
Insights into the Therapeutic Targets and Molecular Mechanisms of Eruca sativa Against Colorectal Cancer: An Integrated Approach Combining Network Pharmacology, Molecular Docking and Dynamics Simulation

 ,
,  ,
,  ,
,  , , ,
, , ,  and
and 
Abstract
1. Introduction
2. Results
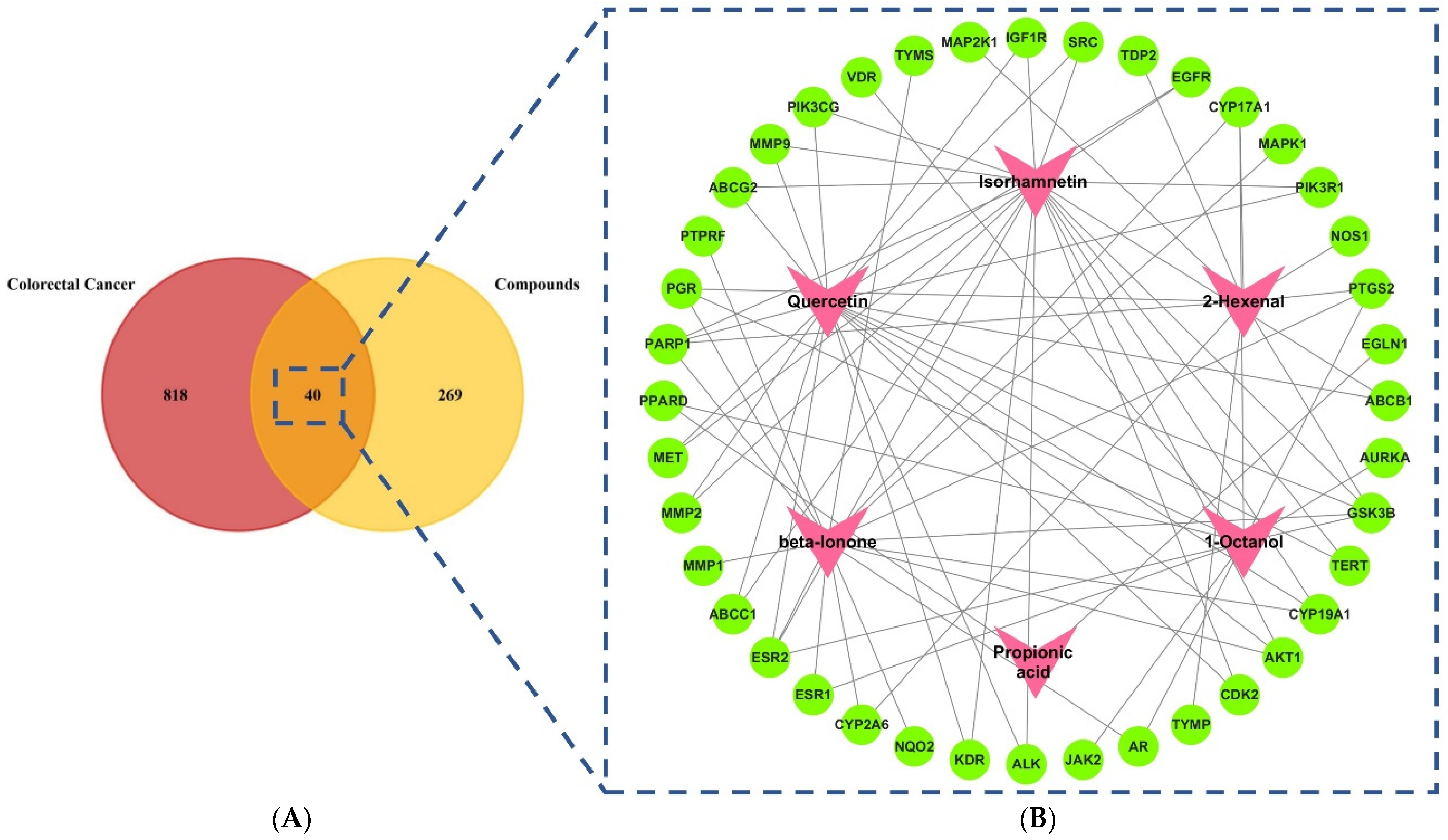
2.1. Screening of Active Compounds and Targets
2.2. Compounds–Target Network Construction
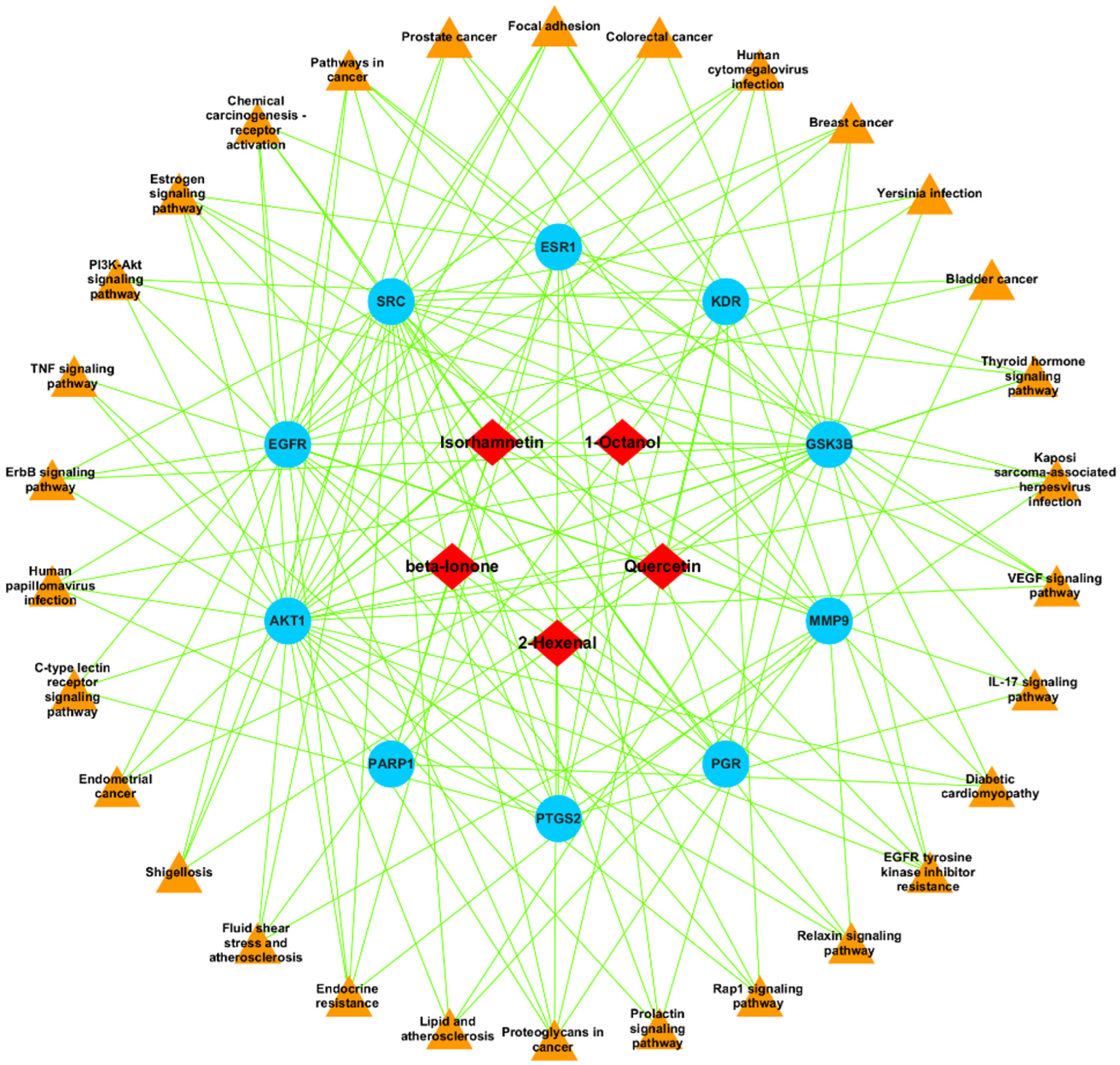
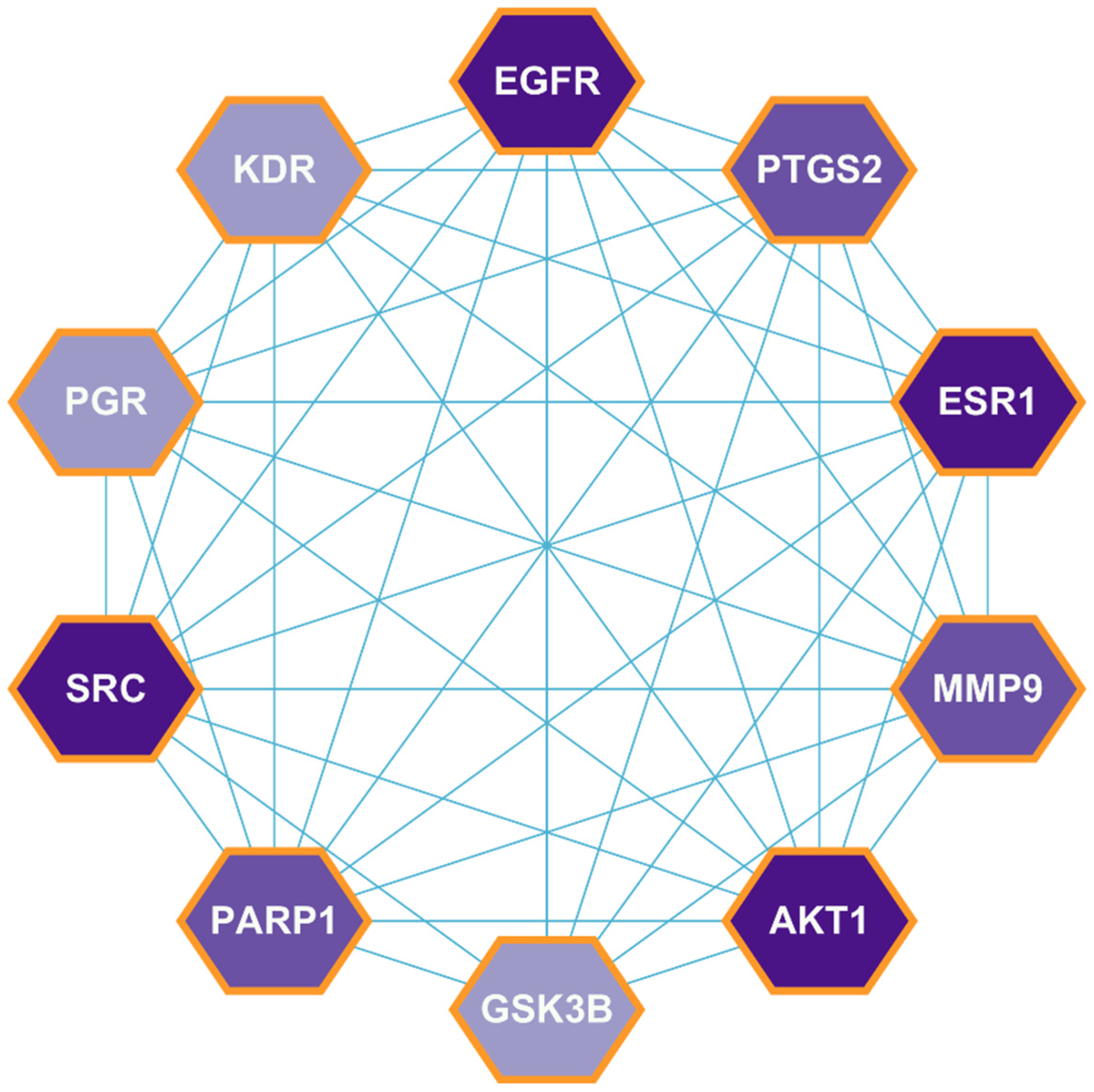
2.3. PPI Network Construction
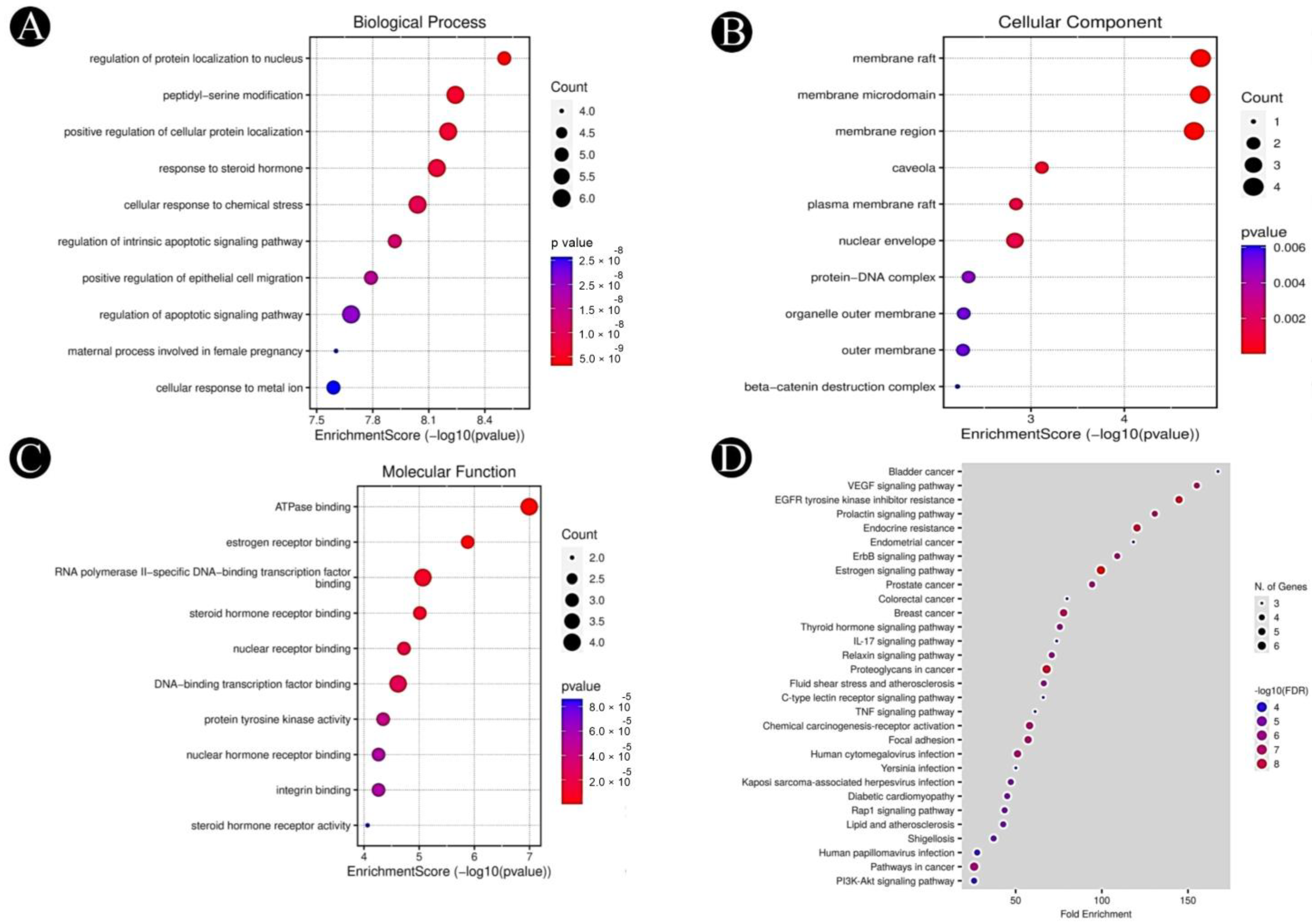
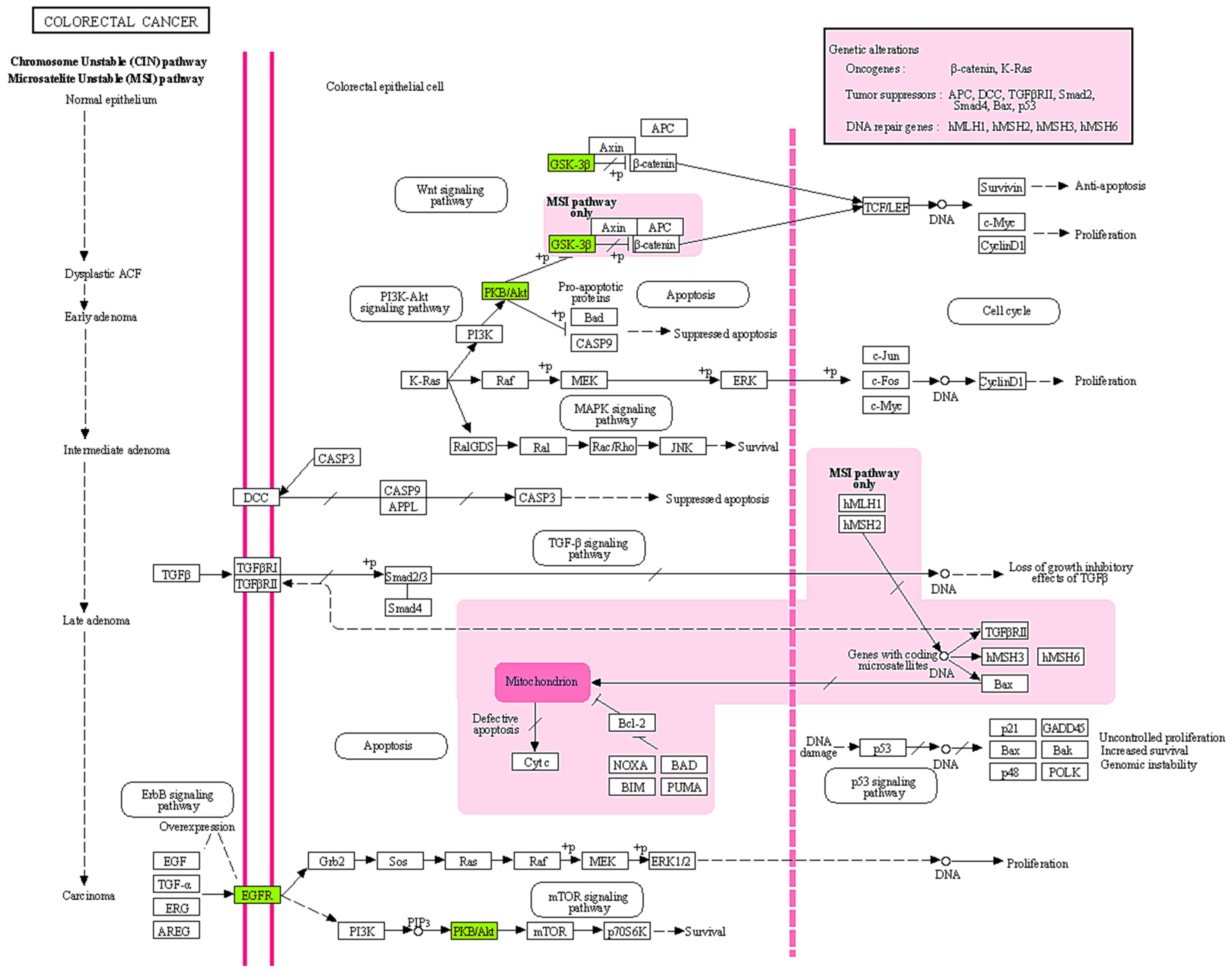
2.4. GO and KEGG Pathway Analysis
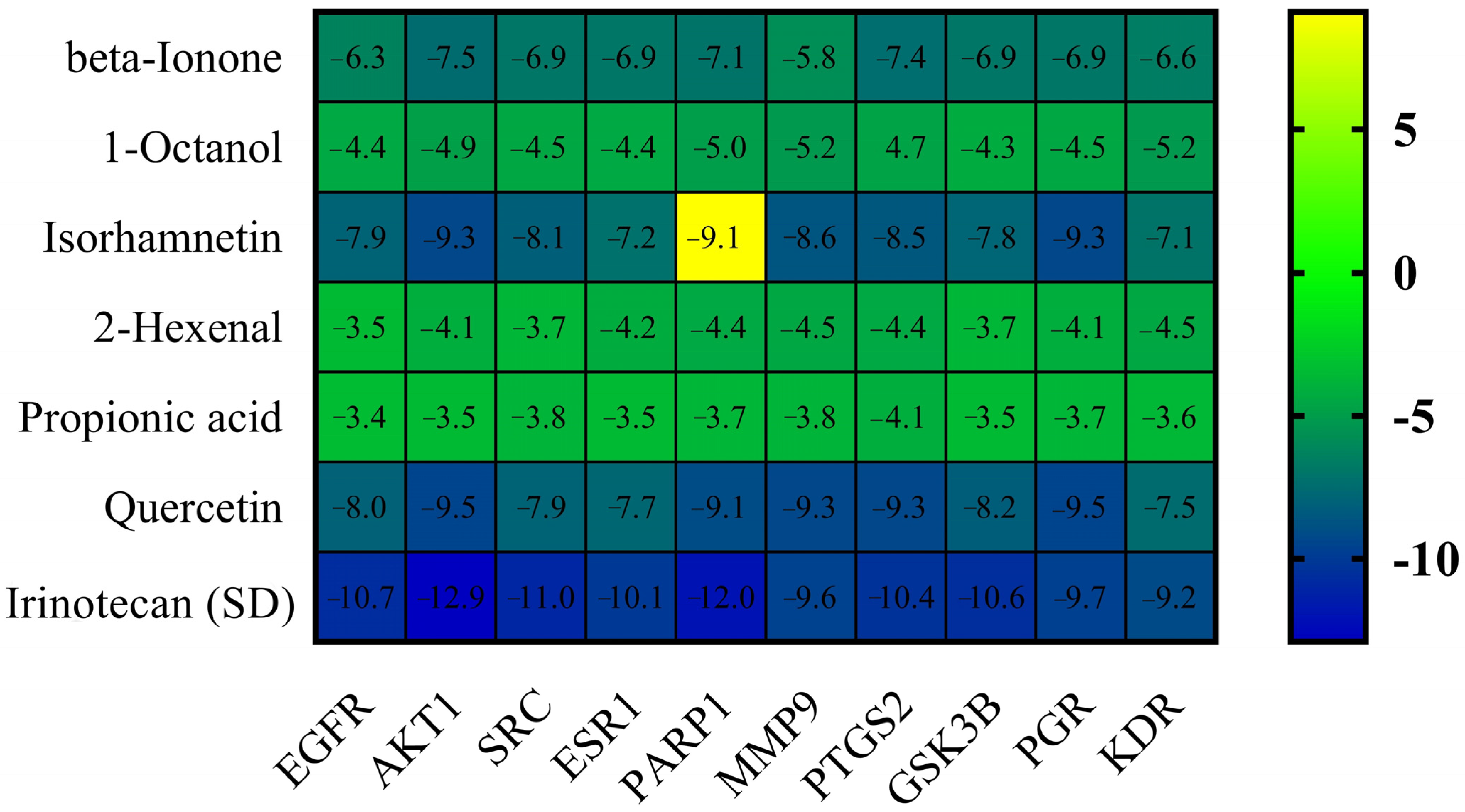
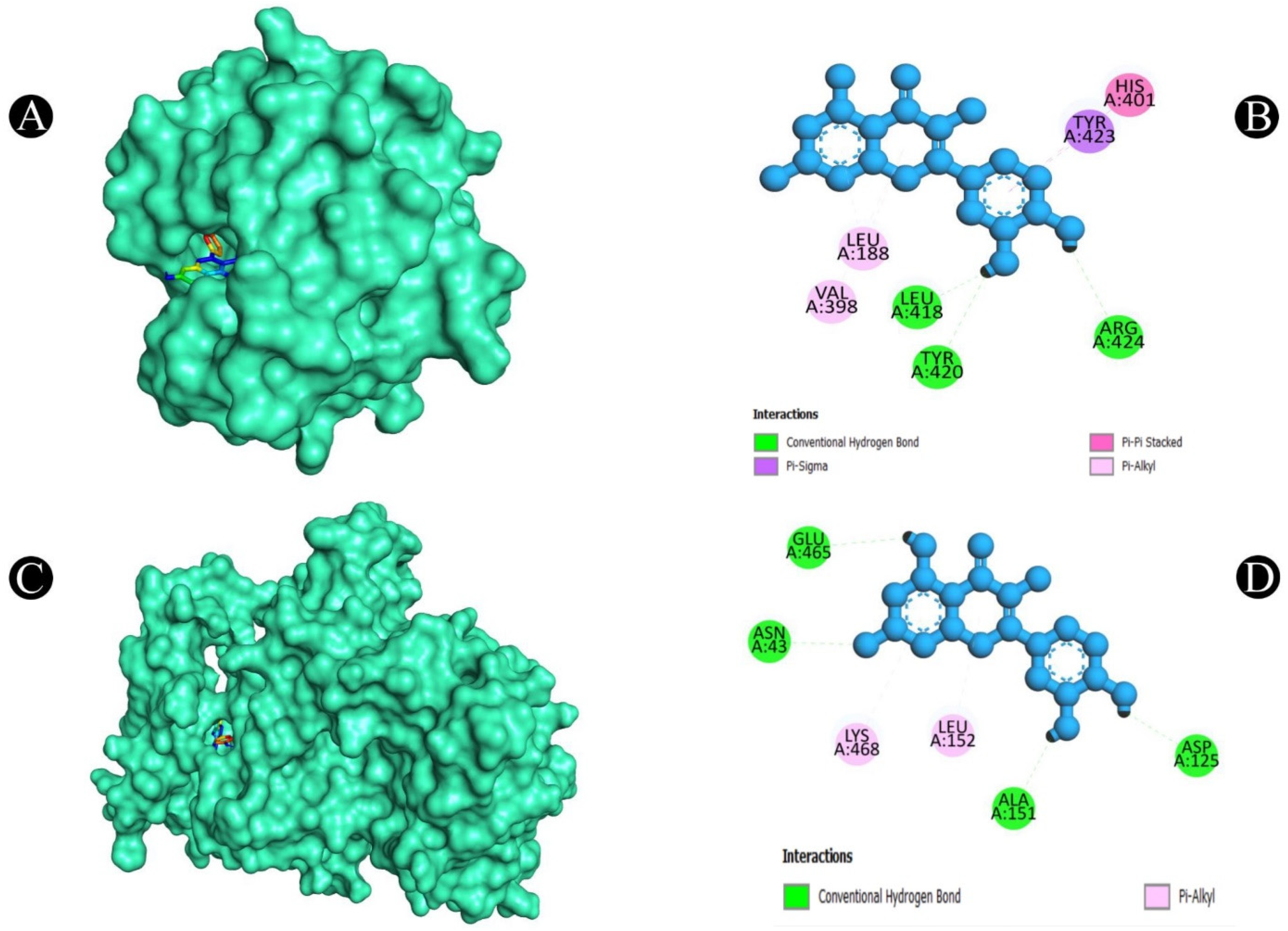
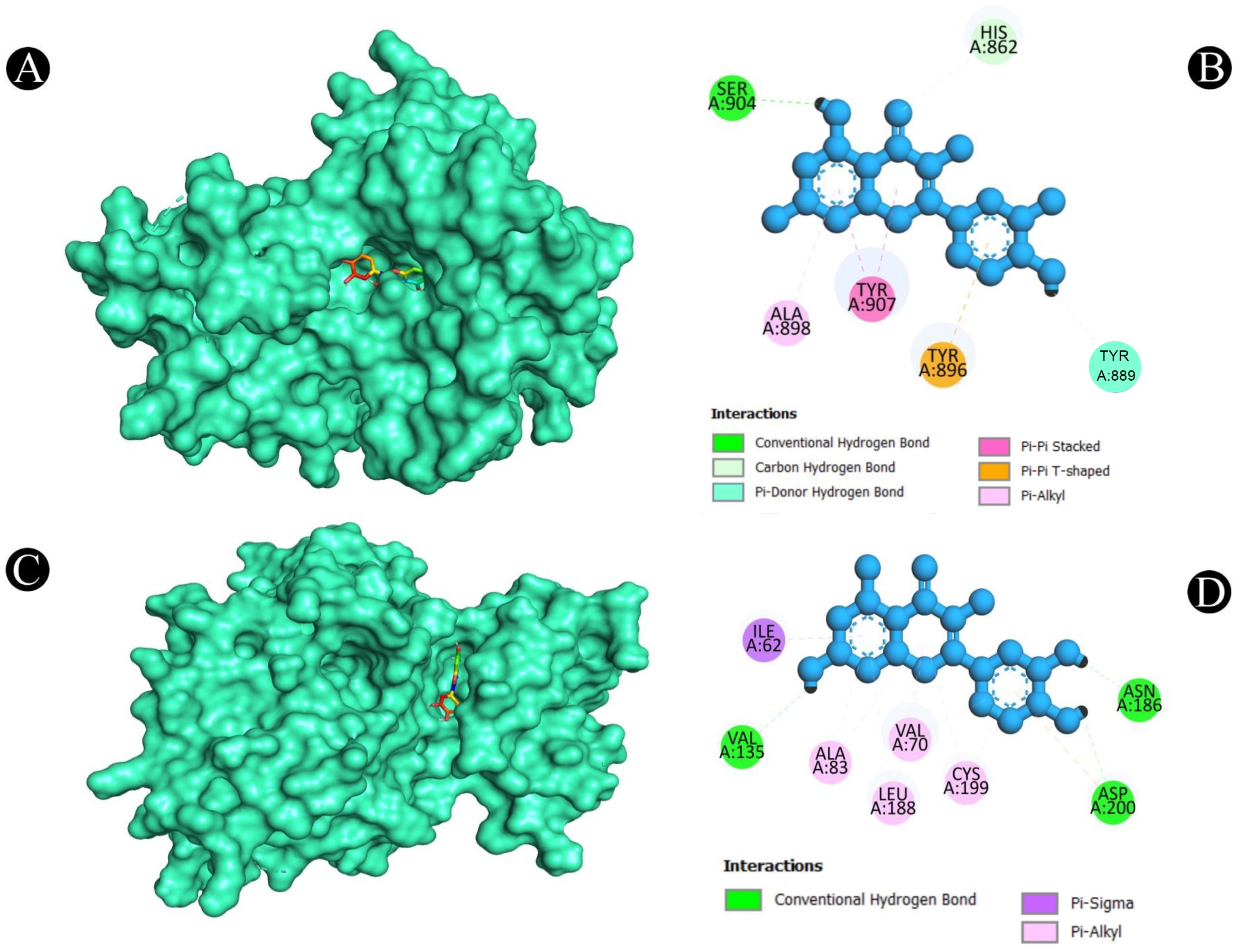
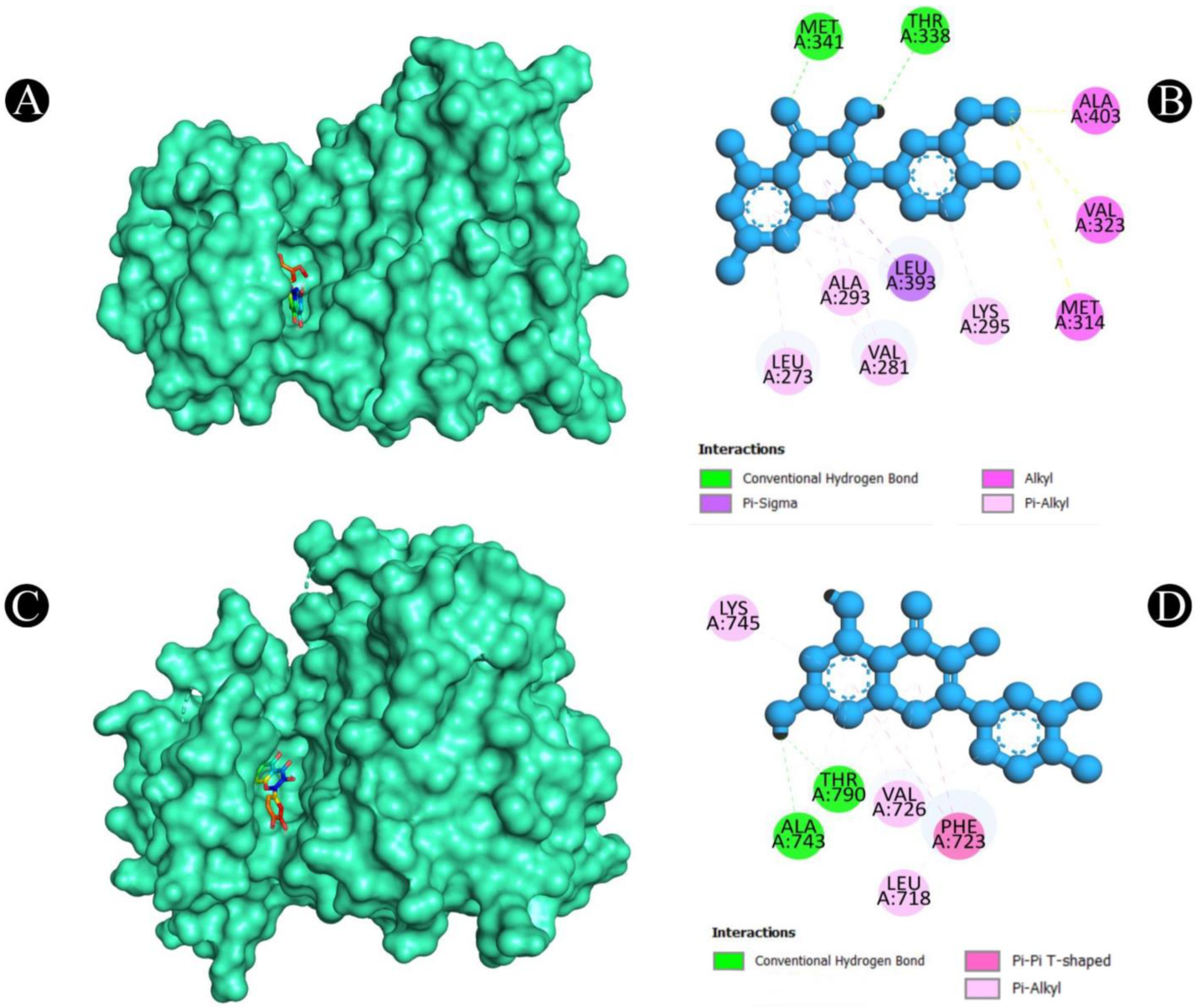
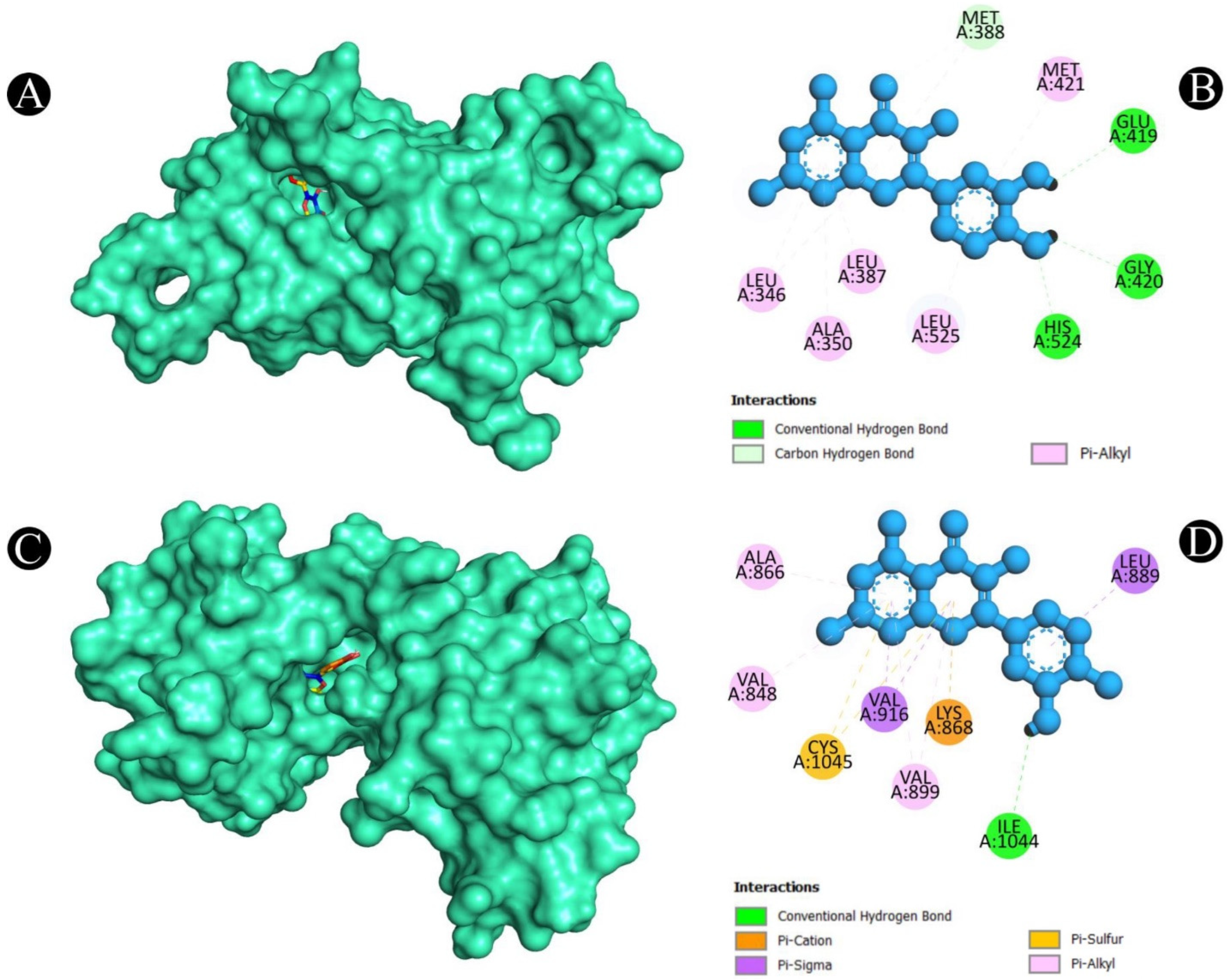
2.5. Molecular Docking
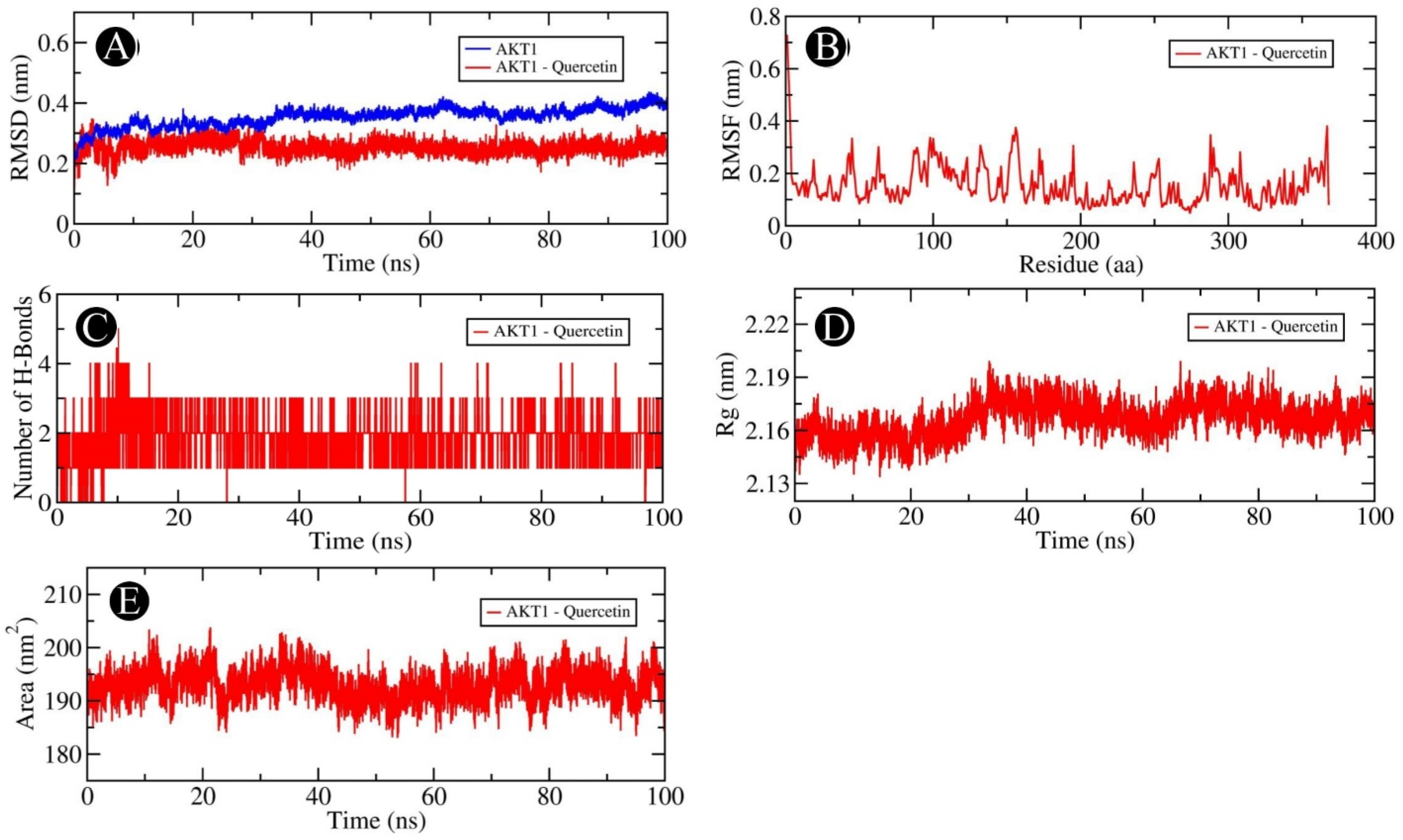
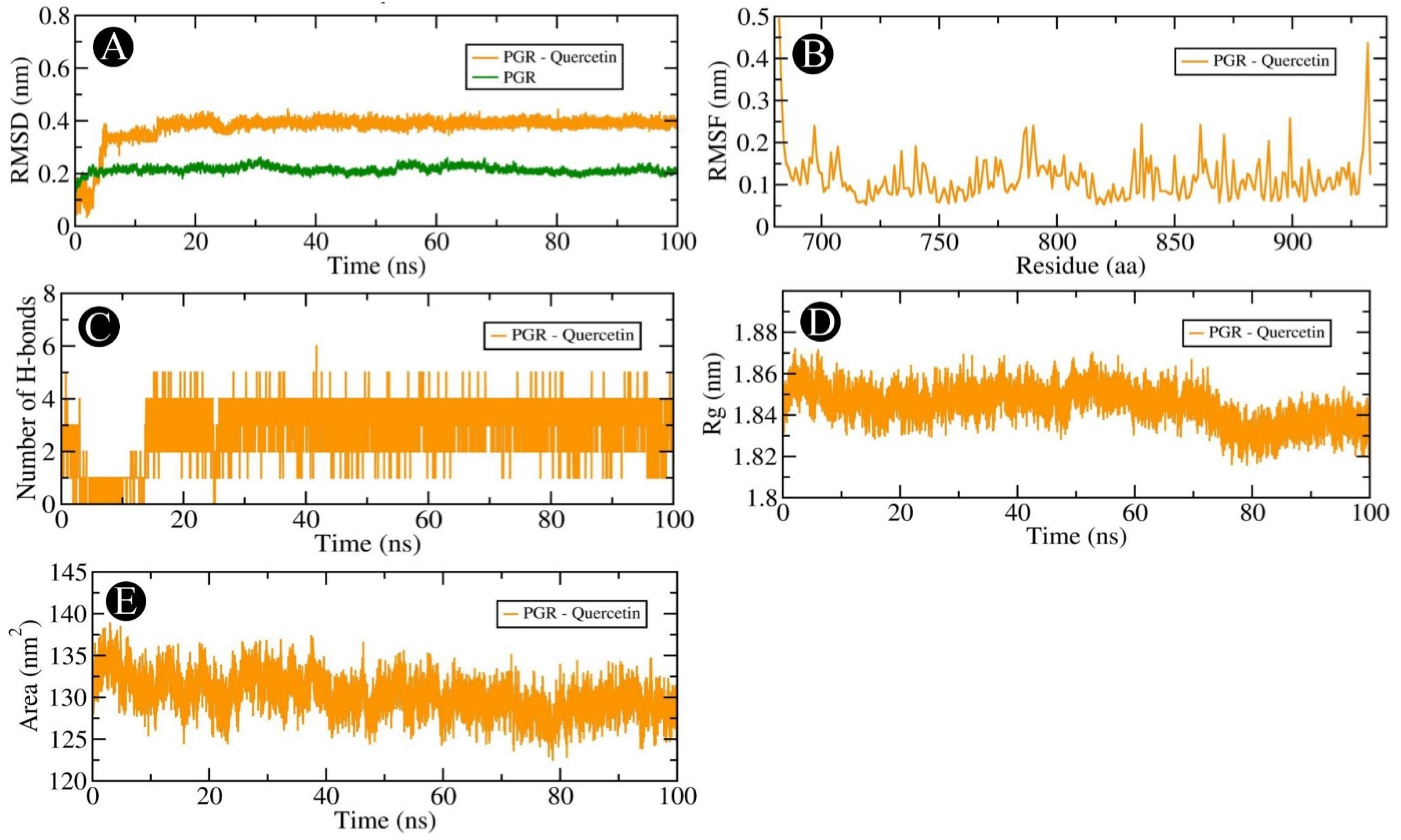
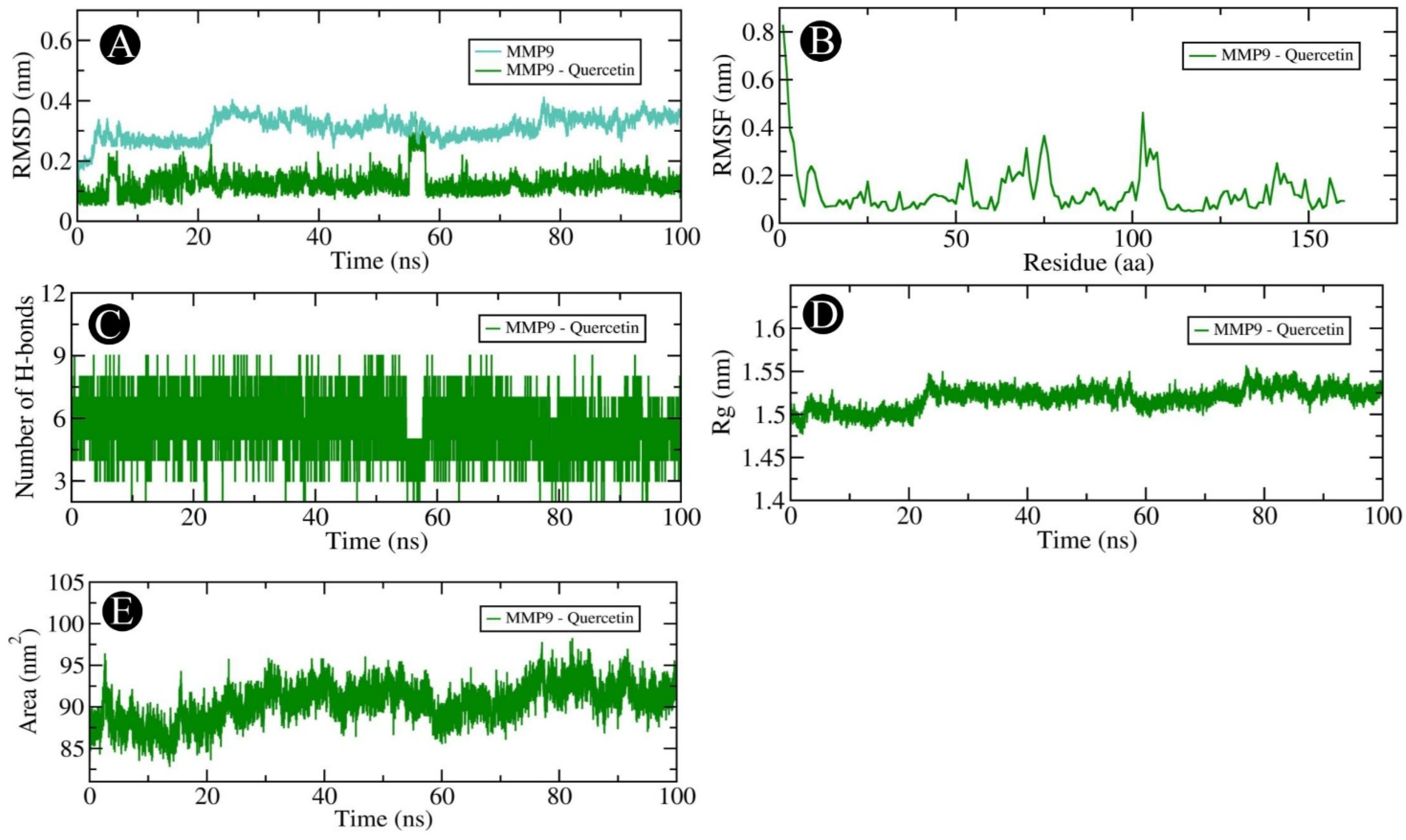
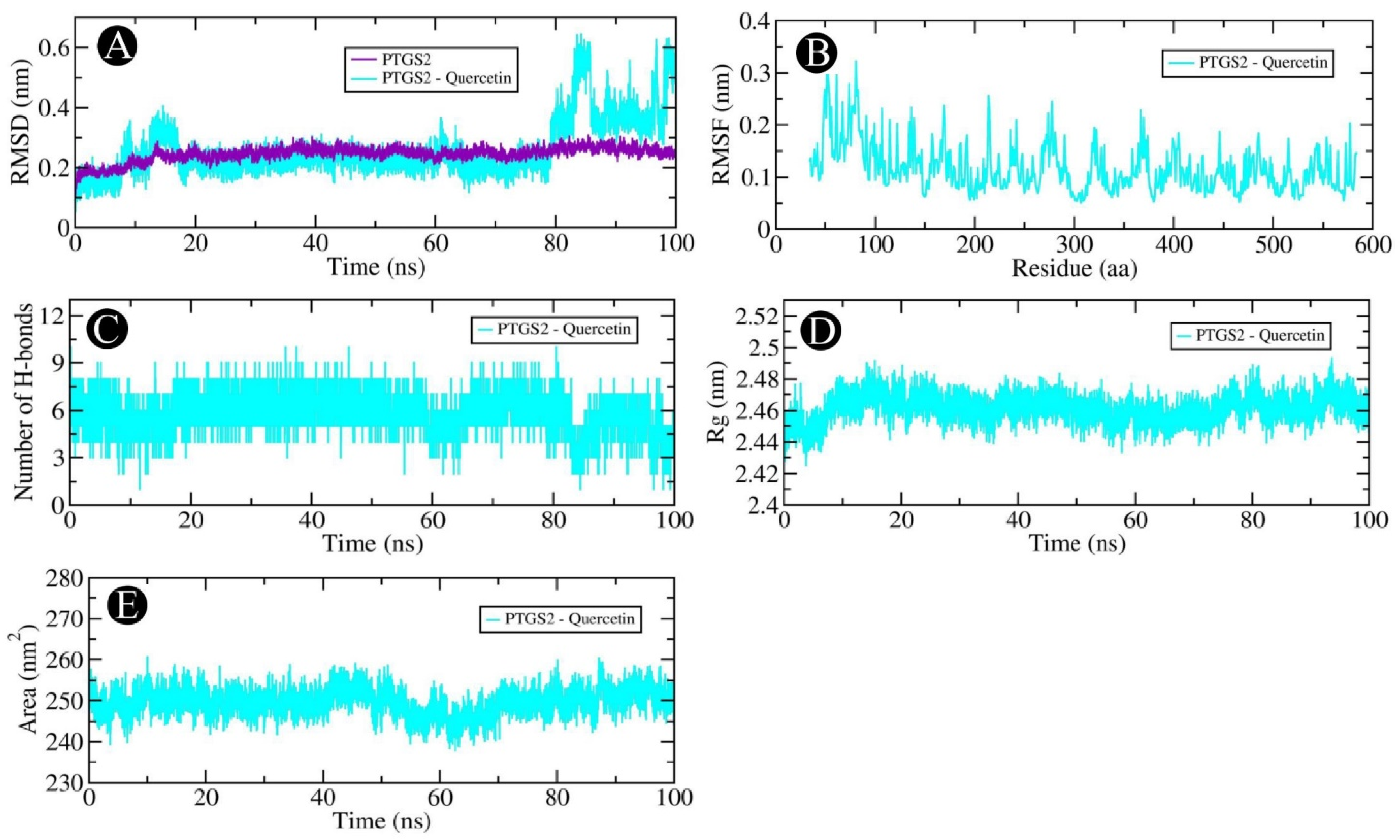
2.6. MD Simulation Analysis
3. Discussion
4. Materials and Methods
4.1. A Virtual Screening of Active Ingredients
4.2. Screening of Target Genes
4.3. Construction of Network
4.4. Pathway and Functional Enrichment Analysis
4.5. PPI Network Construction and Molecular Docking Analysis
4.6. Molecular Dynamics Simulation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Xi, Y.; Xu, P. Global colorectal cancer burden in 2020 and projections to 2040. Transl. Oncol. 2021, 14, 101174. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.S.; Karuniawati, H.; Jairoun, A.A.; Urbi, Z.; Ooi, D.J.; John, A.; Lim, Y.C.; Kibria, K.K.; Mohiuddin, A.K.M.; Ming, L.C.; et al. Colorectal cancer: A review of carcinogenesis, global epidemiology, current challenges, risk factors, preventive and treatment strategies. Cancers 2022, 14, 1732. [Google Scholar] [CrossRef]
- Golshani, G.; Zhang, Y. Advances in immunotherapy for colorectal cancer: A review. Ther. Adv. Gastroenterol. 2020, 13, 1756284820917527. [Google Scholar] [CrossRef]
- Xie, Y.H.; Chen, Y.X.; Fang, J.Y. Comprehensive review of targeted therapy for colorectal cancer. Signal Transduct. Target. Ther. 2020, 5, 22. [Google Scholar] [CrossRef]
- Fadlallah, H.; El Masri, J.; Fakhereddine, H.; Youssef, J.; Chemaly, C.; Doughan, S.; Abou-Kheir, W. Colorectal cancer: Recent advances in management and treatment. World J. Clin. Oncol. 2024, 15, 1136. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, N.; Lanjhiyana, S.K.; Jaiswal, M.; Siddiqui, M.A.; Gupta, S. Colorectal Cancer Spectrum: From Subtypes and Epidemiology to Oncotherapies. Gastroenterol. Endosc. 2025, 3, 55–64. [Google Scholar] [CrossRef]
- Wu, C.W.; Reid, M.; Leedham, S.; Lui, R.N. The emerging era of personalized medicine in advanced colorectal cancer. J. Gastroenterol. Hepatol. 2022, 37, 1411–1425. [Google Scholar] [CrossRef]
- De Visser, K.E.; Joyce, J.A. The evolving tumor microenvironment: From cancer initiation to metastatic outgrowth. Cancer Cell 2023, 41, 374–403. [Google Scholar] [CrossRef]
