

The Role of APOA-I in Alzheimer’s Disease: Bridging Peripheral Tissues and the Central Nervous System

 ,
,
Abstract

1. Introduction
2. APOA-I: Physiological Functions, Transport Pathways, and Association with AD
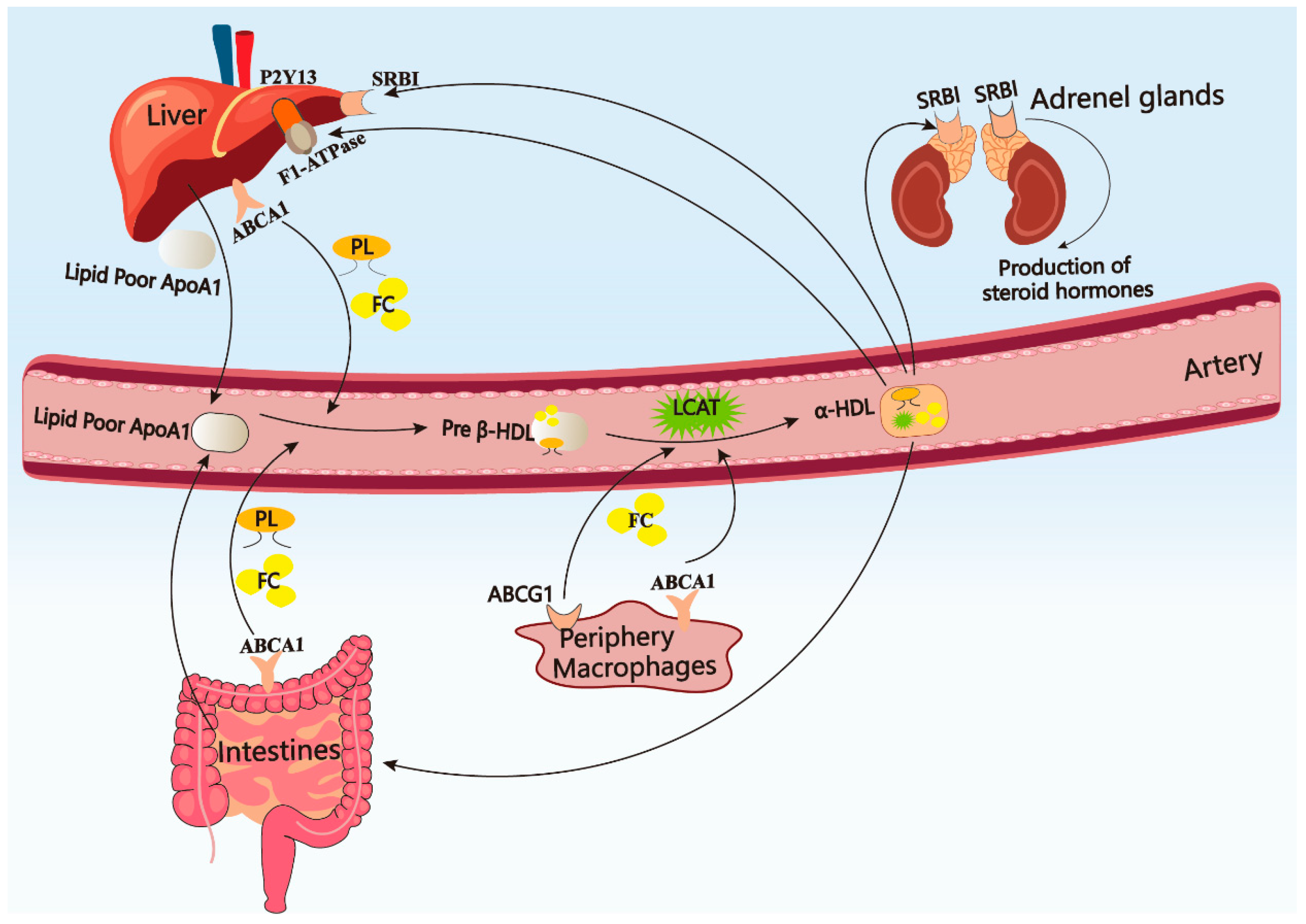
2.1. Metabolism of APOA-I in Peripheral Circulation
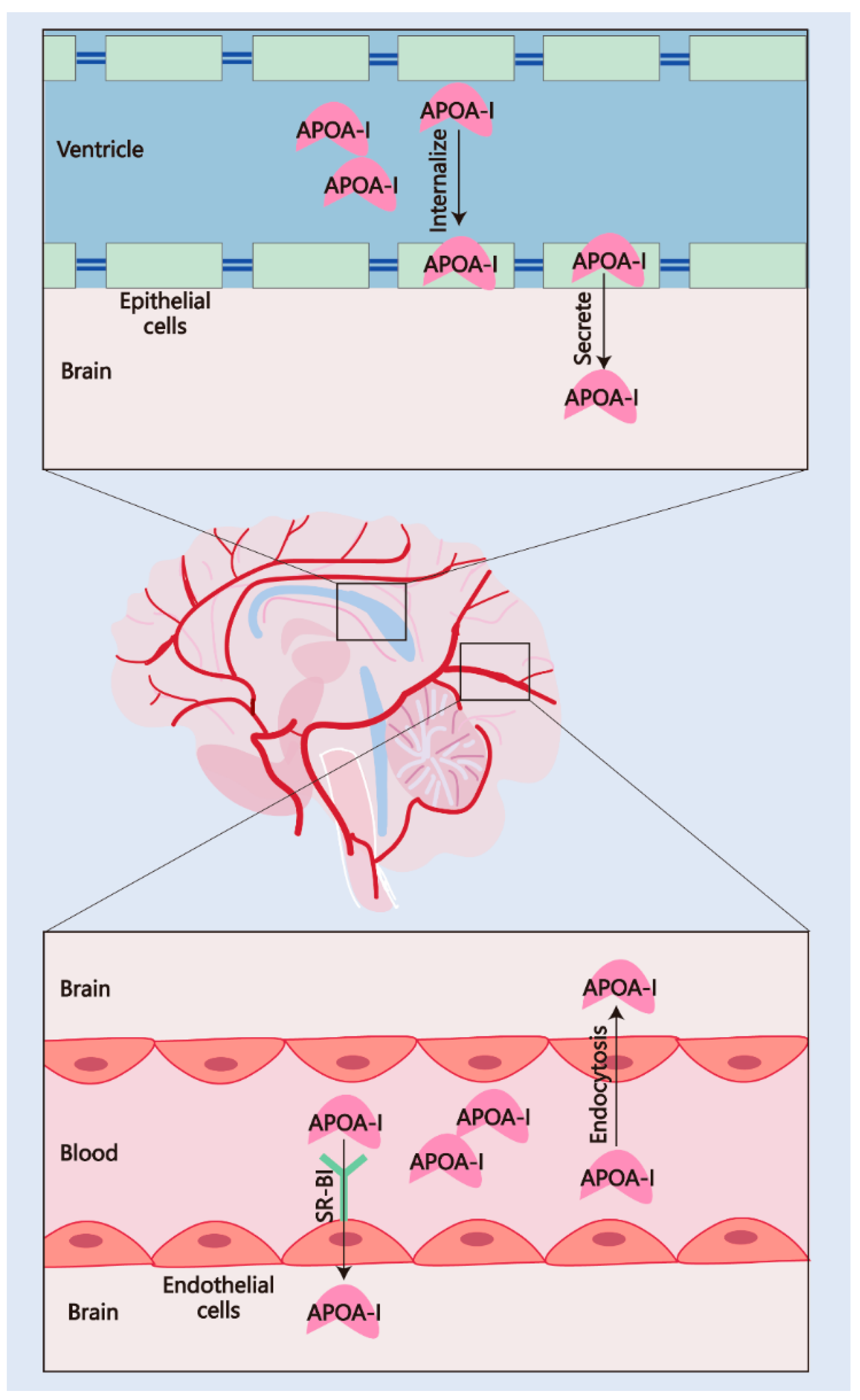
2.2. The Pathways of APOA-I Transport to the CNS
2.2.1. Transport via the BBB
2.2.2. Transport via the BCSFB
2.3. Clinical Evidence Links APOA-I to AD Pathogenesis
2.4. APOA-I Polymorphisms and AD
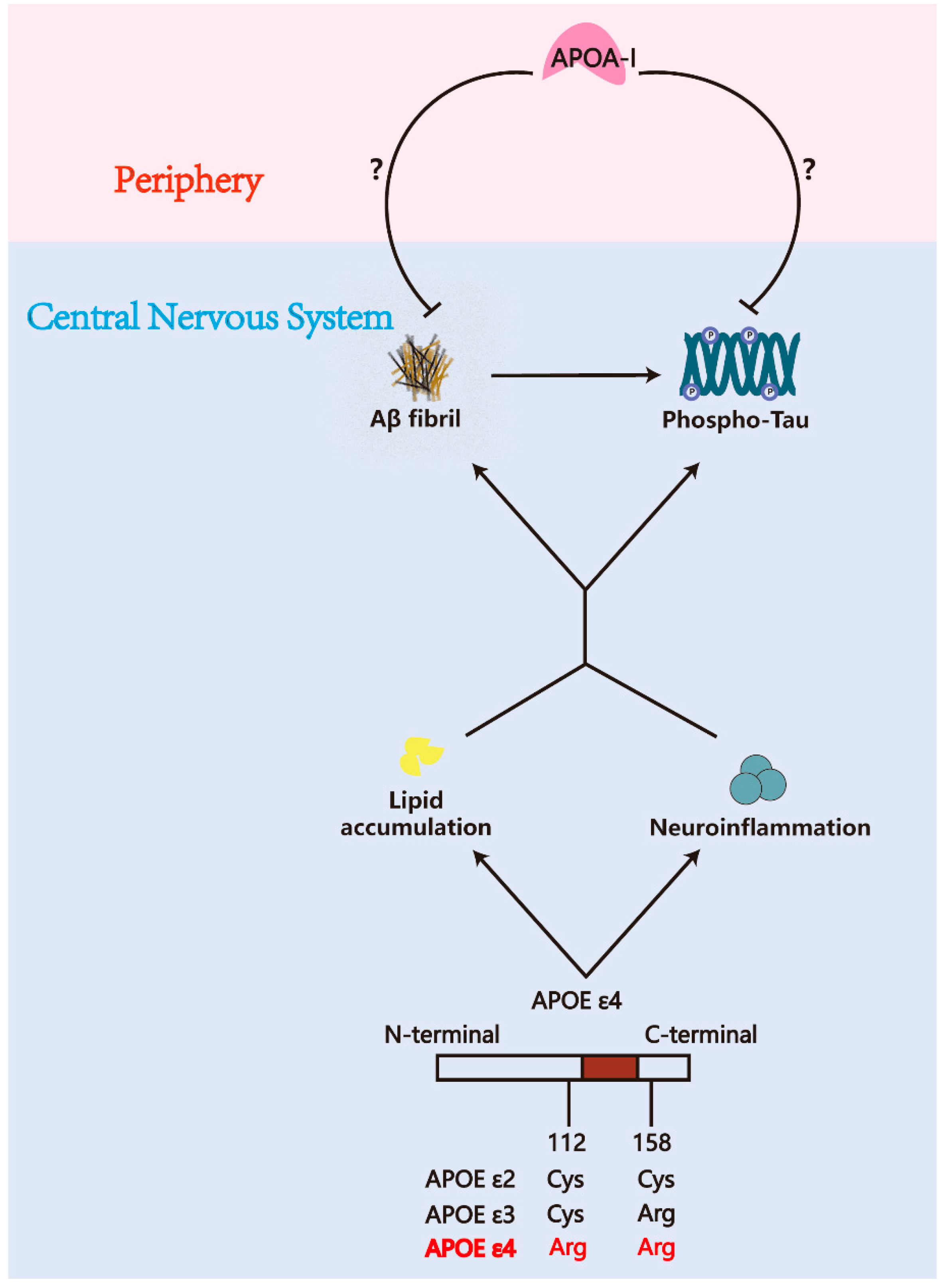
3. APOA-I Modifies the Pathology of AD
3.1. APOA-I and Aβ
3.2. APOA-I and Tauopathy
3.3. APOA-I and Microglia
3.4. APOA-I and Astrocytes
3.5. APOA-I and Oligodendrocytes
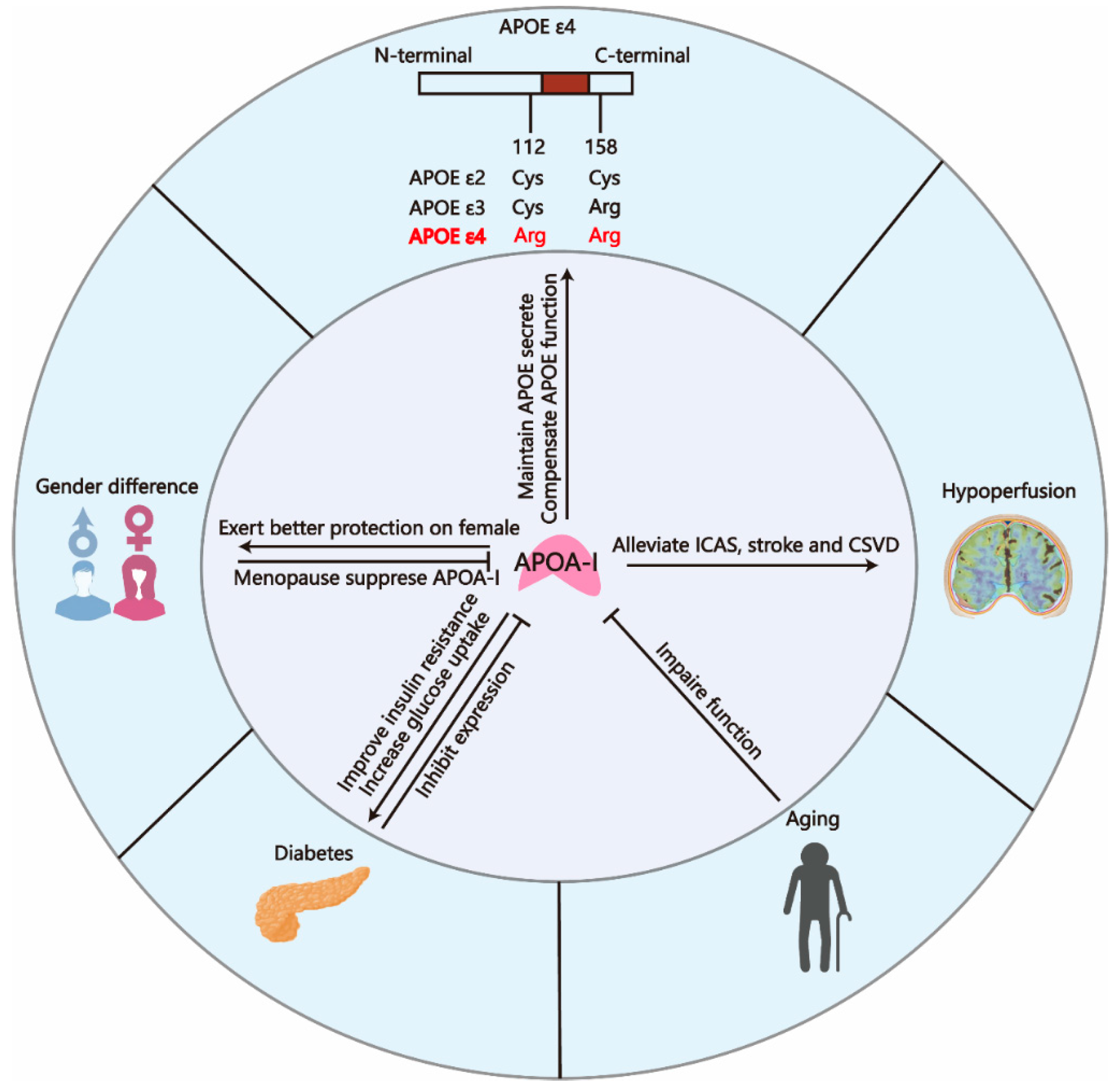
4. APOA-I Is Associated with Multiple Risk Factors for AD
4.1. APOA-I and Gender Difference
4.2. APOA-I and Aging
4.3. APOA-I and APOE ε4
4.4. APOA-I and Diabetes
4.5. APOA-I and Cerebrovascular Diseases
5. APOA-I Based Therapy
6. APOA-I and Other Neurodegenerative Diseases
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Alzheimer’s Association. 2024 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2024, 20, 3708–3821. [Google Scholar] [CrossRef] [PubMed]
- Mateev, E.; Karatchobanov, V.; Dedja, M.; Diamantakos, K.; Mateeva, A.; Muhammed, M.T.; Irfan, A.; Kondeva-Burdina, M.; Valkova, I.; Georgieva, M.; et al. Novel Pyrrole Derivatives as Multi-Target Agents for the Treatment of Alzheimer’s Disease: Microwave-Assisted Synthesis, In Silico Studies and Biological Evaluation. Pharmaceuticals 2024, 17, 1171. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Wang, X. Alzheimer’s disease: Insights into pathology, molecular mechanisms, and therapy. Protein Cell 2025, 16, 83–120. [Google Scholar] [CrossRef]
- Cavazzoni, P. FDA’s Decision to Approve New Treatment for Alzheimer’s Disease [EB/OL]. (2021-06-07). Available online: https://www.fda.gov/drugs/news-events-human-drugs/fdas-decision-approve-new-treatment-alzheimers-disease (accessed on 1 January 2023).
