
IL-1β-Induced CXCL10 Expression in THP-1 Monocytic Cells Involves the JNK/c-Jun and NF-κB-Mediated Signaling
 , , , , , ,
, , , , , ,  ,
,  and
and 
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. IL-1β Treatment Upregulates CXCL10 Gene and Protein Expression in THP-1 Cells
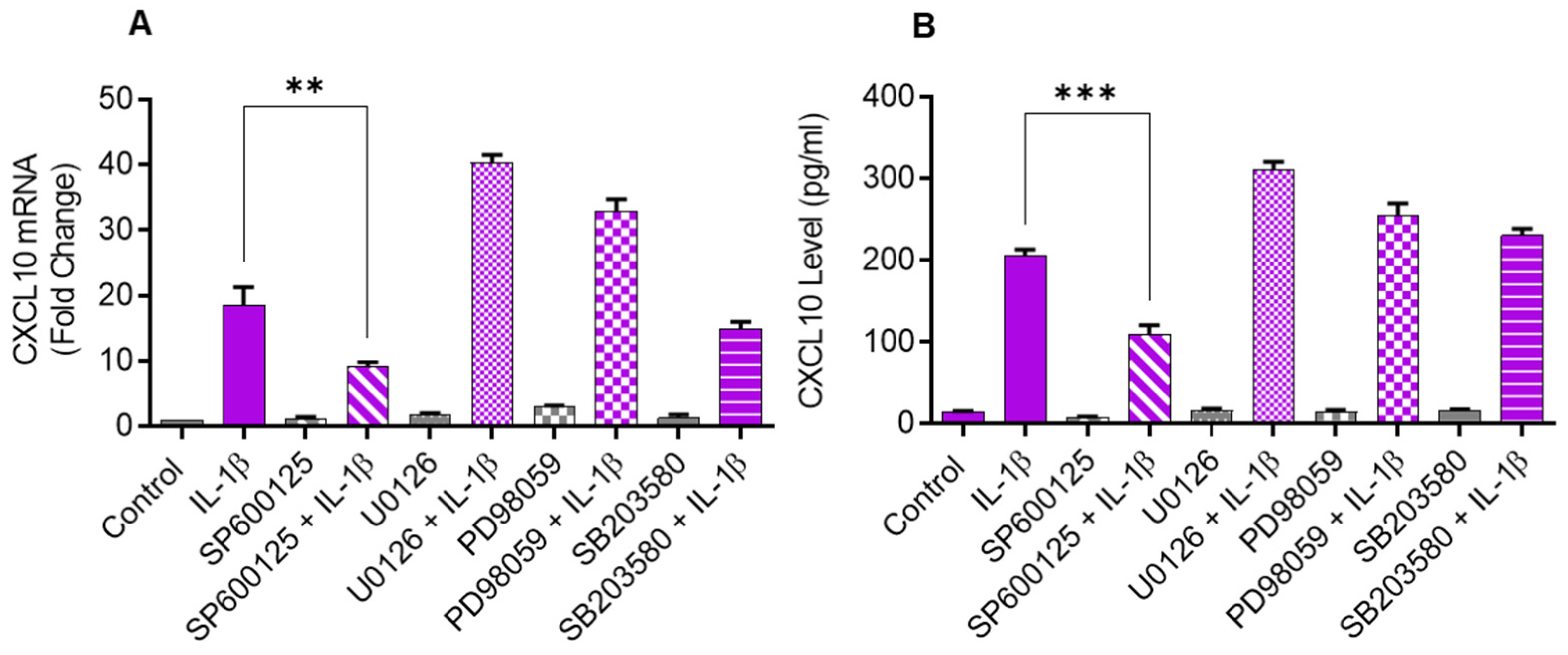
2.2. Inhibiting JNK Suppresses the IL-1β-Induced CXCL10 Expression
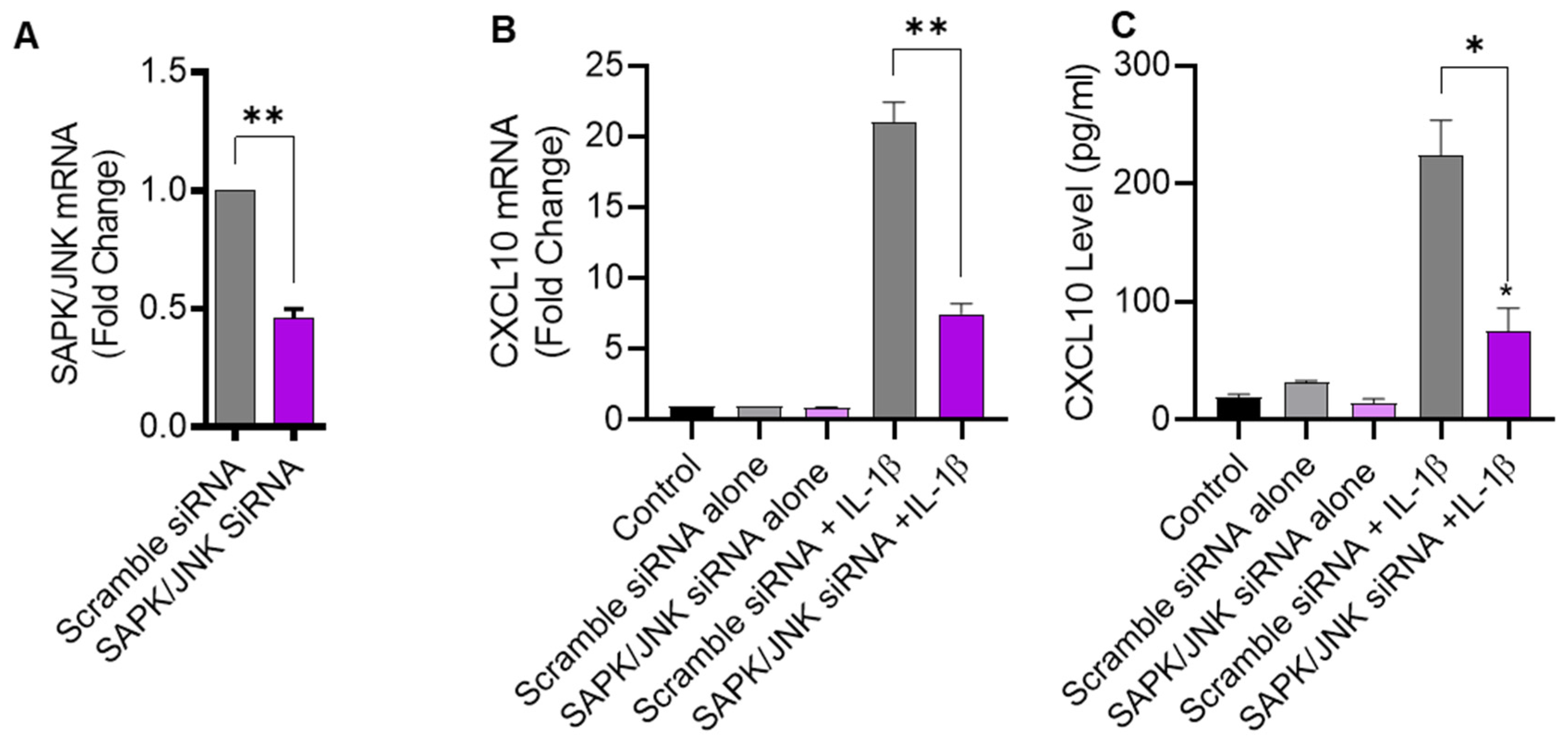
2.3. JNK Deficiency Also Leads to Suppression in IL-1β-Induced CXCL10
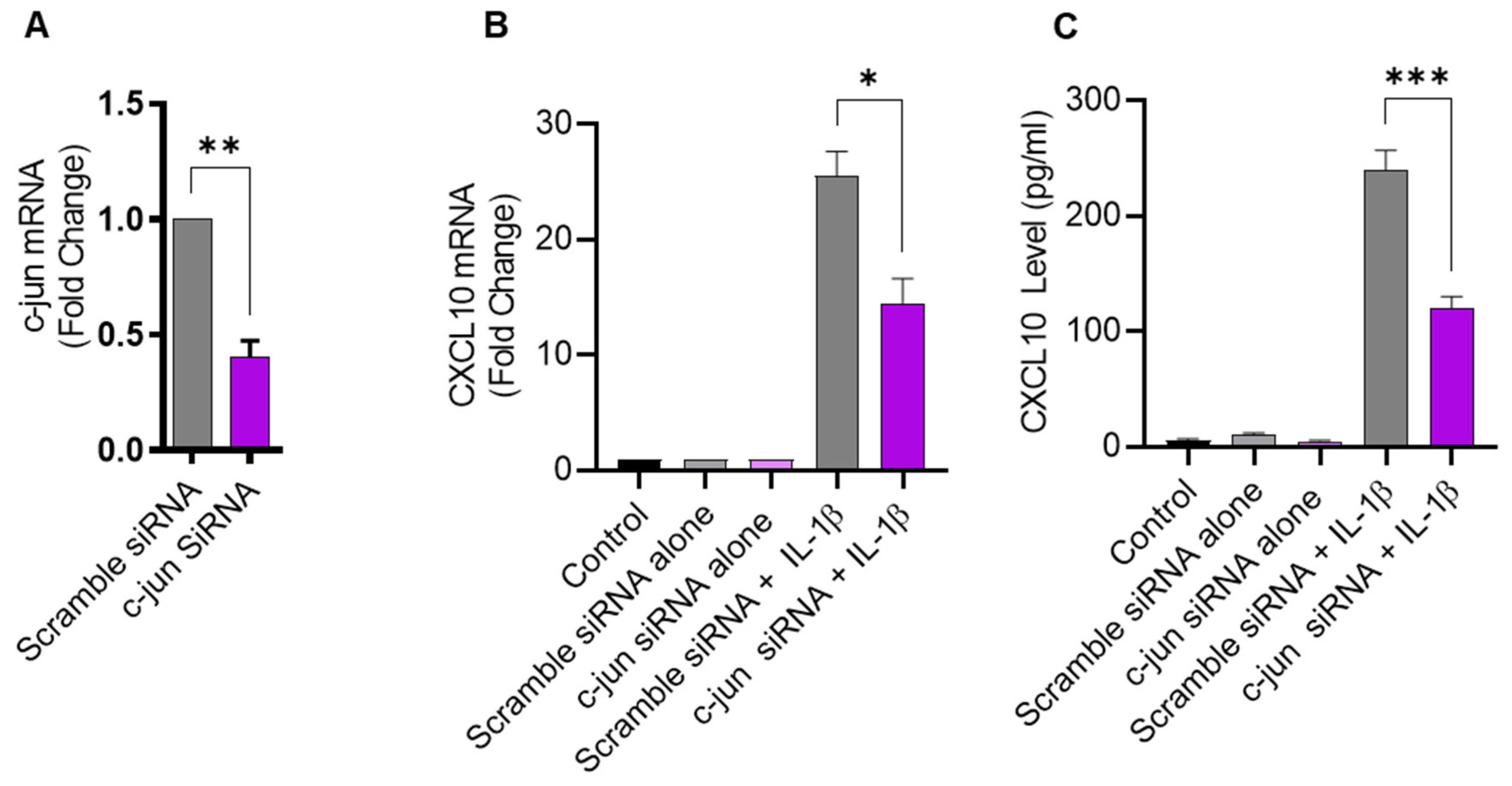
2.4. IL-1β-Induced CXCL10 Expression in THP-1 Cells Involves c-Jun Mediated Signaling
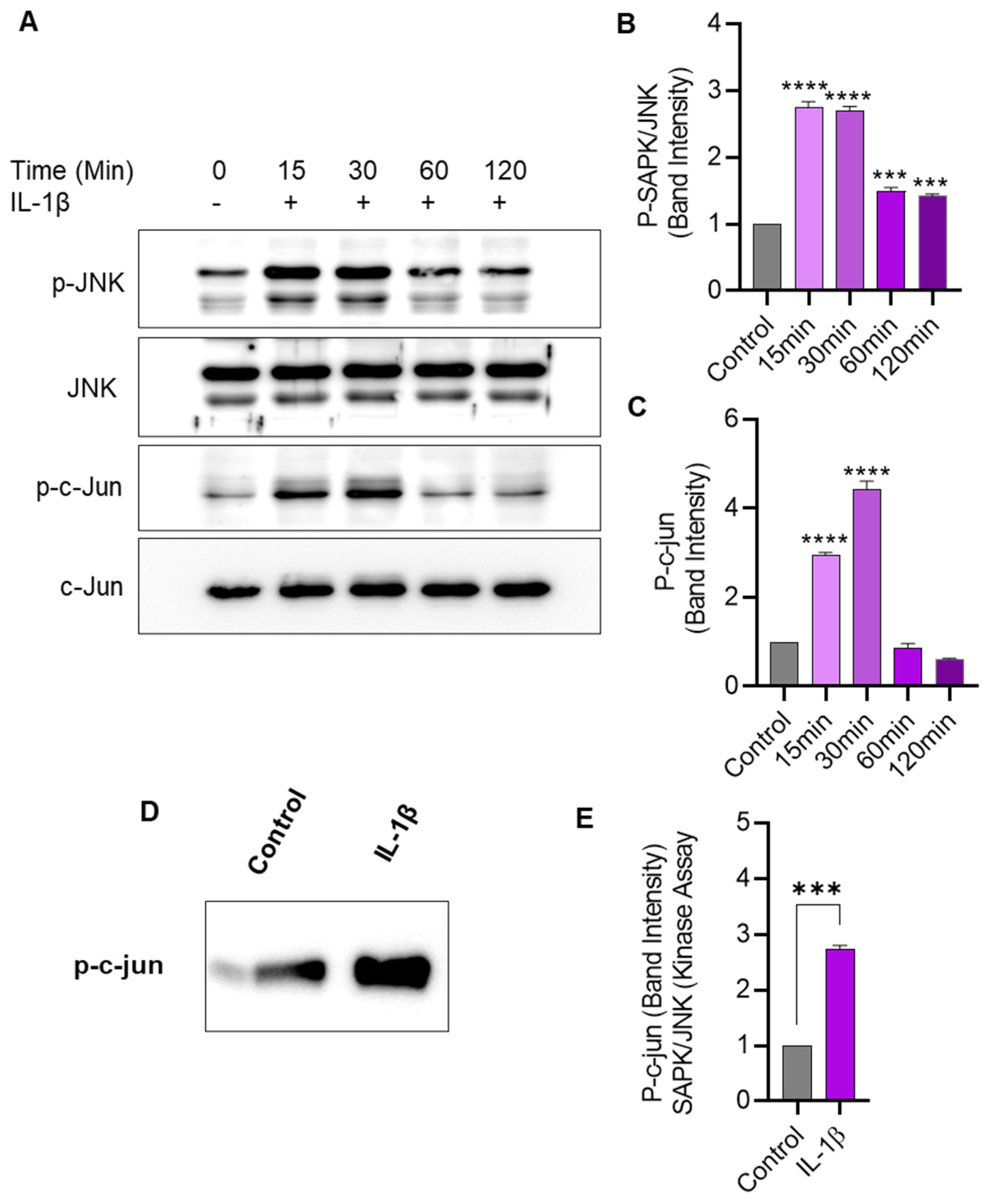
2.5. IL-1β Stimulation Induces JNK/c-Jun Phosphorylation and Enhances the JNK Kinase Activity in THP-1 Cells
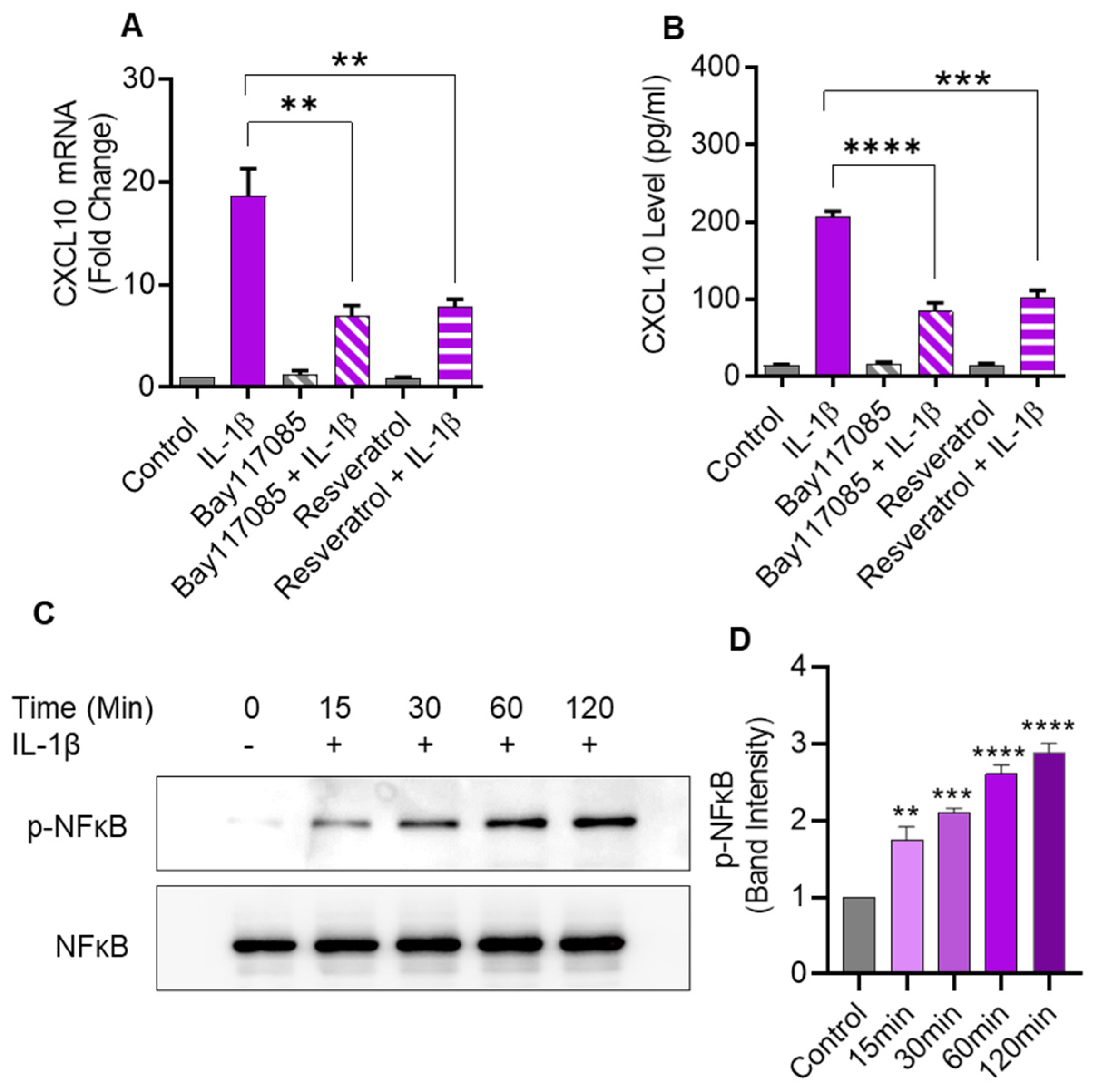
2.6. IL-1β-Induced CXCL10 Expression Also Involves the Canonical NF-κB-Mediated Signaling
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Stimulations
4.2. Small Interfering RNA (siRNA) Transfections
4.3. Real Time Reverse Transcription–Polymerase Chain Reaction (RT-PCR)
4.4. SAPK/JNK Kinase Assay
4.5. Quantification of Secreted CXCL10 Protein
4.6. Western Blotting
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lois, K.; Kumar, S. Obesity and diabetes. Endocrinol. Nutr. 2009, 56S4, 38–42. [Google Scholar] [CrossRef]
- Kalra, S. Diabesity. J. Pak. Med. Assoc. 2013, 63, 532–534. [Google Scholar]
