
Investigating the Antiviral Properties of Nyctanthes arbor-tristis Linn against the Ebola, SARS-CoV-2, Nipah, and Chikungunya Viruses: A Computational Simulation Study

 , ,
, ,  and
and 
Abstract
1. Introduction
2. Results
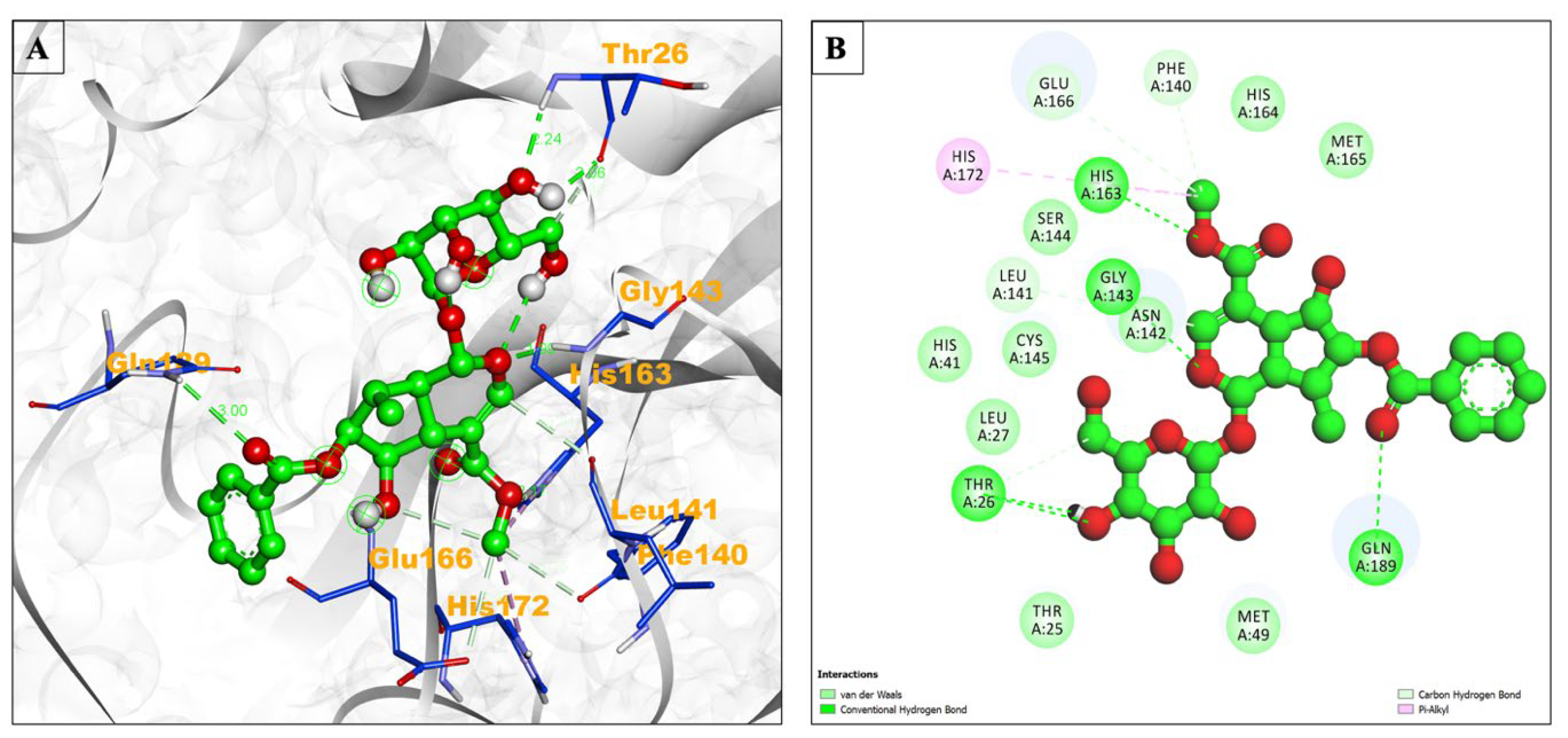
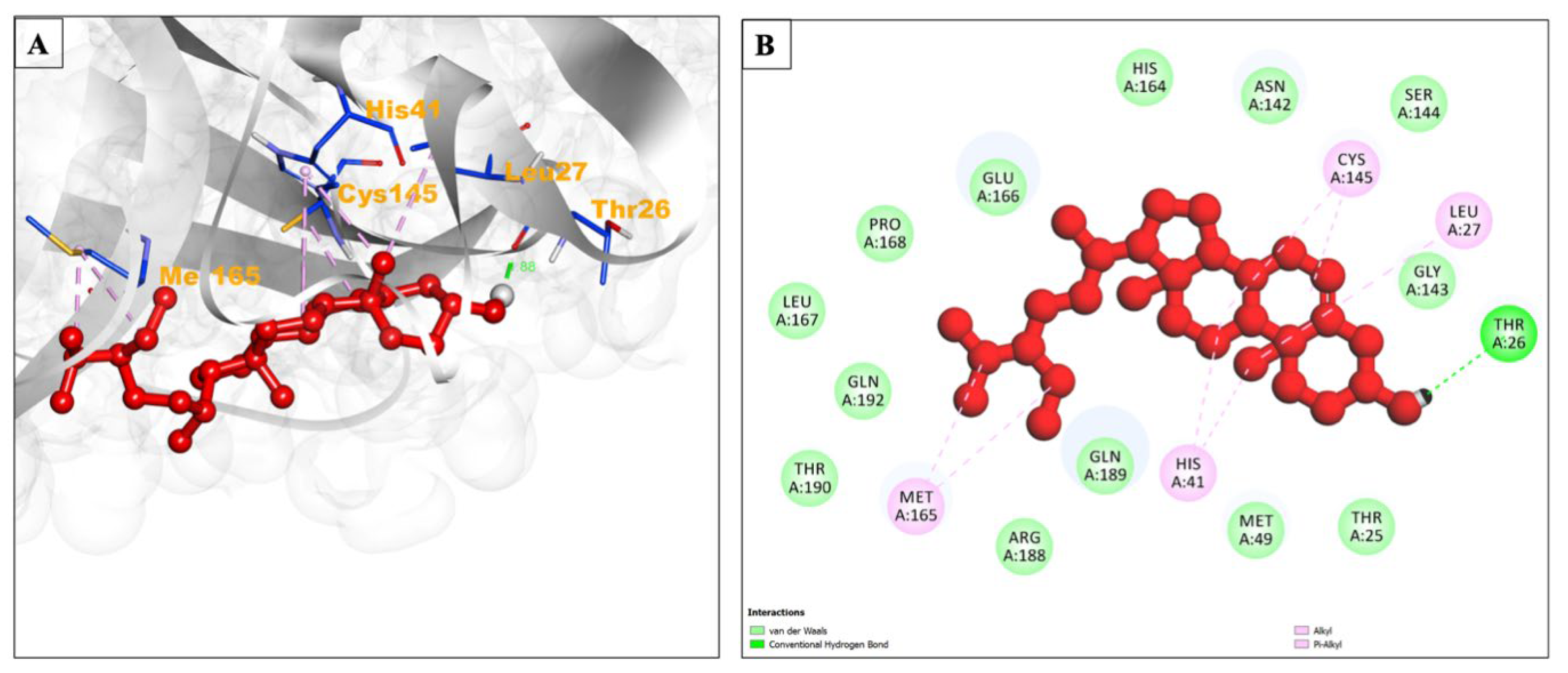
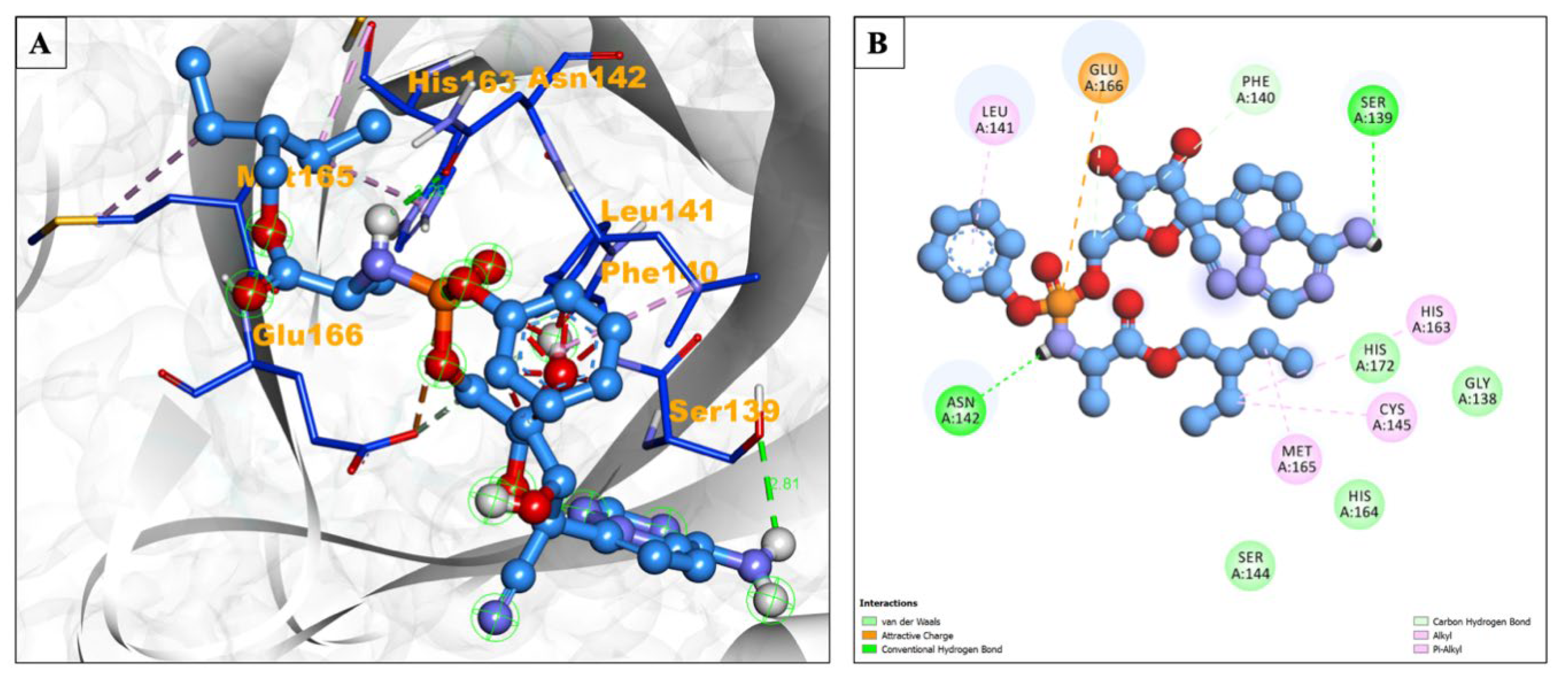
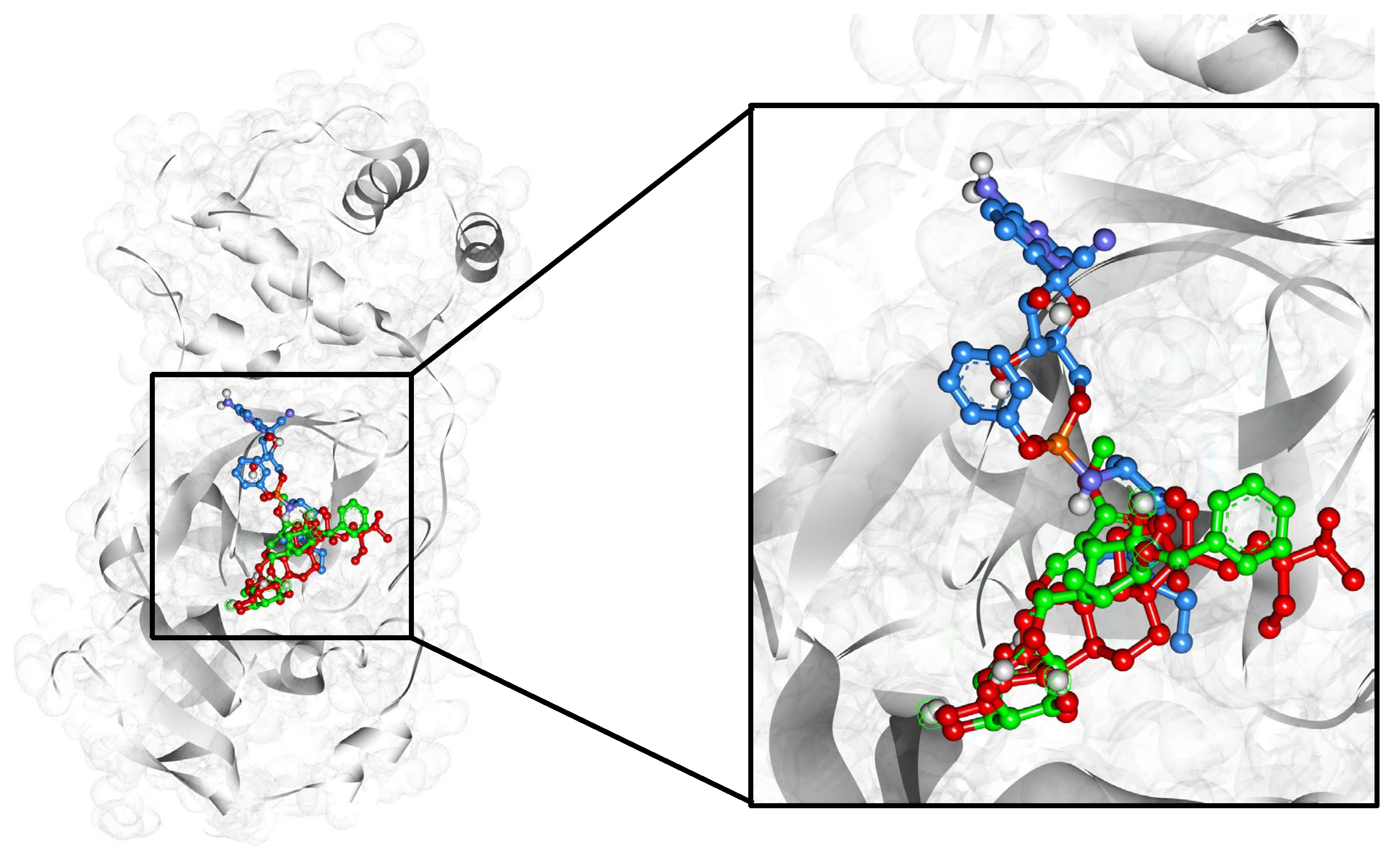
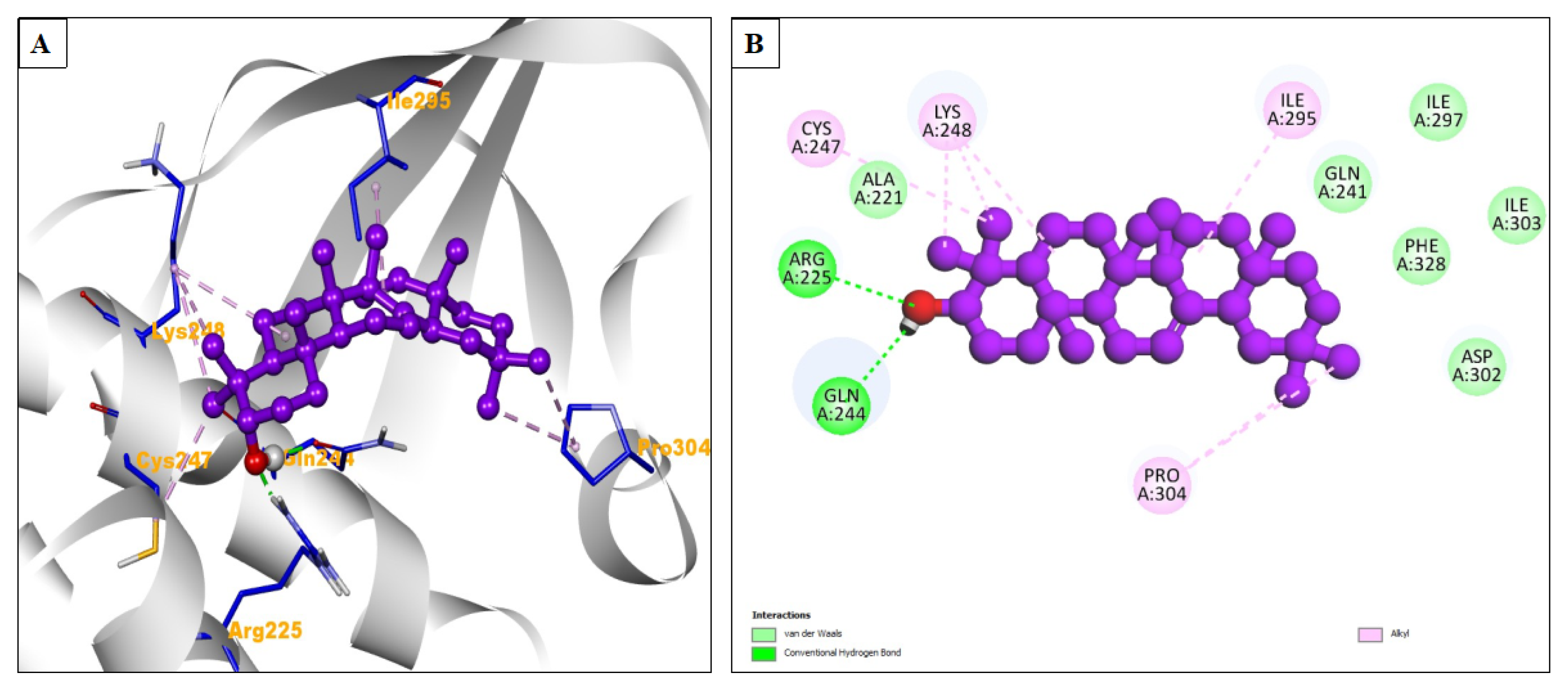
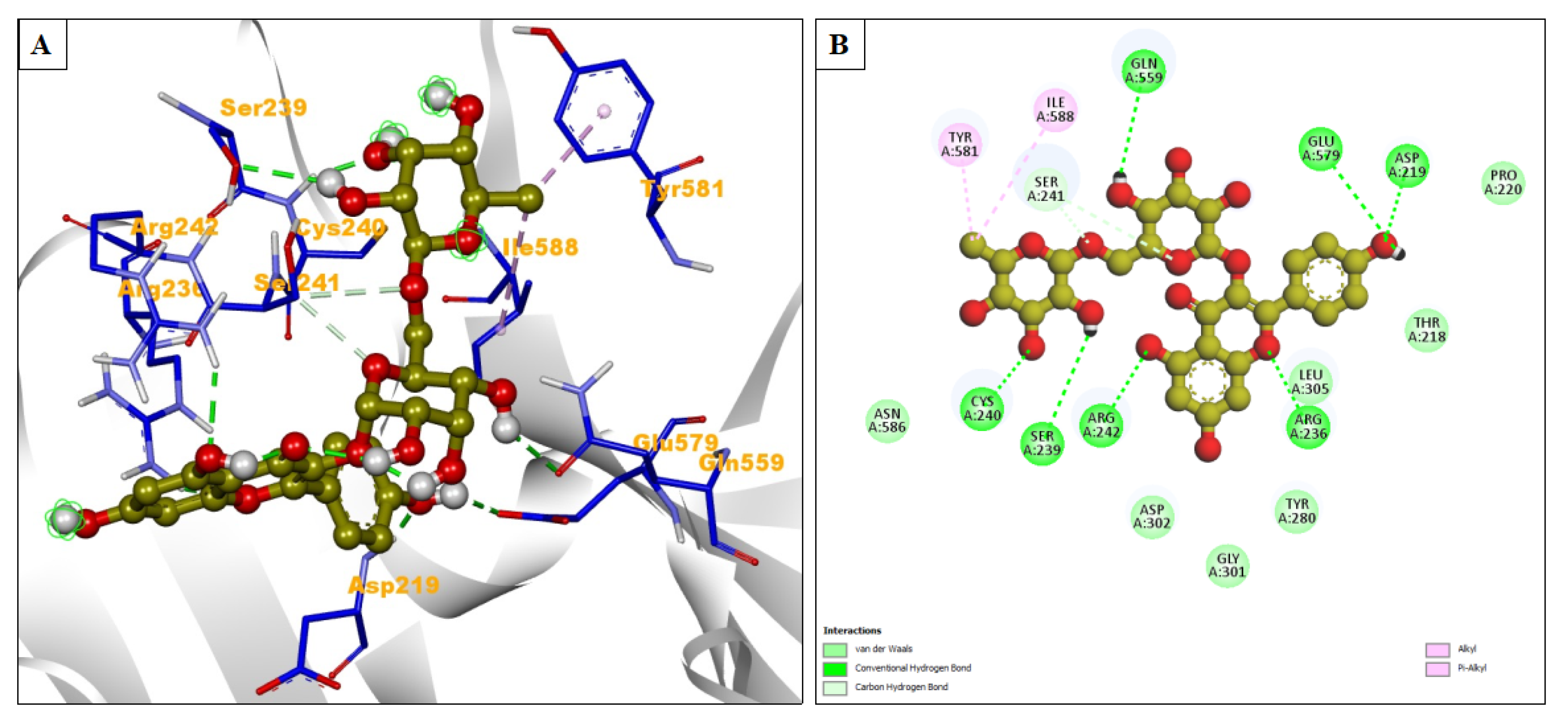
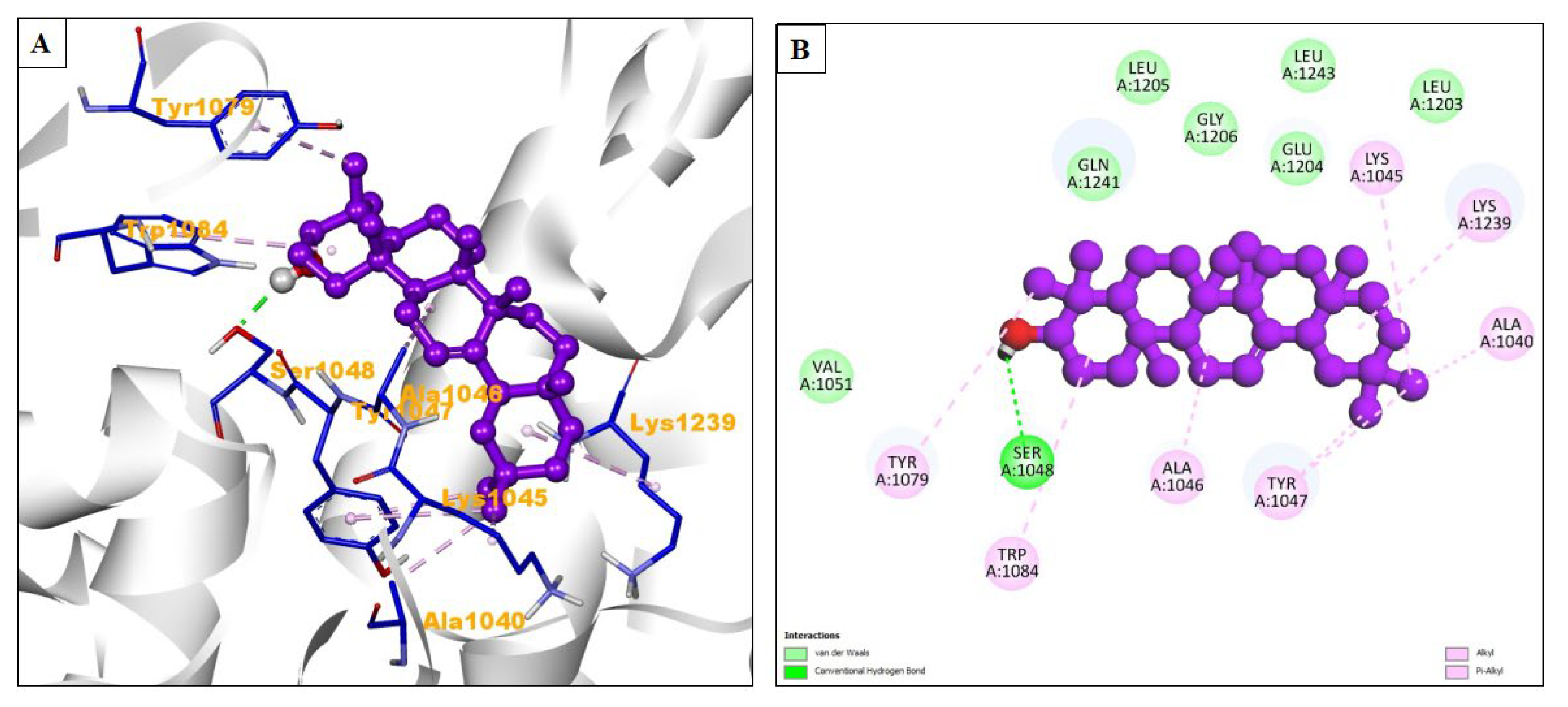
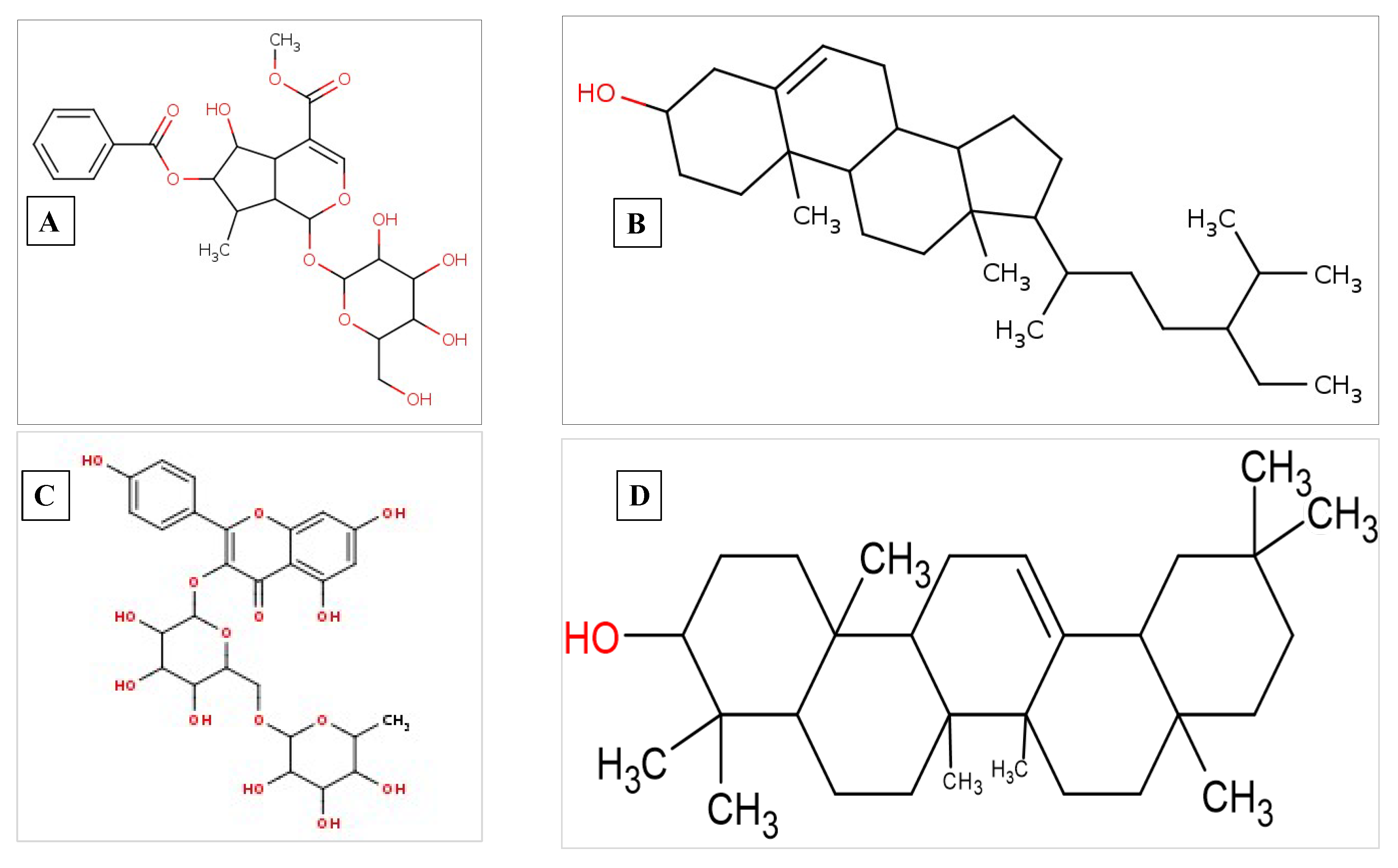
2.1. Docking Data Interpretation
2.2. Drug-Likeness and ADMET Analysis
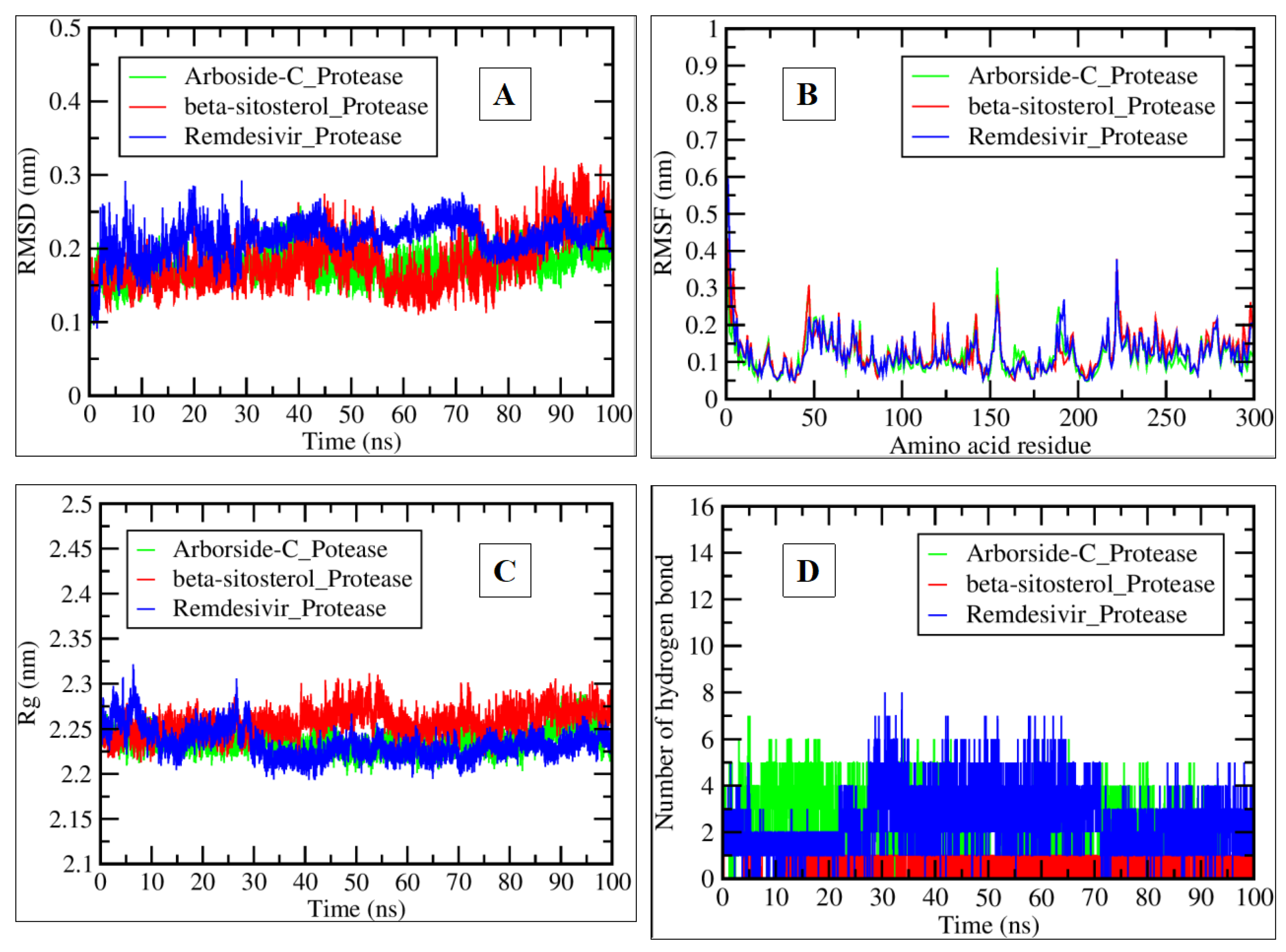
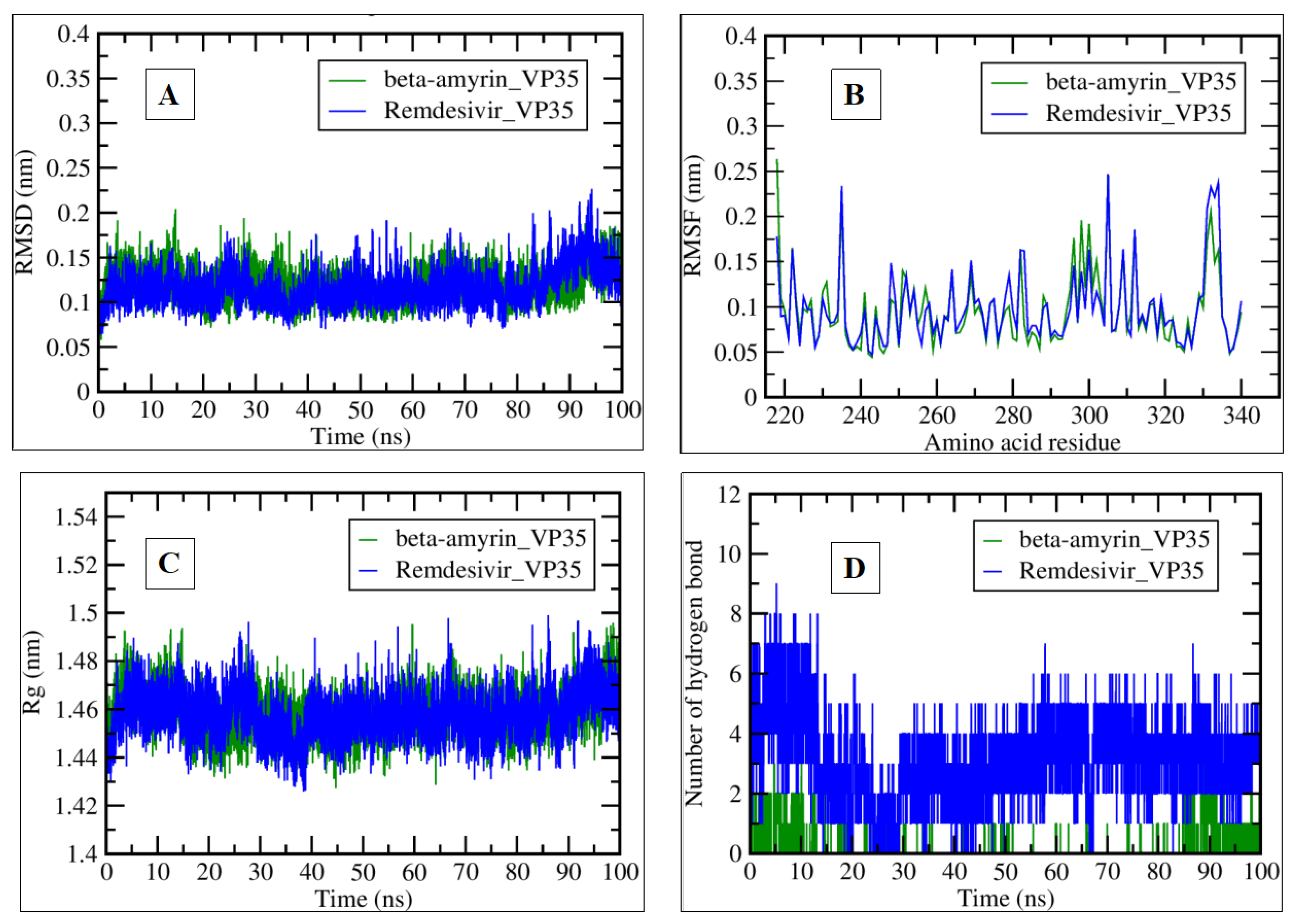
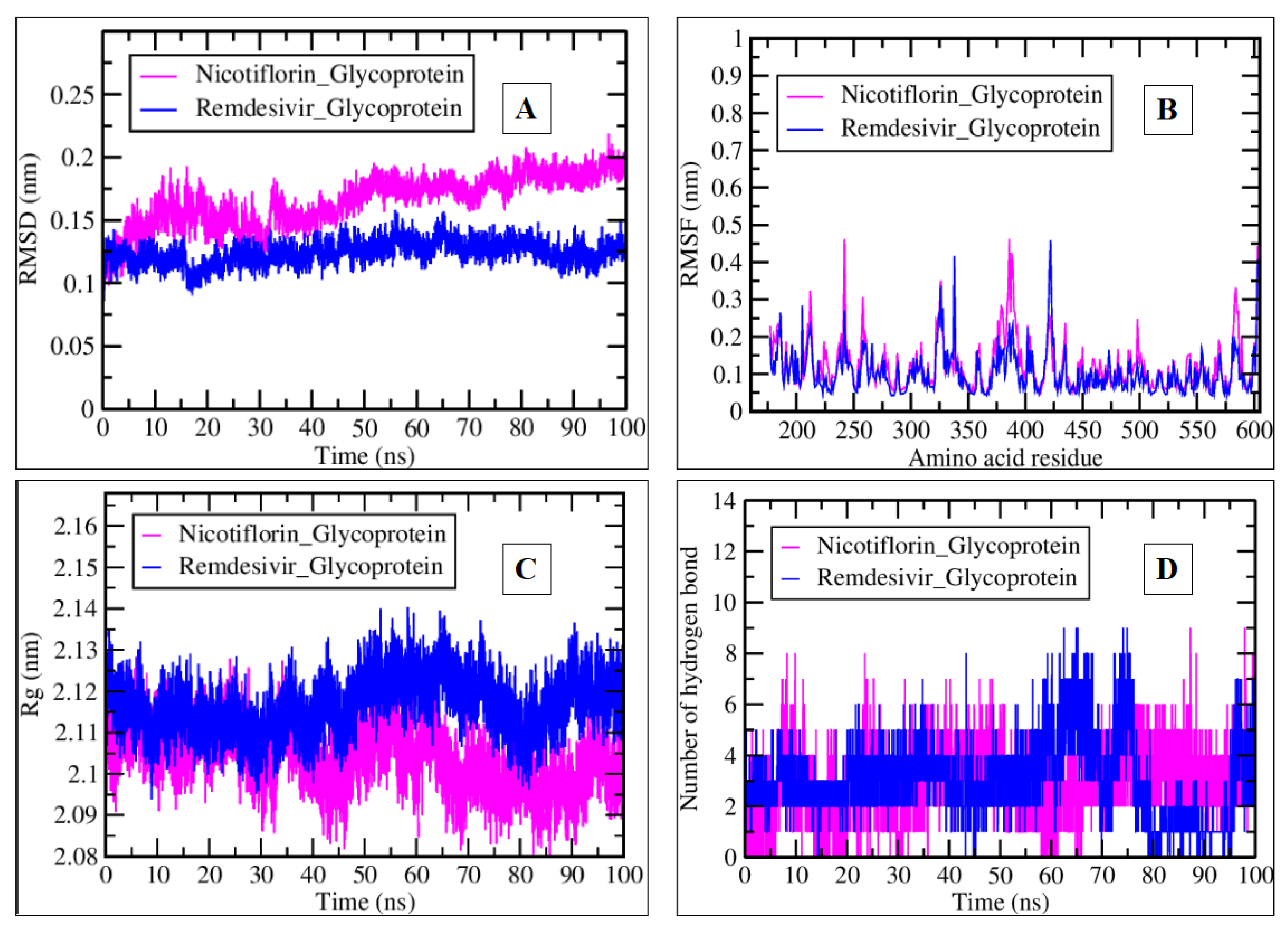
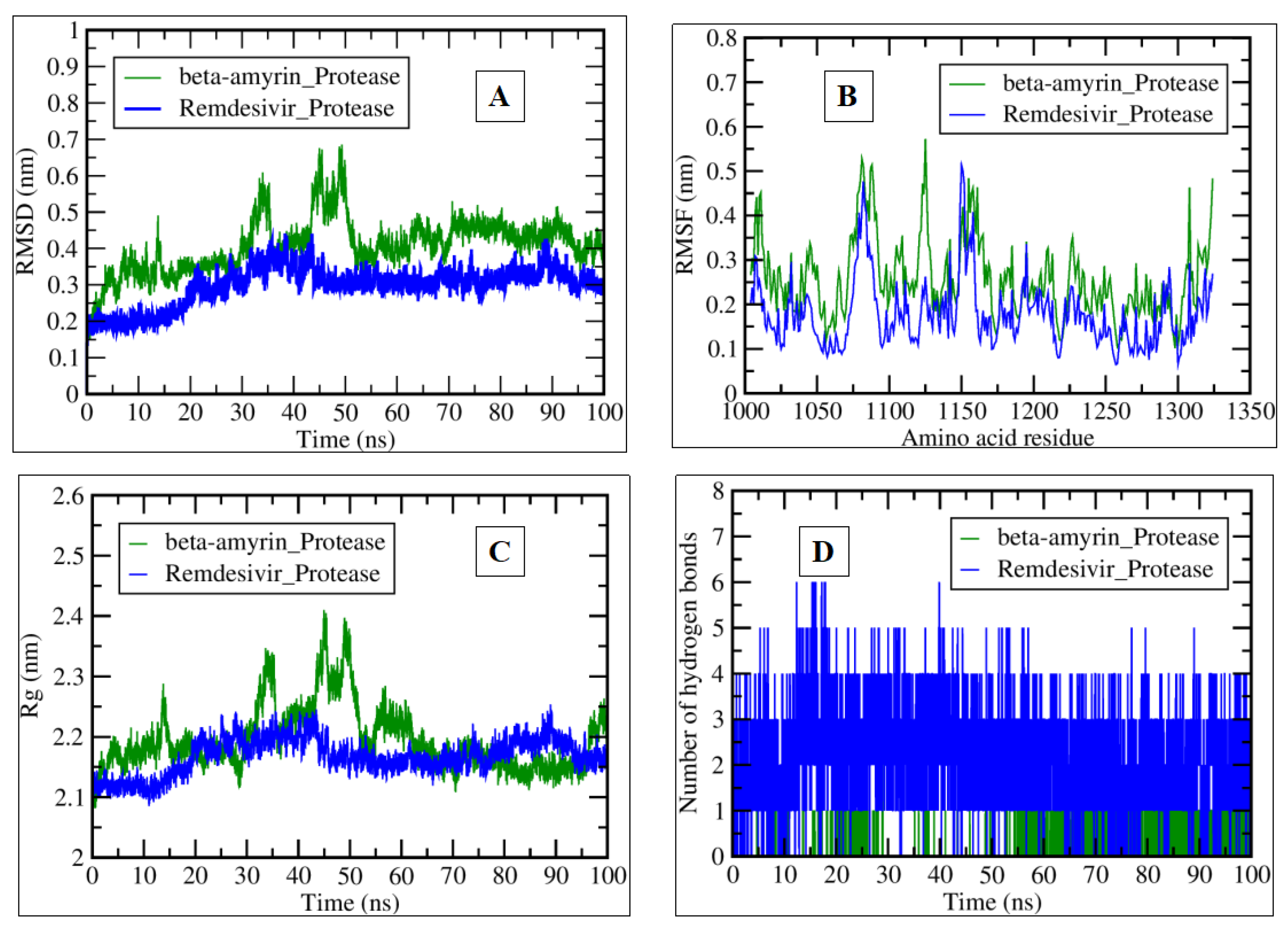
2.3. MDS Analysis
3. Discussion
4. Materials and Methods
4.1. Ligand Preparation
4.2. Viral Receptor Preparation
4.3. Molecular Docking Studies
4.4. Drug-Likeness and ADMET
4.5. MDS
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Barua, A.; Junaid, M.; Shamsuddin, T.; Alam, M.S.; Mouri, N.J.; Akter, R.; Sharmin, T.; Hosen, S. Nyctanthes arbor-tristis Linn.: A Review on its Traditional Uses, Phytochemistry, Pharmacological Activities, and Toxicity. Curr. Tradit. Med. 2023, 9, 10–22. [Google Scholar]
- Paikara, D.; Singh, S.; Pandey, B. Phytochemical analysis of leave extract of Nyctanthes arbortristis. IOSR J. Environ. Sci. Toxicol. Food Technol. 2015, 1, 39–42. [Google Scholar]
