:H2O; (c) TBAF, THF.
Following the protocol previously described by Muchowski et al. [61], brominated pyrrole 106 was converted to boronate ester 107, ready to undergo Suzuki coupling with the suitable aromatic substrate, in this case, nitro compound 109, obtained starting from aryl halide 108. Final TBAF-mediated deprotection afforded pyrrolnitrin [60].
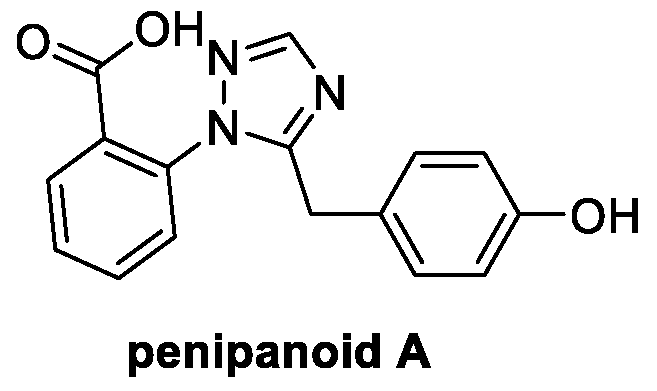
4.2. Triazoles: Penipanoid A (from Bacteria)
Penipanoid A is a natural product isolated from the marine fungus Penicillium paneum SD-44, found in a deep-sea sediment of the South China Sea [62]. Its main structural feature is a 1,2,4-triazole ring, widely present in a huge number of fungicides, such as fluconazole and voriconazole, bearing a benzoic acid and a phenol ring (Figure 10) [63]. To date, cytotoxic and antimicrobial activities have been reported, and further investigations are needed to assess its fungicidal potential.
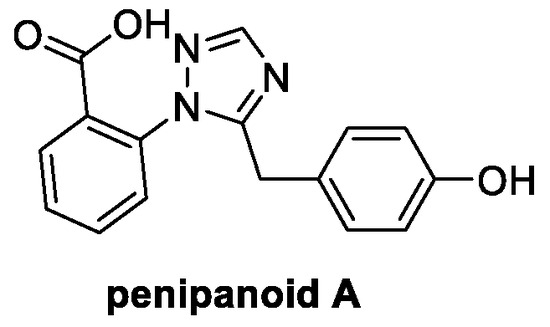
Figure 10.
Chemical structure of penipanoid A.
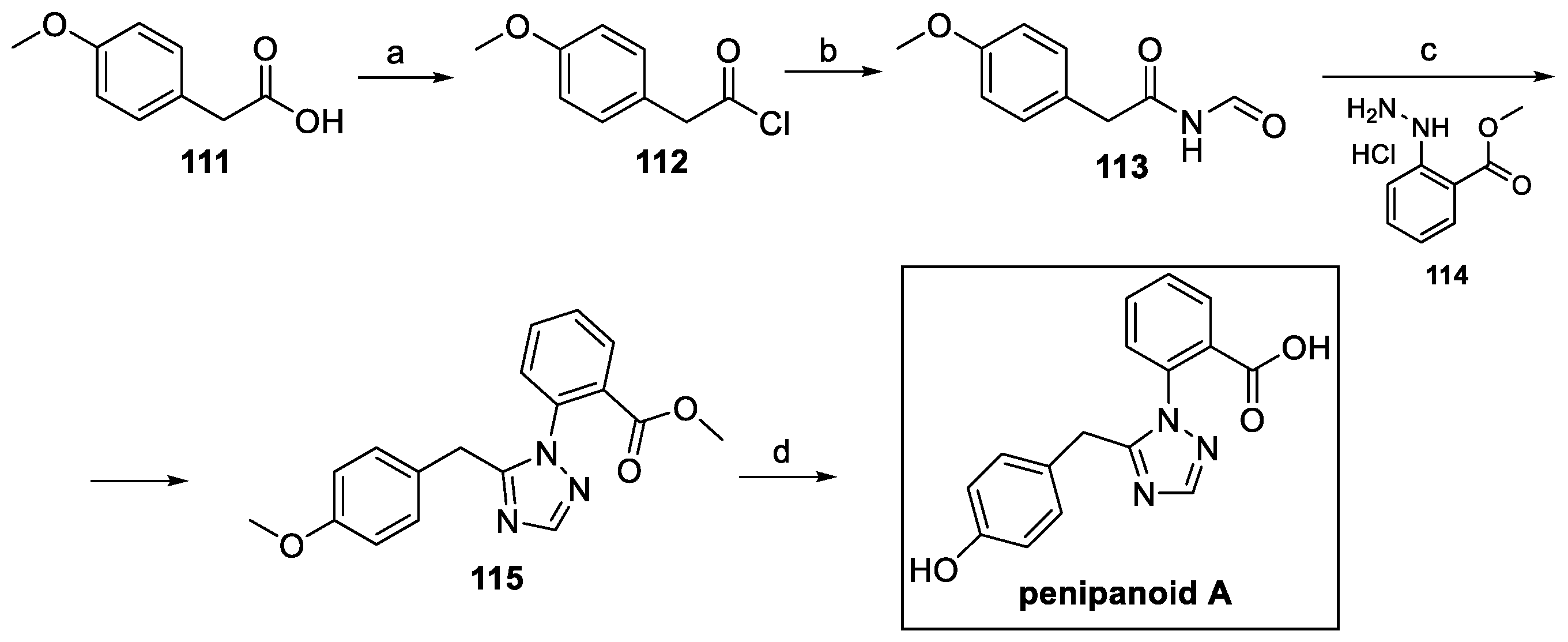
The total synthesis of this compound was reported in 2022 [63]. At first, commercially available phenylacetic acid 111 was activated and reacted with formamide to yield key intermediate 113 (Scheme 26). The triazole moiety of 115 was achieved by cyclization with phenyl hydrazine 114. Finally, ester hydrolysis, followed by methyl ether deprotection, allowed for the obtainment of penipanoid A [63].
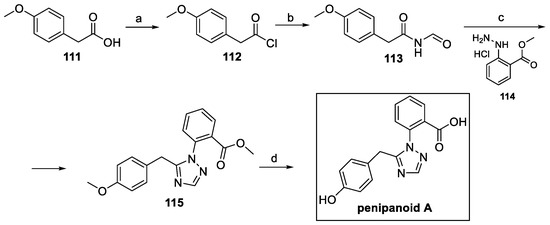
Scheme 26.
Synthesis of penipanoid A, starting from 4-methoxy phenylacetic acid 111. (a) (COCl)2; (b) formamide, Py, acetone; (c) 114, AcOH; (d) BBr3, DCM.
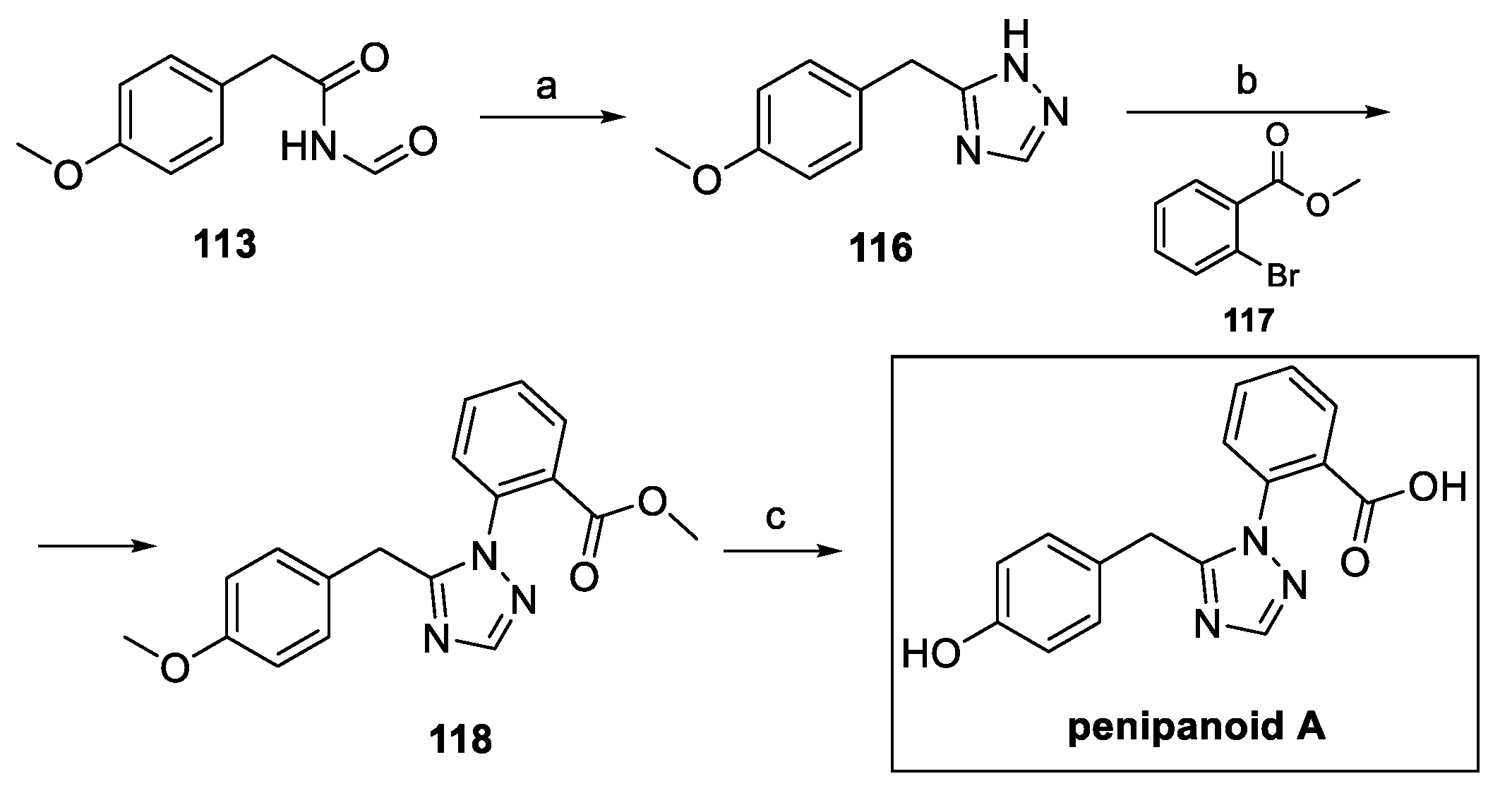
The same authors described further improvements in the synthetic pathway, starting from intermediate 113. Construction of the triazole ring was afforded in the presence of hydrazine. Ullmann reaction with 2-bromomethyl benzoate 117 yielded the 1,5-disubstituted desired moiety 118, which was completely demethylated to obtain pure penipanoid A, with a 60% overall yield, while shortening the synthetic pathway by one step (Scheme 27).
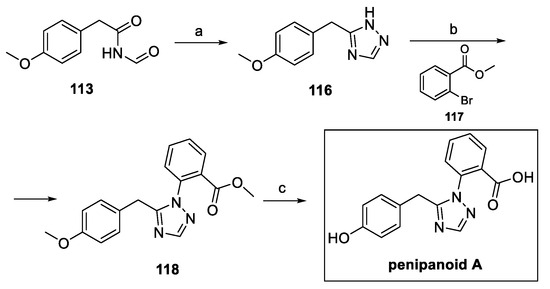
Scheme 27.
Synthesis of penipanoid A, starting from intermediate 113. (a) NH2-NH2, AcOH; (b) 117, CuI, Cs2CO3, L-proline, DMF; (c) BBr3, DCM.