- Anand, U.; Dey, A.; Chandel, A.K.S.; Sanyal, R.; Mishra, A.; Pandey, D.K.; De Falco, V.; Upadhyay, A.; Kandimalla, R.; Chaudhary, A.; et al. Cancer chemotherapy and beyond: Current status, drug candidates, associated risks and progress in targeted therapeutics. Genes Dis. 2023, 10, 1367–1401. [Google Scholar] [CrossRef]
- Awadelkareem, A.M.; Al-Shammari, E.; Elkhalifa, A.O.; Adnan, M.; Siddiqui, A.J.; Mahmood, D.; Azad, Z.A.A.; Patel, M.; Mehmood, K.; Danciu, C.; et al. Anti-adhesion and antibiofilm activity of Eruca sativa miller extract targeting cell adhesion proteins of food-borne bacteria as a potential mechanism: Combined in vitro-in silico approach. Plants 2022, 11, 610. [Google Scholar] [CrossRef]
- Awadelkareem, A.M.; Al-Shammari, E.; Elkhalifa, A.O.; Adnan, M.; Siddiqui, A.J.; Patel, M.; Khan, M.I.; Mehmood, K.; Ashfaq, F.; Badraoui, R.; et al. Biosynthesized silver nanoparticles from Eruca sativa miller leaf extract exhibits antibacterial, antioxidant, anti-quorum-sensing, antibiofilm, and anti-metastatic activities. Antibiotics 2022, 11, 853. [Google Scholar] [CrossRef] [PubMed]
- Jaafar, N.S.; Jaafar, I.S. Eruca sativa Linn.: Pharmacognostical and pharmacological properties and pharmaceutical preparations. Asian J. Pharm. Clin. Res. 2019, 12, 39–45. [Google Scholar] [CrossRef]
- Garg, G.; Sharma, V. Eruca sativa (L.): Botanical description, crop improvement, and medicinal properties. J. Herbs Spices Med. Plants 2014, 20, 171–182. [Google Scholar] [CrossRef]
- Bell, L.; Oruna-Concha, M.J.; Wagstaff, C. Identification and quantification of glucosinolate and flavonol compounds in rocket salad (Eruca sativa, Eruca vesicaria and Diplotaxis tenuifolia) by LC–MS: Highlighting the potential for improving nutritional value of rocket crops. Food Chem. 2015, 172, 852–861. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Koroleva, O.A.; Gibson, T.; Swanston, J.; Magan, J.; Zhang, Y.A.N.; Rowland, I.R.; Wagstaff, C. Analysis of phytochemical composition and chemoprotective capacity of rocket (Eruca sativa and Diplotaxis tenuifolia) leafy salad following cultivation in different environments. J. Agric. Food Chem. 2009, 57, 5227–5234. [Google Scholar] [CrossRef]
- Pagnotta, E.; Ugolini, L.; Matteo, R.; Righetti, L. Bioactive compounds from Eruca sativa seeds. Encyclopedia 2022, 2, 1866–1879. [Google Scholar] [CrossRef]
- Awadelkareem, A.M.; Al-Shammari, E.; Elkhalifa, A.E.O.; Adnan, M.; Siddiqui, A.J.; Snoussi, M.; Khan, M.I.; Azad, Z.A.A.; Patel, M.; Ashraf, S.A. Phytochemical and in silico ADME/Tox analysis of Eruca sativa extract with antioxidant, antibacterial and anticancer potential against Caco-2 and HCT-116 colorectal carcinoma cell lines. Molecules 2022, 27, 1409. [Google Scholar] [CrossRef]
- Singh, S.; Haque, S.; Arora, S. Development and optimization of nanoparticles loaded with erucin, a dietary isothiocyanate isolated from Eruca sativa: Antioxidant and antiproliferative activities in ehrlich-ascites carcinoma cell line. Front. Pharmacol. 2023, 13, 1080977. [Google Scholar] [CrossRef] [PubMed]
