
Synthesis, Molecular Simulation, DFT, and Kinetic Study of Imidazotriazole-Based Thiazolidinone as Dual Inhibitor of Acetylcholinesterase and Butyrylcholinesterase Enzymes

 , ,
, ,
Abstract

1. Introduction
2. Results and Discussion
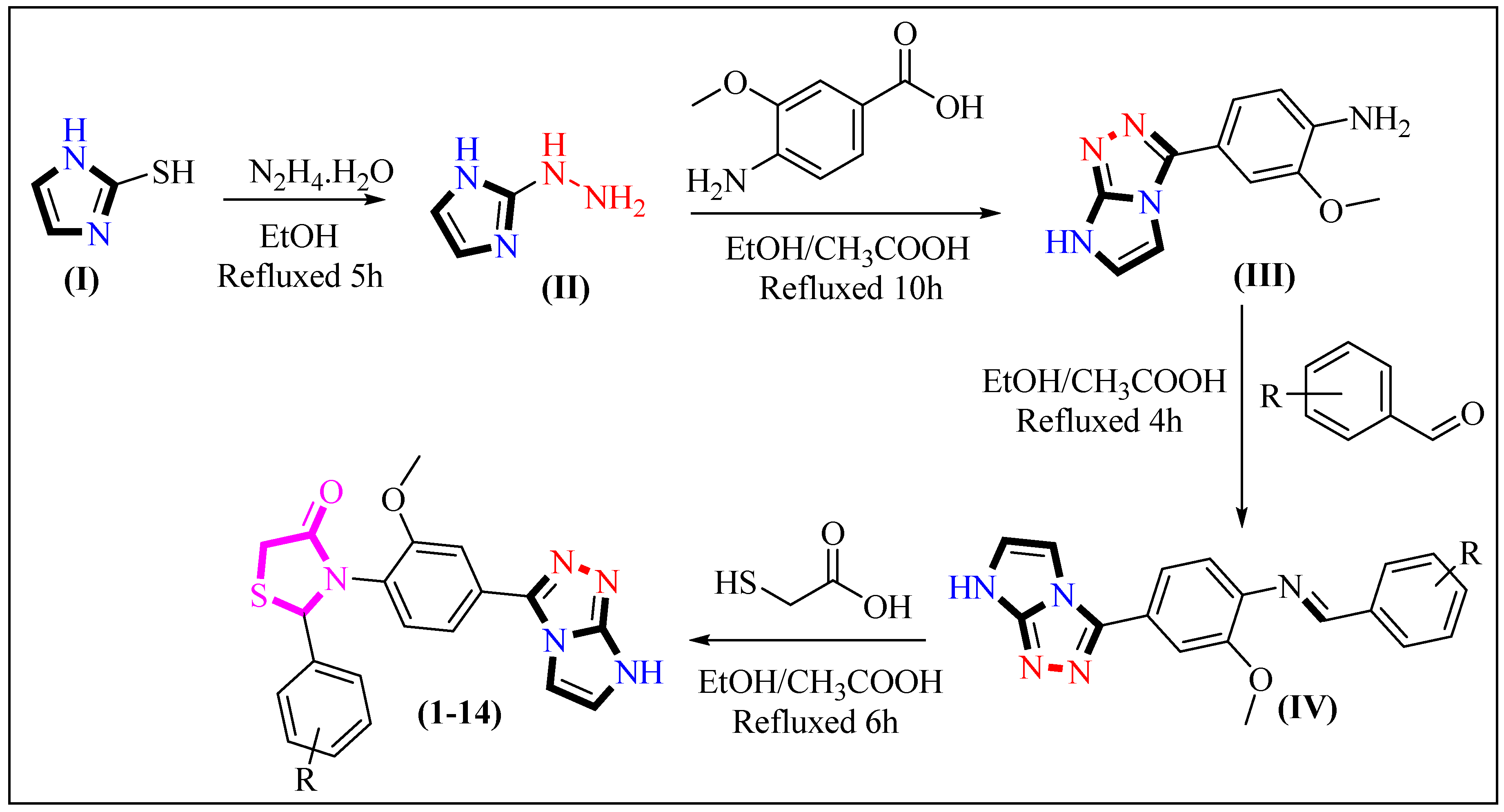
2.1. Chemistry
2.2. Spectral Analysis
2.2.1. Description of 1H-NMR
2.2.2. Description of 13C-NMR
2.3. AChE and BChE Activity Analysis
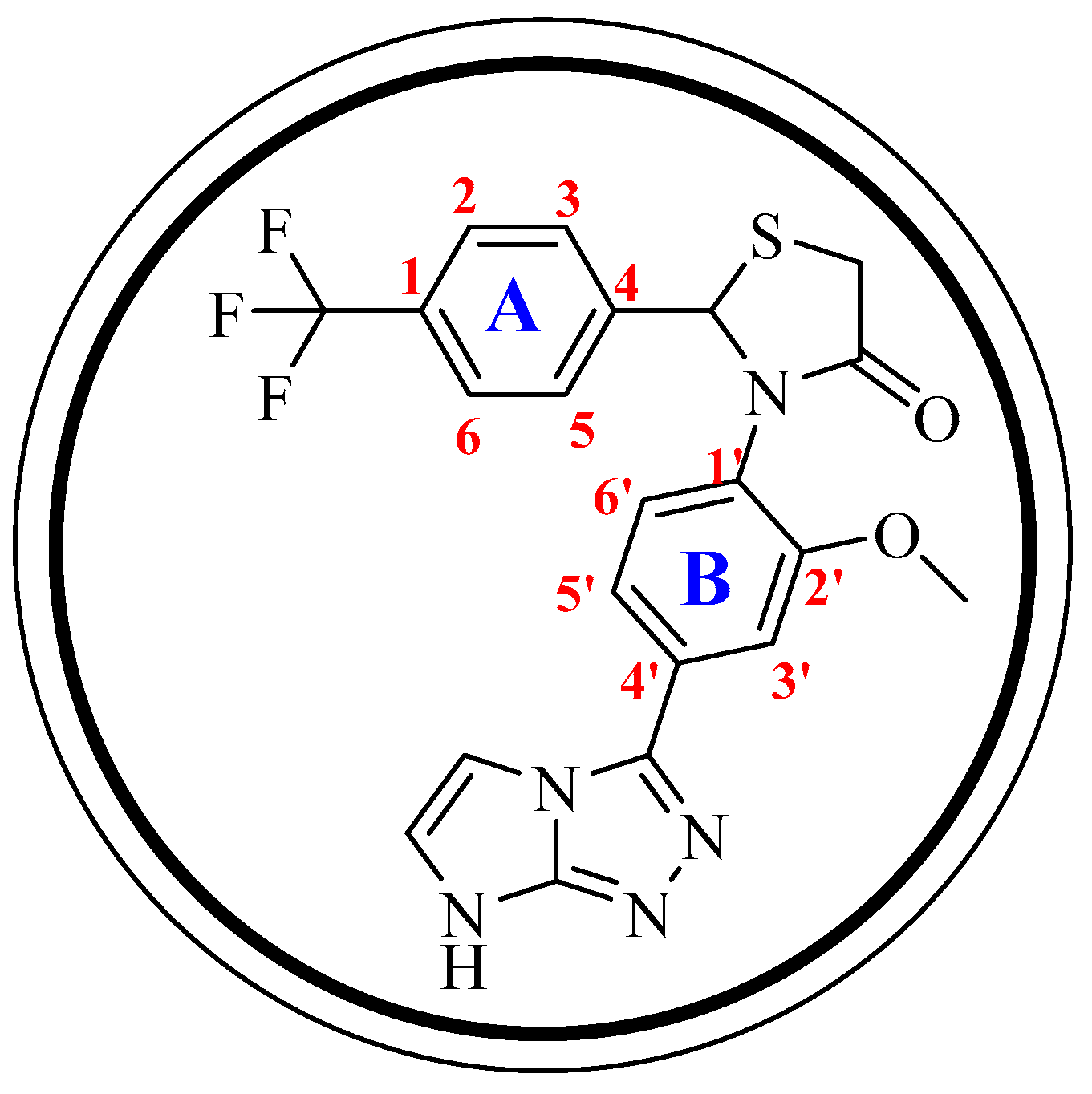
Structure–Activity Relationship (SAR)
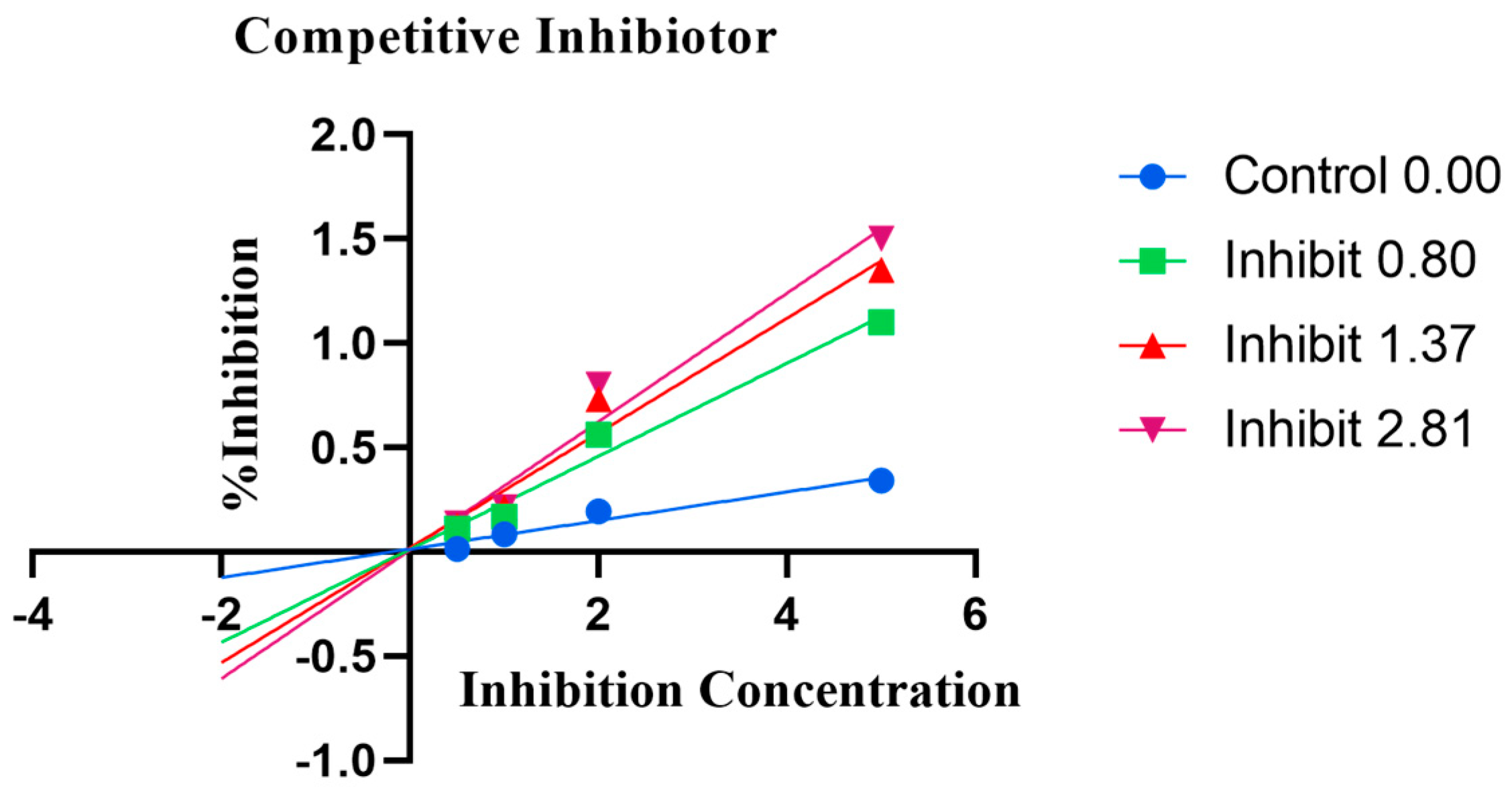
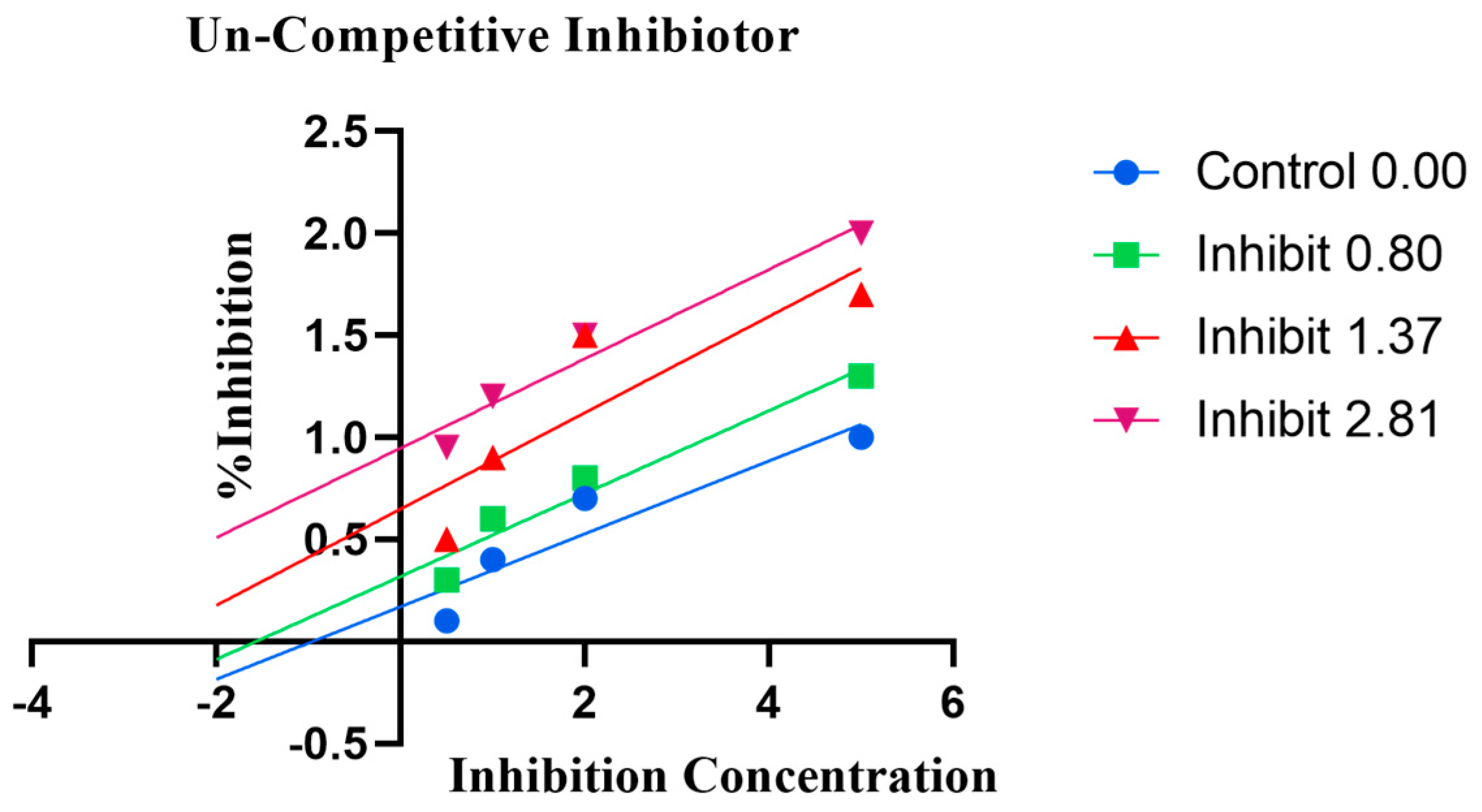
2.4. Enzyme Kinetics
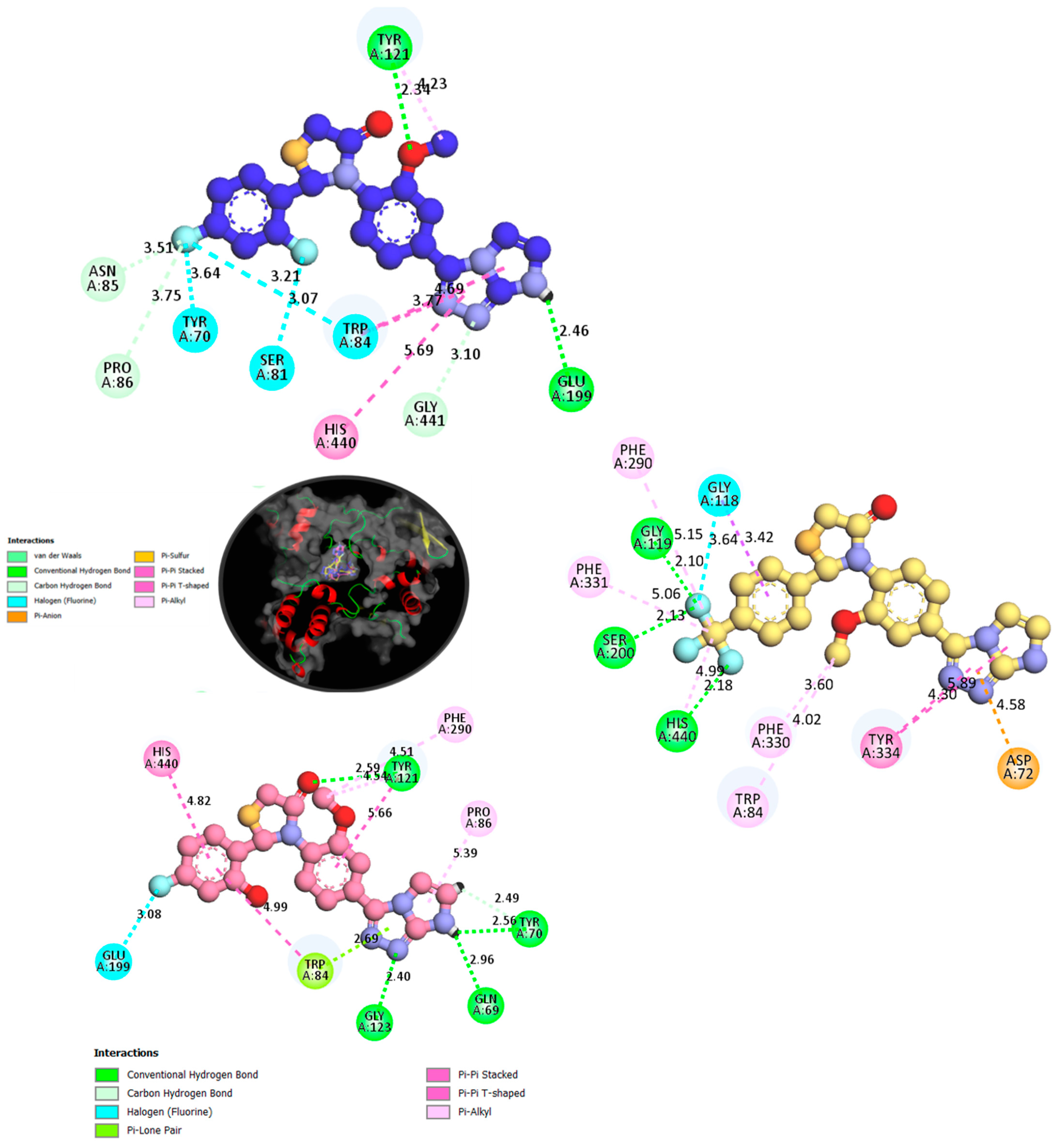
2.5. Molecular Docking
2.6. Pharmacophore Modeling
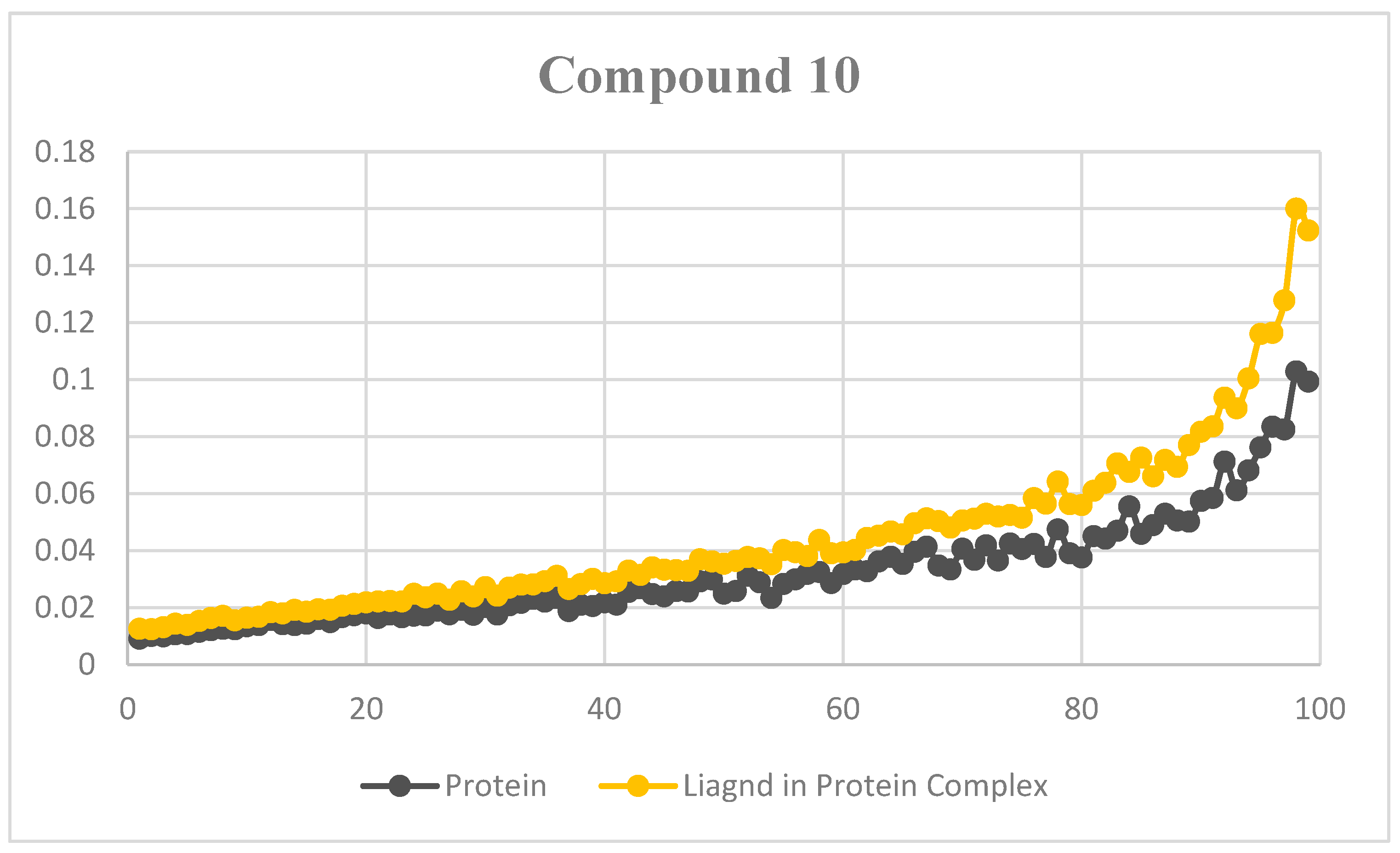
2.7. MD Simulations
2.8. ADMET Profile
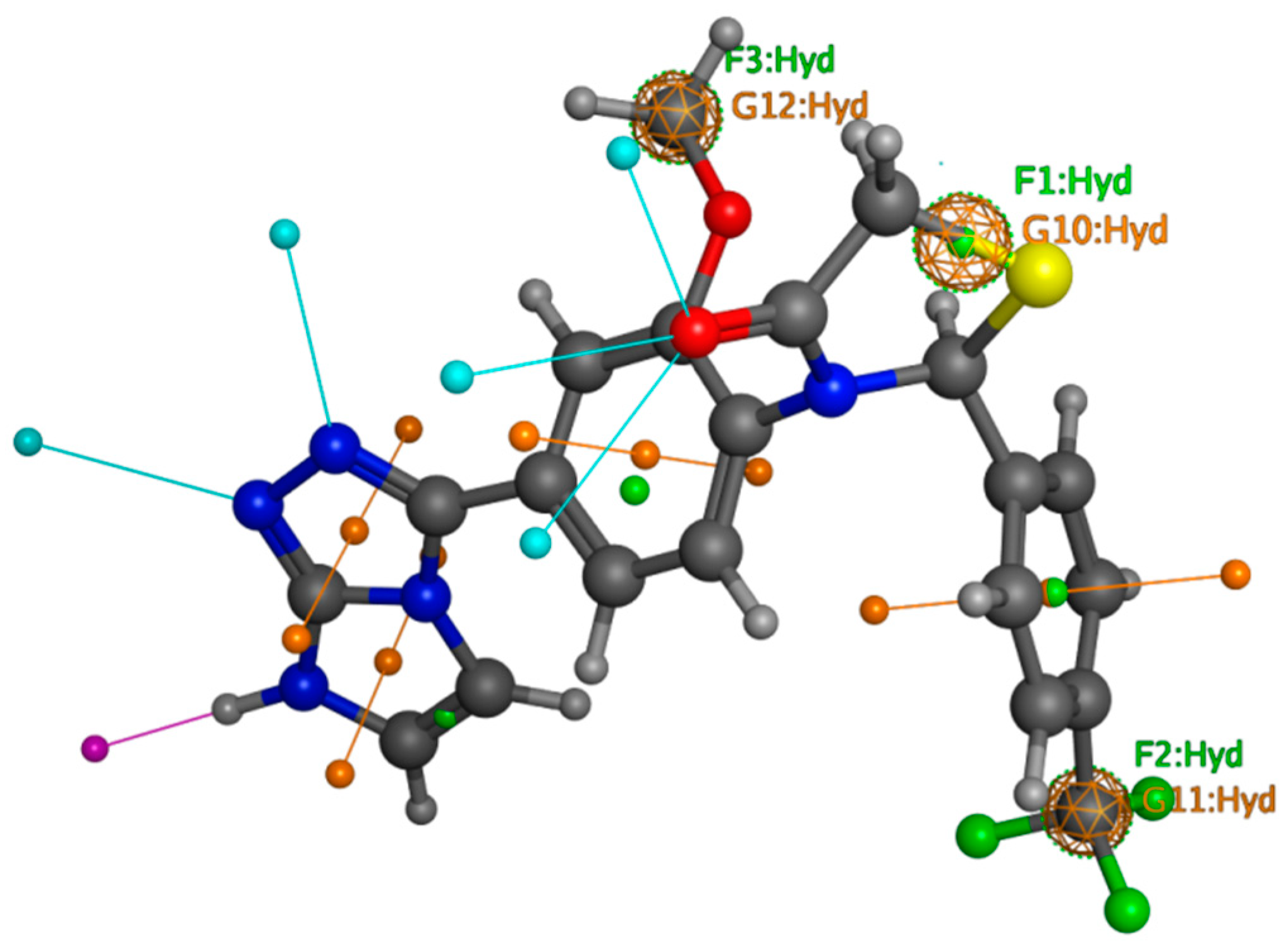
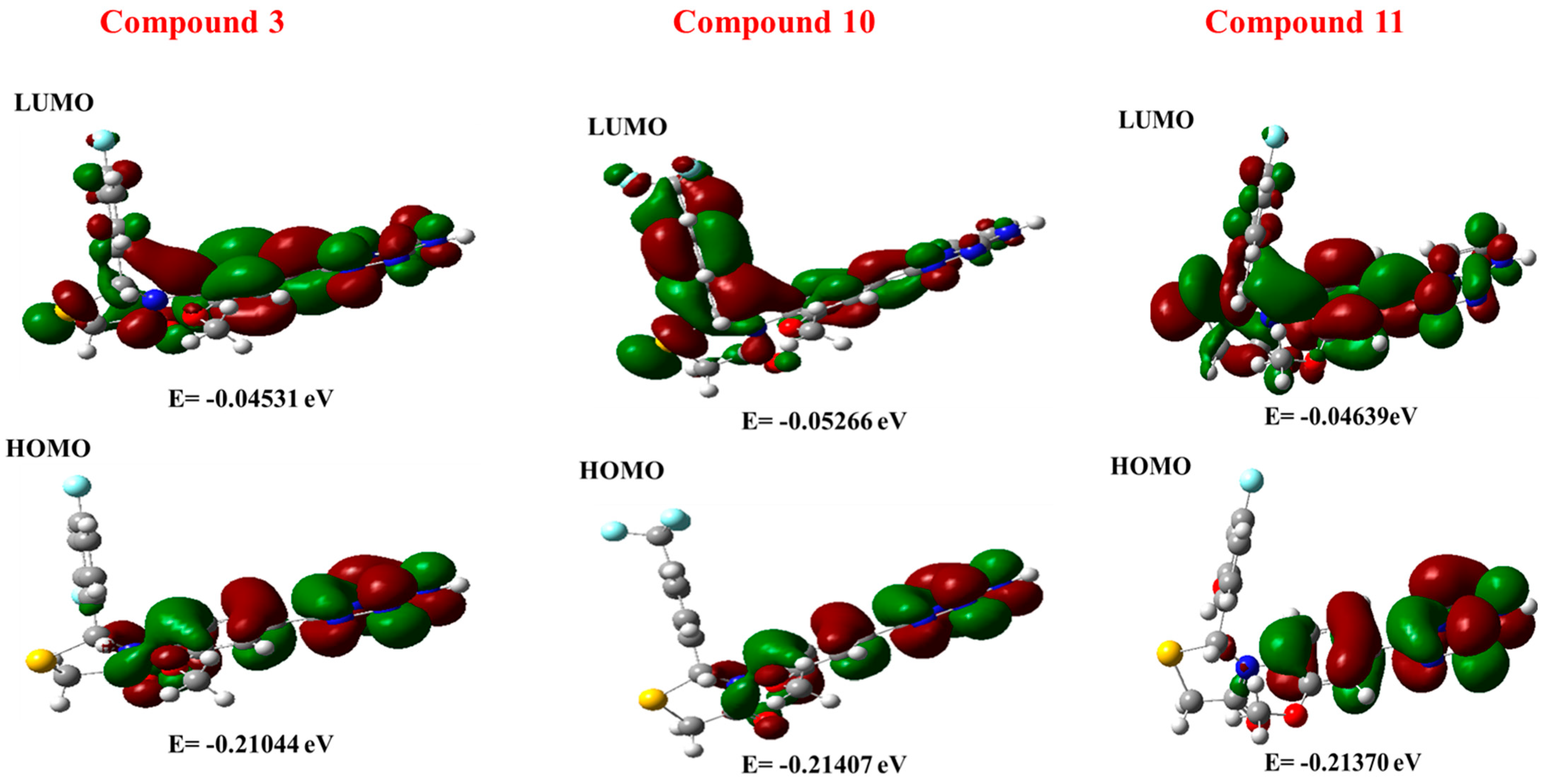
2.9. DFT
2.9.1. MESP Analysis
2.9.2. FMO Analysis
3. Materials and Methods
3.1. Materials
3.2. Methodology for the Synthesis of Imidazotriazole-Based Thiazolidinone Derivatives
3.3. AChE and BuChE Assay Protocols
3.4. Molecular Docking Protocol
3.5. DFT Assay Protocol
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jahn, H. Memory loss in Alzheimer’s disease. Dialogues Clin. Neurosci. 2013, 31, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Tarawneh, R.; Holtzman, D.M. The clinical problem of symptomatic Alzheimer disease and mild cognitive impairment. Cold Spring Harb. Perspect. Med. 2012, 1, 6148. [Google Scholar] [CrossRef]
- McDaniel, K.D.; Edland, S.D.; Heyman, A. Relationship between level of insight and severity of dementia in Alzheimer disease. Alzheimer Dis. Assoc. Disord. 1995, 1, 101–104. [Google Scholar] [CrossRef] [PubMed]
- Ganguli, M.; Dodge, H.H.; Shen, C.; Pandav, R.S.; DeKosky, S.T. Alzheimer disease and mortality: A 15-year epidemiological study. Arch. Neurol. 2005, 1, 779–784. [Google Scholar] [CrossRef] [PubMed]
- Weiner, M.F.; Hynan, L.S.; Bret, M.E.; White, C. Early behavioral symptoms and course of Alzheimer’s disease. Acta Psychiatr. Scand. 2005, 111, 367–371. [Google Scholar] [CrossRef]
- Porsteinsson, A.P.; Isaacson, R.S.; Knox, S.; Sabbagh, M.N.; Rubino, I. Diagnosis of early Alzheimer’s disease: Clinical practice in 2021. J. Prev. Alzheimer’s. Dis. 2021, 8, 371–386. [Google Scholar] [CrossRef]
