
A Hybrid Approach Combining Shape-Based and Docking Methods to Identify Novel Potential P2X7 Antagonists from Natural Product Databases

 , , ,
, , ,  , , ,
, , ,
Abstract

1. Introduction
2. Results and Discussion
2.1. Shape-Based Screening
2.2. Drug-like Filter
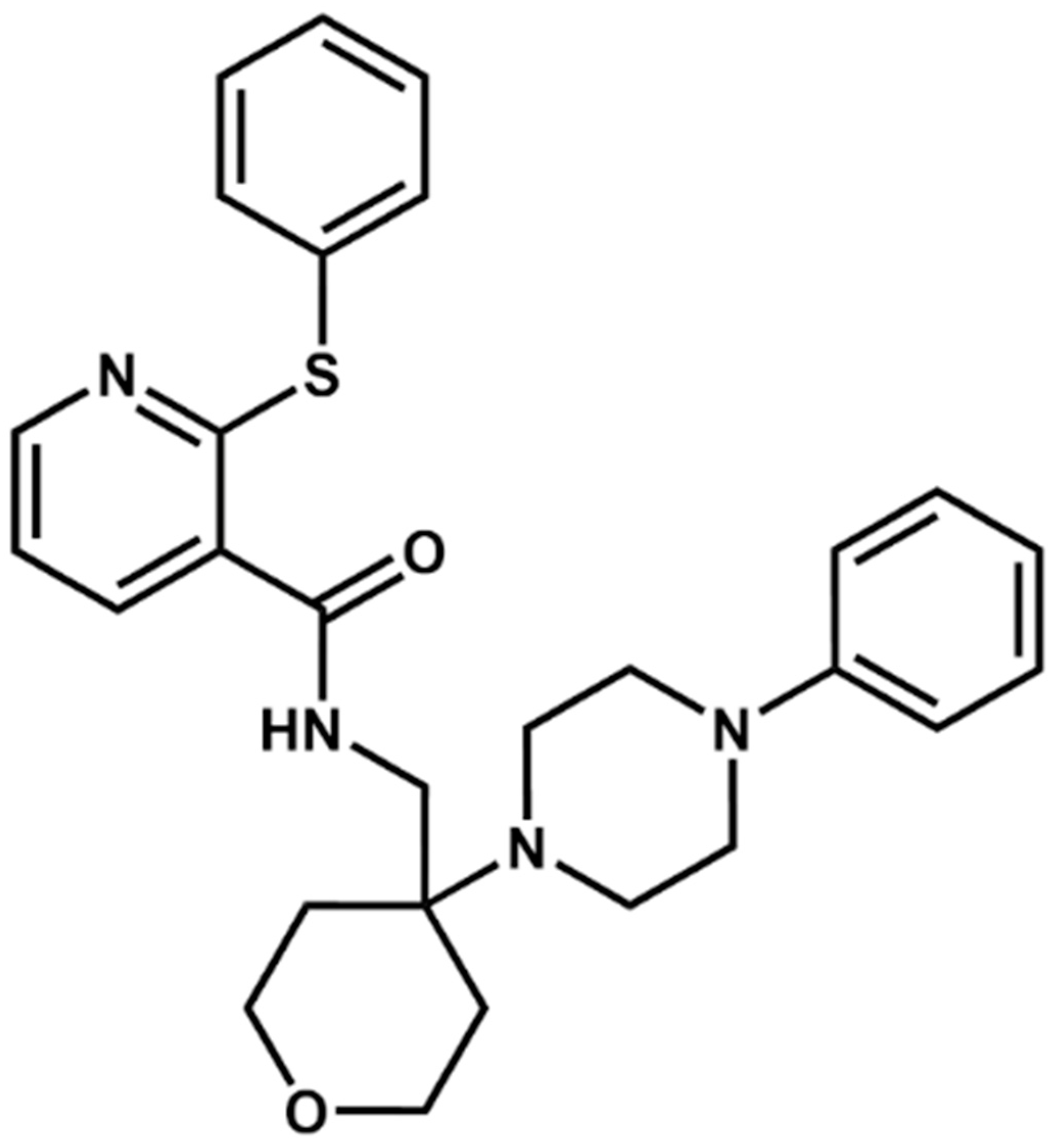
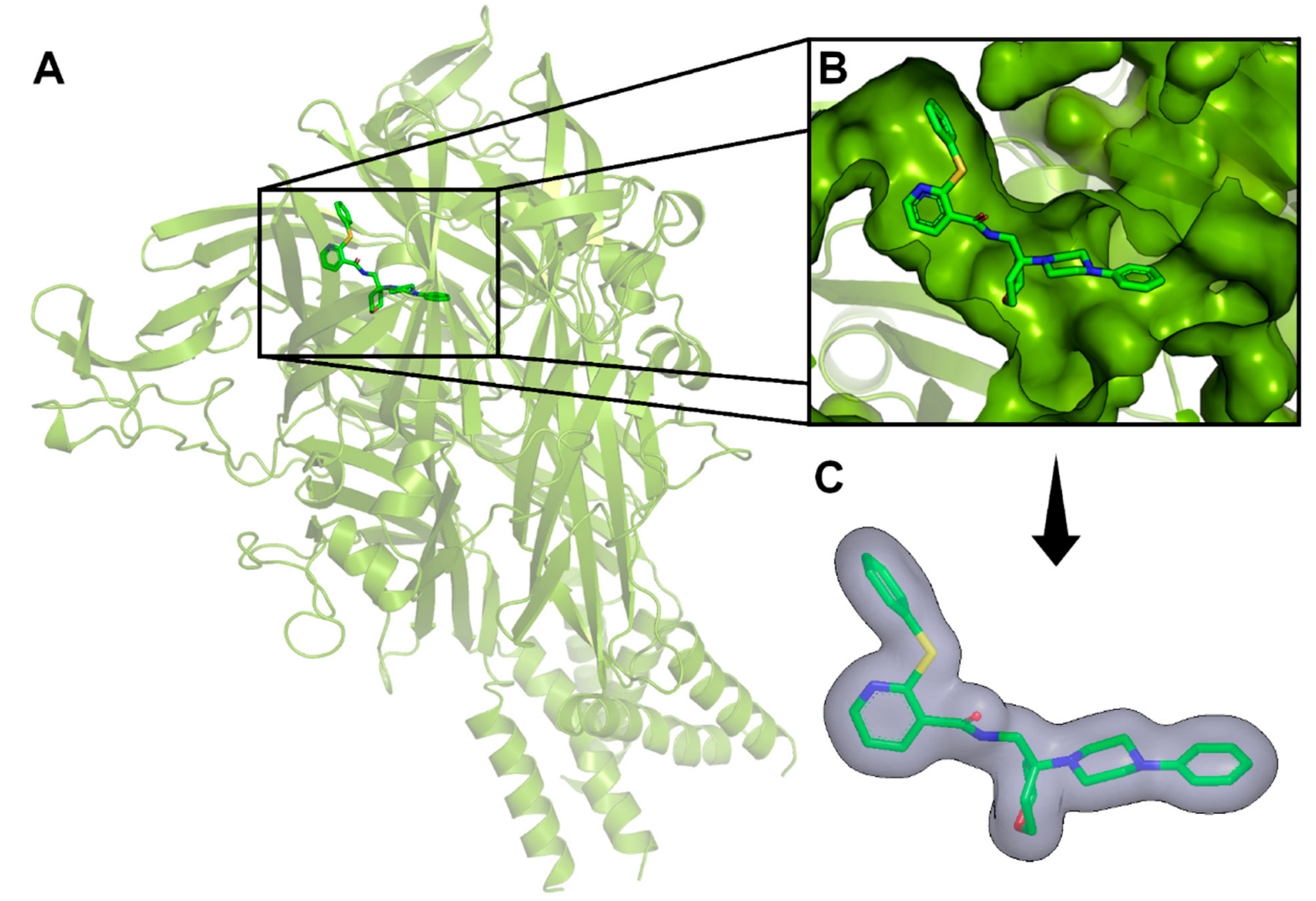
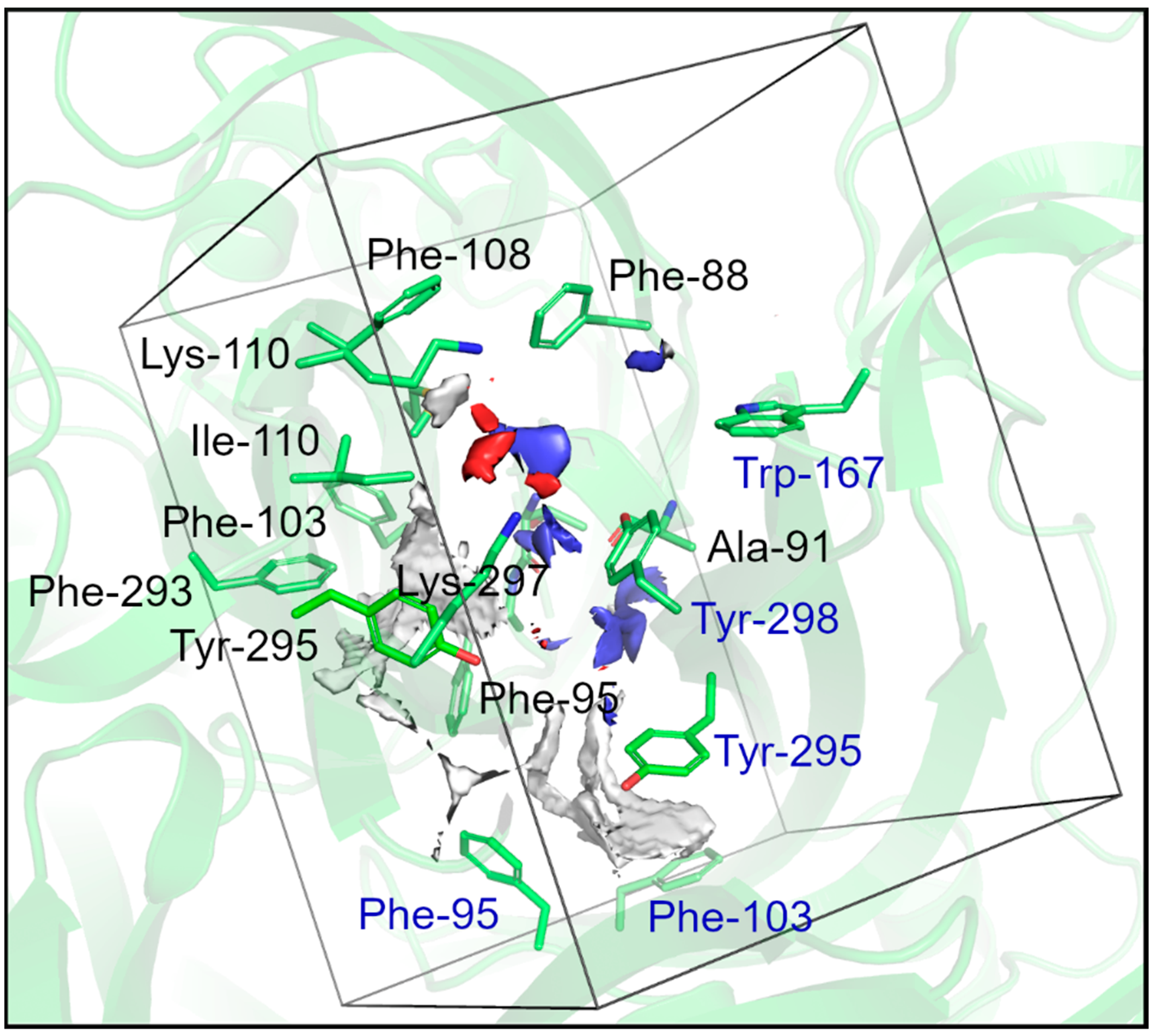
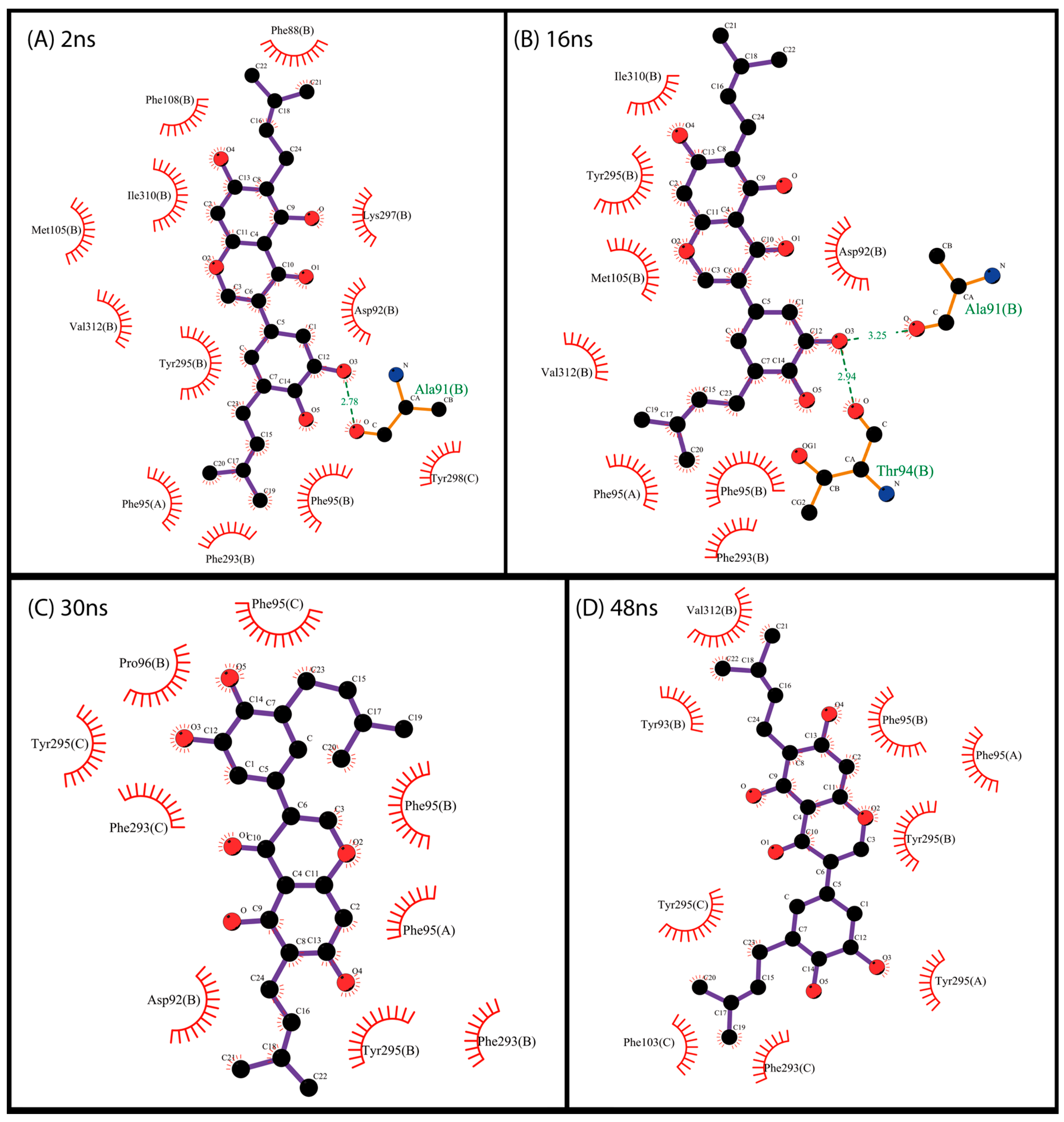
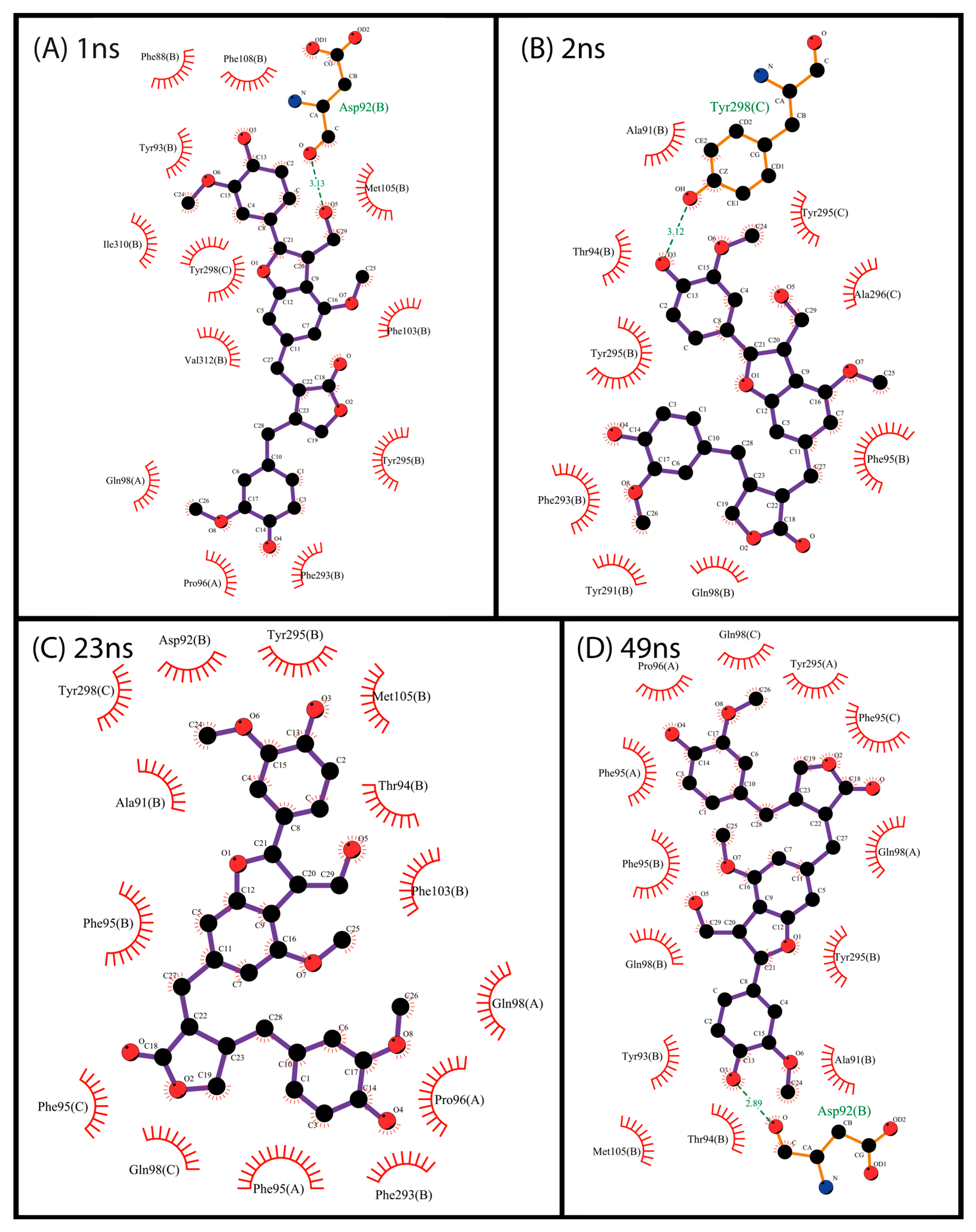
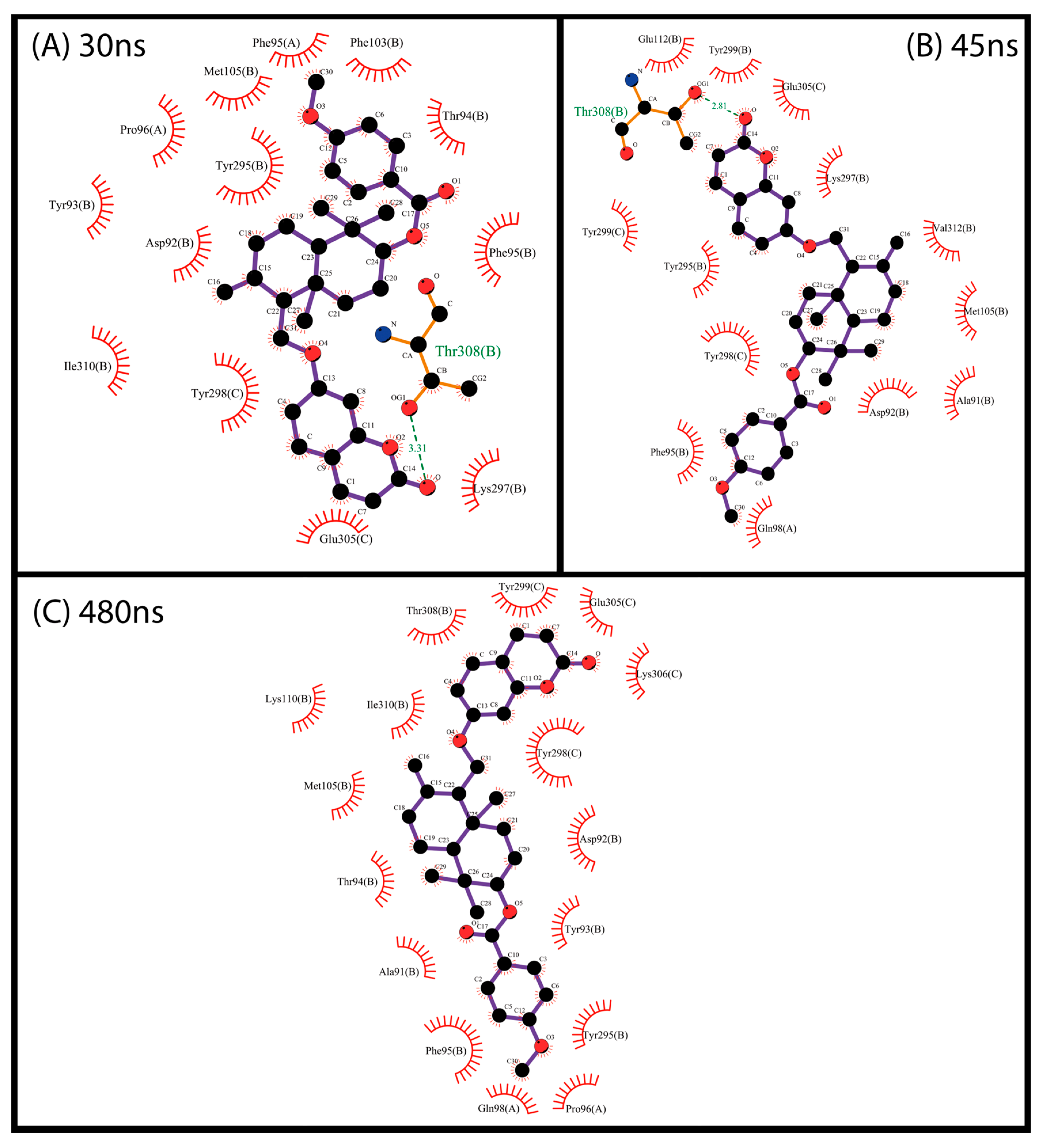
2.3. Docking and Visual Inspection
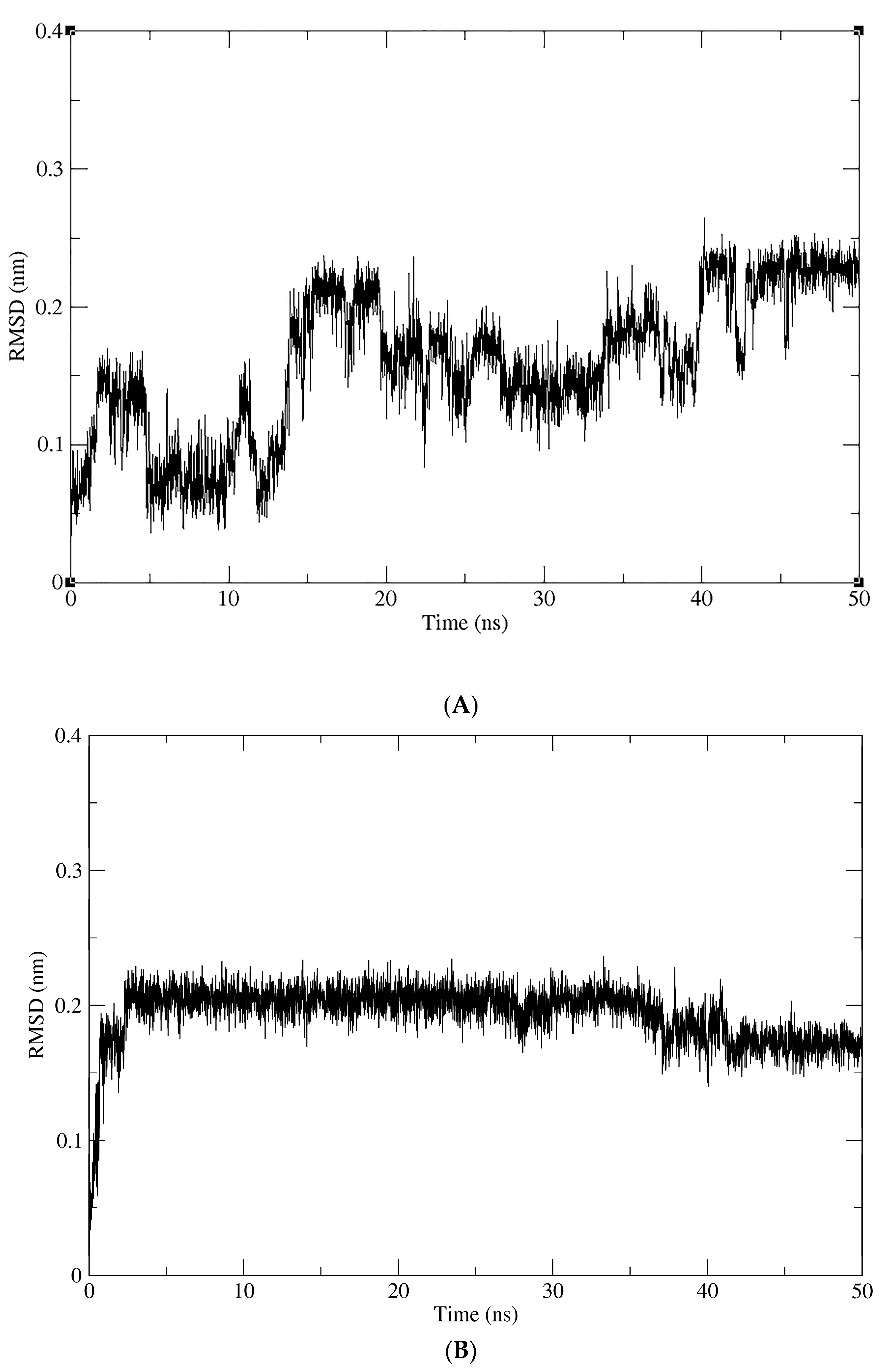
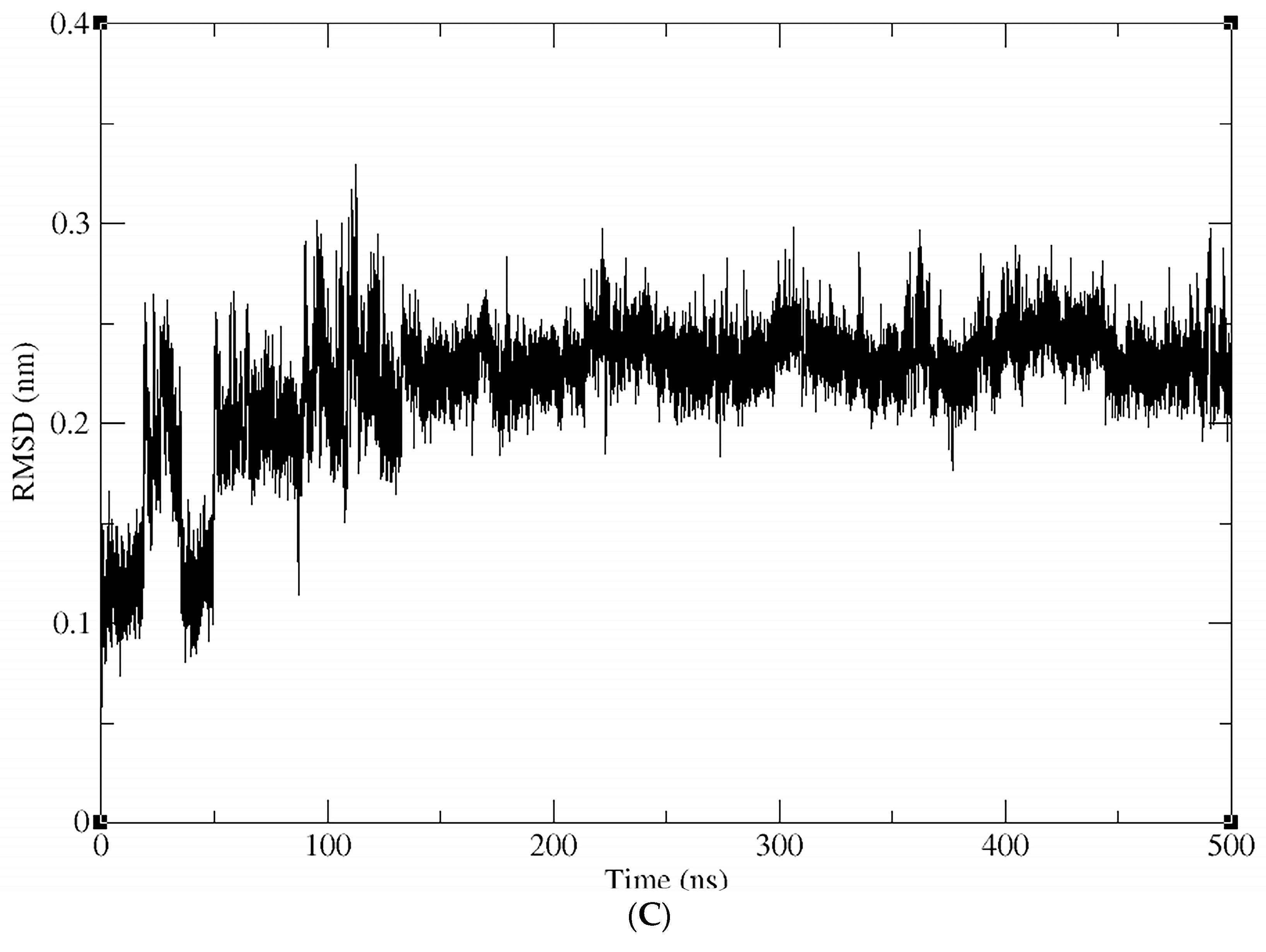
2.4. Molecular Dynamics (MD) Simulations
3. Material and Methods
3.1. Compound Databases’ Preparation
3.2. Shape-Based Screening Procedures
3.3. Drug-like Filter
3.4. Protein Structure Preparation for Docking Procedures
3.5. Docking Procedures and Visual Inspection
3.6. Molecular Dynamics Simulations
3.6.1. Ligand Parameterization
3.6.2. Geometric Configuration of Simulated Systems
3.6.3. Analysis of Trajectories Simulated by Molecular Dynamics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fredholm, B.B.; Abbracchio, M.P.; Burnstock, G.; Daly, J.W.; Harden, T.K.; Jacobson, K.A.; Leff, P.; Williams, M. Nomenclature and Classification of Purinoceptors. Pharmacol. Rev. 1994, 46, 143–156. [Google Scholar] [PubMed]
- Alves, L.A.; de Melo Reis, R.A.; de Souza, C.A.M.; de Freitas, M.S.; Teixeira, P.C.N.; Neto Moreira Ferreira, D.; Xavier, R.F. The P2X7 Receptor: Shifting from a Low- to a High-Conductance Channel—An Enigmatic Phenomenon? Biochim. Biophys. Acta (BBA)—Biomembr. 2014, 1838, 2578–2587. [Google Scholar] [CrossRef] [PubMed]
- Lister, M.F.; Sharkey, J.; Sawatzky, D.A.; Hodgkiss, J.P.; Davidson, D.J.; Rossi, A.G.; Finlayson, K. The Role of the Purinergic P2X7 Receptor in Inflammation. J. Inflamm. 2007, 4, 5. [Google Scholar] [CrossRef] [PubMed]
- Friedle, S.A.; Curet, M.A.; Watters, J.J. Recent Patents on Novel P2X(7) Receptor Antagonists and Their Potential for Reducing Central Nervous System Inflammation. Recent. Pat. CNS Drug Discov. 2010, 5, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, R.; Stokes, L.; Sluyter, R. The P2X7 Receptor Channel: Recent Developments and the Use of P2X7 Antagonists in Models of Disease. Pharmacol. Rev. 2014, 66, 638–675. [Google Scholar] [CrossRef] [PubMed]
- Riteau, N.; Baron, L.; Villeret, B.; Guillou, N.; Savigny, F.; Ryffel, B.; Rassendren, F.; Le Bert, M.; Gombault, A.; Couillin, I. ATP Release and Purinergic Signaling: A Common Pathway for Particle-Mediated Inflammasome Activation. Cell Death Dis. 2012, 3, e403. [Google Scholar] [CrossRef] [PubMed]