- Sujata, M.P. Study of associated fungal pathogens on seeds of Eruca sativa Mill., (Gargeer). Agric. Sci. Dig. 2021, 41, 215–217. [Google Scholar] [CrossRef]
- Fuentes, E.; Alarcón, M.; Fuentes, M.; Carrasco, G.; Palomo, I. A novel role of Eruca sativa Mill. (rocket) extract: Antiplatelet (NF-κB inhibition) and antithrombotic activities. Nutrients 2014, 6, 5839–5852. [Google Scholar] [CrossRef]
- El-Wakeel, M.A.; Ahmed, S.D.G.; El-Desoki, E.R. Allelopathic efficiency of Eruca sativa in controlling two weeds associated with Pisum sativum plants. J. Plant Prot. Res. 2019, 59, 170–179. [Google Scholar] [CrossRef]
- Ahmed, D.F.I. Determination of the effects of Eruca sativa oil, sodium carbonate, lavender oil and Aloe vera oil on lipid profile and breast tumour markers in breast cancer-induced doxorubicin treated female albino rats. Annu. Res. Rev. Biol. 2020, 34, 1–14. [Google Scholar] [CrossRef]
- Bassyouni, R.H.; Kamel, Z.; Algameel, A.A.; Ismail, G.; Gaber, S.N. In-vitro determination of antimicrobial activities of Eruca sativa seed oil against antibiotic-resistant gram-negative clinical isolates from neonates: A future prospect. BMC Complement. Med. Ther. 2022, 22, 229. [Google Scholar] [CrossRef]
- Raouf, C.G.; Noori, A.O.; Salih, K.Y.; Mohammad, T.H.; Mohammed, A.R.; Hadi, J.M. Effect of Eruca sativa on Spermatogenesis in Rats Exposed to Cigarette Smoke. Cureus 2024, 16, e67662. [Google Scholar] [CrossRef]
- Al-Haj, S.A.; Ali, O.H.A.; Alameen, A.O. Effect of Eruca sativa seeds on semen quality in lohmann roosters. Asian J. Res. Anim. Vet. Sci. 2022, 9, 57–64. [Google Scholar] [CrossRef]
- Hou, Y.N.; Deng, G.; Mao, J.J. Practical application of “about herbs” website: Herbs and dietary supplement use in oncology settings. Cancer J. 2019, 25, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Chandra, S.; Gahlot, M.; Choudhary, A.N.; Palai, S.; de Almeida, R.S.; de Vasconcelos, J.E.L.; dos Santos, F.A.V.; de Farias, P.A.M.; Coutinho, H.D.M. Scientific evidences of anticancer potential of medicinal plants. Food Chem. Adv. 2023, 2, 100239. [Google Scholar] [CrossRef]
- Esmeeta, A.; Adhikary, S.; Dharshnaa, V.; Swarnamughi, P.; Maqsummiya, Z.U.; Banerjee, A.; Pathak, S.; Duttaroy, A.K. Plant-derived bioactive compounds in colon cancer treatment: An updated review. Biomed. Pharmacother. 2022, 153, 113384. [Google Scholar] [CrossRef]
- Adnan, M.; Siddiqui, A.J.; Ashraf, S.A.; Bardakci, F.; Alreshidi, M.; Badraoui, R.; Noumi, E.; Tepe, B.; Sachidanandan, M.; Patel, M. Network Pharmacology, Molecular Docking, and Molecular Dynamics Simulation to Elucidate the Molecular Targets and Potential Mechanism of Phoenix dactylifera (Ajwa Dates) against Candidiasis. Pathogens 2023, 12, 1369. [Google Scholar] [CrossRef]
- Alotaibi, N.M.; Alotaibi, M.O.; Alshammari, N.; Adnan, M.; Patel, M. Network Pharmacology Combined with Molecular Docking, Molecular Dynamics, and In Vitro Experimental Validation Reveals the Therapeutic Potential of Thymus vulgaris L. Essential Oil (Thyme Oil) against Human Breast Cancer. ACS Omega 2023, 8, 48344–48359. [Google Scholar] [CrossRef]