- Al Mamun, A.; Uddin, M.S.; Mathew, B.; Ashraf, G.M. Toxic tau: Structural origins of tau aggregation in Alzheimer’s disease. Neural Regen. Res. 2020, 15, 1417–1420. [Google Scholar]
- Hampel, H.; Hardy, J.; Blennow, K.; Chen, C.; Perry, G.; Kim, S.H.; Villemagne, V.L.; Aisen, P.; Vendruscolo, M.; Iwatsubo, T.; et al. The amyloid-β pathway in Alzheimer’s disease. Mol. Psychiatry 2021, 26, 5481–5503. [Google Scholar] [CrossRef]
- Camps, P.; Formosa, X.; Galdeano, C.; Gómez, T.; Munoz-Torrero, D.; Scarpellini, M.; Viayna, E.; Badia, A.; Clos, M.V.; Camins, A.; et al. Novel donepezil-based inhibitors of acetyl-and butyrylcholinesterase and acetylcholinesterase-induced β-amyloid aggregation. J. Med. Chem. 2008, 51, 3588–3598. [Google Scholar] [CrossRef]
- Das, U.N. Acetylcholinesterase and butyrylcholinesterase as markers of low-grade systemic inflammation. Ann. Hepatol. 2012, 3, 409–411. [Google Scholar] [CrossRef]
- Cavalli, A.; Bolognesi, M.L.; Minarini, A.; Rosini, M.; Tumiatti, V.; Recanatini, M.; Melchiorre, C. Multi-target-directed ligands to combat neurodegenerative diseases. J. Med. Chem. 2008, 51, 347–372. [Google Scholar] [CrossRef] [PubMed]
- Rockwood, K.; Mintzer, J.; Truyen, L.; Wessel, T.; Wilkinson, D. Effects of a flexible galantamine dose in Alzheimer’s disease: A randomised, controlled trial. J. Neurol. Neurosurg. Psychiatry 2001, 71, 589–595. [Google Scholar] [CrossRef]
- Zarenezhad, E.; Behrouz, S.; Behrouz, M.; Rad, M.N. Synthesis, Anti-microbial, Antifungal and In Silico Assessment of Some 2, 4, 5-Trisubstituted Imidazole Analogues. J. Mol. Stru 2024, 15, 1296. [Google Scholar] [CrossRef]
- Vitale, R.M.; Rispoli, V.; Desiderio, D.; Sgammato, R.; Thellung, S.; Canale, C.; Vassalli, M.; Carbone, M.; Ciavatta, M.L.; Mollo, E.; et al. In Silico identification and experimental validation of novel anti-alzheimer’s multitargeted ligands from a marine source featuring a “2-aminoimidazole plus aromatic group” scaffold. ACS Chem. Neurosci. 2018, 23, 1290–1303. [Google Scholar] [CrossRef]
- Awasthi, A.; Rahman, M.A.; Bhagavan, R.M. Synthesis, in silico studies, and in vitro anti-inflammatory activity of novel imidazole derivatives targeting P38 MAP kinase. ACS Omega 2023, 12, 17788–17799. [Google Scholar] [CrossRef]
- Sharma, P.; LaRosa, C.; Antwi, J.; Govindarajan, R.; Werbovetz, K.A. Imidazoles as potential anticancer agents: An update on recent studies. Molecules 2021, 11, 4213. [Google Scholar] [CrossRef] [PubMed]
