



Evaluating the Physicochemical Properties–Activity Relationship and Discovering New 1,2-Dihydropyridine Derivatives as Promising Inhibitors for PIM1-Kinase: Evidence from Principal Component Analysis, Molecular Docking, and Molecular Dynamics Studies




Abstract
1. Introduction
2. Results and Discussion
2.1. PCA
2.2. Drug Likeness
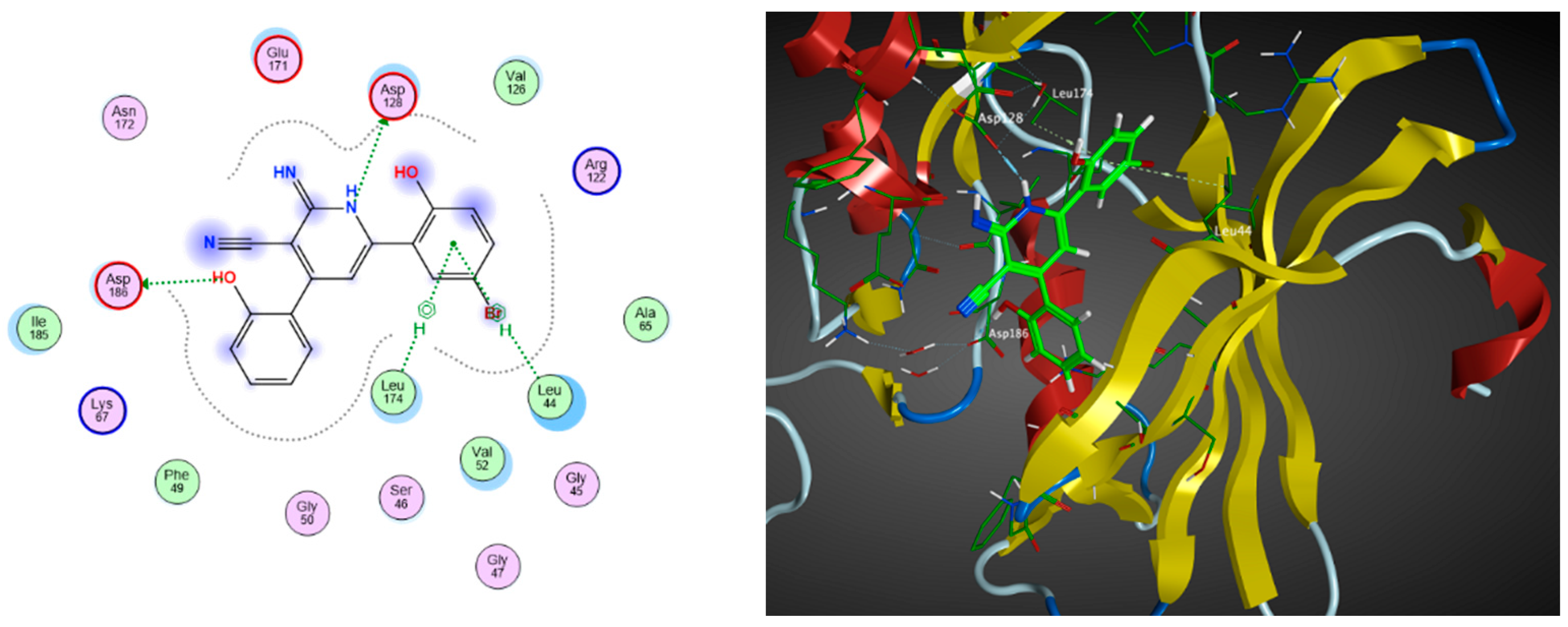
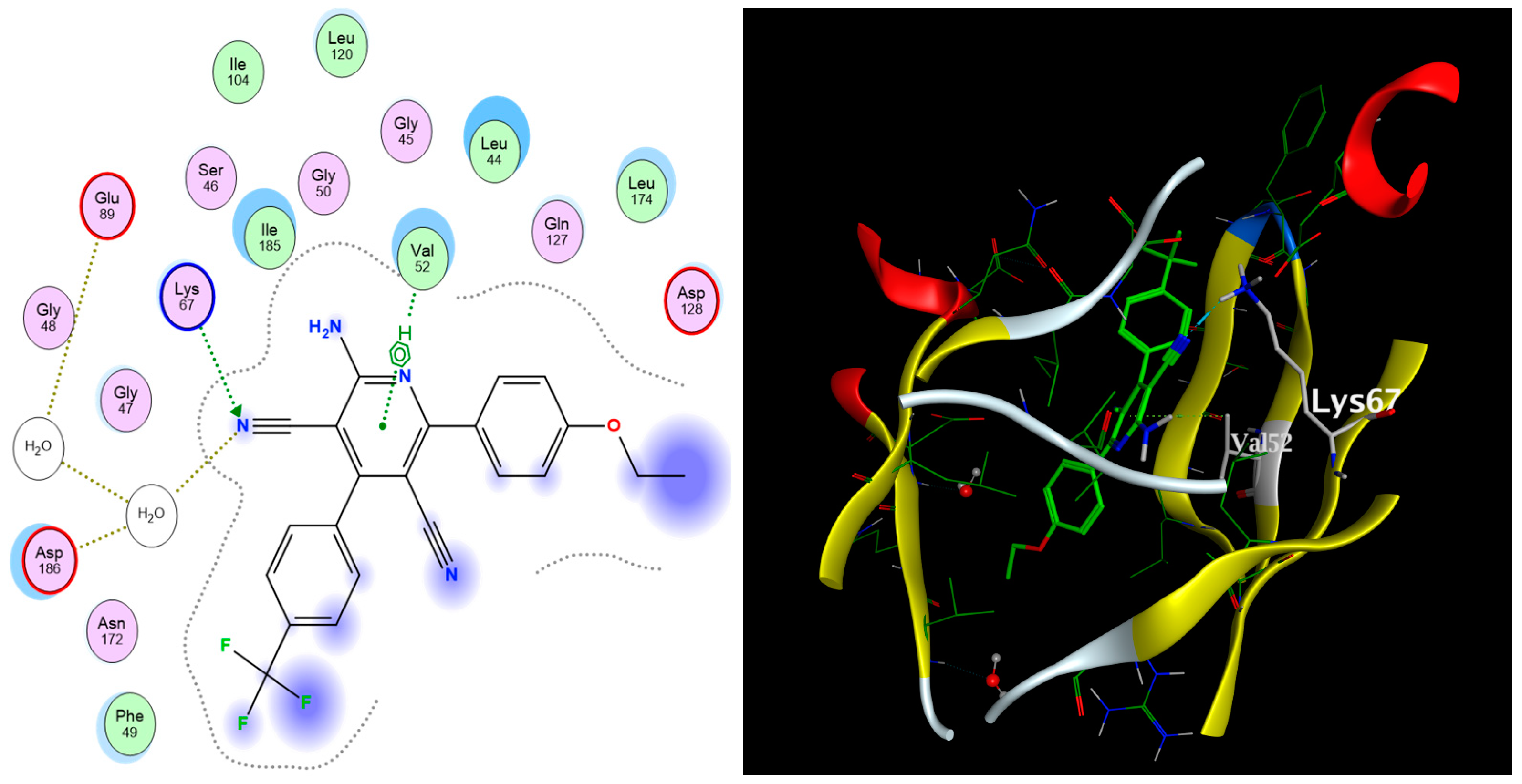
2.3. Molecular Docking
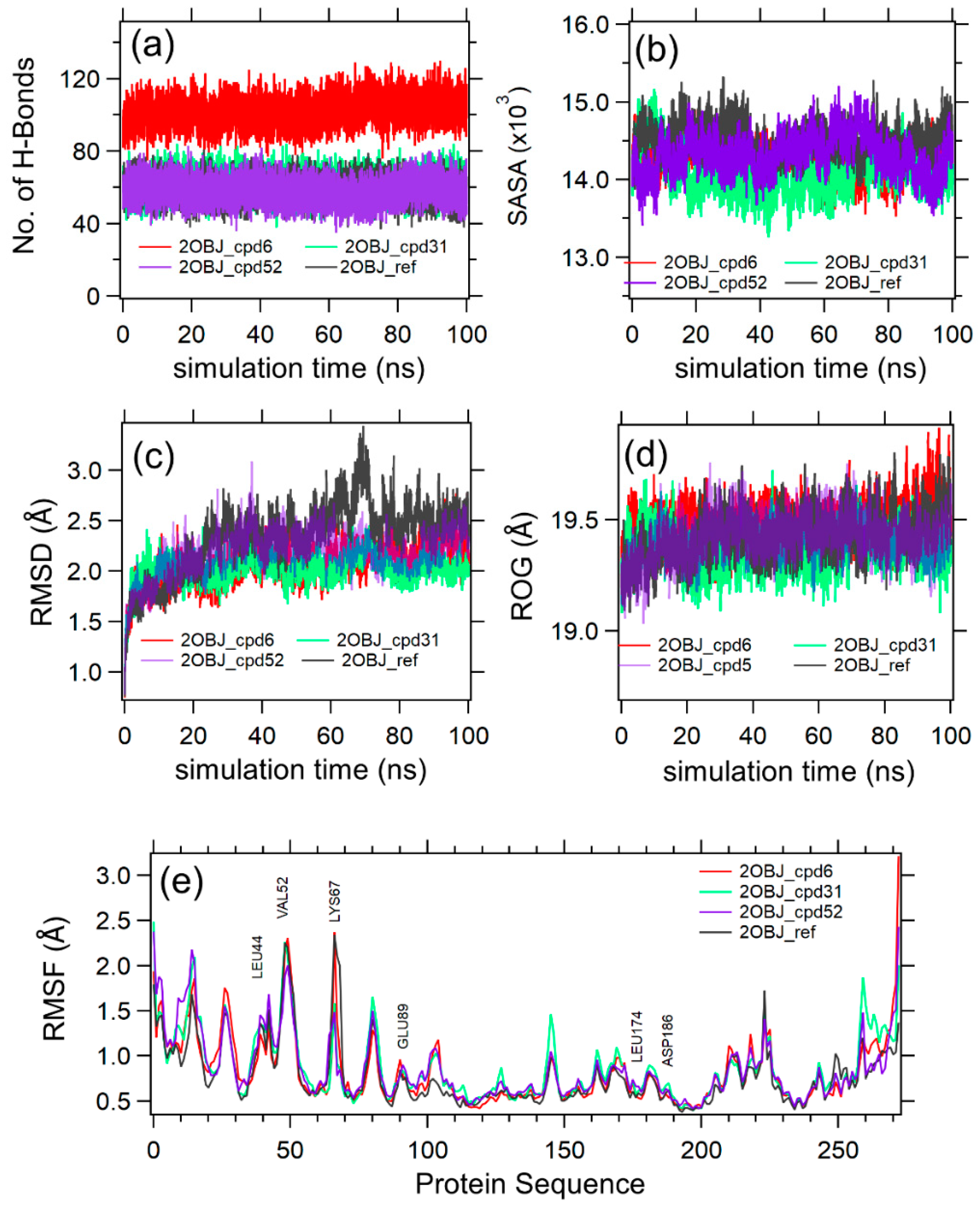
2.4. Molecular Dynamics (MD)
3. Methods
3.1. Computing the Descriptors of the 1,2-Dihydropyridine Derivatives
3.2. Principal Component Analysis (PCA)
3.3. Molecular Docking
3.4. Molecular Dynamics (MD) Simulations
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Drabu, S.; Kumar, N. Synthesis and Biological Screening of Substituted 2-Aminocyano Pyridines. Asian J. Chem. 2007, 19, 4957. [Google Scholar]
- Wang, Y.; Liu, G.; Reyes, J.C.P.; Duverna, R. One-Pot Synthesis of 3-Cyano-2-pyridones. J. Heterocycl. Chem. 2015, 52, 1185–1191. [Google Scholar] [CrossRef]
- Sunderhaus, J.D.; Martin, S.F. Applications of Multicomponent Reactions to the Synthesis of Diverse Heterocyclic Scaffolds. Chem. A Eur. J. 2009, 15, 1300–1308. [Google Scholar] [CrossRef] [PubMed]
- Alaa, A.-M.; El-Subbagh, H.I.; Kunieda, T. Lewis Acid-Promoted Transformation of 2-Alkoxypyridines into 2-Aminopyridines and Their Antibacterial Activity. Part 2: Remarkably Facile C–N Bond Formation. Bioorg. Med. Chem. 2005, 13, 4929–4935. [Google Scholar]
- Bekhit, A.A.; Baraka, A.M. Novel Milrinone Analogs of Pyridine-3-Carbonitrile Derivatives as Promising Cardiotonic Agents. Eur. J. Med. Chem. 2005, 40, 1405–1413. [Google Scholar] [CrossRef]
- Abdel-Latif, N.A. Synthesis and Antidepressant Activity of Some New Coumarin Derivatives. Sci. Pharm. 2005, 73, 193–216. [Google Scholar] [CrossRef]