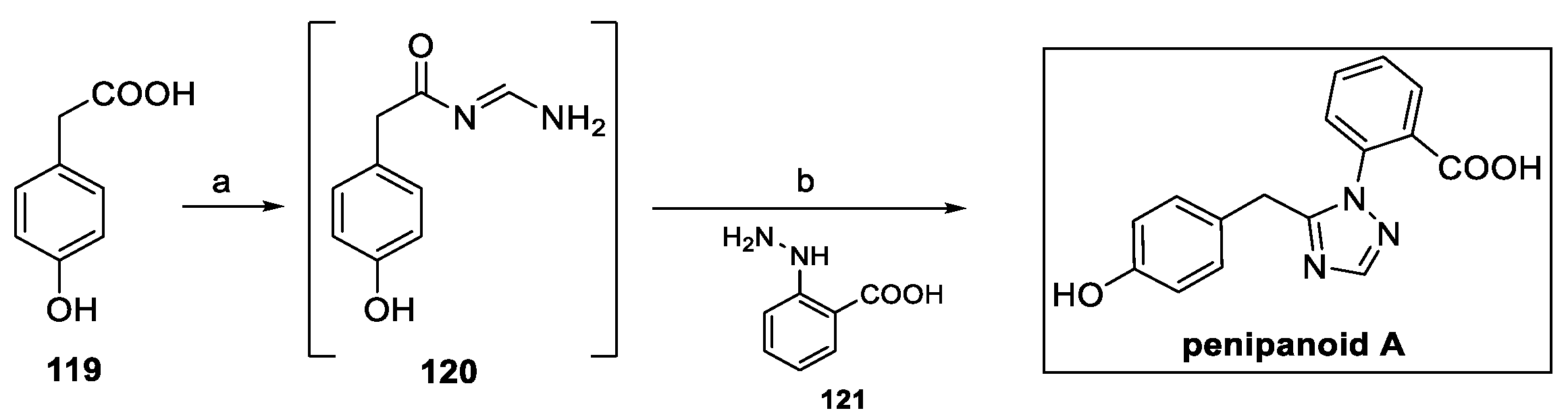
Recently a new one-pot strategy for the preparation of penipanoid A was designed to allow for a more accessible preparation of new analogs and to provide the opportunity to investigate their structure–activity relationships as antifungal agents [64]. Exploiting such an approach, 4-hydroxyphenyl acetic acid 119 was converted to intermediate 120 in the presence of formamide (Scheme 28). The addition of 2-hydrazinylbenzoic acid hydrochloride 121 finally afforded the product in a 74% yield, avoiding any protection–deprotection sequence.
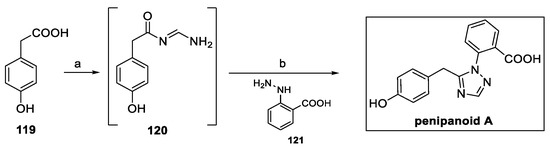
Scheme 28.
Protection-free synthesis of penipanoid A: (a) formamidine hydrochloride, HATU, DIPEA, DMF; (b) 121, AcOH.
4.3. Naphthimidazoles: Kealiinines (from Marine Environment)
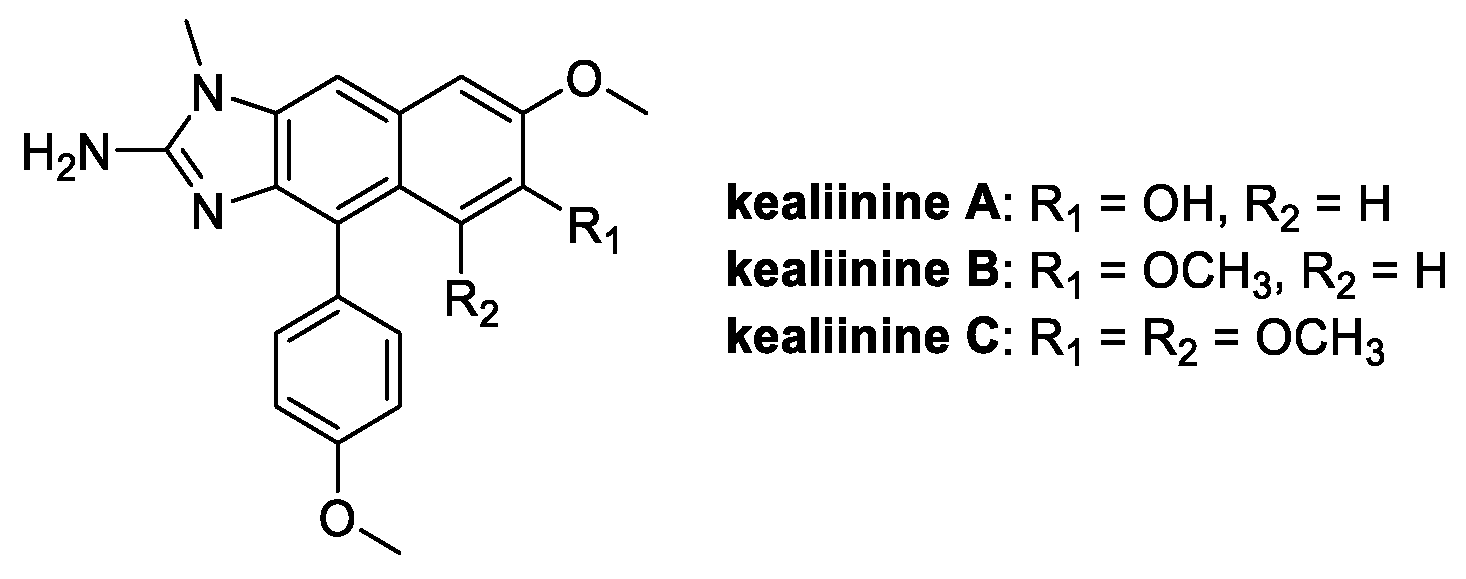
Kealiinines A-C (Figure 11) are alkaloids isolated from the Indonesian sponge Leucetta chagosensis and studied since 2013 for their antitumor properties [65]. Among these, kealiinine B, with its 2-aminonaphthimidazole core, is endowed with the highest antimicrobial activity in the field of crop protection, performing better than ribavirin on tobacco mosaic virus, and suppressing the growth rate of P. capisci (68%), P. piricola (63%), and R. solani (61%) [66].
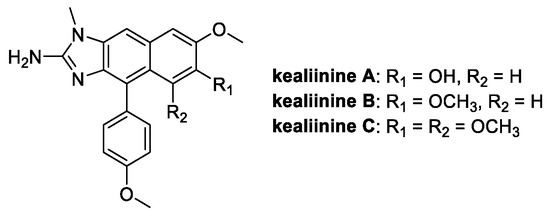
Figure 11.
Chemical structure of kealiinine A–C.
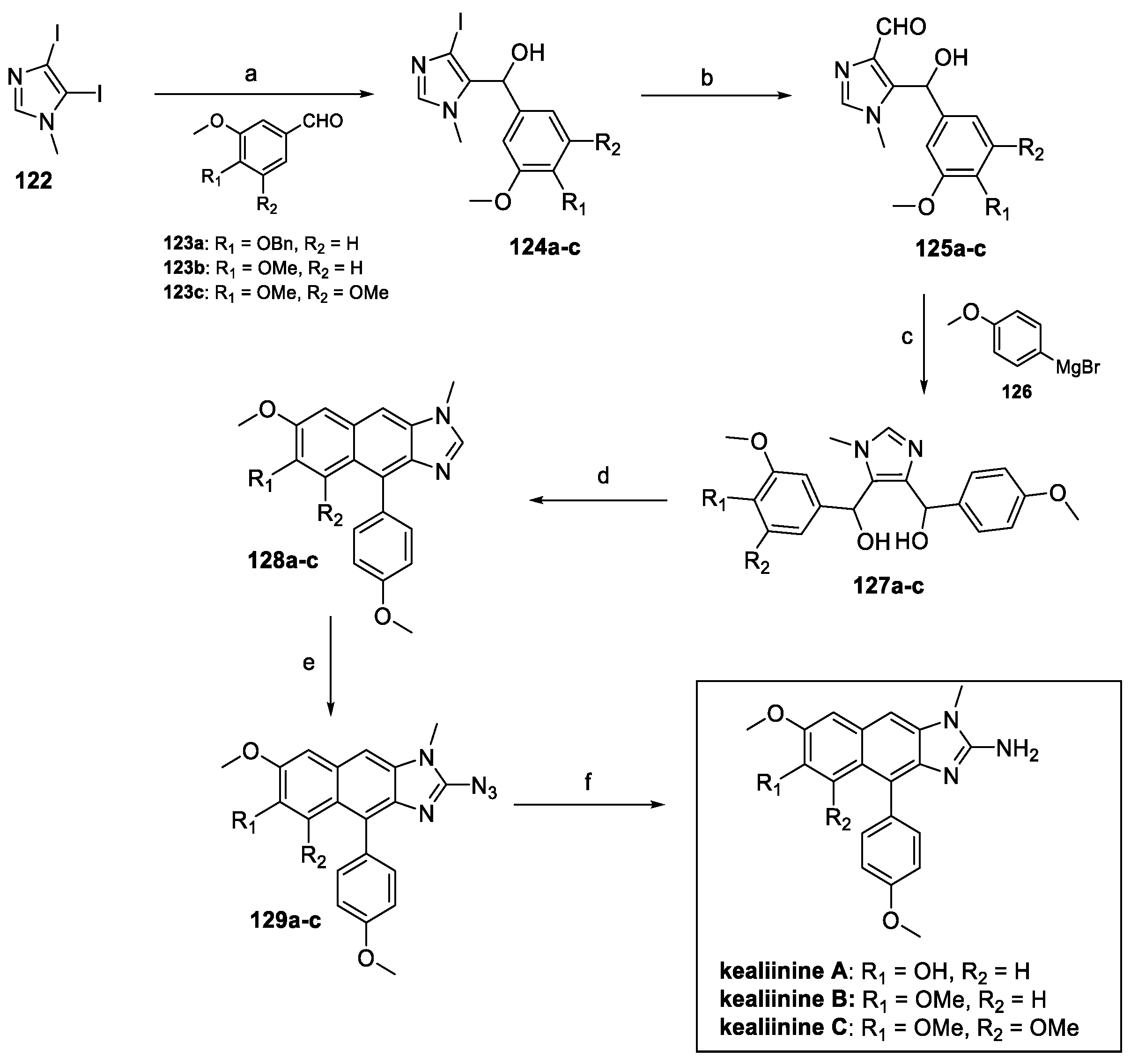
The total synthesis of kealiinine B has been reported by different groups, mainly exploiting the method developed by Das et al [67]. 4,5-diiiodo-1-methyl-1H-imidazole 122 reacted with differently substituted benzaldehydes to afford aryl alcohol 123a–c (Scheme 29). Formylation at C(4) of the imidazole core afforded hydroxy aldehyde 125a–c, ready to undergo nucleophilic addition via Grignard reagent 126. Ring closure and an additional dehydration–aromatization step led to intermediate 128a–c. Lithiation and imidazole azidolysis at C(2) resulted in compound 129a–c, giving access, upon Pd catalyzed hydrogenation, to kealiinine A–C.
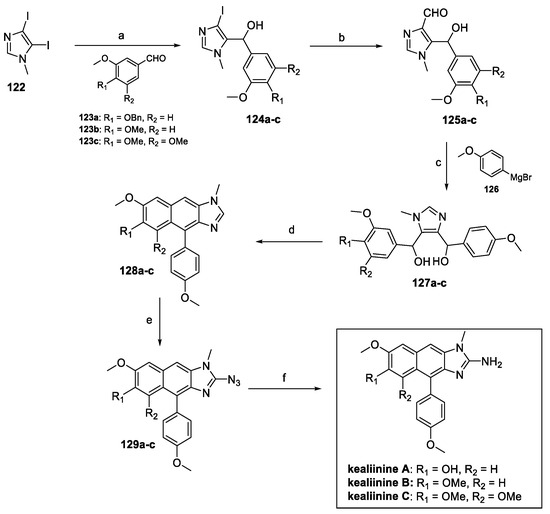
Scheme 29.
(a) EtMgBr/THF; (b) N-methyl formanilide, EtMgBr/THF; (c) 126, THF; (d) HCl, DCM; (e) i n-BuLi, THF; ii TsN3; (f) Pd-C, H2, MeOH/THF.
