
Histone Deacetylase Inhibitors Promote the Anticancer Activity of Cisplatin: Mechanisms and Potential

 , ,
, ,
Abstract

1. Introduction
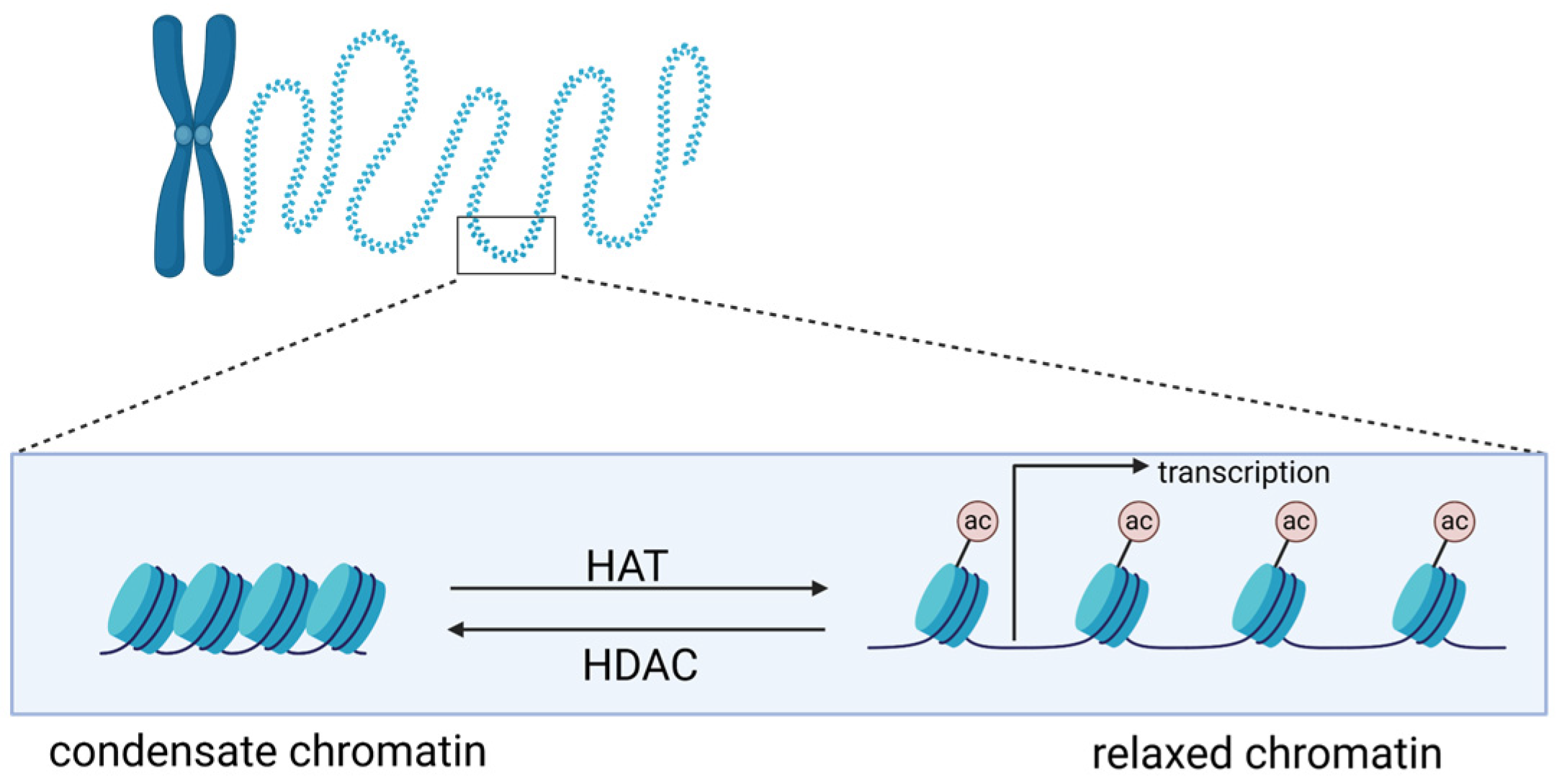
2. Histones and Histone Acetylation
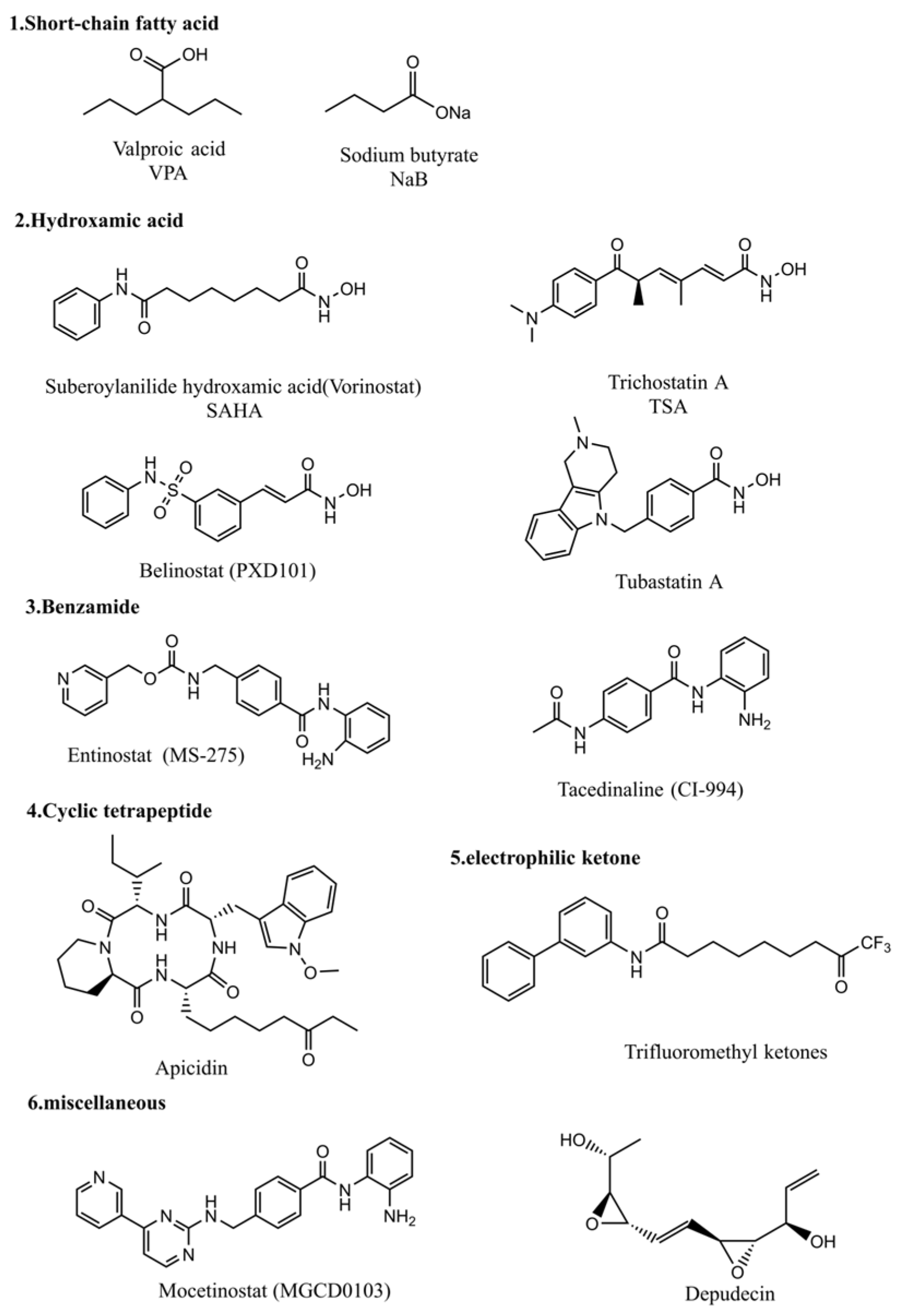
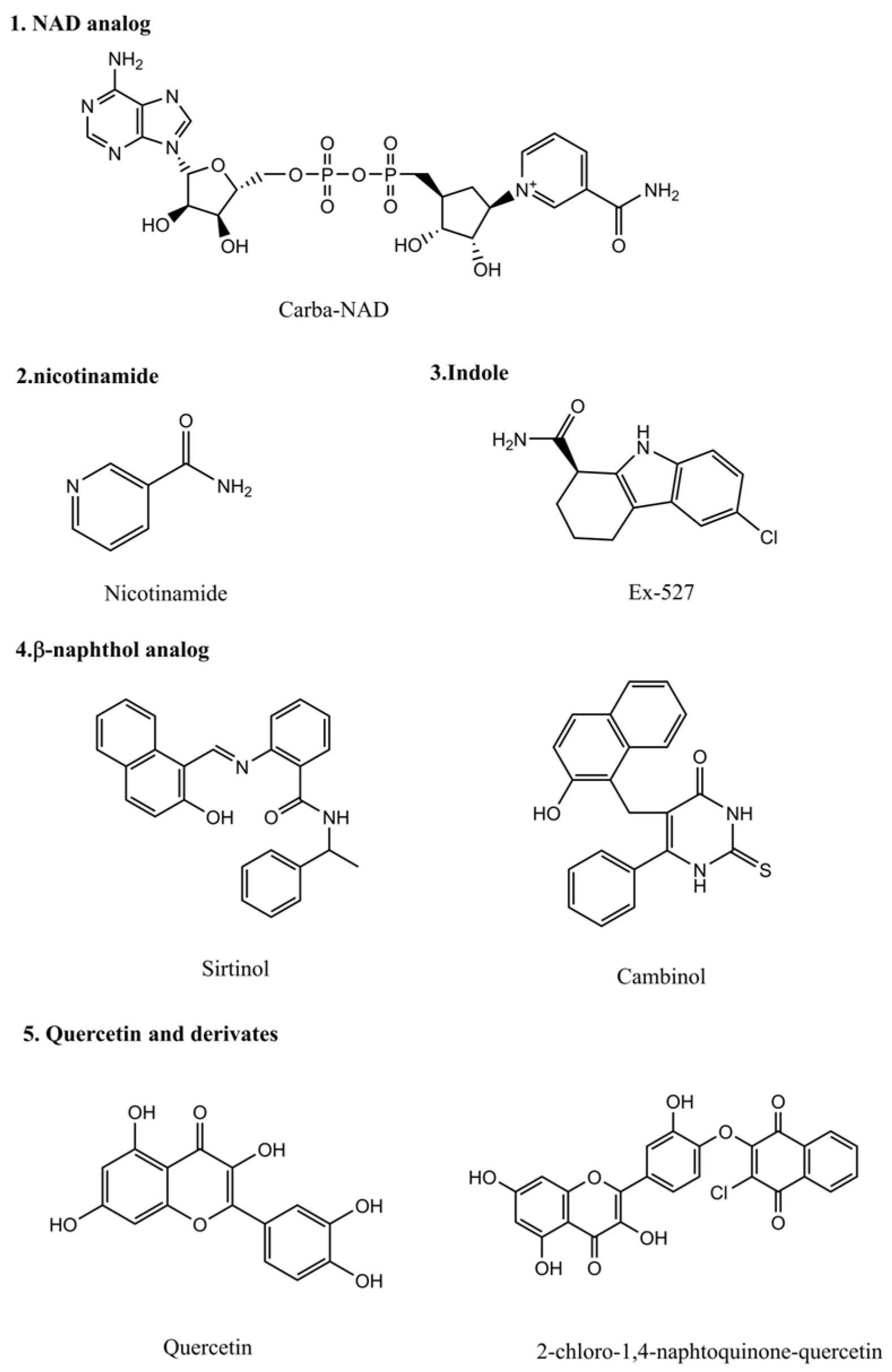
3. Histone Deacetylase Inhibitors
4. HDAC Inhibitors Promote Cisplatin Cytotoxicity
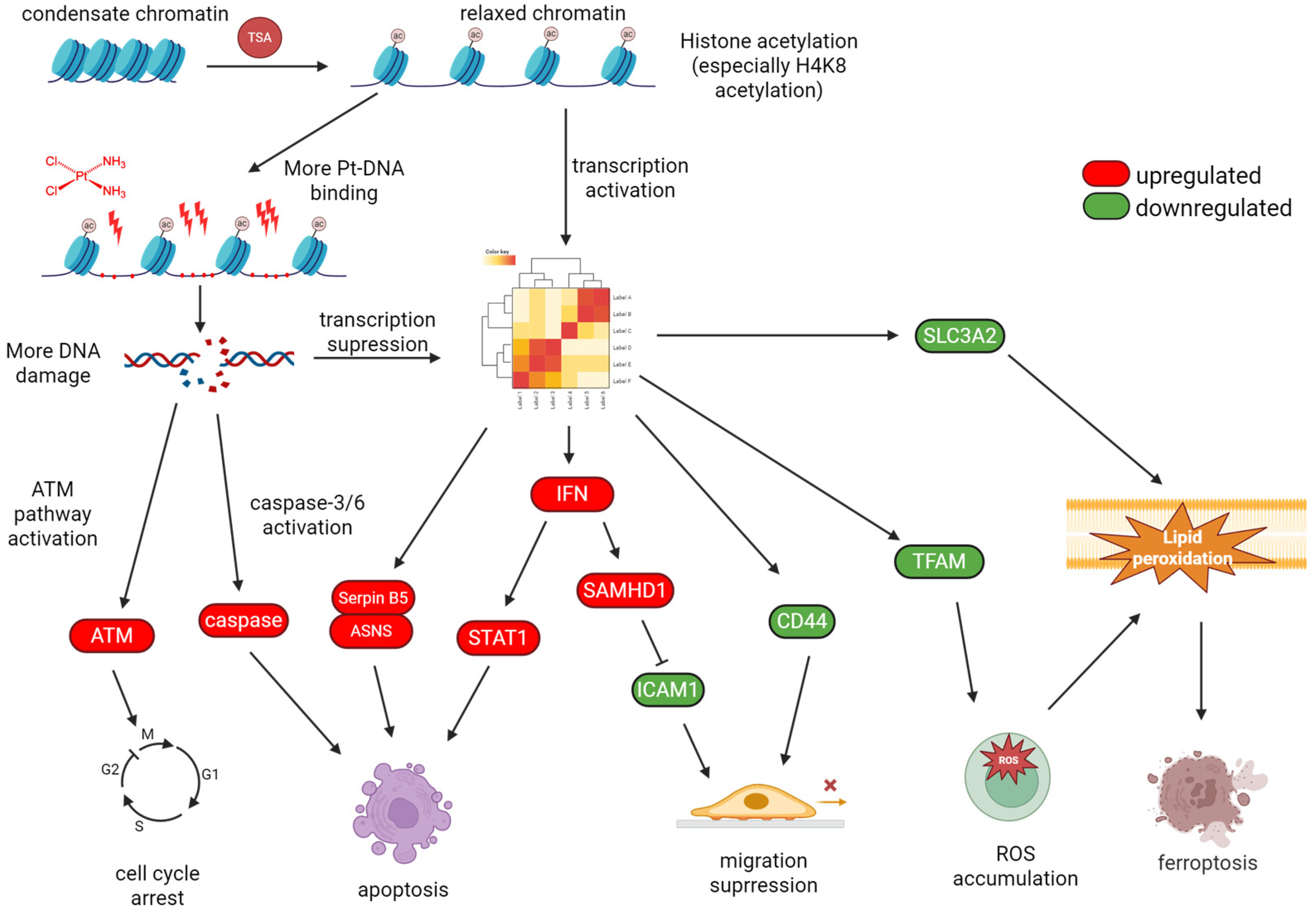
4.1. TSA
4.2. SAHA
4.3. VPA
4.4. Others
5. Perspectives and Remarks
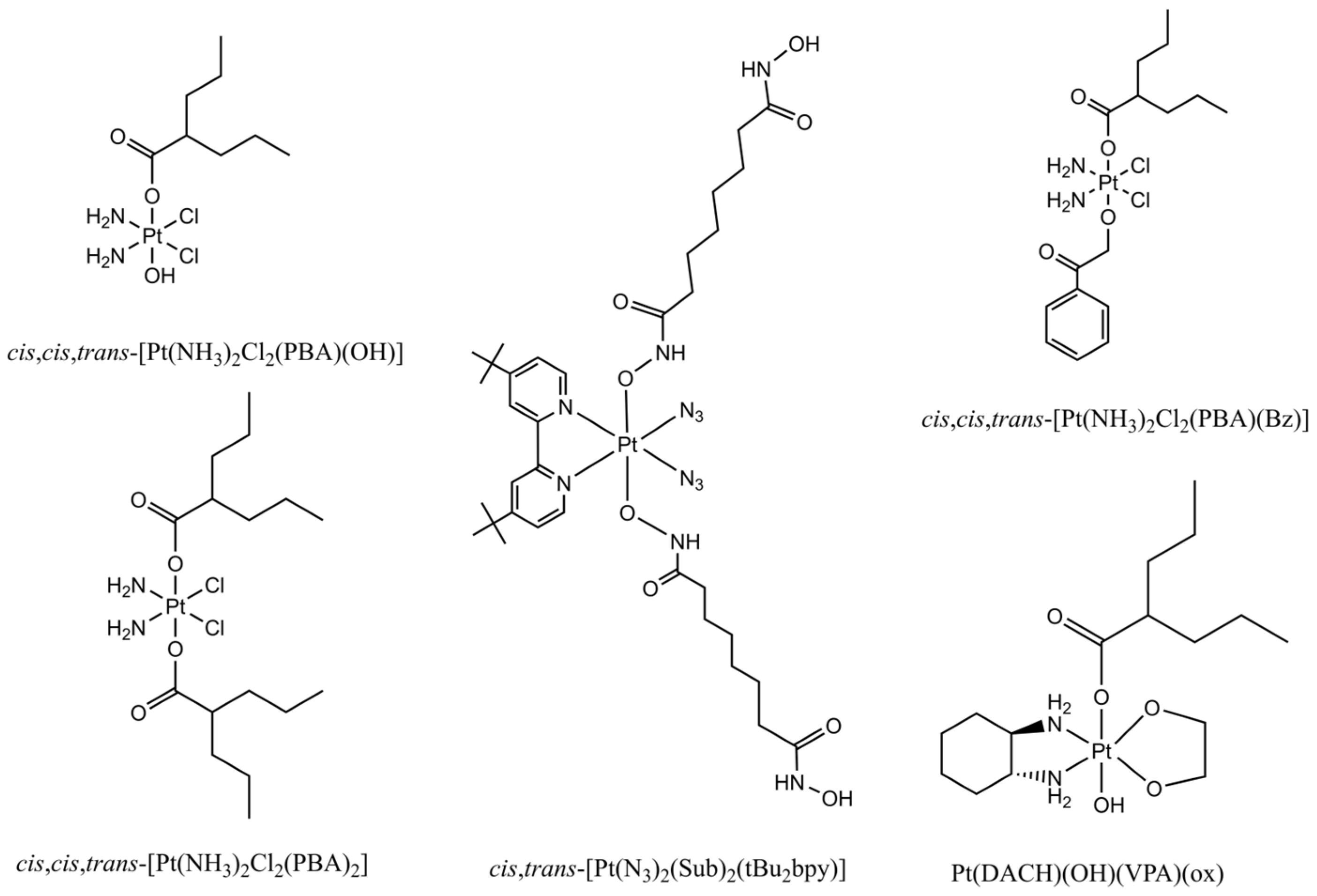
5.1. Conjugation of HDAC Inhibitors with Pt(IV) Prodrugs for Multitargeting Chemotherapy
5.2. HDACi Promote the Effectiveness of Other Chemotherapeutics and Radiotherapy
5.3. HAT Inhibitor Also Enhances Cisplatin Anticancer Effect
6. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Peyrone, M. Ueber die Einwirkung des Ammoniaks auf Platinchlorür. Justus Liebigs Ann. Chem. 1844, 51, 1–29. [Google Scholar] [CrossRef]
- Rosenberg, B.; Van Camp, L.; Krigas, T. Inhibition of Cell Division in Escherichia coli by Electrolysis Products from a Platinum Electrode. Nature 1965, 205, 698–699. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, B.; Vancamp, L.; Trosko, J.E.; Mansour, V.H. Platinum Compounds: A New Class of Potent Antitumour Agents. Nature 1969, 222, 385–386. [Google Scholar] [CrossRef] [PubMed]
- Romani, A.M.P. Cisplatin in cancer treatment. Biochem. Pharmacol. 2022, 206, 115323. [Google Scholar] [CrossRef]
- Cutillas, N.; Martínez, A.; Yellol, G.S.; Rodríguez, V.; Zamora, A.; Pedreño, M.; Donaire, A.; Janiak, C.; Ruiz, J. Anticancer C,N-Cycloplatinated(II) Complexes Containing Fluorinated Phosphine Ligands: Synthesis, Structural Characterization, and Biological Activity. Inorg. Chem. 2013, 52, 13529–13535. [Google Scholar] [CrossRef]
- Jamieson, E.R.; Lippard, S.J. Structure, Recognition, and Processing of Cisplatin−DNA Adducts. Chem. Rev. 1999, 99, 2467–2498. [Google Scholar] [CrossRef]
- Jung, Y.; Lippard, S.J. Direct Cellular Responses to Platinum-Induced DNA Damage. Chem. Rev. 2007, 107, 1387–1407. [Google Scholar] [CrossRef]
- Wang, D.; Lippard, S.J. Cellular processing of platinum anticancer drugs. Nat. Rev. Drug Discov. 2005, 4, 307–320. [Google Scholar] [CrossRef]
- Chu, G. Cellular responses to cisplatin. The roles of DNA-binding proteins and DNA repair. J. Biol. Chem. 1994, 269, 787–790. [Google Scholar] [CrossRef]
- Marullo, R.; Werner, E.; Degtyareva, N.; Moore, B.; Altavilla, G.; Ramalingam, S.S.; Doetsch, P.W. Cisplatin Induces a Mitochondrial-ROS Response That Contributes to Cytotoxicity Depending on Mitochondrial Redox Status and Bioenergetic Functions. PLoS ONE 2013, 8, e81162. [Google Scholar] [CrossRef]
- Qi, L.; Luo, Q.; Zhang, Y.; Jia, F.; Zhao, Y.; Wang, F. Advances in Toxicological Research of the Anticancer Drug Cisplatin. Chem. Res. Toxicol. 2019, 32, 1469–1486. [Google Scholar] [CrossRef]
- Peng, K.; Liang, B.-B.; Liu, W.; Mao, Z.-W. What blocks more anticancer platinum complexes from experiment to clinic: Major problems and potential strategies from drug design perspectives. Coord. Chem. Rev. 2021, 449, 214210. [Google Scholar] [CrossRef]
- Asgar, M.A.; Senawong, G.; Sripa, B.; Senawong, T. Synergistic anticancer effects of cisplatin and histone deacetylase inhibitors (SAHA and TSA) on cholangiocarcinoma cell lines. Int. J. Oncol. 2016, 48, 409–420. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Blake, M.; Baek, J.H.; Kohlhagen, G.; Pommier, Y.; Carrier, F. Inhibition of Histone Deacetylase Increases Cytotoxicity to Anticancer Drugs Targeting DNA. Cancer Res. 2003, 63, 7291–7300. [Google Scholar] [PubMed]
- Williams, R.J. Trichostatin A, an inhibitor of histone deacetylase, inhibits hypoxia-induced angiogenesis. Expert Opin. Investig. Drugs 2001, 10, 1571–1573. [Google Scholar] [CrossRef]
- Schölz, C.; Weinert, B.T.; Wagner, S.A.; Beli, P.; Miyake, Y.; Qi, J.; Jensen, L.J.; Streicher, W.; McCarthy, A.R.; Westwood, N.J.; et al. Acetylation site specificities of lysine deacetylase inhibitors in human cells. Nat. Biotechnol. 2015, 33, 415–423. [Google Scholar] [CrossRef]
- Zhou, Y.; Luo, Q.; Zeng, F.; Liu, X.; Han, J.; Gu, L.; Tian, X.; Zhang, Y.; Zhao, Y.; Wang, F. Trichostatin A Promotes Cytotoxicity of Cisplatin, as Evidenced by Enhanced Apoptosis/Cell Death Markers. Molecules 2024, 29, 2623. [Google Scholar] [CrossRef]
- Kornberg, R.D. Structure of Chromatin. Annu. Rev. Biochem. 1977, 46, 931–954. [Google Scholar] [CrossRef]
- McGhee, J.D.; Felsenfeld, G. Nucleosome Structure. Annu. Rev. Biochem. 1980, 49, 1115–1156. [Google Scholar] [CrossRef]
- Luger, K.; Mäder, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 1997, 389, 251–260. [Google Scholar] [CrossRef]
- Jenuwein, T.; Allis, C.D. Translating the Histone Code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef] [PubMed]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef]
- Roth, S.Y.; Denu, J.M.; Allis, C.D. Histone Acetyltransferases. Annu. Rev. Biochem. 2001, 70, 81–120. [Google Scholar] [CrossRef] [PubMed]
- Thiagalingam, S.A.M.; Cheng, K.-H.; Lee, H.J.; Mineva, N.; Thiagalingam, A.; Ponte, J.F. Histone Deacetylases: Unique Players in Shaping the Epigenetic Histone Code. Ann. N. Y. Acad. Sci. 2003, 983, 84–100. [Google Scholar] [CrossRef] [PubMed]
- Guardiola, A.R.; Yao, T.-P. Molecular Cloning and Characterization of a Novel Histone Deacetylase HDAC10. J. Biol. Chem. 2002, 277, 3350–3356. [Google Scholar] [CrossRef]
- Bhalla, K.N. Epigenetic and Chromatin Modifiers As Targeted Therapy of Hematologic Malignancies. J. Clin. Oncol. 2005, 23, 3971–3993. [Google Scholar] [CrossRef]
- Chang, J.; Varghese, D.S.; Gillam, M.C.; Peyton, M.; Modi, B.; Schiltz, R.L.; Girard, L.; Martinez, E.D. Differential response of cancer cells to HDAC inhibitors trichostatin A and depsipeptide. Br. J. Cancer 2012, 106, 116–125. [Google Scholar] [CrossRef]
- Zhang, Y.; Kwon, S.; Yamaguchi, T.; Cubizolles, F.; Rousseaux, S.; Kneissel, M.; Cao, C.; Li, N.; Cheng, H.-L.; Chua, K.; et al. Mice Lacking Histone Deacetylase 6 Have Hyperacetylated Tubulin but Are Viable and Develop Normally. Mol. Cell. Biol. 2008, 28, 1688–1701. [Google Scholar] [CrossRef]
- Zeyen, P.; Zeyn, Y.; Herp, D.; Mahmoudi, F.; Yesiloglu, T.Z.; Erdmann, F.; Schmidt, M.; Robaa, D.; Romier, C.; Ridinger, J.; et al. Identification of histone deacetylase 10 (HDAC10) inhibitors that modulate autophagy in transformed cells. Eur. J. Med. Chem. 2022, 234, 114272. [Google Scholar] [CrossRef]
- Gao, L.; Cueto, M.A.; Asselbergs, F.; Atadja, P. Cloning and Functional Characterization of HDAC11, a Novel Member of the Human Histone Deacetylase Family. J. Biol. Chem. 2002, 277, 25748–25755. [Google Scholar] [CrossRef]
- Sui, L.; Zhang, S.; Huang, R.; Li, Z. HDAC11 promotes meiotic apparatus assembly during mouse oocyte maturation via decreasing H4K16 and α-tubulin acetylation. Cell Cycle 2020, 19, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Sui, L.; Fu, C.; Zhai, Y.; Dai, X.; Zhang, S.; Li, Z. HDAC11 inhibition disrupts porcine oocyte meiosis via regulating α-tubulin acetylation and histone modifications. Aging 2021, 13, 8849–8864. [Google Scholar] [CrossRef]
- Jin, H.; Liang, L.; Liu, L.; Deng, W.; Liu, J. HDAC inhibitor DWP0016 activates p53 transcription and acetylation to inhibit cell growth in U251 glioblastoma cells. J. Cell. Biochem. 2013, 114, 1498–1509. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Yu, W.; Wu, L.; Yang, W.; Lü, Y. Chitotriosidase attenuates brain inflammation via HDAC3/NF-κB pathway in D-galactose and aluminum-induced rat model with cognitive impairments. Neurosci. Res. 2021, 172, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yamashita, H.; Toyama, T.; Sugiura, H.; Ando, Y.; Mita, K.; Hamaguchi, M.; Hara, Y.; Kobayashi, S.; Iwase, H. Quantitation of HDAC1 mRNA Expression in Invasive Carcinoma of the Breast. Breast Cancer Res. Treat. 2005, 94, 11–16. [Google Scholar] [CrossRef]
