
HDAC/σ1R Dual-Ligand as a Targeted Melanoma Therapeutic

 ,
,  ,
,  , , and
, , and 
Abstract

1. Introduction
2. Results
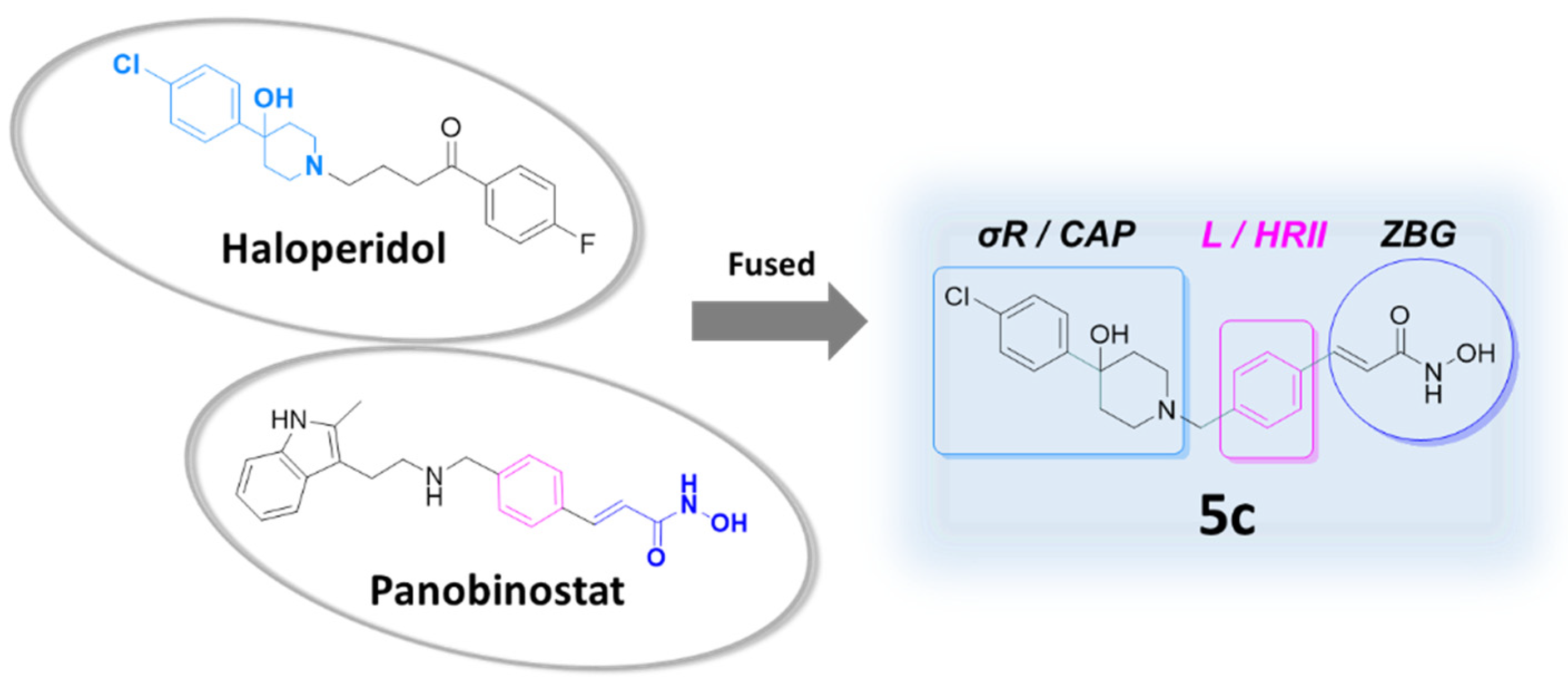
2.1. Design of the Dual-Target Anti-Melanoma Hybrid
2.2. Chemical Synthesis
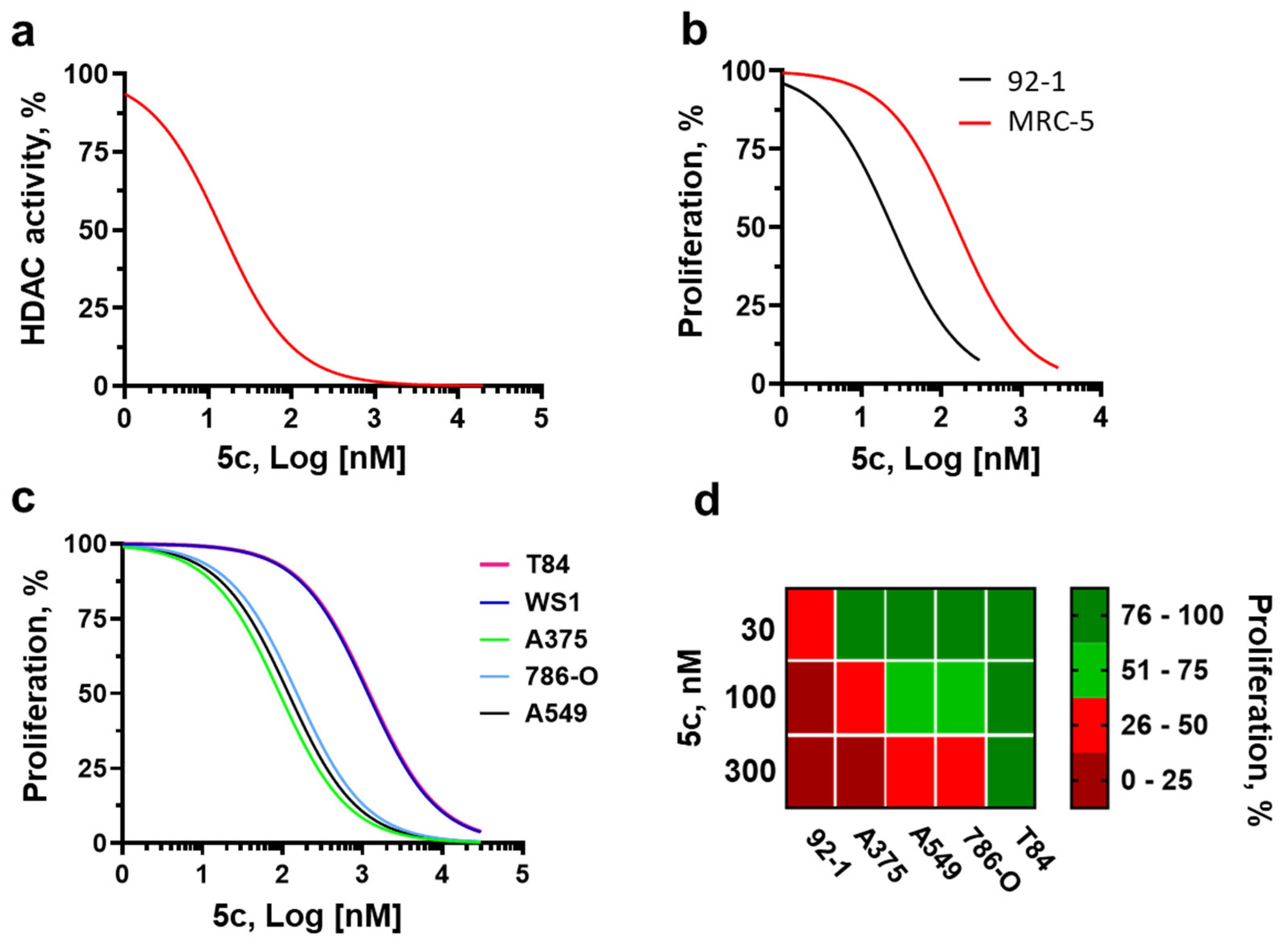
2.3. Molecular Pharmacology and Tumor Selectivity
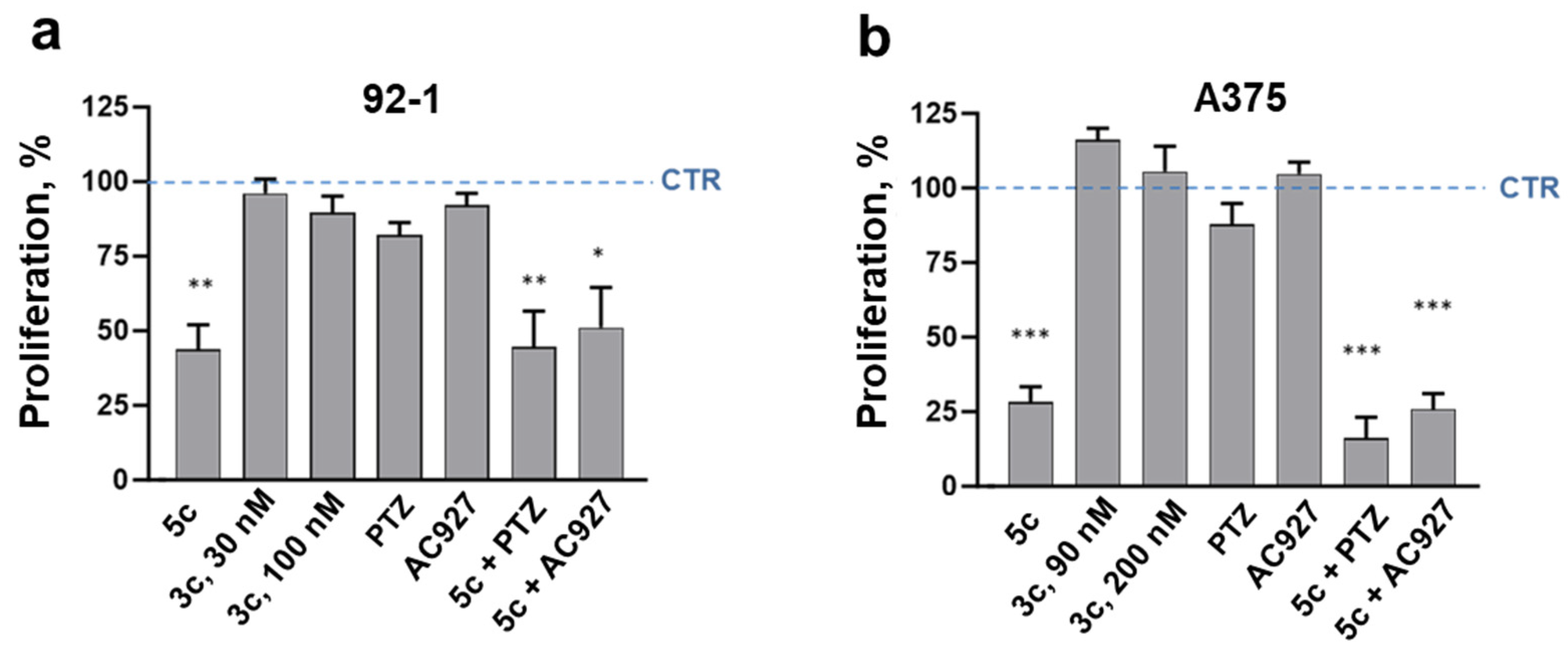
2.4. Cancer Specificity and Antiproliferative Signaling in Melanoma Cells
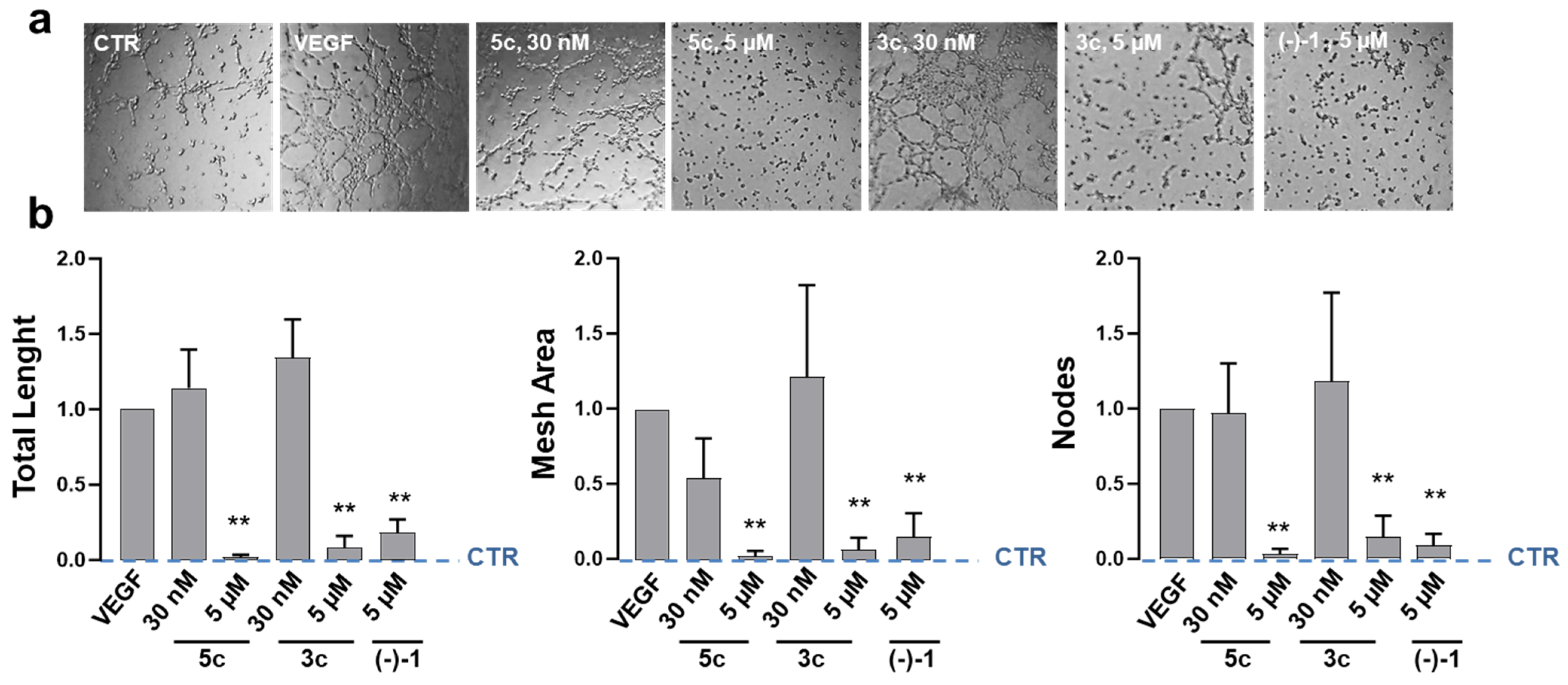
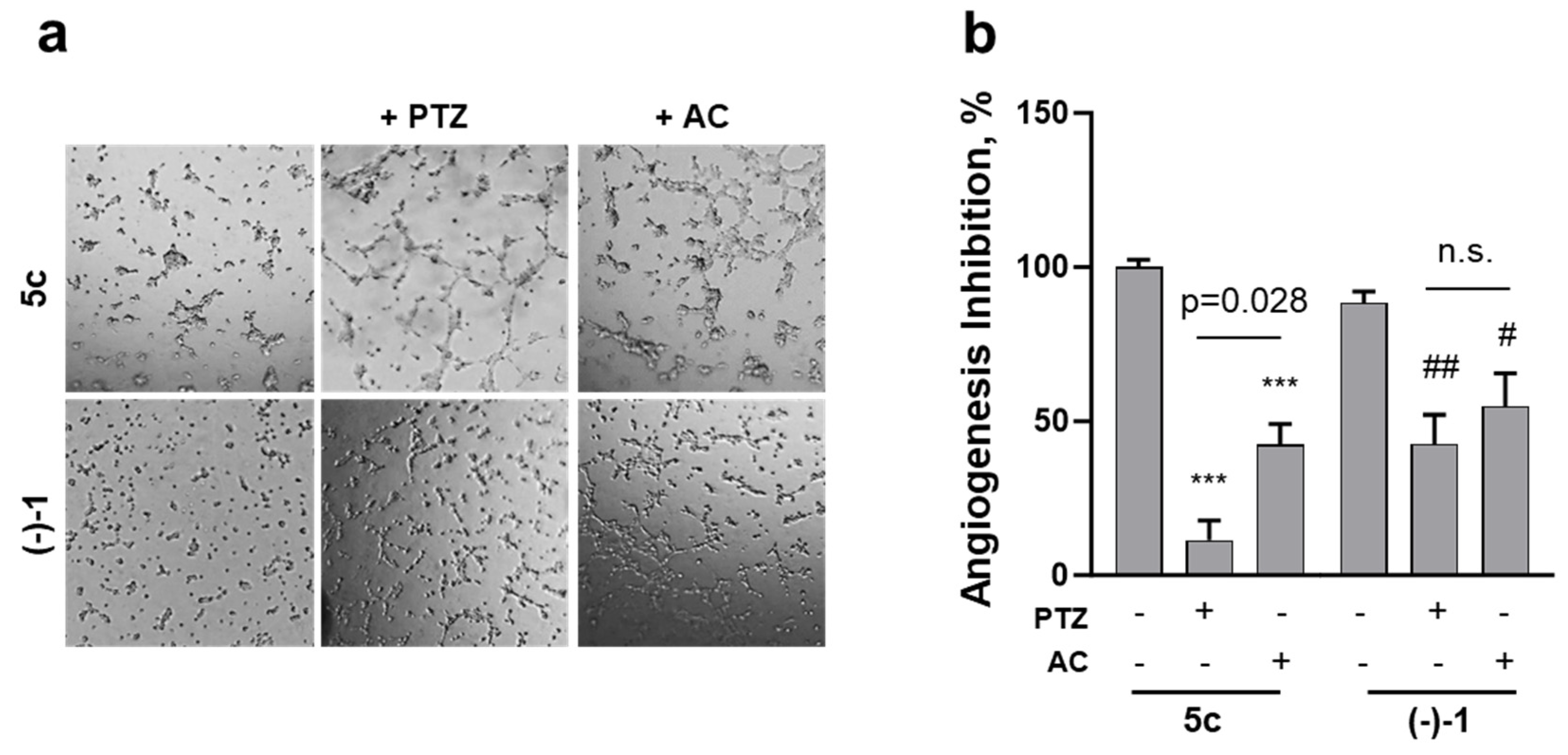
2.5. Effects on Endothelial-Dependent Angiogenic Processes
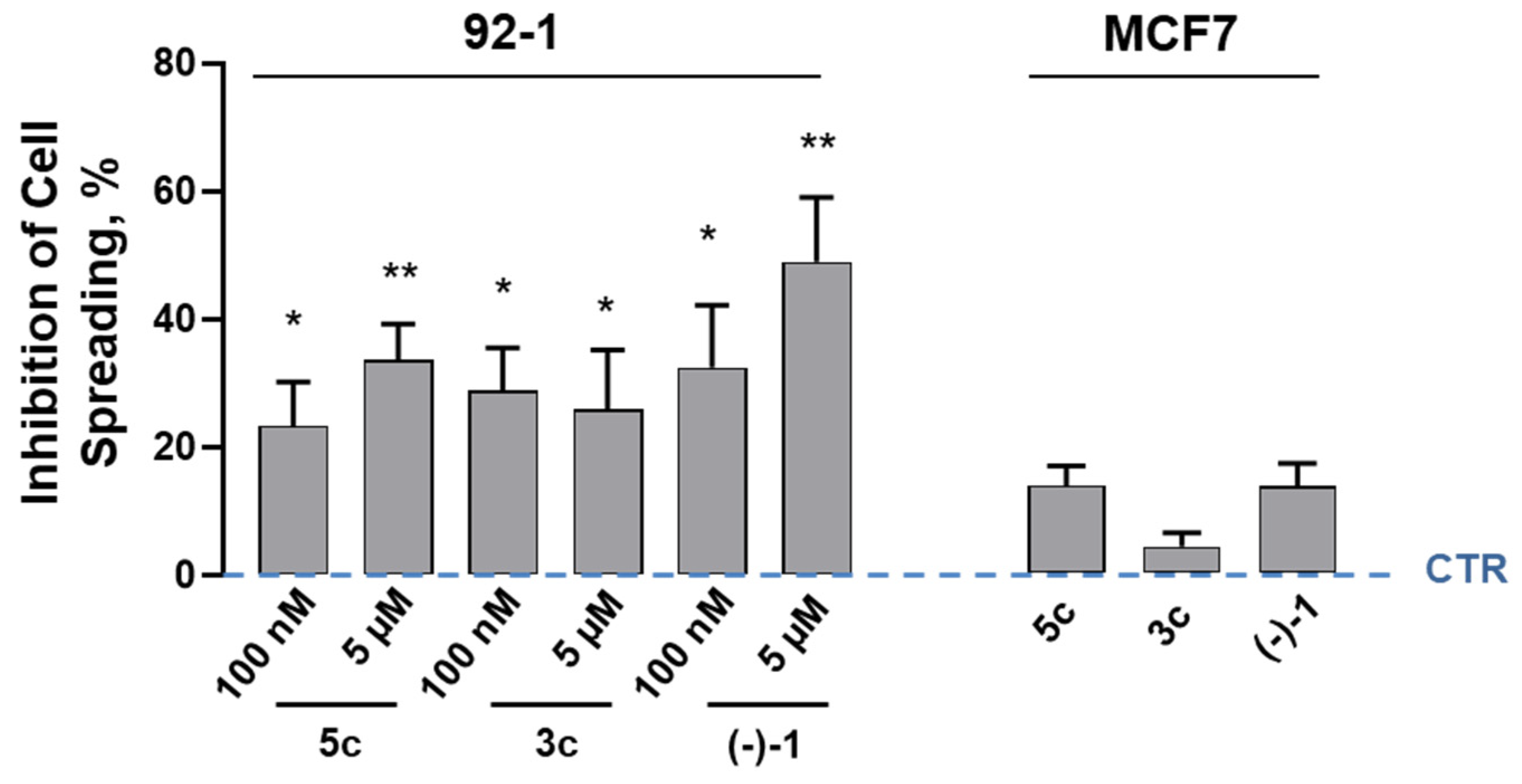
2.6. Receptor Signaling in Melanoma Cell Spreading
3. Discussion
4. Materials and Methods
4.1. Chemistry
4.1.1. General Procedure
4.1.2. Synthesis and Product Characterization
4.2. Cell Cultures and Reagents
4.3. Cell Proliferation
4.4. Radioligand Binding Assay
4.5. HDAC Activity Assay
4.6. Tube Formation Assay
4.7. Cell Spreading
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Bolick, N.L.; Geller, A.C. Epidemiology of Melanoma. Hematol. Oncol. Clin. N. Am. 2021, 35, 57–72. [Google Scholar] [CrossRef] [PubMed]
- Leonardi, G.C.; Falzone, L.; Salemi, R.; Zanghì, A.; Spandidos, D.A.; Mccubrey, J.A.; Candido, S.; Libra, M. Cutaneous melanoma: From pathogenesis to therapy (Review). Int. J. Oncol. 2018, 52, 1071–1080. [Google Scholar] [CrossRef]
- Gilchrest, B.A.; Eller, M.S.; Geller, A.C.; Yaar, M. The pathogenesis of melanoma induced by ultraviolet radiation. N. Engl. J. Med. 1999, 340, 1341–1348. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, B.; Qadir, M.I.; Ghafoor, S. Malignant Melanoma: Skin Cancer-Diagnosis, Prevention, and Treatment. Crit. Rev. Eukaryot. Gene Exp. 2020, 30, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Clark, W.H.J.; From, L.; Bernardino, E.A.; Mihm, M.C. The histogenesis and biologic behavior of primary human malignant melanomas of the skin. Cancer Res. 1969, 29, 705–727. [Google Scholar]
