
Central Insulin-like Growth Factor-1 Treatment Enhances Working and Reference Memory by Reducing Neuroinflammation and Amyloid Beta Deposition in a Rat Model of Sporadic Alzheimer’s Disease


Abstract
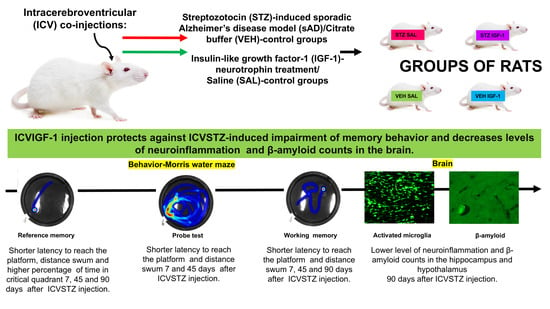
1. Introduction
2. Results
2.1. Spatial Memory Impairments in the Morris Water Maze (MWM) Test
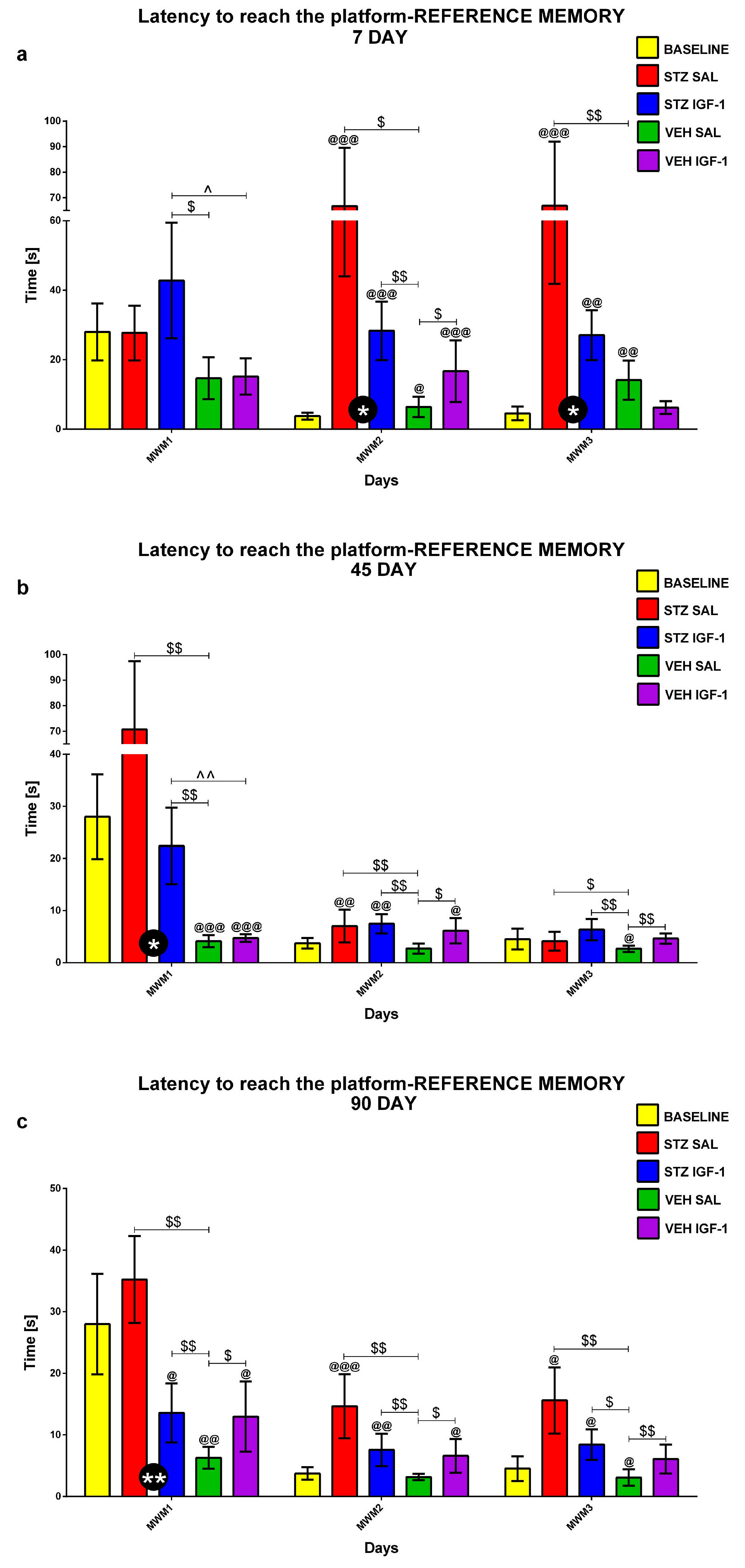
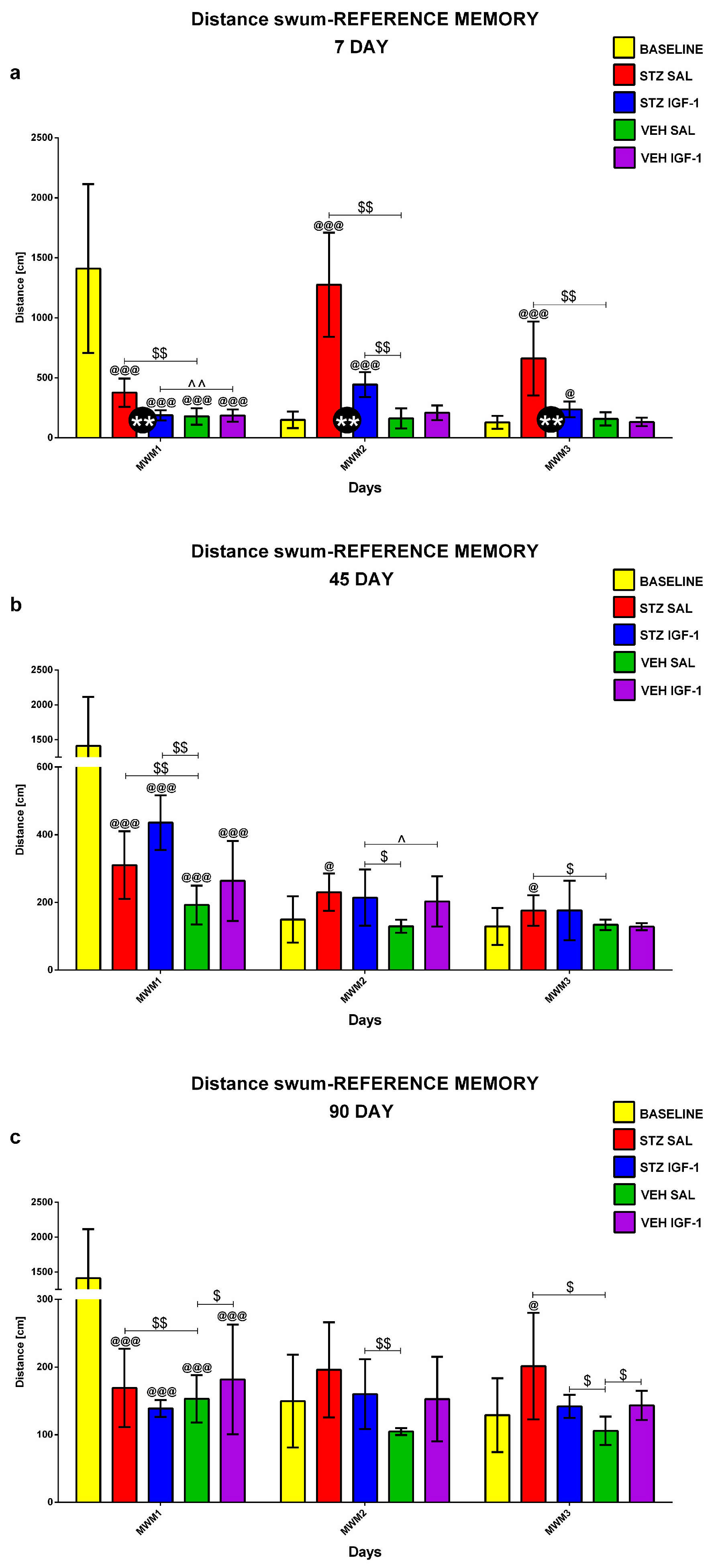
2.1.1. Reference Memory in the Morris Water Maze (MWM) Test
2.1.2. Reference Memory Performance in the Probe Test on Day 4 of the Morris Water Maze (MWM) Test
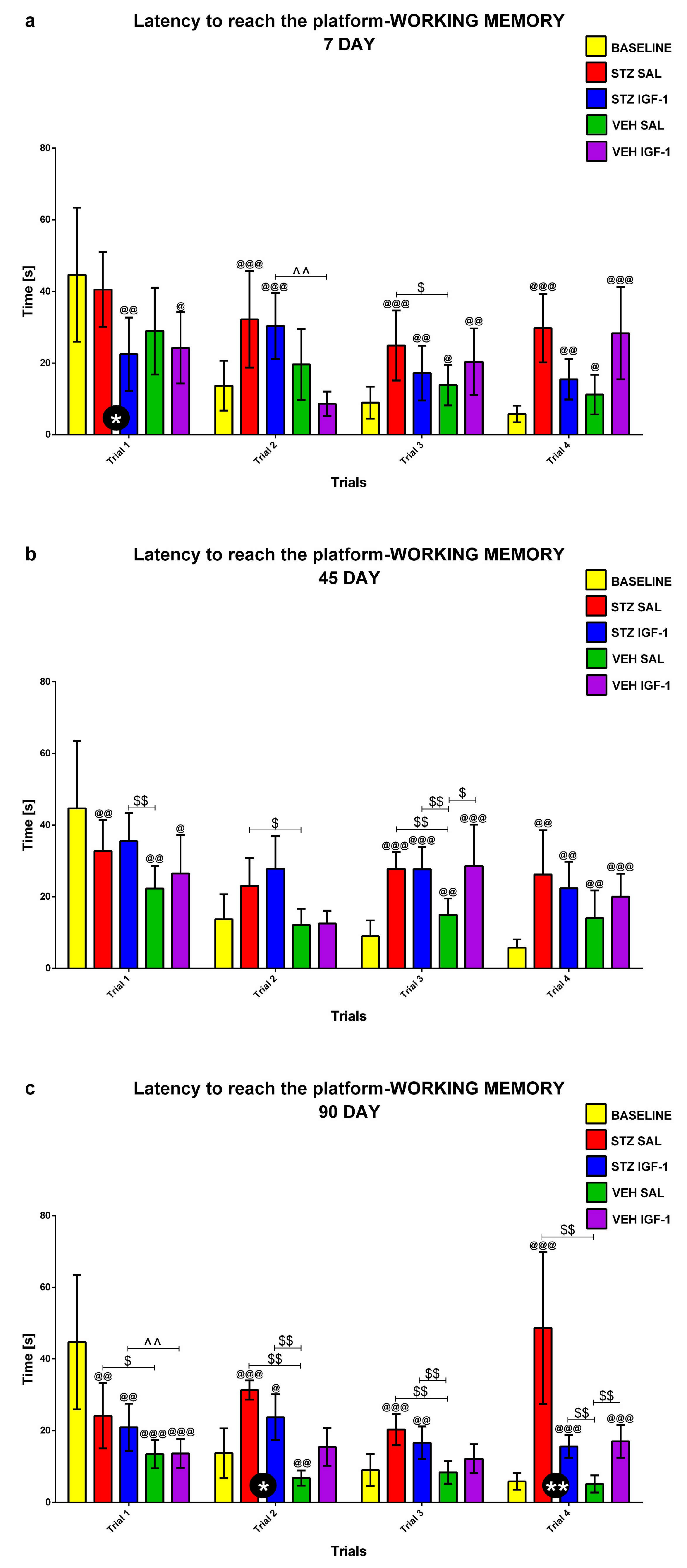
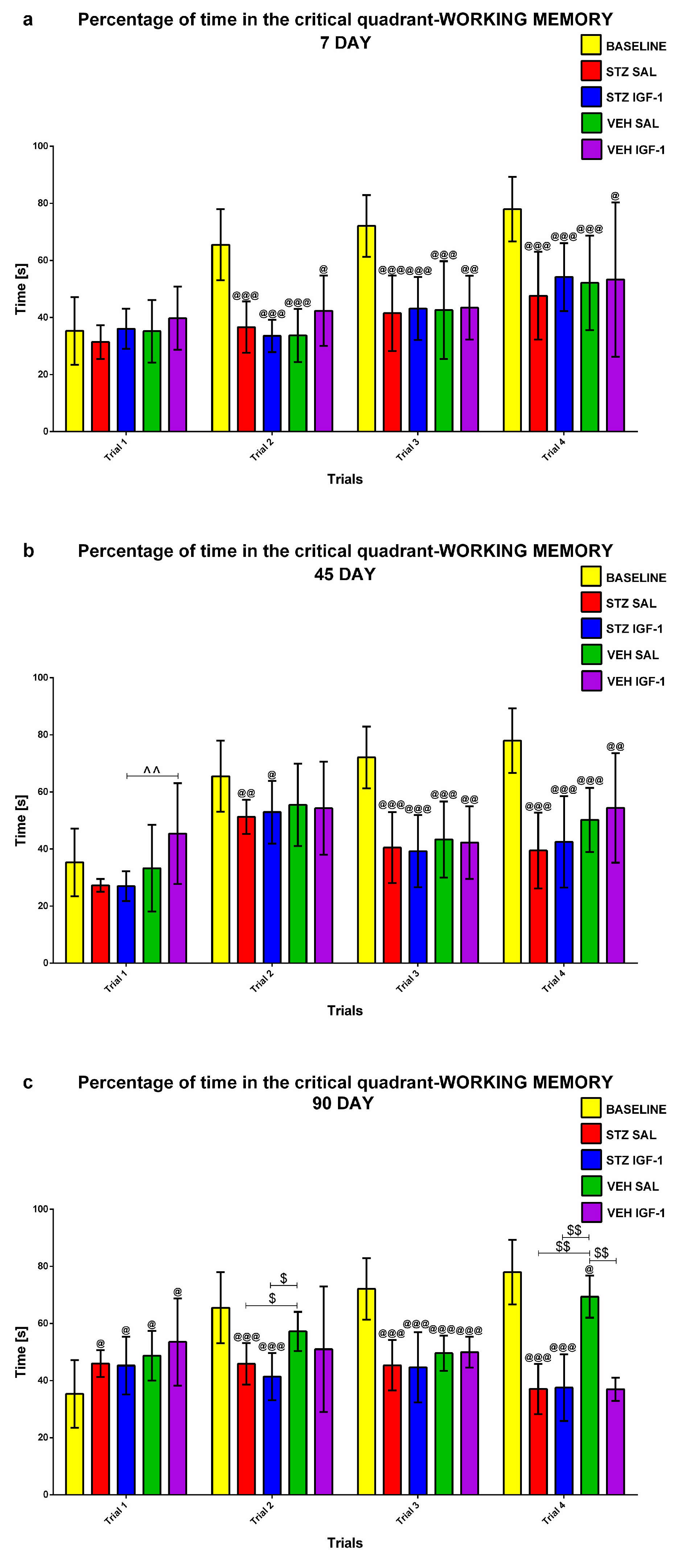
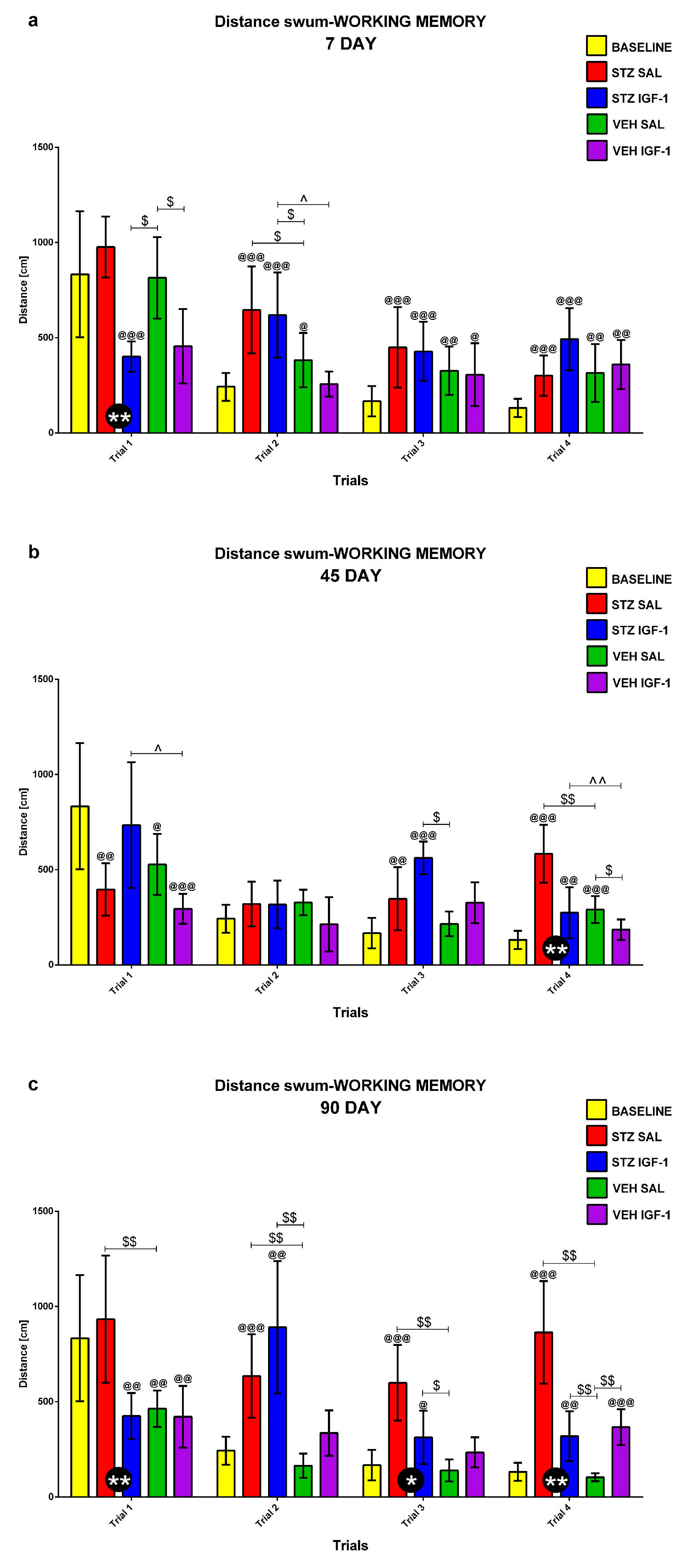
2.1.3. Working Memory During Trials 1–4 of Consecutive Days 5–8 of the Morris Water Maze (MWM) Test
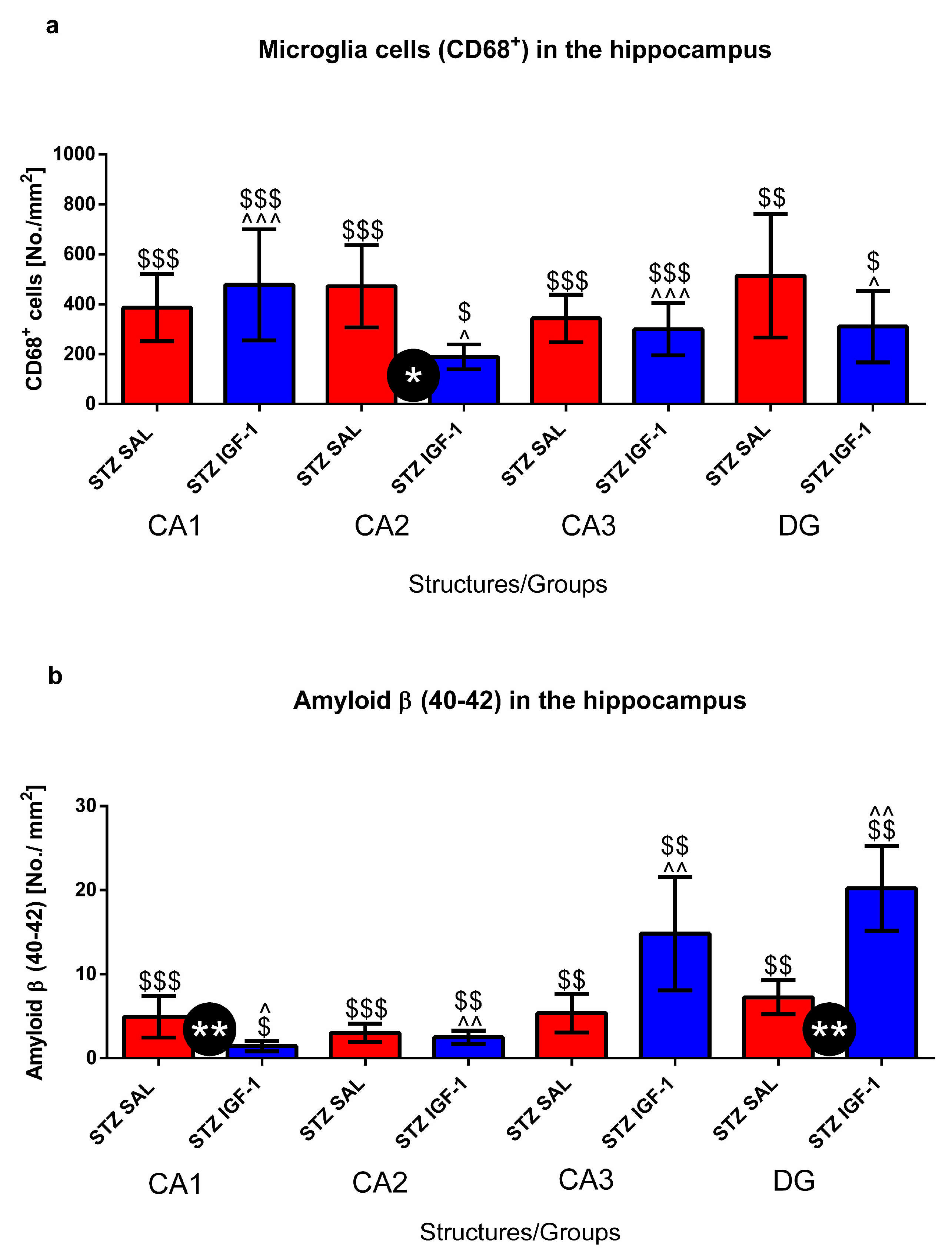
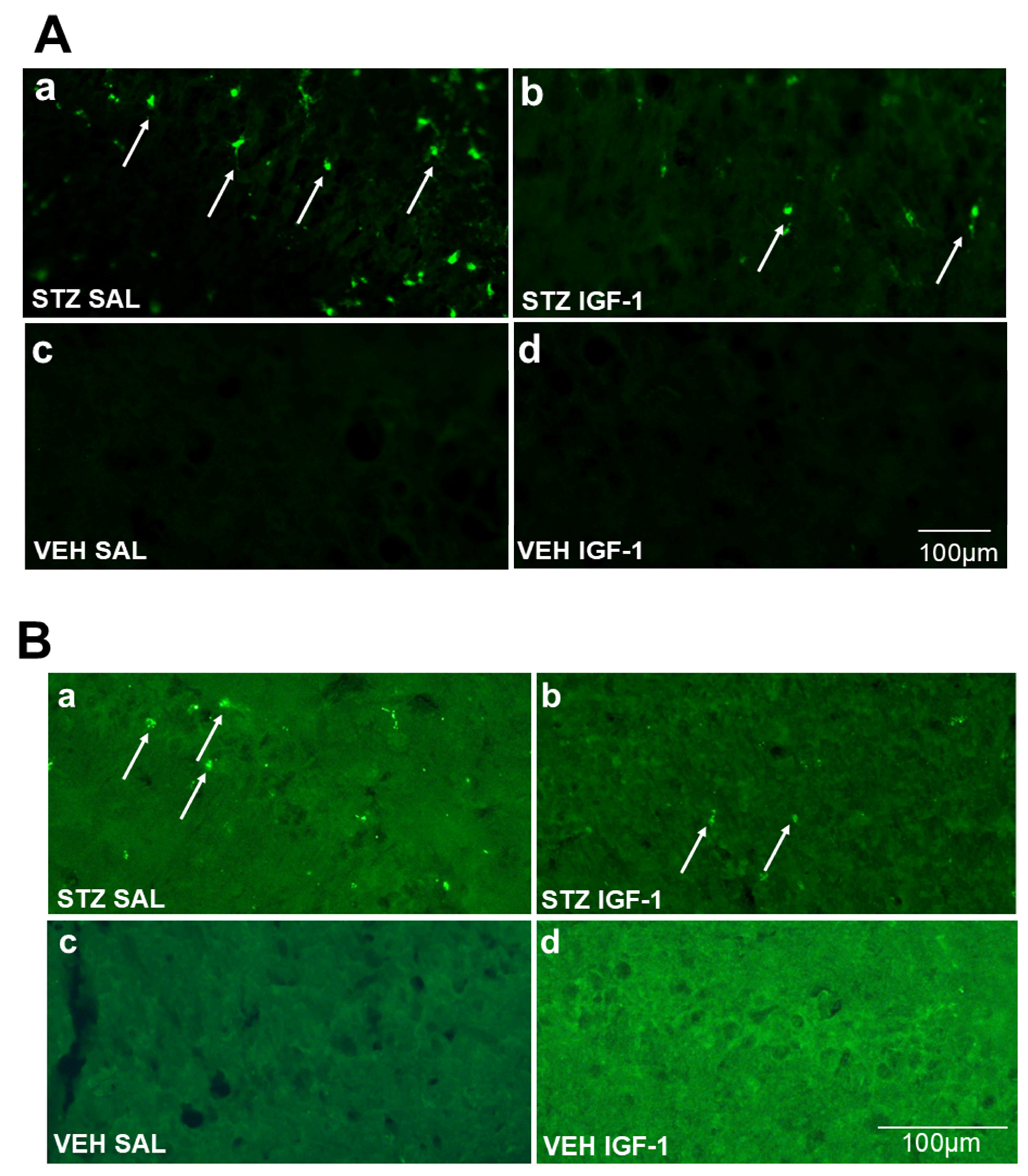
2.2. Microglia Activated Cells (CD68+ Cells) and Amyloid β (Aβ40–42) Deposition in the Hippocampus (CA1, CA2, CA3, DG)
3. Discussion
4. Materials and Methods
4.1. Animals
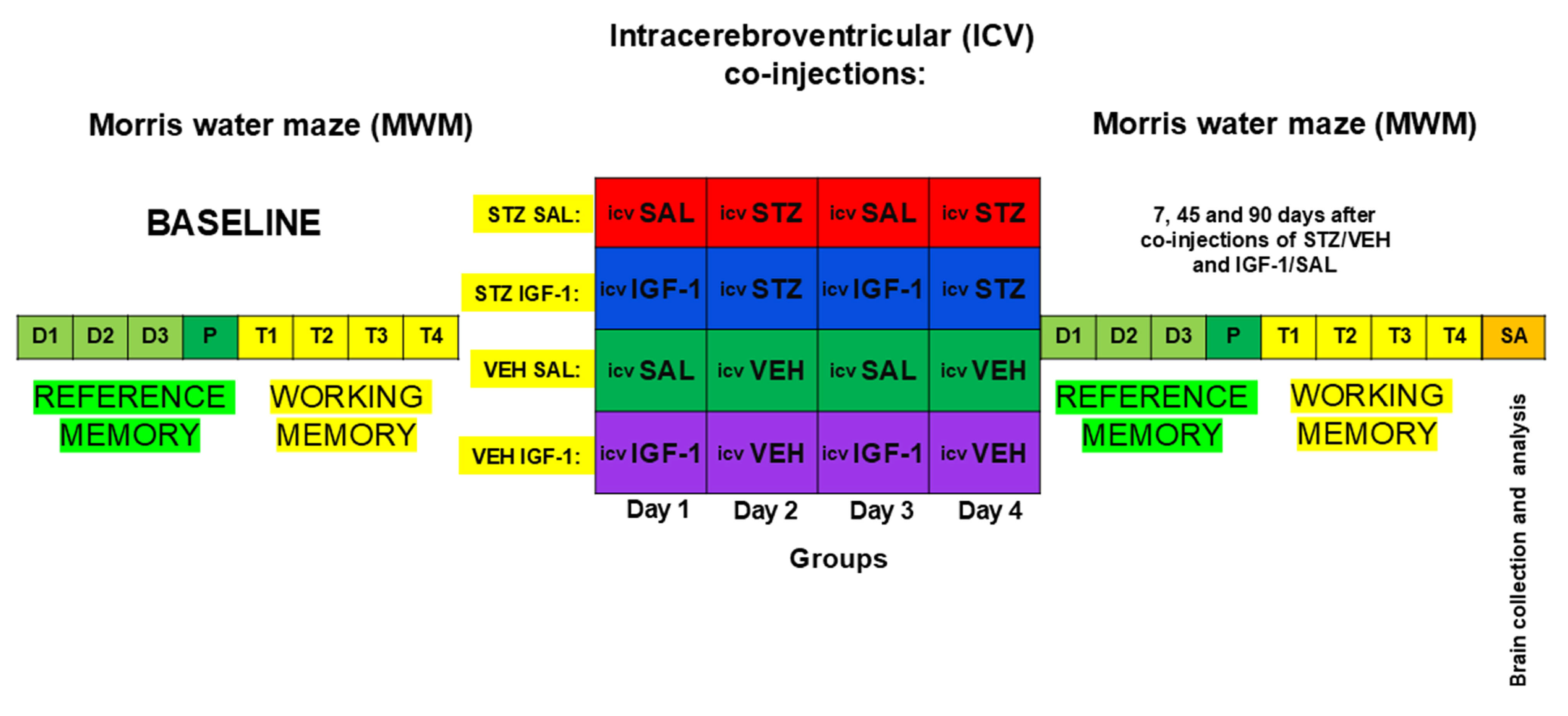
4.2. Experimental Procedure
4.2.1. Intracerebroventricular (ICV) Co-Injections of Insulin-Like Growth Factor-1 (IGF-1) and Streptozotocin (STZ)
4.2.2. Drug Administration
4.2.3. Behavior Associated with Reference and Working Memory in Morris Water Maze (MWM)
4.2.4. Immunohistochemistry
4.2.5. Quantification of Labeled Cells
4.2.6. Slaughtering Animals and Collection of Tissues
4.2.7. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| IGF-1 | Insulin-like growth factor-1 |
| SAL | Saline |
| Aβ | Amyloid βeta |
| STZ | Streptozotocin |
| VEH | Citrate buffer |
| AD | Alzheimer’s disease |
| sAD | Sporadic Alzheimer’s disease |
| CD68+ cells | activated microglia cells |
| ICV | Intracerebroventricular |
| MWM | Morris Water Maze test |
| CA1 CA2 | Cornus ammonis 1 area of the hippocampus Cornus ammonis 2 area of the hippocampus |
| CA3 | Cornus ammonis 3 area of the hippocampus |
| DG | Dentate gyrus area of the hippocampus |
| IL-10 | Interleukin-10 |
| IL-4 | Interleukin-4 |
| TNF-α | Tumor necrosis factor-α |
| IL-1β | Interleukin-1β |
| APP | Amyloid precursor protein |
| BDNF | Brain-derived neurotrophic factor |
| IRS-1 | Insulin receptor substrate 1 |
| IRS-2 | Insulin receptor substrate 2 |
| cAMP | Cyclic adenosine monophosphate |
| PI3K PBS | Phosphoinositide 3-kinase-protein kinase B Phosphate-buffered saline |
References
- Miao, J.; Zhang, Y.; Su, C.; Zheng, Q.; Guo, J. Insulin-like growth factor signaling in Alzheimer’s disease: Pathophysiology and therapeutic strategies. Mol. Neurobiol. 2024. Epub ahead of print. [Google Scholar] [CrossRef]
- Qiao, X.; Yan, J.; Zang, Z.; Xi, L.; Zhu, W.; Zhang, E.; Wu, L. Association between IGF-1 levels and MDD: A case-control and meta-analysis. Front. Psychiatry 2024, 15, 1396938. [Google Scholar] [CrossRef]
- Gray, S.C.; Kinghorn, K.J.; Woodling, N.S. Shifting equilibriums in Alzheimer’s disease: The complex roles of microglia in neuroinflammation, neuronal survival and neurogenesis. Neural Regen. Res. 2020, 15, 1208–1219. [Google Scholar] [CrossRef]