- Halkidou, K.; Gaughan, L.; Cook, S.; Leung, H.Y.; Neal, D.E.; Robson, C.N. Upregulation and Nuclear Recruitment of HDAC1 in Hormone Refractory Prostate Cancer. The Prostate 2004, 59, 177–189. [Google Scholar] [CrossRef]
- Choi, J.-H.; Kwon, H.J.; Yoon, B., II; Kim, J.-H.; Han, S.U.; Joo, H.J.; Kim, D.-Y. Expression Profile of Histone Deacetylase 1 in Gastric Cancer Tissues. Jpn. J. Cancer Res. 2001, 92, 1300–1304. [Google Scholar] [CrossRef]
- Wilson, A.J.; Byun, D.-S.; Popova, N.; Murray, L.B.; L’Italien, K.; Sowa, Y.; Arango, D.; Velcich, A.; Augenlicht, L.H.; Mariadason, J.M. Histone Deacetylase 3 (HDAC3) and Other Class I HDACs Regulate Colon Cell Maturation and p21 Expression and Are Deregulated in Human Colon Cancer. J. Biol. Chem. 2006, 281, 13548–13558. [Google Scholar] [CrossRef]
- Grunstein, M. Histone acetylation in chromatin structure and transcription. Nature 1997, 389, 349–352. [Google Scholar] [CrossRef]
- Cheung, W.L.; Briggs, S.D.; Allis, C.D. Acetylation and chromosomal functions. Curr. Opin. Cell Biol. 2000, 12, 326–333. [Google Scholar] [CrossRef]
- Yang, X.-J.; Seto, E. Lysine Acetylation: Codified Crosstalk with Other Posttranslational Modifications. Mol. Cell 2008, 31, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela-Fernández, A.; Cabrero, J.R.; Serrador, J.M.; Sánchez-Madrid, F. HDAC6: A key regulator of cytoskeleton, cell migration and cell–cell interactions. Trends Cell Biol. 2008, 18, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Witt, O.; Deubzer, H.E.; Milde, T.; Oehme, I. HDAC family: What are the cancer relevant targets? Cancer Lett. 2009, 277, 8–21. [Google Scholar] [CrossRef]
- Senese, S.; Zaragoza, K.; Minardi, S.; Muradore, I.; Ronzoni, S.; Passafaro, A.; Bernard, L.; Draetta, G.F.; Alcalay, M.; Seiser, C.; et al. Role for Histone Deacetylase 1 in Human Tumor Cell Proliferation. Mol. Cell. Biol. 2007, 27, 4784–4795. [Google Scholar] [CrossRef]
- Inoue, S.; Mai, A.; Dyer, M.J.S.; Cohen, G.M. Inhibition of Histone Deacetylase Class I but not Class II Is Critical for the Sensitization of Leukemic Cells to Tumor Necrosis Factor–Related Apoptosis-Inducing Ligand–Induced Apoptosis. Cancer Res. 2006, 66, 6785–6792. [Google Scholar] [CrossRef] [PubMed]
- Harms, K.L.; Chen, X. Histone Deacetylase 2 Modulates p53 Transcriptional Activities through Regulation of p53-DNA Binding Activity. Cancer Res. 2007, 67, 3145–3152. [Google Scholar] [CrossRef]
- Spurling, C.C.; Godman, C.A.; Noonan, E.J.; Rasmussen, T.P.; Rosenberg, D.W.; Giardina, C. HDAC3 overexpression and colon cancer cell proliferation and differentiation. Mol. Carcinog. 2008, 47, 137–147. [Google Scholar] [CrossRef]
- Lagger, G.; O’Carroll, D.; Rembold, M.; Khier, H.; Tischler, J.; Weitzer, G.; Schuettengruber, B.; Hauser, C.; Brunmeir, R.; Jenuwein, T.; et al. Essential function of histone deacetylase 1 in proliferation control and CDK inhibitor repression. EMBO J. 2002, 21, 2672–2681. [Google Scholar] [CrossRef]
- Keshelava, N.; Davicioni, E.; Wan, Z.; Ji, L.; Sposto, R.; Triche, T.J.; Reynolds, C.P. Histone Deacetylase 1 Gene Expression and Sensitization of Multidrug-Resistant Neuroblastoma Cell Lines to Cytotoxic Agents by Depsipeptide. J. Natl. Cancer Inst. 2007, 99, 1107–1119. [Google Scholar] [CrossRef]
- Qian, D.Z.; Kachhap, S.K.; Collis, S.J.; Verheul, H.M.W.; Carducci, M.A.; Atadja, P.; Pili, R. Class II Histone Deacetylases Are Associated with VHL-Independent Regulation of Hypoxia-Inducible Factor 1α. Cancer Res. 2006, 66, 8814–8821. [Google Scholar] [CrossRef]
- Shan, B.; Yao, T.-P.; Nguyen, H.T.; Zhuo, Y.; Levy, D.R.; Klingsberg, R.C.; Tao, H.; Palmer, M.L.; Holder, K.N.; Lasky, J.A. Requirement of HDAC6 for Transforming Growth Factor-β1-induced Epithelial-Mesenchymal Transition. J. Biol. Chem. 2008, 283, 21065–21073. [Google Scholar] [CrossRef] [PubMed]
- Haggarty, S.J.; Koeller, K.M.; Wong, J.C.; Grozinger, C.M.; Schreiber, S.L. Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proc. Natl. Acad. Sci. USA 2003, 100, 4389–4394. [Google Scholar] [CrossRef]
- Watamoto, K.; Towatari, M.; Ozawa, Y.; Miyata, Y.; Okamoto, M.; Abe, A.; Naoe, T.; Saito, H. Altered interaction of HDAC5 with GATA-1 during MEL cell differentiation. Oncogene 2003, 22, 9176–9184. [Google Scholar] [CrossRef]
- Mottet, D.; Bellahcène, A.; Pirotte, S.; Waltregny, D.; Deroanne, C.; Lamour, V.; Lidereau, R.; Castronovo, V. Histone Deacetylase 7 Silencing Alters Endothelial Cell Migration, a Key Step in Angiogenesis. Circ. Res. 2007, 101, 1237–1246. [Google Scholar] [CrossRef]
- Oehme, I.; Deubzer, H.E.; Wegener, D.; Pickert, D.; Linke, J.-P.; Hero, B.; Kopp-Schneider, A.; Westermann, F.; Ulrich, S.M.; von Deimling, A.; et al. Histone Deacetylase 8 in Neuroblastoma Tumorigenesis. Clin. Cancer Res. 2008, 15, 91–99. [Google Scholar] [CrossRef]
- Yeon, M.; Kwon, N.; Jeoung, J.; Jeoung, D. HDAC9 and miR-512 Regulate CAGE-Promoted Anti-Cancer Drug Resistance and Cellular Proliferation. Curr. Issues Mol. Biol. 2024, 46, 5178–5193. [Google Scholar] [CrossRef] [PubMed]
- Salgado, E.; Bian, X.; Feng, A.; Shim, H.; Liang, Z. HDAC9 overexpression confers invasive and angiogenic potential to triple negative breast cancer cells via modulating microRNA-206. Biochem. Biophys. Res. Commun. 2018, 503, 1087–1091. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Piao, C.; Zhang, Z.; Zhu, Y.; Sun, S.; Bi, J.; Kong, C.; Ju, M. Decreased expression and hypomethylation of HDAC9 lead to poor prognosis and inhibit immune cell infiltration in clear cell renal cell carcinoma. Urol. Oncol. 2020, 38, 740.e741–740.e749. [Google Scholar] [CrossRef]
- Shi, Z.; Jiang, T.; Sun, X.; Peng, L.; Cao, B.; Wang, Y. HDAC10 inhibits non-small-cell lung cancer cell ferroptosis through the microRNA-223-5p-SLC7A11 axis. Toxicol. Res. 2024, 13, tfae164. [Google Scholar] [CrossRef]
- Guo, H.; Ren, H.; Han, K.; Li, J.; Dong, Y.; Zhao, X.; Li, C. Knockdown of HDAC10 inhibits POLE2-mediated DNA damage repair in NSCLC cells by increasing SP1 acetylation levels. Pulm. Pharmacol. Ther. 2023, 83, 102250. [Google Scholar] [CrossRef]
- Tao, X.; Yan, Y.; Lu, L.; Chen, B. HDAC10 expression is associated with DNA mismatch repair gene and is a predictor of good prognosis in colon carcinoma. Oncol. Lett. 2017, 14, 4923–4929. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Tong, X.; Hu, W.; Chen, D. HDAC11: A novel target for improved cancer therapy. Biomed. Pharmacother. 2023, 166, 115418. [Google Scholar] [CrossRef]
- Bora-Singhal, N.; Mohankumar, D.; Saha, B.; Colin, C.M.; Lee, J.Y.; Martin, M.W.; Zheng, X.; Coppola, D.; Chellappan, S. Novel HDAC11 inhibitors suppress lung adenocarcinoma stem cell self-renewal and overcome drug resistance by suppressing Sox2. Sci. Rep. 2020, 10, 4722. [Google Scholar] [CrossRef]
- Zhang, L.; Bu, L.; Hu, J.; Xu, Z.; Ruan, L.; Fang, Y.; Wang, P. HDAC1 knockdown inhibits invasion and induces apoptosis in non-small cell lung cancer cells. Biol. Chem. 2018, 399, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Ding, S.; Huang, H.; Luo, P.; Qing, B.; Zhang, S.; Tang, R. HDAC1 triggers the proliferation and migration of breast cancer cells via upregulation of interleukin-8. Biol. Chem. 2017, 398, 1347–1356. [Google Scholar] [CrossRef]
- Darvishi, N.; Rahimi, K.; Mansouri, K.; Fathi, F.; Menbari, M.-N.; Mohammadi, G.; Abdi, M. MiR-646 prevents proliferation and progression of human breast cancer cell lines by suppressing HDAC2 expression. Mol. Cell. Probes 2020, 53, 101649. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Peng, C.; Jin, C.; Wang, Y.; Wang, T.; Yang, P.; Peng, W.; Sun, Q.; Xu, H.; Nie, H.; et al. PJA2 Suppresses Colorectal Cancer Progression by Controlling HDAC2 Degradation and Stability. Adv. Sci. 2025, 12, 2401964. [Google Scholar] [CrossRef]