- Kershaw, E.E.; Flier, J.S. Adipose tissue as an endocrine organ. J. Clin. Endocrinol. Metab. 2004, 89, 2548–2556. [Google Scholar] [CrossRef]
- Griffith, J.W.; Sokol, C.L.; Luster, A.D. Chemokines and chemokine receptors: Positioning cells for host defense and immunity. Annu. Rev. Immunol. 2014, 32, 659–702. [Google Scholar] [CrossRef]
- Ciesielski, C.J.; Andreakos, E.; Foxwell, B.M.; Feldmann, M. Tnfalpha-induced macrophage chemokine secretion is more dependent on nf-kappab expression than lipopolysaccharides-induced macrophage chemokine secretion. Eur. J. Immunol. 2002, 32, 2037–2045. [Google Scholar] [CrossRef]
- Ohmori, Y.; Hamilton, T.A. The interferon-stimulated response element and a kappa b site mediate synergistic induction of murine ip-10 gene transcription by ifn-gamma and tnf-alpha. J. Immunol. 1995, 154, 5235–5244. [Google Scholar] [CrossRef]
- Ohmori, Y.; Hamilton, T.A. Cell type and stimulus specific regulation of chemokine gene expression. Biochem. Biophys. Res. Commun. 1994, 198, 590–596. [Google Scholar] [CrossRef]
- Chang, C.C.; Wu, C.L.; Su, W.W.; Shih, K.L.; Tarng, D.C.; Chou, C.T.; Chen, T.Y.; Kor, C.T.; Wu, H.M. Interferon gamma-induced protein 10 is associated with insulin resistance and incident diabetes in patients with nonalcoholic fatty liver disease. Sci. Rep. 2015, 5, 10096. [Google Scholar] [CrossRef]