- Murata, T.; Shimizu, K.; Narita, M.; Manganiello, V.C.; Tagawa, T. Characterization of Phosphodiesterase 3 in Human Malignant Melanoma Cell Line. Anticancer Res. 2002, 22, 3171–3174. [Google Scholar] [PubMed]
- Murata, T.; Shimada, M.; Sakakibara, S.; Yoshino, T.; Kadono, H.; Masuda, T.; Shimazaki, M.; Shintani, T.; Fuchikami, K.; Sakai, K. Discovery of Novel and Selective IKK-β Serine-Threonine Protein Kinase Inhibitors. Part 1. Bioorg. Med. Chem. Lett. 2003, 13, 913–918. [Google Scholar] [CrossRef]
- Cheney, I.W.; Yan, S.; Appleby, T.; Walker, H.; Vo, T.; Yao, N.; Hamatake, R.; Hong, Z.; Wu, J.Z. Identification and Structure–Activity Relationships of Substituted Pyridones as Inhibitors of Pim-1 Kinase. Bioorg. Med. Chem. Lett. 2007, 17, 1679–1683. [Google Scholar] [CrossRef]
- Wendt, M.D.; Sun, C.; Kunzer, A.; Sauer, D.; Sarris, K.; Hoff, E.; Yu, L.; Nettesheim, D.G.; Chen, J.; Jin, S. Discovery of a Novel Small Molecule Binding Site of Human Survivin. Bioorg. Med. Chem. Lett. 2007, 17, 3122–3129. [Google Scholar] [CrossRef]
- Chen, J.; Tang, G. PIM-1 Kinase: A Potential Biomarker of Triple-Negative Breast Cancer. OncoTargets Ther. 2019, 12, 6267. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, J.; Zupan, J. Neural Networks in Chemistry. Angew. Chem. Int. Ed. Engl. 1993, 32, 503–527. [Google Scholar] [CrossRef]
- Behler, J. First Principles Neural Network Potentials for Reactive Simulations of Large Molecular and Condensed Systems. Angew. Chem. Int. Ed. 2017, 56, 12828–12840. [Google Scholar] [CrossRef] [PubMed]
- Butler, K.T.; Davies, D.W.; Cartwright, H.; Isayev, O.; Walsh, A. Machine Learning for Molecular and Materials Science. Nature 2018, 559, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Deringer, V.L.; Bernstein, N.; Bartók, A.P.; Cliffe, M.J.; Kerber, R.N.; Marbella, L.E.; Grey, C.P.; Elliott, S.R.; Csányi, G. Realistic Atomistic Structure of Amorphous Silicon from Machine-Learning-Driven Molecular Dynamics. J. Phys. Chem. Lett. 2018, 9, 2879–2885. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Lengeling, B.; Aspuru-Guzik, A. Inverse Molecular Design Using Machine Learning: Generative Models for Matter Engineering. Science 2018, 361, 360–365. [Google Scholar] [CrossRef]