- Tong, J.; Shen, Y.; Chen, X.; Wang, R.; Hu, Y.; Zhang, X.; Zhang, Z.; Han, L. FKBP3 mediates oxaliplatin resistance in colorectal cancer cells by regulating HDAC2 expression. Oncol. Rep. 2019, 42, 1404–1412. [Google Scholar] [CrossRef]
- Huang, R.; Langdon, S.P.; Tse, M.; Mullen, P.; Um, I.H.; Faratian, D.; Harrison, D.J. The role of HDAC2 in chromatin remodelling and response to chemotherapy in ovarian cancer. Oncotarget 2015, 7, 4695–4711. [Google Scholar] [CrossRef]
- Zhuang, H.; Zhang, Z.; Wang, W.; Qu, H. RNF144B-mediated p21 degradation regulated by HDAC3 contribute to enhancing ovarian cancer growth and metastasis. Tissue Cell 2024, 86, 102277. [Google Scholar] [CrossRef]
- Chen, D.-Q.; Yu, C.; Zhang, X.-F.; Liu, Z.-F.; Wang, R.; Jiang, M.; Chen, H.; Yan, F.; Tao, M.; Chen, L.-B.; et al. HDAC3-mediated silencing of miR-451 decreases chemosensitivity of patients with metastatic castration-resistant prostate cancer by targeting NEDD9. Ther. Adv. Med. Oncol. 2018, 2018, 1758835918783132. [Google Scholar] [CrossRef]
- Wang, T.; Lu, Z.; Han, T.; Wang, Y.; Gan, M.; Wang, J.-B. Deacetylation of Glutaminase by HDAC4 contributes to Lung Cancer Tumorigenesis. Int. J. Biol. Sci. 2022, 18, 4452–4465. [Google Scholar] [CrossRef] [PubMed]
- Kaowinn, S.; Kaewpiboon, C.; Koh, S.S.; Krämer, O.H.; Chung, Y.H. STAT1-HDAC4 signaling induces epithelial-mesenchymal transition and sphere formation of cancer cells overexpressing the oncogene, CUG2. Oncol. Rep. 2018, 40, 2619–2627. [Google Scholar] [CrossRef]
- OuYang, C.; Shu, G.; Liu, J.; Deng, S.; Lu, P.; Li, Y.; Gan, Y.; Xie, B.; Liu, J.; Yin, G. HDAC5, negatively regulated by miR-148a-3p, promotes colon cancer cell migration. Cancer Sci. 2022, 113, 2560–2574. [Google Scholar] [CrossRef]
- Huang, W.-T.; Tsai, Y.-H.; Chen, S.-H.; Kuo, C.-W.; Kuo, Y.-L.; Lee, K.-T.; Chen, W.-C.; Wu, P.C.; Chuang, C.-Y.; Cheng, S.M.; et al. HDAC2 and HDAC5 Up-Regulations Modulate Survivin and miR-125a-5p Expressions and Promote Hormone Therapy Resistance in Estrogen Receptor Positive Breast Cancer Cells. Front. Pharmacol. 2017, 8, 902. [Google Scholar] [CrossRef]
- Lei, Y.; Liu, L.; Zhang, S.; Guo, S.; Li, X.; Wang, J.; Su, B.; Fang, Y.; Chen, X.; Ke, H.; et al. Hdac7 promotes lung tumorigenesis by inhibiting Stat3 activation. Mol. Cancer 2017, 16, 170. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Liu, H.; Hou, S.; Wu, L.; Yang, Z.; Shen, J.; Zhou, L.; Zheng, S.S.; Jiang, B. MiR-489 suppresses tumor growth and invasion by targeting HDAC7 in colorectal cancer. Clin. Transl. Oncol. 2018, 20, 703–712. [Google Scholar] [CrossRef] [PubMed]
- Deng, Q.; Yang, J.; Chen, Y.; Chen, Z.; Li, J.; Fu, Z. LncRNA HOXB-AS4 promotes proliferation and migration of colorectal cancer via the miR-140-5p/hdac7 axis. Biotechnol. Genet. Eng. Rev. 2024, 40, 1262–1280. [Google Scholar] [CrossRef]
- Wu, J.; Du, C.; Lv, Z.; Ding, C.; Cheng, J.; Xie, H.; Zhou, L.; Zheng, S. The Up-Regulation of Histone Deacetylase 8 Promotes Proliferation and Inhibits Apoptosis in Hepatocellular Carcinoma. Dig. Dis. Sci. 2013, 58, 3545–3553. [Google Scholar] [CrossRef]
- Kang, Y.; Nian, H.; Rajendran, P.; Kim, E.; Dashwood, W.M.; Pinto, J.T.; Boardman, L.A.; Thibodeau, S.N.; Limburg, P.J.; Löhr, C.V.; et al. HDAC8 and STAT3 repress BMF gene activity in colon cancer cells. Cell Death Dis. 2014, 5, e1476. [Google Scholar] [CrossRef]
- Park, S.Y.; Jun, J.A.; Jeong, K.J.; Heo, H.J.; Sohn, J.S.; Lee, H.Y.; Park, C.G.; Kang, J. Histone deacetylases 1, 6 and 8 are critical for invasion in breast cancer. Oncol. Rep. 2011, 25, 1677–1681. [Google Scholar] [CrossRef] [PubMed]
- Ridinger, J.; Koeneke, E.; Kolbinger, F.R.; Koerholz, K.; Mahboobi, S.; Hellweg, L.; Gunkel, N.; Miller, A.K.; Peterziel, H.; Schmezer, P.; et al. Dual role of HDAC10 in lysosomal exocytosis and DNA repair promotes neuroblastoma chemoresistance. Sci. Rep. 2018, 8, 10039. [Google Scholar] [CrossRef]
- Bi, L.; Ren, Y.; Feng, M.; Meng, P.; Wang, Q.; Chen, W.; Jiao, Q.; Wang, Y.; Du, L.; Zhou, F.; et al. HDAC11 Regulates Glycolysis through the LKB1/AMPK Signaling Pathway to Maintain Hepatocellular Carcinoma Stemness. Cancer Res. 2021, 81, 2015–2028. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Zhang, X.; Li, X.; Tian, L.; Shen, S.; Ma, J.; Ai, F. Histone deacetylase (HDAC) 11 inhibits matrix metalloproteinase (MMP) 3 expression to suppress colorectal cancer metastasis. J. Cancer 2022, 13, 1923–1932. [Google Scholar] [CrossRef]
- Leslie, P.L.; Chao, Y.L.; Tsai, Y.-H.; Ghosh, S.K.; Porrello, A.; Van Swearingen, A.E.D.; Harrison, E.B.; Cooley, B.C.; Parker, J.S.; Carey, L.A.; et al. Histone deacetylase 11 inhibition promotes breast cancer metastasis from lymph nodes. Nat. Commun. 2019, 10, 4192. [Google Scholar] [CrossRef]
- Luo, J.; Nikolaev, A.Y.; Imai, S.-I.; Chen, D.; Su, F.; Shiloh, A.; Guarente, L.; Gu, W. Negative Control of p53 by Sir2α Promotes Cell Survival under Stress. Cell 2001, 107, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, H.; Dessain, S.K.; Eaton, E.N.; Imai, S.-I.; Frye, R.A.; Pandita, T.K.; Guarente, L.; Weinberg, R.A. hSIR2SIRT1 Functions as an NAD-Dependent p53 Deacetylase. Cell 2001, 107, 149–159. [Google Scholar] [CrossRef]
- Jin, X.; Wei, Y.; Xu, F.; Zhao, M.; Dai, K.; Shen, R.; Yang, S.; Zhang, N. SIRT1 promotes formation of breast cancer through modulating Akt activity. J. Cancer 2018, 9, 2012–2023. [Google Scholar] [CrossRef]
- Parija, M.; Prakash, S.; Krishna, B.M.; Dash, S.; Mishra, S.K. SIRT1 mediates breast cancer development and tumorigenesis controlled by estrogen-related receptor β. Breast Cancer 2024, 31, 440–455. [Google Scholar] [CrossRef]
- Yu, S.; Zhou, R.; Yang, T.; Liu, S.; Cui, Z.; Qiao, Q.; Zhang, J. Hypoxia promotes colorectal cancer cell migration and invasion in a SIRT1-dependent manner. Cancer Cell Int. 2019, 19, 116. [Google Scholar] [CrossRef]
- Wang, C.; Chen, L.; Hou, X.; Li, Z.; Kabra, N.; Ma, Y.; Nemoto, S.; Finkel, T.; Gu, W.; Cress, W.D.; et al. Interactions between E2F1 and SirT1 regulate apoptotic response to DNA damage. Nat. Cell Biol. 2006, 8, 1025–1031. [Google Scholar] [CrossRef]
- Oberdoerffer, P.; Michan, S.; McVay, M.; Mostoslavsky, R.; Vann, J.; Park, S.-K.; Hartlerode, A.; Stegmuller, J.; Hafner, A.; Loerch, P.; et al. SIRT1 Redistribution on Chromatin Promotes Genomic Stability but Alters Gene Expression during Aging. Cell 2008, 135, 907–918. [Google Scholar] [CrossRef]
- Wang, R.-H.; Zheng, Y.; Kim, H.-S.; Xu, X.; Cao, L.; Lahusen, T.; Lee, M.-H.; Xiao, C.; Vassilopoulos, A.; Chen, W.; et al. Interplay among BRCA1, SIRT1, and Survivin during BRCA1-Associated Tumorigenesis. Mol. Cell 2008, 32, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-S.; Vassilopoulos, A.; Wang, R.-H.; Lahusen, T.; Xiao, Z.; Xu, X.; Li, C.; Veenstra, T.D.; Li, B.; Yu, H.; et al. SIRT2 Maintains Genome Integrity and Suppresses Tumorigenesis through Regulating APC/C Activity. Cancer Cell 2011, 20, 487–499. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Im, J.; Kim, S.; Jang, S.; Han, Y.; Yang, K.-M.; Kim, S.-J.; Dhanasekaran, D.N.; Song, Y.S. ROS-Induced SIRT2 Upregulation Contributes to Cisplatin Sensitivity in Ovarian Cancer. Antioxidants 2020, 9, 1137. [Google Scholar] [CrossRef]
- Wang, B.; Ye, Y.; Yang, X.; Liu, B.; Wang, Z.; Chen, S.; Jiang, K.; Zhang, W.; Jiang, H.; Mustonen, H.; et al. SIRT2‐dependent IDH1 deacetylation inhibits colorectal cancer and liver metastases. EMBO Rep. 2020, 21, e48183. [Google Scholar] [CrossRef]