- El-Hakim, Y.; Mani, K.K.; Pickle, K.A.; Akbari, Z.; Samiya, N.; Pham, C.; Salas, G.; Pilla, R.; Sohrabji, F. Peripheral, but not central, IGF-1 treatment attenuates stroke-induced cognitive impairment in middle-aged female Sprague Dawley rats: The gut as a therapeutic target. Brain Behav. Immun. 2024, 122, 150–166. [Google Scholar] [CrossRef]
- Zhao, F.; Siu, J.J.; Huang, W.; Askwith, C.; Cao, L. Insulin modulates excitatory synaptic transmission and synaptic plasticity in the mouse hippocampus. Neuroscience 2019, 411, 237–254. [Google Scholar] [CrossRef]
- Takeishi, J.; Tatewaki, Y.; Nakase, T.; Takano, Y.; Tomita, N.; Yamamoto, S.; Mutoh, T.; Taki, Y. Alzheimer’s disease and type 2 diabetes mellitus: The use of MCT oil and a ketogenic diet. Int. J. Mol. Sci. 2021, 22, 12310. [Google Scholar] [CrossRef]
- Zappa Villar, M.F.; López Hanotte, J.; Crespo, R.; Pardo, J.; Reggiani, P.C. Insulin-like growth factor 1 gene transfer for sporadic Alzheimer’s disease: New evidence for trophic factor mediated hippocampal neuronal and synaptic recovery-based behavior improvement. Hippocampus 2021, 31, 1137–1153. [Google Scholar] [CrossRef]
- Chami, B.; Steel, A.J.; De La Monte, S.M.; Sutherland, G.T. The rise and fall of insulin signaling in Alzheimer’s disease. Metab. Brain Dis. 2016, 31, 497–515. [Google Scholar] [CrossRef]
- Dineley, K.T.; Jahrling, J.B.; Denner, L. Insulin resistance in Alzheimer’s disease. Neurobiol. Dis. 2014, 72, 92–103. [Google Scholar] [CrossRef]
- Lewitt, M.S.; Boyd, G.W. The role of insulin-like growth factors and insulin-like growth factor-binding proteins in the nervous system. Biochem. Insights 2019, 12, 1–18. [Google Scholar] [CrossRef]
- Fluca, A.L.; Pani, B.; Janjusevic, M.; Zwas, D.R.; Abraham, Y.; Calligaris, M.; Beltrami, A.P.; Campos Corgosinho, F.; Marketou, M.; D’Errico, S.; et al. Unraveling the relationship among insulin resistance, IGF-1, and amyloid-beta 1-40: Is the definition of type 3 diabetes applicable in the cardiovascular field? Life Sci. 2024, 352, 122911. [Google Scholar] [CrossRef] [PubMed]
- Bi, A.; An, W.; Wang, C.; Hua, Y.; Fang, F.; Dong, X.; Chen, R.; Zhang, Z.; Luo, L. SCR-1693 inhibits tau phosphorylation and improves insulin resistance associated cognitive deficits. Neuropharmacology 2020, 168, 108027. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.K.; Singh, S.; Shukla, S.; Shukla, R. Intracerebroventricular streptozotocin impairs adult neurogenesis and cognitive functions via regulating neuroinflammation and insulin signaling in adult rats. Neurochem. Int. 2018, 113, 56–68. [Google Scholar] [CrossRef] [PubMed]
- Ma, N.; Liang, Y.; Yue, L.; Liu, P.; Xu, Y.; Zhu, C. The identities of insulin signaling pathway are affected by overexpression of Tau and its phosphorylation form. Front. Aging Neurosci. 2022, 14, 1057281. [Google Scholar] [CrossRef]
- Hamzé, R.; Delangre, E.; Tolu, S.; Moreau, M.; Janel, N.; Bailbé, D.; Movassat, J. Type 2 diabetes mellitus and Alzheimer’s disease: Shared molecular mechanisms and potential common therapeutic targets. Int. J. Mol. Sci. 2022, 23, 15287. [Google Scholar] [CrossRef]
