
Advances and Challenges in Targeting TGF-β Isoforms for Therapeutic Intervention of Cancer: A Mechanism-Based Perspective

Abstract
1. Introduction
2. Early Times in TGF-β Research
2.1. TGF-β Discovery
2.2. Discovering the Tumor Suppressor and Oncogenic Functions of TGF-β
2.3. Identification of TGF-β Isoforms
2.4. The TGF-β Superfamily
3. TGF-β Ligands, Their Function, Expression, and Regulation
4. TGF-β Biosynthesis and Activation
5. TGF-β Binding Proteins
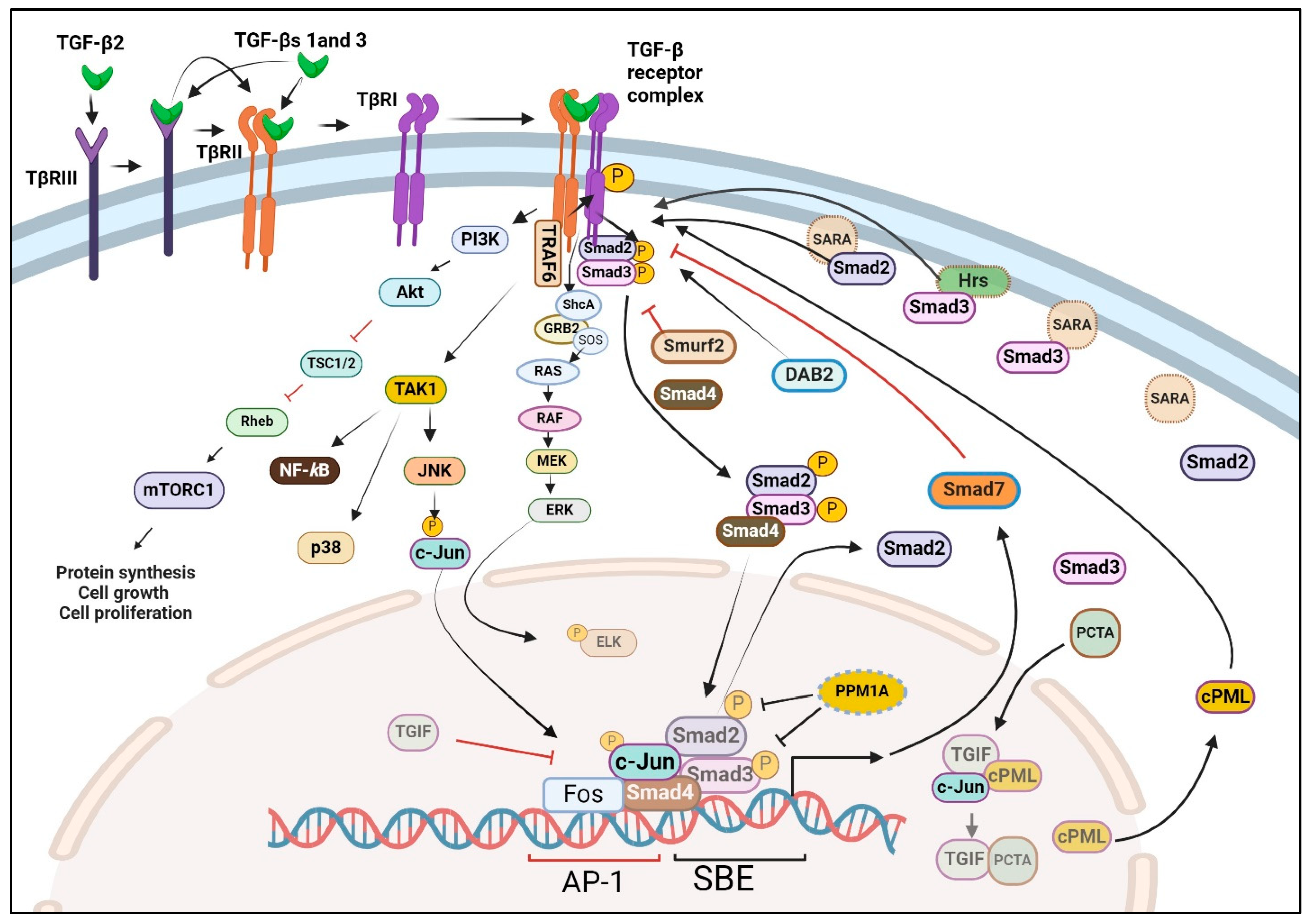
6. TGF-β Receptors
7. Smads and Transcriptional Control
8. Non-Canonical Pathways of TGF-β Signaling
9. Normal Functions of TGF-βs
9.1. Suppression of Proliferation
9.2. Induction of Apoptosis
9.3. Role of TGF-β1 in the Immune System
9.4. Role of TGF-β1 in Wound Healing
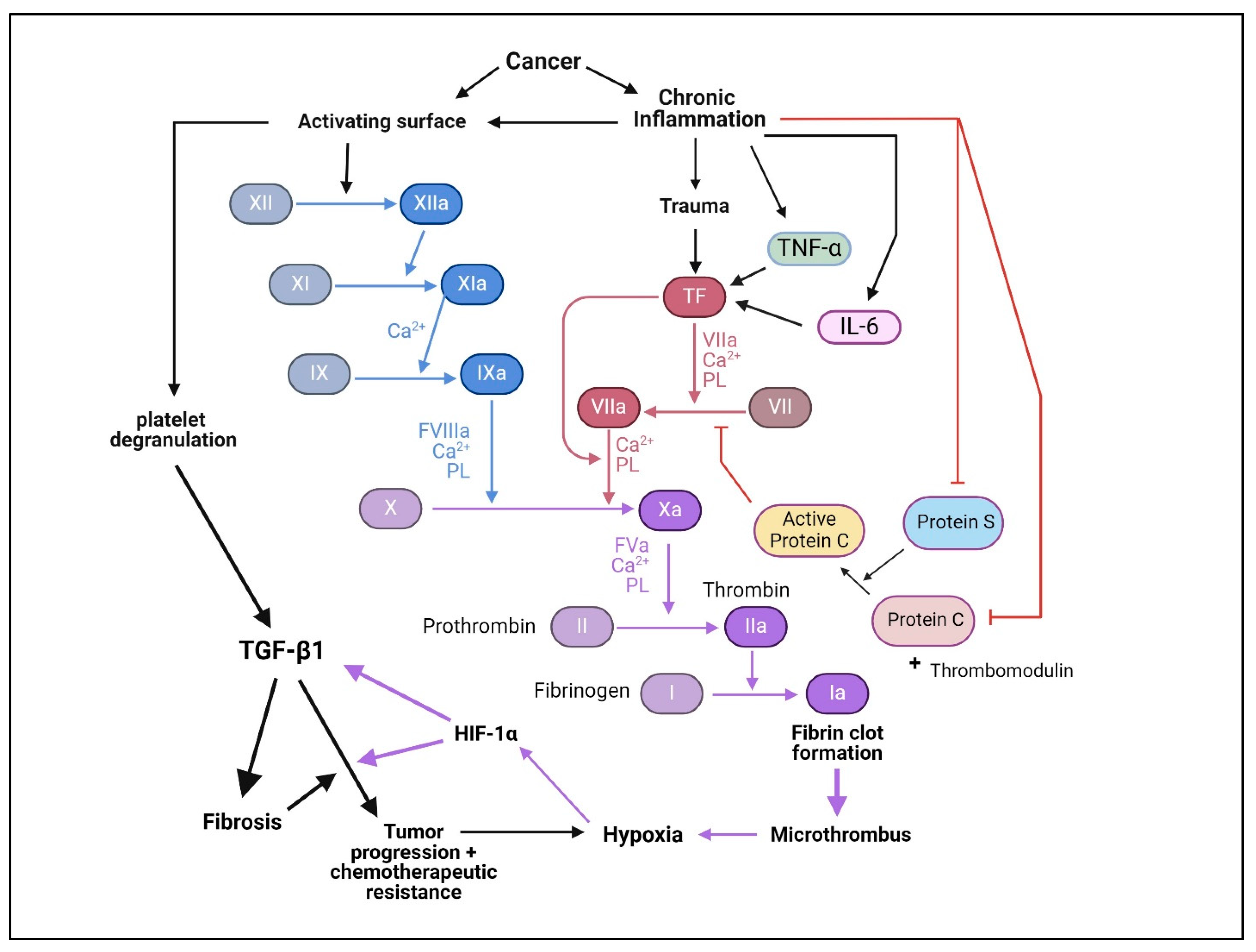
10. Mechanism of TGF-βs-Induced Tumor Progression
10.1. Intrinsic Mechanisms of Tumor Promotion
10.2. Extrinsic Mechanisms of Tumor Promotion
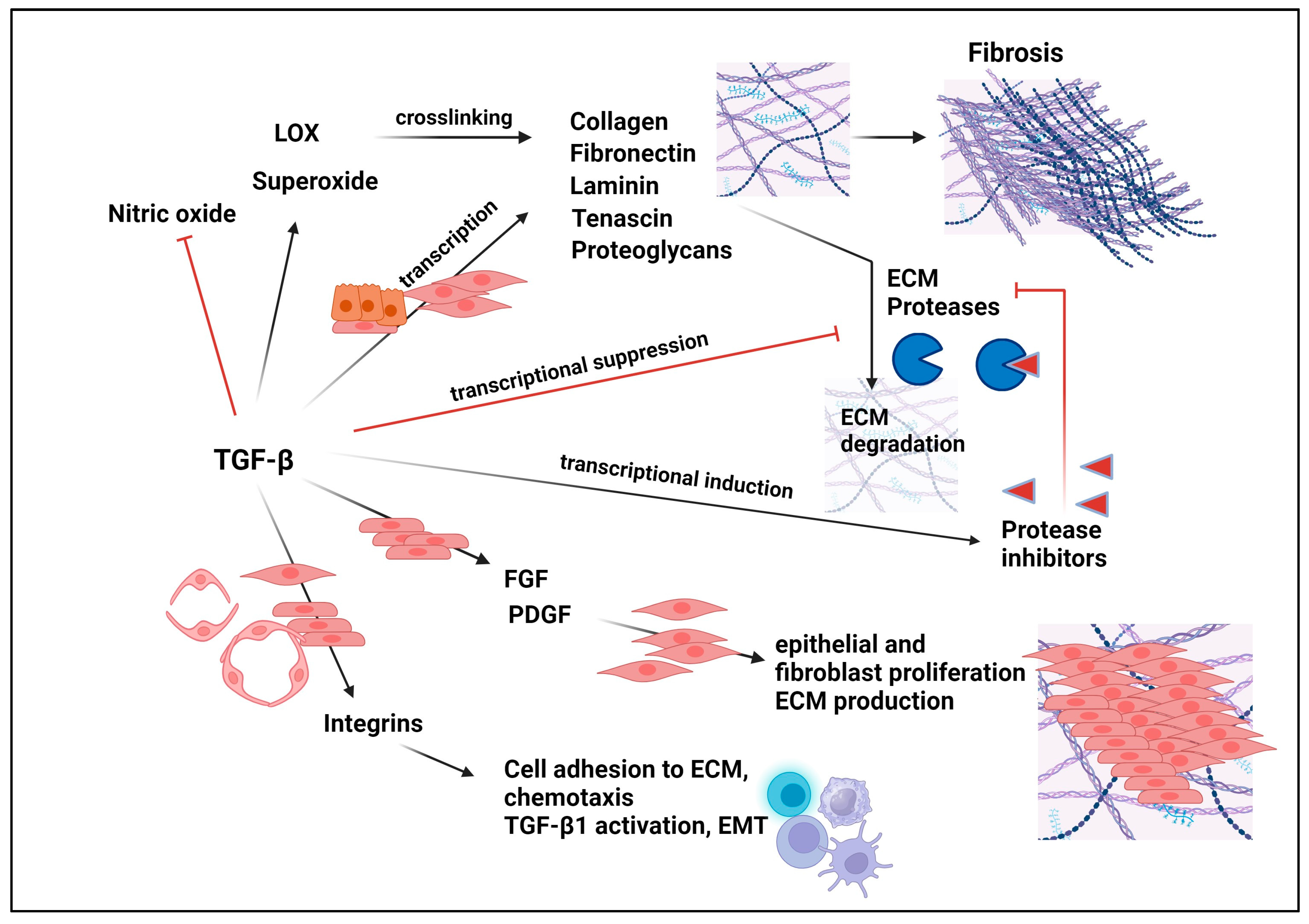
11. Role of TGF-βs in the Pathophysiology of Fibrosis
12. Current Approaches in the Therapeutics for Targeting TGF-β in Cancer
12.1. Important Considerations
12.2. Standalone versus Combination TGF-β Signaling-Blockade Therapies
12.3. Role of TGF-β in the Mechanism of Resistance to Cancer Chemotherapeutic Agents
12.4. Potential Role of TGF-β-Blockade Drugs in Immune Checkpoint Therapy
12.5. Translation of Preclinical Results into Clinical Success
12.6. TGF-β Receptor Kinase Inhibitors

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Target | Class | Pre-Clinical (Ref) | Clinical Phase/(Ref) | NCT Registry # (Drug Combination) | Cancer Types/Indications (Pt Numbers) | Efficacy | Most Frequent Adverse Events (AE) |
|---|---|---|---|---|---|---|---|---|
| SB-431542 | TβRI kinase | SCI | [394] | None | Various cell lines; mouse studies | 0.1–5 μM > p-Smad3 Inhibits ALKs 5, 4 and 7, but not ALKs 1, 2, 3, and 6 and many other kinases. | No signs of toxicity in mice [409]. | |