- Elasbali, A.M.; Al-Soud, W.A.; Mousa Elayyan, A.E.; Alhassan, H.H.; Danciu, C.; Elfaki, E.M.; Alharethi, S.H.; Alharbi, B.; Alanazi, H.H.; Mohtadi, M.E. Antioxidative and ROS-dependent apoptotic effects of Cuscuta reflexa Roxb. stem against human lung cancer: Network pharmacology and in vitro experimental validation. J. Biomol. Struct. Dyn. 2023, 42, 11651–11676. [Google Scholar] [CrossRef] [PubMed]
- Agu, P.; Afiukwa, C.; Orji, O.; Ezeh, E.; Ofoke, I.; Ogbu, C.; Ugwuja, E.; Aja, P. Molecular docking as a tool for the discovery of molecular targets of nutraceuticals in diseases management. Sci. Rep. 2023, 13, 13398. [Google Scholar] [CrossRef]
- Gurung, A.B.; Ali, M.A.; Lee, J.; Farah, M.A.; Al-Anazi, K.M. Molecular docking and dynamics simulation study of bioactive compounds from Ficus carica L. with important anticancer drug targets. PLoS ONE 2021, 16, e0254035. [Google Scholar] [CrossRef]
- Ghalehbandi, S.; Yuzugulen, J.; Pranjol, M.Z.I.; Pourgholami, M.H. The role of VEGF in cancer-induced angiogenesis and research progress of drugs targeting VEGF. Eur. J. Pharmacol. 2023, 949, 175586. [Google Scholar] [CrossRef]
- Levantini, E.; Maroni, G.; Del Re, M.; Tenen, D.G. October. EGFR signaling pathway as therapeutic target in human cancers. In Seminars in Cancer Biology; Academic Press: Cambridge, MA, USA, 2022; Volume 85, pp. 253–275. [Google Scholar]
- Neradugomma, N.K.; Subramaniam, D.; Tawfik, O.W.; Goffin, V.; Kumar, T.R.; Jensen, R.A.; Anant, S. Prolactin signaling enhances colon cancer stemness by modulating Notch signaling in a Jak2-STAT3/ERK manner. Carcinogenesis 2014, 35, 795–806. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Berezov, A.; Wang, Q.; Zhang, G.; Drebin, J.; Murali, R.; Greene, M.I. ErbB receptors: From oncogenes to targeted cancer therapies. J. Clin. Investig. 2007, 117, 2051–2058. [Google Scholar] [CrossRef]
- Barzi, A.; Lenz, A.M.; Labonte, M.J.; Lenz, H.J. Molecular pathways: Estrogen pathway in colorectal cancer. Clin. Cancer Res. 2013, 19, 5842–5848. [Google Scholar] [CrossRef]
- Cicatiello, A.G.; Ambrosio, R.; Dentice, M. Thyroid hormone promotes differentiation of colon cancer stem cells. Mol. Cell. Endocrinol. 2017, 459, 84–89. [Google Scholar] [CrossRef]
- Gorczynski, R.M. IL-17 signaling in the tumor microenvironment. In Tumor Microenvironment; Birbrair, A., Ed.; Advances in Experimental Medicine and Biology; Springer: Cham, Switzerland, 2020; Volume 1240, pp. 47–58. [Google Scholar]
- Ng, H.H.; Shen, M.; Samuel, C.S.; Schlossmann, J.; Bennett, R.G. Relaxin and extracellular matrix remodeling: Mechanisms and signaling pathways. Mol. Cell. Endocrinol. 2019, 487, 59–65. [Google Scholar] [CrossRef]
- Yan, H.; Kamiya, T.; Suabjakyong, P.; Tsuji, N.M. Targeting C-type lectin receptors for cancer immunity. Front. Immunol. 2015, 6, 408. [Google Scholar] [CrossRef]
- Zhang, Y.L.; Wang, R.C.; Cheng, K.; Ring, B.Z.; Su, L. Roles of Rap1 signaling in tumor cell migration and invasion. Cancer Biol. Med. 2017, 14, 90. [Google Scholar]
- Leiphrakpam, P.D.; Are, C. PI3K/Akt/mTOR Signaling Pathway as a Target for Colorectal Cancer Treatment. Int. J. Mol. Sci. 2024, 25, 3178. [Google Scholar] [CrossRef] [PubMed]