- Shields, P.L.; Morland, C.M.; Salmon, M.; Qin, S.; Hubscher, S.G.; Adams, D.H. Chemokine and chemokine receptor interactions provide a mechanism for selective t cell recruitment to specific liver compartments within hepatitis c-infected liver1. J. Immunol. 1999, 163, 6236–6243. [Google Scholar] [CrossRef]
- Chalin, A.; Lefevre, B.; Devisme, C.; Pronier, C.; Carrière, V.; Thibault, V.; Amiot, L.; Samson, M. Serum cxcl10, cxcl11, cxcl12, and cxcl14 chemokine patterns in patients with acute liver injury. Cytokine 2018, 111, 500–504. [Google Scholar] [CrossRef]
- Sorensen, T.L.; Trebst, C.; Kivisakk, P.; Klaege, K.L.; Majmudar, A.; Ravid, R.; Lassmann, H.; Olsen, D.B.; Strieter, R.M.; Ransohoff, R.M.; et al. Multiple sclerosis: A study of cxcl10 and cxcr3 co-localization in the inflamed central nervous system. J. Neuroimmunol. 2002, 127, 59–68. [Google Scholar] [CrossRef]
- Jain, S.K.; Kahlon, G.; Morehead, L.; Lieblong, B.; Stapleton, T.; Hoeldtke, R.; Bass, P.F., 3rd; Levine, S.N. The effect of sleep apnea and insomnia on blood levels of leptin, insulin resistance, ip-10, and hydrogen sulfide in type 2 diabetic patients. Metab. Syndr. Relat. Disord. 2012, 10, 331–336. [Google Scholar] [CrossRef]
- Xu, H.; Nakayama, K.; Ogawa, S.; Sugiura, A.; Kato, T.; Sato, T.; Sato, H.; Ito, S. Elevated plasma concentration of ip-10 in patients with type 2 diabetes mellitus. Nihon Jinzo Gakkai Shi 2005, 47, 524–530. [Google Scholar]