- Latz, E.; Ts, X.; Stutz, A. Activation and Regulation of the Inflammasomes. Nat. Rev. Immunol. 2013, 13, 23702978. [Google Scholar] [CrossRef]
- Honore, P.; Donnelly-Roberts, D.; Namovic, M.T.; Hsieh, G.; Zhu, C.Z.; Mikusa, J.P.; Hernandez, G.; Zhong, C.; Gauvin, D.M.; Chandran, P.; et al. A-740003 [N-(1-{[(Cyanoimino)(5-Quinolinylamino) Methyl]Amino}-2,2-Dimethylpropyl)-2-(3,4-Dimethoxyphenyl)Acetamide], a Novel and Selective P2X7 Receptor Antagonist, Dose-Dependently Reduces Neuropathic Pain in the Rat. J. Pharmacol. Exp. Ther. 2006, 319, 1376–1385. [Google Scholar] [CrossRef]
- Caporali, F.; Capecchi, P.L.; Gamberucci, A.; Lazzerini, P.E.; Pompella, G.; Natale, M.; Lorenzini, S.; Selvi, E.; Galeazzi, M.; Laghi Pasini, F. Human Rheumatoid Synoviocytes Express Functional P2X7 Receptors. J. Mol. Med. 2008, 86, 937–949. [Google Scholar] [CrossRef]
- Diaz-Hernandez, J.I.; Gomez-Villafuertes, R.; León-Otegui, M.; Hontecillas-Prieto, L.; del Puerto, A.; Trejo, J.L.; Lucas, J.J.; Garrido, J.J.; Gualix, J.; Miras-Portugal, M.T.; et al. In Vivo P2X7 Inhibition Reduces Amyloid Plaques in Alzheimer’s Disease through GSK3B and Secretases. Neurobiol. Aging 2012, 33, 1816–1828. [Google Scholar] [CrossRef]
- Graziano, F.; Desdouits, M.; Garzetti, L.; Podini, P.; Alfano, M.; Rubartelli, A.; Furlan, R.; Benaroch, P.; Poli, G. Extracellular ATP Induces the Rapid Release of HIV-1 from Virus Containing Compartments of Human Macrophages. Proc. Natl. Acad. Sci. USA 2015, 112, E3265–E3273. [Google Scholar] [CrossRef]
- Lara, R.; Adinolfi, E.; Harwood, C.A.; Philpott, M.; Barden, J.A.; Di Virgilio, F.; McNulty, S. P2X7 in Cancer: From Molecular Mechanisms to Therapeutics. Front. Pharmacol. 2020, 11, 793. [Google Scholar] [CrossRef] [PubMed]
- Alves, L.; Bezerra, R.; Faria, R.; Ferreira, L.; da Silva Frutuoso, V. Physiological Roles and Potential Therapeutic Applications of the P2X7 Receptor in Inflammation and Pain. Molecules 2013, 18, 10953–10972. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Ruiz, C.; Calzaferri, F.; García, A.G. P2X7 Receptor Antagonism as a Potential Therapy in Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2020, 13, 93. [Google Scholar] [CrossRef]
- Gu, J.; Gui, Y.; Chen, L.; Yuan, G.; Lu, H.-Z.; Xu, X. Use of Natural Products as Chemical Library for Drug Discovery and Network Pharmacology. PLoS ONE 2013, 8, e62839. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef]
- Faria, R.; Ferreira, L.; Bezerra, R.; Frutuoso, V.; Alves, L. Action of Natural Products on P2 Receptors: A Reinvented Era for Drug Discovery. Molecules 2012, 17, 13009–13025. [Google Scholar] [CrossRef] [PubMed]