- Debnath, S.; Hazarika, A.; Sarma, J. Evaluation of Analgesic Activity of Ethanolic, Hydroethanolic, Aqueous and Chloroform Extracts of Nyctanthes arbortristis Leaves. Biol. Forum–Int. J. 2023, 15, 28–35. [Google Scholar]
- Singh, A.K.; Kumar, P.; Rajput, V.D.; Mishra, S.K.; Tiwari, K.N.; Singh, A.K.; Minkina, T.; Pandey, A.K. Phytochemicals, Antioxidant, Anti-inflammatory Studies, and Identification of Bioactive Compounds Using GC–MS of Ethanolic Novel Polyherbal Extract. Appl. Biochem. Biotechnol. 2023, 457, 4447–4468. [Google Scholar] [CrossRef]
- Jamal, Q.M.S. Antiviral Potential of Plants against COVID-19 during Outbreaks-An Update. Int. J. Mol. Sci. 2022, 23, 13564. [Google Scholar] [CrossRef] [PubMed]
- Naidu, S.A.G.; Mustafa, G.; Clemens, R.A.; Naidu, A.S. Plant-Derived Natural Non-Nucleoside Analog Inhibitors (NNAIs) against RNA-Dependent RNA Polymerase Complex (nsp7/nsp8/nsp12) of SARS-CoV-2. J. Diet. Suppl. 2023, 20, 254–283. [Google Scholar] [CrossRef] [PubMed]
- Jalal, K.; Khan, K.; Basharat, Z.; Abbas, M.N.; Uddin, R.; Ali, F.; Khan, S.A.; Hassan, S.S.U. Reverse vaccinology approach for multi-epitope centered vaccine design against delta variant of the SARS-CoV-2. Environ. Sci. Pollut. Res. Int. 2022, 29, 60035–60053. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wu, J.; Nie, J.; Zhang, L.; Hao, H.; Liu, S.; Zhao, C.; Zhang, Q.; Liu, H.; Nie, L. The impact of mutations in SARS-CoV-2 spike on viral infectivity and antigenicity. Cell 2020, 182, 1284–1294.e1289. [Google Scholar] [CrossRef] [PubMed]