- Garbe, C.; Amaral, T.; Peris, K.; Hauschild, A.; Arenberger, P.; Basset-Seguin, N.; Bastholt, L.; Bataille, V.; del Marmol, V.; Dréno, B.; et al. European consensus-based interdisciplinary guideline for melanoma. Part 1: Diagnostics: Update 2022. Eur. J. Cancer 2022, 170, 236–255. [Google Scholar] [CrossRef]
- Kaliki, S.; Shields, C.L. Uveal melanoma: Relatively rare but deadly cancer. Eye 2017, 31, 241–257. [Google Scholar] [CrossRef]
- Andreoli, M.T.; Mieler, W.F.; Leiderman, Y.I. Epidemiological trends in uveal melanoma. Br. J. Ophthalmol. 2015, 99, 1550–1553. [Google Scholar] [CrossRef]
- Palamaris, K.; Moutafi, M.; Gakiopoulou, H.; Theocharis, S. Histone Deacetylase (HDAC) Inhibitors: A Promising Weapon to Tackle Therapy Resistance in Melanoma. Int. J. Mol. Sci. 2022, 23, 3660. [Google Scholar] [CrossRef]
- Park, S.Y.; Kim, J.S. A Short Guide to Histone Deacetylases Including Recent Progress on Class II Enzymes. Exp. Mol. Med. 2020, 52, 204–212. [Google Scholar] [CrossRef]
- Wilmott, J.S.; Colebatch, A.J.; Kakavand, H.; Shang, P.; Carlino, M.S.; Thompson, J.F.; Long, G.V.; Scolyer, R.A.; Hersey, P. Expression of the Class 1 Histone Deacetylases HDAC8 and 3 Are Associated with Improved Survival of Patients with Metastatic Melanoma. Mod. Pathol. 2015, 28, 884–894. [Google Scholar] [CrossRef] [PubMed]
- Goutas, D.; Theocharis, S.; Tsourouflis, G. Unraveling the Epigenetic Role and Clinical Impact of Histone Deacetylases in Neoplasia. Diagnostics 2021, 11, 1346. [Google Scholar] [CrossRef] [PubMed]
- Gatti, L.; Sevko, A.; de Cesare, M.; Arrighetti, N.; Manenti, G.; Ciusani, E.; Verderio, P.; Ciniselli, C.M.; Cominetti, D.; Carenini, N.; et al. Histone Deacetylase Inhibitor-Temozolomide Co-Treatment Inhibits Melanoma Growth through Suppression of Chemokine (C-C Motif) Ligand 2-Driven Signals. Oncotarget 2014, 5, 4516–4528. [Google Scholar] [CrossRef]