- Soares-Bezerra, R.J.; da Silva Ferreira, N.C.; Alves, T.M.A.; Zani, C.L.; Rosa, L.H.; Faria, R.X.; Frutuoso, V.S.; Alves, L.A. A New Insight into Purinergic Pharmacology: Three Fungal Species as Natural P2X7R Antagonists. Phytotherapy 2019, 33, 2319–2328. [Google Scholar] [CrossRef]
- Liu, L.; Zou, J.; Liu, X.; Jiang, L.-H.; Li, J. Inhibition of ATP-Induced Macrophage Death by Emodin via Antagonizing P2X7 Receptor. Eur. J. Pharmacol. 2010, 640, 15–19. [Google Scholar] [CrossRef]
- Jelassi, B.; Anchelin, M.; Chamouton, J.; Cayuela, M.L.; Clarysse, L.; Li, J.; Goré, J.; Jiang, L.-H.; Roger, S. Anthraquinone Emodin Inhibits Human Cancer Cell Invasiveness by Antagonizing P2X7 Receptors. Carcinogenesis 2013, 34, 1487–1496. [Google Scholar] [CrossRef]
- Han, J.-W.; Shim, D.-W.; Shin, W.-Y.; Heo, K.-H.; Kwak, S.-B.; Sim, E.-J.; Jeong, J.-H.; Kang, T.-B.; Lee, K.-H. Anti-Inflammatory Effect of Emodin via Attenuation of NLRP3 Inflammasome Activation. Int. J. Mol. Sci. 2015, 16, 8102–8109. [Google Scholar] [CrossRef] [PubMed]
- Marques-da-Silva, C.; Chaves, M.M.; Castro, N.G.; Coutinho-Silva, R.; Guimaraes, M.Z.P. Colchicine Inhibits Cationic Dye Uptake Induced by ATP in P2X2 and P2X7 Receptor-Expressing Cells: Implications for Its Therapeutic Action. Br. J. Pharmacol. 2011, 163, 912–926. [Google Scholar] [CrossRef]
- Buchanan, M.S.; Carroll, A.R.; Addepalli, R.; Avery, V.M.; Hooper, J.N.A.; Quinn, R.J. Natural Products, Stylissadines A and B, Specific Antagonists of the P2X7 Receptor, an Important Inflammatory Target. J. Org. Chem. 2007, 72, 2309–2317. [Google Scholar] [CrossRef]
- Blay, V.; Tolani, B.; Ho, S.P.; Arkin, M.R. High-Throughput Screening: Today’s Biochemical and Cell-Based Approaches. Drug Discov. Today 2020, 25, 1807–1821. [Google Scholar] [CrossRef] [PubMed]
- Shaker, B.; Ahmad, S.; Lee, J.; Jung, C.; Na, D. In Silico Methods and Tools for Drug Discovery. Comput. Biol. Med. 2021, 137, 104851. [Google Scholar] [CrossRef]
- Grebner, C.; Malmerberg, E.; Shewmaker, A.; Batista, J.; Nicholls, A.; Sadowski, J. Virtual Screening in the Cloud: How Big Is Big Enough? J. Chem. Inf. Model. 2020, 60, 4274–4282. [Google Scholar] [CrossRef]
- Stumpfe, D.; Bajorath, J. Current Trends, Overlooked Issues, and Unmet Challenges in Virtual Screening. J. Chem. Inf. Model. 2020, 60, 4112–4115. [Google Scholar] [CrossRef] [PubMed]
- Piccirillo, E.; do Amaral, A.T. Virtual Screening of Bioactive Compounds: Concepts and Aplications. Quim. Nova 2018, 41, 662–677. [Google Scholar] [CrossRef]
- Malvezzi, A.; Queiroz, R.F.; de Rezende, L.; Augusto, O.; do Amaral, A.T. MPO Inhibitors Selected by Virtual Screening. Mol. Inform. 2011, 30, 605–613. [Google Scholar] [CrossRef]