- FDA Grants Accelerated Approval for Alzheimer’s Disease Treatment [EB/OL]. (2023-01-06). Available online: https://www.fda.gov/drugs/news-events-human-drugs/fda-approves-treatment-adults-alzheimers-disease (accessed on 1 February 2023).
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef]
- Yin, F. Lipid metabolism and Alzheimer’s disease: Clinical evidence, mechanistic link and therapeutic promise. FEBS J. 2023, 290, 1420–1453. [Google Scholar] [CrossRef]
- Ahmed, H.; Wang, Y.; Griffiths, W.J.; Levey, A.I.; Pikuleva, I.; Liang, S.H.; Haider, A. Brain cholesterol and Alzheimer’s disease: Challenges and opportunities in probe and drug development. Brain 2024, 147, 1622–1635. [Google Scholar] [CrossRef]
- Solomon, A.; Kåreholt, I.; Ngandu, T.; Winblad, B.; Nissinen, A.; Tuomilehto, J.; Soininen, H.; Kivipelto, M. Serum cholesterol changes after midlife and late-life cognition: Twenty-one-year follow-up study. Neurology 2007, 68, 751–756. [Google Scholar] [CrossRef]
- Solomon, A.; Kivipelto, M.; Wolozin, B.; Zhou, J.; Whitmer, R.A. Midlife serum cholesterol and increased risk of Alzheimer’s and vascular dementia three decades later. Dement. Geriatr. Cogn. Disord. 2009, 28, 75–80. [Google Scholar] [CrossRef]
- Petek, B.; Häbel, H.; Xu, H.; Villa-Lopez, M.; Kalar, I.; Hoang, M.T.; Maioli, S.; Pereira, J.B.; Mostafaei, S.; Winblad, B.; et al. Statins and cognitive decline in patients with Alzheimer’s and mixed dementia: A longitudinal registry-based cohort study. Alzheimer’s Res. Ther. 2023, 15, 220. [Google Scholar] [CrossRef]
- Vilella, A.; Bodria, M.; Papotti, B.; Zanotti, I.; Zimetti, F.; Remaggi, G.; Elviri, L.; Potì, F.; Ferri, N.; Lupo, M.G.; et al. PCSK9 ablation attenuates Aβ pathology, neuroinflammation and cognitive dysfunctions in 5XFAD mice. Brain Behav. Immun. 2024, 115, 517–534. [Google Scholar] [CrossRef]
- Koch, M.; Jensen, M.K. HDL-cholesterol and apolipoproteins in relation to dementia. Curr. Opin. Lipidol. 2016, 27, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Button, E.B.; Robert, J.; Caffrey, T.M.; Fan, J.; Zhao, W.; Wellington, C.L. HDL from an Alzheimer’s disease perspective. Curr. Opin. Lipidol. 2019, 30, 224–234. [Google Scholar] [CrossRef] [PubMed]
- Swaminathan, S.K.; Zhou, A.L.; Ahlschwede, K.M.; Curran, G.L.; Lowe, V.J.; Li, L.; Kandimalla, K.K. High-Density Lipoprotein Mimetic Peptide 4F Efficiently Crosses the Blood-Brain Barrier and Modulates Amyloid-β Distribution between Brain and Plasma. J. Pharmacol. Exp. Ther. 2020, 375, 308–316. [Google Scholar] [CrossRef]
- Atzmon, G.; Gabriely, I.; Greiner, W.; Davidson, D.; Schechter, C.; Barzilai, N. Plasma HDL levels highly correlate with cognitive function in exceptional longevity. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2002, 57, M712–M715. [Google Scholar] [CrossRef]
- Van Exel, E.; de Craen, A.J.; Gussekloo, J.; Houx, P.; Bootsma-van der Wiel, A.; Macfarlane, P.W.; Blauw, G.J.; Westendorp, R.G. Association between high-density lipoprotein and cognitive impairment in the oldest old. Ann. Neurol. 2002, 51, 716–721. [Google Scholar] [CrossRef]
- Singh-Manoux, A.; Gimeno, D.; Kivimaki, M.; Brunner, E.; Marmot, M.G. Low HDL cholesterol is a risk factor for deficit and decline in memory in midlife: The Whitehall II study. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1556–1562. [Google Scholar] [CrossRef]
- Vitali, C.; Wellington, C.L.; Calabresi, L. HDL and cholesterol handling in the brain. Cardiovasc. Res. 2014, 103, 405–413. [Google Scholar] [CrossRef]
- Haney, M.S.; Pálovics, R.; Munson, C.N.; Long, C.; Johansson, P.K.; Yip, O.; Dong, W.; Rawat, E.; West, E.; Schlachetzki, J.C.M.; et al. APOE4/4 is linked to damaging lipid droplets in Alzheimer’s disease microglia. Nature 2024, 628, 154–161. [Google Scholar] [CrossRef]
- Fortea, J.; Pegueroles, J.; Alcolea, D.; Belbin, O.; Dols-Icardo, O.; Vaqué-Alcázar, L.; Videla, L.; Gispert, J.D.; Suárez-Calvet, M.; Johnson, S.C.; et al. APOE4 homozygozity represents a distinct genetic form of Alzheimer’s disease. Nat. Med. 2024, 30, 1284–1291. [Google Scholar] [CrossRef]
- Rao, A.; Chen, N.; Kim, M.J.; Blumenfeld, J.; Yip, O.; Hao, Y.; Liang, Z.; Nelson, M.R.; Koutsodendris, N.; Grone, B.; et al. Microglia Depletion Reduces Human Neuronal APOE4-Driven Pathologies in a Chimeric Alzheimer’s Disease Model. bioRxiv 2023. [Google Scholar] [CrossRef]
- Dal Magro, R.; Simonelli, S.; Cox, A.; Formicola, B.; Corti, R.; Cassina, V.; Nardo, L.; Mantegazza, F.; Salerno, D.; Grasso, G.; et al. The Extent of Human Apolipoprotein A-I Lipidation Strongly Affects the β-Amyloid Efflux Across the Blood-Brain Barrier in vitro. Front. Neurosci. 2019, 13, 419. [Google Scholar] [CrossRef] [PubMed]
- Koldamova, R.P.; Lefterov, I.M.; Lefterova, M.I.; Lazo, J.S. Apolipoprotein A-I directly interacts with amyloid precursor protein and inhibits A beta aggregation and toxicity. Biochemistry 2001, 40, 3553–3560. [Google Scholar] [CrossRef] [PubMed]
- Lefterov, I.; Fitz, N.F.; Cronican, A.A.; Fogg, A.; Lefterov, P.; Kodali, R.; Wetzel, R.; Koldamova, R. Apolipoprotein A-I deficiency increases cerebral amyloid angiopathy and cognitive deficits in APP/PS1DeltaE9 mice. J. Biol. Chem. 2010, 285, 36945–36957. [Google Scholar] [CrossRef]
- Button, E.B.; Boyce, G.K.; Wilkinson, A.; Stukas, S.; Hayat, A.; Fan, J.; Wadsworth, B.J.; Robert, J.; Martens, K.M.; Wellington, C.L. ApoA-I deficiency increases cortical amyloid deposition, cerebral amyloid angiopathy, cortical and hippocampal astrogliosis, and amyloid-associated astrocyte reactivity in APP/PS1 mice. Alzheimer’s Res. Ther. 2019, 11, 44. [Google Scholar] [CrossRef]
- Slot, R.E.; Van Harten, A.C.; Kester, M.I.; Jongbloed, W.; Bouwman, F.H.; Teunissen, C.E.; Scheltens, P.; Veerhuis, R.; van der Flier, W.M. Apolipoprotein A1 in Cerebrospinal Fluid and Plasma and Progression to Alzheimer’s Disease in Non-Demented Elderly. J. Alzheimer’s Dis. 2017, 56, 687–697. [Google Scholar] [CrossRef]
- Choi, H.J.; Seo, E.H.; Yi, D.; Sohn, B.K.; Choe, Y.M.; Byun, M.S.; Lee, J.M.; Woo, J.I.; Lee, D.Y. Amyloid-Independent Amnestic Mild Cognitive Impairment and Serum Apolipoprotein A1 Levels. Am. J. Geriatr. Psychiatry 2016, 24, 144–153. [Google Scholar] [CrossRef]
- Stukas, S.; Robert, J.; Lee, M.; Kulic, I.; Carr, M.; Tourigny, K.; Fan, J.; Namjoshi, D.; Lemke, K.; DeValle, N.; et al. Intravenously injected human apolipoprotein A-I rapidly enters the central nervous system via the choroid plexus. J. Am. Heart Assoc. 2014, 3, e001156. [Google Scholar] [CrossRef]
- Zhou, A.L.; Swaminathan, S.K.; Curran, G.L.; Poduslo, J.F.; Lowe, V.J.; Li, L.; Kandimalla, K.K. Apolipoprotein A-I Crosses the Blood-Brain Barrier through Clathrin-Independent and Cholesterol-Mediated Endocytosis. J. Pharmacol. Exp. Ther. 2019, 369, 481–488. [Google Scholar] [CrossRef]
- Cheng, Y.; He, C.Y.; Tian, D.Y.; Chen, S.H.; Ren, J.R.; Sun, H.L.; Xu, M.Y.; Tan, C.R.; Fan, D.Y.; Jian, J.M.; et al. Physiological β-amyloid clearance by the liver and its therapeutic potential for Alzheimer’s disease. Acta Neuropathol. 2023, 145, 717–731. [Google Scholar] [CrossRef]
- Chandrashekar, D.V.; Roules, G.C.; Jagadeesan, N.; Panchal, U.R.; Oyegbesan, A.; Imiruaye, O.E.; Zhang, H.; Garcia, J.; Kaur, K.; Win, S.; et al. Hepatic LRP-1 plays an important role in amyloidosis in Alzheimer’s disease mice: Potential role in chronic heavy alcohol feeding. Neurobiol. Dis. 2024, 199, 106570. [Google Scholar] [CrossRef]
- Peng, X.; Zhang, X.; Xu, Z.; Li, L.; Mo, X.; Peng, Z.; Shan, Z.; Yan, H.; Xu, J.; Liu, L. Peripheral amyloid-β clearance mediates cognitive impairment in non-alcoholic fatty liver disease. EBioMedicine 2024, 102, 105079. [Google Scholar] [CrossRef] [PubMed]
- Tian, D.Y.; Cheng, Y.; Zhuang, Z.Q.; He, C.Y.; Pan, Q.G.; Tang, M.Z.; Hu, X.L.; Shen, Y.Y.; Wang, Y.R.; Chen, S.H.; et al. Physiological clearance of amyloid-beta by the kidney and its therapeutic potential for Alzheimer’s disease. Mol. Psychiatry 2021, 26, 6074–6082. [Google Scholar] [CrossRef] [PubMed]
- Sakai, K.; Senda, T.; Hata, R.; Kuroda, M.; Hasegawa, M.; Kato, M.; Abe, M.; Kawaguchi, K.; Nakai, S.; Hiki, Y.; et al. Patients that have Undergone Hemodialysis Exhibit Lower Amyloid Deposition in the Brain: Evidence Supporting a Therapeutic Strategy for Alzheimer’s Disease by Removal of Blood Amyloid. J. Alzheimer’s Dis. 2016, 51, 997–1002. [Google Scholar] [CrossRef]
- Xiang, Y.; Bu, X.L.; Liu, Y.H.; Zhu, C.; Shen, L.L.; Jiao, S.S.; Zhu, X.Y.; Giunta, B.; Tan, J.; Song, W.H.; et al. Physiological amyloid-beta clearance in the periphery and its therapeutic potential for Alzheimer’s disease. Acta Neuropathol. 2015, 130, 487–499. [Google Scholar] [CrossRef]
- Wang, J.; Jin, W.S.; Bu, X.L.; Zeng, F.; Huang, Z.L.; Li, W.W.; Shen, L.L.; Zhuang, Z.Q.; Fang, Y.; Sun, B.L.; et al. Physiological clearance of tau in the periphery and its therapeutic potential for tauopathies. Acta Neuropathol. 2018, 136, 525–536. [Google Scholar] [CrossRef]
- Yuan, X.; Nie, S.; Yang, Y.; Liu, C.; Xia, D.; Meng, L.; Xia, Y.; Su, H.; Zhang, C.; Bu, L.; et al. Propagation of pathologic α-synuclein from kidney to brain may contribute to Parkinson’s disease. Nat. Neurosci. 2025, 28, 577–588. [Google Scholar] [CrossRef]
- Zhu, C.; Zhu, J.; Xiang, Y.; Bu, X.L.; Jin, W.S.; Wang, Y.J. A Conceptual Study on the Peripheral Clearance of Brain-Derived α-Synuclein in Humans. J. Alzheimer’s Dis. 2022, 90, 1485–1492. [Google Scholar] [CrossRef]
- Kane, J.P.; Malloy, M.J. Prebeta-1 HDL and coronary heart disease. Curr. Opin. Lipidol. 2012, 23, 367–371. [Google Scholar] [CrossRef]
- Zannis, V.I.; Chroni, A.; Kypreos, K.E.; Kan, H.Y.; Cesar, T.B.; Zanni, E.E.; Kardassis, D. Probing the pathways of chylomicron and HDL metabolism using adenovirus-mediated gene transfer. Curr. Opin. Lipidol. 2004, 15, 151–166. [Google Scholar] [CrossRef]
- Martín-Campos, J.M.; Julve, J.; Escolà, J.C.; Ordóñez-Llanos, J.; Gómez, J.; Binimelis, J.; González-Sastre, F.; Blanco-Vaca, F. ApoA-I(MALLORCA) impairs LCAT activation and induces dominant familial hypoalphalipoproteinemia. J. Lipid Res. 2002, 43, 115–123. [Google Scholar] [CrossRef]
- Zannis, V.I.; Chroni, A.; Krieger, M. Role of apoA-I, ABCA1, LCAT, and SR-BI in the biogenesis of HDL. J. Mol. Med. 2006, 84, 276–294. [Google Scholar] [CrossRef] [PubMed]