| SB-505124 | TβRI kinase | SCI | [395] | None | Various cell lines; mouse studies | 3- to 5-fold greater potency than SB-431542, similar specificity to SB-431542; 76.4% oral bioavailability. | Tx was not adequately assessed. | |
| SB-525334 | TβRI kinase | SCI | [397] | None | IC50 of 58.5 nM on ALK5, >200 nM on ALK4; effective in mice orally at 1 to 10 mg/kg/d. | No significant toxicity on human peritoneal mesothelial cells up to 10 μM [410]. | ||
| Ki26894 | TβRI kinase | SCI | [398] | None | Tx was not adequately assessed. | |||
| A83-01 | TβRI kinase | SCI | [399] | None | ||||
| SD-208 | TβRI kinase | SCI | [400] | None | Human and murine glioma cells | Inhibited TGF-β-induced growth inhibition, migration, and invasion of glioma cells; extended survival of mice bearing glioma. | Well tolerated, without observable Tx. | |
| LY364947, (HTS466284, SM305) | TβRI kinase | SCI | None | Tx was not adequately assessed. | ||||
| LY2109761 | TβRI and TβRII kinases | SCI | [401] | None | Orthotopic murine models of PC | Effective in inhibiting PC growth in combination with gemcitabine; prolonged mice survival. | Tx was not adequately assessed. | |
| LY3200882 | TβRI kinase | SCI | [402] | |||||
| [403] Phase I | NCT02937272 Single-agent ± anti-cancer agents (gemcitabine, paclitaxel) | Advanced cancers (139 pts): glioma, PC, HNSCC | Durable PP in Glioma. 75% DCR in the combination arm of PC pts. | 39.6% AE; grade 3 only in combination arm; rare CV toxicity (one in 139 pts). | ||||
| Vactosertib (EW-7197) | TβRI kinase | SCI | [404] | IC50 = 12.1 to 16.5 nM on ALK5 and ALK4. Did not inhibit 320 other kinases tested. Inhibitor of fibrosis in animal models. | ||||
| [411] | 4T1 BC | Inhibited cell migration, invasion, and lung metastasis. | Optimal dosing not established. | |||||