- Viviani, L.G.; Piccirillo, E.; Ulrich, H.; do Amaral, A.T. Virtual Screening Approach for the Identification of Hydroxamic Acids as Novel Human Ecto-5′-Nucleotidase Inhibitors. J. Chem. Inf. Model. 2020, 60, 621–630. [Google Scholar] [CrossRef]
- Hawkins, P.C.D.; Skillman, A.G.; Nicholls, A. Comparison of Shape-Matching and Docking as Virtual Screening Tools. J. Med. Chem. 2007, 50, 74–82. [Google Scholar] [CrossRef]
- Nicholls, A.; McGaughey, G.B.; Sheridan, R.P.; Good, A.C.; Warren, G.; Mathieu, M.; Muchmore, S.W.; Brown, S.P.; Grant, J.A.; Haigh, J.A.; et al. Molecular Shape and Medicinal Chemistry: A Perspective. J. Med. Chem. 2010, 53, 3862–3886. [Google Scholar] [CrossRef]
- Kirchmair, J.; Distinto, S.; Markt, P.; Schuster, D.; Spitzer, G.M.; Liedl, K.R.; Wolber, G. How to Optimize Shape-Based Virtual Screening: Choosing the Right Query and Including Chemical Information. J. Chem. Inf. Model. 2009, 49, 678–692. [Google Scholar] [CrossRef] [PubMed]
- Tawada, M.; Fushimi, M.; Masuda, K.; Sun, H.; Uchiyama, N.; Kosugi, Y.; Lane, W.; Tjhen, R.; Endo, S.; Koike, T. Discovery of a Novel and Brain-Penetrant O-GlcNAcase Inhibitor via Virtual Screening, Structure-Based Analysis, and Rational Lead Optimization. J. Med. Chem. 2021, 64, 1103–1115. [Google Scholar] [CrossRef]
- Ferreira De Freitas, R.; Liu, Y.; Szewczyk, M.M.; Mehta, N.; Li, F.; McLeod, D.; Zepeda-Velázquez, C.; Dilworth, D.; Hanley, R.P.; Gibson, E.; et al. Discovery of Small-Molecule Antagonists of the PWWP Domain of NSD2. J. Med. Chem. 2021, 64, 1584–1592. [Google Scholar] [CrossRef]
- Sotriffer, C. Docking of Covalent Ligands: Challenges and Approaches. Mol. Inform. 2018, 37, 1800062. [Google Scholar] [CrossRef]
- Lyu, J.; Irwin, J.J.; Shoichet, B.K. Modeling the Expansion of Virtual Screening Libraries. Nat. Chem. Biol. 2023, 19, 712–718. [Google Scholar] [CrossRef]
- Bender, B.J.; Gahbauer, S.; Luttens, A.; Lyu, J.; Webb, C.M.; Stein, R.M.; Fink, E.A.; Balius, T.E.; Carlsson, J.; Irwin, J.J.; et al. A Practical Guide to Large-Scale Docking. Nat. Protoc. 2021, 16, 4799–4832. [Google Scholar] [CrossRef]
- Kitchen, D.B.; Decornez, H.; Furr, J.R.; Bajorath, J. Docking and Scoring in Virtual Screening for Drug Discovery: Methods and Applications. Nat. Rev. Drug Discov. 2004, 3, 935–949. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Shoichet, B.K. Docking Screens for Novel Ligands Conferring New Biology. J. Med. Chem. 2016, 59, 4103–4120. [Google Scholar] [CrossRef]