- Aspuru-Guzik, A.; Baik, M.-H.; Balasubramanian, S.; Banerjee, R.; Bart, S.; Borduas-Dedekind, N.; Chang, S.; Chen, P.; Corminboeuf, C.; Coudert, F.X. Charting a Course for Chemistry. Nat. Chem. 2019, 11, 286–294. [Google Scholar] [CrossRef]
- Kononova, O.; Huo, H.; He, T.; Rong, Z.; Botari, T.; Sun, W.; Tshitoyan, V.; Ceder, G. Text-Mined Dataset of Inorganic Materials Synthesis Recipes. Sci. Data 2019, 6, 203. [Google Scholar] [CrossRef]
- Joliffe, I.T.; Morgan, B.J.T. Principal Component Analysis and Exploratory Factor Analysis. Stat. Methods Med. Res. 1992, 1, 69–95. [Google Scholar] [CrossRef]
- Younes, K.; Grasset, L. Comparison of Thermochemolysis and Classical Chemical Degradation and Extraction Methods for the Analysis of Carbohydrates, Lignin and Lipids in a Peat Bog. J. Anal. Appl. Pyrolysis 2018, 134, 61–72. [Google Scholar] [CrossRef]
- Younes, K.; Grasset, L. The Application of DFRC Method for the Analysis of Carbohydrates in a Peat Bog: Validation and Comparison with Conventional Chemical and Thermochemical Degradation Techniques. Chem. Geol. 2020, 545, 119644. [Google Scholar] [CrossRef]
- Ibrahimi, M.; Korichi, W.; Loqman, S.; Hafidi, M.; Ouhdouch, Y.; Lemee, L. Thermochemolysis–GC-MS as a Tool for Chemotaxonomy and Predation Monitoring of a Predatory Actinobacteria against a Multidrug Resistant Bacteria. J. Anal. Appl. Pyrolysis 2020, 145, 104740. [Google Scholar] [CrossRef]
- Younes, K.; Grasset, L. Analysis of Molecular Proxies of a Peat Core by Thermally Assisted Hydrolysis and Methylation-Gas Chromatography Combined with Multivariate Analysis. J. Anal. Appl. Pyrolysis 2017, 124, 726–732. [Google Scholar] [CrossRef]