- Al-Sheikh, A.A.; Khan, D.; Naqvi, A.; Al-Blewi, F.F.; Rezki, N.; Aouad, M.R.; Hagar, M. Design, synthesis, molecular modeling, anticancer studies, and density functional theory calculations of 4-(1,2,4-triazol-3-ylsulfanylmethyl)-1,2,3-triazole derivatives. ACS Omega 2020, 31, 301–316. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B. Comprehensive review on the anti-bacterial activity of 1, 2, 3-triazole hybrids. Eur. J. Med. Chem. 2019, 15, 357–372. [Google Scholar] [CrossRef]
- Chinthala, Y.; Thakur, S.; Tirunagari, S.; Chinde, S.; Domatti, A.K.; Arigari, N.K.; Srinivas, K.V.; Alam, S.; Jonnala, K.K.; Khan, F.; et al. Synthesis, docking and ADMET studies of novel chalcone triazoles for anti-cancer and anti-diabetic activity. Eur. J. Med. Chem. 2015, 26, 564–573. [Google Scholar] [CrossRef]
- Khan, B.; Naiyer, A.; Athar, F.; Ali, S.; Thakur, S.C. Synthesis, characterization and anti-inflammatory activity evaluation of 1, 2, 4-triazole and its derivatives as a potential scaffold for the synthesis of drugs against prostaglandin-endoperoxide synthase. J. Biomole. Stru. Dyn. 2021, 22, 457–475. [Google Scholar] [CrossRef]
- Haroun, M.; Petrou, A.; Tratrat, C.; Kositsi, K.; Gavalas, A.; Geronikaki, A.; Venugopala, K.N.; Sreeharsha, N. Discovery of benzothiazole-based thiazolidinones as potential anti-inflammatory agents: Anti-inflammatory activity, soybean lipoxygenase inhibition effect and molecular docking studies. SAR QSAR Environ. Res. 2022, 3, 485–497. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Kader, S.N.; El-Abd Saltani, H.; Abbas Al-Issa, F.; Abul-kasem, E.A.; Gomaa Melad, A.S. Evaluation of Antifungal and Antioxidant Activity of Newly Synthesized 4-Thiazolidinones. J. Chin. Chem. Soc. 2013, 60, 1353–1358. [Google Scholar] [CrossRef]
- Mohamed, F.K. Synthesis, reactions and antimicrobial activity on some novel phthalazinones derivatives. Der. Chemica. Sin. 2010, 1, 20–31. [Google Scholar]
- Khan, Y.; Khan, S.; Hussain, R.; Rehman, W.; Maalik, A.; Gulshan, U.; Attwa, M.W.; Darwish, H.W.; Ghabbour, H.A.; Ali, N. Identification of Indazole-Based Thiadiazole-Bearing Thiazolidinone Hybrid Derivatives: Theoretical and Computational Approaches to Develop Promising Anti-Alzheimer’s Candidates. Pharmaceuticals 2023, 30, 1667. [Google Scholar] [CrossRef] [PubMed]
