
Aronia Berry Extract Modulates MYD88/NF-kB/P-Glycoprotein Axis to Overcome Gemcitabine Resistance in Pancreatic Cancer

 ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
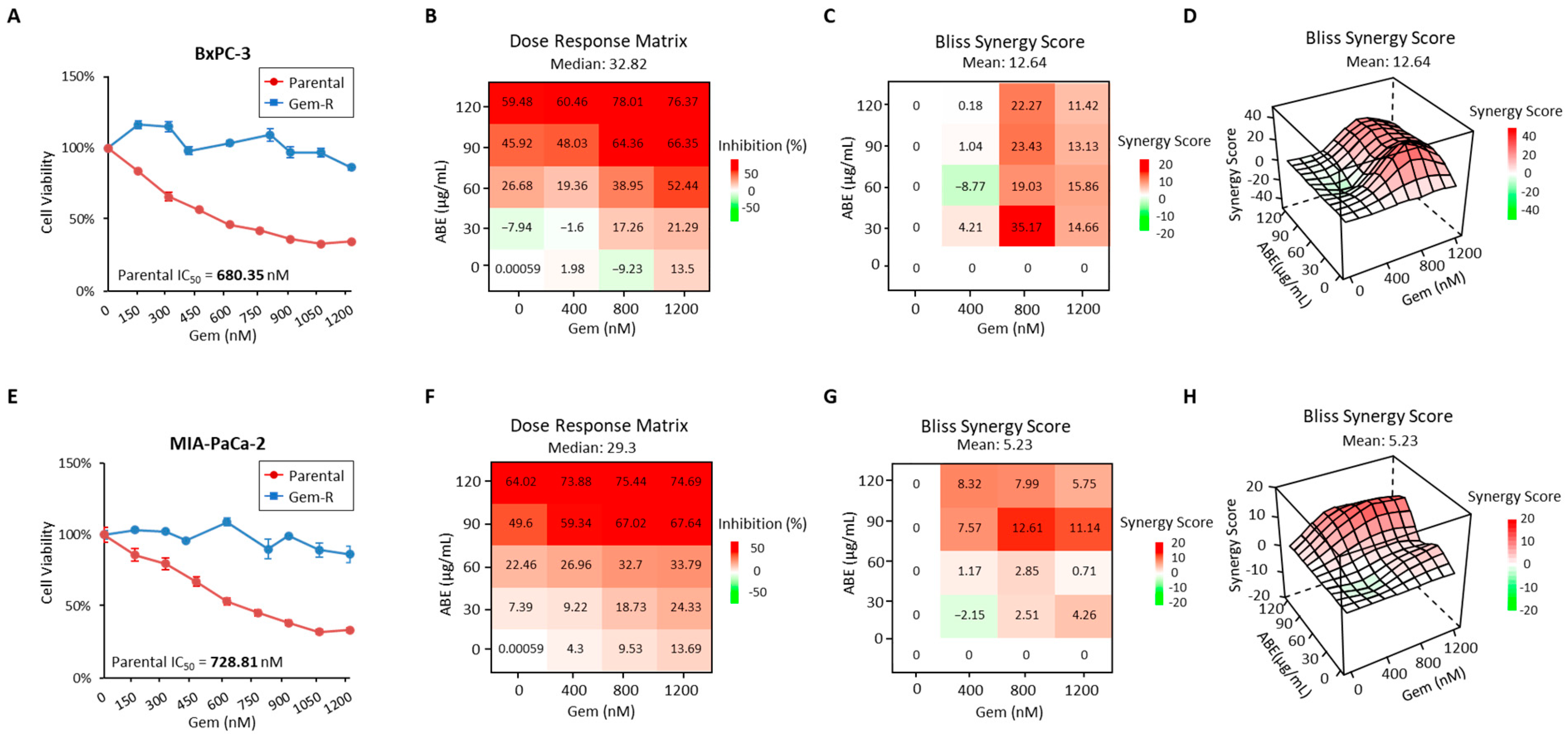
2.1. The Treatment of ABE and Gemcitabine Shows Synergistic Inhibition in Gem-R Pancreatic Cancer Cells
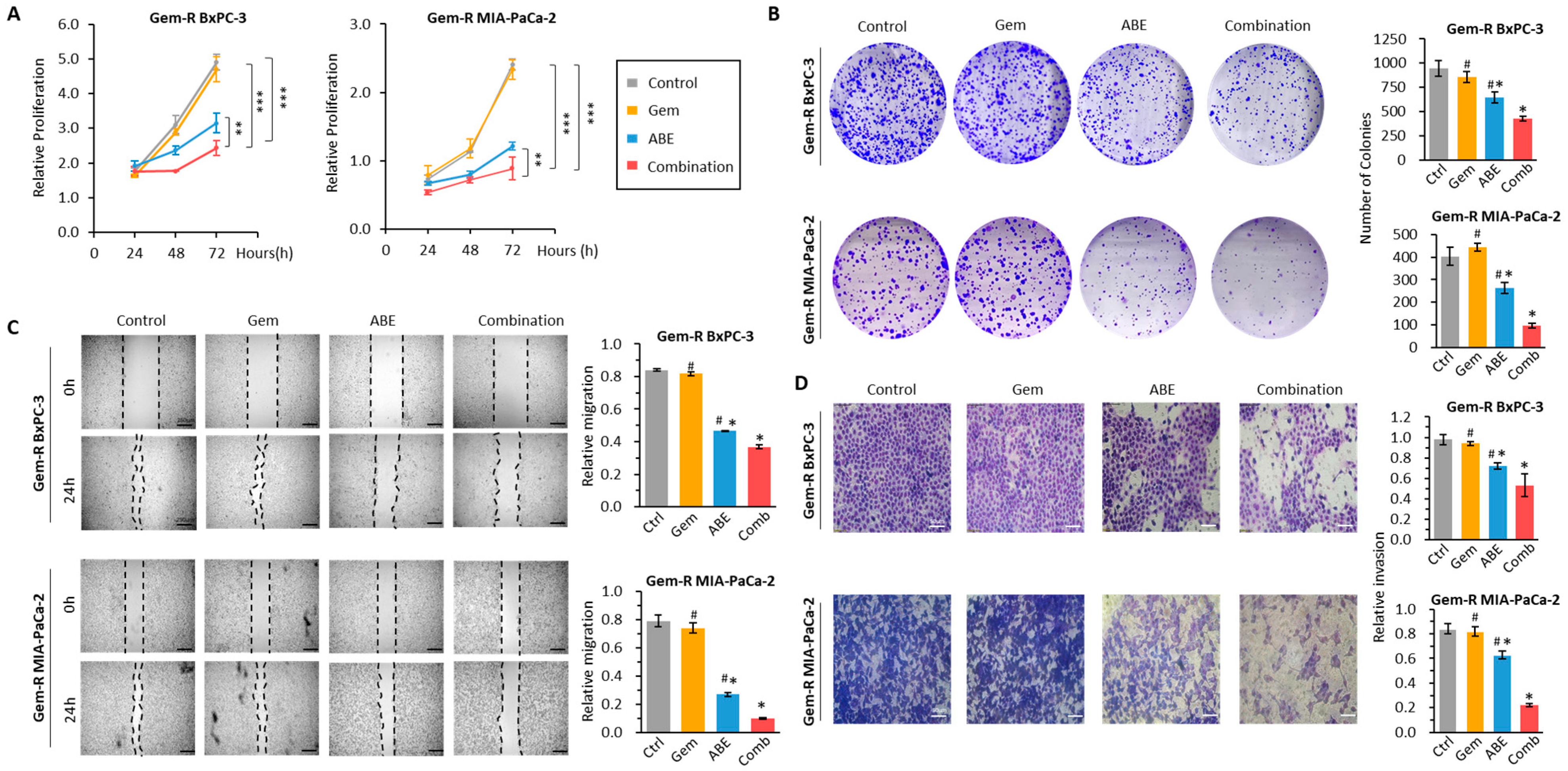
2.2. The Combination of ABE and Gemcitabine Inhibits Cell Proliferation, Colony Formation, Migration, and Invasion in Gem-R PDAC Cell Lines
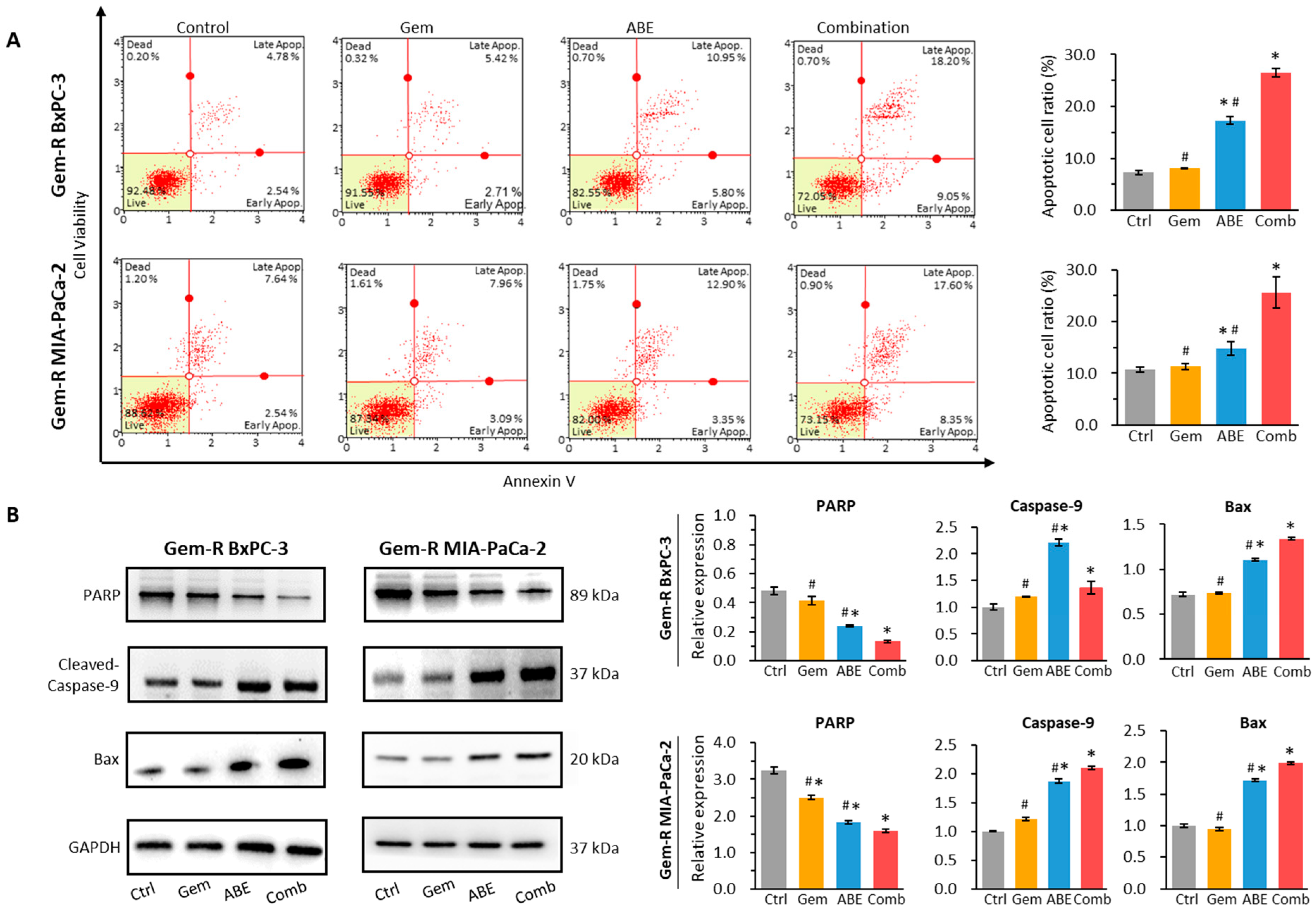
2.3. ABE, in Combination with Gemcitabine, Promotes Cell Apoptosis
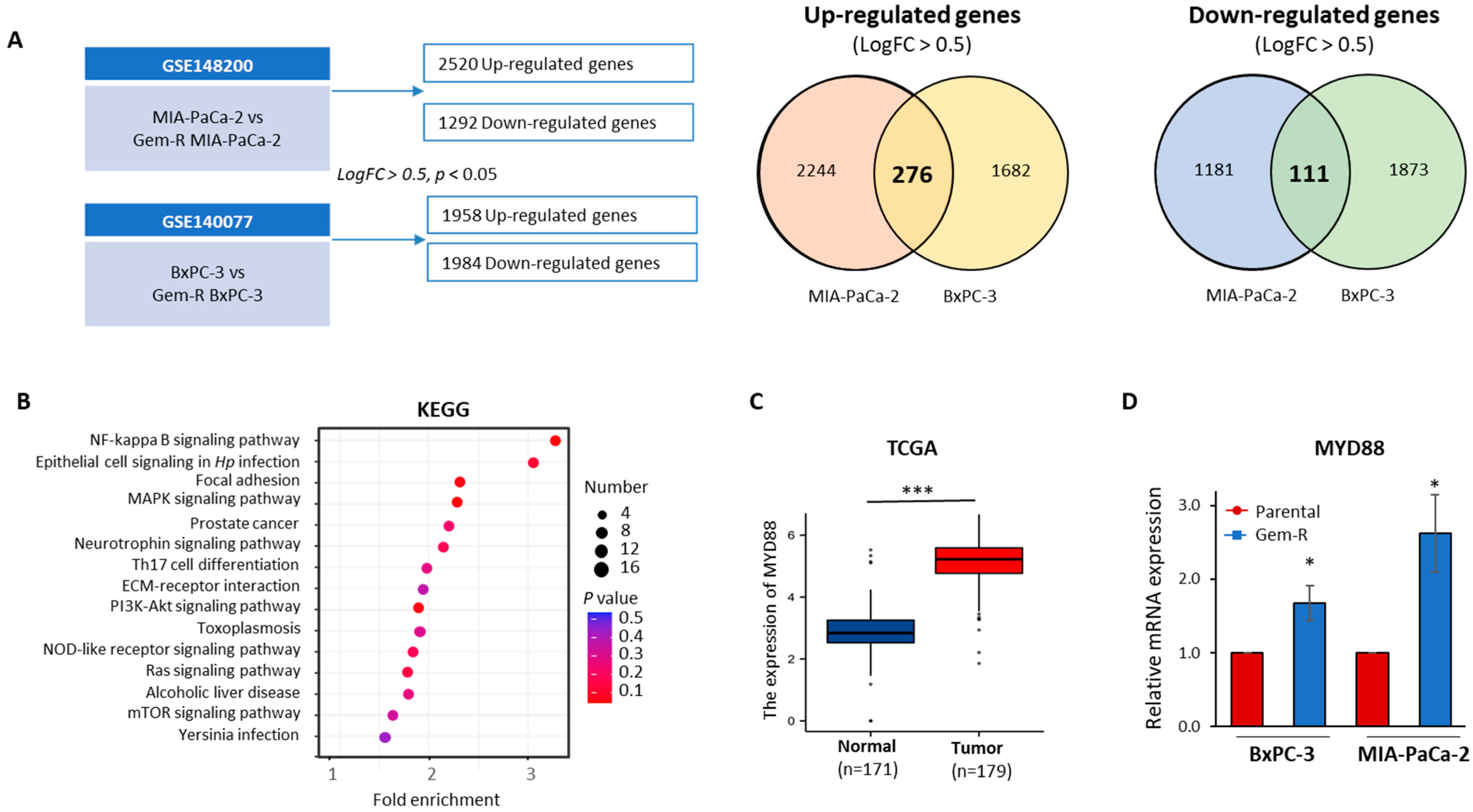
2.4. The MYD88/NF-κB Signaling Pathway Is Aviated in Gem-R PDAC Cells
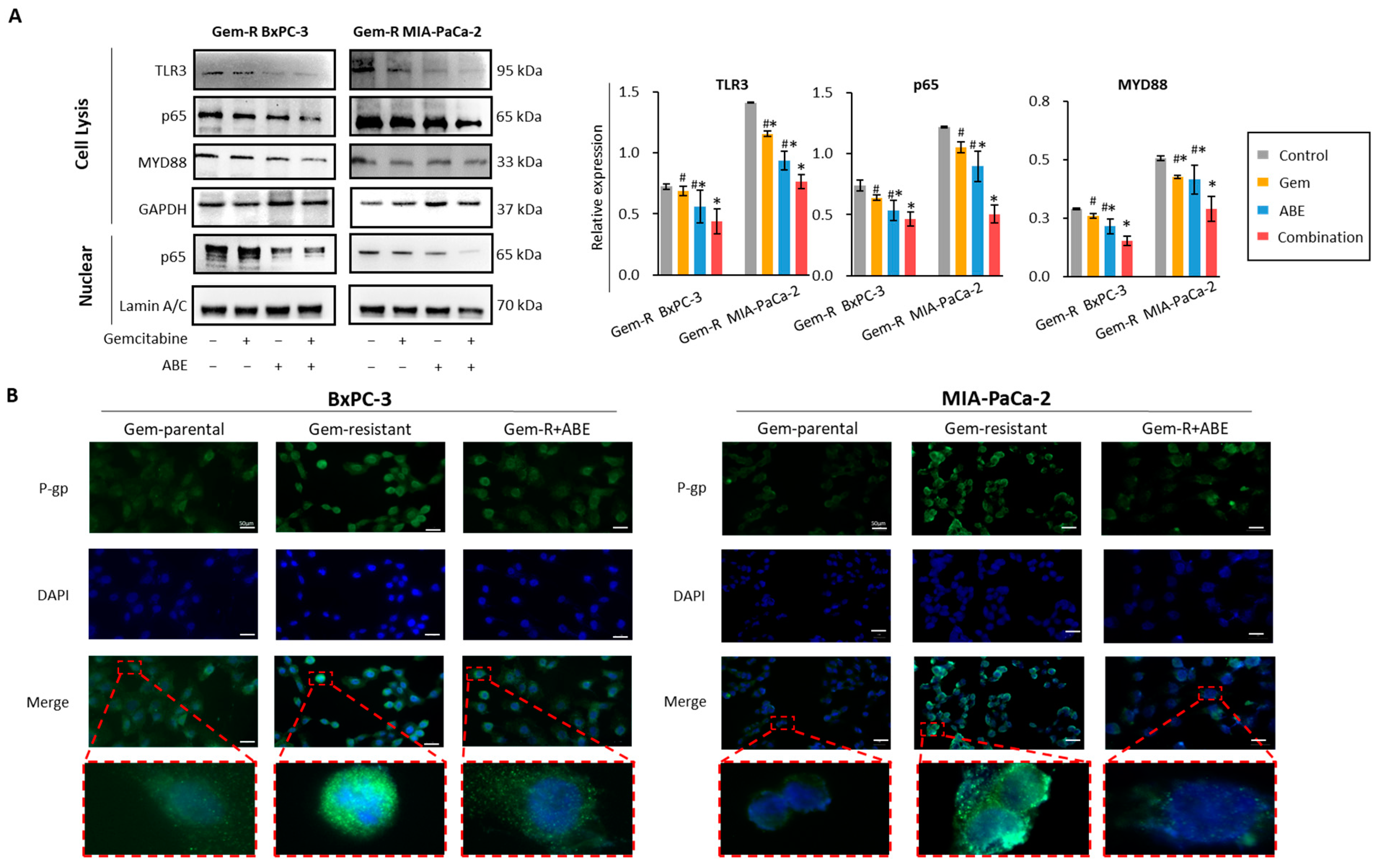
2.5. Combined Treatment with Gemcitabine and ABE Downregulates P-gp through the MYD88/TLR3/NF-κB Signaling Pathway
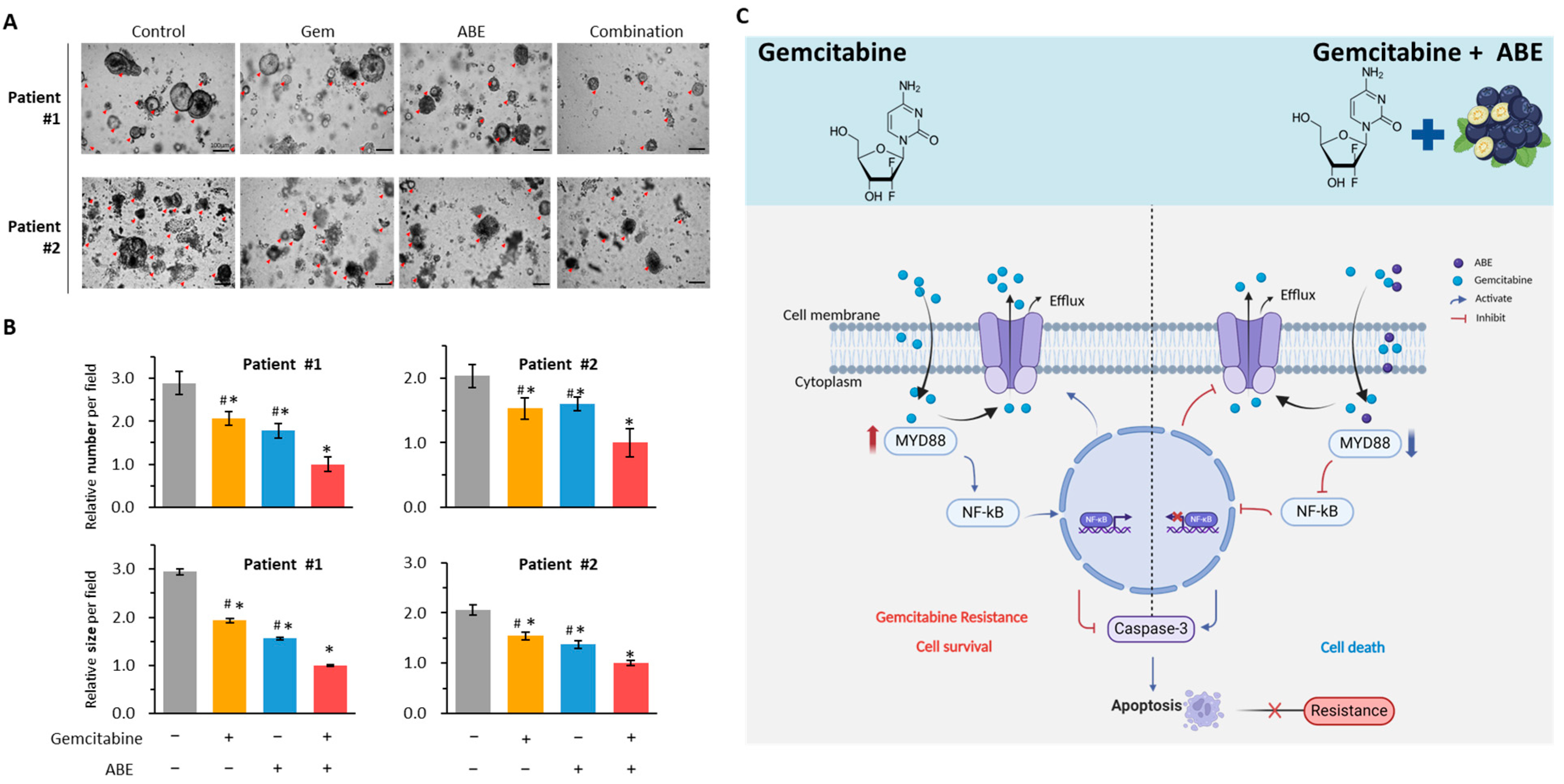
2.6. The Combination of Gemcitabine and ABE Suppressed the Growth of PDOs
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Herbal Preparations
4.3. Reagents
4.4. Cell Counting Kit-8 Assays
4.5. Drug Response Testing
4.6. Colony Formation Assay
4.7. Wound Healing Assay
4.8. Invasion Assays
4.9. Apoptosis Assay
4.10. Gene Enrichment and Pathway Analysis
4.11. Isolation of Cytosolic and Nuclear Extracts
4.12. Protein Isolation and Western Blot
4.13. Quantitative Reverse Transcription PCR (qRT-PCR)
4.14. Immunofluorescence Assay
4.15. Patient-Derived 3-Dimensional Tumor Organoids (PDOs)
4.16. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McGuigan, A.; Kelly, P.; Turkington, R.C.; Jones, C.; Coleman, H.G.; McCain, R.S. Pancreatic cancer: A review of clinical diagnosis, epidemiology, treatment and outcomes. World J. Gastroenterol. 2018, 24, 4846–4861. [Google Scholar] [CrossRef] [PubMed]
- Wood, L.D.; Canto, M.I.; Jaffee, E.M.; Simeone, D.M. Pancreatic Cancer: Pathogenesis, Screening, Diagnosis, and Treatment. Gastroenterology 2022, 163, 386–402.e381. [Google Scholar] [CrossRef] [PubMed]
- Adamska, A.; Domenichini, A.; Falasca, M. Pancreatic Ductal Adenocarcinoma: Current and Evolving Therapies. Int. J. Mol. Sci. 2017, 18, 1338. [Google Scholar] [CrossRef] [PubMed]
- Gourgou-Bourgade, S.; Bascoul-Mollevi, C.; Desseigne, F.; Ychou, M.; Bouche, O.; Guimbaud, R.; Becouarn, Y.; Adenis, A.; Raoul, J.L.; Boige, V.; et al. Impact of FOLFIRINOX compared with gemcitabine on quality of life in patients with metastatic pancreatic cancer: Results from the PRODIGE 4/ACCORD 11 randomized trial. J. Clin. Oncol. 2013, 31, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.J.; Goldstein, D.; Hamm, J.; Figer, A.; Hecht, J.R.; Gallinger, S.; Au, H.J.; Murawa, P.; Walde, D.; Wolff, R.A.; et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: A phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J. Clin. Oncol. 2007, 25, 1960–1966. [Google Scholar] [CrossRef] [PubMed]