- Piccirillo, E.; Alegria, T.G.P.; Discola, K.F.; Cussiol, J.R.R.; Domingos, R.M.; De Oliveira, M.A.; de Rezende, L.; Netto, L.E.S.; do Amaral, A.T. Structural Insights on the Efficient Catalysis of Hydroperoxide Reduction by Ohr: Crystallographic and Molecular Dynamics Approaches. PLoS ONE 2018, 13, e0196918. [Google Scholar] [CrossRef] [PubMed]
- Viviani, L.G.; Kokh, D.B.; Wade, R.C.; do Amaral, A.T. Molecular Dynamics Simulations of the Human Ecto-5′-Nucleotidase (h-Ecto-5′-NT, CD73): Insights into Protein Flexibility and Binding Site Dynamics. J. Chem. Inf. Model. 2023, 63, 4691–4707. [Google Scholar] [CrossRef] [PubMed]
- Kraemer, O.; Hazemann, I.; Podjarny, A.D.; Klebe, G. Virtual Screening for Inhibitors of Human Aldose Reductase. Proteins 2004, 55, 814–823. [Google Scholar] [CrossRef]
- Fischer, A.; Smieško, M.; Sellner, M.; Lill, M.A. Decision Making in Structure-Based Drug Discovery: Visual Inspection of Docking Results. J. Med. Chem. 2021, 64, 2489–2500. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, A.; Wang, Q.; Ao, H.; Shoblock, J.R.; Lord, B.; Aluisio, L.; Fraser, I.; Nepomuceno, D.; Neff, R.A.; Welty, N.; et al. Pharmacological Characterization of a Novel Centrally Permeable P2X7 Receptor Antagonist: JNJ-47965567. Br. J. Pharmacol. 2013, 170, 624–640. [Google Scholar] [CrossRef] [PubMed]
- Karasawa, A.; Kawate, T. Structural Basis for Subtype-Specific Inhibition of the P2X7 Receptor. eLife 2016, 5, e22153. [Google Scholar] [CrossRef]
- Hawkins, P.C.D.; Nicholls, A. Conformer Generation with OMEGA: Learning from the Data Set and the Analysis of Failures. J. Chem. Inf. Model. 2012, 52, 2919–2936. [Google Scholar] [CrossRef]
- Hawkins, P.C.D.; Skillman, A.G.; Warren, G.L.; Ellingson, B.A.; Stahl, M.T. Conformer Generation with OMEGA: Algorithm and Validation Using High Quality Structures from the Protein Databank and Cambridge Structural Database. J. Chem. Inf. Model. 2010, 50, 572–584. [Google Scholar] [CrossRef]
- Goodford, P.J. A Computational Procedure for Determining Energetically Favorable Binding Sites on Biologically Important Macromolecules. J. Med. Chem. 1985, 28, 849–857. [Google Scholar] [CrossRef]
- Wade, R.C.; Goodford, P.J. Further Development of Hydrogen Bond Functions for Use in Determining Energetically Favorable Binding Sites on Molecules of Known Structure. 2. Ligand Probe Groups with the Ability to Form More Than Two Hydrogen Bonds. J. Med. Chem. 1993, 36, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Wade, R.C.; Clark, K.J.; Goodford, P.J. Further Development of Hydrogen Bond Functions for Use in Determining Energetically Favorable Binding Sites on Molecules of Known Structure. 1. Ligand Probe Groups with the Ability to Form Two Hydrogen Bonds. J. Med. Chem. 1993, 36, 140–147. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, A.E.; Yoshioka, C.; Mansoor, S.E. Full-Length P2X7 Structures Reveal How Palmitoylation Prevents Channel Desensitization. Cell 2019, 179, 659–670.e13. [Google Scholar] [CrossRef] [PubMed]