- Abbasi, S.A.; Rehman, W.; Rahim, F.; Hussain, R.; Hawsawi, M.B.; Alluhaibi, M.S.; Alharbi, M.; Taha, M.; Khan, S.; Rasheed, L.; et al. Molecular modeling and synthesis of novel benzimidazole-derived thiazolidinone bearing chalcone derivatives: A promising approach to develop potential anti-diabetic agents. Z. Für Naturforschung C 2024, 20. [Google Scholar] [CrossRef] [PubMed]
- Li, J.C.; Zhang, J.; Rodrigues, M.C.; Ding, D.J.; Longo, J.P.; Azevedo, R.B.; Muehlmann, L.A.; Jiang, C.S. Synthesis and evaluation of novel 1, 2, 3-triazole-based acetylcholinesterase inhibitors with neuroprotective activity. Bioorganic Med. Chem. Lett. 2016, 26, 3881–3885. [Google Scholar] [CrossRef]
- Sıcak, Y.; Aktar, B.S.K.; Yılmaz, G.T.; Öztürk, F.A.; Öztürk, M.; Tok, T.T.; Emre, E.E.O. Design, synthesis, pharmacological activities, structure–activity relationship, and in silico studies of novel 5-Substituted-2-(morpholinoimino)-thiazolidin-4-ones. ACS Omega 2023, 8, 38641–38657. [Google Scholar] [CrossRef]
- Hussain, R.; Khan, S.; Ullah, H.; Ali, F.; Khan, Y.; Sardar, A.; Iqbal, R.; Ataya, F.S.; El-Sabbagh, N.M.; Batiha, G.E.S. Benzimidazole-Based Schiff Base Hybrids Scaffolds: A Promising Approach to Develop Multitarget Drugs for Alzheimer’s disease. Pharmaceuticals 2023, 16, 1278. [Google Scholar] [CrossRef]
- Khan, S.; Hussain, R.; Khan, Y.; Iqbal, T.; Khan, M.B.; Al-Ahmary, K.M.; Mhyawi, S.R.A. Insight into role of triazole derived Schiff base bearing sulfonamide derivatives in targeting Alzheimer’s disease: Synthesis, characterization, in vitro and in silico assessment. J. Mol. Struct. 2024, 1315, 138845. [Google Scholar] [CrossRef]
- Khan, S.; Hussain, R.; Ullah, H.; Shoaib, K.; Iqbal, T.; Khan, M.K.; Akif, M. Exploring effective diagnosis of Alzheimer disease: Experimental and Computational analysis of hybrid benzimidazole based thiazolidinone derivatives. Results Chem. 2024, 9, 101663. [Google Scholar] [CrossRef]
- Khan, S.; Hussain, R.; Khan, Y.; Iqbal, T.; Shoaib, K.; Jehangir, U.; Khowdiary, M.M.; Felemban, S. Developing Correlation Between In Silico Molecular Modeling and In Vitro α-Amylase, Anti-Cancer and SARS-COV-2 Studies of Bis-Indole Derived Triazine-Based Thiazolidinone Hybrid Derivatives. Chem. Sel. 2024, 9, 202400686. [Google Scholar] [CrossRef]
- Ghasemi, M.; Mahdavi, M.; Dehghan, M.; Eftekharian, M.; Mojtabavi, S.; Faramarzi, M.A.; Iraji, A.; Al-Harrasi, A. Substituted piperazine conjugated to quinoline-thiosemicarbazide as potent α-glucosidase inhibitors to target hyperglycemia. Sci. Rep. 2025, 15, 1871. [Google Scholar] [CrossRef]