- Sarvepalli, D.; Rashid, M.U.; Rahman, A.U.; Ullah, W.; Hussain, I.; Hasan, B.; Jehanzeb, S.; Khan, A.K.; Jain, A.G.; Khetpal, N.; et al. Gemcitabine: A Review of Chemoresistance in Pancreatic Cancer. Crit. Rev. Oncog. 2019, 24, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zheng, Y.; Yang, F.; Zhu, L.; Zhu, X.Q.; Wang, Z.F.; Wu, X.L.; Zhou, C.H.; Yan, J.Y.; Hu, B.Y.; et al. The molecular biology of pancreatic adenocarcinoma: Translational challenges and clinical perspectives. Signal Transduct. Target. Ther. 2021, 6, 249. [Google Scholar] [CrossRef] [PubMed]
- Walker, E.J.; Ko, A.H. Beyond first-line chemotherapy for advanced pancreatic cancer: An expanding array of therapeutic options? World J. Gastroenterol. 2014, 20, 2224–2236. [Google Scholar] [CrossRef]
- Wang-Gillam, A.; Hubner, R.A.; Siveke, J.T.; Von Hoff, D.D.; Belanger, B.; de Jong, F.A.; Mirakhur, B.; Chen, L.T. NAPOLI-1 phase 3 study of liposomal irinotecan in metastatic pancreatic cancer: Final overall survival analysis and characteristics of long-term survivors. Eur. J. Cancer 2019, 108, 78–87. [Google Scholar] [CrossRef]
- Gu, J.; Huang, W.; Wang, X.; Zhang, J.; Tao, T.; Zheng, Y.; Liu, S.; Yang, J.; Chen, Z.S.; Cai, C.Y.; et al. Hsa-miR-3178/RhoB/PI3K/Akt, a novel signaling pathway regulates ABC transporters to reverse gemcitabine resistance in pancreatic cancer. Mol. Cancer 2022, 21, 112. [Google Scholar] [CrossRef]
- Liu, J.; Hu, G.; Gong, Y.; Yu, Q.; He, B.; Li, W.; He, Z.; Hao, W.; He, Z.; Liu, Y. Silencing of TRPM8 inhibits aggressive tumor phenotypes and enhances gemcitabine sensitivity in pancreatic cancer. Pancreatology 2018, 18, 935–944. [Google Scholar] [CrossRef]
- Kohan, H.G.; Boroujerdi, M. Time and concentration dependency of P-gp, MRP1 and MRP5 induction in response to gemcitabine uptake in Capan-2 pancreatic cancer cells. Xenobiotica 2015, 45, 642–652. [Google Scholar] [CrossRef]
- Robey, R.W.; Pluchino, K.M.; Hall, M.D.; Fojo, A.T.; Bates, S.E.; Gottesman, M.M. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat. Rev. Cancer 2018, 18, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Elmeliegy, M.; Vourvahis, M.; Guo, C.; Wang, D.D. Effect of P-glycoprotein (P-gp) Inducers on Exposure of P-gp Substrates: Review of Clinical Drug-Drug Interaction Studies. Clin. Pharmacokinet. 2020, 59, 699–714. [Google Scholar] [CrossRef] [PubMed]