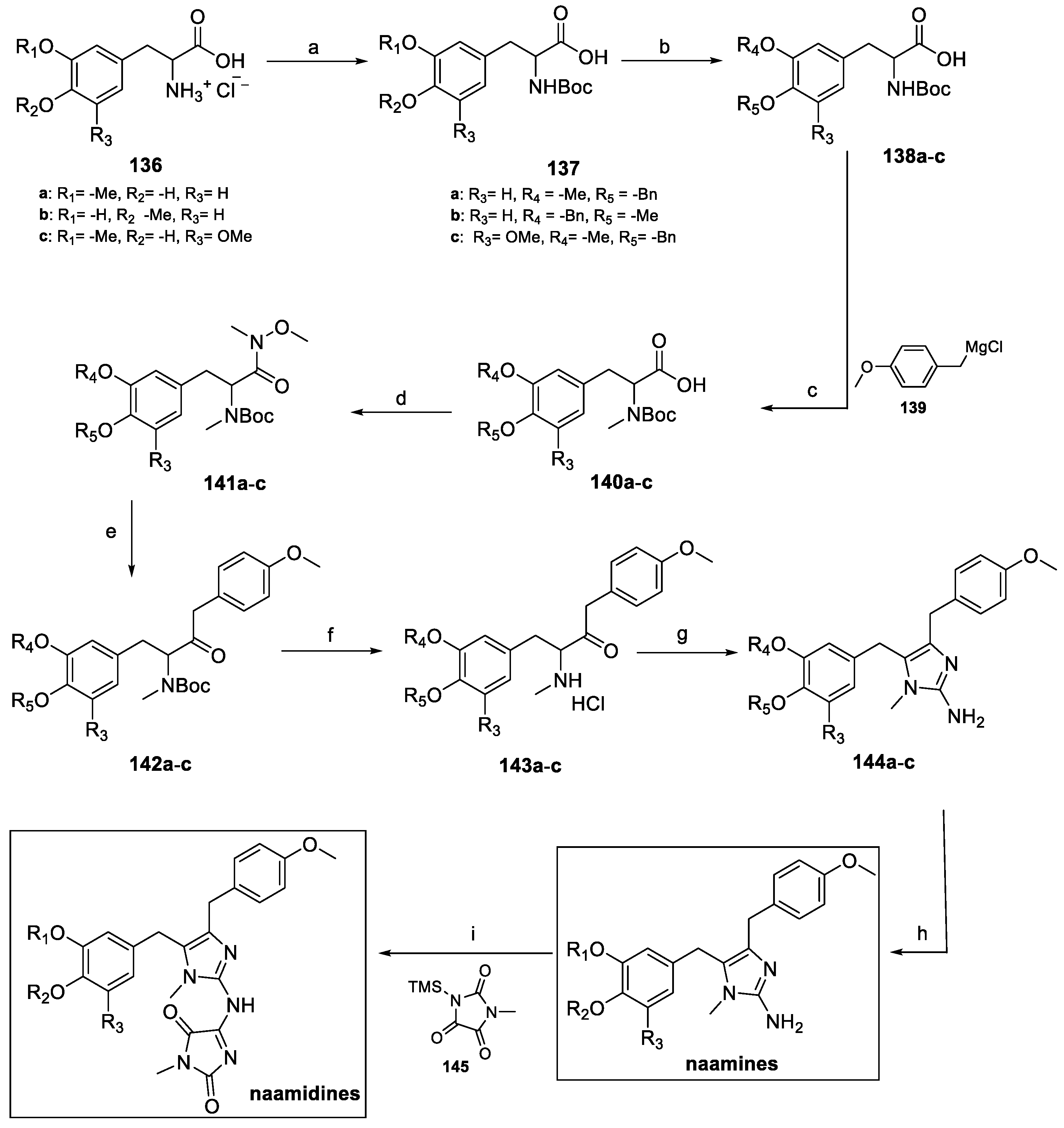
4.4. Aminoimidazoles: Naamines and Naamidines (from Marine Environment)
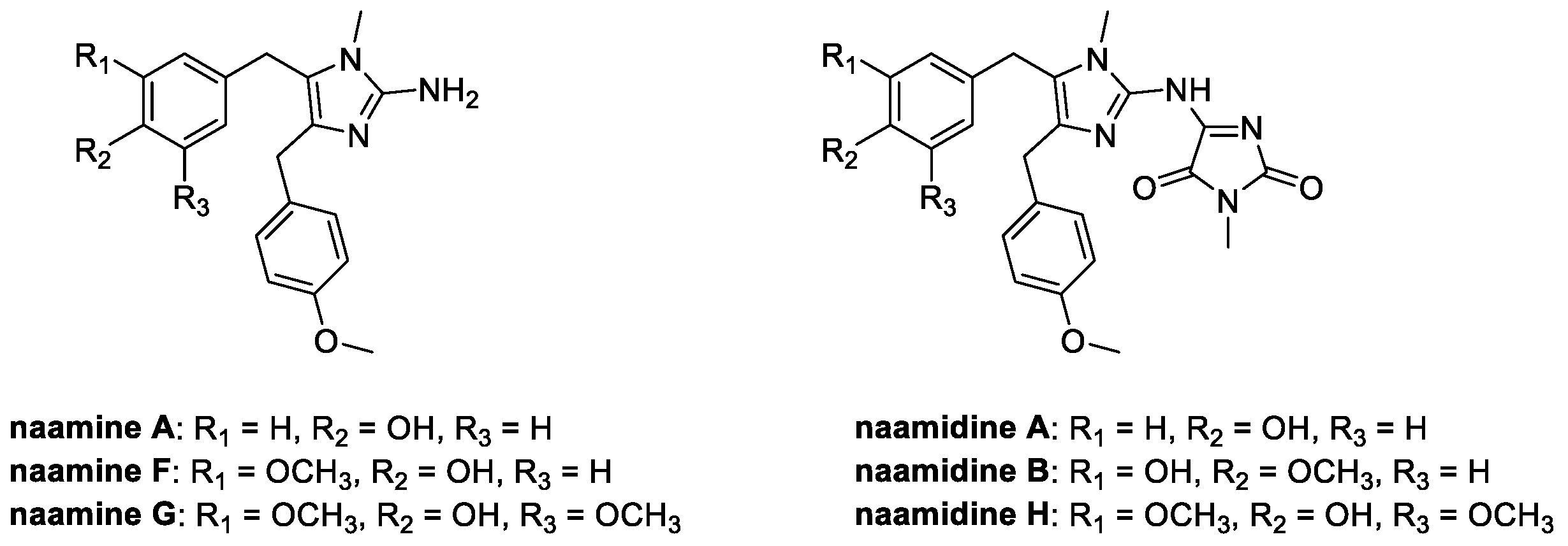
Together with kealiinines, Leucetta chagosensis is also the natural source of a series of alkaloids having the 2-amino imidazole ring in common (Figure 12). These molecules, called naamines and naamidines, are known for their interesting biological activities, ranging from antitumoral to nitric oxide synthase inhibition; moreover, they are characterized by the ability to prevent the spread of plant diseases [68].
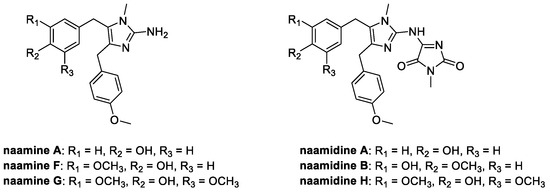
Figure 12.
Chemical structure of naamines and naamidines.
Naamine A and naamidine A were first synthetized in 2000 by Ohta et al [69], while naamines B [70], C, E, G [71], and naamidines G–H [72,73] were prepared by imidazole metalation or alkyne amination (Scheme 30 and Scheme 31).
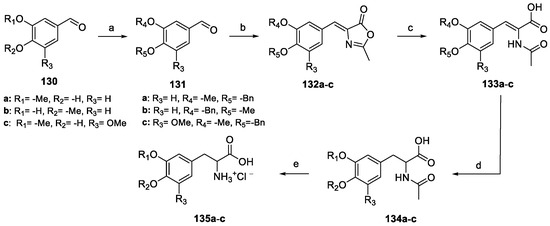
Scheme 30.
Synthesis of amino acids 135a–c. (a) BnBr, MeOH, K2CO3; (b) N-acetylglycine, AcONa, Ac2O; (c) (i) aqueous NaOH; (ii) aqueous HCl; (d) Pd/C, H2; (e) HCl conc.
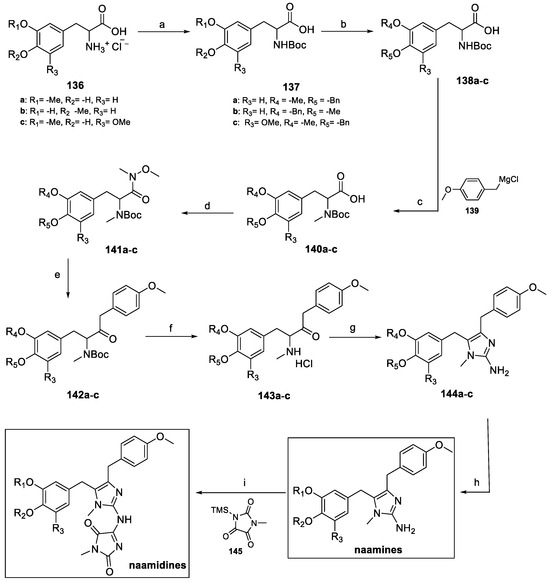
Scheme 31.
Synthetic approaches to naamines and naamidines. (a) Boc2O, NEt3, 1,4-dioxane, H2O; (b) BnBr, K2CO3, MeOH; (c) MeI, NaH, THF; (d) N,O-dimethylhydroxylamine hydrochloride, DIPEA, HOBt, EDCI, DCM; (e) 139, THF, Et2O; (f) HCl, H2O; (g) NH2CN, H2O, EtOH; (h) H2, Pd/C, MeOH, AcOH; (i) toluene.
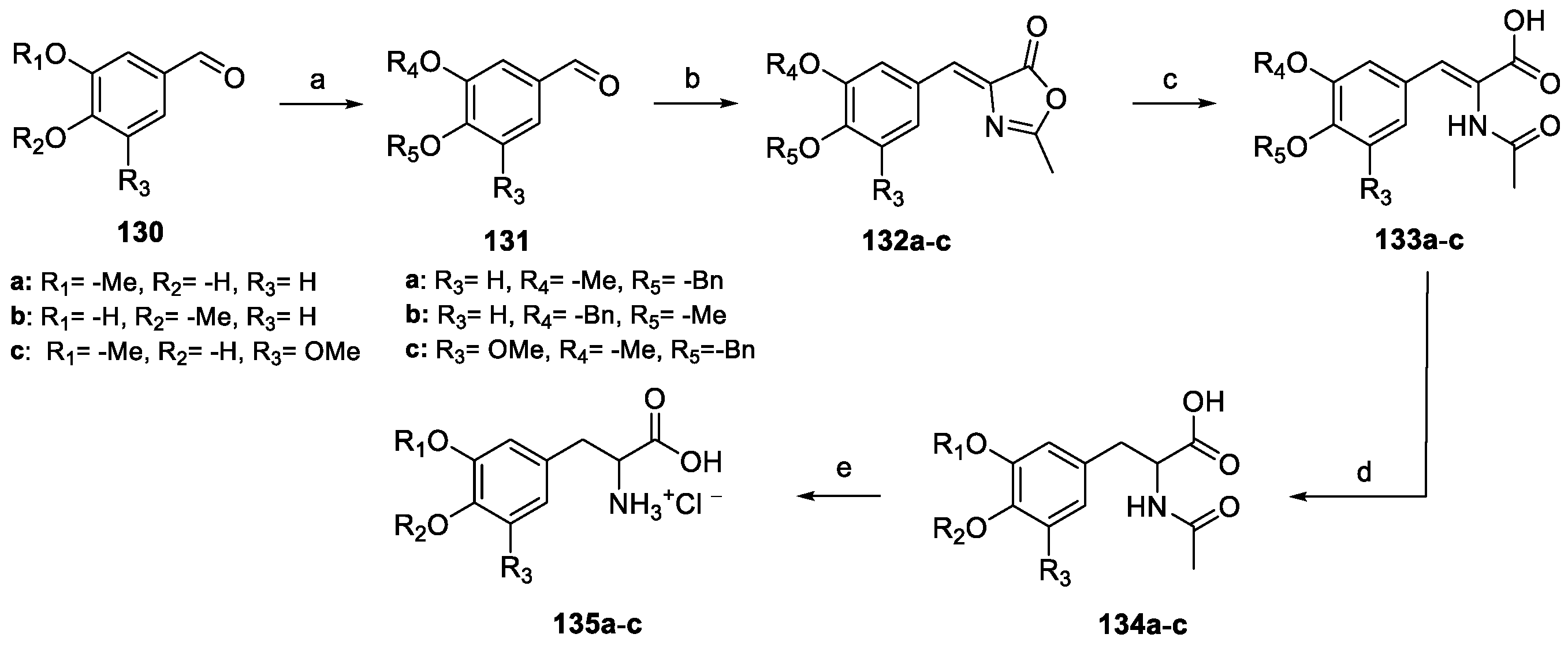
Once protected as benzyl ethers, benzaldehyde 131a–c reacted with N-acetyl glycine to yield oxazolones 132a–c. Alkaline hydrolysis and further catalytic hydrogenation yielded carboxylic acids 134a–c, which were finally deacetylated under acidic conditions to amino acids 135a–c.
