
Plasminogen Activation Inhibitor-1 Promotes Resilience to Acute Oxidative Stress in Cerebral Arteries from Females


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
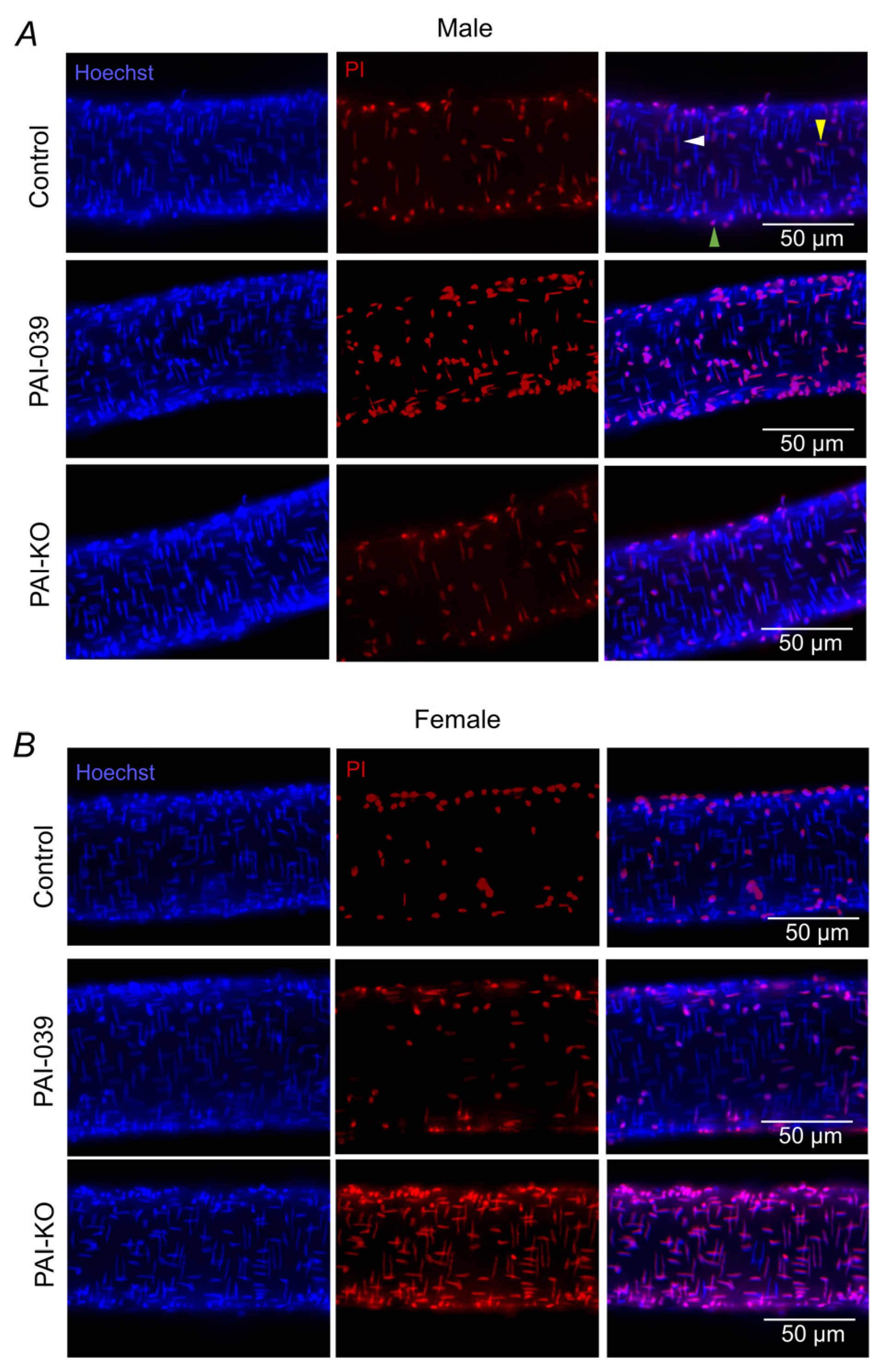
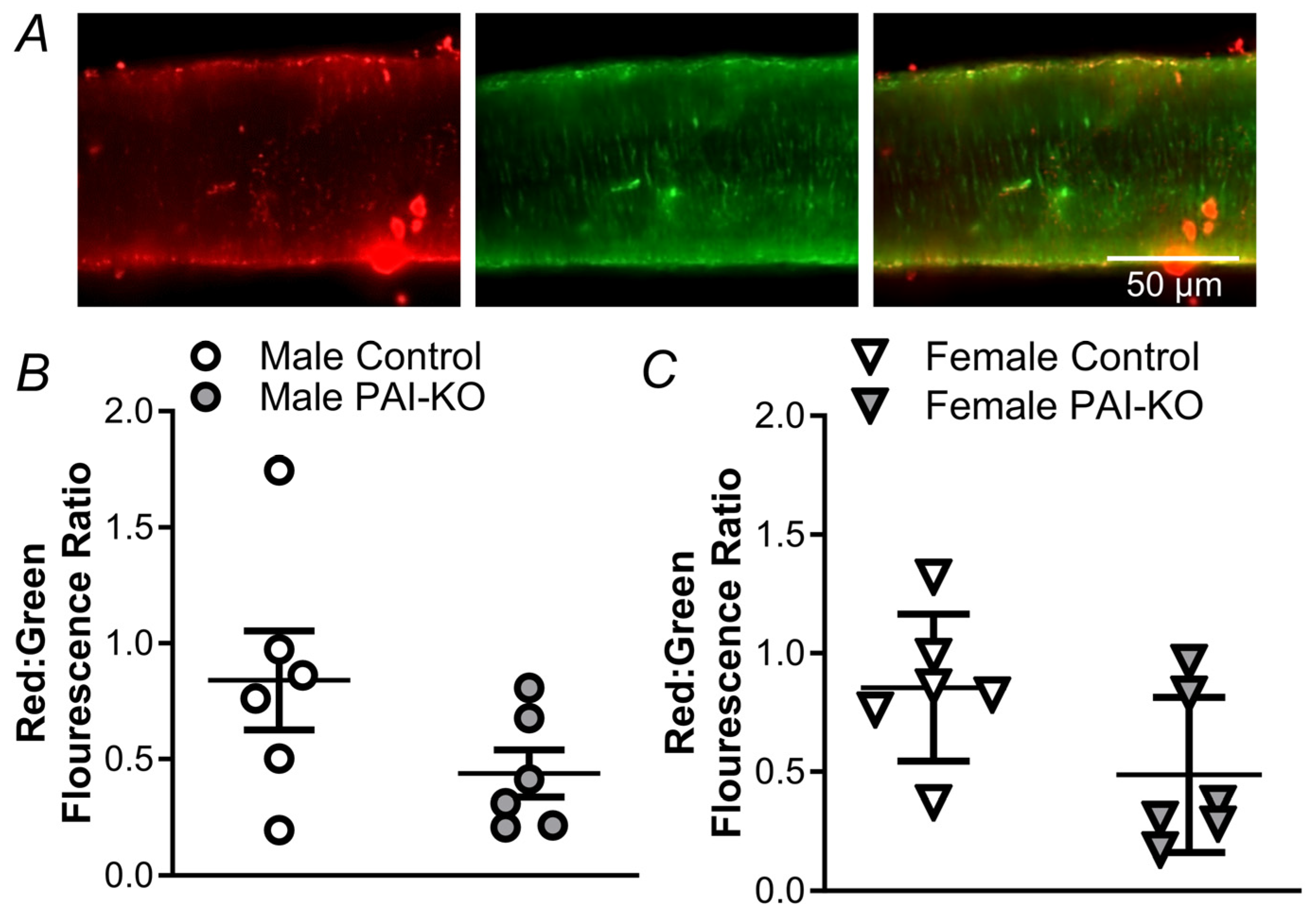
2.1. Genetic, but Not Pharmacological, Loss of PAI-1 Signaling Enhances H2O2-Induced Cell Death in Females
2.2. PAI-1 Levels in Brain and Cerebral Vasculature
2.3. PAI-1 Attenuates the Sustained [Ca2+]i Response Following H2O2 Exposure in Females
2.4. PAI-1 Limits ΔΨm Depolarization to H2O2 in Females
2.5. PAI-1 Limits Mitochondrial O2− Production during Acute Oxidative Stress in Females
3. Discussion
3.1. PAI-1 Enhances Resilience to Oxidative Stress in Cerebral Arteries from Females
3.2. PAI-1 Limits Mitochondrial Disruption during Acute Oxidative Stress
3.3. Differences in Pharmacological and Genetic Targeting of PAI-1 and Other Limitations
3.4. PAI-1 in Stroke
3.5. Conclusions
4. Materials and Methods
4.1. Animal Care and Use
4.2. Preparation of Isolated Posterior Cerebral Arteries
4.3. Quantification of Cell Death
4.4. Immunofluorescence for PAI-1 Expression
4.5. Calcium Photometry
4.6. Mitochondrial Membrane Potential
4.7. Quantification of Mitochondrial ROS Production
4.8. Statistics
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nagai, N.; Suzuki, Y.; Von Hoef, B.; Lijnen, H.R.; Collen, D. Effects of plasminogen activator inhibitor-1 on ischemic brain injury in permanent and thrombotic middle cerebral artery occlusion models in mice. J. Thromb. Haemost. 2005, 3, 1379–1384. [Google Scholar] [CrossRef] [PubMed]
- Morrow, G.B.; Whyte, C.S.; Mutch, N.J. A serpin with a finger in many PAIs: PAI-1’s central function in thromboinflammation and cardiovascular disease. Front. Cardiovasc. Med. 2021, 8, 653655. [Google Scholar] [CrossRef] [PubMed]
- Jung, R.G.; Motazedian, P.; Ramirez, F.D.; Simard, T.; Di Santo, P.; Visintini, S.; Faraz, M.A.; Labinaz, A.; Jung, Y.; Hibbert, B. Association between plasminogen activator inhibitor-1 and cardiovascular events: A systematic review and meta-analysis. Thromb. J. 2018, 16, 12. [Google Scholar] [CrossRef] [PubMed]
- Catanese, L.; Trarsia, J.; Fisher, M. Acute ischemic stroke therapy overview. Circ. Res. 2017, 120, 541–558. [Google Scholar] [CrossRef]