- Zein, A.A.; Kaur, R.; Hussein, T.O.K.; Graf, G.A.; Lee, J.Y. ABCG5/G8: A structural view to pathophysiology of the hepatobiliary cholesterol secretion. Biochem. Soc. Trans. 2019, 47, 1259–1268. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Weng, W.; Breslow, J.L.; Tall, A.R. Scavenger receptor BI (SR-BI) is up-regulated in adrenal gland in apolipoprotein A-I and hepatic lipase knock-out mice as a response to depletion of cholesterol stores. In vivo evidence that SR-BI is a functional high density lipoprotein receptor under feedback control. J. Biol. Chem. 1996, 271, 21001–21004. [Google Scholar] [CrossRef]
- Vantourout, P.; Radojkovic, C.; Lichtenstein, L.; Pons, V.; Champagne, E.; Martinez, L.O. Ecto-F1-ATPase: A moonlighting protein complex and an unexpected apoA-I receptor. World J. Gastroenterol. 2010, 16, 5925–5935. [Google Scholar]
- Van der Velde, A.E.; Brufau, G.; Groen, A.K. Transintestinal cholesterol efflux. Curr. Opin. Lipidol. 2010, 21, 167–171. [Google Scholar] [CrossRef]
- Koch, S.; Donarski, N.; Goetze, K.; Kreckel, M.; Stuerenburg, H.J.; Buhmann, C.; Beisiegel, U. Characterization of four lipoprotein classes in human cerebrospinal fluid. J. Lipid Res. 2001, 42, 1143–1151. [Google Scholar] [CrossRef]
- Li, K.; Xie, X.; Guo, Y. HDL and Therapy. Adv. Exp. Med. Biol. 2022, 1377, 171–187. [Google Scholar] [CrossRef]
- Ganzetti, G.S.; Parolini, C. Microarray analysis identifies human apoA-I(Milano) and apoA-II as determinants of the liver gene expression related to lipid and energy metabolism. Exp. Cell Res. 2023, 433, 113826. [Google Scholar] [CrossRef]
- Castellani, L.W.; Lusis, A.J. ApoA-II versus ApoA-I: Two for one is not always a good deal. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 1870–1872. [Google Scholar] [CrossRef]
- Wang, Y.; Sawashita, J.; Qian, J.; Zhang, B.; Fu, X.; Tian, G.; Chen, L.; Mori, M.; Higuchi, K. ApoA-I deficiency in mice is associated with redistribution of apoA-II and aggravated AApoAII amyloidosis. J. Lipid Res. 2011, 52, 1461–1470. [Google Scholar] [CrossRef]
- Argmann, C.A.; Violante, S.; Dodatko, T.; Amaro, M.P.; Hagen, J.; Gillespie, V.L.; Buettner, C.; Schadt, E.E.; Houten, S.M. Germline deletion of Krüppel-like factor 14 does not increase risk of diet induced metabolic syndrome in male C57BL/6 mice. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2017, 1863, 3277–3285. [Google Scholar] [CrossRef] [PubMed]
- Sniderman, A.D.; Furberg, C.D.; Keech, A.; Roeters van Lennep, J.E.; Frohlich, J.; Jungner, I.; Walldius, G. Apolipoproteins versus lipids as indices of coronary risk and as targets for statin treatment. Lancet 2003, 361, 777–780. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Hong, K.S.; Lee, E.J.; Lee, J.; Kim, D.E. High levels of apolipoprotein B/AI ratio are associated with intracranial atherosclerotic stenosis. Stroke 2011, 42, 3040–3046. [Google Scholar] [CrossRef]
- Ishii, M. Apolipoprotein B as a New Link Between Cholesterol and Alzheimer Disease. JAMA Neurol. 2019, 76, 751–753. [Google Scholar] [CrossRef]
- Martin, L.; Boutwell, B.B.; Messerlian, C.; Adams, C.D. Mendelian randomization reveals apolipoprotein B shortens healthspan and possibly increases risk for Alzheimer’s disease. Commun. Biol. 2024, 7, 230. [Google Scholar] [CrossRef]
- Agarwal, M.; Khan, S. Plasma Lipids as Biomarkers for Alzheimer’s Disease: A Systematic Review. Cureus 2020, 12, e12008. [Google Scholar] [CrossRef]
- Deng, S.; Xu, Y.; Zheng, L. HDL Structure. In HDL Metabolism and Diseases; Advances in Experimental Medicine and Biology; Springer: Singapore, 2022; Volume 1377, pp. 1–11. [Google Scholar] [CrossRef]
- Eichinger, A.; Nasreen, A.; Kim, H.J.; Skerra, A. Structural insight into the dual ligand specificity and mode of high density lipoprotein association of apolipoprotein D. J. Biol. Chem. 2007, 282, 31068–31075. [Google Scholar] [CrossRef]
- Ganfornina, M.D.; Do Carmo, S.; Lora, J.M.; Torres-Schumann, S.; Vogel, M.; Allhorn, M.; González, C.; Bastiani, M.J.; Rassart, E.; Sanchez, D. Apolipoprotein D is involved in the mechanisms regulating protection from oxidative stress. Aging Cell 2008, 7, 506–515. [Google Scholar] [CrossRef]
- Elliott, D.A.; Weickert, C.S.; Garner, B. Apolipoproteins in the brain: Implications for neurological and psychiatric disorders. Clin. Lipidol. 2010, 51, 555–573. [Google Scholar] [CrossRef]
- Rassart, E.; Bedirian, A.; Do Carmo, S.; Guinard, O.; Sirois, J.; Terrisse, L.; Milne, R. Apolipoprotein D. Biochim. Biophys. Acta (BBA) Protein Struct. Mol. Enzymol. 2000, 1482, 185–198. [Google Scholar] [CrossRef]
- Wang, M.; Pan, W.; Xu, Y.; Zhang, J.; Wan, J.; Jiang, H. Microglia-Mediated Neuroinflammation: A Potential Target for the Treatment of Cardiovascular Diseases. J. Inflamm. Res. 2022, 15, 3083–3094. [Google Scholar] [CrossRef] [PubMed]
- Desai, P.P.; Ikonomovic, M.D.; Abrahamson, E.E.; Hamilton, R.L.; Isanski, B.A.; Hope, C.E.; Klunk, W.E.; DeKosky, S.T.; Kamboh, M.I. Apolipoprotein D is a component of compact but not diffuse amyloid-beta plaques in Alzheimer’s disease temporal cortex. Neurobiol. Dis. 2005, 20, 574–582. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, E.; Frangione, B.; Ghiso, J. Characterization of apolipoprotein J-Alzheimer’s A beta interaction. J. Biol. Chem. 1995, 270, 7563–7567. [Google Scholar] [CrossRef]
- Howlett, D.R.; Hortobágyi, T.; Francis, P.T. Clusterin associates specifically with Aβ40 in Alzheimer’s disease brain tissue. Brain Pathol. 2013, 23, 623–632. [Google Scholar] [CrossRef]
- Koudinov, A.; Matsubara, E.; Frangione, B.; Ghiso, J. The soluble form of Alzheimer’s amyloid beta protein is complexed to high density lipoprotein 3 and very high density lipoprotein in normal human plasma. Biochem. Biophys. Res. Commun. 1994, 205, 1164–1171. [Google Scholar] [CrossRef]
- Fernández-de-Retana, S.; Cano-Sarabia, M.; Marazuela, P.; Sánchez-Quesada, J.L.; Garcia-Leon, A.; Montañola, A.; Montaner, J.; Maspoch, D.; Hernández-Guillamon, M. Characterization of ApoJ-reconstituted high-density lipoprotein (rHDL) nanodisc for the potential treatment of cerebral β-amyloidosis. Sci. Rep. 2017, 7, 14637. [Google Scholar] [CrossRef]
- Johansson, P.; Almqvist, E.G.; Bjerke, M.; Wallin, A.; Johansson, J.O.; Andreasson, U.; Blennow, K.; Zetterberg, H.; Svensson, J. Reduced Cerebrospinal Fluid Concentration of Apolipoprotein A-I in Patients with Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 59, 1017–1026. [Google Scholar] [CrossRef]
- Fania, C.; Arosio, B.; Capitanio, D.; Torretta, E.; Gussago, C.; Ferri, E.; Mari, D.; Gelfi, C. Protein signature in cerebrospinal fluid and serum of Alzheimer’s disease patients: The case of apolipoprotein A-1 proteoforms. PLoS ONE 2017, 12, e0179280. [Google Scholar] [CrossRef]
- Tong, J.H.; Gong, S.Q.; Zhang, Y.S.; Dong, J.R.; Zhong, X.; Wei, M.J.; Liu, M.Y. Association of Circulating Apolipoprotein AI Levels in Patients With Alzheimer’s Disease: A Systematic Review and Meta-Analysis. Front. Aging Neurosci. 2022, 14, 899175. [Google Scholar] [CrossRef]
- Balazs, Z.; Panzenboeck, U.; Hammer, A.; Sovic, A.; Quehenberger, O.; Malle, E.; Sattler, W. Uptake and transport of high-density lipoprotein (HDL) and HDL-associated alpha-tocopherol by an in vitro blood-brain barrier model. J. Neurochem. 2004, 89, 939–950. [Google Scholar] [CrossRef]
- Grifoni, L.; Landucci, E.; Pieraccini, G.; Mazzantini, C.; Bergonzi, M.C.; Pellegrini-Giampietro, D.E.; Bilia, A.R. Development and Blood–Brain Barrier Penetration of Nanovesicles Loaded with Cannabidiol. Pharmaceuticals 2025, 18, 160. [Google Scholar] [CrossRef] [PubMed]
- Fung, K.Y.; Wang, C.; Nyegaard, S.; Heit, B.; Fairn, G.D.; Lee, W.L. SR-BI Mediated Transcytosis of HDL in Brain Microvascular Endothelial Cells Is Independent of Caveolin, Clathrin, and PDZK1. Front. Physiol. 2017, 8, 841. [Google Scholar] [CrossRef] [PubMed]
- Khalil, A.; Berrougui, H.; Pawelec, G.; Fulop, T. Impairment of the ABCA1 and SR-BI-mediated cholesterol efflux pathways and HDL anti-inflammatory activity in Alzheimer’s disease. Mech. Ageing Dev. 2012, 133, 20–29. [Google Scholar] [CrossRef]
- Thanopoulou, K.; Fragkouli, A.; Stylianopoulou, F.; Georgopoulos, S. Scavenger receptor class B type I (SR-BI) regulates perivascular macrophages and modifies amyloid pathology in an Alzheimer mouse model. Proc. Natl. Acad. Sci. USA 2010, 107, 20816–20821. [Google Scholar] [CrossRef]
- Johanson, C.E.; Stopa, E.G.; McMillan, P.N. The blood-cerebrospinal fluid barrier: Structure and functional significance. Methods Mol. Biol. 2011, 686, 101–131. [Google Scholar] [CrossRef]
- Zlokovic, B.V. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat. Rev. Neurosci. 2011, 12, 723–738. [Google Scholar] [CrossRef]
- Johanson, C.E.; Duncan, J.A., 3rd; Klinge, P.M.; Brinker, T.; Stopa, E.G.; Silverberg, G.D. Multiplicity of cerebrospinal fluid functions: New challenges in health and disease. Cerebrospinal Fluid Res. 2008, 5, 10. [Google Scholar] [CrossRef]
- Kim, K.; Abramishvili, D.; Du, S.; Papadopoulos, Z.; Cao, J.; Herz, J.; Smirnov, I.; Thomas, J.L.; Colonna, M.; Kipnis, J. Meningeal lymphatics-microglia axis regulates synaptic physiology. Cell 2025, 188, 2705–2719.e23. [Google Scholar] [CrossRef]
- Kalayci, A.; Gibson, C.M.; Ridker, P.M.; Wright, S.D.; Kingwell, B.A.; Korjian, S.; Chi, G.; Lee, J.J.; Tricoci, P.; Kazmi, S.H.; et al. ApoA-I Infusion Therapies Following Acute Coronary Syndrome: Past, Present, and Future. Curr. Atheroscler. Rep. 2022, 24, 585–597. [Google Scholar] [CrossRef]
- Kuriyama, M.; Takahashi, K.; Yamano, T.; Hokezu, Y.; Togo, S.; Osame, M.; Igakura, T. Low levels of serum apolipoprotein A I and A II in senile dementia. Jpn. J. Psychiatry Neurol. 1994, 48, 589–593. [Google Scholar] [CrossRef]
- Kawano, M.; Kawakami, M.; Otsuka, M.; Yashima, H.; Yaginuma, T.; Ueki, A. Marked decrease of plasma apolipoprotein AI and AII in Japanese patients with late-onset non-familial Alzheimer’s disease. Clin. Chim. Acta Int. J. Clin. Chem. 1995, 239, 209–211. [Google Scholar] [CrossRef] [PubMed]
- Song, F.; Poljak, A.; Crawford, J.; Kochan, N.A.; Wen, W.; Cameron, B.; Lux, O.; Brodaty, H.; Mather, K.; Smythe, G.A.; et al. Plasma apolipoprotein levels are associated with cognitive status and decline in a community cohort of older individuals. PLoS ONE 2012, 7, e34078. [Google Scholar] [CrossRef] [PubMed]
- Reitz, C.; Tang, M.X.; Schupf, N.; Manly, J.J.; Mayeux, R.; Luchsinger, J.A. Association of higher levels of high-density lipoprotein cholesterol in elderly individuals and lower risk of late-onset Alzheimer disease. Arch. Neurol. 2010, 67, 1491–1497. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.Y.; Shan, C.L.; Qing, H.; Wang, W.; Zhu, Y.; Yin, M.M.; Machado, S.; Yuan, T.F.; Wu, T. The Effects of Aerobic Exercise on Cognitive Function of Alzheimer’s Disease Patients. CNS Neurol. Disord. Drug Targets 2015, 14, 1292–1297. [Google Scholar] [CrossRef]