- Bell, E.L.; Emerling, B.M.; Ricoult, S.J.H.; Guarente, L. SirT3 suppresses hypoxia inducible factor 1α and tumor growth by inhibiting mitochondrial ROS production. Oncogene 2011, 30, 2986–2996. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-S.; Patel, K.; Muldoon-Jacobs, K.; Bisht, K.S.; Aykin-Burns, N.; Pennington, J.D.; van der Meer, R.; Nguyen, P.; Savage, J.; Owens, K.M.; et al. SIRT3 Is a Mitochondria-Localized Tumor Suppressor Required for Maintenance of Mitochondrial Integrity and Metabolism during Stress. Cancer Cell 2010, 17, 41–52. [Google Scholar] [CrossRef]
- Jeong, S.M.; Xiao, C.; Finley, L.W.S.; Lahusen, T.; Souza, A.L.; Pierce, K.; Li, Y.-H.; Wang, X.; Laurent, G.; German, N.J.; et al. SIRT4 Has Tumor-Suppressive Activity and Regulates the Cellular Metabolic Response to DNA Damage by Inhibiting Mitochondrial Glutamine Metabolism. Cancer Cell 2013, 23, 450–463. [Google Scholar] [CrossRef]
- Miyo, M.; Yamamoto, H.; Konno, M.; Colvin, H.; Nishida, N.; Koseki, J.; Kawamoto, K.; Ogawa, H.; Hamabe, A.; Uemura, M.; et al. Tumour-suppressive function of SIRT4 in human colorectal cancer. Br. J. Cancer 2015, 113, 492–499. [Google Scholar] [CrossRef]
- Du, L.; Liu, X.; Ren, Y.; Li, J.; Li, P.; Jiao, Q.; Meng, P.; Wang, F.; Wang, Y.; Wang, Y.-S.; et al. Loss of SIRT4 promotes the self-renewal of Breast Cancer Stem Cells. Theranostics 2020, 10, 9458–9476. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Zuo, Y.; Feng, Y.; Zhang, M. SIRT5 facilitates cancer cell growth and drug resistance in non-small cell lung cancer. Tumor Biol. 2014, 35, 10699–10705. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Yang, Y.; Pang, X.; Shi, J.; Jiang, T.; Zheng, X. Quercetin inhibits DNA damage responses to induce apoptosis via SIRT5/PI3K/AKT pathway in non-small cell lung cancer. Biomed. Pharmacother. 2023, 165, 115071. [Google Scholar] [CrossRef]
- Guo, D.; Song, X.; Guo, T.; Gu, S.; Chang, X.; Su, T.; Yang, X.; Liang, B.; Huang, D. Vimentin acetylation is involved in SIRT5-mediated hepatocellular carcinoma migration. Am. J. Cancer Res. 2018, 8, 2453–2466. [Google Scholar]
- Sun, X.; Wang, S.; Gai, J.; Guan, J.; Li, J.; Li, Y.; Zhao, J.; Zhao, C.; Fu, L.; Li, Q. SIRT5 Promotes Cisplatin Resistance in Ovarian Cancer by Suppressing DNA Damage in a ROS-Dependent Manner via Regulation of the Nrf2/HO-1 Pathway. Front. Oncol. 2019, 9, 754. [Google Scholar] [CrossRef] [PubMed]
- Van Meter, M.; Zhiyong, M.; Vera, G.; Seluanov, A. SIRT6 overexpression induces massive apoptosis in cancer cells but not in normal cells. Cell Cycle 2011, 10, 3153–3158. [Google Scholar] [CrossRef]
- Zhang, Z.-G.; Qin, C.-Y. Sirt6 suppresses hepatocellular carcinoma cell growth via inhibiting the extracellular signal-regulated kinase signaling pathway. Mol. Med. Rep. 2014, 9, 882–888. [Google Scholar] [CrossRef]
- Fukuda, T.; Wada-Hiraike, O.; Oda, K.; Tanikawa, M.; Makii, C.; Inaba, K.; Miyasaka, A.; Miyamoto, Y.; Yano, T.; Maeda, D.; et al. Putative tumor suppression function of SIRT6 in endometrial cancer. FEBS Lett. 2015, 589, 2274–2281. [Google Scholar] [CrossRef]
- Barber, M.F.; Michishita-Kioi, E.; Xi, Y.; Tasselli, L.; Kioi, M.; Moqtaderi, Z.; Tennen, R.I.; Paredes, S.; Young, N.L.; Chen, K.; et al. SIRT7 links H3K18 deacetylation to maintenance of oncogenic transformation. Nature 2012, 487, 114–118. [Google Scholar] [CrossRef]
- Kumari, P.; Tarighi, S.; Fuchshuber, E.; Li, L.; Fernández-Duran, I.; Wang, M.; Ayoson, J.; Castelló-García, J.M.; Gámez-García, A.; Espinosa-Alcantud, M.; et al. SIRT7 promotes lung cancer progression by destabilizing the tumor suppressor ARF. Proc. Natl. Acad. Sci. USA 2024, 121, e2409269121. [Google Scholar] [CrossRef]
- Finkel, T.; Deng, C.-X.; Mostoslavsky, R. Recent progress in the biology and physiology of sirtuins. Nature 2009, 460, 587–591. [Google Scholar] [CrossRef] [PubMed]
- Sebastián, C.; Zwaans, B.M.M.; Silberman, D.M.; Gymrek, M.; Goren, A.; Zhong, L.; Ram, O.; Truelove, J.; Guimaraes, A.R.; Toiber, D.; et al. The Histone Deacetylase SIRT6 Is a Tumor Suppressor that Controls Cancer Metabolism. Cell 2012, 151, 1185–1199. [Google Scholar] [CrossRef]
- Vaquero, A.; Scher, M.B.; Lee, D.H.; Sutton, A.; Cheng, H.-L.; Alt, F.W.; Serrano, L.; Sternglanz, R.; Reinberg, D. SirT2 is a histone deacetylase with preference for histone H4 Lys 16 during mitosis. Genes Dev. 2006, 20, 1256–1261. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, T.L.A.; Michishita, E.; Adler, A.S.; Damian, M.; Berber, E.; Lin, M.; McCord, R.A.; Ongaigui, K.C.L.; Boxer, L.D.; Chang, H.Y.; et al. SIRT6 Links Histone H3 Lysine 9 Deacetylation to NF-κB-Dependent Gene Expression and Organismal Life Span. Cell 2009, 136, 62–74. [Google Scholar] [CrossRef]
- Min, L.; Ji, Y.; Bakiri, L.; Qiu, Z.; Cen, J.; Chen, X.; Chen, L.; Scheuch, H.; Zheng, H.; Qin, L.; et al. Liver cancer initiation is controlled by AP-1 through SIRT6-dependent inhibition of survivin. Nat. Cell Biol. 2012, 14, 1203–1211. [Google Scholar] [CrossRef]
- Michishita, E.; McCord, R.A.; Berber, E.; Kioi, M.; Padilla-Nash, H.; Damian, M.; Cheung, P.; Kusumoto, R.; Kawahara, T.L.A.; Barrett, J.C.; et al. SIRT6 is a histone H3 lysine 9 deacetylase that modulates telomeric chromatin. Nature 2008, 452, 492–496. [Google Scholar] [CrossRef] [PubMed]
- Michishita, E.; McCord, R.A.; Boxer, L.D.; Barber, M.F.; Hong, T.; Gozani, O.; Chua, K.F. Cell cycle-dependent deacetylation of telomeric histone H3 lysine K56 by human SIRT6. Cell Cycle 2009, 8, 2664–2666. [Google Scholar] [CrossRef]
- Kugel, S.; Mostoslavsky, R. Chromatin and beyond: The multitasking roles for SIRT6. Trends Biochem. Sci. 2014, 39, 72–81. [Google Scholar] [CrossRef]
- Yang, B.; Zwaans, B.M.M.; Eckersdorff, M.; Lombard, D.B. The sirtuin SIRT6 deacetylates H3 K56Ac in vivo to promote genomic stability. Cell Cycle 2009, 8, 2662–2663. [Google Scholar] [CrossRef]
- Jeong, S.M.; Lee, A.; Lee, J.; Haigis, M.C. SIRT4 Protein Suppresses Tumor Formation in Genetic Models of Myc-induced B Cell Lymphoma. J. Biol. Chem. 2014, 289, 4135–4144. [Google Scholar] [CrossRef]
- Riggs, M.G.; Whittaker, R.G.; Neumann, J.R.; Ingram, V.M. n-Butyrate causes histone modification in HeLa and Friend erythroleukaemia cells. Nature 1977, 268, 462–464. [Google Scholar] [CrossRef]
- Bolden, J.E.; Peart, M.J.; Johnstone, R.W. Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discov. 2006, 5, 769–784. [Google Scholar] [CrossRef] [PubMed]
- Bieliauskas, A.V.; Pflum, M.K.H. Isoform-selective histone deacetylase inhibitors. Chem. Soc. Rev. 2008, 37, 1402–1413. [Google Scholar] [CrossRef]
- Mariadason, J.M.; Corner, G.A.; Augenlicht, L.H. Genetic Reprogramming in Pathways of Colonic Cell Maturation Induced by Short Chain Fatty Acids: Comparison with Trichostatin A, Sulindac, and Curcumin and Implications for Chemoprevention of Colon Cancer1. Cancer Res. 2000, 60, 4561–4572. [Google Scholar] [PubMed]
- Glaser, K.B.; Staver, M.J.; Waring, J.F.; Stender, J.; Ulrich, R.G.; Davidsen, S.K. Gene Expression Profiling of Multiple Histone Deacetylase (HDAC) Inhibitors: Defining a Common Gene Set Produced by HDAC Inhibition in T24 and MDA Carcinoma Cell Lines. Mol. Cancer Ther. 2003, 2, 151–163. [Google Scholar] [PubMed]
- Yamashita, K.; Upadhyay, S.; Osada, M.; Hoque, M.O.; Xiao, Y.; Mori, M.; Sato, F.; Meltzer, S.J.; Sidransky, D. Pharmacologic unmasking of epigenetically silenced tumor suppressor genes in esophageal squamous cell carcinoma. Cancer Cell 2002, 2, 485–495. [Google Scholar] [CrossRef]
- Insinga, A.; Monestiroli, S.; Ronzoni, S.; Gelmetti, V.; Marchesi, F.; Viale, A.; Altucci, L.; Nervi, C.; Minucci, S.; Pelicci, P.G. Inhibitors of histone deacetylases induce tumor-selective apoptosis through activation of the death receptor pathway. Nat. Med. 2005, 11, 71–76. [Google Scholar] [CrossRef]
- Kwon, S.H.; Ahn, S.H.; Kim, Y.K.; Bae, G.-U.; Yoon, J.W.; Hong, S.; Lee, H.Y.; Lee, Y.-W.; Lee, H.-W.; Han, J.-W. Apicidin, a Histone Deacetylase Inhibitor, Induces Apoptosis and Fas/Fas Ligand Expression in Human Acute Promyelocytic Leukemia Cells. J. Biol. Chem. 2002, 277, 2073–2080. [Google Scholar] [CrossRef]