- Li, J.; Li, W.; Sua, J.; Liu, W.; Altura, B.T.; Altura, B.M. Hydrogen peroxide induces apoptosis in cerebral vascular smooth muscle cells: Possible relation to neurodegenerative diseases and strokes. Brain Res. Bull. 2003, 62, 101–106. [Google Scholar] [CrossRef]
- Dalkara, T.; Arsava, E.M. Can restoring incomplete microcirculatory reperfusion improve stroke outcome after thrombolysis? J. Cereb. Blood Flow. Metab. 2012, 32, 2091–2099. [Google Scholar] [CrossRef]
- Norton, C.E.; Shaw, R.L.; Mittler, R.; Segal, S.S. Endothelial cells promote smooth muscle cell resilience to H2O2-induced cell death in mouse cerebral arteries. Acta Physiol. 2022, 235, e13819. [Google Scholar] [CrossRef]
- Singh, M.; Sharma, H.; Singh, N. Hydrogen peroxide induces apoptosis in HeLa cells through mitochondrial pathway. Mitochondrion 2007, 7, 367–373. [Google Scholar] [CrossRef]
- Humphries, B.A.; Buschhaus, J.M.; Chen, Y.C.; Haley, H.R.; Qyli, T.; Chiang, B.; Shen, N.; Rajendran, S.; Cutter, A.; Cheng, Y.H.; et al. Plasminogen activator inhibitor 1 (PAI-1) promotes actin cytoskeleton reorganization and glycolytic metabolism in triple negative breast cancer. Mol. Cancer Res. 2019, 17, 1142–1154. [Google Scholar] [CrossRef]
- Schneider, D.J.; Chen, Y.; Sobel, B.E. The effect of plasminogen activator inhibitor type 1 on apoptosis. Thromb. Haemost. 2008, 100, 1037–1040. [Google Scholar]
- Kissela, B.M.; Khoury, J.C.; Alwell, K.; Moomaw, C.J.; Woo, D.; Adeoye, O.; Flaherty, M.L.; Khatri, P.; Ferioli, S.; De Los Rios La Rosa, F.; et al. Age at stroke: Temporal trends in stroke incidence in a large, biracial population. Neurology 2012, 79, 1781–1787. [Google Scholar] [CrossRef]
- Petrea, R.E.; Beiser, A.S.; Seshadri, S.; Kelly-Hayes, M.; Kase, C.S.; Wolf, P.A. Gender differences in stroke incidence and poststroke disability in the Framingham heart study. Stroke 2009, 40, 1032–1037. [Google Scholar] [CrossRef] [PubMed]
- Norton, C.E.; Shaw, R.L.; Safa; Dockery, B.; Domeier, T.L.; Segal, S.S. Advanced age and female sex protect cerebral arteries from mitochondrial depolarization and apoptosis during acute oxidative stress. Aging Cell 2024, 23, e14110. [Google Scholar] [CrossRef] [PubMed]