- D’Esposito, V.; Ambrosio, M.R.; Liguoro, D.; Perruolo, G.; Lecce, M.; Cabaro, S.; Aprile, M.; Marino, A.; Pilone, V.; Forestieri, P.; et al. In severe obesity, subcutaneous adipose tissue cell-derived cytokines are early markers of impaired glucose tolerance and are modulated by quercetin. Int. J. Obes. 2021, 45, 1811–1820. [Google Scholar] [CrossRef]
- Alomar, S.Y.; Gentili, A.; Zaibi, M.S.; Kepczynska, M.A.; Trayhurn, P. Il-1beta (interleukin-1beta) stimulates the production and release of multiple cytokines and chemokines by human preadipocytes. Arch. Physiol. Biochem. 2016, 122, 117–122. [Google Scholar] [CrossRef]
- Yoshimatsu, G.; Kunnathodi, F.; Saravanan, P.B.; Shahbazov, R.; Chang, C.; Darden, C.M.; Zurawski, S.; Boyuk, G.; Kanak, M.A.; Levy, M.F.; et al. Pancreatic beta-cell-derived ip-10/cxcl10 isletokine mediates early loss of graft function in islet cell transplantation. Diabetes 2017, 66, 2857–2867. [Google Scholar] [CrossRef]
- Kai, K.; Nasu, K.; Nakamura, S.; Fukuda, J.; Nishida, M.; Miyakawa, I. Expression of interferon-gamma-inducible protein-10 in human endometrial stromal cells. Mol. Hum. Reprod. 2002, 8, 176–180. [Google Scholar] [CrossRef]
- Smeehuijzen, L.; Gijbels, A.; Nugteren-Boogaard, J.P.; Vrieling, F.; Boudjadja, M.B.; Trouwborst, I.; Jardon, K.M.; Hul, G.B.; Feskens, E.J.M.; Blaak, E.E.; et al. Immunometabolic signatures of circulating monocytes in humans with obesity and insulin resistance. Diabetes 2024, 24, db230970. [Google Scholar] [CrossRef]