- Plante, J.A.; Liu, Y.; Liu, J.; Xia, H.; Johnson, B.A.; Lokugamage, K.G.; Zhang, X.; Muruato, A.E.; Zou, J.; Fontes-Garfias, C.R.; et al. Spike mutation D614G alters SARS-CoV-2 fitness. Nature 2021, 592, 116–121. [Google Scholar] [CrossRef]
- Darko, L.K.; Broni, E.; Amuzu, D.S.; Wilson, M.D.; Parry, C.S.; Kwofie, S.K. Computational study on potential novel anti-Ebola virus protein VP35 natural compounds. Biomedicines 2021, 9, 1796. [Google Scholar] [CrossRef]
- Biering, S.B.; Huang, A.; Vu, A.T.; Robinson, L.R.; Bradel-Tretheway, B.; Choi, E.; Lee, B.; Aguilar, H.C. N-Glycans on the Nipah virus attachment glycoprotein modulate fusion and viral entry as they protect against antibody neutralization. J. Virol. 2012, 86, 11991–12002. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Oezguen, N.; Urvil, P.; Ferguson, C.; Dann, S.M.; Savidge, T.C. Regulation of protein-ligand binding affinity by hydrogen bond pairing. Sci. Adv. 2016, 2, e1501240. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.S.; Jacobsen, E.N. Asymmetric catalysis by chiral hydrogen-bond donors. Angew. Chem. Int. Ed. 2006, 45, 1520–1543. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.H.; Linstead, E.; Swamidass, S.J.; Wang, D.; Baldi, P. ChemDB update--full-text search and virtual chemical space. Bioinformatics 2007, 23, 2348–2351. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2012, 64, 4–17. [Google Scholar] [CrossRef]