- Bhalla, S.; Mehan, S.; Khan, A.; Rehman, M.U. Protective role of IGF-1 and GLP-1 signaling activation in neurological dysfunctions. Neurosci. Biobehav. Rev. 2022, 142, 104896. [Google Scholar] [CrossRef]
- Huffman, D.M.; Farias Quipildor, G.; Mao, K.; Zhang, X.; Wan, J.; Apontes, P.; Cohen, P.; Barzilai, N. Central insulin-like growth factor-1 (IGF-1) restores whole-body insulin action in a model of age-related insulin resistance and IGF-1 decline. Aging Cell 2016, 15, 181–186. [Google Scholar] [CrossRef]
- Farias Quipildor, G.E.; Mao, K.; Hu, Z.; Novaj, A.; Cui, M.H.; Gulinello, M.; Branch, C.A.; Gubbi, S.; Patel, K.; Moellering, D.R.; et al. Central IGF-1 protects against features of cognitive and sensorimotor decline with aging in male mice. Geroscience 2019, 41, 185–208. [Google Scholar] [CrossRef]
- Gubbi, S.; Quipildor, G.F.; Barzilai, N.; Huffman, D.M.; Milman, S. 40 years of IGF1: IGF1: The Jekyll and Hyde of the aging brain. J. Mol. Endocrinol. 2018, 61, T171–T185. [Google Scholar] [CrossRef]
- Ashpole, N.M.; Herron, J.C.; Mitschelen, M.C.; Farley, J.A.; Logan, S.; Yan, H.; Ungvari, Z.; Hodges, E.L.; Csiszar, A.; Ikeno, Y.; et al. IGF-1 regulates vertebral bone aging through sex-specific and time-dependent mechanisms. J. Bone Miner. Res. 2016, 31, 443–454. [Google Scholar] [CrossRef]
- Hossain, M.A.; Adithan, A.; Alam, M.J.; Kopalli, S.R.; Kim, B.; Kang, C.W.; Hwang, K.C.; Kim, J.H. IGF-1 facilitates cartilage reconstruction by regulating PI3K/AKT, MAPK, and NF-kB signaling in rabbit osteoarthritis. J. Inflamm. Res. 2021, 14, 3555–3568. [Google Scholar] [CrossRef] [PubMed]
- Herrera, M.L.; Camparini, L.G.; Oliveros, A.L.; Bellini, M.J.; Herenu, C.B. Potentialities of IGF-1 for regulating oxidative stress in neuroinflammation and neurodegeneration: Theoretical review. Explor. Neuroprot. Ther. 2024, 4, 442–458. [Google Scholar] [CrossRef]
- Salkovic-Petrisic, M.; Knezovic, A.; Hoyer, S.; Riederer, P. What have we learned from the streptozotocin-induced animal model of sporadic Alzheimer’s disease, about the therapeutic strategies in Alzheimer’s research. J. Neural. Transm. 2013, 120, 233–252. [Google Scholar] [CrossRef]
- Hoyer, S.; Lannert, H. Long-term abnormalities in brain glucose/energy metabolism after inhibition of the neuronal insulin receptor: Implication of tau-protein. J. Neural. Transm. Suppl. 2007, 72, 195–202. [Google Scholar] [CrossRef]
- Agrawal, R.; Tyagi, E.; Shukla, R.; Nath, C. Insulin receptor signaling in rat hippocampus: A study in STZ (ICV) induced memory deficit model. Eur. Neuropsychopharmacol. 2011, 21, 261–273. [Google Scholar] [CrossRef]
- Rai, S.; Kamat, P.K.; Nath, C.; Shukla, R. Glial activation and post-synaptic neurotoxicity: The key events in streptozotocin (ICV) induced memory impairment in rats. Pharmacol. Biochem. Behav. 2014, 117, 104–117. [Google Scholar] [CrossRef]
- Fuster-Matanzo, A.; Llorens-Martín, M.; Hernández, F.; Avila, J. Role of neuroinflammation in adult neurogenesis and Alzheimer disease: Therapeutic approaches. Mediat. Inflamm. 2013, 2013, 260925. [Google Scholar] [CrossRef]