- Merched, A.; Xia, Y.; Visvikis, S.; Serot, J.M.; Siest, G. Decreased high-density lipoprotein cholesterol and serum apolipoprotein AI concentrations are highly correlated with the severity of Alzheimer’s disease. Neurobiol. Aging 2000, 21, 27–30. [Google Scholar] [CrossRef]
- Harr, S.D.; Uint, L.; Hollister, R.; Hyman, B.T.; Mendez, A.J. Brain expression of apolipoproteins E, J, and A-I in Alzheimer’s disease. J. Neurochem. 1996, 66, 2429–2435. [Google Scholar] [CrossRef]
- Song, H.; Saito, K.; Seishima, M.; Noma, A.; Urakami, K.; Nakashima, K. Cerebrospinal fluid apo E and apo A-I concentrations in early- and late-onset Alzheimer’s disease. Neurosci. Lett. 1997, 231, 175–178. [Google Scholar] [CrossRef]
- Sagare, A.P.; Bell, R.D.; Zlokovic, B.V. Neurovascular dysfunction and faulty amyloid β-peptide clearance in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2, a011452. [Google Scholar] [CrossRef]
- Jin, Y.; Chifodya, K.; Han, G.; Jiang, W.; Chen, Y.; Shi, Y.; Xu, Q.; Xi, Y.; Wang, J.; Zhou, J.; et al. High-density lipoprotein in Alzheimer’s disease: From potential biomarkers to therapeutics. J. Control. Release 2021, 338, 56–70. [Google Scholar] [CrossRef]
- Helbecque, N.; Codron, V.; Cottel, D.; Amouyel, P. An apolipoprotein A-I gene promoter polymorphism associated with cognitive decline, but not with Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 2008, 25, 97–102. [Google Scholar] [CrossRef]
- Kamboh, M.I.; Aston, C.E.; Nestlerode, C.M.; McAllister, A.E.; Hamman, R.F. Haplotype analysis of two APOA1/MspI polymorphisms in relation to plasma levels of apo A-I and HDL-cholesterol. Atherosclerosis 1996, 127, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Tuteja, R.; Tuteja, N.; Melo, C.; Casari, G.; Baralle, F.E. Transcription efficiency of human apolipoprotein A-I promoter varies with naturally occurring A to G transition. FEBS Lett. 1992, 304, 98–101. [Google Scholar] [CrossRef] [PubMed]
- Angotti, E.; Mele, E.; Costanzo, F.; Avvedimento, E.V. A polymorphism (G-->A transition) in the -78 position of the apolipoprotein A-I promoter increases transcription efficiency. J. Biol. Chem. 1994, 269, 17371–17374. [Google Scholar] [CrossRef]
- Wang, X.L.; Liu, S.X.; McCredie, R.M.; Wilcken, D.E. Polymorphisms at the 5′-end of the apolipoprotein AI gene and severity of coronary artery disease. J. Clin. Investig. 1996, 98, 372–377. [Google Scholar] [CrossRef]
- Lopez-Miranda, J.; Ordovas, J.M.; Espino, A.; Marin, C.; Salas, J.; Lopez-Segura, F.; Jimenez-Pereperez, J.; Perez-Jimenez, F. Influence of mutation in human apolipoprotein A-1 gene promoter on plasma LDL cholesterol response to dietary fat. Lancet 1994, 343, 1246–1249. [Google Scholar] [CrossRef]
- Vollbach, H.; Heun, R.; Morris, C.M.; Edwardson, J.A.; McKeith, I.G.; Jessen, F.; Schulz, A.; Maier, W.; Kölsch, H. APOA1 polymorphism influences risk for early-onset nonfamiliar AD. Ann. Neurol. 2005, 58, 436–441. [Google Scholar] [CrossRef]
- Shibata, N.; Nagata, T.; Shinagawa, S.; Ohnuma, T.; Shimazaki, H.; Komatsu, M.; Kuerban, B.; Tomson, K.; Nakayama, K.; Yamada, H.; et al. Genetic association between APOA1 and APOD polymorphisms and Alzheimer’s disease in a Japanese population. J. Neural Transm. 2013, 120, 1599–1603. [Google Scholar] [CrossRef]
- Smach, M.A.; Edziri, H.; Charfeddine, B.; Ben Othman, L.; Lammouchi, T.; Ltaief, A.; Nafati, S.; Dridi, H.; Bennamou, S.; Limem, K. Polymorphism in apoA1 Influences High-Density Lipoprotein Cholesterol Levels but Is Not a Major Risk Factor of Alzheimer’s Disease. Dement. Geriatr. Cogn. Disord. Extra 2011, 1, 249–257. [Google Scholar] [CrossRef]
- Fanaee-Danesh, E.; Gali, C.C.; Tadic, J.; Zandl-Lang, M.; Carmen Kober, A.; Agujetas, V.R.; de Dios, C.; Tam-Amersdorfer, C.; Stracke, A.; Albrecher, N.M.; et al. Astaxanthin exerts protective effects similar to bexarotene in Alzheimer’s disease by modulating amyloid-beta and cholesterol homeostasis in blood-brain barrier endothelial cells. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2019, 1865, 2224–2245. [Google Scholar] [CrossRef]
- Wang, X.; Li, R.; Zacharek, A.; Landschoot-Ward, J.; Chopp, M.; Chen, J.; Cui, X. ApoA-I Mimetic Peptide Reduces Vascular and White Matter Damage After Stroke in Type-2 Diabetic Mice. Front. Neurosci. 2019, 13, 1127. [Google Scholar] [CrossRef]
- Paula-Lima, A.C.; Tricerri, M.A.; Brito-Moreira, J.; Bomfim, T.R.; Oliveira, F.F.; Magdesian, M.H.; Grinberg, L.T.; Panizzutti, R.; Ferreira, S.T. Human apolipoprotein A-I binds amyloid-beta and prevents Abeta-induced neurotoxicity. Int. J. Biochem. Cell Biol. 2009, 41, 1361–1370. [Google Scholar] [CrossRef] [PubMed]
- Robert, J.; Stukas, S.; Button, E.; Cheng, W.H.; Lee, M.; Fan, J.; Wilkinson, A.; Kulic, I.; Wright, S.D.; Wellington, C.L. Reconstituted high-density lipoproteins acutely reduce soluble brain Aβ levels in symptomatic APP/PS1 mice. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2016, 1862, 1027–1036. [Google Scholar] [CrossRef] [PubMed]
- Robert, J.; Button, E.B.; Stukas, S.; Boyce, G.K.; Gibbs, E.; Cowan, C.M.; Gilmour, M.; Cheng, W.H.; Soo, S.K.; Yuen, B.; et al. High-density lipoproteins suppress Aβ-induced PBMC adhesion to human endothelial cells in bioengineered vessels and in monoculture. Mol. Neurodegener. 2017, 12, 60. [Google Scholar] [CrossRef]
- Frankel, R.; Sparr, E.; Linse, S. Retardation of Aβ42 fibril formation by apolipoprotein A-I and recombinant HDL particles. J. Biol. Chem. 2023, 299, 105273. [Google Scholar] [CrossRef]
- Malajczuk, C.J.; Mancera, R.L. Molecular Simulation of the Binding of Amyloid Beta to Apolipoprotein A-I in High-Density Lipoproteins. Int. J. Mol. Sci. 2025, 26, 1380. [Google Scholar] [CrossRef]
- Malajczuk, C.J.; Mancera, R.L. An atomistic characterization of high-density lipoproteins and the conserved “LN” region of apoA-I. Biophys. J. 2024, 123, 1116–1128. [Google Scholar] [CrossRef]
- Navia-Pelaez, J.M.; Choi, S.H.; Dos Santos Aggum Capettini, L.; Xia, Y.; Gonen, A.; Agatisa-Boyle, C.; Delay, L.; Gonçalves Dos Santos, G.; Catroli, G.F.; Kim, J.; et al. Normalization of cholesterol metabolism in spinal microglia alleviates neuropathic pain. J. Exp. Med. 2021, 218, e20202059. [Google Scholar] [CrossRef]
- Kim, Y.S.; Choi, S.H.; Kim, K.Y.; Navia-Pelaez, J.M.; Perkins, G.A.; Choi, S.; Kim, J.; Nazarenkov, N.; Rissman, R.A.; Ju, W.K.; et al. AIBP controls TLR4 inflammarafts and mitochondrial dysfunction in a mouse model of Alzheimer’s disease. J. Neuroinflamm. 2024, 21, 245. [Google Scholar] [CrossRef]
- Lewis, T.L.; Cao, D.; Lu, H.; Mans, R.A.; Su, Y.R.; Jungbauer, L.; Linton, M.F.; Fazio, S.; LaDu, M.J.; Li, L. Overexpression of human apolipoprotein A-I preserves cognitive function and attenuates neuroinflammation and cerebral amyloid angiopathy in a mouse model of Alzheimer disease. J. Biol. Chem. 2010, 285, 36958–36968. [Google Scholar] [CrossRef]
- Handattu, S.P.; Garber, D.W.; Monroe, C.E.; van Groen, T.; Kadish, I.; Nayyar, G.; Cao, D.; Palgunachari, M.N.; Li, L.; Anantharamaiah, G.M. Oral apolipoprotein A-I mimetic peptide improves cognitive function and reduces amyloid burden in a mouse model of Alzheimer’s disease. Neurobiol. Dis. 2009, 34, 525–534. [Google Scholar] [CrossRef]
- Fernández-de Retana, S.; Montañola, A.; Marazuela, P.; De La Cuesta, M.; Batlle, A.; Fatar, M.; Grudzenski, S.; Montaner, J.; Hernández-Guillamon, M. Intravenous treatment with human recombinant ApoA-I Milano reduces beta amyloid cerebral deposition in the APP23-transgenic mouse model of Alzheimer’s disease. Neurobiol. Aging 2017, 60, 116–128. [Google Scholar] [CrossRef] [PubMed]
- Vanherle, S.; Jorissen, W.; Dierckx, T.; Loix, M.; Grajchen, E.; Mingneau, F.; Guns, J.; Gervois, P.; Lambrichts, I.; Dehairs, J.; et al. The ApoA-I mimetic peptide 5A enhances remyelination by promoting clearance and degradation of myelin debris. Cell Rep. 2022, 41, 111591. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhu, Z.; Li, Y.; Chen, Y.; Hu, X.; Liu, Y.; Shi, Y.; Hu, Y.; Bi, Y.; Xu, X.; et al. D-4F, an apolipoprotein A-I mimetic, promotes the clearance of myelin debris and the reduction of foamy macrophages after spinal cord injury. Bioengineered 2022, 13, 11794–11809. [Google Scholar] [CrossRef]
- Cui, X.; Chopp, M.; Zacharek, A.; Cui, C.; Yan, T.; Ning, R.; Chen, J. D-4F Decreases White Matter Damage After Stroke in Mice. Stroke 2016, 47, 214–220. [Google Scholar] [CrossRef]
- Boyles, J.K.; Zoellner, C.D.; Anderson, L.J.; Kosik, L.M.; Pitas, R.E.; Weisgraber, K.H.; Hui, D.Y.; Mahley, R.W.; Gebicke-Haerter, P.J.; Ignatius, M.J.; et al. A role for apolipoprotein E, apolipoprotein A-I, and low density lipoprotein receptors in cholesterol transport during regeneration and remyelination of the rat sciatic nerve. J. Clin. Investig. 1989, 83, 1015–1031. [Google Scholar] [CrossRef]
- Boyles, J.K.; Notterpek, L.M.; Anderson, L.J. Accumulation of apolipoproteins in the regenerating and remyelinating mammalian peripheral nerve. Identification of apolipoprotein D, apolipoprotein A-IV, apolipoprotein E, and apolipoprotein A-I. J. Biol. Chem. 1990, 265, 17805–17815. [Google Scholar] [CrossRef]
- Li, Q.; Shi, Y.; Li, X.; Yang, Y.; Zhang, X.; Xu, L.; Ma, Z.; Wang, J.; Fan, L.; Wu, L. Proteomic-Based Approach Reveals the Involvement of Apolipoprotein A-I in Related Phenotypes of Autism Spectrum Disorder in the BTBR Mouse Model. Int. J. Mol. Sci. 2022, 23, 15290. [Google Scholar] [CrossRef]
- Haenisch, F.; Alsaif, M.; Guest, P.C.; Rahmoune, H.; Dickerson, F.; Yolken, R.; Bahn, S. Multiplex immunoassay analysis of plasma shows prominent upregulation of growth factor activity pathways linked to GSK3β signaling in bipolar patients. J. Affect. Disord. 2014, 156, 139–143. [Google Scholar] [CrossRef]
- Xiong, J.; Zhang, Z.; Ye, K. C/EBPβ/AEP Signaling Drives Alzheimer’s Disease Pathogenesis. Neurosci. Bull. 2023, 39, 1173–1185. [Google Scholar] [CrossRef]
- Sharma, H.S.; Castellani, R.J.; Smith, M.A.; Sharma, A. The blood-brain barrier in Alzheimer’s disease: Novel therapeutic targets and nanodrug delivery. Int. Rev. Neurobiol. 2012, 102, 47–90. [Google Scholar] [CrossRef]
- Provias, J.; Jeynes, B. The role of the blood-brain barrier in the pathogenesis of senile plaques in Alzheimer’s disease. Int. J. Alzheimer’s Dis. 2014, 2014, 191863. [Google Scholar] [CrossRef] [PubMed]