- Younes, K.; Grasset, L. Carbohydrates as Proxies in Ombrotrophic Peatland: DFRC Molecular Method Coupled with PCA. Chem. Geol. 2022, 606, 120994. [Google Scholar] [CrossRef]
- Younes, K.; Laduranty, J.; Descostes, M.; Grasset, L. Molecular Biomarkers Study of an Ombrotrophic Peatland Impacted by an Anthropogenic Clay Deposit. Org. Geochem. 2017, 105, 20–32. [Google Scholar] [CrossRef]
- Korichi, W.; Ibrahimi, M.; Loqman, S.; Ouhdouch, Y.; Younes, K.; Lemée, L. Assessment of Actinobacteria Use in the Elimination of Multidrug-Resistant Bacteria of Ibn Tofail Hospital Wastewater (Marrakesh, Morocco): A Chemometric Data Analysis Approach. Environ. Sci. Pollut. Res. 2021, 28, 26840–26848. [Google Scholar] [CrossRef]
- Abdelf-Fattah, M.A.O.; El-Naggar, M.A.M.; Rashied, R.M.H.; Gary, B.D.; Piazza, G.A.; Abadi, A.H. Four-Component Synthesis of 1,2-Dihydropyridine Derivatives and Their Evaluation as Anticancer Agents. Med. Chem. 2012, 8, 392–400. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev. 2012, 64, 4–17. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Wei, W.; Cherukupalli, S.; Jing, L.; Liu, X.; Zhan, P. Fsp3: A New Parameter for Drug-Likeness. Drug Discov. Today 2020, 25, 1839–1845. [Google Scholar] [CrossRef]
- Sorkun, M.C.; Khetan, A.; Er, S. AqSolDB, a Curated Reference Set of Aqueous Solubility and 2D Descriptors for a Diverse Set of Compounds. Sci. Data 2019, 6, 143. [Google Scholar] [CrossRef]
- Method of Using Aminocyanopyridine Compounds as Mitogen Activated Protein Kinase-Activated Protein Kinase-2 Inhibitors—Patent EP-1569645-A2—PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/patent/EP-1569645-A2 (accessed on 23 June 2024).
- AID 540256—qHTS for Inhibitors of Binding or Entry into Cells for Lassa Virus—PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/bioassay/540256 (accessed on 23 June 2024).