- Sepehrmansourie, H.; Azimi, M.; Ebadi, A.; Chehardoli, G.; Zolfigol, M.A.; Amanlou, M.; Montazer, M.N.; Mahdavi, M.; Najafi, Z. Novel coumarin-based acetohydrazide-1, 2, 3-triazole derivatives as urease enzyme inhibitors: Synthesis, in vitro evaluation, and molecular dynamics simulation studies. Heliyon 2025, 11, e41321. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Revision D. 01; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Khan, A.U.; Muhammad, S.; Khera, R.A.; Shehzad, R.A.; Ayub, K.; Iqbal, J. DFT study of superhalogen (AlF4) doped boron nitride for tuning their nonlinear optical properties. Optik 2021, 231, 166464. [Google Scholar] [CrossRef]
- Esrafili, M.D.; Saeidi, N. DFT calculations on the catalytic oxidation of CO over Si-doped (6,0) boron nitride nanotubes. Struct. Chem. 2015, 27, 595–604. [Google Scholar] [CrossRef]
- Khan, S.; Yar, M.; Kosar, N.; Ayub, K.; Arshad, M.; Zahid, M.N.; Mahmood, T. First-principles study for exploring the adsorption behavior of G-series nerve agents on graphdyine surface. Comput. Theor. Chem. 2020, 1191, 113043. [Google Scholar] [CrossRef]
- Prasad, O.; Sinha, L.; Kumar, N. Theoretical Raman and IR spectra of tegafur and comparison of molecular electrostatic potential surfaces, polarizability and hyerpolarizability of tegafur with 5-fluoro-uracil by density functional theory. J. At. Mol. Sci. 2010, 1, 201–214. [Google Scholar]
- Hagar, M.; Ahmed, H.A.; Aljohani, G.; Alhaddad, O.A. Investigation of Some Antiviral N-Heterocycles as COVID-19 Drug: Molecular Docking and DFT Calculations. Int. J. Mol. Sci. 2020, 21, 3922. [Google Scholar] [CrossRef]
- Liu, H.B.; Gao, W.W.; Tangadanchu, V.K.; Zhou, C.H.; Geng, R.X. Novel aminopyrimidinyl benzimidazoles as potentially antimicrobial agents: Design, synthesis and biological evaluation. Eur. J. Med. Chem. 2018, 143, 66–84. [Google Scholar] [CrossRef]
- Marinho, E.S.; Marinho, M.M. A DFT study of synthetic drug topiroxostat: MEP, HOMO, LUMO. Int. J. Sci. Eng. Res. 2016, 7, 1264–1270. [Google Scholar]
- Ellman, G.L.; Courtney, K.D.; Andres, V., Jr.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Korb, O.; Stutzle, T.; Exner, T.E. Empirical scoring functions for advanced protein− ligand docking with PLANTS. J. Chem. Inform. Model. 2009, 49, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J. Gaussian 98, Revision A.9; Gaussian Inc.: Pittsburgh, PA, USA, 1998. [Google Scholar]