- Hueso, L.; Ortega, R.; Selles, F.; Wu-Xiong, N.Y.; Ortega, J.; Civera, M.; Ascaso, J.F.; Sanz, M.J.; Real, J.T.; Piqueras, L. Upregulation of angiostatic chemokines ip-10/cxcl10 and i-tac/cxcl11 in human obesity and their implication for adipose tissue angiogenesis. Int. J. Obes. 2018, 42, 1406–1417. [Google Scholar] [CrossRef]
- Kochumon, S.; Madhoun, A.A.; Al-Rashed, F.; Azim, R.; Al-Ozairi, E.; Al-Mulla, F.; Ahmad, R. Adipose tissue gene expression of cxcl10 and cxcl11 modulates inflammatory markers in obesity: Implications for metabolic inflammation and insulin resistance. Ther. Adv. Endocrinol. Metab. 2020, 11, 2042018820930902. [Google Scholar] [CrossRef]
- Deiuliis, J.A.; Oghumu, S.; Duggineni, D.; Zhong, J.; Rutsky, J.; Banerjee, A.; Needleman, B.; Mikami, D.; Narula, V.; Hazey, J.; et al. Cxcr3 modulates obesity-induced visceral adipose inflammation and systemic insulin resistance. Obesity 2014, 22, 1264–1274. [Google Scholar] [CrossRef]
- Burke, S.J.; Goff, M.R.; Lu, D.; Proud, D.; Karlstad, M.D.; Collier, J.J. Synergistic expression of the cxcl10 gene in response to il-1β and ifn-γ involves nf-κb, phosphorylation of stat1 at tyr701, and acetylation of histones h3 and h4. J. Immunol. 2013, 191, 323–336. [Google Scholar] [CrossRef]
- Korpi-Steiner, N.L.; Valkenaar, S.M.; Bates, M.E.; Evans, M.D.; Gern, J.E.; Bertics, P.J. Human monocytic cells direct the robust release of cxcl10 by bronchial epithelial cells during rhinovirus infection. Clin. Exp. Allergy J. Br. Soc. Allergy Clin. Immunol. 2010, 40, 1203–1213. [Google Scholar] [CrossRef]