- Use of Aminocyanopyridines as Mitogen-Activated Protein Kinase-Activated Protein Kinase-2 Inhibitors—Patent JP-2006512338-A—PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/patent/JP-2006512338-A (accessed on 23 June 2024).
- Coburn, C.A.; Holloway, M.K.; Stachel, S.J. 2-Aminopyridine Compounds Useful as Beta-Secretase Inhibitors for the Treatment of Alzheimer’s Disease. EP1807396A1, 26 June 2013. [Google Scholar]
- Murata, T.; Umeda, M.; Sakakibara, S.; Yoshiro, T.; Sato, H.; Masuda, T.; Koriyama, Y.; Shimada, M.; Shintani, T.; Kadono, H.; et al. Pyridine Derivatives. EA200700099A1, 31 August 2007. [Google Scholar]
- Boehringer, M.; Loeffler, B.M.; Peters, J.-U.; Riemer, C.; Weiss, P. Pyridine and Quinoline Derivatives. US20030195188A1, 5 October 2004. [Google Scholar]
- AID 1671174—Cornell—Phenotypic Assay to Identify Agents That Inhibit Growth of Mycobacterium Tuberculosis—PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/bioassay/1671174 (accessed on 23 June 2024).
- Albert, J.S.; Callaghan, O.; Campbell, J.; Carr, R.A.E.; Chessari, G.; Cowan, S.; Congreve, M.S.; Edwards, P.; Frederickson, M.; Murray, C.W.; et al. Substituted Aminopyridines and Uses Thereof 2008 US20080287399A1. Available online: https://patents.google.com/patent/US20080287399A1/en?oq=Patent:+US-20080287399-A1 (accessed on 23 June 2024).
- Shao, B.; Victory, S.; Ilyin, V.I.; Goehring, R.R.; Sun, Q.; Hogenkamp, D.; Hodges, D.D.; Islam, K.; Sha, D.; Zhang, C.; et al. Phenoxyphenyl Pyridines as Novel State-Dependent, High-Potency Sodium Channel Inhibitors. J. Med. Chem. 2004, 47, 4277–4285. [Google Scholar] [CrossRef]
- Amberg, W.; Netz, A.; Kling, A.; Ochse, M.; Lange, U.; Hutchins, C.W.; Garcia-Ladona, F.J.; Wernet, W. Heterocyclische Verbindungen und Ihre Verwendung als Bindungspartner Für 5-ht5-Rezeptoren. WO2007022946A1, 1 March 2007. [Google Scholar]
- Lowinger, T.B.; Murata, T.; Umeda, M.; Sakakibara, S.; Yoshino, T.; Sato, H.; Masuda, T.; Koriyama, Y.; Shimada, M.; Shintani, T.; et al. Pyridine Derivatives with Ikb-Kinase (Ikk-Beta) Inhibiting Activity. WO2002024679A1, 19 December 2007. [Google Scholar]
- Schirok, H.; Redlich, G.; Grande, Y.C.; Leineweber, K.; Bender, E.; Tinel, H.; Münter, K.; Kolkhof, P.; Himmel, H.; Straub, A. Substituierte 2-amino-3-Cyanopyridine als Inhibitoren des Natrium Calcium Austausches und Ihre Verwendung bei Kardiovaskulären Erkrankungen. WO2013144191A1, 27 March 2013. [Google Scholar]
- Calderone, V.; Minutolo, F.; Tuccinardi, T.; Testai, L.; Granchi, C.; Martelli, A.; Citi, V.; Gardinal, V.D.L.; Lenzi, G.; Leo, F.; et al. Fnew Activators of Sirt1 Enzyme for the Treatment of Cardiovascular and Cardiometabolic Pathologies 2019. WO2019162911A1. Available online: https://patents.google.com/patent/WO2019162911A1/en?oq=WO-2019162911-A1 (accessed on 23 June 2024).