- Miyagawa, R.; Asakura, T.; Nakamura, T.; Okada, H.; Iwaki, S.; Sobel, B.E.; Fujii, S. Increased expression of plasminogen activator inhibitor type-1 (PAI-1) in HEPG2 cells induced by insulin mediated by the 3’ untranslated region of the PAI-1 gene and its pharmacologic implications. Coron. Artery Dis. 2010, 21, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Roy-O’Reilly, M.; McCullough, L.D. Age and sex are critical factors in ischemic stroke pathology. Endocrinology 2018, 159, 3120–3131. [Google Scholar] [CrossRef] [PubMed]
- Rutten-Jacobs, L.C.A.; Arntz, R.M.; Maaijwee, N.A.; Schoodnerwaldt, H.C.; Dorresteihn, L.D.; van Dijk, E.J.; de Leeuw, R.E. Long-term mortality after stroke among adults aged 18 to 50 years. JAMA 2013, 309, 1136–1144. [Google Scholar] [CrossRef]
- Maselli, A.; Matarrese, P.; Straface, E.; Canu, S.; Franconi, F.; Malorni, W. Cell sex: A new look at cell fate studies. FASEB J. 2009, 23, 978–984. [Google Scholar] [CrossRef]
- Vina, J.; Borras, C.; Gambini, J.; Sastre, J.; Pallardo, F.V. Why females live longer than males? Importance of the upregulation of longevity-associated genes by oestrogenic compounds. FEBS Lett. 2005, 579, 2541–2545. [Google Scholar] [CrossRef]
- Shaw, R.L.; Norton, C.E.; Segal, S.S. Apoptosis in resistance arteries induced by hydrogen peroxide: Greater resilience of endothelium versus smooth muscle. Am. J. Physiol. Heart Circ. Physiol. 2021, 320, H1625–H1633. [Google Scholar] [CrossRef]
- Chen, Y.; Kelm, R.J.; Budd, R.C.; Sobel, B.E.; Schneider, D.J. Inhibition of apoptosis and caspase-3 in vascular smooth muscle cells by plasminogen activator inhibitor Type-1. J. Cell Biochem. 2004, 92, 178–188. [Google Scholar] [CrossRef]
- Soeda, S.; Koyanagi, S.; Kuramoto, Y.; Kimura, M.; Oda, M.; Kozako, T.; Hayashida, S.; Shimeno, H. Anti-apoptotic roles of plasminogen activator inhibitor-1 as a neurotrophic factor in the central nervous system. Thromb. Haemost. 2008, 100, 1014–1020. [Google Scholar] [PubMed]
- Zhao, H.; Morimoto, T.; Sasa, M.; Tanaka, T.; Izumi, K. Immunnohistochemical expression of UPA, PAI-1, cathepsin D and apoptotic cells in ductal carcinoma in situ of the breast. Breast Cancer 2002, 9, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, P.; Ho-Tin-Noe, B.; Vranckx, R.; Bouton, M.C.; Meilac, O.; Lijnen, R.R.; Buillin, M.C.; Michel, J.B.; Angeles-Cano, E. Protease nexin-1 inhibits plasminogen action-induced apoptosis of adherent cells. J. Biol. Chem. 2004, 279, 10346–10356. [Google Scholar] [CrossRef] [PubMed]