BOC protection of 137a–c was required to methylate the imidazole NH group, while Weinreb amides 141a–c were obtained by treatment with N,O-dimethylhydroxylamine hydrochloride (Scheme 32). Nucleophilic substitution with the Grignard reagent 139, followed by BOC deprotection afforded the methylamino hydrochloride salts 143a–c. Treatment with cyanamide allowed for the formation of the aminoimidazole moiety (compound 144a–c), which was finally debenzylated to yield naamines. Further functionalization with methyl parabanic acid 145 afforded naamidines.
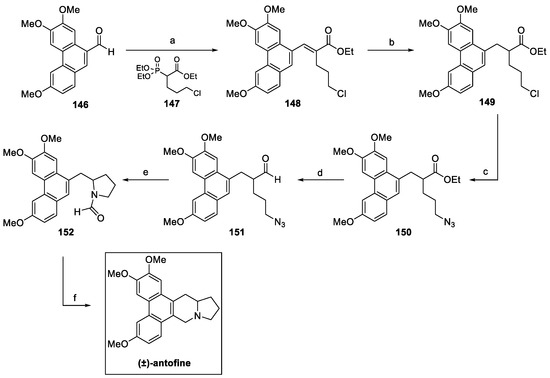
Scheme 32.
Synthesis of (±)-antofine. (a) NaH, THF; (b) H2, Pd/C, EtOAc, then (c) NaN3, TBAI, DMF; (d) DIBAL-H, DCM; (e) TFA, DCM, then (f) HCHO, HCl EtOH.
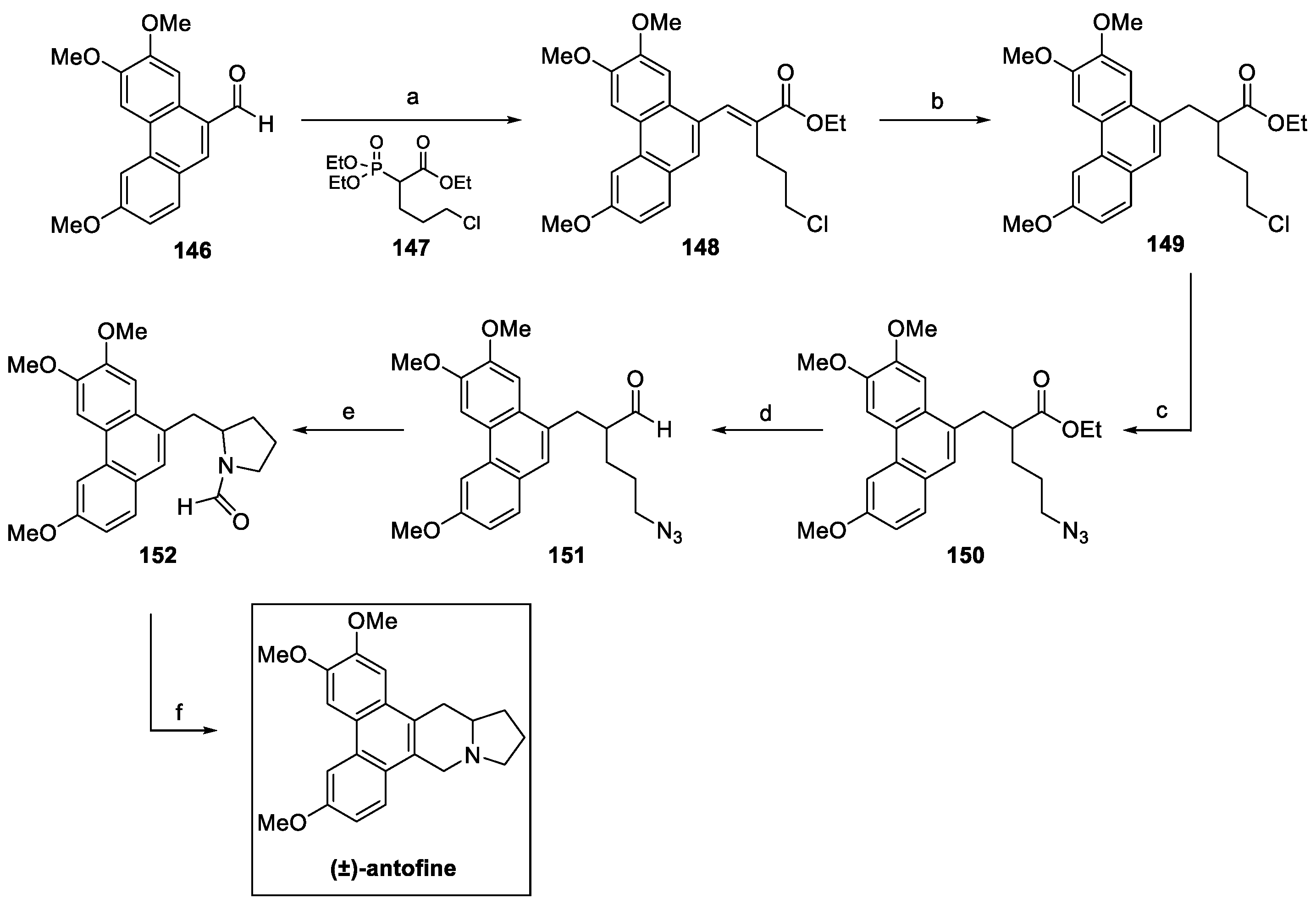
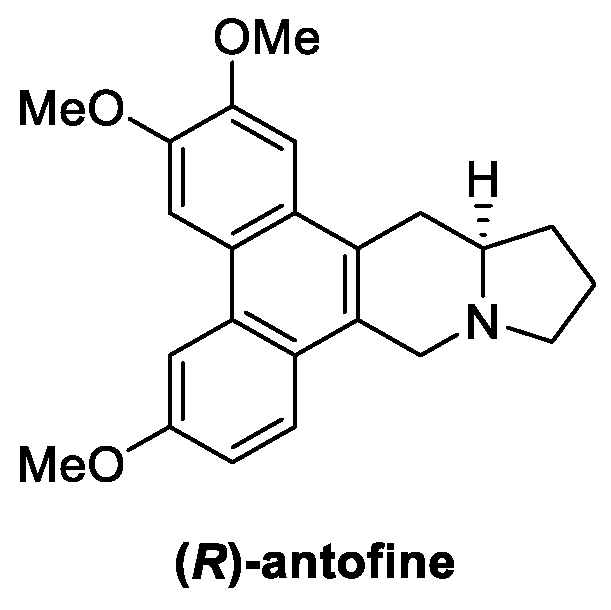
4.5. Indolizidines: Antofine (from Plants)
Antofine (Figure 13) is a bioactive alkaloid extracted from Cynanchum atratum (Bunge), belonging to the family Asclepiadaceae and widely used in Chinese traditional medicine for its antipyretic, antioxidant, anti-inflammatory, and antiviral properties [74,75,76,77]. In the last decade, antifungal potential of antofine was investigated by Wang et al., revealing significant activity, with inhibition rates up to 100%, against Alternaria solani, Cercospora arachidicola, Physalospora piricola, and Cladosporium cucumerium at 50 μg/mL [78]. Furthermore, a dose-dependent growth inhibition of Penicillium italicum (MIC = 1.56 µg/mL) was recently reported as result of plasma membrane perturbation, the reduction of ergosterol biosynthesis, and the disruption of the Krebs cycle in the mycelia [74,79].
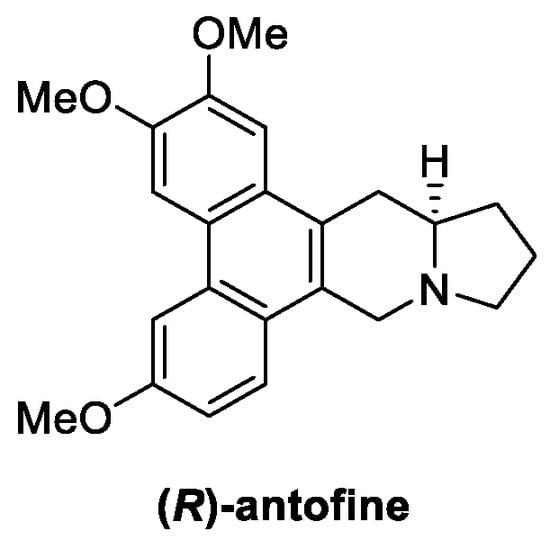
Figure 13.
Chemical structure of (R)-antofine.
In 2014, M. Yi et al. reported an efficient preparation of (±)-antofine in six steps from phenanthryl aldehyde 146, with an overall yield of 35%, as shown in Scheme 32 [80]. The Horner–Wadsworth–Emmons reaction with phosphonate 147 yielded the α,β-unsaturated ester 148, further hydrogenated and converted to terminal azide 150. Reduction of the ester to aldehyde 151 was followed by a TFA-promoted intramolecular Schmidt reaction to yield the desired formamide 152, which underwent intramolecular ring closure in the presence of formaldehyde, according to the Pictet–Spengler protocol, to furnish (±)-antofine.