- Souri, Z.; Jochemsen, A.G.; Versluis, M.; Wierenga, A.P.A.; Nemati, F.; van der Velden, P.A.; Kroes, W.G.M.; Verdijk, R.M.; Luyten, G.P.M.; Jager, M.J. HDAC Inhibition Increases HLA Class I Expression in Uveal Melanoma. Cancers 2020, 12, 3690. [Google Scholar] [CrossRef]
- Ny, L.; Jespersen, H.; Karlsson, J.; Alsén, S.; Filges, S.; All-Eriksson, C.; Andersson, B.; Carneiro, A.; Helgadottir, H.; Levin, M.; et al. The PEMDAC Phase 2 Study of Pembrolizumab and Entinostat in Patients with Metastatic Uveal Melanoma. Nat. Commun. 2021, 12, 5155. [Google Scholar] [CrossRef]
- Ibrahim, N.; Buchbinder, E.I.; Granter, S.R.; Rodig, S.J.; Giobbie-Hurder, A.; Becerra, C.; Tsiaras, A.; Gjini, E.; Fisher, D.E.; Hodi, F.S. A Phase I Trial of Panobinostat (LBH589) in Patients with Metastatic Melanoma. Cancer Med. 2016, 5, 3041–3050. [Google Scholar] [CrossRef]
- Gallinari, P.; Marco, S.; Jones, P.; Pallaoro, M.; Steinkühler, C. HDACs, histone deacetylation and gene transcription: From molecular biology to cancer therapeutics. Cell Res. 2007, 17, 195–211. [Google Scholar] [CrossRef]
- Souri, Z.; Jochemsen, A.G.; Wierenga, A.P.A.; Kroes, W.G.M.; Verdijk, R.M.; van der Velden, P.A.; Luyten, G.P.M.; Jager, M.J. Expression of Hdacs 1, 3 and 8 Is Upregulated in the Presence of Infiltrating Lymphocytes in Uveal Melanoma. Cancers 2021, 13, 4146. [Google Scholar] [CrossRef]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int. J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef]
- Venugopal, B.; Baird, R.; Kristeleit, R.S.; Plummer, R.; Cowan, R.; Stewart, A.; Fourneau, N.; Hellemans, P.; Elsayed, Y.; McClue, S.; et al. A phase I Study of Quisinostat (JNJ-26481585), an Oral Hydroxamate Histone Deacetylase Inhibitor with Evidence of Target Modulation and Antitumor Activity, in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2013, 19, 4262–4272. [Google Scholar] [CrossRef]