- Joshi, P.; Vishwakarma, R.A.; Bharate, S.B. Natural alkaloids as P-gp inhibitors for multidrug resistance reversal in cancer. Eur. J. Med. Chem. 2017, 138, 273–292. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Li, M.; Lin, Y.; Zhan, X. Encapsulation of verapamil and doxorubicin by MPEG-PLA to reverse drug resistance in ovarian cancer. Biomed. Pharmacother. 2018, 108, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Goel, A.; Boland, C.R.; Chauhan, D.P. Specific inhibition of cyclooxygenase-2 (COX-2) expression by dietary curcumin in HT-29 human colon cancer cells. Cancer Lett. 2001, 172, 111–118. [Google Scholar] [CrossRef]
- Goel, A.; Jhurani, S.; Aggarwal, B.B. Multi-targeted therapy by curcumin: How spicy is it? Mol. Nutr. Food Res. 2008, 52, 1010–1030. [Google Scholar] [CrossRef] [PubMed]
- Goel, A.; Kunnumakkara, A.B.; Aggarwal, B.B. Curcumin as “Curecumin”: From kitchen to clinic. Biochem. Pharmacol. 2008, 75, 787–809. [Google Scholar] [CrossRef]
- Goel, A.; Aggarwal, B.B. Curcumin, the golden spice from Indian saffron, is a chemosensitizer and radiosensitizer for tumors and chemoprotector and radioprotector for normal organs. Nutr. Cancer 2010, 62, 919–930. [Google Scholar] [CrossRef] [PubMed]
- Link, A.; Balaguer, F.; Goel, A. Cancer chemoprevention by dietary polyphenols: Promising role for epigenetics. Biochem. Pharmacol. 2010, 80, 1771–1792. [Google Scholar] [CrossRef]
- Toden, S.; Goel, A. The Holy Grail of Curcumin and its Efficacy in Various Diseases: Is Bioavailability Truly a Big Concern? J. Restor. Med. 2017, 6, 27–36. [Google Scholar] [CrossRef]
- Toden, S.; Theiss, A.L.; Wang, X.; Goel, A. Essential turmeric oils enhance anti-inflammatory efficacy of curcumin in dextran sulfate sodium-induced colitis. Sci. Rep. 2017, 7, 814. [Google Scholar] [CrossRef]
- Weng, W.; Goel, A. Curcumin and colorectal cancer: An update and current perspective on this natural medicine. Semin. Cancer Biol. 2022, 80, 73–86. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.; Ali, S.; Ashraf, G.M.; Bilgrami, A.L.; Yadav, D.K.; Hassan, M.I. Epigallocatechin 3-gallate: From green tea to cancer therapeutics. Food Chem. 2022, 379, 132135. [Google Scholar] [CrossRef]
- Efferth, T.; Oesch, F. Anti-inflammatory and anti-cancer activities of frankincense: Targets, treatments and toxicities. Semin. Cancer Biol. 2022, 80, 39–57. [Google Scholar] [CrossRef]
- Naeem, M.; Iqbal, M.O.; Khan, H.; Ahmed, M.M.; Farooq, M.; Aadil, M.M.; Jamaludin, M.I.; Hazafa, A.; Tsai, W.C. A Review of Twenty Years of Research on the Regulation of Signaling Pathways by Natural Products in Breast Cancer. Molecules 2022, 27, 3412. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Feng, F.; Wu, J.; Fan, S.; Han, J.; Wang, S.; Yang, L.; Liu, W.; Wang, C.; Xu, K. Demethylzeylasteral targets lactate by inhibiting histone lactylation to suppress the tumorigenicity of liver cancer stem cells. Pharmacol. Res. 2022, 181, 106270. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, C.; Goel, A. A combined treatment with melatonin and andrographis promotes autophagy and anticancer activity in colorectal cancer. Carcinogenesis 2022, 43, 217–230. [Google Scholar] [CrossRef]