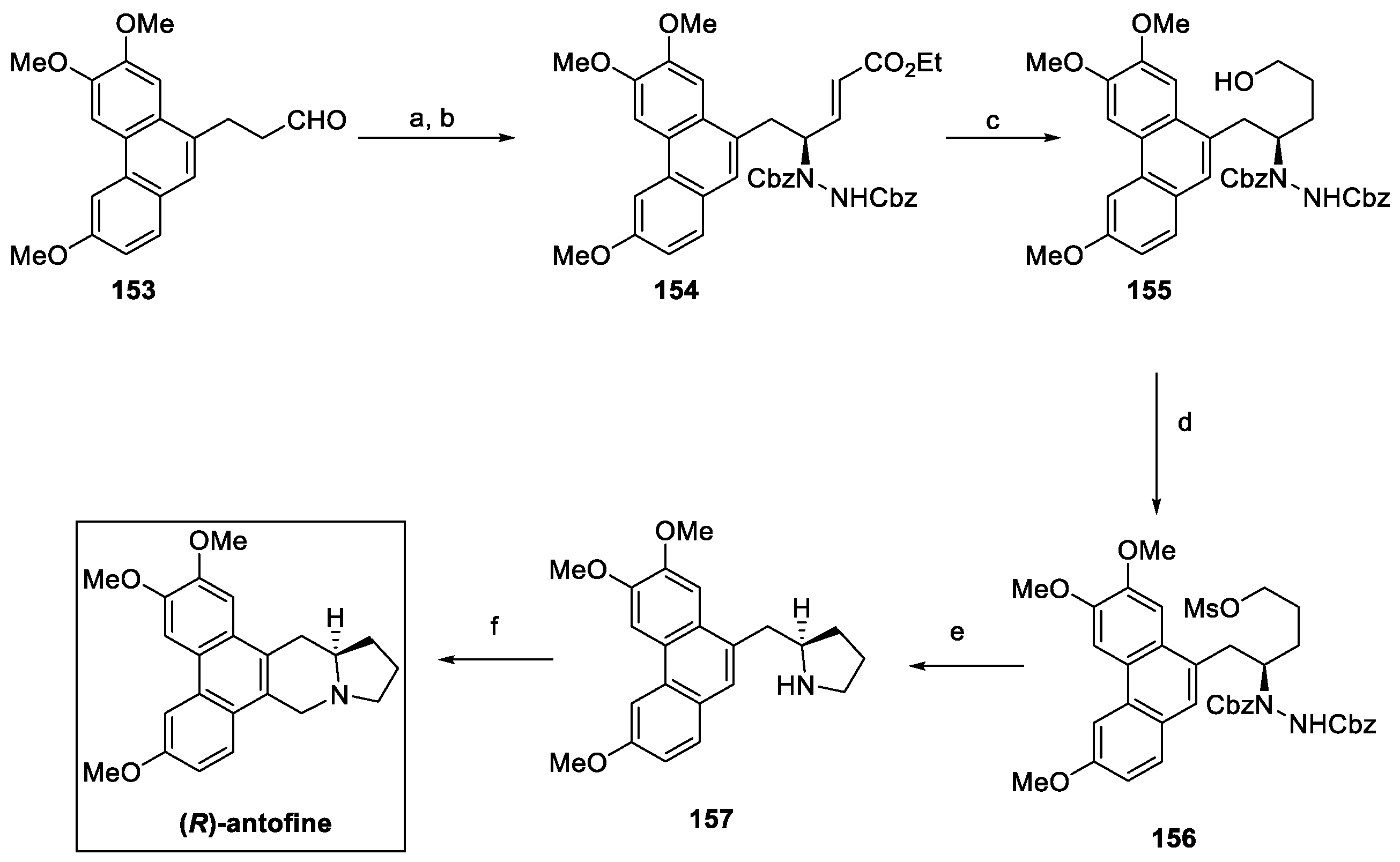
In 2018, an enantioselective approach to (R)-antofine was reported, starting from aldehyde 153 (Scheme 33) [81]. Asymmetric α-hydrazination with dibenzyl azodicarboxylate (DBAD) in the presence of D-proline was followed by reaction with the suitable phosphorane to yield the α,β-unsaturated ester 154, with a 94% enantiomeric excess. Reduction of 154 with LiBH4 resulted in the primary alcohol 155, which was further converted to mesylate 156. Cbz deprotection by hydrogenolysis over a Raney Ni catalyst favored the intramolecular substitution to the enantiomerically pure pyrrolidine derivative 157. The Pictet–Spengler reaction finally yielded (R)-antofine, with an overall yield of 58% in five steps.
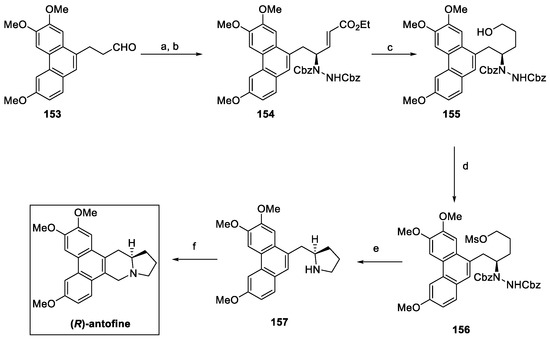
Scheme 33.
Synthesis of enantiopure (R)-antofine. (a) CbzN=NCbz, D-Proline, CH3CN, DCM, then (b) Ph3P=CHCOOEt, 94% ee; (c) LiBH4, THF; (d) MsCl, Et3N, DMAP, DCM; (e) H2, Raney Ni, MeOH; (f) HCHO, HCl, EtOH.
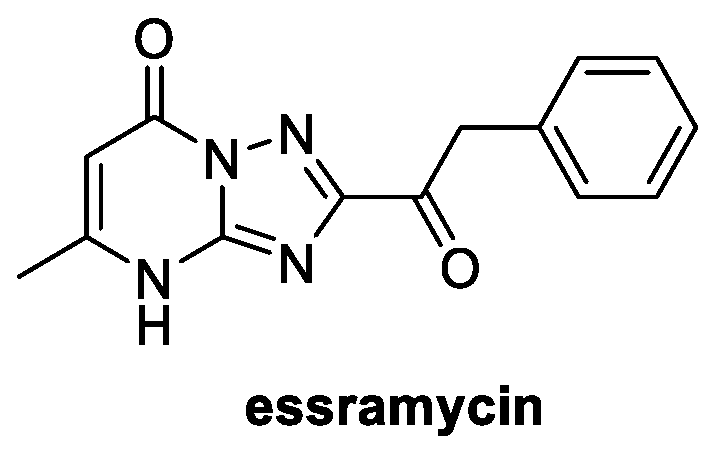
4.6. Pyrimidines: Essramycin (from Marine Environment)
Essramycin was isolated from the fermentation broth of Streptomyces sp. Merv8102, a marine organism found in sediments from the Mediterranean Sea off the Egyptian coast [82]. Its structure is quite unique for a natural product, since it is claimed to be the first one assigned with a 1,2,4-triazolo[1,5-α]pyrimidine core (Figure 14) [83].
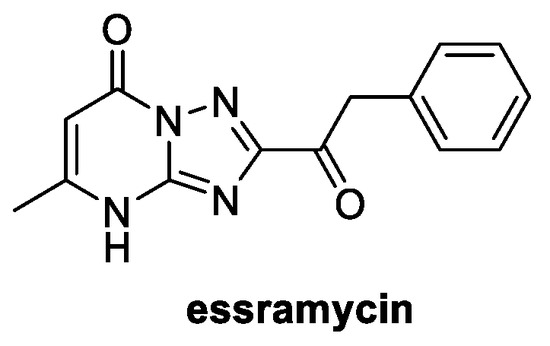
Figure 14.
Structure of essramycin.
Pyrimidoquinolines are endowed with important biological properties such as anticancer, phosphodiesterase inhibiting, antitubercular, antiepileptic, antimalarial, and antibacterial activities [84]. As for essramycin, promising results were observed against E. coli, P. aeruginosa, B. subtilis, S. aureus, and M. luteus; however, its structural similarity with ribavirin suggested its possible application as an antiviral compound for crop protection against tobacco mosaic virus [85]. Recently, essramycin was screened as a fungicide against a set of phytopathogenic fungi, showing inhibition rates between 45 and 60% against B. cinerea, P. infestans, and R. cerealis at a 50 µg/mL concentration [86].
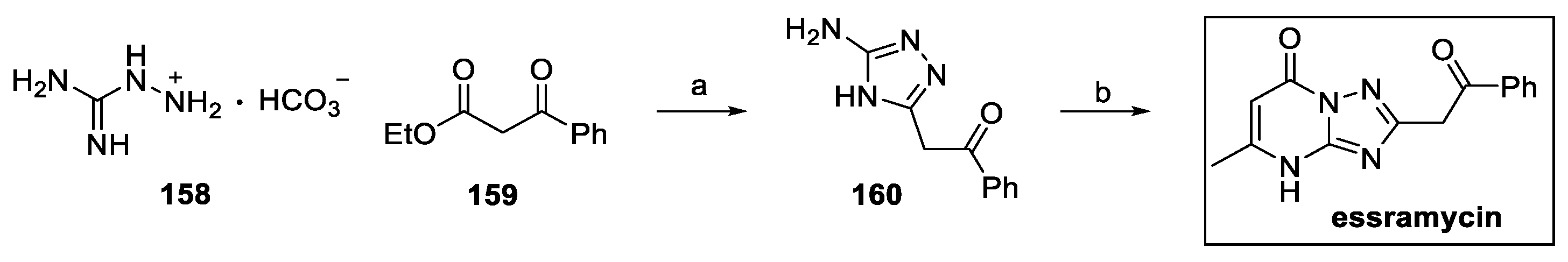
To date, the only protocol reported for the preparation of essramycin consists of a two-step synthesis, starting from aminoguanidine bicarbonate 158 and ethyl benzoylacetate 159 (Scheme 34) [83]. Cyclization to 1,2,4-triazole intermediate 160 was followed by condensation with ethyl acetoacetate, affording the pyrimidoquinoline core of essramycin in a 35% yield.
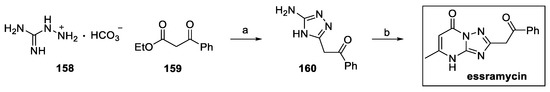
Scheme 34.
Synthesis of essramycin: (a) n-butanol; (b) ethyl acetoacetate, glacial AcOH.
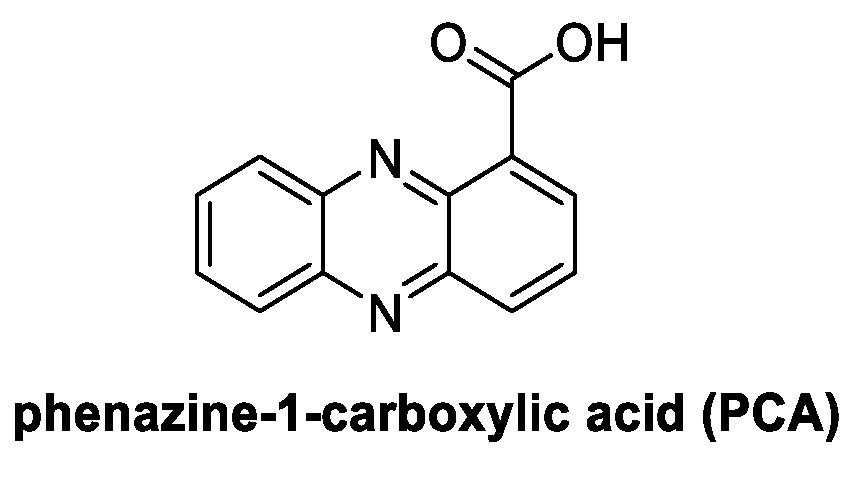
4.7. Phenazines: Phenazine-1-Carboylic Acid, PCA (from Bacteria)
Phenazine-1-carboxylic acid (PCA) is a relevant bacterium-derived natural compound which is well known for its antifungal activity and applications in crop protection (Figure 15) [87]. Isolated as a metabolite from bacteria of the genera Pseudomonas and Streptomyces, it is currently registered in China as Shenqinbactin. It acts as a biofungicide characterized by high efficacy (related to the disruption of redox homeostasis in fungal cells) [88] and associated with low human, animal, and environmental toxicity [88].
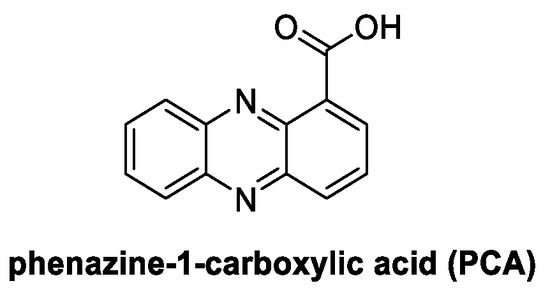
Figure 15.
Chemical structure of phenazine-1-carboxylic acid (PCA).