- Majkutewicz, I.; Kurowska, E.; Podlacha, M.; Myślińska, D.; Grembecka, B.; Ruciński, J.; Plucińska, K.; Jerzemowska, G.; Wrona, D. Dimethyl fumarate attenuates intracerebroventricular streptozotocin-induced spatial memory impairment and hippocampal neurodegeneration in rats. Behav. Brain Res. 2016, 308, 24–37. [Google Scholar] [CrossRef]
- Majkutewicz, I.; Kurowska, E.; Podlacha, M.; Myślińska, D.; Grembecka, B.; Ruciński, J.; Pierzynowska, K.; Wrona, D. Age-dependent effects of dimethyl fumarate on cognitive and neuropathological features in the streptozotocin-induced rat model of Alzheimer’s disease. Brain Res. 2018, 1686, 19–33. [Google Scholar] [CrossRef]
- Kurowska-Rucińska, E.; Ruciński, J.; Myślińska, D.; Grembecka, B.; Wrona, D.; Majkutewicz, I. Dimethyl fumarate alleviates adult neurogenesis disruption in hippocampus and olfactory bulb and spatial cognitive deficits induced by intracerebroventricular streptozotocin injection in young and aged rats. Int. J. Mol. Sci. 2022, 23, 15449. [Google Scholar] [CrossRef]
- Wrona, D.; Majkutewicz, I.; Świątek, G.; Dunacka, J.; Grembecka, B.; Glac, W. Dimethyl fumarate as the peripheral blood inflammatory mediators inhibitor in prevention of streptozotocin-induced neuroinflammation in aged rats. J. Inflamm. Res. 2022, 15, 33–52. [Google Scholar] [CrossRef] [PubMed]
- Dunacka, J.; Świątek, G.; Wrona, D. High behavioral reactivity to novelty as a susceptibility factor for memory and anxiety disorders in streptozotocin-induced neuroinflammation as a rat model of Alzheimer’s disease. Int. J. Mol. Sci. 2024, 25, 11562. [Google Scholar] [CrossRef] [PubMed]
- Talbot, K.; Wang, H.Y.; Kazi, H.; Han, L.Y.; Bakshi, K.P.; Stucky, A.; Fuino, R.L.; Kawaguchi, K.R.; Samoyedny, A.J.; Wilson, R.S.; et al. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J. Clin. Investig. 2012, 122, 1316–1338. [Google Scholar] [CrossRef] [PubMed]
- Moloney, A.M.; Griffin, R.J.; Timmons, S.; O’Connor, R.; Ravid, R.; O’Neill, C. Defects in IGF-1 receptor, insulin receptor and IRS-1/2 in Alzheimer’s disease indicate possible resistance to IGF-1 and insulin signaling. Neurobiol. Aging 2010, 31, 224–243. [Google Scholar] [CrossRef]
- Reger, M.A.; Watson, G.S.; Green, P.S.; Baker, L.D.; Cholerton, B.; Fishel, M.A.; Plymate, S.R.; Cherrier, M.M.; Schellenberg, G.D.; Frey, W.H., II; et al. Intranasal insulin administration dose-dependently modulates verbal memory and plasma amyloid-β in memory-impaired older adults. J. Alzheimers Dis. 2008, 13, 323–331. [Google Scholar] [CrossRef]
- Craft, S.; Baker, L.D.; Montine, T.J.; Minoshima, S.; Watson, G.S.; Claxton, A.; Arbuckle, M.; Callaghan, M.; Tsai, E.; Plymate, S.R.; et al. Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment, a pilot clinical trial. Arch. Neurol. 2012, 69, 29–38. [Google Scholar] [CrossRef]
- Shoham, S.; Bejar, C.; Kovalev, E.; Weinstock, M. Intracerebroventricular injection of streptozotocin causes neurotoxicity to myelin that contributes to spatial memory deficits in rats. Exp. Neurol. 2023, 184, 1043–1052. [Google Scholar] [CrossRef]