- El-Baz, A.F.; Sorour, N.M.; Shetaia, Y.M. Trichosporon jirovecii-mediated synthesis of cadmium sulfide nanoparticles. J. Basic. Microbiol. 2016, 56, 520–530. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Flurry, R.L., Jr. Molecular Orbital Theory of Bonding in Organic Molecules; Marcel Dekker: New York, NY, USA, 1968. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. No | Molecular Formula | Color | Yield | Melting Point (°C) | Retardation Factor (Rf) | ||
|---|---|---|---|---|---|---|---|
| Substance Distance (cm) | Solvent Distance (cm) | Rf | |||||
| 1 | C20H16FN5O2S | Light green | 80% | 220–224 | 4 | 6 | 0.66 |
| 2 | C20H15F2N5O2S | White | 73% | 222–226 | 3.5 | 6 | 0.58 |
| 3 | C20H15F2N5O2S | Light yellow | 76% | 228–232 | 4.5 | 6 | 0.75 |
| 4 | C20H17N5O3S | Brown | 70% | 221–225 | 4.8 | 6 | 0.8 |
| 5 | C20H16N6O4S | Green | 81% | 227–231 | 5 | 6 | 0.83 |
| 6 | C20H16ClN5O2S | White | 82% | 230–234 | 4.6 | 6 | 0.76 |
| 7 | C20H15Cl2N5O2S | Light brown | 68% | 231–235 | 3.8 | 6 | 0.63 |
| 8 | C20H15ClN6O4S | Light green | 74% | 223–227 | 3.7 | 6 | 0.61 |
| 9 | C20H16FN5O3S | Green | 78% | 222–226 | 4.5 | 6 | 0.75 |
| 10 | C21H16F3N5O2S | Yellow | 80% | 226–230 | 4.2 | 6 | 0.7 |
| 11 | C20H16FN5O3S | White | 71% | 233–237 | 4 | 6 | 0.66 |
| 12 | C20H16FN5O3S | Green | 67% | 234–238 | 4.6 | 6 | 0.76 |
| 13 | C20H15FN6O4S | White | 73% | 228–232 | 5 | 6 | 0.83 |
| 14 | C21H19N5O3S | Light yellow | 78% | 225–229 | 3.8 | 6 | 0.63 |
| S/No | R | AchE IC50 ± SEM (µM) | BChE IC50 ± SEM (µM) |
|---|---|---|---|
| 1 |  | 12.50 ± 0.10 | 13.10 ± 0.20 |
| 2 | 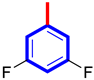 | 10.30 ± 0.20 | 11.60 ± 0.10 |
| 3 | 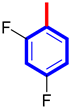 | 8.10 ± 0.40 | 8.50 ± 0.30 |
| 4 | 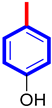 | 11.50 ± 0.20 | 12.10 ± 0.30 |
| 5 |  | 21.40 ± 0.10 | 22.10 ± 0.10 |
| 6 | 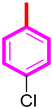 | 17.40 ± 0.20 | 18.10 ± 0.30 |
| 7 | 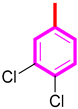 | 18.10 ± 0.10 | 18.70 ± 0.40 |
| 8 |  | 20.20 ± 0.30 | 21.30 ± 0.10 |
| 9 | 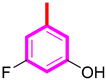 | 9.70 ± 0.20 | 10.10 ± 0.30 |
| 10 |  | 6.70 ± 0.20 | 7.10 ± 0.10 |
| 11 |  | 8.30 ± 0.20 | 8.60 ± 0.30 |