- Turner, P. XMGRACE; Version 5.1. 19; Center for Coastal and Land-Margin Research, Oregon Graduate Institute of Science and Technology: Beaverton, OR, USA, 2005; p. 2. [Google Scholar]
- Hubbard, R.E.; Kamran Haider, M. Hydrogen Bonds in Proteins: Role and Strength. In Encyclopedia of Life Sciences; Wiley: Hoboken, NJ, USA, 2010. [Google Scholar]
- Mahmood, N.; Nasir, S.B.; Hefferon, K. Plant-based drugs and vaccines for COVID-19. Vaccines 2020, 9, 15. [Google Scholar] [CrossRef] [PubMed]
- Anywar, G.; Akram, M.; Chishti, M.A. African and asian medicinal plants as a repository for prospective antiviral metabolites against HIV-1 and SARS-CoV-2: A mini review. Front. Pharmacol. 2021, 12, 703837. [Google Scholar] [CrossRef] [PubMed]
- Beressa, T.B.; Deyno, S.; Mtewa, A.G.; Aidah, N.; Tuyiringire, N.; Lukubye, B.; Weisheit, A.; Tolo, C.U.; Ogwang, P.E. Potential benefits of antiviral African medicinal plants in the management of viral infections: Systematic review. Front. Pharmacol. 2021, 12, 682794. [Google Scholar] [CrossRef] [PubMed]
- Aschale, Y.; Tegegne, B.A.; Yihunie, W. Medicinal Plants Used for the Management of Hepatitis Over the Past 15 Years in Ethiopia: A Systematic Review. Hepatic Med. Evid. Res. 2023, 15, 11–19. [Google Scholar] [CrossRef]