- Schann, S.; Fer, M.; Mayer, S.; Doebelin, C. 5-Heteroaryl-Pyridin-2-Amine Confounds as Neuropeptide Ff Receptor Antagonists. EP4010328A1, 11 February 2021. [Google Scholar]
- Oluwafisayo Akintemi, E.; Kuben Govender, K.; Singh, T. Molecular Dynamics and Docking Investigation of Flavonol Aglycones against Sulfonylurea Receptor 1 (SUR1) for Anti–Diabetic Drug Design. ChemistrySelect 2024, 9, e202302488. [Google Scholar] [CrossRef]
- Bai, Q.; Tan, S.; Xu, T.; Liu, H.; Huang, J.; Yao, X. MolAICal: A Soft Tool for 3D Drug Design of Protein Targets by Artificial Intelligence and Classical Algorithm. Brief. Bioinform. 2020, 22, bbaa161. [Google Scholar] [CrossRef]
- Bai, Q. Research and Development of MolAICal for Drug Design via Deep Learning and Classical Programming. arXiv 2020, arXiv:2006.09747. [Google Scholar] [CrossRef]
- Kim, M.; Chang, J.W.; Park, K.; Yang, D.R. Comprehensive Assessment of the Effects of Operating Conditions on Membrane Intrinsic Parameters of Forward Osmosis (FO) Based on Principal Component Analysis (PCA). J. Membr. Sci. 2022, 641, 119909. [Google Scholar] [CrossRef]
- Hawkins, P.C.D.; Nicholls, A. Conformer Generation with OMEGA: Learning from the Data Set and the Analysis of Failures. J. Chem. Inf. Model. 2012, 52, 2919–2936. [Google Scholar] [CrossRef]
- Harrach, M.F.; Drossel, B. Structure and Dynamics of TIP3P, TIP4P, and TIP5P water near smooth and atomistic walls of different hydroaffinity. J. Chem. Phys. 2014, 140, 174501. [Google Scholar] [CrossRef]
- Mark, P.; Nilsson, L. Structure and Dynamics of the TIP3P, SPC, and SPC/E Water Models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compound | Fluoro Position | R | X | HT-29 Growth Inhibition IC50 ± SD (μM) | Compound | Fluoro Position | R | X | HT-29 Growth Inhibition IC50 ± SD (μM) |
| 1 | 4-F | 2-methoxyphenyl | O | >50 | 16 | 3-F | 2-ethoxyphenyl | NH | 1.50 ± 0.1 |
| 2 | 4-F | 2-methoxyphenyl | NH | 6.30 ± 0.8 | 17 | 3-F | 2-furanyl | O | >50 |
| 3 | 4-F | 2-hydroxyphenyl | O | 42.5 ± 1 | 18 | 3-F | 2-furanyl | NH | 1.26 ± 0.2 |
| 4 | 4-F | 2-hydroxyphenyl | NH | 12.70 ± 0.7 | 19 | 3-F | 3,4-dichlorophenyl | O | >50 |
| 5 | 4-F | 2-ethoxyphenyl | O | >50 | 20 | 3-F | 3,4-dichlorophenyl | NH | 3.96 ± 0.5 |
| 6 | 4-F | 2-ethoxyphenyl | NH | 0.70 ± 0.1 | 21 | 2-F | 2-hydroxyphenyl | O | >50 |
| 7 | 4-F | 2-furanyl | O | >50 | 22 | 2-F | 2-hydroxyphenyl | NH | >50 |
| 8 | 4-F | 2-furanyl | NH | 3.46 ± 0.2 | 23 | 2-F | 2-methoxyphenyl | O | >50 |
| 9 | 4-F | 3,4-dichlorophenyl | O | >50 | 24 | 2-F | 2-methoxyphenyl | NH | 10.50 ± 0.4 |
| 10 | 4-F | 3,4-dichlorophenyl | NH | 2.18 ± 0.1 | 25 | 2-F | 2-ethoxyphenyl | O | 12.30 ± 1.0 |
| 11 | 3-F | 2-hydroxyphenyl | O | >50 | 26 | 2-F | 2-ethoxyphenyl | NH | 2.50 ± 0.3 |
| 12 | 3-F | 2-hydroxyphenyl | NH | >50 | 27 | 2-F | 2-furanyl | O | >50 |
| 13 | 3-F | 2-methoxyphenyl | O | 12.70 ± 1.2 | 28 | 2-F | 2-furanyl | NH | 8.82 |
| 14 | 3-F | 2-methoxyphenyl | NH | 9.30 ± 0.8 | 29 | 2-F | 3,4-dichlorophenyl | O | >50 |
| 15 | 3-F | 2-ethoxyphenyl | O | 10.20 ± 1.2 | 30 | 2-F | 3,4-dichlorophenyl | NH | 5.74 ± 0.4 |
| Compound | Drug Likeness-Related Descriptors | |||||
|---|---|---|---|---|---|---|
| LogP | Csp3 | b_1rotN | LogS | TPSA | lip_don | |
| 2 | 4.095 | 0.05 | 3 | −5.467 | 68.9 | 2 |
| 6 | 4.436 | 0.1 | 4 | −5.794 | 68.9 | 2 |
| 8 | 2.856 | 0 | 2 | −5.168 | 72.81 | 2 |
| 10 | 5.360 | 0 | 2 | −6.885 | 59.67 | 2 |
| 14 | 4.132 | 0.05 | 3 | −5.467 | 68.9 | 2 |
| 16 | 4.473 | 0.1 | 4 | −5.794 | 68.9 | 2 |
| 18 | 2.893 | 0 | 2 | −5.168 | 72.81 | 2 |