| 12 |  | 10.40 ± 0.10 | 10.90 ± 0.40 |
| 13 |  | 16.20 ± 0.20 | 16.70 ± 0.80 |
| 14 | 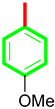 | 19.20 ± 0.10 | 19.90 ± 0.60 |
| Donepezil | 8.50 ± 0.20 | 8.90 ± 0.10 | |
| Receptor Amino Acid | Interactions Mode | Binding Distance (Å) | Docking Score |
|---|---|---|---|
| Compound 3 in AChE Complex | |||
| GLU;A:199 | Pi-Anion | 5.14 | −11.45 |
| GLU;A:199 | Pi-Anion | 6.59 | |
| PHE;A:331 | Pi-R | 6.35 | |
| PHE;A:290 | Pi-R | 6.18 | |
| TYR;A:121 | Pi-R | 5.93 | |
| TYR;A:121 | H-Bond | 6.08 | |
| TYR;A:334 | Pi-S | 3.67 | |
| ASP;A:72 | Pi-Anion | 5.10 | |
| SER;A:81 | H-F | 5.00 | |
| ASN;A:85 | H-F | 3.46 | |
| TRP;A:84 | H-F | 5.34 | |
| TRP;A:84 | Pi-Pi Stacked | 5.58 | |
| TRP;A:84 | Pi-Pi Stacked | 4.61 | |
| TRP;A:84 | Pi-Pi Stacked | 4.90 | |
| HIS;A:440 | Pi-Pi Stacked | 5.05 | |
| GLY;A:441 | C-H Bond | 3.47 | |
| Compound 3 in BChE Complex | |||
| TYR;A:332 | Pi-Pi Stacked | 6.78 | −10.67 |
| TYR;A:332 | Pi-Pi Stacked | 7.70 | |
| SER;A:198 | H-Bond | 6.31 | |
| HIS;A:438 | Pi-R | 6.31 | |
| HIS;A:438 | H-Bond | 5.25 | |
| PHE;A:329 | Pi-S | 7.23 | |
| LEU;A:285 | Pi-S | 4.09 | |
| GLY;A:117 | C-H Bond | 4.26 | |
| GLY;A:119 | H-F | 4.27 | |
| SER;A:287 | H-Bond | 4.46 | |
| TRP;A:82 | Pi-Pi Stacked | 4.71 | |
| TRP;A:82 | Pi-Pi Stacked | 5.03 | |
| Compound 10 in BChE Complex | |||
| ASP;A:72 | H-Bond | 4.07 | −12.14 |
| PHE;A:330 | Pi-R | 5.22 | |
| TYR;A:377 | Pi-R | 3.77 | |
| TYR;A:121 | Pi-Pi Stacked | 5.89 | |
| TYR;A:121 | H-Bond | 6.49 | |
| GLY;A:117 | H-F | 3.44 | |
| GLY;A:117 | H-Bond | 2.82 | |
| SER;A:200 | C-H Bond | 4.25 | |
| GLU;A:199 | H-F | 3.82 | |
| GLU;A:199 | H-F | 4.27 | |
| TYR;A:130 | H-Bond | 6.38 | |
| HIS;A:440 | Pi-Pi Stacked | 5.84 | |
| TRP;A:84 | Pi-R | 5.03 | |
| TRP;A:84 | Pi-Pi Stacked | 5.83 | |
| TRP;A:84 | C-H Bond | 5.15 | |
| PRO;A;86 | Pi-R | 5.85 | |
| GLN;A:69 | C-H Bond | 5.06 | |
| GLN;A:69 | H-Bond | 4.07 | |
| TYR;A:70 | H-Bond | 5.34 | |
| Compound 10 in BChE Complex | |||
| ASP;A:70 | C-H Bond | 4.55 | −11.95 |
| PHE;A:329 | Pi-S | 4.06 | |
| HIS;A:439 | Pi-S | 4.66 | |
| HIS;A:439 | Pi-Pi Stacked | 5.50 | |
| TYR;A:128 | H-Bond | 7.33 | |
| GLU;A:197 | H-F | 5.29 | |
| GLU;A:197 | H-F | 4.93 | |
| GLY;A:115 | H-F | 3.18 | |
| GLY;A:115 | H-F | 3.84 | |
| TRP;A:82 | Pi-R | 5.27 | |
| TRP;A:82 | Pi-Pi Stacked | 5.87 | |
| TRP;A:82 | Pi-R | 4.95 | |