- Fairley, L.H.; Lai, K.O.; Wong, J.H.; Chong, W.J.; Vincent, A.S.; D’Agostino, G.; Wu, X.; Naik, R.R.; Jayaraman, A.; Langley, S.R.; et al. Mitochondrial control of microglial phagocytosis by the translocator protein and hexokinase 2 in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2023, 120, e2209177120. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Chun, H.; Kim, Y.; Kim, Y.; Park, U.; Chu, J.; Bhalla, M.; Choi, S.H.; Yousefian-Jazi, A.; Kim, S.; et al. Astrocytic autophagy plasticity modulates Aβ clearance and cognitive function in Alzheimer’s disease. Mol. Neurodegener. 2024, 19, 55. [Google Scholar] [CrossRef]
- Kim, E.; Kim, H.; Jedrychowski, M.P.; Bakiasi, G.; Park, J.; Kruskop, J.; Choi, Y.; Kwak, S.S.; Quinti, L.; Kim, D.Y.; et al. Irisin reduces amyloid-β by inducing the release of neprilysin from astrocytes following downregulation of ERK-STAT3 signaling. Neuron 2023, 111, 3619–3633.e8. [Google Scholar] [CrossRef]
- Da Mesquita, S.; Louveau, A.; Vaccari, A.; Smirnov, I.; Cornelison, R.C.; Kingsmore, K.M.; Contarino, C.; Onengut-Gumuscu, S.; Farber, E.; Raper, D.; et al. Functional aspects of meningeal lymphatics in ageing and Alzheimer’s disease. Nature 2018, 560, 185–191. [Google Scholar] [CrossRef]
- Rego, S.; Sanchez, G.; Da Mesquita, S. Current views on meningeal lymphatics and immunity in aging and Alzheimer’s disease. Mol. Neurodegener. 2023, 18, 55. [Google Scholar] [CrossRef]
- Montañola, A.; de Retana, S.F.; López-Rueda, A.; Merino-Zamorano, C.; Penalba, A.; Fernández-Álvarez, P.; Rodríguez-Luna, D.; Malagelada, A.; Pujadas, F.; Montaner, J.; et al. ApoA1, ApoJ and ApoE Plasma Levels and Genotype Frequencies in Cerebral Amyloid Angiopathy. Neuromol. Med. 2016, 18, 99–108. [Google Scholar] [CrossRef]
- Liu, S.; Suzuki, H.; Ito, H.; Korenaga, T.; Akatsu, H.; Meno, K.; Uchida, K. Serum levels of proteins involved in amyloid-β clearance are related to cognitive decline and neuroimaging changes in mild cognitive impairment. Alzheimer’s Dement. 2019, 11, 85–97. [Google Scholar] [CrossRef]
- Marsillach, J.; Adorni, M.P.; Zimetti, F.; Papotti, B.; Zuliani, G.; Cervellati, C. HDL Proteome and Alzheimer’s Disease: Evidence of a Link. Antioxidants 2020, 9, 1224. [Google Scholar] [CrossRef]
- Zuin, M.; Cervellati, C.; Trentini, A.; Passaro, A.; Rosta, V.; Zimetti, F.; Zuliani, G. Association between Serum Concentrations of Apolipoprotein A-I (ApoA-I) and Alzheimer’s Disease: Systematic Review and Meta-Analysis. Diagnostics 2021, 11, 984. [Google Scholar] [CrossRef]
- Solé, M.; Marazuela, P.; Castellote, L.; Bonaterra-Pastra, A.; Giménez-Llort, L.; Hernández-Guillamon, M. Therapeutic effect of human ApoA-I-Milano variant in aged transgenic mouse model of Alzheimer’s disease. Br. J. Pharmacol. 2023, 180, 1999–2017. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, J.; Zhang, Z.; Ye, K. Tau in neurodegenerative diseases: Molecular mechanisms, biomarkers, and therapeutic strategies. Transl. Neurodegener. 2024, 13, 40. [Google Scholar] [CrossRef] [PubMed]
- Congdon, E.E.; Ji, C.; Tetlow, A.M.; Jiang, Y.; Sigurdsson, E.M. Tau-targeting therapies for Alzheimer disease: Current status and future directions. Nat. Rev. Neurol. 2023, 19, 715–736. [Google Scholar] [CrossRef]
- Zou, S.; Zhang, J.; Chen, W. Subtypes Based on Six Apolipoproteins in Non-Demented Elderly Are Associated with Cognitive Decline and Subsequent Tau Accumulation in Cerebrospinal Fluid. J. Alzheimer’s Dis. 2019, 72, 413–423. [Google Scholar] [CrossRef]
- Han, G.; Bai, K.; Yang, X.; Sun, C.; Ji, Y.; Zhou, J.; Zhang, H.; Ding, Y. “Drug-Carrier” Synergy Therapy for Amyloid-β Clearance and Inhibition of Tau Phosphorylation via Biomimetic Lipid Nanocomposite Assembly. Adv. Sci. 2022, 9, e2106072. [Google Scholar] [CrossRef]
- Kapic, A.; Sabnis, N.; Dossou, A.S.; Chavez, J.; Ceresa, L.; Gryczynski, Z.; Fudala, R.; Dickerman, R.; Bunnell, B.A.; Lacko, A.G. Photophysical Characterization and In Vitro Evaluation of α-Mangostin-Loaded HDL Mimetic Nano-Complex in LN-229 Glioblastoma Spheroid Model. Int. J. Mol. Sci. 2024, 25, 7378. [Google Scholar] [CrossRef]
- Bellaver, B.; Povala, G.; Ferreira, P.C.L.; Ferrari-Souza, J.P.; Leffa, D.T.; Lussier, F.Z.; Benedet, A.L.; Ashton, N.J.; Triana-Baltzer, G.; Kolb, H.C.; et al. Astrocyte reactivity influences amyloid-β effects on tau pathology in preclinical Alzheimer’s disease. Nat. Med. 2023, 29, 1775–1781. [Google Scholar] [CrossRef]
- Ciccone, L.; Shi, C.; di Lorenzo, D.; Van Baelen, A.C.; Tonali, N. The Positive Side of the Alzheimer’s Disease Amyloid Cross-Interactions: The Case of the Aβ 1-42 Peptide with Tau, TTR, CysC, and ApoA1. Molecules 2020, 25, 2439. [Google Scholar] [CrossRef]
- Ye, J.; Wan, H.; Chen, S.; Liu, G.P. Targeting tau in Alzheimer’s disease: From mechanisms to clinical therapy. Neural Regen. Res. 2024, 19, 1489–1498. [Google Scholar] [CrossRef]
- Walker, J.M.; Orr, M.E.; Orr, T.C.; Thorn, E.L.; Christie, T.D.; Yokoda, R.T.; Vij, M.; Ehrenberg, A.J.; Marx, G.A.; McKenzie, A.T.; et al. Spatial proteomics of hippocampal subfield-specific pathology in Alzheimer’s disease and primary age-related tauopathy. Alzheimer’s Dement. 2024, 20, 783–797. [Google Scholar] [CrossRef]
- Arnsten, A.F.T.; Datta, D.; Del Tredici, K.; Braak, H. Hypothesis: Tau pathology is an initiating factor in sporadic Alzheimer’s disease. Alzheimer’s Dement. 2021, 17, 115–124. [Google Scholar] [CrossRef]
- Takahashi, M.; Iseki, E.; Kosaka, K. Cdk5 and munc-18/p67 co-localization in early stage neurofibrillary tangles-bearing neurons in Alzheimer type dementia brains. J. Neurol. Sci. 2000, 172, 63–69. [Google Scholar] [CrossRef]
- Ferrer, I.; Barrachina, M.; Puig, B. Glycogen synthase kinase-3 is associated with neuronal and glial hyperphosphorylated tau deposits in Alzheimer’s disease, Pick’s disease, progressive supranuclear palsy and corticobasal degeneration. Acta Neuropathol. 2002, 104, 583–591. [Google Scholar] [CrossRef]
- Wegmann, S.; Biernat, J.; Mandelkow, E. A current view on Tau protein phosphorylation in Alzheimer’s disease. Curr. Opin. Neurobiol. 2021, 69, 131–138. [Google Scholar] [CrossRef]
- Kalakech, H.; Hibert, P.; Prunier-Mirebeau, D.; Tamareille, S.; Letournel, F.; Macchi, L.; Pinet, F.; Furber, A.; Prunier, F. RISK and SAFE signaling pathway involvement in apolipoprotein A-I-induced cardioprotection. PLoS ONE 2014, 9, e107950. [Google Scholar] [CrossRef]
- Lee, S.; Shea, T.B. Caspase-mediated truncation of tau potentiates aggregation. Int. J. Alzheimer’s Dis. 2012, 2012, 731063. [Google Scholar] [CrossRef]
- Arvanitakis, Z.; Capuano, A.W.; Leurgans, S.E.; Bennett, D.A.; Schneider, J.A. Relation of cerebral vessel disease to Alzheimer’s disease dementia and cognitive function in elderly people: A cross-sectional study. Lancet Neurol. 2016, 15, 934–943. [Google Scholar] [CrossRef]
- Gibson, C.M.; Duffy, D.; Korjian, S.; Bahit, M.C.; Chi, G.; Alexander, J.H.; Lincoff, A.M.; Heise, M.; Tricoci, P.; Deckelbaum, L.I.; et al. Apolipoprotein A1 Infusions and Cardiovascular Outcomes after Acute Myocardial Infarction. N. Engl. J. Med. 2024, 390, 1560–1571. [Google Scholar] [CrossRef]
- Michael Gibson, C.; Korjian, S.; Tricoci, P.; Daaboul, Y.; Yee, M.; Jain, P.; Alexander, J.H.; Steg, P.G.; Lincoff, A.M.; Kastelein, J.J.; et al. Safety and Tolerability of CSL112, a Reconstituted, Infusible, Plasma-Derived Apolipoprotein A-I, After Acute Myocardial Infarction: The AEGIS-I Trial (ApoA-I Event Reducing in Ischemic Syndromes I). Circulation 2016, 134, 1918–1930. [Google Scholar] [CrossRef]
- Lee, M.; Lim, J.S.; Kim, Y.; Park, S.H.; Lee, S.H.; Kim, C.; Lee, B.C.; Yu, K.H.; Lee, J.J.; Oh, M.S. High ApoB/ApoA-I Ratio Predicts Post-Stroke Cognitive Impairment in Acute Ischemic Stroke Patients with Large Artery Atherosclerosis. Nutrients 2023, 15, 4670. [Google Scholar] [CrossRef]
- Cserép, C.; Pósfai, B.; Lénárt, N.; Fekete, R.; László, Z.I.; Lele, Z.; Orsolits, B.; Molnár, G.; Heindl, S.; Schwarcz, A.D.; et al. Microglia monitor and protect neuronal function through specialized somatic purinergic junctions. Science 2020, 367, 528–537. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.D.; Abdalla, S.; Eissa, N.; Akour, A.; Jha, N.K.; Ojha, S.; Sadek, B. Targeting Microglia in Neuroinflammation: H3 Receptor Antagonists as a Novel Therapeutic Approach for Alzheimer’s Disease, Parkinson’s Disease, and Autism Spectrum Disorder. Pharmaceuticals 2024, 17, 831. [Google Scholar] [CrossRef]
- Wu, X.; Miller, J.A.; Lee, B.T.K.; Wang, Y.; Ruedl, C. Reducing microglial lipid load enhances β amyloid phagocytosis in an Alzheimer’s disease mouse model. Sci. Adv. 2025, 11, eadq6038. [Google Scholar] [CrossRef]
- Yang, D.; Wang, X.; Zhang, L.; Fang, Y.; Zheng, Q.; Liu, X.; Yu, W.; Chen, S.; Ying, J.; Hua, F. Lipid metabolism and storage in neuroglia: Role in brain development and neurodegenerative diseases. Cell Biosci. 2022, 12, 106. [Google Scholar] [CrossRef]
- Choi, M.; Kim, D.; Youn, Y.J.; Ryu, J.; Jeong, Y.H. Effect of Obesity and High-Density Lipoprotein Concentration on the Pathological Characteristics of Alzheimer’s Disease in High-Fat Diet-Fed Mice. Int. J. Mol. Sci. 2022, 23, 12296. [Google Scholar] [CrossRef]
- Endo, F.; Kasai, A.; Soto, J.S.; Yu, X.; Qu, Z.; Hashimoto, H.; Gradinaru, V.; Kawaguchi, R.; Khakh, B.S. Molecular basis of astrocyte diversity and morphology across the CNS in health and disease. Science 2022, 378, eadc9020. [Google Scholar] [CrossRef]
- Cai, Z.; Wan, C.Q.; Liu, Z. Astrocyte and Alzheimer’s disease. J. Neurol. 2017, 264, 2068–2074. [Google Scholar] [CrossRef]
- Allaman, I.; Gavillet, M.; Bélanger, M.; Laroche, T.; Viertl, D.; Lashuel, H.A.; Magistretti, P.J. Amyloid-beta aggregates cause alterations of astrocytic metabolic phenotype: Impact on neuronal viability. J. Neurosci. 2010, 30, 3326–3338. [Google Scholar] [CrossRef]
- Kim, J.; Yoo, I.D.; Lim, J.; Moon, J.S. Pathological phenotypes of astrocytes in Alzheimer’s disease. Exp. Mol. Med. 2024, 56, 95–99. [Google Scholar] [CrossRef]
- Litvinchuk, A.; Suh, J.H.; Guo, J.L.; Lin, K.; Davis, S.S.; Bien-Ly, N.; Tycksen, E.; Tabor, G.T.; Remolina Serrano, J.; Manis, M.; et al. Amelioration of Tau and ApoE4-linked glial lipid accumulation and neurodegeneration with an LXR agonist. Neuron 2024, 112, 384–403.e8. [Google Scholar] [CrossRef]
- Stukas, S.; May, S.; Wilkinson, A.; Chan, J.; Donkin, J.; Wellington, C.L. The LXR agonist GW3965 increases apoA-I protein levels in the central nervous system independent of ABCA1. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2012, 1821, 536–546. [Google Scholar] [CrossRef]