- Hino, H.; Akiyama, H.; Iseki, E.; Kato, M.; Ikeda, K.; Kosaka, K. Immunohistochemical localization of plasminogen activator inhibitor-1 in rat and human brain tissues. Neurosci. Lett. 2001, 297, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Sawdy, M.S.; Loskutoff, D.J. Regulation of murine type 1 plasminogen activator inhibitor gene expression in vivo. Tissue specificity and induction by lipopolysaccharide, tumor necrosis factor-alpha, and transforming growth factor-beta. J. Clin. Investig. 1991, 88, 1346–1353. [Google Scholar] [CrossRef]
- Krishnamurti, C.; Tang, D.B.; Barr, C.F.; Alving, B.M. Plasminogen activator and plasminogen activator inhibition activities in a reference population. Am. J. Clin. Pathol. 1988, 89, 747–752. [Google Scholar] [CrossRef]
- Tantral, L.; Malathi, K.; Kohyama, S.; Silane, M.; Berenstein, A.; Thottala, J. Intracellular calcium release is required for caspase-3 and -9 activation. Cell Biochem. Funct. 2004, 22, 35–40. [Google Scholar]
- Norton, C.E.; Sinkler, S.Y.; Jacobsen, N.L.; Segal, S.S. Advanced age protects resistance arteries of mouse skeletal muscle from oxidative stress through attenuating apoptosis induced by hydrogen peroxide. J. Physiol. 2019, 597, 3801–3816. [Google Scholar] [CrossRef]
- Jezek, P.; Holendova, B.; Garlid, K.D.; Jaburek, M. Mitochondrial uncoupling proteins: Subtle regulators of cellular redox signaling. Antioxid. Redox Signal 2018, 29, 667–714. [Google Scholar]
- Ma, L.J.; Mao, S.L.; Taylor, K.L.; Kanjanabuch, T.; Guan, Y.F.; Zhang, Y.H.; Brown, N.J.; Swift, L.L.; McGuinness, O.P.; Wasserman, D.H.; et al. Prevention of obesity and insulin resistance in mice lacking plasminogen activator inhibitor 1. Diabetes 2004, 53, 336–346. [Google Scholar] [CrossRef]
- Piao, L.; Jung, I.; Huh, J.Y.; Miyata, T.; Ha, H. A novel plasminogen activator inhibitor, TM5441, protects against high-fat diet-induced obesity and adipocyte injury in mice. Br. J. Pharmacol. 2016, 173, 2622–2632. [Google Scholar] [CrossRef] [PubMed]
- Mailloux, R.J.; Harper, M.E. Uncoupling proteins and the control of mitochondrial reactive oxygen species production. Free Radic. Biol. Med. 2011, 51, 1106–1115. [Google Scholar] [CrossRef] [PubMed]