- Grűnblatt, E.; Salkovic-Petrisic, M.; Osmanovic, J.; Riederer, P.; Hoyer, S. Brain insulin system dysfunction in streptozotocin intracerebroventriculary treated rats generates hyperphosphorylated tau protein. J. Neurochem. 2007, 101, 757–770. [Google Scholar] [CrossRef]
- Salkovic-Petrisic, M.; Tribl, F.; Schmidt, M.; Hoyer, S.; Riederer, P. Alzheimer-like changes in protein kinase B and glycogen synthase kinase-3 in rat frontal cortex and hippocampus after damage to the insulin signaling pathway. J. Neurochem. 2006, 96, 1005–1015. [Google Scholar] [CrossRef]
- Salkovic-Petrisic, M.; Osmanovic-Barilar, J.; Bruckner, M.K.; Hoyer, S.; Arendt, T.; Riederer, P. Cerebral amyloid angiopathy in streptozotocin rat model of sporadic Alzhimer’s disease: A long-term follow up study. J. Neural. Transm. 2011, 118, 765–772. [Google Scholar] [CrossRef]
- Grűnblatt, E.; Hoyer, S.; Riederer, P. Gene expression profile in streptozotocin rat model for sporadic Alzheimer’s disease. J. Neural Transm. 2004, 111, 367–386. [Google Scholar] [CrossRef] [PubMed]
- Aguado-Llera, D.; Canelles, S.; Frago, L.M.; Chowen, J.A.; Argente, J.; Arilla, E.; Barrios, V. The protective effects of IGF-I against β-amyloid-related downregulation of hippocampal somatostatinergic system involve activation of Akt and protein kinase A. Neuroscience 2018, 374, 104–118. [Google Scholar] [CrossRef] [PubMed]
- Ghaffari, M.K.; Rafati, A.; Karbalaei, N.; Haghani, M.; Nemati, M.; Sefati, N.; Namavar, M.R. The effect of intra-nasal co-treatment with insulin and growth factor-rich serum on behavioral defects, hippocampal oxidative-nitrosative stress, and histological changes induced by icv-STZ in a rat model. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2024, 397, 4833–4849. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Wu, K.; Gu, L.; Huang, L.; Zhuge, Q.; Yang, S.; Wang, Z. IGF-1R stimulation alters microglial polarization via TLR4/NF-κB pathway after cerebral hemorrhage in mice. Brain Res. Bull. 2020, 164, 221–234. [Google Scholar] [CrossRef]
- Toth, L.; Czigler, A.; Hegedus, E.; Komaromy, H.; Amrein, K.; Czeiter, E.; Yabluchanskiy, A.; Koller, A.; Orsi, G.; Perlaki, G.; et al. Age-related decline in circulating IGF-1 associates with impaired neurovascular coupling responses in older adults. GeroScience 2022, 44, 2771–2783. [Google Scholar] [CrossRef]
- Pinto-Benito, D.; Paradela-Leal, C.; Ganchala, D.; de Castro-Molina, P.; Arevalo, M.A. IGF-1 regulates astrocytic phagocytosis and inflammation through the p110α isoform of PI3K in a sex-specific manner. Glia 2022, 70, 1153–1169. [Google Scholar] [CrossRef]
- Fernandez, A.M.; Hernandez-Garzón, E.; Perez-Domper, P.; Perez-Alvarez, A.; Mederos, S.; Matsui, T.; Santi, A.; Trueba-Saiz, A.; García-Guerra, L.; Pose-Utrilla, J.; et al. Insulin regulates astrocytic glucose handling through cooperation with IGF-I. Diabetes 2017, 1, 64–74. [Google Scholar] [CrossRef]