- Reuter, S.; Gupta, S.C.; Park, B.; Goel, A.; Aggarwal, B.B. Epigenetic changes induced by curcumin and other natural compounds. Genes Nutr. 2011, 6, 93–108. [Google Scholar] [CrossRef]
- Chandran, B.; Goel, A. A randomized, pilot study to assess the efficacy and safety of curcumin in patients with active rheumatoid arthritis. Phytother. Res. 2012, 26, 1719–1725. [Google Scholar] [CrossRef]
- Shakibaei, M.; Mobasheri, A.; Lueders, C.; Busch, F.; Shayan, P.; Goel, A. Curcumin enhances the effect of chemotherapy against colorectal cancer cells by inhibition of NF-κB and Src protein kinase signaling pathways. PLoS ONE 2013, 8, e57218. [Google Scholar] [CrossRef]
- Toden, S.; Okugawa, Y.; Buhrmann, C.; Nattamai, D.; Anguiano, E.; Baldwin, N.; Shakibaei, M.; Boland, C.R.; Goel, A. Novel Evidence for Curcumin and Boswellic Acid-Induced Chemoprevention through Regulation of miR-34a and miR-27a in Colorectal Cancer. Cancer Prev. Res. 2015, 8, 431–443. [Google Scholar] [CrossRef]
- Yoshida, K.; Toden, S.; Ravindranathan, P.; Han, H.; Goel, A. Curcumin sensitizes pancreatic cancer cells to gemcitabine by attenuating PRC2 subunit EZH2, and the lncRNA PVT1 expression. Carcinogenesis 2017, 38, 1036–1046. [Google Scholar] [CrossRef]
- Miyazaki, K.; Xu, C.; Shimada, M.; Goel, A. Curcumin and Andrographis Exhibit Anti-Tumor Effects in Colorectal Cancer via Activation of Ferroptosis and Dual Suppression of Glutathione Peroxidase-4 and Ferroptosis Suppressor Protein-1. Pharmaceuticals 2023, 16, 383. [Google Scholar] [CrossRef]
- Miyazaki, K.; Morine, Y.; Xu, C.; Nakasu, C.; Wada, Y.; Teraoku, H.; Yamada, S.; Saito, Y.; Ikemoto, T.; Shimada, M.; et al. Curcumin-Mediated Resistance to Lenvatinib via EGFR Signaling Pathway in Hepatocellular Carcinoma. Cells 2023, 12, 612. [Google Scholar] [CrossRef]
- Sharma, P.; Shimura, T.; Banwait, J.K.; Goel, A. Andrographis-mediated chemosensitization through activation of ferroptosis and suppression of β-catenin/Wnt-signaling pathways in colorectal cancer. Carcinogenesis 2020, 41, 1385–1394. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wang, C.; Goel, A. Andrographis overcomes 5-fluorouracil-associated chemoresistance through inhibition of DKK1 in colorectal cancer. Carcinogenesis 2021, 42, 814–825. [Google Scholar] [CrossRef]