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved Protein—Ligand Docking Using GOLD. Proteins Struct. Funct. Bioinform. 2003, 623, 609–623. [Google Scholar] [CrossRef] [PubMed]
- Korb, O.; Stützle, T.; Exner, T.E. Empirical Scoring Functions for Advanced Protein-Ligand Docking with PLANTS. J. Chem. Inf. Model. 2009, 49, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Guedes, I.A.; Costa, L.S.C.; dos Santos, K.B.; Karl, A.L.M.; Rocha, G.K.; Teixeira, I.M.; Galheigo, M.M.; Medeiros, V.; Krempser, E.; Custódio, F.L.; et al. Drug Design and Repurposing with DockThor-VS Web Server Focusing on SARS-CoV-2 Therapeutic Targets and Their Non-Synonym Variants. Sci. Rep. 2021, 11, 5543. [Google Scholar] [CrossRef] [PubMed]
- Wolber, G.; Langer, T. LigandScout: 3-D Pharmacophores Derived from Protein-Bound Ligands and Their Use as Virtual Screening Filters. J. Chem. Inf. Model. 2008, 45, 160–169. [Google Scholar] [CrossRef]
- Salentin, S.; Schreiber, S.; Haupt, V.J.; Adasme, M.F.; Schroeder, M. PLIP: Fully Automated Protein-Ligand Interaction Profiler. Nucleic Acids Res. 2015, 43, W443–W447. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Antechamber, an Accessory Software Package for Molecular Mechanical Calculations. J. Chem. Inf. Comput. Sci. 2001, 222, U403. [Google Scholar]
- Silva, D.; Vranken, B.F. ACPYPE-AnteChamber PYthon Parser InterfacE. BMC Res. Notes 2012, 5, 367. [Google Scholar]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Jakalian, A.; Jack, D.B.; Bayly, C.I. Fast, Efficient Generation of High-quality Atomic Charges. AM1-BCC Model: II. Parameterization and Validation. J. Comput. Chem. 2002, 23, 1623–1641. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Showalter, S.A.; Brüschweiler, R. Validation of Molecular Dynamics Simulations of Biomolecules Using NMR Spin Relaxation as Benchmarks: Application to the AMBER99SB Force Field. J. Chem. Theory Comput. 2007, 3, 961–975. [Google Scholar] [CrossRef] [PubMed]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald: An N⋅ Log (N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Grubmüller, H.; Heller, H.; Windemuth, A.; Schulten, K. Generalized Verlet Algorithm for Efficient Molecular Dynamics Simulations with Long-Range Interactions. Mol. Simul. 1991, 6, 121–142. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A Linear Constraint Solver for Molecular Simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Hess, B. P-LINCS: A Parallel Linear Constraint Solver for Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 116–122. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DB | Compound (Structure) | Molecular Formula | MW (g∙mol−1) | clogP | TPSA (Å) | HA | HD | GOLD ChemPLP Score | DockThor Score |