- Cho, W.C.; Jour, G.; Aung, P.P. Role of angiogenesis in melanoma progression: Update on key angiogenic mechanisms and other associated components. Semin. Cancer Biol. 2019, 59, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Tarhini, A.A.; Frankel, P.; Margolin, K.A.; Christensen, S.; Ruel, C.; Shipe-Spotloe, J.; Gandara, D.R.; Chen, A.; Kirkwood, J.M. Aflibercept (VEGF Trap) in inoperable stage III or stage IV melanoma of cutaneous or uveal origin. Clin. Cancer Res. 2011, 17, 6574–6581. [Google Scholar] [CrossRef] [PubMed]
- Barbaraci, C.; di Giacomo, V.; Maruca, A.; Patamia, V.; Rocca, R.; Dichiara, M.; Di Rienzo, A.; Cacciatore, I.; Cataldi, A.; Balaha, M.; et al. Discovery of first novel sigma/HDACi dual-ligands with a potent in vitro antiproliferative activity. Bioorg. Chem. 2023, 140, 106794. [Google Scholar] [CrossRef] [PubMed]
- Rousseaux, C.G.; Greene, S.F. Sigma receptors [σRs]: Biology in normal and diseased states. J. Recept. Signal Transduct. Res. 2016, 36, 327–388. [Google Scholar] [CrossRef]
- Hanner, M.; Moebius, F.F.; Flandorfer, A.; Knaus, H.G.; Striessnig, J.; Kempner, E.; Glossmann, H. Purification, molecular cloning, and expression of the mammalian sigma1-binding site. Proc. Natl. Acad. Sci. USA 1996, 93, 8072–8077. [Google Scholar] [CrossRef]
- Alon, A.; Schmidt, H.R.; Wood, M.D.; Sahn, J.J.; Martin, S.F.; Kruse, A.C. Identification of the gene that codes for the σ 2 receptor. Proc. Natl. Acad. Sci. USA. 2017, 114, 7160–7165. [Google Scholar] [CrossRef]
- Borde, P.; Cosgrove, N.; Charmsaz, S.; Safrany, S.T.; Young, L. An investigation of Sigma-1 receptor expression and ligand-induced endoplasmic reticulum stress in breast cancer. Cancer Gene Ther. 2023, 30, 368–374. [Google Scholar] [CrossRef]
- Yang, K.; Zeng, C.; Wang, C.; Sun, M.; Yin, D.; Sun, T. Sigma-2 Receptor—A Potential Target for Cancer/Alzheimer’s Disease Treatment via Its Regulation of Cholesterol Homeostasis. Molecules 2020, 25, 5439. [Google Scholar] [CrossRef]
- Georgiadis, M.-O.; Karoutzou, O.; Foscolos, A.-S.; Papanastasiou, I. Sigma Receptor (σR) Ligands with Antiproliferative and Anticancer Activity. Molecules 2017, 22, 1408. [Google Scholar] [CrossRef]