- Pantsar, T.; Poso, A. Binding affinity via docking: Fact and fiction. Molecules 2018, 23, 1899. [Google Scholar] [CrossRef]
- Felice, M.R.; Maugeri, A.; De Sarro, G.; Navarra, M.; Barreca, D. Molecular pathways involved in the anti-cancer activity of flavonols: A focus on myricetin and kaempferol. Int. J. Mol. Sci. 2022, 23, 4411. [Google Scholar] [CrossRef]
- Ullah, H.; Majeed, A.; Rahim, F.; Hussain, A.; Khan, F.; Nawaz, H.; Khan, M.S.; Umar, A.; Wadood, A.; Samad, A. Synthesis of benzimidazole-thiosemicarbazone hybrid derivatives, in vitro..-glucosidase and..-amylase activities, and an in silico molecular docking study. Chem. Data Collect. 2023, 45, 101027. [Google Scholar] [CrossRef]
- Umar, A.; Khan, M.S.; Wajid, M.; Ullah, H. Biocompatibility, antimicrobial efficacy, and therapeutic potential of cobalt carbonate nanoparticles in wound healing, sex hormones, and metabolic regulation in diabetic albino mice. Biochem. Biophys. Res. Commun. 2024, 734, 150773. [Google Scholar] [CrossRef]
- Khan, T.; Umar, A.; Subhan, Z.; Khan, M.S.; Ali, H.Z.; Ullah, H.; Sabri, S.; Wajid, M.; Iqbal, R.; Bhat, M.A.; et al. Therapeutic Potential of Novel Silver Carbonate Nanostructures in Wound Healing and Antibacterial Activity Against Pseudomonas chengduensis and Staphylococcus aureus. Pharmaceuticals 2024, 17, 1471. [Google Scholar] [CrossRef] [PubMed]
- Aslam, M.W.; Umar, A.; Khan, M.S.; Wajid, M.; Khan, M.U. Impact of Copper Carbonate Nanoparticles on Hematological, Liver, and Kidney Function, Lipid Profile, and Hormonal Regulation in Albino Mice: A Combined Experimental and Computational Analysis. BioNanoScience 2025, 15, 120. [Google Scholar] [CrossRef]
- Kciuk, M.; Marciniak, B.; Kontek, R. Irinotecan—Still an important player in cancer chemotherapy: A comprehensive overview. Int. J. Mol. Sci. 2020, 21, 4919. [Google Scholar] [CrossRef]
- Michael, H.; Shafik, R.; Rasmy, G. Studies on the chemical constituents of fresh leaf of Eruca sativa extract and its biological activity as anticancer agent in vitro. J. Med. Plants Res. 2011, 5, 1184–1191. [Google Scholar]
- Khoobchandani, M.; Ojeswi, B.; Ganesh, N.; Srivastava, M.; Gabbanini, S.; Matera, R.; Iori, R.; Valgimigli, L. Antimicrobial properties and analytical profile of traditional Eruca sativa seed oil: Comparison with various aerial and root plant extracts. Food Chem. 2010, 120, 217–224. [Google Scholar] [CrossRef]
- Kim, H.J.; Kim, S.K.; Kim, B.S.; Lee, S.H.; Park, Y.S.; Park, B.K.; Kim, S.J.; Kim, J.; Choi, C.; Kim, J.S.; et al. Apoptotic effect of quercetin on HT-29 colon cancer cells via the AMPK signaling pathway. J. Agric. Food Chem. 2010, 58, 8643–8650. [Google Scholar] [CrossRef] [PubMed]
- Darband, S.G.; Kaviani, M.; Yousefi, B.; Sadighparvar, S.; Pakdel, F.G.; Attari, J.A.; Mohebbi, I.; Naderi, S.; Majidinia, M. Quercetin: A functional dietary flavonoid with potential chemo-preventive properties in colorectal cancer. J. Cell. Physiol. 2018, 233, 6544–6560. [Google Scholar] [CrossRef] [PubMed]