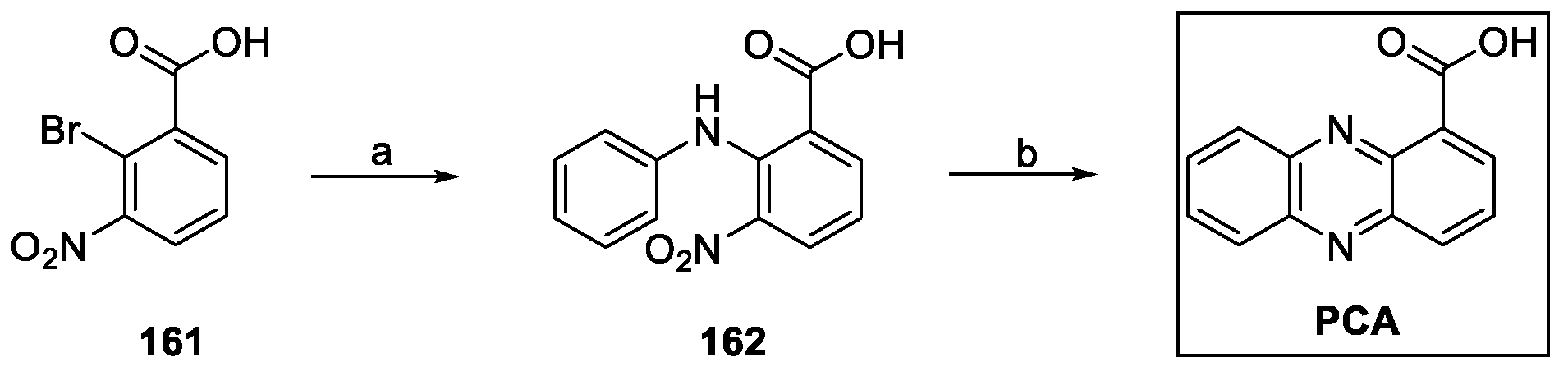
Xiong et al. described the synthesis of PCA, starting with an Ullmann reaction between aniline and the bromo benzoate 161 to afford 162. Reduction of the nitro group readily produced ring closure, with subsequent formation of phenazine-1-carboxamide (Scheme 35) [89,90].
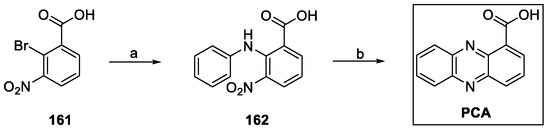
Scheme 35.
Synthesis of phenazine-1-carboxylic acid (PCA): (a) aniline, CuCl2, copper powder, N-ethyl morpholine, 2,3-butanediol; (b) NaBH4, EtONa, EtOH.
5. Antifungal Activity
All the described natural compounds were assayed in terms of antifungal activity on several fungal strains. The data discussed in the previous paragraphs have been summarized in Table 1, indicating the phytopathogen and the tested alkaloid, together with the percentage of mycelia growth inhibition, the minimum inhibitory concentration (MIC), or the half maximal effective concentration (EC50).

Table 1.
Antifungal activity of all the synthesized compounds reported. All the compounds were evaluated using the mycelium growth rate method (procedures in the corresponding references), except for PCA, which was evaluated via the hyphae growth velocity assay.
6. Conclusions
Alkaloids represent a promising class of natural antifungal agents for crop protection. With the increasing demand for eco-friendly agricultural practices, research into the development and commercialization of alkaloid-based fungicides could provide a valuable tool for managing fungal diseases in crops.
Synthesizing alkaloids for agricultural use can be beneficial for several reasons, including the overcoming of supply and extraction limitations and improving cost-effectiveness and scalability.
Over the last ten years, numerous methods have been exploited for the synthesis of polycyclic nitrogen-containing moieties, usually with good overall yields and a reasonable number of steps. Moreover, remarkable advances have been reported, especially for the synthesis of enantiomerically pure compounds, exploiting chiral species in catalytic amounts and cascade/domino reactions. Such efforts allowed for the efficient customization and optimization of chemical structures, which is both advantageous and convenient for the preparation of new nature-derived chemotypes, potentially useful as valuable tools in advancing agricultural practices while safeguarding ecological balance. However, as for the synthesis of other natural compounds, the chemical complexity of alkaloids (e.g., multiple stereogenic centers, fused rings), in most cases, still requires multistep processes affecting the overall yield of the synthetic approach. Moreover, the catalysts, reagents, and reaction conditions required could be cost-intensive and must be followed by purification and isolations steps, which are not always scalable for an industrial process.
The fascinating complexity of natural biosynthesis is still difficult to reproduce in a laboratory, but since natural compounds represent the most replenished arsenal against pathogens, researchers from all over the world will continue researching innovative and revolutionary paths to mimic what nature has mastered over millennia.
Author Contributions
Conceptualization, A.P., S.D., F.A. and S.P.; investigation, D.D., F.S., F.A. and S.P.; data curation, D.D., F.S., F.A. and S.P.; writing—original draft preparation, D.D., F.S., F.A. and S.P.; writing—review and editing, D.D., F.S., A.P., S.D., F.A. and S.P.; supervision, A.P., S.D., F.A. and S.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the Italian MIUR Project PRIN202223SDALL_01 and received no external funding.
Acknowledgments
The authors acknowledge the support of the APC central fund of the University of Milan.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- FAO. Achieving Zero Hunger: The Critical Role of Investments in Social Protection and Agriculture; FAO: Rome, Italy, 2015. [Google Scholar]
- Peng, Y.; Li, S.J.; Yan, J.; Tang, Y.; Cheng, J.P.; Gao, A.J.; Yao, X.; Ruan, J.J.; Xu, B.L. Research Progress on Phytopathogenic Fungi and Their Role as Biocontrol Agents. Front. Microbiol. 2021, 12, 670135. [Google Scholar] [CrossRef]
- Zubrod, J.P.; Bundschuh, M.; Arts, G.; Brühl, C.A.; Imfeld, G.; Knäbel, A.; Payraudeau, S.; Rasmussen, J.J.; Rohr, J.; Scharmüller, A.; et al. Fungicides: An Overlooked Pesticide Class? Environ. Sci. Technol. 2019, 53, 3347–3365. [Google Scholar] [CrossRef]
- Pérez-Pizá, M.C.; Sautua, F.J.; Szparaga, A.; Bohata, A.; Kocira, S.; Carmona, M.A. New Tools for the Management of Fungal Pathogens in Extensive Cropping Systems for Friendly Environments. CRC Crit. Rev. Plant Sci. 2024, 43, 63–93. [Google Scholar] [CrossRef]
- Tsalidis, G.A. Human Health and Ecosystem Quality Benefits with Life Cycle Assessment Due to Fungicides Elimination in Agriculture. Sustainability 2022, 14, 846. [Google Scholar] [CrossRef]
- Yeshi, K.; Crayn, D.; Ritmejerytė, E.; Wangchuk, P. Plant Secondary Metabolites Produced in Response to Abiotic Stresses Has Potential Application in Pharmaceutical Product Development. Molecules 2022, 27, 313. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Grijalva, E.P.; López-Martínez, L.X.; Contreras-Angulo, L.A.; Elizalde-Romero, C.A.; Heredia, J.B. Plant Alkaloids: Structures and Bioactive Properties. In Plant-Derived Bioactives; Springer: Singapore, 2020; pp. 85–117. [Google Scholar]
- Mazid, M.; Khan, T.A.; Mohammad, F. Role of Secondary Metabolites in Defense Mechanisms of Plants. Biol. Med. 2011, 3, 232–249. [Google Scholar]
- Thawabteh, A.; Juma, S.; Bader, M.; Karaman, D.; Scrano, L.; Bufo, S.; Karaman, R. The Biological Activity of Natural Alkaloids against Herbivores, Cancerous Cells and Pathogens. Toxins 2019, 11, 656. [Google Scholar] [CrossRef]
- Youssef, F.S.; Simal-Gandara, J. Comprehensive Overview on the Chemistry and Biological Activities of Selected Alkaloid Producing Marine-Derived Fungi as a Valuable Reservoir of Drug Entities. Biomedicines 2021, 9, 485. [Google Scholar] [CrossRef]
- Hashim, S.E.; Basar, N.; Abd Samad, A.; Jamil, S.; Bakar, M.B.; Jamalis, J.; Abd-Aziz, N.; Wagiran, A.; Razak, M.R.M.A.; Samad, A.F.A. Advancing Alkaloid-Based Medicines: Medical Applications, Scalable Production and Synthetic Innovations. Phytochem. Rev. 2024. [Google Scholar] [CrossRef]
- Wu, Y.; Ren, D.; Gao, C.; Li, J.; Du, B.; Wang, Z.; Qian, S. Recent Advances for Alkaloids as Botanical Pesticides for Use in Organic Agriculture. Int. J. Pest. Manag. 2023, 69, 288–298. [Google Scholar] [CrossRef]
- Bhambhani, S.; Kondhare, K.R.; Giri, A.P. Diversity in Chemical Structures and Biological Properties of Plant Alkaloids. Molecules 2021, 26, 3374. [Google Scholar] [CrossRef] [PubMed]
- Yahyazadeh, M.; Nowak, M.; Kima, H.; Selmar, D. Horizontal Natural Product Transfer: A Potential Source of Alkaloidal Contaminants in Phytopharmaceuticals. Phytomedicine 2017, 34, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.Z.; Zhu, J.K.; Yin, X.D.; Yan, Y.F.; Wang, Y.L.; Shang, X.F.; Liu, Y.Q.; Zhao, Z.M.; Peng, J.W.; Liu, H. Design, Synthesis, and Antifungal Evaluation of Novel Quinoline Derivatives Inspired from Natural Quinine Alkaloids. J. Agric. Food Chem. 2019, 67, 11340–11353. [Google Scholar] [CrossRef]
- Antika, L.D.; Triana, D.; Ernawati, T. Antimicrobial Activity of Quinine Derivatives against Human Pathogenic Bacteria. IOP Conf. Ser. Earth Environ. Sci. 2020, 462, 012006. [Google Scholar] [CrossRef]
- Stork, G.; Niu, D.; Fujimoto, A.; Koft, E.R.; Balkovec, J.M.; Tata, J.R.; Dake, G.R. The First Stereoselective Total Synthesis of Quinine. J. Am. Chem. Soc. 2001, 123, 3239–3242. [Google Scholar] [CrossRef]
- Shiomi, S.; Misaka, R.; Kaneko, M.; Ishikawa, H. Enantioselective Total Synthesis of the Unnatural Enantiomer of Quinine. Chem. Sci. 2019, 10, 9433–9437. [Google Scholar] [CrossRef]
- Nair, V.; Menon, R.S.; Vellalath, S. Asymmetric Synthesis of Quinine: A Landmark in Organic Synthesis. Nat. Prod. Comm. 2006, 1, 1934578X0600101009. [Google Scholar] [CrossRef]
- O’Donovan, D.H.; Aillard, P.; Berger, M.; de la Torre, A.; Petkova, D.; Knittl-Frank, C.; Geerdink, D.; Kaiser, M.; Maulide, N. C−H Activation Enables a Concise Total Synthesis of Quinine and Analogues with Enhanced Antimalarial Activity. Angew. Chem. Int. Ed. 2018, 57, 10737–10741. [Google Scholar] [CrossRef]