- Okuno, K.; Xu, C.; Pascual-Sabater, S.; Tokunaga, M.; Takayama, T.; Han, H.; Fillat, C.; Kinugasa, Y.; Goel, A. Andrographis Reverses Gemcitabine Resistance through Regulation of ERBB3 and Calcium Signaling Pathway in Pancreatic Ductal Adenocarcinoma. Biomedicines 2023, 11, 119. [Google Scholar] [CrossRef]
- Buhrmann, C.; Shayan, P.; Kraehe, P.; Popper, B.; Goel, A.; Shakibaei, M. Resveratrol induces chemosensitization to 5-fluorouracil through up-regulation of intercellular junctions, Epithelial-to-mesenchymal transition and apoptosis in colorectal cancer. Biochem. Pharmacol. 2015, 98, 51–68. [Google Scholar] [CrossRef] [PubMed]
- Buhrmann, C.; Yazdi, M.; Popper, B.; Shayan, P.; Goel, A.; Aggarwal, B.B.; Shakibaei, M. Evidence that TNF-β induces proliferation in colorectal cancer cells and resveratrol can down-modulate it. Exp. Biol. Med. 2019, 244, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Okuno, K.; Pratama, M.Y.; Li, J.; Tokunaga, M.; Wang, X.; Kinugasa, Y.; Goel, A. Ginseng mediates its anticancer activity by inhibiting the expression of DNMTs and reactivating methylation-silenced genes in colorectal cancer. Carcinogenesis 2023, 44, 394–403. [Google Scholar] [CrossRef] [PubMed]
- Dewanjee, S.; Dua, T.K.; Bhattacharjee, N.; Das, A.; Gangopadhyay, M.; Khanra, R.; Joardar, S.; Riaz, M.; Feo, V.; Zia-Ul-Haq, M. Natural Products as Alternative Choices for P-Glycoprotein (P-gp) Inhibition. Molecules 2017, 22, 871. [Google Scholar] [CrossRef]
- Ren, Y.; Frank, T.; Meyer, G.; Lei, J.; Grebenc, J.R.; Slaughter, R.; Gao, Y.G.; Kinghorn, A.D. Potential Benefits of Black Chokeberry (Aronia melanocarpa) Fruits and Their Constituents in Improving Human Health. Molecules 2022, 27, 7823. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.S.; Kim, J.H.; Kim, S.L.; Deng, H.Y.; Lee, D.; Kim, C.S.; Yun, B.S.; Lee, D.S. Catechol derived from aronia juice through lactic acid bacteria fermentation inhibits breast cancer stem cell formation via modulation Stat3/IL-6 signaling pathway. Mol. Carcinog. 2018, 57, 1467–1479. [Google Scholar] [CrossRef] [PubMed]
- Thani, N.A.; Keshavarz, S.; Lwaleed, B.A.; Cooper, A.J.; Rooprai, H.K. Cytotoxicity of gemcitabine enhanced by polyphenolics from Aronia melanocarpa in pancreatic cancer cell line AsPC-1. J. Clin. Pathol. 2014, 67, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Gourley, C.; Balmana, J.; Ledermann, J.A.; Serra, V.; Dent, R.; Loibl, S.; Pujade-Lauraine, E.; Boulton, S.J. Moving From Poly (ADP-Ribose) Polymerase Inhibition to Targeting DNA Repair and DNA Damage Response in Cancer Therapy. J. Clin. Oncol. 2019, 37, 2257–2269. [Google Scholar] [CrossRef] [PubMed]
- Jenner, A.; Pena-Blanco, A.; Salvador-Gallego, R.; Ugarte-Uribe, B.; Zollo, C.; Ganief, T.; Bierlmeier, J.; Mund, M.; Lee, J.E.; Ries, J.; et al. DRP1 interacts directly with BAX to induce its activation and apoptosis. EMBO J. 2022, 41, e108587. [Google Scholar] [CrossRef] [PubMed]