- Nebbioso, A.; Clarke, N.; Voltz, E.; Germain, E.; Ambrosino, C.; Bontempo, P.; Alvarez, R.; Schiavone, E.M.; Ferrara, F.; Bresciani, F.; et al. Tumor-selective action of HDAC inhibitors involves TRAIL induction in acute myeloid leukemia cells. Nat. Med. 2005, 11, 77–84. [Google Scholar] [CrossRef]
- Shimizu, R.; Kikuchi, J.; Wada, T.; Ozawa, K.; Kano, Y.; Furukawa, Y. HDAC inhibitors augment cytotoxic activity of rituximab by upregulating CD20 expression on lymphoma cells. Leukemia 2010, 24, 1760–1768. [Google Scholar] [CrossRef]
- Kelly, W.K.; Marks, P.A. Drug Insight: Histone deacetylase inhibitors—Development of the new targeted anticancer agent suberoylanilide hydroxamic acid. Nat. Clin. Pract. Oncol. 2005, 2, 150–157. [Google Scholar] [CrossRef]
- Ryu, J.K.; Lee, W.J.; Lee, K.H.; Hwang, J.-H.; Kim, Y.-T.; Yoon, Y.B.; Kim, C.Y. SK-7041, a new histone deacetylase inhibitor, induces G2-M cell cycle arrest and apoptosis in pancreatic cancer cell lines. Cancer Lett. 2006, 237, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Rosato, R.R.; Almenara, J.A.; Grant, S. The Histone Deacetylase Inhibitor MS-275 Promotes Differentiation or Apoptosis in Human Leukemia Cells through a Process Regulated by Generation of Reactive Oxygen Species and Induction of p21CIP1/WAF11. Cancer Res. 2003, 63, 3637–3645. [Google Scholar]
- Deroanne, C.F.; Bonjean, K.; Servotte, S.; Devy, L.; Colige, A.; Clausse, N.; Blacher, S.; Verdin, E.; Foidart, J.-M.; Nusgens, B.V.; et al. Histone deacetylases inhibitors as anti-angiogenic agents altering vascular endothelial growth factor signaling. Oncogene 2002, 21, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Coradini, D.; Zorzet, S.; Rossin, R.; Scarlata, I.; Pellizzaro, C.; Turrin, C.; Bello, M.; Cantoni, S.; Speranza, A.; Sava, G.; et al. Inhibition of Hepatocellular Carcinomas in vitro and Hepatic Metastases in vivo in Mice by the Histone Deacetylase Inhibitor HA-But. Clin. Cancer Res. 2004, 10, 4822–4830. [Google Scholar] [CrossRef]
- Nusinzon, I.; Horvath, C.M. Interferon-stimulated transcription and innate antiviral immunity require deacetylase activity and histone deacetylase 1. Proc. Natl. Acad. Sci. USA 2003, 100, 14742–14747. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-R.; Kim, E.-J.; Yang, S.-H.; Jeong, E.-T.; Park, C.; Lee, J.-H.; Youn, M.-J.; So, H.-S.; Park, R. Trichostatin A induces apoptosis in lung cancer cells via simultaneous activation of the death receptor-mediated and mitochondrial pathway. Exp. Mol. Med. 2006, 38, 616–624. [Google Scholar] [CrossRef]
- Gilardini Montani, M.S.; Granato, M.; Santoni, C.; Del Porto, P.; Merendino, N.; D’Orazi, G.; Faggioni, A.; Cirone, M. Histone deacetylase inhibitors VPA and TSA induce apoptosis and autophagy in pancreatic cancer cells. Cell. Oncol. 2017, 40, 167–180. [Google Scholar] [CrossRef]
- You, B.R.; Park, W.H. Trichostatin A induces apoptotic cell death of HeLa cells in a Bcl-2 and oxidative stress-dependent manner. Int. J. Oncol. 2013, 42, 359–366. [Google Scholar] [CrossRef]
- Francisco, R.; Pérez-Perarnau, A.; Cortés, C.; Gil, J.; Tauler, A.; Ambrosio, S. Histone deacetylase inhibition induces apoptosis and autophagy in human neuroblastoma cells. Cancer Lett. 2012, 318, 42–52. [Google Scholar] [CrossRef]
- Bai, Y.; Chen, Y.; Chen, X.; Jiang, J.; Wang, X.; Wang, L.; Wang, J.; Zhang, J.; Gao, L. Trichostatin A activates FOXO1 and induces autophagy in osteosarcoma. Arch. Med. Sci. 2019, 15, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.-H.; Cao, Y.-M.; Rong, Z.-P.; Ding, J.; Pan, X. Trichostatin A Induces Autophagy in Cervical Cancer Cells by Regulating the PRMT5-STC1-TRPV6-JNK Pathway. Pharmacology 2020, 106, 60–69. [Google Scholar] [CrossRef]
- Subramanian, C.; Jarzembowski, J.A.; Opipari, A.W.; Castle, V.P.; Kwok, R.P.S. CREB-Binding Protein Is a Mediator of Neuroblastoma Cell Death Induced By the Histone Deacetylase Inhibitor Trichostatin A. Neoplasia 2007, 9, 495–503. [Google Scholar] [CrossRef]
- Ding, H.; Peterson, K.L.; Correia, C.; Koh, B.; Schneider, P.A.; Nowakowski, G.S.; Kaufmann, S.H. Histone deacetylase inhibitors interrupt HSP90•RASGRP1 and HSP90•CRAF interactions to upregulate BIM and circumvent drug resistance in lymphoma cells. Leukemia 2017, 31, 1593–1602. [Google Scholar] [CrossRef]
- Yang, L.; Qu, M.; Wang, Y.; Duan, H.; Chen, P.; Wang, Y.; Shi, W.; Danielson, P.; Zhou, Q. Trichostatin A Inhibits Transforming Growth Factor-β-Induced Reactive Oxygen Species Accumulation and Myofibroblast Differentiation via Enhanced NF-E2-Related Factor 2-Antioxidant Response Element Signaling. Mol. Pharmacol. 2013, 83, 671–680. [Google Scholar] [CrossRef] [PubMed]
- Kang, F.-W.; Que, L.; Wu, M.; Wang, Z.-L.; Sun, J. Effects of trichostatin A on HIF-1α and VEGF expression in human tongue squamous cell carcinoma cells in vitro. Oncol. Rep. 2012, 28, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Park, I.-H.; Kang, J.-H.; Shin, J.-M.; Lee, H.-M. Trichostatin A Inhibits Epithelial Mesenchymal Transition Induced by TGF-β1 in Airway Epithelium. PLoS ONE 2016, 11, e0162058. [Google Scholar] [CrossRef]
- Wall, K.A.; Klis, M.; Kornet, J.; Coyle, D.; AmÉ, J.-C.; Jacobson, M.K.; Slama, J.T. Inhibition of the intrinsic NAD+ glycohydrolase activity of CD38 by carbocyclic NAD analogues. Biochem. J. 1998, 335, 631–636. [Google Scholar] [CrossRef]
- Jackson, M.D.; Schmidt, M.T.; Oppenheimer, N.J.; Denu, J.M. Mechanism of Nicotinamide Inhibition and Transglycosidation by Sir2 Histone/Protein Deacetylases. J. Biol. Chem. 2003, 278, 50985–50998. [Google Scholar] [CrossRef]
- Yasuda, M.; Wilson, D.R.; Fugmann, S.D.; Moaddel, R. Synthesis and Characterization of SIRT6 Protein Coated Magnetic Beads: Identification of a Novel Inhibitor of SIRT6 Deacetylase from Medicinal Plant Extracts. Anal. Chem. 2011, 83, 7400–7407. [Google Scholar] [CrossRef]
- Singh, N.; Ravichandran, S.; Spelman, K.; Fugmann, S.D.; Moaddel, R. The identification of a novel SIRT6 modulator from Trigonella foenum-graecum using ligand fishing with protein coated magnetic beads. J. Chromatogr. B 2014, 968, 105–111. [Google Scholar] [CrossRef]
- Heger, V.; Tyni, J.; Hunyadi, A.; Horáková, L.; Lahtela-Kakkonen, M.; Rahnasto-Rilla, M. Quercetin based derivatives as sirtuin inhibitors. Biomed. Pharmacother. 2019, 111, 1326–1333. [Google Scholar] [CrossRef]
- Napper, A.D.; Hixon, J.; McDonagh, T.; Keavey, K.; Pons, J.-F.; Barker, J.; Yau, W.T.; Amouzegh, P.; Flegg, A.; Hamelin, E.; et al. Discovery of Indoles as Potent and Selective Inhibitors of the Deacetylase SIRT1. J. Med. Chem. 2005, 48, 8045–8054. [Google Scholar] [CrossRef] [PubMed]
- Grozinger, C.M.; Chao, E.D.; Blackwell, H.E.; Moazed, D.; Schreiber, S.L. Identification of a Class of Small Molecule Inhibitors of the Sirtuin Family of NAD-dependent Deacetylases by Phenotypic Screening. J. Biol. Chem. 2001, 276, 38837–38843. [Google Scholar] [CrossRef] [PubMed]
- Zentner, G.E.; Henikoff, S. Regulation of nucleosome dynamics by histone modifications. Nat. Struct. Mol. Biol. 2013, 20, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.; Huang, C.; Xu, Q.; Yang, Y.; Liu, Y.; Meng, X.; Li, J.; Ye, M.; Liang, H. Suppression of BMP-7 by histone deacetylase 2 promoted apoptosis of renal tubular epithelial cells in acute kidney injury. Cell Death Dis. 2017, 8, e3139. [Google Scholar] [CrossRef]
- Li, Y.; Li, K.; Zhao, W.; Wang, H.; Xue, X.; Chen, X.; Li, W.; Xu, P.; Wang, K.; Liu, P.; et al. VPA improves ferroptosis in tubular epithelial cells after cisplatin-induced acute kidney injury. Front. Pharmacol. 2023, 14, 1147772. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M.; Nomura, S.; Beppu, T. Effects of Trichostatins on Differentiation of Murine Erythroleukemia Cells. Cancer Res. 1987, 47, 3688–3691. [Google Scholar]