|---|---|---|---|---|---|---|---|---|---|
| MegX | 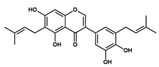 NP-016468 | C25H26O6 | 422.47 | 6.23 | 107.22 | 6 | 4 | 89.99 | −10.924 |
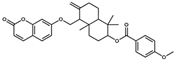 NP-025047 | C32H36O6 | 516.63 | 6.67 | 71.06 | 4 | 0 | 94.67 | −12.169 | |
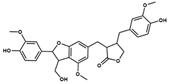 NP-025357 | C30H32O9 | 536.57 | 3.77 | 123.91 | 8 | 3 | 101.02 | −11.800 | |
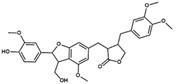 NP-025358 | C31H34O9 | 550.60 | 3.91 | 112.91 | 8 | 2 | 99.42 | −11.984 | |
| NatX | 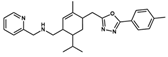 NAT28-412055 | C27H34N4O | 430.59 | 4.66 | 63.84 | 4 | 1 | 99.19 | −11.061 |
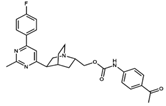
 NAT28-416626 | C26H29FN4O2 | 448.54 | 3.72 | 80.91 | 4 | 1 | 101.31 | −11.352 | |
 NAT14-350419 | C33H41N5O3 | 555.72 | 4.03 | 90.71 | 4 | 2 | 99.97 | −11.554 | |
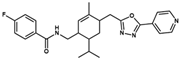
 NAT13-340161 | C28H29FN4O3 | 488.56 | 4.62 | 84.42 | 6 | 1 | 99.96 | −11.028 |
| Compound | ||||||||
|---|---|---|---|---|---|---|---|---|
| NP-016468 | NP-025047 | NP-025357 | NP-025358 | |||||
| Residues Subunit A | GOLD | DockThor | GOLD | DockThor | GOLD | DockThor | GOLD | DockThor |
| F88 | HD | HD | HD | HD | HD | HD | ||
| A91 | HB | HB | ||||||
| D92 | HD | |||||||
| Y93 | HB | |||||||
| T94 | HB | HB | ||||||
| F95 | HD | HD | HD | HD | HD | HD | HD | HD |
| P96 | HD | |||||||
| F103 | HD | HD | HD | |||||
| M105 | HD | HD | HD | |||||
| F108 | HD | |||||||
| K110 | HB | HB | HB | HB | HB | |||
| F293 | HD | HD | HD | HD | HD | |||
| Y295 | HD | HD | HB | HB, π-π | HB, HB | HD, HB, π-π | ||
| K297 | HB | HB | π-C, SB | HB | π-π | |||
| I310 | HD | HD | HD | HD | HD | HD | ||
| V312 | HD | HD | HD | |||||
| Residues Subunit B | GOLD | DockThor | GOLD | DockThor | GOLD | DockThor | GOLD | DockThor |
| W167 | HD | HD | HD | |||||
| F293 | HD | |||||||
| Y295 | HD | HD | ||||||
| A296 | HB | |||||||
| Y298 | HB | HD, HB | HD | HD, HB | HD | HD | ||
| Residues Subunit C | GOLD | DockThor | GOLD | DockThor | GOLD | DockThor | GOLD | DockThor |
| F95 | HD | |||||||
| Compound ID | ||||||||
|---|---|---|---|---|---|---|---|---|
| NAT28-412055 | NAT28-416626 | NAT14-350419 | NAT13-340161 | |||||
| Residues Subunit A | GOLD | DockThor | GOLD | DockThor | GOLD | DockThor | GOLD | DockThor |