- Semenov, V.A.; Krivdin, L.B. Combined computational NMR and molecular docking scrutiny of potential natural SARS-CoV-2 Mpro inhibitors. J. Phys. Chem. B 2022, 126, 2173–2187. [Google Scholar] [CrossRef]
- Verma, S.; Patel, C.N.; Chandra, M. Identification of novel inhibitors of SARS-CoV-2 main protease (Mpro) from Withania sp. by molecular docking and molecular dynamics simulation. J. Comput. Chem. 2021, 42, 1861–1872. [Google Scholar] [CrossRef] [PubMed]
- Patel, C.N.; Jani, S.P.; Jaiswal, D.G.; Kumar, S.P.; Mangukia, N.; Parmar, R.M.; Rawal, R.M.; Pandya, H.A. Identification of antiviral phytochemicals as a potential SARS-CoV-2 main protease (Mpro) inhibitor using docking and molecular dynamics simulations. Sci. Rep. 2021, 11, 20295. [Google Scholar] [CrossRef] [PubMed]
- Saxena, R.S.; Gupta, B.; Saxena, K.K.; Singh, R.C.; Prasad, D.M. Study of anti-inflammatory activity in the leaves of Nyctanthes arbor tristis Linn—An Indian medicinal plant. J. Ethnopharmacol. 1984, 11, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Gahtori, R.; Tripathi, A.H.; Chand, G.; Pande, A.; Joshi, P.; Rai, R.C.; Upadhyay, S.K. Phytochemical Screening of Nyctanthes arbor-tristis Plant Extracts and Their Antioxidant and Antibacterial Activity Analysis. Appl. Biochem. Biotechnol. 2024, 196, 436–456. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, J.; Pal, A. Nyctanthes arbor-tristis Linn--a critical ethnopharmacological review. J. Ethnopharmacol. 