- Barbaraci, C.; Giurdanella, G.; Leotta, C.G.; Longo, A.; Amata, E.; Dichiara, M.; Pasquinucci, L.; Turnaturi, R.; Prezzavento, O.; Cacciatore, I.; et al. Haloperidol Metabolite II Valproate Ester (S)-(-)-MRJF22: Preliminary Studies as a Potential Multifunctional Agent Against Uveal Melanoma. J. Med. Chem. 2021, 64, 13622–13632. [Google Scholar] [CrossRef]
- Longhitano, L.; Castracani, C.C.; Tibullo, D.; Avola, R.; Viola, M.; Russo, G.; Prezzavento, O.; Marrazzo, A.; Amata, E.; Reibaldi, M.; et al. Sigma-1 and Sigma-2 receptor ligands induce apoptosis and autophagy but have opposite effect on cell proliferation in uveal melanoma. Oncotarget 2017, 8, 91099–91111. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.A.; Witter, D.J.; Belvedere, S. Histone deacetylase inhibitors. J. Med. Chem. 2003, 46, 5097–5116. [Google Scholar] [CrossRef] [PubMed]
- Mai, A.; Massa, S.; Rotili, D.; Pezzi, R.; Bottoni, P.; Scatena, R.; Meraner, J.; Brosch, G. Exploring the connection unit in the HDAC inhibitor pharmacophore model: Novel uracil-based hydroxamates. Bioorg. Med. Chem. Lett. 2005, 15, 4656–4661. [Google Scholar] [CrossRef] [PubMed]
- Glennon, R.A. Pharmacophore identification for sigma-1 (sigma1) receptor binding: Application of the “deconstruction-reconstruction-elaboration” approach. Mini Rev. Med. Chem. 2005, 5, 927–940. [Google Scholar]
- Gasparre, G.; Abate, C.; Carlucci, R.; Berardi, F.; Cassano, G. The σ1 receptor agonist (+)-pentazocine increases store-operated Ca2+ entry in MCF7σ1 and SK-N-SH cell lines. Pharmacol. Rep. 2017, 69, 542–545. [Google Scholar] [CrossRef]
- Vigushin, D.M.; Ali, S.; Pace, P.E.; Mirsaidi, N.; Ito, K.; Adcock, I.; Coombes, R.C. Trichostatin A is a histone deacetylase inhibitor with potent antitumor activity against breast cancer in vivo. Clin. Cancer Res. 2001, 7, 971–976. [Google Scholar]