- Park, S.E.; Dantzer, R.; Kelley, K.W.; McCusker, R.H. Central administration of insulin-like growth factor-I decreases depressive-like behavior and brain cytokine expression in mice. J. Neuroinflammation 2011, 8, 12. [Google Scholar] [CrossRef]
- Carro, E.; Spuch, C.; Trejo, J.L.; Antequera, D.; Torres-Aleman, I. Choroid plexus megalin is involved in neuroprotection by serum insulin-like growth factor I. J. Neurosci. 2005, 25, 10884–10893. [Google Scholar] [CrossRef]
- Ma, Q.L.; Yang, F.; Rosario, E.R.; Ubeda, O.J.; Beech, W.; Gant, D.J.; Chen, P.P.; Hudspeth, B.; Chen, C.; Zhao, Y.; et al. β-amyloid oligomers induce phosphorylation of tau and inactivation of insulin receptor substrate via c-Jun N-terminal kinase signaling: Suppression by omega-3 fatty acids and curcumin. J. Neurosci. 2009, 29, 9078–9089. [Google Scholar] [CrossRef]
- Bomfim, T.R.; Forny-Germano, L.; Sathler, L.B.; Brito-Moreira, J.; Houzel, J.C.; Decker, H.; Silverman, M.A.; Kazi, H.; Melo, H.M.; McClean, P.L.; et al. An anti-diabetes agent protects the mouse brain from defective insulin signaling caused by Alzheimer’s disease–associated Aβ oligomers. J. Clin. Investig. 2012, 122, 1339–1353. [Google Scholar] [CrossRef]
- Freude, S.; Hettich, M.M.; Schumann, C.; Stöhr, O.; Koch, L.; Köhler, C.; Udelhoven, M.; Leeser, U.; Müller, M.; Kubota, N.; et al. Neuronal IGF-1 resistance reduces Abeta accumulation and protects against premature death in a model of Alzheimer’s disease. FASEB J. 2009, 23, 3315–3324. [Google Scholar] [CrossRef]
- Ziegler, A.N.; Levison, S.W.; Wood, T.L. Insulin and IGF-1 receptor signaling in neural-stem-cell homeostasis. Nat. Rev. Endocrinol. 2015, 11, 161–170. [Google Scholar] [CrossRef]
- Kondo, M. Molecular mechanisms of exercise-induced hippocampal neurogenesis and antidepressant effects. JMA J. 2023, 6, 114–119. [Google Scholar] [CrossRef]
- Mir, S.; Cai, W.; Carlson, S.W.; Saatman, K.E.; Andres, D.A. IGF-1 mediated neurogenesis involves a Novel RIT1/Akt/Sox2 Cascade. Sci. Rep. 2017, 7, 3283. [Google Scholar] [CrossRef]
- Liu, W.; Ye, P.; O’Kusky, J.R.; D’Ercole, A.J. Type 1 insulin-like growth factor receptor signaling is essential for the development of the hippocampal formation and dentate gyrus. J. Neurosci. Res. 2009, 87, 2821–2832. [Google Scholar] [CrossRef]
- Soto, M.; Cai, W.; Konishi, M.; Kahn, C.R. Insulin signaling in the hippocampus and amygdala regulates metabolism and neurobehavior. Proc. Natl. Acad. Sci. USA 2019, 116, 6379–6384. [Google Scholar] [CrossRef]
- Hayes, C.A.; Wilson, D.; De Leon, M.A.; Jolayemi Mustapha, M.; Morale, S.; Odden, M.C.; Ashpole, N.M. Insulin-like growth factor-1 and cognitive health: Exploring cellular, preclinical, and clinical dimensions. Front. Neuroendocrinol. 2025, 76, 101161. [Google Scholar] [CrossRef]
- Blume, G.R.; Royes, L.F.F. Peripheral to brain and hippocampus crosstalk induced by exercise mediates cognitive and structural hippocampal adaptations. Life Sci. 2024, 352, 122799. [Google Scholar] [CrossRef]
- Kim, Y.K.; Jo, D.; Arjunan, A.; Ryu, Y.; Lim, Y.H.; Choi, S.Y.; Kim, H.K.; Song, J. Identification of IGF-1 effects on white adipose tissue and hippocampus in Alzheimer’s disease mice via transcriptomic and cellular analysis. Int. J. Mol. Sci. 2024, 25, 2567. [Google Scholar] [CrossRef]