- Rauf, A.; Imran, M.; Khan, I.A.; Ur-Rehman, M.; Gilani, S.A.; Mehmood, Z.; Mubarak, M.S. Anticancer potential of quercetin: A comprehensive review. Phytother. Res. 2018, 32, 2109–2130. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Xu, W.; Liu, Q.; Gao, L.; Wang, X.; Li, X. Quercetin induces apoptosis and enhances gemcitabine therapeutic efficacy against gemcitabine-resistant cancer cells. Anti-Cancer Drugs 2020, 31, 684–692. [Google Scholar] [CrossRef]
- Jaramillo, S.; Lopez, S.; Varela, L.M.; Rodriguez-Arcos, R.; Jimenez, A.; Abia, R.; Guillen, R.; Muriana, F.J. The flavonol isorhamnetin exhibits cytotoxic effects on human colon cancer cells. J. Agric. Food Chem. 2010, 58, 10869–10875. [Google Scholar] [CrossRef]
- Dwikarina, A.; Bayati, M.; Efrat, N.; Roy, A.; Lei, Z.; Ho, K.V.; Sumner, L.; Greenlief, M.; Thomas, A.L.; Applequist, W.; et al. Exploring American Elderberry Compounds for Antioxidant, Antiviral, and Antibacterial Properties Through High-Throughput Screening Assays Combined with Untargeted Metabolomics. bioRxiv 2024. [Google Scholar] [CrossRef]
- Frenț, O.D.; Stefan, L.; Morgovan, C.M.; Duteanu, N.; Dejeu, I.L.; Marian, E.; Vicaș, L.; Manole, F. A Systematic Review: Quercetin—Secondary Metabolite of the Flavonol Class, with Multiple Health Benefits and Low Bioavailability. Int. J. Mol. Sci. 2024, 25, 12091. [Google Scholar] [CrossRef]
- Kalai, F.Z.; Boulaaba, M.; Ferdousi, F.; Isoda, H. Effects of isorhamnetin on diabetes and its associated complications: A review of in vitro and in vivo studies and a post hoc transcriptome analysis of involved molecular pathways. Int. J. Mol. Sci. 2022, 23, 704. [Google Scholar] [CrossRef]
- Xie, J.; Wu, J.; Yang, S.; Zhou, H. Network Pharmacology-Based Study on the Mechanism of Aloe Vera for Treating Cancer. Evid.-Based Complement. Altern. Med. 2021, 2021, 6077698. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, Y.; Yang, Y.; Gu, L. Network pharmacology and molecular docking to explore the mechanism of kangxian decoction for epilepsy. Evid.-Based Complement. Altern. Med. 2022, 2022, 3333878. [Google Scholar] [CrossRef]
- Lu, J.; Wang, Z.; Li, S.; Xin, Q.; Yuan, M.; Li, H.; Song, X.; Gao, H.; Pervaiz, N.; Sun, X.; et al. Quercetin inhibits the migration and invasion of HCCLM3 cells by suppressing the expression of p-Akt1, matrix metalloproteinase (MMP) MMP-2, and MMP-9. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2018, 24, 2583. [Google Scholar] [CrossRef]
- Yang, L.; Hu, Z.; Zhu, J.; Liang, Q.; Zhou, H.; Li, J.; Fan, X.; Zhao, Z.; Pan, H.; Fei, B. Systematic elucidation of the mechanism of quercetin against gastric cancer via network pharmacology approach. BioMed Res. Int. 2020, 2020, 3860213. [Google Scholar] [CrossRef]
- Russo, M.; Milito, A.; Spagnuolo, C.; Carbone, V.; Rosén, A.; Minasi, P.; Lauria, F.; Russo, G.L. CK2 and PI3K are direct molecular targets of quercetin in chronic lymphocytic leukaemia. Oncotarget 2017, 8, 42571. [Google Scholar] [CrossRef] [PubMed]