- Levinzon, L.; Madigan, M.; Nguyen, V.; Hasic, E.; Conway, M.; Cherepanoff, S. Tumour Expression of Histone Deacetylases in Uveal Melanoma. Ocul. Oncol. Pathol. 2019, 5, 153–161. [Google Scholar] [CrossRef]
- Crawford, K.W.; Coop, A.; Bowen, W.D. Sigma(2)Receptors regulate changes in sphingolipid levels in breast tumor cells. Eur. J. Pharmacol. 2002, 443, 207–209. [Google Scholar] [CrossRef]
- McCollum, C.W.; Conde-Vancells, J.; Hans, C.; Vazquez-Chantada, M.; Kleinstreuer, N.; Tal, T.; Knudsen, T.; Shah, S.S.; Merchant, F.A.; Finnell, R.H.; et al. Identification of vascular disruptor compounds by analysis in zebrafish embryos and mouse embryonic endothelial cells. Reprod. Toxicol. 2017, 70, 60–69. [Google Scholar] [CrossRef]
- Ikawa, T.; Miyagawa, T.; Fukui, Y.; Toyama, S.; Omatsu, J.; Awaji, K.; Norimatsu, Y.; Watanabe, Y.; Yoshizaki, A.; Sato, S.; et al. Endothelial CCR6 expression due to FLI1 deficiency contributes to vasculopathy associated with systemic sclerosis. Arthritis Res. Ther. 2021, 23, 283. [Google Scholar] [CrossRef]
- Flavahan, W.A.; Gaskell, E.; Bernstein, B.E. Epigenetic Plasticity and the Hallmarks of Cancer. Science 2017, 357, eaal2380. [Google Scholar] [CrossRef] [PubMed]
- Lubbe, W.J.; Zhou, Z.Y.; Fu, W.; Zuzga, D.; Schulz, S.; Fridman, R.; Muschel, R.J.; Waldman, S.A.; Pitari, G.M. Tumor Epithelial Cell Matrix Metalloproteinase 9 Is a Target for Antimetastatic Therapy in Colorectal Cancer. Clin. Cancer Res. 2006, 12, 1876–1882. [Google Scholar] [CrossRef] [PubMed]
- Pihl, E.; Hughes, E.S.; McDermott, F.T.; Milne, B.J.; Price, A.B. Disease-free survival and recurrence after resection of colorectal carcinoma. J. Surg. Oncol. 1981, 16, 333–341. [Google Scholar] [CrossRef] [PubMed]
- McCawley, L.J.; Matrisian, L.M. Matrix metalloproteinases: They’re not just for matrix anymore! Curr. Opin. Cell Biol. 2001, 13, 534–540. [Google Scholar] [CrossRef]
- Wu, Y.; Bai, X.; Li, X.; Zhu, C.; Wu, Z.P. Overexpression of sigma-1 receptor in MCF-7 cells enhances proliferation via the classic protein kinase C subtype signaling pathway. Oncol. Lett. 2018, 16, 6763–6769. [Google Scholar] [CrossRef]