- Jo, D.; Choi, S.Y.; Ahn, S.Y.; Song, J. IGF1 enhances memory function in obese mice and stabilizes the neural structure under insulin resistance via AKT-GSK3β-BDNF signaling. Biomed. Pharmacother. 2025, 183, 117846. [Google Scholar] [CrossRef]
- Akbari, S.; Haghani, M.; Ghobadi, M.; Hooshmandi, E.; Haghighi, A.B.; Salehi, M.S.; Pandamooz, S.; Azarpira, N.; Afshari, A.; Zabihi, S.; et al. Combination therapy with platelet-rich plasma and epidermal neural crest stem cells increases treatment efficacy in vascular dementia. Stem Cells Int. 2023, 2023, 3784843. [Google Scholar] [CrossRef]
- Kim, S.H.; Kim, C.H. Neuronal IGF-1 overexpression restores hippocampal newborn cell survival and recent CFC memory consolidation in Cav1.3 knock-out mice. Brain Res. 2024, 1825, 148712. [Google Scholar] [CrossRef]
- Azargoonjahromi, A. Serotonin enhances neurogenesis biomarkers, hippocampal volumes, and cognitive functions in Alzheimer’s disease. Mol. Brain 2024, 17, 93. [Google Scholar] [CrossRef]
- Kondo, M.; Koyama, Y.; Nakamura, Y.; Shimada, S. A novel 5HT3 receptor-IGF1 mechanism distinct from SSRI-induced antidepressant effects. Mol. Psychiatry 2018, 23, 833–842. [Google Scholar] [CrossRef]
- van Berlo, D.; Woutersen, M.; Muller, A.; Pronk, M.; Vriend, J.; Hakkert, B. 10% Body weight (gain) change as criterion for the maximum tolerated dose: A critical analysis. Regul. Toxicol. Pharmacol. 2022, 134, 105235. [Google Scholar] [CrossRef]
- Paxinos, G.; Watson, C. The Rat Brain in Stereotaxic Coordinates, 6th ed.; Academic Press: San Diego, CA, USA, 2007; ISBN 978-0-0804-7515-8. [Google Scholar]
- Malberg, J.E.; Platt, B.; Sukoff Rizzo, S.J.; Ring, R.H.; Lucki, I.; Schechter, L.E.; Rosenzweig-Lipson, S. Increasing the levels of insulin-like growth factor-1 by an IGF binding protein inhibitor produces anxiolytic and antidepressant-like effects. Neuropsychopharmacology 2007, 32, 2360–2368. [Google Scholar] [CrossRef]
- Basta-Kaim, A.; Szczesny, E.; Glombik, K.; Stachowicz, K.; Slusarczyk, J.; Nalepa, I.; Zelek-Molik, A.; Rafa-Zablocka, K.; Budziszewska, B.; Kubera, M.; et al. Prenatal stress affects insulin-like growth factor-1 (IGF-1) level and IGF-1 receptor phosphorylation in the brain of adult rats. Eur. Neuropsychopharmacol. 2014, 24, 1546–1556. [Google Scholar] [CrossRef]
- Schnieder, T.P.; Trenceska, I.; Rosoklia, G.; Stankov, A.; Mann, J.J.; Smiley, J.; Dwork, A.J. Microglia of prefrontal white matter in suicide. J. Neuropathol. Exp. 2014, 73, 880–890. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Microglia Cells (CD68+) No./mm2 | |||||
| a | Structures/Groups | STZ SAL | STZ IGF-1 | VEH SAL | VEH IGF-1 |
| Prefrontal cortex | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 | |
| Corpus calosum | 560 ± 151.66 | 475 ± 103.51 | 300 ± 115 | 371.43 ± 131.39 | |
| Caudate putamen | 242.86 ± 85.91 $ | 287.5 ± 105.01 $$$^^ | 0 ± 0 | 22.22 ± 11.03 | |
| Medial septal nucleus | 142.86 ± 61.00 | 0 ± 0 | 0 ± 0 | 0 ± 0 | |