- Ghasemi, J.B.; Abdolmaleki, A.; Shiri, F. Molecular docking challenges and limitations. In Pharmaceutical Sciences: Breakthroughs in Research and Practice; IGI Global: New York, NY, USA, 2017; pp. 770–794. [Google Scholar]
- Ahmed, M.; Maldonado, A.M.; Durrant, J.D. From byte to bench to bedside: Molecular dynamics simulations and drug discovery. BMC Biol. 2023, 21, 299. [Google Scholar] [CrossRef]
- Ilyin, S.E.; Belkowski, S.M.; Plata-Salamán, C.R. Biomarker discovery and validation: Technologies and integrative approaches. Trends Biotechnol. 2004, 22, 411–416. [Google Scholar] [CrossRef]
- López-Lázaro, M. Two preclinical tests to evaluate anticancer activity and to help validate drug candidates for clinical trials. Oncoscience 2015, 2, 91. [Google Scholar] [CrossRef] [PubMed]
- Harloff-Helleberg, S.; Nielsen, L.H.; Nielsen, H.M. Animal models for evaluation of oral delivery of biopharmaceuticals. J. Control. Release 2017, 268, 57–71. [Google Scholar] [CrossRef]
- Pezzani, R.; Salehi, B.; Vitalini, S.; Iriti, M.; Zuñiga, F.A.; Sharifi-Rad, J.; Martorell, M.; Martins, N. Synergistic effects of plant derivatives and conventional chemotherapeutic agents: An update on the cancer perspective. Medicina 2019, 55, 110. [Google Scholar] [CrossRef]
- Abdul, W.M.; Razvi, S.S. Eruca sativa LA promising source of drug lead for antimicrobial, neuroprotective and anticancer treatment regimens: Pharmacological properties of medicinal plant “Eruca sativa”. Eur. J. Cell Sci. 2019, 17–21. [Google Scholar] [CrossRef]
- Sadiq, A.; Hayat, M.Q.; Mall, S.M. Qualitative and quantitative determination of secondary metabolites and antioxidant potential of Eruca sativa. Nat. Prod. Chem. Res. 2014, 2, 137. [Google Scholar] [CrossRef]
- Kim, S. Exploring chemical information in PubChem. Curr. Protoc. 2021, 1, e217. [Google Scholar] [CrossRef]
- James, T.; Hsieh, M.L.; Knipling, L.; Hinton, D. Determining the architecture of a protein–DNA complex by combining FeBABE cleavage analyses, 3-D printed structures, and the ICM Molsoft program. In DNA-Protein Interactions: Principles and Protocols; Humana Press: New York, NY, USA, 2015; pp. 29–40. [Google Scholar]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Pathan, M.; Keerthikumar, S.; Ang, C.S.; Gangoda, L.; Quek, C.Y.; Williamson, N.A.; Mouradov, D.; Sieber, O.M.; Simpson, R.J.; Salim, A.; et al. FunRich: An open access standalone functional enrichment and interaction network analysis tool. Proteomics 2015, 15, 2597–2601. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Dennis, G.; Sherman, B.T.; Hosack, D.A.; Yang, J.; Gao, W.; Lane, H.C.; Lempicki, R.A. DAVID: Database for annotation, visualization, and integrated discovery. Genome Biol. 2003, 4, R60. [Google Scholar] [CrossRef]
- Mering Cv Huynen, M.; Jaeggi, D.; Schmidt, S.; Bork, P.; Snel, B. STRING: A database of predicted functional associations between proteins. Nucleic Acids Res. 2003, 31, 258–261. [Google Scholar] [CrossRef]
- Kouranov, A.; Xie, L.; de la Cruz, J.; Chen, L.; Westbrook, J.; Bourne, P.E.; Berman, H.M. The RCSB PDB information portal for structural genomics. Nucleic Acids Res. 2006, 34, D302–D305. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Olson, A.J. Using autodock for ligand-receptor docking. Curr. Protoc. Bioinform. 2008, 24, 8–14. [Google Scholar] [CrossRef]