| [412] | Osteosarcoma | Tumor regression, blocked tumor invasion, and prolonged survival [413]. | No severe toxicity [404,411]. | |||||
| [414] Phase I | NCT02160106 Dose escalation. 30 mg–340 mg/d for 5 d, 2 d off. | PC (29 pts) | T1/2 = 3.2 h 6 pts of 16 treated pts achieved stable disease at ≥ 140 mg/d | Excellent overall safety. Most common: fatigue. One pt of 16 pts had abdominal pain, pulmonary edema, and liver enzyme elevation. One pt with stroke at 100 mg/d. | ||||
| Galunisertib (LY2157299) | TβRI kinase | SCI | Phase II [415] | NCT01246986 (+ Sorafenib) (47 pts) | Advanced HHC | Prolonged OS (18.8 m), PSF (4.1 m), and PR in 2 pts, SD in 2 pts. | Acceptable safety profile. 1 pt grade 4 renal injury. Diarrhea (43.2%) and pruritis (25%). 59.6% pts with a serious EV. | |
| Phase II [416] | NCT02008318 (41 pts) | MDS | Hematologic improvements in 24.4% pts; 44% pts had reduced fatigue. | Acceptable safety profile. EV included fatigue (20%), diarrhea (17%), pyrexia (12%), and vomiting (12%). | ||||
| Phase II [417] | NCT01220271 (+ Temozolomide-radiation therapy) (56 pts) | Malignant gliomas | Improved DRR (80%). | Fatigue, nausea, and constipation. | ||||
| Phase II [418] | NCT02688712 (+ Radiotherapy) (38 pts) | Localized CRC | Complete response in 38% of pt at 1 yr. | Grade 3 EV (diarrhea in 16%, hematological Tx in 18%). Two pts had grade 4 EV related to radiotherapy and ischemia. | ||||
| Phase II [419] | NCT02734160 (+ Durvalumab) (32 pts) | Metastatic PC | 1 pt PR, 7 pts SD, and 15 pts had objective progressive disease. 25% DCR. Median OS (5.72 m), and PRS (1.87 m). | No dose-limiting toxicity was recorded. | ||||
| Phase II [420] | NCT01246986 (+ Ramucirumab) (8 pts) | Advanced HCC | MTD was established at 150 mg/d/twice daily with 8 mg/kg ramucirumab every 2 wks. | No dose-limiting Tx was observed. EV included nausea in 25% pts, and vomiting in 25% pts. One pt cerebrovascular accident. | ||||