- Kheirollah, A.; Ito, J.; Nagayasu, Y.; Lu, R.; Yokoyama, S. Cyclosporin A inhibits apolipoprotein A-I-induced early events in cellular cholesterol homeostasis in rat astrocytes. Neuropharmacology 2006, 51, 693–700. [Google Scholar] [CrossRef]
- Victor, M.B.; Leary, N.; Luna, X.; Meharena, H.S.; Scannail, A.N.; Bozzelli, P.L.; Samaan, G.; Murdock, M.H.; von Maydell, D.; Effenberger, A.H.; et al. Lipid accumulation induced by APOE4 impairs microglial surveillance of neuronal-network activity. Cell Stem Cell 2022, 29, 1197–1212.e8. [Google Scholar] [CrossRef]
- Blanchard, J.W.; Akay, L.A.; Davila-Velderrain, J.; von Maydell, D.; Mathys, H.; Davidson, S.M.; Effenberger, A.; Chen, C.Y.; Maner-Smith, K.; Hajjar, I.; et al. APOE4 impairs myelination via cholesterol dysregulation in oligodendrocytes. Nature 2022, 611, 769–779. [Google Scholar] [CrossRef]
- Yeung, M.S.Y.; Djelloul, M.; Steiner, E.; Bernard, S.; Salehpour, M.; Possnert, G.; Brundin, L.; Frisén, J. Dynamics of oligodendrocyte generation in multiple sclerosis. Nature 2019, 566, 538–542. [Google Scholar] [CrossRef]
- Lopez-Lee, C.; Kodama, L.; Fan, L.; Zhu, D.; Zhu, J.; Wong, M.Y.; Ye, P.; Norman, K.; Foxe, N.R.; Ijaz, L.; et al. Tlr7 drives sex differences in age- and Alzheimer’s disease-related demyelination. Science 2024, 386, eadk7844. [Google Scholar] [CrossRef]
- Sasmita, A.O.; Depp, C.; Nazarenko, T.; Sun, T.; Siems, S.B.; Ong, E.C.; Nkeh, Y.B.; Böhler, C.; Yu, X.; Bues, B.; et al. Oligodendrocytes produce amyloid-β and contribute to plaque formation alongside neurons in Alzheimer’s disease model mice. Nat. Neurosci. 2024, 27, 1668–1674. [Google Scholar] [CrossRef]
- Berghoff, S.A.; Spieth, L.; Sun, T.; Hosang, L.; Schlaphoff, L.; Depp, C.; Düking, T.; Winchenbach, J.; Neuber, J.; Ewers, D.; et al. Microglia facilitate repair of demyelinated lesions via post-squalene sterol synthesis. Nat. Neurosci. 2021, 24, 47–60. [Google Scholar] [CrossRef]
- Wang, X.; Magkos, F.; Mittendorfer, B. Sex differences in lipid and lipoprotein metabolism: It’s not just about sex hormones. J. Clin. Endocrinol. Metab. 2011, 96, 885–893. [Google Scholar] [CrossRef]
- Palmisano, B.T.; Zhu, L.; Eckel, R.H.; Stafford, J.M. Sex differences in lipid and lipoprotein metabolism. Mol. Metab. 2018, 15, 45–55. [Google Scholar] [CrossRef]
- Yin, Z.G.; Wang, Q.S.; Yu, K.; Wang, W.W.; Lin, H.; Yang, Z.H. Sex differences in associations between blood lipids and cerebral small vessel disease. Nutr. Metab. Cardiovasc. Dis. NMCD 2018, 28, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Siddiqi, H.K.; Kiss, D.; Rader, D. HDL-cholesterol and cardiovascular disease: Rethinking our approach. Curr. Opin. Cardiol. 2015, 30, 536–542. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Hou, X.H.; Wang, D.D.; Ma, Y.H.; Tan, C.C.; Sun, F.R.; Cui, M.; Dong, Q.; Tan, L.; Yu, J.T. Apolipoprotein B/AI ratio as an independent risk factor for intracranial atherosclerotic stenosis. Aging 2019, 11, 6851–6862. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Liu, S.; Wang, X.; Huang, H.; Wang, M.; Qu, W.; Yu, Z.; Wang, W.; Luo, X. Lipids, Apolipoproteins, Lipid-Lowering Drugs, and the Risk of Cerebral Small Vessel Disease: A Mendelian Randomization Study. J. Am. Heart Assoc. 2024, 13, e032409. [Google Scholar] [CrossRef]
- Karavia, E.A.; Hatziri, A.; Kalogeropoulou, C.; Papachristou, N.I.; Xepapadaki, E.; Constantinou, C.; Natsos, A.; Petropoulou, P.I.; Sasson, S.; Papachristou, D.J.; et al. Deficiency in apolipoprotein A-I ablates the pharmacological effects of metformin on plasma glucose homeostasis and hepatic lipid deposition. Eur. J. Pharmacol. 2015, 766, 76–85. [Google Scholar] [CrossRef]
- Stenkula, K.G.; Lindahl, M.; Petrlova, J.; Dalla-Riva, J.; Göransson, O.; Cushman, S.W.; Krupinska, E.; Jones, H.A.; Lagerstedt, J.O. Single injections of apoA-I acutely improve in vivo glucose tolerance in insulin-resistant mice. Diabetologia 2014, 57, 797–800. [Google Scholar] [CrossRef]
- Drew, B.G.; Duffy, S.J.; Formosa, M.F.; Natoli, A.K.; Henstridge, D.C.; Penfold, S.A.; Thomas, W.G.; Mukhamedova, N.; de Courten, B.; Forbes, J.M.; et al. High-density lipoprotein modulates glucose metabolism in patients with type 2 diabetes mellitus. Circulation 2009, 119, 2103–2111. [Google Scholar] [CrossRef]
- Nasr, A.; Matthews, K.A.; Brooks, M.M.; Barinas-Mitchell, E.; Orchard, T.; Billheimer, J.; Wang, N.C.; McConnell, D.; Rader, D.J.; El Khoudary, S.R. Early Midlife Cardiovascular Health Influences Future HDL Metrics in Women: The SWAN HDL Study. J. Am. Heart Assoc. 2022, 11, e026243. [Google Scholar] [CrossRef]
- Qi, M.; Billheimer, J.; Chang, C.H.; Janssen, I.; Brooks, M.M.; Orchard, T.; Karlamangla, A.S.; Barinas-Mitchell, E.; Derby, C.A.; McConnell, D.; et al. High-density lipoprotein over midlife and future cognition in women: The SWAN HDL ancillary study. J. Clin. Endocrinol. Metab. 2024, dgae697. [Google Scholar] [CrossRef]
- Holzer, M.; Trieb, M.; Konya, V.; Wadsack, C.; Heinemann, A.; Marsche, G. Aging affects high-density lipoprotein composition and function. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2013, 1831, 1442–1448. [Google Scholar] [CrossRef]
- Zhu, D.; Montagne, A.; Zhao, Z. Alzheimer’s pathogenic mechanisms and underlying sex difference. Cell. Mol. Life Sci. CMLS 2021, 78, 4907–4920. [Google Scholar] [CrossRef]
- Breeze, B.; Connell, E.; Wileman, T.; Muller, M.; Vauzour, D.; Pontifex, M.G. Menopause and Alzheimer’s disease susceptibility: Exploring the potential mechanisms. Brain Res. 2024, 1844, 149170. [Google Scholar] [CrossRef]
- Scheyer, O.; Rahman, A.; Hristov, H.; Berkowitz, C.; Isaacson, R.S.; Diaz Brinton, R.; Mosconi, L. Female Sex and Alzheimer’s Risk: The Menopause Connection. J. Prev. Alzheimer’s Dis. 2018, 5, 225–230. [Google Scholar] [CrossRef]
- Xiong, J.; Kang, S.S.; Wang, Z.; Liu, X.; Kuo, T.C.; Korkmaz, F.; Padilla, A.; Miyashita, S.; Chan, P.; Zhang, Z.; et al. FSH blockade improves cognition in mice with Alzheimer’s disease. Nature 2022, 603, 470–476. [Google Scholar] [CrossRef]
- Xiong, J.; Kang, S.S.; Wang, M.; Wang, Z.; Xia, Y.; Liao, J.; Liu, X.; Yu, S.P.; Zhang, Z.; Ryu, V.; et al. FSH and ApoE4 contribute to Alzheimer’s disease-like pathogenesis via C/EBPβ/δ-secretase in female mice. Nat. Commun. 2023, 14, 6577. [Google Scholar] [CrossRef]
- Jin, Y.; Topaloudi, A.; Shekhar, S.; Chen, G.; Scott, A.N.; Colon, B.D.; Drineas, P.; Rochet, C.; Paschou, P. Neuropathology-based approach reveals novel Alzheimer’s Disease genes and highlights female-specific pathways and causal links to disrupted lipid metabolism: Insights into a vicious cycle. Acta Neuropathol. Commun. 2025, 13, 1. [Google Scholar] [CrossRef]
- Hye, A.; Riddoch-Contreras, J.; Baird, A.L.; Ashton, N.J.; Bazenet, C.; Leung, R.; Westman, E.; Simmons, A.; Dobson, R.; Sattlecker, M.; et al. Plasma proteins predict conversion to dementia from prodromal disease. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2014, 10, 799–807.e2. [Google Scholar] [CrossRef]
- Liu, Y.; Tan, Y.; Zhang, Z.; Yi, M.; Zhu, L.; Peng, W. The interaction between ageing and Alzheimer’s disease: Insights from the hallmarks of ageing. Transl. Neurodegener. 2024, 13, 7. [Google Scholar] [CrossRef]
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef]
- Zhao, J.; Huai, J. Role of primary aging hallmarks in Alzheimer’s disease. Theranostics 2023, 13, 197–230. [Google Scholar] [CrossRef]
- Guo, J.; Huang, X.; Dou, L.; Yan, M.; Shen, T.; Tang, W.; Li, J. Aging and aging-related diseases: From molecular mechanisms to interventions and treatments. Signal Transduct. Target. Ther. 2022, 7, 391. [Google Scholar] [CrossRef] [PubMed]
- Maity, S.; Farrell, K.; Navabpour, S.; Narayanan, S.N.; Jarome, T.J. Epigenetic Mechanisms in Memory and Cognitive Decline Associated with Aging and Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 12280. [Google Scholar] [CrossRef] [PubMed]
- Hipp, M.S.; Kasturi, P.; Hartl, F.U. The proteostasis network and its decline in ageing. Nat. Rev. Mol. Cell Biol. 2019, 20, 421–435. [Google Scholar] [CrossRef]
- Lu, J.; Han, G.; Liu, X.; Chen, B.; Peng, K.; Shi, Y.; Zhang, M.; Yang, Y.; Cui, J.; Song, L.; et al. Association of high-density lipoprotein cholesterol with all-cause and cause-specific mortality in a Chinese population of 3.3 million adults: A prospective cohort study. Lancet Reg. Health West. Pac. 2024, 42, 100874. [Google Scholar] [CrossRef]
- Farrer, L.A.; Cupples, L.A.; Haines, J.L.; Hyman, B.; Kukull, W.A.; Mayeux, R.; Myers, R.H.; Pericak-Vance, M.A.; Risch, N.; van Duijn, C.M. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA 1997, 278, 1349–1356. [Google Scholar] [CrossRef]
- Neu, S.C.; Pa, J.; Kukull, W.; Beekly, D.; Kuzma, A.; Gangadharan, P.; Wang, L.S.; Romero, K.; Arneric, S.P.; Redolfi, A.; et al. Apolipoprotein E Genotype and Sex Risk Factors for Alzheimer Disease: A Meta-analysis. JAMA Neurol. 2017, 74, 1178–1189. [Google Scholar] [CrossRef]
- Genin, E.; Hannequin, D.; Wallon, D.; Sleegers, K.; Hiltunen, M.; Combarros, O.; Bullido, M.J.; Engelborghs, S.; De Deyn, P.; Berr, C.; et al. APOE and Alzheimer disease: A major gene with semi-dominant inheritance. Mol. Psychiatry 2011, 16, 903–907. [Google Scholar] [CrossRef]
- Wahrle, S.E.; Jiang, H.; Parsadanian, M.; Legleiter, J.; Han, X.; Fryer, J.D.; Kowalewski, T.; Holtzman, D.M. ABCA1 is required for normal central nervous system ApoE levels and for lipidation of astrocyte-secreted apoE. J. Biol. Chem. 2004, 279, 40987–40993. [Google Scholar] [CrossRef]
- Karten, B.; Campenot, R.B.; Vance, D.E.; Vance, J.E. Expression of ABCG1, but not ABCA1, correlates with cholesterol release by cerebellar astroglia. J. Biol. Chem. 2006, 281, 4049–4057. [Google Scholar] [CrossRef]
- Huang, Y.A.; Zhou, B.; Wernig, M.; Südhof, T.C. ApoE2, ApoE3, and ApoE4 Differentially Stimulate APP Transcription and Aβ Secretion. Cell 2017, 168, 427–441.e21. [Google Scholar] [CrossRef]
- Wang, C.; Najm, R.; Xu, Q.; Jeong, D.E.; Walker, D.; Balestra, M.E.; Yoon, S.Y.; Yuan, H.; Li, G.; Miller, Z.A.; et al. Gain of toxic apolipoprotein E4 effects in human iPSC-derived neurons is ameliorated by a small-molecule structure corrector. Nat. Med. 2018, 24, 647–657. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.C.; Zhao, N.; Fu, Y.; Wang, N.; Linares, C.; Tsai, C.W.; Bu, G. ApoE4 Accelerates Early Seeding of Amyloid Pathology. Neuron 2017, 96, 1024–1032.e3. [Google Scholar] [CrossRef] [PubMed]