- Perfettini, J.L.; Roumier, T.; Kroemer, G. Mitochondrial fusion and fission in the control of apoptosis. Trends Cell Biol. 2005, 15, 179–183. [Google Scholar] [CrossRef]
- Szabadkai, G.; Simoni, A.M.; Chami, M.; Chami, M.; Wieckowski, M.R.; Youle, R.J.; Rizzuto, R. Drp-1-dependent division of the mitochondrial network blocks intraorganellar Ca2+ waves and protects against Ca2+-mediated apoptosis. Mol. Cell 2004, 16, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Arago, M.; Formentinim, L.; Cuezva, J.M. Mitochondria-mediated energy adaption in cancer: The H+-ATP synthase-geared switch of metabolism in human tumors. Antioxid. Redox Signal 2013, 19, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Kushnareva, Y.E.; Sokolove, P.M. Prooxidants open both the mitochondrial permeability transition pore and a low-conductance channel in the inner mitochondrial membrane. Arch. Biochem. Biophys. 2000, 376, 377–388. [Google Scholar] [CrossRef]
- Fraser, P.A. The role of free radical generation in increasing cerebrovascular permeability. Free Radic. Biol. Med. 2011, 51, 967–977. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef]
- Gorlatova, N.V.; Cale, J.M.; Elokdah, H.; Li, D.; Fan, K.; Warnock, M.; Crandall, D.L.; Lawrence, D.A. Mechanism of inactivatin of plasminogen actiator inhibitor-1 by a small molecule inhibitor. J. Biol. Chem. 2007, 282, 9288–9296. [Google Scholar] [CrossRef]
- Zhou, A.Y.; Huntington, J.A.; Pannu, N.S.; Carrell, R.W.; Read, R.J. How vitronectin binds PAI-1 to modulate fibrinolysis and cell migration. Nat. Struct. Biol. 2003, 10, 541–544. [Google Scholar] [CrossRef]
- Furuya, H.; Sasaki, Y.; Chen, R.; Peres, R.; Hokutan, K.; Murakami, K.; Kim, N.; Chan, O.T.M.; Pagano, I.; Dyrskjøt, L.; et al. PAI-1 is a potential transcriptional silencer that supports bladder cancer cell activity. Sci. Rep. 2022, 12, 12186. [Google Scholar] [CrossRef] [PubMed]