| GFH018 | TβRI kinase | SCI | [406] Phase I | NCT05051241 | ASTS (50 pts) | MTD = mg BID, 14 d Stable disease (9 pts), tumor shrinkage (1 pt). | Mostly Grade 1 and 2, proteinuria, anemia, and increased liver enzymes. | |
| YL-13027 | TβRI kinase | SCI | [407] Phase I | NCT03869632 60–300 mg/day | ASTS (13 pts) | MTD not reached T1/2 = 4.2 h | Anemia, + GGT. | |
| Long-Acting Tumor-Activated Prodrug | TβRI kinase | SC | [421] | None | Long-acting. | No mortality in tox studies. Valvulopathy (50% rats). | ||
| Fresolimumab (GC-1008 | TGF-β1, TGF-β2, TGF-β3 | mAb | [422] Phase I (29 pt) | NCT00356460 Dose escalation 0.01 to 15 mg/kg every 2 wks | RCC (28 pts), MM (1 pt). | 1 pt PR, 6 pts SD, 24 wks PFS, no DLT up to 15 mg/kg. T1/2 = 21.7 d | Hyperkeratosis, non-malignant keratoacanthomas at high drug doses | |
| [423] Phase II | NCT01401062 (+ radiotherapy) (23 pts) | Metastatic BC | Longer mean survival and improved PMC count at 10 mg/kg than at 1 mg/kg | Well tolerated; 7 grade ¾ AE in 5 of 11 pts in 1 mg arm and 2 of 12 in 10 mg arm. | ||||
| Phase II | NCT00923169 | Advanced MM | Results pending. | |||||
| Phase II | NCT01291784 | MF | Results pending. | |||||
| TβM1 (LY2382770) | TGF-β1 | mAb | [424] | Advanced MM (18 pts), 20 to 240 mg/m | T1/2 = 9 days SD; no significant response; discontinued. | Generally safe; nausea, diarrhea, & fatigue in 15% pts. | ||
| NIS793 | TGF-β1, TGF-β2, TGF-β3 | mAb | [425] | NCT02947165 + Spartalizumab | AST (60 pts), MSS-CRC, anti-PD1-resistant NSCLC | Target engagement and TGF-β inhibition. | No DLT up to 30 mg/kg NIS797 +300 mg/kg Spartalizumab every 3 wks. | |
| SAR439459 | TGF-β1, TGF-β2, TGF-β3 | mAb | [426] | NCT03192345 | Analysis of 1000 pts’ tumors | Achieved significant correlation of high TGF-β pathway with resistance to anti-PD-1. | Not applicable. | |