- Yoshida, M.; Kijima, M.; Akita, M.; Beppu, T. Potent and specific inhibition of mammalian histone deacetylase both in vivo and in vitro by trichostatin A. J. Biol. Chem. 1990, 265, 17174–17179. [Google Scholar] [CrossRef]
- Zhang, X.; Jiang, S.-J.; Shang, B.; Jiang, H.-J. Effects of histone deacetylase inhibitor trichostatin A combined with cisplatin on apoptosis of A549 cell line. Thorac. Cancer 2015, 6, 202–208. [Google Scholar] [CrossRef]
- Yoon, C.Y.; Park, M.J.; Lee, J.S.; Lee, S.C.; Oh, J.J.; Park, H.; Chung, C.W.; Abdullajanov, M.M.; Jeong, S.J.; Hong, S.K.; et al. The Histone Deacetylase Inhibitor Trichostatin A Synergistically Resensitizes a Cisplatin Resistant Human Bladder Cancer Cell Line. J. Urol. 2011, 185, 1102–1111. [Google Scholar] [CrossRef]
- Muscolini, M.; Cianfrocca, R.; Sajeva, A.; Mozzetti, S.; Ferrandina, G.; Costanzo, A.; Tuosto, L. Trichostatin A up-regulates p73 and induces Bax-dependent apoptosis in cisplatin-resistant ovarian cancer cells. Mol. Cancer Ther. 2008, 7, 1410–1419. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Hu, C.-P.; Gu, Q.-H.; Li, Y.-P.; Song, M. Trichostatin A sensitizes cisplatin-resistant A549 cells to apoptosis by up-regulating death-associated protein kinase. Acta Pharmacol. Sin. 2010, 31, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, G.; Zhao, J.; Ding, H.; Cunningham, C.; Chen, F.; Flynn, D.C.; Reed, E.; Li, Q.Q. Antiproliferative effect of β-elemene in chemoresistant ovarian carcinoma cells is mediated through arrest of the cell cycle at the G2-M phase. Cell. Mol. Life Sci. 2005, 62, 894–904. [Google Scholar] [CrossRef]
- Mueller, S.; Schittenhelm, M.; Honecker, F.; Malenke, E.; Lauber, K.; Wesselborg, S.; Hartmann, J.T.; Bokemeyer, C.; Mayer, F. Cell-cycle progression and response of germ cell tumors to cisplatin in vitro. Int. J. Oncol. 2006, 29, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.L.; Shimizu, T.; Ise, T.; Numata, T.; Kohno, K.; Okada, Y. Impaired activity of volume-sensitive Cl− channel is involved in cisplatin resistance of cancer cells. J. Cell. Physiol. 2007, 211, 513–521. [Google Scholar] [CrossRef]
- Lambert, I.H.; Nielsen, D.; Stürup, S. Impact of the histone deacetylase inhibitor trichostatin A on active uptake, volume-sensitive release of taurine, and cell fate in human ovarian cancer cells. Am. J. Physiol. Cell Physiol. 2020, 318, C581–C597. [Google Scholar] [CrossRef]
- Tang, J.; Shi, Y.; Liu, N.; Xu, L.; Zang, X.; Li, P.; Zhang, J.; Zheng, X.; Qiu, A.; Zhuang, S. Blockade of histone deacetylase 6 protects against cisplatin-induced acute kidney injury. Clin. Sci. 2018, 132, 339–359. [Google Scholar] [CrossRef]
- Liu, J.; Livingston, M.J.; Dong, G.; Tang, C.; Su, Y.; Wu, G.; Yin, X.-M.; Dong, Z. Histone deacetylase inhibitors protect against cisplatin-induced acute kidney injury by activating autophagy in proximal tubular cells. Cell Death Dis. 2018, 9, 322. [Google Scholar] [CrossRef]
- Ranganathan, P.; Hamad, R.; Mohamed, R.; Jayakumar, C.; Muthusamy, T.; Ramesh, G. Histone deacetylase–mediated silencing of AMWAP expression contributes to cisplatin nephrotoxicity. Kidney Int. 2016, 89, 317–326. [Google Scholar] [CrossRef]
- Zhu, H.; Jiang, W.; Zhao, H.; He, C.; Tang, X.; Xu, S.; Xu, C.; Feng, R.; Li, J.; Ma, T.; et al. PSTPIP2 inhibits cisplatin-induced acute kidney injury by suppressing apoptosis of renal tubular epithelial cells. Cell Death Dis. 2020, 11, 1057. [Google Scholar] [CrossRef] [PubMed]
- Richon, V.M.; Emiliani, S.; Verdin, E.; Webb, Y.; Breslow, R.; Rifkind, R.A.; Marks, P.A. A class of hybrid polar inducers of transformed cell differentiation inhibits histone deacetylases. Proc. Natl. Acad. Sci. USA 1998, 95, 3003–3007. [Google Scholar] [CrossRef]
- Suzuki, M.; Endo, M.; Shinohara, F.; Echigo, S.; Rikiishi, H. Enhancement of cisplatin cytotoxicity by SAHA involves endoplasmic reticulum stress-mediated apoptosis in oral squamous cell carcinoma cells. Cancer Chemother. Pharmacol. 2009, 64, 1115–1122. [Google Scholar] [CrossRef]
- Jin, K.L.; Park, J.Y.; Noh, E.J.; Hoe, K.L.; Lee, J.H.; Kim, J.H.; Nam, J.H. The effect of combined treatment with cisplatin and histone deacetylase inhibitors on HeLa cells. J. Gynecol. Oncol. 2010, 21, 262–268. [Google Scholar] [CrossRef]
- Zacharioudakis, E.; Agarwal, P.; Bartoli, A.; Abell, N.; Kunalingam, L.; Bergoglio, V.; Xhemalce, B.; Miller, K.M.; Rodriguez, R. Chromatin Regulates Genome Targeting with Cisplatin. Angew. Chem. Int. Ed. 2017, 56, 6483–6487. [Google Scholar] [CrossRef]
- Rikiishi, H.; Shinohara, F.; Sato, T.; Sato, Y.; Suzuki, M.; Echigo, S. Chemosensitization of oral squamous cell carcinoma cells to cisplatin by histone deacetylase inhibitor, suberoylanilide hydroxamic acid. Int. J. Oncol. 2007, 30, 1181–1188. [Google Scholar] [CrossRef]
- Sato, T.; Suzuki, M.; Sato, Y.; Echigo, S.; Rikiishi, H. Sequence-dependent interaction between cisplatin and histone deacetylase inhibitors in human oral squamous cell carcinoma cells. Int. J. Oncol. 2006, 28, 1233–1241. [Google Scholar] [CrossRef] [PubMed]
- Hegedűs, L.; Rittler, D.; Garay, T.; Stockhammer, P.; Kovács, I.; Döme, B.; Theurer, S.; Hager, T.; Herold, T.; Kalbourtzis, S.; et al. HDAC Inhibition Induces PD-L1 Expression in a Novel Anaplastic Thyroid Cancer Cell Line. Pathol. Oncol. Res. 2020, 26, 2523–2535. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.-H.; Chang, Y.-F.; Lee, M.-S.; Wen, B.C.; Ko, J.-C.; Liang, S.-K.; Liang, M.-C. Vorinostat enhances the cisplatin-mediated anticancer effects in small cell lung cancer cells. BMC Cancer 2016, 16, 857. [Google Scholar] [CrossRef]
- Venkatasamy, A.; Guerin, E.; Blanchet, A.; Orvain, C.; Devignot, V.; Jung, M.; Jung, A.C.; Chenard, M.-P.; Romain, B.; Gaiddon, C.; et al. Ultrasound and Transcriptomics Identify a Differential Impact of Cisplatin and Histone Deacetylation on Tumor Structure and Microenvironment in a Patient-Derived In Vivo Model of Gastric Cancer. Pharmaceutics 2021, 13, 1485. [Google Scholar] [CrossRef]
- Bruni, J.; Wilder, B.J. Valproic Acid: Review of a New Antiepileptic Drug. Arch. Neurol. 1979, 36, 393–398. [Google Scholar] [CrossRef]
- Maemoto, Y.; Shimizu, Y.; Katoh, R.; Ito, A. Naturally occurring small molecule compounds that target histone deacetylases and their potential applications in cancer therapy. J. Antibiot. 2021, 74, 667–676. [Google Scholar] [CrossRef]
- Groh, T.; Hrabeta, J.; Khalil, M.A.; Doktorova, H.; Eckschlager, T.; Stiborova, M. The synergistic effects of DNA-damaging drugs cisplatin and etoposide with a histone deacetylase inhibitor valproate in high-risk neuroblastoma cells. Int. J. Oncol. 2015, 47, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-T.; Lai, H.-C.; Lee, H.-Y.; Lin, W.-H.; Chang, C.-C.; Chu, T.-Y.; Lin, Y.-W.; Lee, K.-D.; Yu, M.-H. Valproic acid resensitizes cisplatin-resistant ovarian cancer cells. Cancer Sci. 2008, 99, 1218–1226. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Jing, Y.; Ouyang, S.; Liu, B.; Zhu, T.; Niu, H.; Tian, Y. Inhibitory effect of valproic acid on bladder cancer in combination with chemotherapeutic agents in vitro and in vivo. Oncol. Lett. 2013, 6, 1492–1498. [Google Scholar] [CrossRef]
- Granit, A.; Mishra, K.; Barasch, D.; Peretz-Yablonsky, T.; Eyal, S.; Kakhlon, O. Metabolomic profiling of triple negative breast cancer cells suggests that valproic acid can enhance the anticancer effect of cisplatin. Front. Cell. Dev. Biol. 2022, 10, 1014798. [Google Scholar] [CrossRef]
- Krieger, V.; Hamacher, A.; Cao, F.; Stenzel, K.; Gertzen, C.G.W.; Schäker-Hübner, L.; Kurz, T.; Gohlke, H.; Dekker, F.J.; Kassack, M.U.; et al. Synthesis of Peptoid-Based Class I-Selective Histone Deacetylase Inhibitors with Chemosensitizing Properties. J. Med. Chem. 2019, 62, 11260–11279. [Google Scholar] [CrossRef]