- Lee, J.; Chen, D.Y.-K. A Local-Desymmetrization-Based Divergent Synthesis of Quinine and Quinidine. Angew. Chem. Int. Ed. 2019, 58, 488–493. [Google Scholar] [CrossRef]
- Jiang, Y.; Deiana, L.; Zhang, K.; Lin, S.; Córdova, A. Total Asymmetric Synthesis of Quinine, Quinidine, and Analogues via Catalytic Enantioselective Cascade Transformations. Eur. J. Org. Chem. 2019, 2019, 6016–6023. [Google Scholar] [CrossRef]
- Cagir, A.; Jones, S.H.; Gao, R.; Eisenhauer, B.M.; Hecht, S.M. Luotonin A. A naturally occurring human DNA topoisomerase I poison. J. Am. Chem. Soc. 2003, 125, 13628–13629. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.Z.; Zhang, J.; Peng, J.W.; Zhang, Z.J.; Zhao, W.B.; Wang, R.X.; Ma, K.Y.; Li, J.C.; Liu, Y.Q.; Zhao, Z.M.; et al. Discovery of Luotonin A Analogues as Potent Fungicides and Insecticides: Design, Synthesis and Biological Evaluation Inspired by Natural Alkaloid. Eur. J. Med. Chem. 2020, 194, 112253. [Google Scholar] [CrossRef] [PubMed]
- Kwon, S.H.; Seo, H.A.; Cheon, C.H. Total Synthesis of Luotonin A and Rutaecarpine from an Aldimine via the Designed Cyclization. Org. Lett. 2016, 18, 5280–5283. [Google Scholar] [CrossRef] [PubMed]
- Baguia, H.; Deldaele, C.; Romero, E.; Michelet, B.; Evano, G. Copper-Catalyzed Photoinduced Radical Domino Cyclization of Ynamides and Cyanamides: A Unified Entry to Rosettacin, Luotonin A, and Deoxyvasicinone. Synthesis 2018, 50, 3022–3030. [Google Scholar] [CrossRef]
- Maiti, M.; Nandi, R.; Chaudhuri, K. The effect of ph on the absorption and fluorescence spectra of sanguinarine. Photochem. Photobiol. 1983, 38, 245–249. [Google Scholar] [CrossRef]
- Croaker, A.; King, G.J.; Pyne, J.H.; Anoopkumar-Dukie, S.; Liu, L. Sanguinaria Canadensis: Traditional Medicine, Phytochemical Composition, Biological Activities and Current Uses. Int. J. Mol. Sci. 2016, 17, 1414. [Google Scholar] [CrossRef]
- Zhao, Z.M.; Shang, X.F.; Lawoe, R.K.; Liu, Y.Q.; Zhou, R.; Sun, Y.; Yan, Y.F.; Li, J.C.; Yang, G.Z.; Yang, C.J. Anti-Phytopathogenic Activity and the Possible Mechanisms of Action of Isoquinoline Alkaloid Sanguinarine. Pestic. Biochem. Physiol. 2019, 159, 51–58. [Google Scholar] [CrossRef]
- Tatton, M.R.; Simpson, I.; Donohoe, T.J. New Methods for the Synthesis of Naphthyl Amines; Application to the Synthesis of Dihydrosanguinarine, Sanguinarine, Oxysanguinarine and (±)-Maclekarpines B and C. Chem. Commun. 2014, 50, 11314–11316. [Google Scholar] [CrossRef]
- Parvatkar, P.T.; Parameswaran, P.S. Indoloquinolines: Possible Biogenesis from Common Indole Precursors and Their Synthesis Using Domino Strategies. Curr. Org. Synth. 2016, 15, 58–72. [Google Scholar] [CrossRef]
- Chen, Y.J.; Liu, H.; Zhang, S.Y.; Li, H.; Ma, K.Y.; Liu, Y.Q.; Yin, X.D.; Zhou, R.; Yan, Y.F.; Wang, R.X.; et al. Design, Synthesis, and Antifungal Evaluation of Cryptolepine Derivatives against Phytopathogenic Fungi. J. Agric. Food Chem. 2021, 69, 1259–1271. [Google Scholar] [CrossRef]
- Shang, X.F.; Dai, L.X.; Zhang, Z.J.; Yang, C.J.; Du, S.S.; Wu, T.L.; He, Y.H.; Zhu, J.K.; Liu, Y.Q.; Yan, Y.F.; et al. Integrated Proteomics and Transcriptomics Analyses Reveals the Possible Antifungal Mechanism of an Indoloquinoline Alkaloid Neocryptolepine against Rhizoctonia Solani. J. Agric. Food Chem. 2021, 69, 6455–6464. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.Y.; Jin, Y.R.; Luo, X.F.; Zhang, S.Y.; Dai, T.L.; Ma, L.; Zhang, Z.J.; Wu, Z.R.; Jin, C.X.; Liu, Y.Q. Design, Synthesis, and Antimicrobial Activity Evaluation of Novel Isocryptolepine Derivatives against Phytopathogenic Fungi and Bacteria. J. Agric. Food Chem. 2024, 72, 20831–20841. [Google Scholar] [CrossRef]
- Abe, T.; Suzuki, T.; Anada, M.; Matsunaga, S.; Yamada, K. 2-Hydroxyindoline-3-Triethylammonium Bromide: A Reagent for Formal C3-Electrophilic Reactions of Indoles. Org. Lett. 2017, 19, 4275–4278. [Google Scholar] [CrossRef] [PubMed]
- Mudududdla, R.; Mohanakrishnan, D.; Bharate, S.S.; Vishwakarma, R.A.; Sahal, D.; Bharate, S.B. Orally Effective Aminoalkyl 10H-Indolo[3,2-b]Quinoline-11-Carboxamide Kills the Malaria Parasite by Inhibiting Host Hemoglobin Uptake. ChemMedChem 2018, 13, 2581–2598. [Google Scholar] [CrossRef]
- Majhi, B.; Ganguly, S.; Palit, S.; Parwez, A.; Saha, R.; Basu, G.; Dutta, S. Sequence-Specific Dual DNA Binding Modes and Cytotoxicities of N-6-Functionalized Norcryptotackieine Alkaloids. J. Nat. Prod. 2023, 86, 1667–1676. [Google Scholar] [CrossRef]
- Zhu, J.K.; Gao, J.M.; Yang, C.J.; Shang, X.F.; Zhao, Z.M.; Lawoe, R.K.; Zhou, R.; Sun, Y.; Yin, X.D.; Liu, Y.Q. Design, Synthesis, and Antifungal Evaluation of Neocryptolepine Derivatives against Phytopathogenic Fungi. J. Agric. Food Chem. 2020, 68, 2306–2315. [Google Scholar] [CrossRef]
- Thobokholt, E.N.; Larghi, E.L.; Bracca, A.B.J.; Kaufman, T.S. Isolation and Synthesis of Cryptosanguinolentine (Isocryptolepine), a Naturally-Occurring Bioactive Indoloquinoline Alkaloid. RSC Adv. 2020, 10, 18978–19002. [Google Scholar] [CrossRef]
- Akitake, M.; Noda, S.; Miyoshi, K.; Sonoda, M.; Tanimori, S. Access to γ-Carbolines: Synthesis of Isocryptolepine. J. Org. Chem. 2021, 86, 17727–17737. [Google Scholar] [CrossRef]
- Olmedo, G.M.; Cerioni, L.; González, M.M.; Cabrerizo, F.M.; Rapisarda, V.A.; Volentini, S.I. Antifungal Activity of β-Carbolines on Penicillium Digitatum and Botrytis Cinerea. Food Microbiol. 2017, 62, 9–14. [Google Scholar] [CrossRef]
- Kuang, Y.; Huang, J.; Chen, F. Practical and Phase Transfer–Catalyzed Synthesis of 6-Methoxytryptamine. Synth. Commun. 2006, 36, 1515–1519. [Google Scholar] [CrossRef]
- Du, H.; Tian, S.; Chen, J.; Gu, H.; Li, N.; Wang, J. Synthesis and Biological Evaluation of N9-Substituted Harmine Derivatives as Potential Anticancer Agents. Bioorg Med. Chem. Lett. 2016, 26, 4015–4019. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Liu, X.; Tian, L.; Gao, Y.; Liu, W.; Chen, H.; Jiang, X.; Xu, Z.; Ding, H.; Zhao, Q. Design, Synthesis and Biological Evaluation of Harmine Derivatives as Potent GSK-3β/DYRK1A Dual Inhibitors for the Treatment of Alzheimer’s Disease. Eur. J. Med. Chem. 2021, 222, 113554. [Google Scholar] [CrossRef] [PubMed]
- Sathiyalingam, S.; Roesner, S. Synthesis of A- and Β-Carbolines by a Metalation/Negishi Cross-Coupling/S N Ar Reaction Sequence. Adv. Synth. Catal. 2022, 364, 1769–1774. [Google Scholar] [CrossRef]
- Zhang, P.; Sun, X.; Xu, B.; Bijian, K.; Wan, S.; Li, G.; Alaoui-Jamali, M.; Jiang, T. Total Synthesis and Bioactivity of the Marine Alkaloid Pityriacitrin and Some of Its Derivatives. Eur. J. Med. Chem. 2011, 46, 6089–6097. [Google Scholar] [CrossRef]
- Wang, T.N.; Yang, S.; Shi, S.Y.; Yuan, W.Y.; Chen, J.X.; Duan, Z.Y.; Lu, A.D.; Wang, Z.W.; Wang, Q.M. Pityriacitrin Marine Alkaloids as Novel Antiviral and Anti-Phytopathogenic-Fungus Agents. Pest. Manag. Sci. 2021, 77, 4691–4700. [Google Scholar] [CrossRef]
- Zhu, Y.; Liu, M.; Cai, Q.; Jia, F.; Wu, A. A Cascade Coupling Strategy for One-Pot Total Synthesis of Β-Carboline and Isoquinoline-Containing Natural Products and Derivatives. Chem.—Eur. J. 2013, 19, 10132–10137. [Google Scholar] [CrossRef]
- Huang, D.; Zhang, Z.; Li, Y.; Liu, F.; Huang, W.; Min, Y.; Wang, K.; Yang, J.; Cao, C.; Gong, Y.; et al. Carboline Derivatives Based on Natural Pityriacitrin as Potential Antifungal Agents. Phytochem. Lett. 2022, 48, 100–105. [Google Scholar] [CrossRef]
- Franco, L.H.; Joffé, E.B.D.K.; Puricelli, L.; Tatian, M.; Seldes, A.M.; Palermo, J.A. Indole Alkaloids from the Tunicate Aplidium meridianum. J. Nat. Prod. 1998, 61, 1130–1132. [Google Scholar] [CrossRef]
- Dong, J.; Huang, S.S.; Hao, Y.N.; Wang, Z.W.; Liu, Y.X.; Li, Y.Q.; Wang, Q.M. Marine-Natural-Products for Biocides Development: First Discovery of Meridianin Alkaloids as Antiviral and Anti-Phytopathogenic-Fungus Agents. Pest. Manag. Sci. 2020, 76, 3369–3376. [Google Scholar] [CrossRef]
- Xiao, L. A Review: Meridianins and Meridianins Derivatives. Molecules 2022, 27, 8714. [Google Scholar] [CrossRef]
- Jiang, B.; Yang, C.G. Synthesis of Indolylpyrimidines via Cross-Coupling of Indolylboronic Acid with Chloropyrimidines: Facile Synthesis of Meridianin D. Heterocycles 2000, 53, 1489–1498. [Google Scholar] [CrossRef]
- Fresneda, P.M.; Molina, P.; Bleda, J.A. Synthesis of the Indole Alkaloids Meridianins from the Tunicate Aplidium Meridianum. Tetrahedron 2001, 57, 2355–2363. [Google Scholar] [CrossRef]