| TRP;A:82 | C-H Bond | 6.41 | |
| ILE;A:69 | Pi-R | 4.58 | |
| THR;A:120 | Pi-Sigma | 5.21 | |
| THR;A:120 | Pi-Sigma | 4.45 | |
| ASN;A:68 | H-Bond | 6.04 | |
| Compound 11 in AChE Complex | |||
| TYR;A:130 | C-H Bond | 5.82 | −9.87 |
| TYR;A:121 | Pi-R | 5.68 | |
| TYR;A:121 | Unfavorable donor-donor | 5.41 | |
| TYR;A:121 | C-H Bond | 5.62 | |
| ASP;A:72 | Pi-Anion | 4.45 | |
| SER;A:81 | H-F | 4.94 | |
| ASN;A:85 | H-F | 3.17 | |
| TRP;A:84 | H-F | 5.17 | |
| TRP;A:84 | Pi-Pi Stacked | 7.14 | |
| TRP;A:84 | Pi-Pi Stacked | 5.13 | |
| HIS;A:440 | H-Bond | 5.75 | |
| HIS;A:440 | Pi-Pi Stacked | 5.75 | |
| GLU;A:199 | Pi-Anion | 5.09 | |
| GLY;A:117 | Unfavorable donor-donor | 3.10 | |
| Compound 11 in BChE Complex | |||
| ASP;A:70 | C-H Bond | 3.49 | −8.65 |
| THR;A:120 | H-Bond | 4.08 | |
| TRP;A:82 | Pi-R | 5.13 | |
| TRP;A:82 | Pi-Pi Stacked | 4.37 | |
| HIS;A:438 | H-F | 4.45 | |
| GLU;A:197 | H-Bond | 4.74 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khowdiary, M.M.; Khan, S.; Iqbal, T.; Rehman, W.; Khan, M.B.; Rehman, M.U.; Fiaz, Z.; Hakimullah. Synthesis, Molecular Simulation, DFT, and Kinetic Study of Imidazotriazole-Based Thiazolidinone as Dual Inhibitor of Acetylcholinesterase and Butyrylcholinesterase Enzymes. Pharmaceuticals 2025, 18, 415. https://doi.org/10.3390/ph18030415
Khowdiary MM, Khan S, Iqbal T, Rehman W, Khan MB, Rehman MU, Fiaz Z, Hakimullah. Synthesis, Molecular Simulation, DFT, and Kinetic Study of Imidazotriazole-Based Thiazolidinone as Dual Inhibitor of Acetylcholinesterase and Butyrylcholinesterase Enzymes. Pharmaceuticals. 2025; 18(3):415. https://doi.org/10.3390/ph18030415
Chicago/Turabian StyleKhowdiary, Manal M., Shoaib Khan, Tayyiaba Iqbal, Wajid Rehman, Muhammad Bilal Khan, Mujaddad Ur Rehman, Zanib Fiaz, and Hakimullah. 2025. "Synthesis, Molecular Simulation, DFT, and Kinetic Study of Imidazotriazole-Based Thiazolidinone as Dual Inhibitor of Acetylcholinesterase and Butyrylcholinesterase Enzymes" Pharmaceuticals 18, no. 3: 415. https://doi.org/10.3390/ph18030415
APA StyleKhowdiary, M. M., Khan, S., Iqbal, T., Rehman, W., Khan, M. B., Rehman, M. U., Fiaz, Z., & Hakimullah. (2025). Synthesis, Molecular Simulation, DFT, and Kinetic Study of Imidazotriazole-Based Thiazolidinone as Dual Inhibitor of Acetylcholinesterase and Butyrylcholinesterase Enzymes. Pharmaceuticals, 18(3), 415. https://doi.org/10.3390/ph18030415