- Koprinarova, M.; Markovska, P.; Iliev, I.; Anachkova, B.; Russev, G. Sodium butyrate enhances the cytotoxic effect of cisplatin by abrogating the cisplatin imposed cell cycle arrest. BMC Mol. Biol. 2010, 11, 49. [Google Scholar] [CrossRef]
- Zhou, Y.; Pan, D.-S.; Shan, S.; Zhu, J.-Z.; Zhang, K.; Yue, X.-P.; Nie, L.-P.; Wan, J.; Lu, X.-P.; Zhang, W.; et al. Non-toxic dose chidamide synergistically enhances platinum-induced DNA damage responses and apoptosis in Non-Small-Cell lung cancer cells. Biomed. Pharmacother. 2014, 68, 483–491. [Google Scholar] [CrossRef]
- Yan, W.; Wu, T.H.Y.; Leung, S.S.Y.; To, K.K.W. Flavonoids potentiated anticancer activity of cisplatin in non-small cell lung cancer cells in vitro by inhibiting histone deacetylases. Life Sci. 2020, 258, 118211. [Google Scholar] [CrossRef]
- Huang, W.-J.; Tang, Y.-A.; Chen, M.-Y.; Wang, Y.-J.; Hu, F.-H.; Wang, T.-W.; Chao, S.-W.; Chiu, H.-W.; Yeh, Y.-L.; Chang, H.-Y.; et al. A histone deacetylase inhibitor YCW1 with antitumor and antimetastasis properties enhances cisplatin activity against non-small cell lung cancer in preclinical studies. Cancer Lett. 2014, 346, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Luchenko, V.L.; Salcido, C.D.; Zhang, Y.; Agama, K.; Komlodi-Pasztor, E.; Murphy, R.F.; Giaccone, G.; Pommier, Y.; Bates, S.E.; Varticovski, L. Schedule-dependent synergy of histone deacetylase inhibitors with DNA damaging agents in small cell lung cancer. Cell Cycle 2011, 10, 3119–3128. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Bao, X.; Ren, Y.; Jia, L.; Zou, S.; Han, J.; Zhao, M.; Han, M.; Li, H.; Hua, Q.; et al. Targeting HDAC/OAZ1 axis with a novel inhibitor effectively reverses cisplatin resistance in non-small cell lung cancer. Cell Death Dis. 2019, 10, 400. [Google Scholar] [CrossRef]
- He, Q.; Yu, C.; Li, Y.; Hao, P.; Mai, H.; Guo, R.; Zhong, G.; Zhang, K.; Wong, C.; Chen, Q.; et al. ERRα contributes to HDAC6-induced chemoresistance of osteosarcoma cells. Cell Biol. Toxicol. 2023, 39, 813–825. [Google Scholar] [CrossRef]
- Wang, X.-W.; Guo, Q.-Q.; Yu, Y.; Zhou, T.-T.; Zhang, S.-Y.; Wang, Z.; Liu, J.-W.; Tang, J.; Jiang, X.-Y.; Wang, S.-S.; et al. The deacetylation of Foxk2 by Sirt1 reduces chemosensitivity to cisplatin. J. Cell. Mol. Med. 2022, 26, 491–506. [Google Scholar] [CrossRef]
- Wang, S.-H.; Tsai, K.-L.; Chou, W.-C.; Cheng, H.-C.; Huang, Y.-T.; Ou, H.-C.; Chang, Y.-C. Quercetin Mitigates Cisplatin-Induced Oxidative Damage and Apoptosis in Cardiomyocytes through Nrf2/HO-1 Signaling Pathway. Am. J. Chin. Med. 2022, 50, 1281–1298. [Google Scholar] [CrossRef]
- Huang, J.; Ding, W.; Zhu, X.; Li, B.; Zeng, F.; Wu, K.; Wu, X.; Wang, F. Ligand Evolution in the Photoactivatable Platinum(IV) Anticancer Prodrugs. Front. Chem. 2022, 10, 876410. [Google Scholar] [CrossRef]
- Deng, Z.; Zhu, G. Beyond mere DNA damage: Recent progress in platinum(IV) anticancer complexes containing multi-functional axial ligands. Curr. Opin. Chem. Biol. 2023, 74, 102303. [Google Scholar] [CrossRef]
- Novohradsky, V.; Zerzankova, L.; Stepankova, J.; Vrana, O.; Raveendran, R.; Gibson, D.; Kasparkova, J.; Brabec, V. New insights into the molecular and epigenetic effects of antitumor Pt(IV)-valproic acid conjugates in human ovarian cancer cells. Biochem. Pharmacol. 2015, 95, 133–144. [Google Scholar] [CrossRef]
- Novohradsky, V.; Zerzankova, L.; Stepankova, J.; Vrana, O.; Raveendran, R.; Gibson, D.; Kasparkova, J.; Brabec, V. Antitumor platinum(IV) derivatives of oxaliplatin with axial valproato ligands. J. Inorg. Biochem. 2014, 140, 72–79. [Google Scholar] [CrossRef]
- Almotairy, A.R.Z.; Montagner, D.; Morrison, L.; Devereux, M.; Howe, O.; Erxleben, A. Pt(IV) pro-drugs with an axial HDAC inhibitor demonstrate multimodal mechanisms involving DNA damage and apoptosis independent of cisplatin resistance in A2780/A2780cis cells. J. Inorg. Biochem. 2020, 210, 111125. [Google Scholar] [CrossRef]
- Kasparkova, J.; Kostrhunova, H.; Novakova, O.; Křikavová, R.; Vančo, J.; Trávníček, Z.; Brabec, V. A Photoactivatable Platinum(IV) Complex Targeting Genomic DNA and Histone Deacetylases. Angew. Chem. Int. Ed. 2015, 54, 14478–14482. [Google Scholar] [CrossRef] [PubMed]
- Rebbaa, A.; Zheng, X.; Chu, F.; Mirkin, B.L. The role of histone acetylation versus DNA damage in drug-induced senescence and apoptosis. Cell Death Differ. 2006, 13, 1960–1967. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Lu, J.; Wang, X.; Han, L.; Zhang, Y.; Han, S.; Huang, B. Histone deacetylase inhibitor trichostatin a potentiates doxorubicin-induced apoptosis by up-regulating PTEN expression. Cancer 2007, 109, 1676–1688. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.Z.Y.; Zhang, H.T.; Chen, G.G.; Lai, P.B.S. Trichostatin A sensitizes HBx-expressing liver cancer cells to etoposide treatment. Apoptosis 2011, 16, 683–695. [Google Scholar] [CrossRef]
- Zhang, X.; Yashiro, M.; Ren, J.; Hirakawa, K. Histone deacetylase inhibitor, trichostatin A, increases the chemosensitivity of anticancer drugs in gastric cancer cell lines. Oncol. Rep. 2006, 16, 563–568. [Google Scholar] [CrossRef]
- Fazzone, W.; Wilson, P.M.; LaBonte, M.J.; Lenz, H.-J.; Ladner, R.D. Histone deacetylase inhibitors suppress thymidylate synthase gene expression and synergize with the fluoropyrimidines in colon cancer cells. Int. J. Cancer 2009, 125, 463–473. [Google Scholar] [CrossRef]
- Lee, C.-K.; Wang, S.; Huang, X.; Ryder, J.; Liu, B. HDAC inhibition synergistically enhances alkylator-induced DNA damage responses and apoptosis in multiple myeloma cells. Cancer Lett. 2010, 296, 233–240. [Google Scholar] [CrossRef]
- Poljakova, J.; Hrebackova, J.; Dvorakova, M.; Moserova, M.; Eckschlager, T.; Hrabeta, J.; Göttlicherova, M.; Kopejtkova, B.; Frei, E.; Kizek, R.; et al. Anticancer agent ellipticine combined with histone deacetylase inhibitors, valproic acid and trichostatin A, is an effective DNA damage strategy in human neuroblastoma. Neuro Endocrinol. Lett. 2011, 32 (Suppl. S1), 101–116. [Google Scholar]
- Zhang, Q.C.; Jiang, S.J.; Zhang, S.; Ma, X.B. Histone Deacetylase Inhibitor Trichostatin A Enhances Anti-tumor Effects of Docetaxel or Erlotinib in A549 Cell Line. Asian Pac. J. Cancer Prev. 2012, 13, 3471–3476. [Google Scholar] [CrossRef]
- Karagiannis, T.; El-Osta, A. Clinical Potential of Histone Deacetylase Inhibitors as Stand Alone Therapeutics and in Combination with other Chemotherapeutics or Radiotherapy for Cancer. Epigenetics 2006, 1, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Munshi, A.; Kurland, J.F.; Nishikawa, T.; Tanaka, T.; Hobbs, M.L.; Tucker, S.L.; Ismail, S.; Stevens, C.; Meyn, R.E. Histone Deacetylase Inhibitors Radiosensitize Human Melanoma Cells by Suppressing DNA Repair Activity. Clin. Cancer Res. 2005, 11, 4912–4922. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wong, P.; Radany, E.; Wong, J.Y.C. HDAC Inhibitor, Valproic Acid, Induces p53-Dependent Radiosensitization of Colon Cancer Cells. Cancer Biother. Radiopharm. 2009, 24, 689–699. [Google Scholar] [CrossRef]
- He, G.; Wang, Y.; Pang, X.; Zhang, B. Inhibition of autophagy induced by TSA sensitizes colon cancer cell to radiation. Tumor Biol. 2014, 35, 1003–1011. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.L.; Wang, A.-J.; Yao, L.-F. Antitumor histone deacetylase inhibitors suppress cutaneous radiation syndrome: Implications for increasing therapeutic gain in cancer radiotherapy. Mol. Cancer Ther. 2004, 3, 317–325. [Google Scholar] [CrossRef]
- Jin, X.; Tian, S.; Li, P. Histone Acetyltransferase 1 Promotes Cell Proliferation and Induces Cisplatin Resistance in Hepatocellular Carcinoma. Oncol. Res. 2017, 25, 939–946. [Google Scholar] [CrossRef]