- Studio, D. Discovery studio. Accelrys 2008, 420, 1–9. [Google Scholar]
- Alhomrani, M.; Alsanie, W.F.; Alamri, A.S.; Alyami, H.; Habeeballah, H.; Alkhatabi, H.A.; Felimban, R.I.; Haynes, J.M.; Shakya, S.; Raafat, B.M.; et al. Enhancing the antipsychotic effect of risperidone by increasing its binding affinity to serotonin receptor via picric acid: A molecular dynamics simulation. Pharmaceuticals 2022, 15, 285. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sr.No. | Protein-Compound | Grid Size | Grid Centre | Grid Space | ||||
|---|---|---|---|---|---|---|---|---|
| X | Y | Z | X | Y | Z | |||
| 1 | EGFR | 40 | 40 | 40 | −1.672 | −48.365 | 18.337 | 0.375 Å |
| 2 | AKT1 | 40 | 44 | 40 | 9.511 | −7.594 | 11.38 | 0.375 Å |
| 3 | SRC | 44 | 40 | 40 | 12.833 | −36.083 | −5.417 | 0.375 Å |
| 4 | ESR1 | 36 | 30 | 36 | 30.153 | −0.06 | 24.412 | 0.375 Å |
| 5 | PARP1 | 40 | 40 | 40 | −37.474 | 6.782 | −7.052 | 0.375 Å |
| 6 | MMP9 | 40 | 40 | 40 | 67.77 | 31.04 | 115.898 | 0.375 Å |
| 7 | PTGS2 | 40 | 40 | 40 | 24.507 | 50.313 | 20.363 | 0.375 Å |
| 8 | GSK3B | 40 | 40 | 40 | 34.101 | 7.396 | 32.472 | 0.375 Å |
| 9 | PGR | 40 | 40 | 40 | 27.176 | 13.149 | 63.279 | 0.375 Å |
| 10 | KDR | 40 | 40 | 40 | 19.275 | 7.327 | 11.715 | 0.375 Å |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Banu, H.; Al-Shammari, E.; Shahanawaz, S.; Azam, F.; Patel, M.; Alarifi, N.A.; Ahmad, M.F.; Adnan, M.; Ashraf, S.A. Insights into the Therapeutic Targets and Molecular Mechanisms of Eruca sativa Against Colorectal Cancer: An Integrated Approach Combining Network Pharmacology, Molecular Docking and Dynamics Simulation. Pharmaceuticals 2025, 18, 453. https://doi.org/10.3390/ph18040453
Banu H, Al-Shammari E, Shahanawaz S, Azam F, Patel M, Alarifi NA, Ahmad MF, Adnan M, Ashraf SA. Insights into the Therapeutic Targets and Molecular Mechanisms of Eruca sativa Against Colorectal Cancer: An Integrated Approach Combining Network Pharmacology, Molecular Docking and Dynamics Simulation. Pharmaceuticals. 2025; 18(4):453. https://doi.org/10.3390/ph18040453
Chicago/Turabian StyleBanu, Humera, Eyad Al-Shammari, Syed Shahanawaz, Faizul Azam, Mitesh Patel, Naif Abdulrahman Alarifi, Md Faruque Ahmad, Mohd Adnan, and Syed Amir Ashraf. 2025. "Insights into the Therapeutic Targets and Molecular Mechanisms of Eruca sativa Against Colorectal Cancer: An Integrated Approach Combining Network Pharmacology, Molecular Docking and Dynamics Simulation" Pharmaceuticals 18, no. 4: 453. https://doi.org/10.3390/ph18040453
APA StyleBanu, H., Al-Shammari, E., Shahanawaz, S., Azam, F., Patel, M., Alarifi, N. A., Ahmad, M. F., Adnan, M., & Ashraf, S. A. (2025). Insights into the Therapeutic Targets and Molecular Mechanisms of Eruca sativa Against Colorectal Cancer: An Integrated Approach Combining Network Pharmacology, Molecular Docking and Dynamics Simulation. Pharmaceuticals, 18(4), 453. https://doi.org/10.3390/ph18040453