- Deane, R.; Sagare, A.; Hamm, K.; Parisi, M.; Lane, S.; Finn, M.B.; Holtzman, D.M.; Zlokovic, B.V. apoE isoform-specific disruption of amyloid beta peptide clearance from mouse brain. J. Clin. Investig. 2008, 118, 4002–4013. [Google Scholar] [CrossRef]
- Robert, J.; Button, E.B.; Yuen, B.; Gilmour, M.; Kang, K.; Bahrabadi, A.; Stukas, S.; Zhao, W.; Kulic, I.; Wellington, C.L. Clearance of beta-amyloid is facilitated by apolipoprotein E and circulating high-density lipoproteins in bioengineered human vessels. eLife 2017, 6, e29595. [Google Scholar] [CrossRef]
- Ma, Q.; Zhao, Z.; Sagare, A.P.; Wu, Y.; Wang, M.; Owens, N.C.; Verghese, P.B.; Herz, J.; Holtzman, D.M.; Zlokovic, B.V. Blood-brain barrier-associated pericytes internalize and clear aggregated amyloid-β42 by LRP1-dependent apolipoprotein E isoform-specific mechanism. Mol. Neurodegener. 2018, 13, 57. [Google Scholar] [CrossRef]
- Tcw, J.; Qian, L.; Pipalia, N.H.; Chao, M.J.; Liang, S.A.; Shi, Y.; Jain, B.R.; Bertelsen, S.E.; Kapoor, M.; Marcora, E.; et al. Cholesterol and matrisome pathways dysregulated in astrocytes and microglia. Cell 2022, 185, 2213–2233.e25. [Google Scholar] [CrossRef]
- Wang, S.; Li, B.; Li, J.; Cai, Z.; Hugo, C.; Sun, Y.; Qian, L.; Tcw, J.; Chui, H.C.; Dikeman, D.; et al. Cellular senescence induced by cholesterol accumulation is mediated by lysosomal ABCA1 in APOE4 and AD. Mol. Neurodegener. 2025, 20, 15. [Google Scholar] [CrossRef]
- Gorska-Ciebiada, M.; Saryusz-Wolska, M.; Ciebiada, M.; Loba, J. Mild cognitive impairment and depressive symptoms in elderly patients with diabetes: Prevalence, risk factors, and comorbidity. J. Diabetes Res. 2014, 2014, 179648. [Google Scholar] [CrossRef]
- Janson, J.; Laedtke, T.; Parisi, J.E.; O’Brien, P.; Petersen, R.C.; Butler, P.C. Increased risk of type 2 diabetes in Alzheimer disease. Diabetes 2004, 53, 474–481. [Google Scholar] [CrossRef]
- De la Monte, S.M.; Wands, J.R. Alzheimer’s disease is type 3 diabetes-evidence reviewed. J. Diabetes Sci. Technol. 2008, 2, 1101–1113. [Google Scholar] [CrossRef]
- De la Monte, S.M.; Tong, M.; Wands, J.R. The 20-Year Voyage Aboard the Journal of Alzheimer’s Disease: Docking at ‘Type 3 Diabetes’, Environmental/Exposure Factors, Pathogenic Mechanisms, and Potential Treatments. J. Alzheimer’s Dis. 2018, 62, 1381–1390. [Google Scholar] [CrossRef] [PubMed]
- Kandimalla, R.; Thirumala, V.; Reddy, P.H. Is Alzheimer’s disease a Type 3 Diabetes? A critical appraisal. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2017, 1863, 1078–1089. [Google Scholar] [CrossRef] [PubMed]
- Michailidis, M.; Moraitou, D.; Tata, D.A.; Kalinderi, K.; Papamitsou, T.; Papaliagkas, V. Alzheimer’s Disease as Type 3 Diabetes: Common Pathophysiological Mechanisms between Alzheimer’s Disease and Type 2 Diabetes. Int. J. Mol. Sci. 2022, 23, 2687. [Google Scholar] [CrossRef]
- Mangat, R.; Borthwick, F.; Haase, T.; Jacome, M.; Nelson, R.; Kontush, A.; Vine, D.F.; Proctor, S.D. Intestinal lymphatic HDL miR-223 and ApoA-I are reduced during insulin resistance and restored with niacin. FASEB J. 2018, 32, 1602–1612. [Google Scholar] [CrossRef]
- Feng, X.; Gao, X.; Yao, Z.; Xu, Y. Low apoA-I is associated with insulin resistance in patients with impaired glucose tolerance: A cross-sectional study. Lipids Health Dis. 2017, 16, 69. [Google Scholar] [CrossRef]
- Barter, P.J.; Rye, K.A.; Tardif, J.C.; Waters, D.D.; Boekholdt, S.M.; Breazna, A.; Kastelein, J.J. Effect of torcetrapib on glucose, insulin, and hemoglobin A1c in subjects in the Investigation of Lipid Level Management to Understand its Impact in Atherosclerotic Events (ILLUMINATE) trial. Circulation 2011, 124, 555–562. [Google Scholar] [CrossRef]
- Menon, V.; Kumar, A.; Patel, D.R.; St John, J.; Riesmeyer, J.; Weerakkody, G.; Ruotolo, G.; Wolski, K.E.; McErlean, E.; Cremer, P.C.; et al. Effect of CETP inhibition with evacetrapib in patients with diabetes mellitus enrolled in the ACCELERATE trial. BMJ Open Diabetes Res. Care 2020, 8, e000943. [Google Scholar] [CrossRef]
- Cochran, B.J.; Ryder, W.J.; Parmar, A.; Tang, S.; Reilhac, A.; Arthur, A.; Charil, A.; Hamze, H.; Barter, P.J.; Kritharides, L.; et al. In vivo PET imaging with [(18)F]FDG to explain improved glucose uptake in an apolipoprotein A-I treated mouse model of diabetes. Diabetologia 2016, 59, 1977–1984. [Google Scholar] [CrossRef]
- Tang, S.; Tabet, F.; Cochran, B.J.; Cuesta Torres, L.F.; Wu, B.J.; Barter, P.J.; Rye, K.A. Apolipoprotein A-I enhances insulin-dependent and insulin-independent glucose uptake by skeletal muscle. Sci. Rep. 2019, 9, 1350. [Google Scholar] [CrossRef]
- Wu, B.J.; Sun, Y.; Ong, K.L.; Li, Y.; Tang, S.; Barter, P.J.; Rye, K.A. Apolipoprotein A-I Protects Against Pregnancy-Induced Insulin Resistance in Rats. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1160–1171. [Google Scholar] [CrossRef]
- Dewanjee, S.; Chakraborty, P.; Bhattacharya, H.; Chacko, L.; Singh, B.; Chaudhary, A.; Javvaji, K.; Pradhan, S.R.; Vallamkondu, J.; Dey, A.; et al. Altered glucose metabolism in Alzheimer’s disease: Role of mitochondrial dysfunction and oxidative stress. Free Radic. Biol. Med. 2022, 193, 134–157. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, A.; Sah, S.P. Insulin signaling pathway and related molecules: Role in neurodegeneration and Alzheimer’s disease. Neurochem. Int. 2020, 135, 104707. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.J.; Li, X.R.; Chai, S.F.; Li, W.R.; Li, S.; Hou, M.; Li, J.L.; Ye, Y.C.; Cai, H.Y.; Hölscher, C.; et al. Semaglutide ameliorates cognition and glucose metabolism dysfunction in the 3xTg mouse model of Alzheimer’s disease via the GLP-1R/SIRT1/GLUT4 pathway. Neuropharmacology 2023, 240, 109716. [Google Scholar] [CrossRef]
- Ramírez-Expósito, M.J.; Martínez-Martos, J.M.; Cantón-Habas, V.; Del Pilar Carrera-González, M. Putative Involvement of Endocrine Disruptors in the Alzheimer’s Disease Via the Insulin-Regulated Aminopeptidase/GLUT4 Pathway. Curr. Neuropharmacol. 2021, 19, 939–956. [Google Scholar] [CrossRef]
- Nam, M.H.; Ko, H.Y.; Kim, D.; Lee, S.; Park, Y.M.; Hyeon, S.J.; Won, W.; Chung, J.I.; Kim, S.Y.; Jo, H.H.; et al. Visualizing reactive astrocyte-neuron interaction in Alzheimer’s disease using 11C-acetate and 18F-FDG. Brain 2023, 146, 2957–2974. [Google Scholar] [CrossRef]
- Chen, M.; Huang, N.; Liu, J.; Huang, J.; Shi, J.; Jin, F. AMPK: A bridge between diabetes mellitus and Alzheimer’s disease. Behav. Brain Res. 2021, 400, 113043. [Google Scholar] [CrossRef]
- Pétremand, J.; Puyal, J.; Chatton, J.Y.; Duprez, J.; Allagnat, F.; Frias, M.; James, R.W.; Waeber, G.; Jonas, J.C.; Widmann, C. HDLs protect pancreatic β-cells against ER stress by restoring protein folding and trafficking. Diabetes 2012, 61, 1100–1111. [Google Scholar] [CrossRef]
- Rütti, S.; Ehses, J.A.; Sibler, R.A.; Prazak, R.; Rohrer, L.; Georgopoulos, S.; Meier, D.T.; Niclauss, N.; Berney, T.; Donath, M.Y.; et al. Low- and high-density lipoproteins modulate function, apoptosis, and proliferation of primary human and murine pancreatic beta-cells. Endocrinology 2009, 150, 4521–4530. [Google Scholar] [CrossRef]
- Kellar, D.; Craft, S. Brain insulin resistance in Alzheimer’s disease and related disorders: Mechanisms and therapeutic approaches. Lancet Neurol. 2020, 19, 758–766. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Halliwell, B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat. reviews. Neurosci. 2019, 20, 148–160. [Google Scholar] [CrossRef]
- Wang, Z.H.; Xiang, J.; Liu, X.; Yu, S.P.; Manfredsson, F.P.; Sandoval, I.M.; Wu, S.; Wang, J.Z.; Ye, K. Deficiency in BDNF/TrkB Neurotrophic Activity Stimulates δ-Secretase by Upregulating C/EBPβ in Alzheimer’s Disease. Cell Rep. 2019, 28, 655–669.e5. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, J.T.; Thomas, A. Vascular dementia. Lancet 2015, 386, 1698–1706. [Google Scholar] [CrossRef] [PubMed]
- Caballero MÁ, A.; Song, Z.; Rubinski, A.; Duering, M.; Dichgans, M.; Park, D.C.; Ewers, M. Age-dependent amyloid deposition is associated with white matter alterations in cognitively normal adults during the adult life span. Alzheimer’s Dement. 2020, 16, 651–661. [Google Scholar] [CrossRef]
- Attems, J.; Jellinger, K.A. The overlap between vascular disease and Alzheimer’s disease—lessons from pathology. BMC Med. 2014, 12, 206. [Google Scholar] [CrossRef]
- Gannon, O.J.; Robison, L.S.; Salinero, A.E.; Abi-Ghanem, C.; Mansour, F.M.; Kelly, R.D.; Tyagi, A.; Brawley, R.R.; Ogg, J.D.; Zuloaga, K.L. High-fat diet exacerbates cognitive decline in mouse models of Alzheimer’s disease and mixed dementia in a sex-dependent manner. J. Neuroinflamm. 2022, 19, 110. [Google Scholar] [CrossRef]
- Wang, X.; Xing, A.; Xu, C.; Cai, Q.; Liu, H.; Li, L. Cerebrovascular hypoperfusion induces spatial memory impairment, synaptic changes, and amyloid-β oligomerization in rats. J. Alzheimer’s Dis. 2010, 21, 813–822. [Google Scholar] [CrossRef]
- Sun, X.; He, G.; Qing, H.; Zhou, W.; Dobie, F.; Cai, F.; Staufenbiel, M.; Huang, L.E.; Song, W. Hypoxia facilitates Alzheimer’s disease pathogenesis by up-regulating BACE1 gene expression. Proc. Natl. Acad. Sci. USA 2006, 103, 18727–18732. [Google Scholar] [CrossRef]
- Zhang, X.; Zhou, K.; Wang, R.; Cui, J.; Lipton, S.A.; Liao, F.F.; Xu, H.; Zhang, Y.W. Hypoxia-inducible factor 1alpha (HIF-1alpha)-mediated hypoxia increases BACE1 expression and beta-amyloid generation. J. Biol. Chem. 2007, 282, 10873–10880. [Google Scholar] [CrossRef]
- Guglielmotto, M.; Aragno, M.; Autelli, R.; Giliberto, L.; Novo, E.; Colombatto, S.; Danni, O.; Parola, M.; Smith, M.A.; Perry, G.; et al. The up-regulation of BACE1 mediated by hypoxia and ischemic injury: Role of oxidative stress and HIF1alpha. J. Neurochem. 2009, 108, 1045–1056. [Google Scholar] [CrossRef]
- Li, L.; Zhang, X.; Yang, D.; Luo, G.; Chen, S.; Le, W. Hypoxia increases Abeta generation by altering beta- and gamma-cleavage of APP. Neurobiol. Aging 2009, 30, 1091–1098. [Google Scholar] [CrossRef]
- Fang, H.; Zhang, L.F.; Meng, F.T.; Du, X.; Zhou, J.N. Acute hypoxia promote the phosphorylation of tau via ERK pathway. Neurosci. Lett. 2010, 474, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Ridker, P.M.; Hansson, G.K. Progress and challenges in translating the biology of atherosclerosis. Nature 2011, 473, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Chernick, D.; Ortiz-Valle, S.; Jeong, A.; Swaminathan, S.K.; Kandimalla, K.K.; Rebeck, G.W.; Li, L. High-density lipoprotein mimetic peptide 4F mitigates amyloid-β-induced inhibition of apolipoprotein E secretion and lipidation in primary astrocytes and microglia. J. Neurochem. 2018, 147, 647–662. [Google Scholar] [CrossRef]