- Miyamoto, N.; Izumi, H.; Noguchi, T.; Nakajima, Y.; Ohmiya, Y.; Shiota, M.; Kidani, A.; Tawara, A.; Kohno, K. Tip60 Is Regulated by Circadian Transcription Factor Clock and Is Involved in Cisplatin Resistance. J. Biol. Chem. 2008, 283, 18218–18226. [Google Scholar] [CrossRef]
- Bandyopadhyay, K.; Banères, J.-L.; Martin, A.; Blonski, C.; Parello, J.; Gjerset, R. Spermidinyl-CoA-based HAT inhibitors block DNA repair and provide cancer- specific chemo-and radiosensitization. Cell Cycle 2009, 8, 2779–2788. [Google Scholar] [CrossRef]
- Di Martile, M.; Del Bufalo, D.; Trisciuoglio, D. The multifaceted role of lysine acetylation in cancer: Prognostic biomarker and therapeutic target. Oncotarget 2016, 7, 55789–55810. [Google Scholar] [CrossRef]
- Trisciuoglio, D.; Di Martile, M.; Del Bufalo, D. Emerging Role of Histone Acetyltransferase in Stem Cells and Cancer. Stem Cells Int. 2018, 2018, 8908751. [Google Scholar] [CrossRef]
- Hirano, G.; Izumi, H.; Kidani, A.; Yasuniwa, Y.; Han, B.; Kusaba, H.; Akashi, K.; Kuwano, M.; Kohno, K. Enhanced Expression of PCAF Endows Apoptosis Resistance in Cisplatin-Resistant Cells. Mol. Cancer Res. 2010, 8, 864–872. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Classification | Location | Main Functions | Cancer-Related Progress | Type of Cancer Cells | Refs. |
|---|---|---|---|---|---|---|
| HDAC1 | Class I | Nucleus | cell cycle, proliferation and apoptosis regulation, and multidrug resistance | triggering proliferation, angiogenesis, invasion, migration, and apoptosis suppression | lung cancer and breast cancer | [64,65] |
| Multidrug resistance | neuroblastoma | [49] | ||||
| HDAC2 | Class I | Nucleus | proliferation and apoptosis regulation | Cancer proliferation and progression | breast cancer | [66] |
| Cancer progression and resistance | colorectal cancer | [67,68] | ||||
| Higher order chromatin changes and cellular DNA damage responses in chemotherapy | ovarian cancer | [69] | ||||
| HDAC3 | Class I | Nucleus | proliferation and apoptosis regulation | Proliferation and differentiation | colon cancer | [47] |
| Growth and metastasis | ovarian cancer | [70] | ||||
| Chemosensitivity decrease | prostate cancer | [71] | ||||
| HDAC4 | class IIa | Cytoplasm and nucleus | angiogenesis and differentiation | Tumorigenesis, EMT, and angiogenesis | lung cancer | [50,72,73] |
| Angiogenesis | acute T cell leukemia | [50] | ||||
| HDAC5 | class IIa | Cytoplasm and nucleus | differentiation | MEL cell differentiation | erythroleukemia | [53] |
| Migration promotion | colon cancer | [74] | ||||
| Hormone therapy resistance | ER + breast cancer | [75] | ||||
| HDAC6 | class IIb | Cytoplasm | angiogenesis, EMT, and migration | Angiogenesis | renal cell carcinoma | [50] |
| EMT and migration | lung cancer | [51] | ||||
| Migration | acute T cell leukemia | [52] | ||||
| HDAC7 | class IIa | Cytoplasm and nucleus | morphology, migration, and forming capillary tube-like structures | Tumorigenesis | lung cancer | [76] |
| Proliferation, invasion, and migration | colorectal cancer | [77,78] | ||||
| HDAC8 | Class I | Nucleus | cell proliferation, cell cycle, and differentiation | Proliferation promotion and apoptosis inhibition | hepatocellular carcinoma | [79] |
| Antiapoptosis | colon cancer | [80] | ||||
| Cell cycle control, differentiation, and apoptosis | neuroblastoma | [55] | ||||
| Invasion | breast cancer | [81] | ||||
| HDAC9 | class IIa | Cytoplasm and nucleus | cell proliferation, chemoresistance, invasion, and angiogenesis | Invasion and angiogenesis | triple-negative breast cancer | [57] |
| Poor prognosis and immune cell infiltration inhibition | renal cell carcinoma | [58] | ||||
| Drug resistance, proliferation, and autophagic flux | gastric cancer | [56] | ||||
| HDAC10 | class IIb | Nucleus and cytoplasm | ferroptosis inhibition and DNA damage repair | Ferroptosis inhibition and DNA damage repair | lung cancer | [59,60,61] |
| Chemoresistance: drug efflux promotion and DNA damage repair | neuroblastoma | [82] | ||||
| HDAC11 | class IV | Nucleus and cytoplasm | proliferation and apoptosis regulation, stemness enhancement, and resistance | Stemness and drug resistance | hepatocellular carcinoma and lung cancer | [63,83] |
| Metastasis | colorectal cancer and breast cancer | [84,85] | ||||
| SIRT1 | class III | Nucleus | apoptosis attenuation, cancer formation and development, migration and invasion promotion (oncogene), DNA repair promotion, and antiapoptotic protein upregulation (tumor suppressor) | Apoptosis attenuation | lung and breast cancer | [86,87] |
| Cancer formation and development | Breast cancer | [88,89] | ||||
| Migration and invasion | colorectal cancer | [90] | ||||
| DNA repair promotion | osteosarcoma and lung cancer | [91,92] | ||||
| Antiapoptotic protein upregulation | breast cancer | [93] | ||||
| SIRT2 | class III | Cytoplasm | regulate mitosis and genome integrity (tumor suppressor) | Regulating mitosis and genome integrity | hepatocellular carcinoma | [94] |
| Cisplatin sensitivity | ovarian cancer | [95] | ||||
| Regulating metabolism and inhibiting metastases | colorectal cancer | [96] | ||||
| SIRT3 | class III | Mitochondrion | reduce superoxide levels and maintain genomic stability (tumor suppressor) | Tumor growth suppression by inhibiting ROS production | colon cancer | [97] |
| Decreasing mitochondrial superoxide levels | mammary tumor | [98] | ||||
| SIRT4 | class III | Mitochondrion | glutamine metabolism block and DNA damage response (tumor suppressor) | Glutamine metabolism block and DNA damage response | lung cancer | [99] |
| Suppressed proliferation, migration, and invasion | colorectal cancer | [100] | ||||
| self-renewal promotion | breast cancer | [101] | ||||
| SIRT5 | class III | Mitochondrion | cell growth and drug resistance | Cancer cell growth, transformation, and drug resistance | lung cancer | [102,103] |
| Migration | hepatocellular carcinoma | [104] | ||||
| Suppressing DNA damage and cisplatin resistance | ovarian cancer | [105] | ||||
| SIRT6 | class III | Nucleus | gene expression and metabolic process regulation, telomeric chromatin modulation, DNA repair regulation, and inflammation attenuation (tumor suppressor) | p53 and p73 apoptotic signaling activation | cervical carcinoma and breast tumor | [106] |
| Cell growth suppression | hepatocellular carcinoma | [107] | ||||
| Apoptosis induction | endometrial cancer | [108] | ||||
| SIRT7 | class III | Nucleus | transformed state of cancer cell stabilization, tumor aggressiveness, and poor prognosis | Oncogenic transformation, aggressive tumor phenotypes, and poor prognosis | fibrosarcoma | [109] |
| Cancer progression promotion | lung cancer | [110] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, Y.; Luo, Q.; Gu, L.; Tian, X.; Zhao, Y.; Zhang, Y.; Wang, F. Histone Deacetylase Inhibitors Promote the Anticancer Activity of Cisplatin: Mechanisms and Potential. Pharmaceuticals 2025, 18, 563. https://doi.org/10.3390/ph18040563
Zhou Y, Luo Q, Gu L, Tian X, Zhao Y, Zhang Y, Wang F. Histone Deacetylase Inhibitors Promote the Anticancer Activity of Cisplatin: Mechanisms and Potential. Pharmaceuticals. 2025; 18(4):563. https://doi.org/10.3390/ph18040563
Chicago/Turabian StyleZhou, Yang, Qun Luo, Liangzhen Gu, Xiao Tian, Yao Zhao, Yanyan Zhang, and Fuyi Wang. 2025. "Histone Deacetylase Inhibitors Promote the Anticancer Activity of Cisplatin: Mechanisms and Potential" Pharmaceuticals 18, no. 4: 563. https://doi.org/10.3390/ph18040563
APA StyleZhou, Y., Luo, Q., Gu, L., Tian, X., Zhao, Y., Zhang, Y., & Wang, F. (2025). Histone Deacetylase Inhibitors Promote the Anticancer Activity of Cisplatin: Mechanisms and Potential. Pharmaceuticals, 18(4), 563. https://doi.org/10.3390/ph18040563