- Rose, M.; Burgess, J.T.; O’Byrne, K.; Richard, D.J.; Bolderson, E. PARP Inhibitors: Clinical Relevance, Mechanisms of Action and Tumor Resistance. Front. Cell Dev. Biol. 2020, 8, 564601. [Google Scholar] [CrossRef]
- Spitz, A.Z.; Gavathiotis, E. Physiological and pharmacological modulation of BAX. Trends Pharmacol. Sci. 2022, 43, 206–220. [Google Scholar] [CrossRef]
- Wei, S.; Peng, L.; Yang, J.; Sang, H.; Jin, D.; Li, X.; Chen, M.; Zhang, W.; Dang, Y.; Zhang, G. Exosomal transfer of miR-15b-3p enhances tumorigenesis and malignant transformation through the DYNLT1/Caspase-3/Caspase-9 signaling pathway in gastric cancer. J. Exp. Clin. Cancer Res. 2020, 39, 32. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Sherman, B.T.; Hao, M.; Qiu, J.; Jiao, X.; Baseler, M.W.; Lane, H.C.; Imamichi, T.; Chang, W. DAVID: A web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 2022, 50, W216–W221. [Google Scholar] [CrossRef]
- Chaudhary, P.; Guragain, D.; Chang, J.H.; Kim, J.A. TPH1 and 5-HT7 Receptor Overexpression Leading to Gemcitabine-Resistance Requires Non-Canonical Permissive Action of EZH2 in Pancreatic Ductal Adenocarcinoma. Cancers 2021, 13, 5305. [Google Scholar] [CrossRef]
- Lou, C.; Lu, H.; Ma, Z.; Liu, C.; Zhang, Y. Ginkgolide B enhances gemcitabine sensitivity in pancreatic cancer cell lines via inhibiting PAFR/NF-кB pathway. Biomed. Pharmacother. 2019, 109, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Meng, Q.; Liang, C.; Hua, J.; Zhang, B.; Liu, J.; Zhang, Y.; Wei, M.; Yu, X.; Xu, J.; Shi, S. A miR-146a-5p/TRAF6/NF-kB p65 axis regulates pancreatic cancer chemoresistance: Functional validation and clinical significance. Theranostics 2020, 10, 3967–3979. [Google Scholar] [CrossRef]
- Yao, N.; Chen, Q.; Shi, W.; Tang, L.; Fu, Y. PARP14 promotes the proliferation and gemcitabine chemoresistance of pancreatic cancer cells through activation of NF-κB pathway. Mol. Carcinog. 2019, 58, 1291–1302. [Google Scholar] [CrossRef]
- Bates, M.; Spillane, C.D.; Gallagher, M.F.; McCann, A.; Martin, C.; Blackshields, G.; Keegan, H.; Gubbins, L.; Brooks, R.; Brooks, D.; et al. The role of the MAD2-TLR4-MyD88 axis in paclitaxel resistance in ovarian cancer. PLoS ONE 2020, 15, e0243715. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Luo, J.; Wu, C.; Lu, H.; Cai, S.; Bao, C.; Liu, D.; Kong, J. The miRNA-149-5p/MyD88 axis is responsible for ursolic acid-mediated attenuation of the stemness and chemoresistance of non-small cell lung cancer cells. Environ. Toxicol. 2020, 35, 561–569. [Google Scholar] [CrossRef]