- National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group. Tissue plasminogen activator for acute ischemic stroke. N. Engl. J. Med. 1995, 333, 1581–1587. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wang, C.; Zhang, Y.; Alsrouji, O.K.; Chebl, A.B.; Ding, G.; Jiang, Q.; Mayer, S.A.; Lu, M.; Kole, M.K.; et al. Cerebral endothelial cell-derived small extracellular vesicles enhance neurovascular function and neurological recovery in rat acute ischemic stroke models of mechanical thrombectomy and embolic stroke treatment with tPA. J. Cereb. Blood Flow. Metab. 2021, 41, 2090–2104. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Rosell, A.; Lo, E.H. Targeting extracellular matrix proteolysis for hemorrhagic complications of tPA stroke therapy. CNS Neurol. Disord. Drug Targets 2008, 7, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Dohgu, S.; Takata, F.; Matsumoto, J.; Oda, M.; Harada, E.; Watanabe, T.; Nishioku, T.; Shuto, H.; Tamauchi, A.; Kataoka, Y. Autocrine and paracrine up-regulation of blood–brain barrier function by plasminogen activator inhibitor-1. Microvasc. Res. 2011, 81, 103–107. [Google Scholar] [CrossRef]
- Coco, D.L.; Lopez, G.; Corrao, S. Cognitive impairment and stroke in elderly patients. Vasc. Health Risk Manag. 2016, 12, 105–116. [Google Scholar]
- Kernan, W.N.; Inzucchi, S.E.; Sawan, C.; Macko, R.F.; Furie, K.L. Obesity: A stubbornly obvious target for stroke prevention. Stroke 2012, 44, 278–286. [Google Scholar] [CrossRef]
- Skurk, T.; Hauner, H. Obesity and impaired fibrinolysis: Role of adipose production of plasminogen activator inhibitor-1. Int. J. Obes. 2004, 28, 1357–1364. [Google Scholar] [CrossRef]
- Norton, C.E.; Shaw, R.L.; Segal, S.S. Differential effects of high fat diets on resilience to H2O2-induced cell ceath in mouse cerebral arteries: Role for processed carbohydrates. Antioxidants 2023, 12, 1433. [Google Scholar] [CrossRef]
- Chan, S.L.; Bishop, N.; Li, Z.; Cipolla, M.J. Inhibition of PAI (plasminogen activator inhibitor)-1 improves brain collateral perfusion and injury after acute ischemic stroke in aged hypertensive rats. Stroke 2018, 49, 1969–1976. [Google Scholar] [CrossRef]
- Juhan-Vague, I.; Pyke, S.D.; Alessi, M.C.; Jespersen, J.; Haverkate, F.; Thompson, S.G. Fibrinolytic factors and the risk of myocardial infarction or sudden death in patients with angina pectoris. ECAT Study Group. European concerted action on thrombosis and disabilities. Circulation 1996, 94, 2057–2063. [Google Scholar] [CrossRef]