| F88 | HD | HD | HD | HD | HD | HD | HD | |
| A91 | ||||||||
| D92 | HD | HD | ||||||
| T94 | ||||||||
| F95 | HD | HD | HD | HD | HD | HD | HD | HD |
| F103 | HD | HD | HD | HD | HD | HD | ||
| M105 | HD | HD | HD | |||||
| F108 | π-π | HD | ||||||
| K110 | HB | HD | HB | |||||
| Y291 | HB | |||||||
| F293 | HD | HD | HD | HD | HD | HD | HD | |
| Y295 | HD | HD | π-π | HB | HD | HD | HB | |
| K297 | HBA | HBA | π-C | π -C | π-C | HB | HB | |
| I310 | HD | HD | HD | HD | HD | HD | HD | |
| V312 | HD | HD | ||||||
| Residues Subunit B | GOLD | DockThor | GOLD | DockThor | GOLD | DockThor | GOLD | DockThor |
| F95 | HD | HD | ||||||
| W167 | HD | HD | HD | |||||
| Y293 | HD | HD | ||||||
| Y295 | HD | HD | HD | |||||
| A296 | HD | HD | ||||||
| Y298 | HD | HD | HD | HD | HD, HB | HB | HB | |
| Residues Subunit C | GOLD | DockThor | GOLD | DockThor | GOLD | DockThor | GOLD | DockThor |
| F95 | HD | |||||||
| P96 | HD | HD | HD | |||||
| Q98 | HB | HB | ||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferreira, N.C.d.S.; Viviani, L.G.; Lima, L.M.; Amaral, A.T.d.; Romano, J.V.P.; Fortunato, A.L.; Soares, R.F.; Alberto, A.V.P.; Coelho Neto, J.A.; Alves, L.A. A Hybrid Approach Combining Shape-Based and Docking Methods to Identify Novel Potential P2X7 Antagonists from Natural Product Databases. Pharmaceuticals 2024, 17, 592. https://doi.org/10.3390/ph17050592
Ferreira NCdS, Viviani LG, Lima LM, Amaral ATd, Romano JVP, Fortunato AL, Soares RF, Alberto AVP, Coelho Neto JA, Alves LA. A Hybrid Approach Combining Shape-Based and Docking Methods to Identify Novel Potential P2X7 Antagonists from Natural Product Databases. Pharmaceuticals. 2024; 17(5):592. https://doi.org/10.3390/ph17050592
Chicago/Turabian StyleFerreira, Natiele Carla da Silva, Lucas Gasparello Viviani, Lauro Miranda Lima, Antonia Tavares do Amaral, João Victor Paiva Romano, Anderson Lage Fortunato, Rafael Ferreira Soares, Anael Viana Pinto Alberto, Jose Aguiar Coelho Neto, and Luiz Anastacio Alves. 2024. "A Hybrid Approach Combining Shape-Based and Docking Methods to Identify Novel Potential P2X7 Antagonists from Natural Product Databases" Pharmaceuticals 17, no. 5: 592. https://doi.org/10.3390/ph17050592
APA StyleFerreira, N. C. d. S., Viviani, L. G., Lima, L. M., Amaral, A. T. d., Romano, J. V. P., Fortunato, A. L., Soares, R. F., Alberto, A. V. P., Coelho Neto, J. A., & Alves, L. A. (2024). A Hybrid Approach Combining Shape-Based and Docking Methods to Identify Novel Potential P2X7 Antagonists from Natural Product Databases. Pharmaceuticals, 17(5), 592. https://doi.org/10.3390/ph17050592