- Bonechi, E.; Aldinucci, A.; Mazzanti, B.; di Gioia, M.; Repice, A.M.; Manuelli, C.; Saccardi, R.; Massacesi, L.; Ballerini, C. Increased cxcl10 expression in ms mscs and monocytes is unaffected by ahsct. Ann. Clin. Transl. Neurol. 2014, 1, 650–658. [Google Scholar] [CrossRef]
- Alrashdan, Y.A.; Alkhouri, H.; Chen, E.; Lalor, D.J.; Poniris, M.; Henness, S.; Brightling, C.E.; Burgess, J.K.; Armour, C.L.; Ammit, A.J.; et al. Asthmatic airway smooth muscle cxcl10 production: Mitogen-activated protein kinase jnk involvement. Am. J. Physiol. Lung Cell Mol. Physiol. 2012, 302, L1118–L1127. [Google Scholar] [CrossRef]
- Sunil, Y.; Ramadori, G.; Raddatzc, D. Influence of nfkappab inhibitors on il-1beta-induced chemokine cxcl8 and -10 expression levels in intestinal epithelial cell lines: Glucocorticoid ineffectiveness and paradoxical effect of pdtc. Int. J. Color. Dis. 2010, 25, 323–333. [Google Scholar] [CrossRef]
- David, H.; Knight, C.G.; Oliver, D.-B.; Heike, S.; Anneke, D.; Elke, H.; Klaus, R.; Michael, K. Disruption of the c-jun-jnk complex by a cell-permeable peptide containing the c-jun δ domain induces apoptosis and affects a distinct set of interleukin-1-induced inflammatory genes*. J. Biol. Chem. 2003, 278, 40213–40223. [Google Scholar]
- Derijard, B.; Hibi, M.; Wu, I.H.; Barrett, T.; Su, B.; Deng, T.; Karin, M.; Davis, R.J. Jnk1: A protein kinase stimulated by uv light and ha-ras that binds and phosphorylates the c-jun activation domain. Cell 1994, 76, 1025–1037. [Google Scholar] [CrossRef]