- Hamsten, A.; Wiman, B.; de Faire, U.; Blomack, M. Increased plasma levels of a rapid inhibitor of tissue plasminogen activator in young survivors of myocardial infarction. N. Engl. J. Med. 1985, 313, 1557–1563. [Google Scholar] [CrossRef] [PubMed]
- Badran, M.; Gozal, D. PAI-1: A major player in the vascular dysfunction in obstructive sleep apnea. Int. J. Mol. Sci. 2022, 23, 5516. [Google Scholar] [CrossRef] [PubMed]
- Hyslop, P.A.; Zhang, Z.; Pearson, D.V.; Phebus, L.A. Measurement of striatal H2O2 by microdialysis following global forebrain ischemia and reperfusion in the rat: Correlation with the cytotoxic potential of H2O2 in vitro. Brain Res. 1995, 671, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Weng, Z.; Fish, P.; Goyal, N.; Luo, M.; Myears, S.P.; Strawn, T.L.; Chandrasekar, B.; Wu, J.; Fay, W.P. Pharmacological targeting of plasminogen activator inhibitor-1 decreases vascular smooth muscle cell migration and neointima formation. Arter. Thromb. Vasc. Biol. 2016, 36, 2167–2175. [Google Scholar] [CrossRef]
- Rahman, F.A.; Angus, S.A.; Stokes, K.; Karpowicz, P.; Krause, M.P. Impaired ECM remodeling and macrophage activity define necrosis and regeneration following damage in aged skeletal muscle. Int. J. Mol. Sci. 2020, 21, 4575. [Google Scholar] [CrossRef]
- Sivandzade, F.; Bhalerao, A.; Cucullo, L. Analysis of the mitochondrial membrane potential using the cationic JC-1 dye as a sensitive fluorescent probe. Bio. Protoc. 2019, 9, e3128. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Safa; Norton, C.E. Plasminogen Activation Inhibitor-1 Promotes Resilience to Acute Oxidative Stress in Cerebral Arteries from Females. Pharmaceuticals 2024, 17, 1210. https://doi.org/10.3390/ph17091210
Safa, Norton CE. Plasminogen Activation Inhibitor-1 Promotes Resilience to Acute Oxidative Stress in Cerebral Arteries from Females. Pharmaceuticals. 2024; 17(9):1210. https://doi.org/10.3390/ph17091210
Chicago/Turabian StyleSafa, and Charles E. Norton. 2024. "Plasminogen Activation Inhibitor-1 Promotes Resilience to Acute Oxidative Stress in Cerebral Arteries from Females" Pharmaceuticals 17, no. 9: 1210. https://doi.org/10.3390/ph17091210
APA StyleSafa, & Norton, C. E. (2024). Plasminogen Activation Inhibitor-1 Promotes Resilience to Acute Oxidative Stress in Cerebral Arteries from Females. Pharmaceuticals, 17(9), 1210. https://doi.org/10.3390/ph17091210