- Chen, X.; Zhao, F.; Zhang, H.; Zhu, Y.; Wu, K.; Tan, G. Significance of TLR4/MyD88 expression in breast cancer. Int. J. Clin. Exp. Pathol. 2015, 8, 7034–7039. [Google Scholar]
- Zhu, X.; Burfeind, K.G.; Michaelis, K.A.; Braun, T.P.; Olson, B.; Pelz, K.R.; Morgan, T.K.; Marks, D.L. MyD88 signalling is critical in the development of pancreatic cancer cachexia. J. Cachexia Sarcopenia Muscle 2019, 10, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, J.M.; Minogue, E.; Curham, L.; Tyrrell, H.; Gavigan, P.; Hind, W.; Downer, E.J. MyD88-dependent and -independent signalling via TLR3 and TLR4 are differentially modulated by Δ9-tetrahydrocannabinol and cannabidiol in human macrophages. J. Neuroimmunol. 2020, 343, 577217. [Google Scholar] [CrossRef]
- Haddad, J.J.; Abdel-Karim, N.E. NF-κB cellular and molecular regulatory mechanisms and pathways: Therapeutic pattern or pseudoregulation? Cell Immunol. 2011, 271, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Scagliotti, G.V.; Parikh, P.; von Pawel, J.; Biesma, B.; Vansteenkiste, J.; Manegold, C.; Serwatowski, P.; Gatzemeier, U.; Digumarti, R.; Zukin, M.; et al. Phase III Study Comparing Cisplatin Plus Gemcitabine with Cisplatin Plus Pemetrexed in Chemotherapy-Naive Patients with Advanced-Stage Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2023, 41, 2458–2466. [Google Scholar] [CrossRef] [PubMed]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Geng, Y.; Wang, L.; Xu, H.; Zou, M.; Li, Y.; Zhao, Z.; Chen, T.; Xu, F.; Sun, L.; et al. Systematic exploration of the underlying mechanism of gemcitabine resistance in pancreatic adenocarcinoma. Mol. Oncol. 2022, 16, 3034–3051. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.H.; Yu, A.M. ABC transporters in multidrug resistance and pharmacokinetics, and strategies for drug development. Curr. Pharm. Des. 2014, 20, 793–807. [Google Scholar] [CrossRef] [PubMed]
- Hashem, S.; Ali, T.A.; Akhtar, S.; Nisar, S.; Sageena, G.; Ali, S.; Al-Mannai, S.; Therachiyil, L.; Mir, R.; Elfaki, I.; et al. Targeting cancer signaling pathways by natural products: Exploring promising anti-cancer agents. Biomed. Pharmacother. 2022, 150, 113054. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef]
- Lingel, A.; Ehlers, E.; Wang, Q.; Cao, M.; Wood, C.; Lin, R.; Zhang, L. Kaposi’s Sarcoma-Associated Herpesvirus Reduces Cellular Myeloid Differentiation Primary-Response Gene 88 (MyD88) Expression via Modulation of Its RNA. J. Virol. 2016, 90, 180–188. [Google Scholar] [CrossRef]