- May, G.H.; Allen, K.E.; Clark, W.; Funk, M.; Gillespie, D.A. Analysis of the interaction between c-jun and c-jun n-terminal kinase in vivo. J. Biol. Chem. 1998, 273, 33429–33435. [Google Scholar] [CrossRef]
- Johnson, G.L.; Nakamura, K. The c-jun kinase/stress-activated pathway: Regulation, function and role in human disease. Biochim. Biophys. Acta 2007, 1773, 1341–1348. [Google Scholar] [CrossRef]
- Hah, Y.S.; Kang, H.G.; Cho, H.Y.; Shin, S.H.; Kim, U.K.; Park, B.W.; Lee, S.I.; Rho, G.J.; Kim, J.R.; Byun, J.H. Jnk signaling plays an important role in the effects of tnf-alpha and il-1beta on in vitro osteoblastic differentiation of cultured human periosteal-derived cells. Mol. Biol. Rep. 2013, 40, 4869–4881. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. Nf-κb signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed]
- Pikarsky, E.; Porat, R.M.; Stein, I.; Abramovitch, R.; Amit, S.; Kasem, S.; Gutkovich-Pyest, E.; Urieli-Shoval, S.; Galun, E.; Ben-Neriah, Y. Nf-kappab functions as a tumour promoter in inflammation-associated cancer. Nature 2004, 431, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Oeckinghaus, A.; Ghosh, S. The nf-kappab family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef]