- Sahoo, B.R.; Bekier, M.E., 2nd; Liu, Z.; Kocman, V.; Stoddard, A.K.; Anantharamaiah, G.M.; Nowick, J.; Fierke, C.A.; Wang, Y.; Ramamoorthy, A. Structural Interaction of Apolipoprotein A-I Mimetic Peptide with Amyloid-β Generates Toxic Hetero-oligomers. J. Mol. Biol. 2020, 432, 1020–1034. [Google Scholar] [CrossRef]
- Wang, Z.; Zhong, R.; Curran, G.L.; Min, P.; Lowe, V.J.; Li, L.; Kandimalla, K.K. High-Density Lipoprotein Mimetic Peptide 4F Reduces Toxic Amyloid-Beta Exposure to the Blood-Brain Barrier Endothelium in Alzheimer’s Disease Transgenic Mice. Mol. Pharm. 2024, 21, 5661–5671. [Google Scholar] [CrossRef]
- Zhong, R.; Chernick, D.; Hottman, D.; Tan, Y.; Kim, M.; Narayanan, M.; Li, L. The HDL-Mimetic Peptide 4F Mitigates Vascular and Cortical Amyloid Pathology and Associated Neuroinflammation in a Transgenic Mouse Model of Cerebral Amyloid Angiopathy and Alzheimer’s Disease. Mol. Neurobiol. 2025. Published Online. [Google Scholar] [CrossRef]
- Anantharamaiah, G.M.; Garber, D.W.; White, C.R. Apolipoprotein Mimetic Peptides as Modulators of Lipoprotein Function. Protein Pept. Lett. 2016, 23, 1024–1031. [Google Scholar] [CrossRef]
- Yancey, P.G.; Bielicki, J.K.; Johnson, W.J.; Lund-Katz, S.; Palgunachari, M.N.; Anantharamaiah, G.M.; Segrest, J.P.; Phillips, M.C.; Rothblat, G.H. Efflux of cellular cholesterol and phospholipid to lipid-free apolipoproteins and class A amphipathic peptides. Biochemistry 1995, 34, 7955–7965. [Google Scholar] [CrossRef]
- Wang, W.; Zhu, X. HDL mimetic peptides affect apolipoprotein E metabolism: Equal supplement or functional enhancer?: An Editorial for ‘High-density lipoprotein mimetic peptide 4F mitigates amyloid-β-induced inhibition of apolipoprotein E secretion and lipidation in primary astrocytes and microglia’ on page 647. J. Neurochem. 2018, 147, 580–583. [Google Scholar] [CrossRef]
- Ascherio, A.; Schwarzschild, M.A. The epidemiology of Parkinson’s disease: Risk factors and prevention. Lancet Neurol. 2016, 15, 1257–1272. [Google Scholar] [CrossRef]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [PubMed]
- Swanson, C.R.; Berlyand, Y.; Xie, S.X.; Alcalay, R.N.; Chahine, L.M.; Chen-Plotkin, A.S. Plasma apolipoprotein A1 associates with age at onset and motor severity in early Parkinson’s disease patients. Mov. Disord. 2015, 30, 1648–1656. [Google Scholar] [CrossRef] [PubMed]
- Swanson, C.R.; Li, K.; Unger, T.L.; Gallagher, M.D.; Van Deerlin, V.M.; Agarwal, P.; Leverenz, J.; Roberts, J.; Samii, A.; Gross, R.G.; et al. Lower plasma apolipoprotein A1 levels are found in Parkinson’s disease and associate with apolipoprotein A1 genotype. Mov. Disord. 2015, 30, 805–812. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.C.; Lin, C.H.; Wu, R.M.; Lin, M.S.; Lin, J.W.; Chang, C.H.; Lai, M.S. Discontinuation of statin therapy associates with Parkinson disease: A population-based study. Neurology 2013, 81, 410–416. [Google Scholar] [CrossRef]
- Gao, X.; Simon, K.C.; Schwarzschild, M.A.; Ascherio, A. Prospective study of statin use and risk of Parkinson disease. Arch. Neurol. 2012, 69, 380–384. [Google Scholar] [CrossRef]
- Cho, K.H. Structural and Functional Changes of Reconstituted High-Density Lipoprotein (HDL) by Incorporation of α-synuclein: A Potent Antioxidant and Anti-Glycation Activity of α-synuclein and apoA-I in HDL at High Molar Ratio of α-synuclein. Molecules 2021, 26, 7485. [Google Scholar] [CrossRef]
- Keeney, J.T.; Swomley, A.M.; Förster, S.; Harris, J.L.; Sultana, R.; Butterfield, D.A. Apolipoprotein A-I: Insights from redox proteomics for its role in neurodegeneration. Proteomics. Clin. Appl. 2013, 7, 109–122. [Google Scholar] [CrossRef]
- Ikeda, K.; Nakamura, Y.; Kiyozuka, T.; Aoyagi, J.; Hirayama, T.; Nagata, R.; Ito, H.; Iwamoto, K.; Murata, K.; Yoshii, Y.; et al. Serological profiles of urate, paraoxonase-1, ferritin and lipid in Parkinson’s disease: Changes linked to disease progression. Neuro-Degener. Dis. 2011, 8, 252–258. [Google Scholar] [CrossRef]
- Piguet, O.; Hornberger, M.; Mioshi, E.; Hodges, J.R. Behavioural-variant frontotemporal dementia: Diagnosis, clinical staging, and management. Lancet Neurol. 2011, 10, 162–172. [Google Scholar] [CrossRef]
- Kim, W.S.; He, Y.; Phan, K.; Ahmed, R.M.; Rye, K.A.; Piguet, O.; Hodges, J.R.; Halliday, G.M. Altered High Density Lipoprotein Composition in Behavioral Variant Frontotemporal Dementia. Front. Neurosci. 2018, 12, 847. [Google Scholar] [CrossRef]
- Ahmed, R.M.; MacMillan, M.; Bartley, L.; Halliday, G.M.; Kiernan, M.C.; Hodges, J.R.; Piguet, O. Systemic metabolism in frontotemporal dementia. Neurology 2014, 83, 1812–1818. [Google Scholar] [CrossRef] [PubMed]
- Feldman, E.L.; Goutman, S.A.; Petri, S.; Mazzini, L.; Savelieff, M.G.; Shaw, P.J.; Sobue, G. Amyotrophic lateral sclerosis. Lancet 2022, 400, 1363–1380. [Google Scholar] [CrossRef] [PubMed]
- Xia, K.; Klose, V.; Högel, J.; Huang, T.; Zhang, L.; Dorst, J.; Fan, D.; Ludolph, A.C. Lipids and amyotrophic lateral sclerosis: A two-sample Mendelian randomization study. Eur. J. Neurol. 2023, 30, 1899–1906. [Google Scholar] [CrossRef]
- Chalitsios, C.V.; Ley, H.; Gao, J.; Turner, M.R.; Thompson, A.G. Apolipoproteins, lipids, lipid-lowering drugs and risk of amyotrophic lateral sclerosis and frontotemporal dementia: A meta-analysis and Mendelian randomisation study. J. Neurol. 2024, 271, 6956–6969. [Google Scholar] [CrossRef]
- Huang, R.; Guo, X.; Chen, X.; Zheng, Z.; Wei, Q.; Cao, B.; Zeng, Y.; Shang, H. The serum lipid profiles of amyotrophic lateral sclerosis patients: A study from south-west China and a meta-analysis. Amyotroph. Lateral Scler. Front. Degener. 2015, 16, 359–365. [Google Scholar] [CrossRef]
- Mariosa, D.; Hammar, N.; Malmström, H.; Ingre, C.; Jungner, I.; Ye, W.; Fang, F.; Walldius, G. Blood biomarkers of carbohydrate, lipid, and apolipoprotein metabolisms and risk of amyotrophic lateral sclerosis: A more than 20-year follow-up of the Swedish AMORIS cohort. Ann. Neurol. 2017, 81, 718–728. [Google Scholar] [CrossRef]
- Bjornevik, K.; O’Reilly, É.J.; Cortese, M.; Furtado, J.D.; Kolonel, L.N.; Le Marchand, L.; McCullough, M.L.; Paganoni, S.; Schwarzschild, M.A.; Shadyab, A.H.; et al. Pre-diagnostic plasma lipid levels and the risk of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2021, 22, 133–143. [Google Scholar] [CrossRef]
- Barros, A.; Dourado, M.E.T., Jr.; Pedrosa, L.F.C.; Leite-Lais, L. Association of Copper Status with Lipid Profile and Functional Status in Patients with Amyotrophic Lateral Sclerosis. J. Nutr. Metab. 2018, 2018, 5678698. [Google Scholar] [CrossRef]
- Boxer, A.L.; Sperling, R. Accelerating Alzheimer’s therapeutic development: The past and future of clinical trials. Cell 2023, 186, 4757–4772. [Google Scholar] [CrossRef]
- Huynh, T.V.; Wang, C.; Tran, A.C.; Tabor, G.T.; Mahan, T.E.; Francis, C.M.; Finn, M.B.; Spellman, R.; Manis, M.; Tanzi, R.E.; et al. Lack of hepatic apoE does not influence early Aβ deposition: Observations from a new APOE knock-in model. Mol. Neurodegener. 2019, 14, 37. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| APOA-I Modulation | Model | Effects | Reference |
|---|---|---|---|
| Peptide D-4F + statin | APP/PS1 transgenic mice | Improves cognitive function; ameliorates Aβ burden, suppresses microglial and astrocytic activation, and downregulates pro-inflammatory cytokines IL-1β and TNF-α | [113] |
| Peptide 4F | Human astrocytes; primary mouse astrocytes and microglia | Enhances glial APOE secretion and lipidation, attenuates Aβ-induced APOE dysfunction, activates ABCA1 to maintain APOE functionality | [246] |
| D-4F labeled with 125I | B6SJLF1/J mice injected with Aβ42 and Aβ40; hCMEC/D3 cell line | Increases brain efflux of Aβ42, decreases brain influx of Aβ42 but not Aβ40, decreases Aβ42 accumulation in hCMEC/D3 | [15] |
| Peptide 4F | In vitro binding interaction between Aβ fragments and 4F peptide | Slows down the aggregation kinetics of Aβ (1–42), constrains the structural plasticity of Aβ | [247] |
| Peptide 4F | APP/PS1 transgenic mice | Decreased Aβ42-induced p38 activation in BBB endothelial cells | [248] |
| Peptide 4F | Tg-SwDI mouse | Attenuated CAA-associated microgliosis, reduced CAA, mitigated inflammation from vascular smooth muscle cells | [249] |
| Human APOA-I gene knock-in | APP/PS1/AI triple Tg mice | Exhibits a 2-fold increase in plasma HDL cholesterol levels, reduces cerebral amyloid angiopathy, decreases Aβ-induced neuroinflammation | [112] |
| Purified human APOA-I extraction and lipidation induce | In vitro binding interaction between purified human APOA-I and Aβ1–42, hCMEC/D3 cell line | The initially lipidated human APOA-I demonstrates superior BBB penetrability and most effectively mediates Aβ efflux | [23] |
| Human recombinant APOA-I Milano | APP23-transgenic mouse | The APOA-I Milano shows more powerful neuroprotection and anti-inflammation ability compared to wild-type APOA-I | [114] |
| Human recombinant APOA-I Milano | APP23-transgenic mouse | Improves cognitive function and anxiety behavior, reduces Aβ40 in the brain and increases Aβ40 in CSF (promotes efflux), ameliorates endothelial damage | [134] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, G.; Jiang, G.; Huang, L.; Sun, S.; Wan, Y.; Li, F.; Wu, B.; Zhang, Y.; Li, X.; Xiong, B.; et al. The Role of APOA-I in Alzheimer’s Disease: Bridging Peripheral Tissues and the Central Nervous System. Pharmaceuticals 2025, 18, 790. https://doi.org/10.3390/ph18060790
Xie G, Jiang G, Huang L, Sun S, Wan Y, Li F, Wu B, Zhang Y, Li X, Xiong B, et al. The Role of APOA-I in Alzheimer’s Disease: Bridging Peripheral Tissues and the Central Nervous System. Pharmaceuticals. 2025; 18(6):790. https://doi.org/10.3390/ph18060790
Chicago/Turabian StyleXie, Guanfeng, Gege Jiang, Liqin Huang, Shangqi Sun, Yuwei Wan, Fang Li, Bingjie Wu, Ying Zhang, Xiaoyi Li, Bingwan Xiong, and et al. 2025. "The Role of APOA-I in Alzheimer’s Disease: Bridging Peripheral Tissues and the Central Nervous System" Pharmaceuticals 18, no. 6: 790. https://doi.org/10.3390/ph18060790
APA StyleXie, G., Jiang, G., Huang, L., Sun, S., Wan, Y., Li, F., Wu, B., Zhang, Y., Li, X., Xiong, B., & Xiong, J. (2025). The Role of APOA-I in Alzheimer’s Disease: Bridging Peripheral Tissues and the Central Nervous System. Pharmaceuticals, 18(6), 790. https://doi.org/10.3390/ph18060790