- Yuan, Q.; Gu, J.; Zhang, J.; Liu, S.; Wang, Q.; Tian, T.; Chen, Z.; Zhang, J. MyD88 in myofibroblasts enhances colitis-associated tumorigenesis via promoting macrophage M2 polarization. Cell Rep. 2021, 34, 108724. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Q.; Zhang, J.; Liu, Y.; Chen, H.; Liu, H.; Wang, J.; Niu, M.; Hou, L.; Wu, Z.; Chen, Z.; et al. MyD88 in myofibroblasts regulates aerobic glycolysis-driven hepatocarcinogenesis via ERK-dependent PKM2 nuclear relocalization and activation. J. Pathol. 2022, 256, 414–426. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Cheng, Z.; Huang, Y.; Zheng, W.; Yang, S.; Lin, C.; Ye, J. MyD88 mediates colorectal cancer cell proliferation, migration and invasion via NF-κB/AP-1 signaling pathway. Int. J. Mol. Med. 2020, 45, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.; Kim, J.S.; Kim, W.K.; Oh, K.J.; Kim, J.M.; Lee, H.J.; Han, B.S.; Kim, D.S.; Seo, Y.S.; Lee, S.C.; et al. Intracellular annexin A2 regulates NF-κB signaling by binding to the p50 subunit: Implications for gemcitabine resistance in pancreatic cancer. Cell Death Dis. 2015, 6, e1606. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Chen, S.; Guo, Y.; Sun, C. Oncogenic TRIM31 confers gemcitabine resistance in pancreatic cancer via activating the NF-κB signaling pathway. Theranostics 2018, 8, 3224–3236. [Google Scholar] [CrossRef]
- Ianevski, A.; Giri, A.K.; Aittokallio, T. SynergyFinder 3.0: An interactive analysis and consensus interpretation of multi-drug synergies across multiple samples. Nucleic Acids Res. 2022, 50, W739–W743. [Google Scholar] [CrossRef]
- Raimondi, G.; Mato-Berciano, A.; Pascual-Sabater, S.; Rovira-Rigau, M.; Cuatrecasas, M.; Fondevila, C.; Sanchez-Cabus, S.; Begthel, H.; Boj, S.F.; Clevers, H.; et al. Patient-derived pancreatic tumour organoids identify therapeutic responses to oncolytic adenoviruses. EBioMedicine 2020, 56, 102786. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Xu, C.; Han, H.; Pascual-Sabater, S.; Fillat, C.; Goel, A. Aronia Berry Extract Modulates MYD88/NF-kB/P-Glycoprotein Axis to Overcome Gemcitabine Resistance in Pancreatic Cancer. Pharmaceuticals 2024, 17, 911. https://doi.org/10.3390/ph17070911
Li Y, Xu C, Han H, Pascual-Sabater S, Fillat C, Goel A. Aronia Berry Extract Modulates MYD88/NF-kB/P-Glycoprotein Axis to Overcome Gemcitabine Resistance in Pancreatic Cancer. Pharmaceuticals. 2024; 17(7):911. https://doi.org/10.3390/ph17070911
Chicago/Turabian StyleLi, Yuan, Caiming Xu, Haiyong Han, Silvia Pascual-Sabater, Cristina Fillat, and Ajay Goel. 2024. "Aronia Berry Extract Modulates MYD88/NF-kB/P-Glycoprotein Axis to Overcome Gemcitabine Resistance in Pancreatic Cancer" Pharmaceuticals 17, no. 7: 911. https://doi.org/10.3390/ph17070911
APA StyleLi, Y., Xu, C., Han, H., Pascual-Sabater, S., Fillat, C., & Goel, A. (2024). Aronia Berry Extract Modulates MYD88/NF-kB/P-Glycoprotein Axis to Overcome Gemcitabine Resistance in Pancreatic Cancer. Pharmaceuticals, 17(7), 911. https://doi.org/10.3390/ph17070911