| 20 | 5.397 | 0 | 2 | −6.885 | 59.67 | 2 |
| 24 | 4.093 | 0.05 | 3 | −5.467 | 68.9 | 2 |
| 26 | 4.434 | 0.1 | 4 | −5.794 | 68.9 | 2 |
| 28 | 2.854 | 0 | 2 | −5.168 | 72.81 | 2 |
| 30 | 5.358 | 0 | 2 | −6.885 | 59.67 | 2 |
| Compound | Docking Score (kcal/mol) | Types of Interactions |
|---|---|---|
| Co-crystallized ligand | −12.08 | Val52 (arene–H) Lys67 (H-bond) Ile185 (arene–H) Glu89 (H-bond) Asp186 (H-bond) |
| 6 | −11.77 | Val52 (arene–H) Lys67 (H-bond) |
| 14 | −11.33 | Leu44 (arene–H) Leu174 (arene–H) Glu89 (H-bond) Asp186 (H-bond) |
| 16 | −11.26 | Val52 (arene–H) Lys67 (H-bond) Leu174 (arene–H) |
| 24 | −11.06 | Val52 (H-bond) Lys67 (H-bond) Leu174 (arene–H) |
| 2 | −10.99 | Val52 (arene–H) Lys67 (H-bond) Leu174 (arene–H) |
| 26 | −10.41 | Leu44 (arene–H) Val52 (H-bond) Lys67 (H-bond) Leu174 (arene–H) |
| 18 | −9.83 | Leu44 (arene–H) Val52 (H-bond) Lys67 (H-bond) |
| 28 | −9.44 | Leu44 (arene–H) Val52 (arene–H) Lys67 (H-bond) |
| 8 | −9.27 | Leu44 (arene–H) Val52 (H-bond) Lys67 (H-bond) |
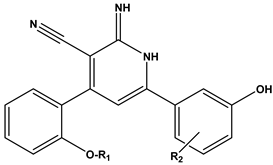 | ||
|---|---|---|
| Compound | R1 | R2 |
| 31 | H | 5-Br |
| 32 | CH3 | 5-Br |
| 33 | CH2CH3 | 5-Br |
| 34 | H | 5-F |
| 35 | CH3 | 5-F |
| 36 | CH2CH3 | 5-F |
| 37 | H | 4-F |
| 38 | CH3 | 4-F |
| 39 | CH2CH3 | 4-F |
| Compound | Docking Score (kcal/mol) | Types of Interactions |
|---|---|---|
| 31 | −13.11 | Leu44 (arene–H) Asp128 (H-bond) Leu174 (H-bond) Asp186 (H-bond) |
| 32 | −11.97 | Val52 (arene–H) Asp128 (H-bond) Lys67 (H-bond) |
| 33 | −12.75 | VAl52 (arene–H) Phe49 (H-bond) |
| 34 | −11.79 | Val52 (arene–H) Lys67 (H-bond) Asp128 (H-bond) Asn172 (H-bond) |
| 35 | −12.43 | Val52 (arene–H) Lys67 (H-bond) Asp128 (H-bond) Leu174 (arene–H) |
| 36 | −11.73 | Leu44 (H-bond) Val52 (arene–H) Lys67 (H-bond) |
| 37 | −12.17 | Val52 (arene–H) Lys67 (H-bond) Asp128 (H-bond) Leu174 (arene–H) |
| 38 | −12.05 | Val52 (arene–H) Lys67 (H-bond) Asp128 (H-bond) Leu174 (arene–H) |
| 39 | −12.34 | Val52 (arene–H) Lys67 (H-bond) Asp128 (H-bond) Leu174 (arene–H) |
| Compound | Structure | Curation Effect | Reference |
|---|---|---|---|
| 40 |  | Mitogen-activated protein Kinase-activated protein Kinase-2 inhibitors. | [32] |
| 41 | 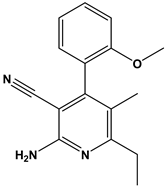 | Inhibitors of Lassa virus’ entry into cells. | [33] |
| 42 | 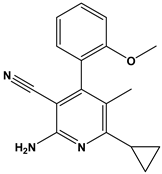 | Mitogen-activated protein Kinase-activated protein Kinase-2 inhibitors. | [34] |
| 43 | 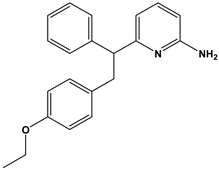 | Beta-secretase inhibitors for the treatment of Alzheimer’s disease. | [35] |
| 44 | 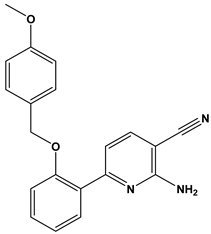 | Treatment of diseases associated with NF-kB activity, in particular for the treatment of inflammatory diseases. | [36] |
| 45 | 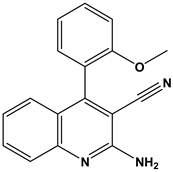 | Treatment and/or prophylaxis of diseases which are associated with DPP IV, such as diabetes, particularly non-insulin-dependent diabetes mellitus, and impaired glucose tolerance. | [37] |
| 46 | 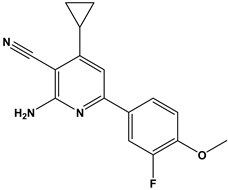 | Inhibitor of nuclear factor kappa-B kinase subunit beta, and inhibitor of Mycobacterium tuberculosis growth. | [38] |
| 47 | 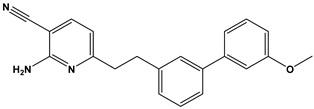 | Treatment or prophylaxis of cognitive impairment, Alzheimer’s disease, neurodegeneration, and dementia. | [39] |
| 48 |  | Sodium channel blockers. | [40] |
| 49 |  | Treating neuro-degenerative and neuropsychiatric disorders. | [41] |
| 50 |  | Treating neuro-degenerative and neuropsychiatric disorders. | [41] |
| 51 | 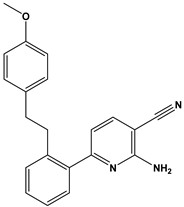 | Anti-inflammatory activity. | [42] |