| [427] Phase I | ±Cemiplimab | AST (52 pts) | Reduced plasma TGF-β1; induced immune cell activation. | DLT observed, MTD not achieved Acceptable tolerability profile. | ||||
| XPA-42-089 | TGF-β1, TGF-β2, TGF-β3 | mAb | [428] | None | ±anti-DP-1 in SSC syngenic mice | 10–20% complete tumor regression. | ||
| Pan-TGFβ mAb | TGF-β1, TGF-β2, TGF-β3 | mAb | [102] | None | Toxicology studies in mice and monkeys. | Significant toxicities: systemic bleeding, CV effects after 5 weeks IV administration of 30 or 100 mg/kg. | ||
| IMC-TR1 (LY3022859) | TβRII | mAb | [429] Phase I | NCT01646203 | Standard chemotherapy-resistant ASD (14 pt) | Primary objective of safe effective dose not achieved. | Cytokine syndrome, infusion-related reactions. | |
| Anti-LAP | TGF-β1 LAP | mAb | [430] | None | Mouse models of MM, CRC, GBM | 10 mg/kg every 3 days decreased tumor growth, LAP +Treg and tolerogenic DC. | Not assessed. | |
| SRK-181 | Latent TGF-β1 | mAb | [431] | Syngeneic mouse models of UC, MM, BC | SRK-181 + anti-PD-1 mAb induced robust antitumor responses, and improved survival of animals bearing anti-PD1-resistant tumors. Restored sensitivity to anti-PD-1 mAb. | No CV Tx. | ||
| 4-week Tx study in rats and monkeys. | Well tolerated, no treatment AE at 200 mg/kg in rats and 300 mg/kg in monkeys. | |||||||
| Phase I | NCT04291079 ± anti-PD-L)1 | ASTS | No results yet. | No results yet. | ||||
| ABBV-151 | GARP | mAb | [432] | NCT03821935 + Budigalima (248 Pt) | Locally advanced or metastatic solid tumors | Enhanced response in anti-DP-1-resistant UC. ORR = 10% | 17% pts ≥ grade 3 AE. | |
| HCC | Safety concerns—discontinued. | |||||||
| PIIO-1 | GARP | mAb | [407] | Murine cancer models | Reduced thrombocytopenia, enhanced CD8+ T cells function, reduced TGF-β signaling. | |||
| DS-1055a | GARP | mAb | [433] | HT-29 CRC in humanized mice | Robustly blocked GARP in the TME, suppressed tumor growth. | |||