2013, 146, 645–658. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.; Singh, A.P.; Singh, A.P. Nyctanthes arbor-tristis: A comprehensive review. World J. Curr. Med. Pharm. Res. 2021, 3, 74–78. [Google Scholar] [CrossRef]
- Naidu, S.A.; Tripathi, Y.B.; Shree, P.; Clemens, R.A.; Naidu, A.S. Phytonutrient inhibitors of SARS-CoV-2/NSP5-encoded main protease (Mpro) autocleavage enzyme critical for COVID-19 pathogenesis. J. Diet. Suppl. 2023, 20, 284–311. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Guo, Q.; Jia, T.; Zhang, X.; Guo, D.; Jia, Y.; Li, J.; Sun, J. Mechanism of Action of Nicotiflorin from Tricyrtis maculata in the Treatment of Acute Myocardial Infarction: From Network Pharmacology to Experimental Pharmacology. Drug Des. Devel Ther. 2021, 15, 2179–2191. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Guo, M.; Zhang, G.; Xu, X.; Li, Q. Nicotiflorin reduces cerebral ischemic damage and upregulates endothelial nitric oxide synthase in primarily cultured rat cerebral blood vessel endothelial cells. J. Ethnopharmacol. 2006, 107, 143–150. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B. PubChem in 2021: New data content and improved web interfaces. Nucleic Acids Res. 2021, 49, D1388–D1395. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef]
- Biovia, D.S. Discovery Studio, Version 21.1. 0; Dassault Systèmes: San Diego, CA, USA, 2021; Volume 627.