- Karpov, A.S.; Merkul, E.; Rominger, F.; Müller, T.J.J. Concise Syntheses of Meridianins by Carbonylative Alkynylation and a Four-Component Pyrimidine Synthesis. Angew. Chem. Int. Ed. 2005, 44, 6951–6956. [Google Scholar] [CrossRef] [PubMed]
- Linares-Otoya, L.; Liu, Y.; Linares-Otoya, V.; Armas-Mantilla, L.; Crüsemann, M.; Ganoza-Yupanqui, M.L.; Campos-Florian, J.; König, G.M.; Schäberle, T.F. Biosynthetic Basis for Structural Diversity of Aminophenylpyrrole-Derived Alkaloids. ACS Chem. Biol. 2019, 14, 176–181. [Google Scholar] [CrossRef]
- Kilani, J.; Fillinger, S. Phenylpyrroles: 30 Years, Two Molecules and (Nearly) No Resistance. Front. Microbiol. 2016, 7, 2014. [Google Scholar] [CrossRef]
- Pawar, S.; Chaudhari, A.; Prabha, R.; Shukla, R.; Singh, D.P. Microbial Pyrrolnitrin: Natural Metabolite with Immense Practical Utility. Biomolecules 2019, 9, 443. [Google Scholar] [CrossRef]
- Nakano, H.; Umio, S.; Kariyone, K.; Tanaka, K.; Kishimoto, T.; Noguchi, H.; Ueda, I.; Nakamura, H.; Morimoto, T. Total Synthesis of Pyrrolnitrin, a New Antibiotic. Tetrahedron Lett. 1966, 7, 737–740. [Google Scholar] [CrossRef]
- Morrison, M.D.; Hanthorn, J.J.; Pratt, D.A. Synthesis of Pyrrolnitrin and Related Halogenated Phenylpyrroles. Org. Lett. 2009, 11, 1051–1054. [Google Scholar] [CrossRef]
- Bray, B.L.; Mathies, P.H.; Naef, R.; Solas, D.R.; Tidwell, T.T.; Artis, D.R.; Muchowski, J.M. N-(Triisopropylsilyl)Pyrrole. A Progenitor “Par Excellence” of 3-Substituted Pyrroles. J. Org. Chem. 1990, 55, 6317–6328. [Google Scholar] [CrossRef]
- Li, C.-S.; An, C.-Y.; Li, X.-M.; Gao, S.-S.; Cui, C.-M.; Sun, H.-F.; Wang, B.-G. Triazole and Dihydroimidazole Alkaloids from the Marine Sediment-Derived Fungus Penicillium Paneum SD-44. J. Nat. Prod. 2011, 74, 1331–1334. [Google Scholar] [CrossRef]
- Reddy, A.S.; Reddy, V.K.; Rao, G.N.; Basavaiah, K. An Efficient Total Synthesis of a Natural Penipanoid A. Tetrahedron Lett. 2022, 90, 153612. [Google Scholar] [CrossRef]
- Reddy, A.S.; Reddy, V.K.; Rao, G.N.; Basavaiah, K.; Nagulapalli Venkata, K.C. A Novel and Efficient One-Pot Strategy for the Synthesis of 1,2,4-Triazoles: Access to Synthesis of Penipanoid A and Its Analogues. RSC Adv. 2023, 13, 2680–2682. [Google Scholar] [CrossRef] [PubMed]
- Das, J.; Bhan, A.; Mandal, S.S.; Lovely, C.J. Total Syntheses and Cytotoxicity of Kealiiquinone, 2-Deoxy-2-Aminokealiiquinone and Analogs. Bioorg Med. Chem. Lett. 2013, 23, 6183–6187. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Guo, J.; Wang, Z.; Liu, Y.; Song, H.; Wang, Q. Marine Natural Products for Drug Discovery: First Discovery of Kealiinines A-C and Their Derivatives as Novel Antiviral and Antiphytopathogenic Fungus Agents. J. Agric. Food Chem. 2018, 66, 7310–7318. [Google Scholar] [CrossRef]
- Das, J.; Koswatta, P.B.; Jones, J.D.; Yousufuddin, M.; Lovely, C.J. Total Syntheses of Kealiinines A–C. Org. Lett. 2012, 14, 6210–6213. [Google Scholar] [CrossRef]
- Guo, P.; Li, G.; Liu, Y.; Lu, A.; Wang, Z.; Wang, Q. Naamines and Naamidines as Novel Agents against a Plant Virus and Phytopathogenic Fungi. Mar. Drugs 2018, 16, 311. [Google Scholar] [CrossRef]
- Ohta, S.; Tsuno, N.; Nakamura, S.; Taguchi, N.; Yamashita, M.; Kawasaki, I.; Fujieda, M. Total Syntheses of Naamine A and Naamidine A, Marine Imidazole Alkaloids. Heterocycles-Sendai Inst. Heterocycl. Chem. 2000, 53, 1939–1956. [Google Scholar] [CrossRef]
- Kawasaki, I.; Nakamura, S.; Yanagitani, S.; Kakuno, A.; Yamashita, M.; Ohta, S. New Access to 1,3-Dialkyl-2,3-Dihydro-2-Imino-1H-Imidazoles and Their Application to the First Total Synthesis of Naamine B, a Marine 2,3-Dihydro-2-Imino-1,3-Dimethyl-1H-Imidazole Alkaloid. J. Chem. Soc. Perkin Trans. 2001, 1, 3095–3099. [Google Scholar] [CrossRef]
- Ermolat’ev, D.S.; Bariwal, J.B.; Steenackers, H.P.L.; De Keersmaecker, S.C.J.; Van der Eycken, E.V. Concise and Diversity-Oriented Route toward Polysubstituted 2-Aminoimidazole Alkaloids and Their Analogues. Angew. Chem. 2010, 122, 9655–9658. [Google Scholar] [CrossRef]
- Koswatta, P.B.; Lovely, C.J. Total Syntheses of Naamidine G and 14-Methoxynaamidine G. Tetrahedron Lett. 2010, 51, 164–166. [Google Scholar] [CrossRef]
- Koswatta, P.B.; Lovely, C.J. Concise Total Synthesis of Naamine G and Naamidine H. Chem. Commun. 2010, 46, 2148. [Google Scholar] [CrossRef] [PubMed]
- Xin, Z.; OuYang, Q.; Wan, C.; Che, J.; Li, L.; Chen, J.; Tao, N. Isolation of Antofine from Cynanchum Atratum BUNGE (Asclepiadaceae) and Its Antifungal Activity against Penicillium Digitatum. Postharvest. Biol. Technol. 2019, 157, 110961. [Google Scholar] [CrossRef]
- Oh, J.; Kim, G.D.; Kim, S.; Lee, S.K. Antofine, a Natural Phenanthroindolizidine Alkaloid, Suppresses Angiogenesis via Regulation of AKT/MTOR and AMPK Pathway in Endothelial Cells and Endothelial Progenitor Cells Derived from Mouse Embryonic Stem Cells. Food Chem. Toxicol. 2017, 107, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Jiang, Y.; Liu, J.; Kang, Y.; Li, R.; Wang, J. The Anti-Tumor Activity and Mechanism of Alkaloids from Aconitum Szechenyianum Gay. Bioorg Med. Chem. Lett. 2016, 26, 380–387. [Google Scholar] [CrossRef]
- Liu, Y.; Cui, Y.; Lu, L.; Gong, Y.; Han, W.; Piao, G. Natural Indole-containing Alkaloids and Their Antibacterial Activities. Arch. Pharm. 2020, 353, 2000120. [Google Scholar] [CrossRef]
- Wang, M.; Zhang, W.; Wang, J.; Lu, J.; Huo, Y. Extraction Separation and Antifungal Activities of Cynanchum Komarovii Al. Iljinski. World Sci. Res. J. 2023, 9, 52–59. [Google Scholar]
- Chen, C.; Qi, W.; Peng, X.; Chen, J.; Wan, C. Inhibitory Effect of 7-Demethoxytylophorine on Penicillium Italicum and Its Possible Mechanism. Microorganisms 2019, 7, 36. [Google Scholar] [CrossRef]
- Yi, M.; Gu, P.; Kang, X.Y.; Sun, J.; Li, R.; Li, X.Q. Total Synthesis of (±)-Antofine. Tetrahedron Lett. 2014, 55, 105–107. [Google Scholar] [CrossRef]
- Ansari, A.; Ramapanicker, R. Enantioselective Synthesis of (R)-Antofine and (R)-Cryptopleurine. ChemistrySelect 2018, 3, 12591–12594. [Google Scholar] [CrossRef]
- El-Gendy, M.M.A.; Shaaban, M.; Shaaban, K.A.; El-Bondkly, A.M.; Laatsch, H. Essramycin: A First Triazolopyrimidine Antibiotic Isolated from Nature†. J. Antibiot. 2008, 61, 149–157. [Google Scholar] [CrossRef]
- Battaglia, U.; Moody, C.J. A Short Synthesis of the Triazolopyrimidine Antibiotic Essramycin. J. Nat. Prod. 2010, 73, 1938–1939. [Google Scholar] [CrossRef] [PubMed]
- Abu-Hashem, A.A.; Gouda, M.A. Synthesis and Antimicrobial Activity of Some Novel Quinoline, Chromene, Pyrazole Derivatives Bearing Triazolopyrimidine Moiety. J. Heterocycl. Chem. 2017, 54, 850–858. [Google Scholar] [CrossRef]
- Tee, E.H.L.; Karoli, T.; Ramu, S.; Huang, J.X.; Butler, M.S.; Cooper, M.A. Synthesis of Essramycin and Comparison of Its Antibacterial Activity. J. Nat. Prod. 2010, 73, 1940–1942. [Google Scholar] [CrossRef]
- Wang, T.; Yang, S.; Li, H.; Lu, A.; Wang, Z.; Yao, Y.; Wang, Q. Discovery, Structural Optimization, and Mode of Action of Essramycin Alkaloid and Its Derivatives as Anti-Tobacco Mosaic Virus and Anti-Phytopathogenic Fungus Agents. J. Agric. Food Chem. 2020, 68, 471–484. [Google Scholar] [CrossRef]
- Shtark, O.Y.; Shaposhnikov, A.I.; Kravchenko, L.V. The Production of Antifungal Metabolites by Pseudomonas Chlororaphis Grown on Different Nutrient Sources. Microbiology 2003, 72, 574–578. [Google Scholar] [CrossRef]
- Niu, J.; Chen, J.; Xu, Z.; Zhu, X.; Wu, Q.; Li, J. Synthesis and Bioactivities of Amino Acid Ester Conjugates of Phenazine-1-Carboxylic Acid. Bioorg Med. Chem. Lett. 2016, 26, 5384–5386. [Google Scholar] [CrossRef]
- Xiong, Y.; Zhu, X.; Hu, J.; Wang, Y.; Du, X.; Li, J.; Wu, Q. Effect of Introducing Amino Acids into Phenazine-1-Carboxylic Acid on Phloem Mobility. Nat. Prod. Res. 2021, 35, 4373–4379. [Google Scholar] [CrossRef]
- Zhong, Y.; Liu, J.; Cheng, X.; Zhang, H.; Zhang, C.; Xia, Z.; Wu, Z.; Zhang, L.; Zheng, Y.; Gao, Z.; et al. Design, synthesis and biological evaluations of diverse Michael acceptor-based phenazine hybrid molecules as TrxR1 inhibitors. Bioorg Chem. 2021, 109, 104736. [Google Scholar] [CrossRef]
- Lv, P.; Chen, Y.; Shi, T.; Wu, X.; Li, Q.X.; Hua, R. Synthesis and fungicidal activities of sanguinarine derivatives. Pestic. Biochem. Physiol. 2018, 147, 3–10. [Google Scholar] [CrossRef]
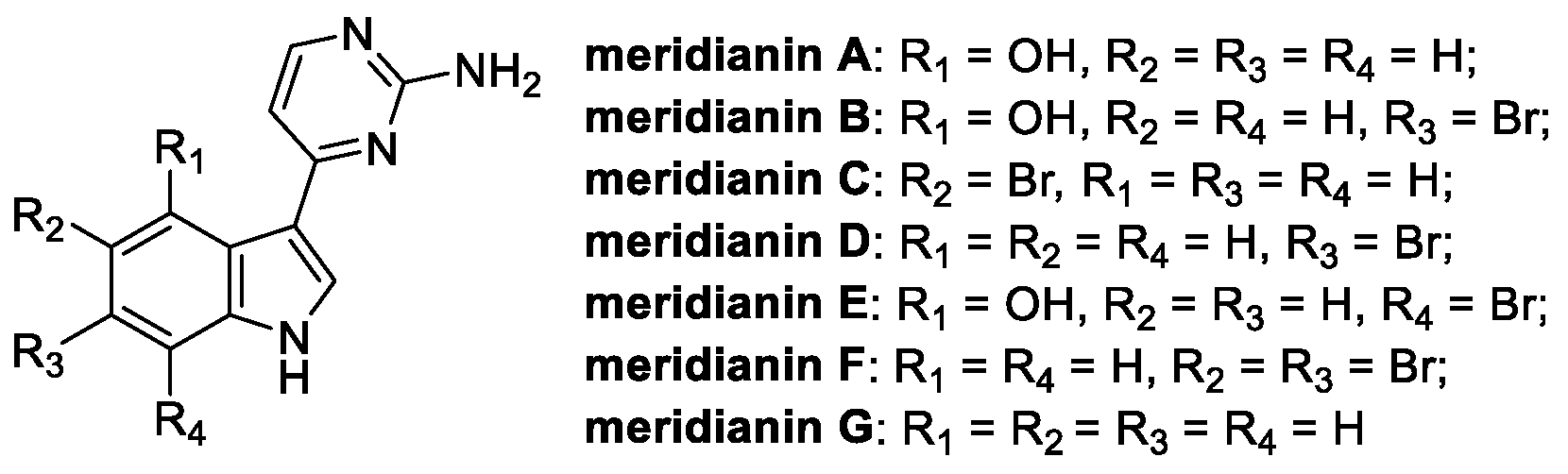
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).