- Glozak, M.; Seto, E. Histone deacetylases and cancer. Oncogene 2007, 26, 5420–5432. [Google Scholar] [CrossRef]
- Oyer, H.M.; Sanders, C.M.; Kim, F.J. Small-Molecule Modulators of Sigma1 and Sigma2/TMEM97 in the Context of Cancer: Foundational Concepts and Emerging Themes. Front. Pharmacol. 2019, 10, 1141. [Google Scholar] [CrossRef]
- Crottès, D.; Rapetti-Mauss, R.; Alcaraz-Perez, F.; Tichet, M.; Gariano, G.; Martial, S.; Guizouarn, H.; Pellissier, B.; Loubat, A.; Popa, A.; et al. SIGMAR1 Regulates Membrane Electrical Activity in Response to Extracellular Matrix Stimulation to Drive Cancer Cell Invasiveness. Cancer Res. 2016, 76, 607–618. [Google Scholar] [CrossRef]
- Xu, Q.X.; Li, E.M.; Zhang, Y.F.; Liao, L.D.; Xu, X.E.; Wu, Z.Y.; Shen, J.H.; Xu, L.Y. Overexpression of sigma1 receptor and its positive associations with pathologic TNM classification in esophageal squamous cell carcinoma. J. Histochem. Cytochem. 2012, 60, 457–466. [Google Scholar] [CrossRef]
- Moody, T.W.; Leyton, J.; John, C. Sigma ligands inhibit the growth of small cell lung cancer cells. Life Sci. 2000, 66, 1979–1986. [Google Scholar] [CrossRef]
- Matsumoto, R.R.; Bowen, W.D.; Tom, M.A.; Vo, V.N.; Truong, D.D.; De Costa, B.R. Characterization of two novel sigma receptor ligands: Antidystonic effects in rats suggest sigma receptor antagonism. Eur. J. Pharmacol. 1995, 280, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Anfuso, C.D.; Longo, A.; Distefano, A.; Amorini, A.M.; Salmeri, M.; Zanghi, G.; Giallongo, C.; Giurdanella, G.; Lupo, G. Uveal Melanoma Cells Elicit Retinal Pericyte Phenotypical and Biochemical Changes in an in Vitro Model of Coculture. Int. J. Mol. Sci. 2020, 21, 5557. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.; Petralla, S.; Protti, M.; Baiula, M.; Kobrlova, T.; Soukup, O.; Spampinato, S.M.; Mercolini, L.; Monti, B.; Bolognesi, M.L. Alpha-linolenic acid-valproic acid conjugates: Toward single-molecule polypharmacology for multiple sclerosis. ACS Med. Chem. Lett. 2020, 11, 2406–2413. [Google Scholar] [CrossRef] [PubMed]