- Abuzinadah, M.F.; Ahmad, V.; Al-Thawdi, S.; Zakai, S.A.; Jamal, Q.M.S. Exploring the Binding Interaction of Active Compound of Pineapple against Foodborne Bacteria and Novel Coronavirus (SARS-CoV-2) Based on Molecular Docking and Simulation Studies. Nutrients 2022, 14, 3045. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.R.; Bruccoleri, R.E.; Olafson, B.D.; States, D.J.; Swaminathan, S.A.; Karplus, M. CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 1983, 4, 187–217. [Google Scholar] [CrossRef]
- Bjelkmar, P.; Larsson, P.; Cuendet, M.A.; Hess, B.; Lindahl, E. Implementation of the CHARMM force field in GROMACS: Analysis of protein stability effects from correction maps, virtual interaction sites, and water models. J. Chem. Theory Comput. 2010, 6, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef]
- Brown, C.S.; Lee, M.S.; Leung, D.W.; Wang, T.; Xu, W.; Luthra, P.; Anantpadma, M.; Shabman, R.S.; Melito, L.M.; MacMillan, K.S.; et al. In Silico Derived Small Molecules Bind the Filovirus VP35 Protein and Inhibit Its Polymerase Cofactor Activity. J. Mol. Biol. 2014, 426, 2045–2058. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Rajashankar, K.R.; Chan, Y.-P.; Himanen, J.P.; Broder, C.C.; Nikolov, D.B. Host cell recognition by the henipaviruses: Crystal structures of the Nipah G attachment glycoprotein and its complex with ephrin-B3. Proc. Natl. Acad. Sci. USA 2008, 105, 9953–9958. [Google Scholar] [CrossRef]
- Cheung, J.; Franklin, M.; Mancia, F.; Rudolph, M.; Cassidy, M.; Gary, E.; Burshteyn, F.; Love, J. Structure of the Chikungunya Virus nsP2 Protease; Worldwide Protein Data Bank: Piscataway, NJ, USA, 2011. [Google Scholar] [CrossRef]
- Burley, S.K.; Bhikadiya, C.; Bi, C.; Bittrich, S.; Chao, H.; Chen, L.; Craig, P.A.; Crichlow, G.V.; Dalenberg, K.; Duarte, J.M.; et al. RCSB Protein Data Bank (RCSB.org): Delivery of experimentally-determined PDB structures alongside one million computed structure models of proteins from artificial intelligence/machine learning. Nucleic Acids Res. 2023, 51, D488–D508. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Jamal, Q.M.S.; Khan, M.I.; Alharbi, A.H.; Ahmad, V.; Yadav, B.S. Identification of Natural Compounds of the Apple as Inhibitors against Cholinesterase for the Treatment of Alzheimer’s Disease: An In Silico Molecular Docking Simulation and ADMET Study. Nutrients 2023, 15, 1579. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.E.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Zoete, V.; Cuendet, M.A.; Grosdidier, A.; Michielin, O. SwissParam: A fast force field generation tool for small organic molecules. J. Comput. Chem. 2011, 32, 2359–2368. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Tiwari, N.; Verma, J.; Waseem, M.; Subbarao, N.; Munde, M. Estimation of a stronger heparin binding locus in fibronectin domain III 14 using thermodynamics and molecular dynamics. RSC Adv. 2020, 10, 20288–20301. [Google Scholar] [CrossRef]
- Kufareva, I.; Abagyan, R. Methods of protein structure comparison. Homol. Model. Methods Protoc. 2012, 857, 231–257. [Google Scholar]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef]
- Miller, B.R., III; McGee, T.D., Jr.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]
- Valdés-Tresanco, M.S.; Valdés-Tresanco, M.E.; Valiente, P.A.; Moreno, E. gmx_MMPBSA: A New Tool to Perform End-State Free Energy Calculations with GROMACS. J. Chem. Theory Comput. 2021, 17, 6281–6291. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Name | Residues Involved in Hydrophobic Interactions | Inhibition Constant (Ki) | Hydrogen Bond Length (Angstrom) | No. of Hydrogen Bonds | Binding Energy (kcal/mol) | Compound Name |
|---|---|---|---|---|---|---|
| Remdesivir | Gly138, Ser144, His164, His172 | 166.35 μM | 2.81 | :UNK1:H70 - A:SER139:OG | −6.18 | Alkyl/Pi-Alkyl = Leu141, Cys145, His163, Met165 attractive charge Glu166 |
| 2.28 | :UNK1:H60 - A:ASN142:OD1 | |||||
| 3.15 | :UNK1:C16 - A:PHE140:O | |||||
| 2.84 | :UNK1:C16 - A:GLU166:OE2 | |||||
| 2.81 | :UNK1:H70 - A:SER139:OG | |||||
| Arborside-C | Thr25, Leu27, His41, Met49, Ser144, Cys145, His164, Met165 | 456.39 nM | 2.24 | A:THR26:HN - :UNK1:O30 | −8.65 | Pi-Alkyl = His172 |
| 1.98 | A:GLY143:HN - :UNK1:O19 | |||||
| 2.16 | A:HIS163:HE2 - :UNK1:O35 | |||||
| 2.99 | A:GLN189:HE21 - :UNK1:O9 | |||||
| 2.06 | :UNK1:H61 - A:THR26:O | |||||
| 3.4 | :UNK1:C18 - A:LEU141:O | |||||
| 2.86 | :UNK1:C28 - A:THR26:O | |||||
| 3.14 | :UNK1:C36 - A:PHE140:O | |||||
| 3.47 | :UNK1:C36 - A:GLU166:OE2 | |||||
| Beta-sitosterol | Thr25, Met49, Asn142, Gly143, Ser144, His164, Glu166, Pro168, Leu167, Gln189, Arg188, Thr190, Gln192 | 210.29 nM | 1.88 | :UNK1:H67 - A:THR26:O | −9.11 | Alkyl/Pi-Alkyl Leu27, His41, Cys145, Met165 |
| Compound Name | Residues Involved in Hydrophobic Interactions | Inhibition Constant (Ki) | Hydrogen Bond Length (Angstrom) | No. of Hydrogen Bonds | Binding Energy (kcal/mol) | Residues Involved in Other Interactions |
|---|---|---|---|---|---|---|
| Remdesivir | Val221, Val245, Gln244, Val294, Pro293, Asp302, Phe328 | 1.21 mM | 2.33 | A:GLN241:HE22 - A:UNK0:N23 | −3.98 | Pi-Sigma = Lys248 Alkyl/Pi-Alkyl = Ile295, Ile297 |
| 2.09 | A:UNK0:H60 - A:HIS296:O | |||||
| 3.11 | A:HIS296:HN - A:UNK0 | |||||
| Beta-amyrin | Ala221, Gln241, Phe328, Asp302, Ile303, Ile297 | 3.08 μM | 2.05 | A:ARG225:HH11 - :UNK1:O24 | −7.52 | Alkyl = Cys247, Lys248, Ile295, Pro304 |
| Compound Name | Residues Involved in Hydrophobic Interactions | Inhibition Constant (Ki) | Hydrogen Bond Length (Angstrom) | No. of Hydrogen Bonds | Binding Energy (kcal/mol) | Residues Involved in Other Interactions |
|---|---|---|---|---|---|---|
| Remdesivir | Pro353, Gly352, Cys282, Tyr351, Phe458, Trp504, Leu305, Arg242, Asp219, Pro220, Val507, Thr218, Ser241, Leu234 | 25.29 μM | 2.65 | A:ARG236:HH22 - A:UNK0:O9 | −6.27 | Pi-Alkyl = Tyr280, Pro441 Pi-Sigma = His281, Pi-Cation = Lys560, Pi-Anion = Asp302 Attractive charge = Asp302 |
| 2.28 | A:UNK0:H71 - A:GLU579:OE1 | |||||
| 2.2 | A:UNK0:H72 - A:GLU579:OE2 | |||||
| 3.39 | A:UNK0:C18 - A:GLN559:OE1 | |||||
| 2.26 | A:LYS560:HZ2 - A:UNK0 | |||||