| Lateral preoptic area | 57.14 ± 26.73 $ | 0 ± 0 * | 0 ± 0 | 0 ± 0 | |
| Medial preoptic nucleus | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 | |
| Supraoptic nucleus | 550 ± 207.36 $$ | 0 ± 0 *** | 0 ± 0 | 37.5 ± 12.93 | |
| Arcuate hypothalamic nucleus | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 | |
| Dorsomedial hypothalamic nucleus | 42.86 ± 13.37 | 0 ± 0 | 0 ± 0 | 0 ± 0 | |
| Lateral hypothalamus | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 | |
| Paraventricular hypothalamic nucleus | 114.29 ± 36.60 | 0 ± 0 | 0 ± 0 | 0 ± 0 | |
| Ventromedial hypothalamic nucleus | 0 ± 0 | 60 ± 29.44 | 0 ± 0 | 0 ± 0 | |
| Nucleus accumbens-Shell | 71.42 ± 37.40 | 0 ± 0 | 0 ± 0 | 0 ± 0 | |
| Nucleus accumbens-Core | 57.14 ± 19.67 | 0 ± 0 | 0 ± 0 | 0 ± 0 | |
| b | Structures/Groups | VEH SAL | VEH IGF-1 | ||
| Hippocampus CA1 | 0 ± 0 | 0 ± 0 | |||
| Hippocampus CA2 | 0 ± 0 | 0 ± 0 | |||
| Hippocampus CA3 | 0 ± 0 | 0 ± 0 | |||
| Dentate gyrus | 0 ± 0 | 0 ± 0 | |||
| Amyloid β (40–42)/No./mm2 | |||||
| c | Structures/Groups | STZ SAL | STZ IGF-1 | VEH SAL | VEH IGF-1 |
| Prefrontal cortex | 3 ± 1.06 $ | 03.06 ± 1.72 | 0 ± 0 | 0 ± 0 | |
| Lateral preoptic area | 3 ± 1.13 $$ | 2.65 ± 0.97 $$^^ | 0 ± 0 | 0 ± 0 | |
| Medial preoptic nucleus | 5.38 ± 2.36 $$ | 1.99 ± 0.63 $$^^ | 0 ± 0 | 0 ± 0 | |
| Nucleus accumbens-Shell | 2.35 ± 1.5 $ | 3.27 ± 1.34 $$^^^ | 0 ± 0 | 0 ± 0 | |
| Nucleus accumbens-Core | 2.75 ± 0.86 $$$ | 02.16 ± 0.96 $$^^^ | 0 ± 0 | 0 ± 0 | |
| d | Structures/Groups | VEH SAL | VEH IGF-1 | ||
| Hippocampus CA1 | 0 ± 0 | 0 ± 0 | |||
| Hippocampus CA2 | 0 ± 0 | 0 ± 0 | |||
| Hippocampus CA3 | 0 ± 0 | 0 ± 0 | |||
| Dentate gyrus | 0 ± 0 | 0 ± 0 | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dunacka, J.; Grembecka, B.; Majkutewicz, I.; Wrona, D. Central Insulin-like Growth Factor-1 Treatment Enhances Working and Reference Memory by Reducing Neuroinflammation and Amyloid Beta Deposition in a Rat Model of Sporadic Alzheimer’s Disease. Pharmaceuticals 2025, 18, 527. https://doi.org/10.3390/ph18040527
Dunacka J, Grembecka B, Majkutewicz I, Wrona D. Central Insulin-like Growth Factor-1 Treatment Enhances Working and Reference Memory by Reducing Neuroinflammation and Amyloid Beta Deposition in a Rat Model of Sporadic Alzheimer’s Disease. Pharmaceuticals. 2025; 18(4):527. https://doi.org/10.3390/ph18040527
Chicago/Turabian StyleDunacka, Joanna, Beata Grembecka, Irena Majkutewicz, and Danuta Wrona. 2025. "Central Insulin-like Growth Factor-1 Treatment Enhances Working and Reference Memory by Reducing Neuroinflammation and Amyloid Beta Deposition in a Rat Model of Sporadic Alzheimer’s Disease" Pharmaceuticals 18, no. 4: 527. https://doi.org/10.3390/ph18040527
APA StyleDunacka, J., Grembecka, B., Majkutewicz, I., & Wrona, D. (2025). Central Insulin-like Growth Factor-1 Treatment Enhances Working and Reference Memory by Reducing Neuroinflammation and Amyloid Beta Deposition in a Rat Model of Sporadic Alzheimer’s Disease. Pharmaceuticals, 18(4), 527. https://doi.org/10.3390/ph18040527