| 52 | 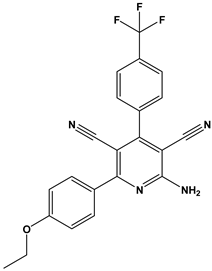 | Inhibitors of sodium–calcium exchange to be used for the prevention and/or management of cardiovascular diseases. | [43] |
| 53 | 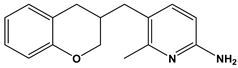 | Inhibit the growth of Mycobacterium tuberculosis. | [38] |
| 54 |  | Treatment of cardiovascular and cardio-metabolic pathologies. | [44] |
| 55 | 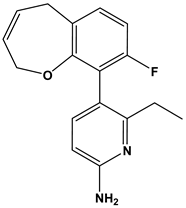 | Neuropeptide FF receptor antagonists | [45] |
| Compound | Docking Score (kcal/mol) | Types of Interactions |
|---|---|---|
| 40 | −10.33 | Val52 (arene–H) Lys 67 (H-bond) |
| 41 | −10.69 | Val52 (arene–H) Lys 67 (H-bond) |
| 42 | −8.634 | Val52 (arene–H) Lys 67 (H-bond) |
| 43 | −9.87 | Val52 (arene–H) Asp128 (H-bond) |
| 44 | −10.47 | Leu44 (arene–H) Val52 (arene–H) Lys 67 (H-bond) |
| 45 | −9.45 | Val52 (arene–H) Ile185 (arene–H) |
| 46 | −9.51 | Val52 (arene–H) Lys67 (H-bond) Leu174 (arene–H) |
| 47 | −11.62 | Lys67 (H-bond) |
| 48 | −9.73 | Leu174 (arene–H) Asp186 (H-bond) |
| 49 | −9.64 | Val52 (arene–H) |
| 50 | −9.82 | Leu174 (arene–H) Ile185 (arene–H) |
| 51 | −11.03 | Val52 (arene–H) |
| 52 | −13.03 | Val52 (arene–H) Lys67 (H-bond) Leu174 (arene–H) |
| 53 | −8.28 | Val52 (arene–H) Leu174 (arene–H) |
| 54 | −9.38 | Val52 (arene–H) Asp128 (H-bond) Ile185 (arene–H) |
| 55 | −9.06 | Val52 (arene–H) |
| Complex Name | Complex E(vdW) | Complex E(Elec) 1 | Protein E(vdW) | Protein E(Elec) 1 | Ligand E(vdW) | Ligand E(Elec) 1 | ΔG (kcal/mol) |
|---|---|---|---|---|---|---|---|
| 2OBJ-cpd31 | −1309.626 | −11,259.7506 | −1269.751 | −11,125.7145 | 9.8251 | −119.6795 | −64.06 |
| 2OBJ-cpd6 | −1298.2813 | −11,220.477 | −1272.5471 | −11,127.3604 | 10.6892 | −106.0379 | −23.5 |
| 2OBJ-ref | −1274.8976 | −11,169.5821 | −1280.6223 | −11,071.7319 | 12.1326 | −98.3834 | −5.87 |
| 2OBJ-cpd52 | −1285.4504 | −11,176.3037 | −1269.4016 | −11,088.5128 | 11.7883 | −100.4193 | −15.21 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dib, H.; Abu-Samha, M.; Younes, K.; Abdelfattah, M.A.O. Evaluating the Physicochemical Properties–Activity Relationship and Discovering New 1,2-Dihydropyridine Derivatives as Promising Inhibitors for PIM1-Kinase: Evidence from Principal Component Analysis, Molecular Docking, and Molecular Dynamics Studies. Pharmaceuticals 2024, 17, 880. https://doi.org/10.3390/ph17070880
Dib H, Abu-Samha M, Younes K, Abdelfattah MAO. Evaluating the Physicochemical Properties–Activity Relationship and Discovering New 1,2-Dihydropyridine Derivatives as Promising Inhibitors for PIM1-Kinase: Evidence from Principal Component Analysis, Molecular Docking, and Molecular Dynamics Studies. Pharmaceuticals. 2024; 17(7):880. https://doi.org/10.3390/ph17070880
Chicago/Turabian StyleDib, Hanna, Mahmoud Abu-Samha, Khaled Younes, and Mohamed A. O. Abdelfattah. 2024. "Evaluating the Physicochemical Properties–Activity Relationship and Discovering New 1,2-Dihydropyridine Derivatives as Promising Inhibitors for PIM1-Kinase: Evidence from Principal Component Analysis, Molecular Docking, and Molecular Dynamics Studies" Pharmaceuticals 17, no. 7: 880. https://doi.org/10.3390/ph17070880
APA StyleDib, H., Abu-Samha, M., Younes, K., & Abdelfattah, M. A. O. (2024). Evaluating the Physicochemical Properties–Activity Relationship and Discovering New 1,2-Dihydropyridine Derivatives as Promising Inhibitors for PIM1-Kinase: Evidence from Principal Component Analysis, Molecular Docking, and Molecular Dynamics Studies. Pharmaceuticals, 17(7), 880. https://doi.org/10.3390/ph17070880