| Nicotiflorin | Asn586, Asp302, Gly301, Tyr280, Leu305, Thr218, Pro220 | 20.63 μM | 1.87 | A:ASP219:HN - :LIG1:O36 | −6.39 | Alkyl/Pi-Alkyl = Ile588, Tyr581 |
| 2.85 | A:ARG236:HH21 - :LIG1:O19 | |||||
| 1.93 | A:CYS240:HN - :LIG1:O41 | |||||
| 2.55 | A:ARG242:HH22 - :LIG1:O28 | |||||
| 2.69 | :LIG1:H66 - A:GLU579:OE2 | |||||
| 2.66 | :LIG1:H70 - A:SER239:OG | |||||
| 2.7 | :LIG1:H69 - A:GLN559:OE1 | |||||
| 2.9 | A:SER241:CB - :LIG1:O15 | |||||
| 3.4 | A:SER241:CB - :LIG1:O8 |
| Compound Name | Residues Involved in Hydrophobic Interactions | Inhibition Constant (Ki) | Hydrogen Bond Length (Angstrom) | No. of hydrogen bonds | Binding Energy (kcal/mol) | Residues Involved in Other Interactions |
|---|---|---|---|---|---|---|
| Remdesivir | His1236, Ala1237, Lys1045, Tyr1047, Tyr1079, Leu1205, Trp1084, Pro1049, Lys1091, Glu1050, Arg1271, Thr1268, Leu1243, Glu1204 | 95.82 μM | 2.62 | A:GLN1241:HE21 - A:UNK0:O15 | −5.48 | Alkyl/Pi-alkyl = Lys1239, Ala1046, Val1051 |
| 2.1 | A:UNK0:H70 - A:ASP1235:O | |||||
| 3.05 | A:SER1048:CB - A:UNK0:O36 | |||||
| Beta-amyrin | Val1051, Gln1241, Leu1205, Gly1206, Glu1204, Leu1243, Leu1203 | 551.06 nM | 2.11 | :UNK1:H63 - A:SER1048:OG | −8.54 | Alkyl/Pi-alkyl = Tyr1079, Trp1084, Al1046, Tyr1047, Ala1040, Lys1239, Lys1045 |
| Complex Free Energy Calculation Components | ||||||||
|---|---|---|---|---|---|---|---|---|
| Complex | ΔVdwaals | ΔEEL | ΔEPB | ΔENPOLAR | ΔEDISPER | ∆GGas | ∆GSolv | ∆GTotal |
| Remdesivir_MERS−CoV−2_Protease | −2176.97 (±17.11) | −18,897.97 (±39.08) | −3003.96 (±33.10) | 74.28 (±0.37) | 0.00 (±0.0) | −1869.88 (±59.08) | −2929.68 (±32.79) | −4799.56 (±26.97) |
| Arborside−C_Protease | −2149.23 (±5.80) | −18,756.90 (±54.05) | −2945.80 (±29.29) | 73.80 (±0.34) | 0.00 (±0.0) | −2007.92 (±43.40) | −2872.00 (±29.07) | −4879.92 (±21.24) |
| Beta−Sitosterol_Protease | −2111.24 (±18.17) | −18,839.26 (±75.99) | −2927.12 (±34.55) | 73.58 (±0.51) | 0.00 (±0.00) | −2049.11 (±72.59) | −2853.54 (±34.05) | −4902.66 (±41.16) |
| Remdesivir_VP35 (Ebola) | −847.72 (±0.31) | −6939.06 (±1.89) | −1966.78 (±1.71) | 36.88 (±0.01) | 0.00 (±0.00) | −532.75 (±1.89) | −1929.90 (±1.71) | −2462.65 (±0.74) |
| Beta−Amyrin_VP35 (Ebola) | −820.95 (±0.33) | −6792.64 (±2.04) | −1874.47 (±1.85) | 35.83 (±0.01) | 0.00 (±0.00) | −567.62 (±2.15) | −18.38.63 (±1.85) | −2406.25 (±0.78) |
| Remdesivir_Glycoprotein (Nipah) | −3049.62 (±0.60) | −26,201.89 (±2.98) | −4002.48 (±2.22) | 94.81 (±0.02) | 0.00 (±0.00) | −3052.14 (±2.79) | −3907.67 (±2.21) | −6959.81 (±1.38) |
| Nicotiflorin_Glycoprotein (Nipah) | −3527.83 (±0.69) | −30,610.79 (±2.23) | −3952.60 (±1.65) | 90.44 (±0.03) | 0.00 (±0.00) | −8915.81 (±2.31) | −3862.16 (±1.64) | −12,777.97 (±1.46) |
| Remdesivir_Protease (Chikungunya) | −2147.94 (±0.52) | −16,269.22 (±2.11) | −4631.77 (±1.59) | 85.68 (±0.03) | 0.00 (±0.00) | −3332.91 (±2.08) | −4546.08 (±2.08) | −7879.00 (±1.23) |
| Beta−Amyrin_Protease (Chikungunya) | −2108.04 (±0.54) | −15,915.65 (±2.21) | −4760.74 (±1.75) | 87.71 (±0.03) | 0.00 (±0.00) | −3218.38 (±2.20) | −4673.03 (±1.74) | −7891.41 (±1.21) |
| Ligand–Receptor Free Energy Calculation Components | ||||||||
|---|---|---|---|---|---|---|---|---|
| Complex | ΔVdwaals | ΔEEL | ΔEPB | ΔENPOLAR | ΔEDISPER | ∆GGas | ∆GSolv | ∆GTotal |
| Remdesivir_MERS-CoV-2_Protease | −43.38 (±0.54) | −10.79 (±3.74) | 36.02 (±3.17) | −4.47 (±0.04) | 0.00 (±0.00) | −54.18 (±3.51) | 31.55 (±3.14) | −22.63 (±0.74) |
| Arborside-C_Protease | −34.44 (±0.93) | −11.90 (±2.06) | 34.33 (±2.08) | −4.30 (±0.14) | 0.00 (±0.00) | −46.34 (±2.50) | 30.04 (±1.98) | −16.30 (±1.16) |
| Beta-Sitosterol_Protease | −31.78 (±0.64) | −5.10 (±0.41) | 18.60 (±0.19) | −3.80 (±0.07) | 0.00 (±0.00) | −36.89 (±0.64) | 14.80 (±0.19) | −22.09 (±0.67) |
| Remdesivir_VP35 (Ebola) | −34.01 (±0.08) | −17.07 (±0.14) | 28.55 (±0.12) | −3.69 (±0.01) | 0.00 (±0.00) | −51.08 (±0.16) | 24.85 (±0.12) | −26.23 (±0.12) |
| Beta-Amyrin_VP35 (Ebola) | −34.93 (±0.07) | −5.54 (±0.15) | 16.04 (±0.08) | −3.66 (±0.00) | 0.00 (±0.00) | −40.47 (±0.16) | 12.38 (±0.18) | −28.10 (±0.10) |
| Remdesivir_Glycoprotein (Nipah) | −26.61 (±0.10) | −21.42 (±0.47) | 32.99 (±0.39) | −3.09 (±0.01) | 0.00 (±0.00) | −48.03 (±0.45) | 29.90 (±0.38) | −18.13 (±0.11) |
| Nicotiflorin_Glycoprotein (Nipah) | −35.37 (±0.06) | −28.22 (±0.24) | 48.94 (±0.23) | −3.96 (±0.00) | 0.00 (±0.00) | −63.59 (±0.24) | 44.98 (±0.23) | −18.16 (±0.12) |
| Remdesivir_Protease (Chikungunya) | −52.91 (±0.09) | 43.85 (±0.21) | 71.67 (±0.16) | −5.25 (±0.01) | 0.00 (±0.00) | −96.76 (±0.22) | 66.42 (±0.16) | −30.34 (±0.12) |
| Beta-Amyrin_Protease (Chikungunya) | −39.15 (±0.10) | −6.18 (±0.06) | 20.25 (±0.06) | −4.06 (±0.01) | 0.00 (±0.00) | −45.33 (±0.11) | 16.19 (±0.06) | −29.14 (±0.09) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Albiheyri, R.; Ahmad, V.; Khan, M.I.; Alzahrani, F.A.; Jamal, Q.M.S. Investigating the Antiviral Properties of Nyctanthes arbor-tristis Linn against the Ebola, SARS-CoV-2, Nipah, and Chikungunya Viruses: A Computational Simulation Study. Pharmaceuticals 2024, 17, 581. https://doi.org/10.3390/ph17050581
Albiheyri R, Ahmad V, Khan MI, Alzahrani FA, Jamal QMS. Investigating the Antiviral Properties of Nyctanthes arbor-tristis Linn against the Ebola, SARS-CoV-2, Nipah, and Chikungunya Viruses: A Computational Simulation Study. Pharmaceuticals. 2024; 17(5):581. https://doi.org/10.3390/ph17050581
Chicago/Turabian StyleAlbiheyri, Raed, Varish Ahmad, Mohammad Imran Khan, Faisal A. Alzahrani, and Qazi Mohammad Sajid Jamal. 2024. "Investigating the Antiviral Properties of Nyctanthes arbor-tristis Linn against the Ebola, SARS-CoV-2, Nipah, and Chikungunya Viruses: A Computational Simulation Study" Pharmaceuticals 17, no. 5: 581. https://doi.org/10.3390/ph17050581
APA StyleAlbiheyri, R., Ahmad, V., Khan, M. I., Alzahrani, F. A., & Jamal, Q. M. S. (2024). Investigating the Antiviral Properties of Nyctanthes arbor-tristis Linn against the Ebola, SARS-CoV-2, Nipah, and Chikungunya Viruses: A Computational Simulation Study. Pharmaceuticals, 17(5), 581. https://doi.org/10.3390/ph17050581