- Qi, X.F.; Kim, D.H.; Yoon, Y.S.; Jin, D.; Huang, X.Z.; Li, J.H.; Deung, Y.K.; Lee, K.J. Essential involvement of cross-talk between ifn-gamma and tnf-alpha in cxcl10 production in human thp-1 monocytes. J. Cell. Physiol. 2009, 220, 690–697. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kochumon, S.; Al-Sayyar, A.; Jacob, T.; Arefanian, H.; Bahman, F.; Almansour, N.; Alzaid, F.; Al-Mulla, F.; Sindhu, S.; Ahmad, R. IL-1β-Induced CXCL10 Expression in THP-1 Monocytic Cells Involves the JNK/c-Jun and NF-κB-Mediated Signaling. Pharmaceuticals 2024, 17, 823. https://doi.org/10.3390/ph17070823
Kochumon S, Al-Sayyar A, Jacob T, Arefanian H, Bahman F, Almansour N, Alzaid F, Al-Mulla F, Sindhu S, Ahmad R. IL-1β-Induced CXCL10 Expression in THP-1 Monocytic Cells Involves the JNK/c-Jun and NF-κB-Mediated Signaling. Pharmaceuticals. 2024; 17(7):823. https://doi.org/10.3390/ph17070823
Chicago/Turabian StyleKochumon, Shihab, Amnah Al-Sayyar, Texy Jacob, Hossein Arefanian, Fatemah Bahman, Nourah Almansour, Fawaz Alzaid, Fahd Al-Mulla, Sardar Sindhu, and Rasheed Ahmad. 2024. "IL-1β-Induced CXCL10 Expression in THP-1 Monocytic Cells Involves the JNK/c-Jun and NF-κB-Mediated Signaling" Pharmaceuticals 17, no. 7: 823. https://doi.org/10.3390/ph17070823
APA StyleKochumon, S., Al-Sayyar, A., Jacob, T., Arefanian, H., Bahman, F., Almansour, N., Alzaid, F., Al-Mulla, F., Sindhu, S., & Ahmad, R. (2024). IL-1β-Induced CXCL10 Expression in THP-1 Monocytic Cells Involves the JNK/c-Jun and NF-κB-Mediated Signaling. Pharmaceuticals, 17(7), 823. https://doi.org/10.3390/ph17070823