- Mach, R.H.; Smith, C.R.; Childers, S.R. Ibogaine possesses a selective affinity for sigma 2 receptors. Life Sci. 1995, 57, PL57–PL62. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Ki (nM) ± SD | |
|---|---|---|
| σ1R | σ2R | |
| 5c | 19.0 ± 1.6 | 2051.2 ± 737.1 |
| 3c | 28.0 ± 4.4 | 1330.0 ± 286.0 |
| HP | 2.6 ± 0.4 | 77.0 ± 18.0 |
| PTZ | 4.3 ± 0.5 | 1465.0 ± 224.0 |
| Human Cells | 5c, IC50 (nM) |
|---|---|
| 92.1 (Uveal Melanoma) | 24.8 ± 5.8 |
| A375 (Cutaneous Melanoma, CM) | 91.5 ± 11.4 |
| A549 (Lung Carcinoma) | 118.1 ± 13.0 |
| 786-O (Renal Adenocarcinoma) | 150.4 ± 24.0 |
| MRC-5 (Normal Skin Fibroblast) | 156.7 ± 55.2 |
| T84 (Colorectal Carcinoma) | 1198.0 ± 380.6 |
| WS1 (Normal Skin Fibroblast) | 1162.0 ± 186.7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leotta, C.G.; Barbaraci, C.; Fiorito, J.; Coco, A.; di Giacomo, V.; Amata, E.; Marrazzo, A.; Pitari, G.M. HDAC/σ1R Dual-Ligand as a Targeted Melanoma Therapeutic. Pharmaceuticals 2025, 18, 179. https://doi.org/10.3390/ph18020179
Leotta CG, Barbaraci C, Fiorito J, Coco A, di Giacomo V, Amata E, Marrazzo A, Pitari GM. HDAC/σ1R Dual-Ligand as a Targeted Melanoma Therapeutic. Pharmaceuticals. 2025; 18(2):179. https://doi.org/10.3390/ph18020179
Chicago/Turabian StyleLeotta, Claudia Giovanna, Carla Barbaraci, Jole Fiorito, Alessandro Coco, Viviana di Giacomo, Emanuele Amata, Agostino Marrazzo, and Giovanni Mario Pitari. 2025. "HDAC/σ1R Dual-Ligand as a Targeted Melanoma Therapeutic" Pharmaceuticals 18, no. 2: 179. https://doi.org/10.3390/ph18020179
APA StyleLeotta, C. G., Barbaraci, C., Fiorito, J., Coco, A., di Giacomo, V., Amata, E., Marrazzo, A., & Pitari, G. M. (2025). HDAC/σ1R Dual-Ligand as a Targeted Melanoma Therapeutic. Pharmaceuticals, 18(2), 179. https://doi.org/10.3390/ph18020179