| C6D4 | αVβ8 | mAb | [434] | C6D4 (10 mg/kg, once to twice weekly) can significantly reduce tumor growth and improve survival. | ||||
| ADWA-11 | αVβ8 | mAb | [435] | SCC, BC, CRC, and PCa in syngeneic mice ± radiotherapy ± immunotherapy | Suppression or complete regression of tumor growth; enhanced expression of gene linked to cell tumor killing in CD8+ T cells. | |||
| AVID200 (Fc-TβRII) | TGF-β1, TGF-β3 | Ligand traps | [436] Phase I | NCT03895112 | MF (21 pts) | Two pts met clinical benefit with improvement of symptoms; improvement of platelet counts in 81% of pts. Two patients attained clinical benefit with spleen and symptom improvement. | No DLT. Grade 3/4 anemia and thrombocytopenia in a subset. | |
| 4T-Trap | TGF-β1, TGF-β3, CD4 | Ligand trap-mAb bifunctional protein | [437] | Twice weekly IV administration inhibits Th cell TGF-β signaling in CD+ lymph nodes. Improved tumor killing. | Induced tumor hypoxia. | |||
| Bintrafusp Alfa (M7824) | TGF-β1, TGF-β3, PD-1 | Ligand trap-mAb bifunctional protein | [438,439] | A range of human cancers. | Reduce Treg on human CD4+ T-cell proliferation. | |||
| Phase I [440] | NCT02517398 | NSCLC (80 pts) with disease progression after platinum-based therapy. Pt were randomized to receive 500 mg/d or 1200 mg/d every 4 wk. | ORR = 21.3% at 500 mg dose. Tumors with higher PD-L1 levels showed higher response rates. | Treatment-related AE in 69% pt; 29% pts grade 3 or higher AE; 10% pt discontinue treatment; no treatment-related deaths. | ||||
| Phase I [441,442] | NCT02699515 | Advanced gastric/gastroesophageal junction cancer 1200 mg/2 wks (31 pts) | ORR = 16%; DCR = 26% | 19% treatment-related grade 3 AE; no grade 4 EA. 19% immune-related EA. | ||||
| Phase I [443] | NCT02517398 | SCCHN (32 pts) | ORR = 13%; PR = 29% pt; DCC = 34% pt. | 23% pt grade 3 AE; grade 3 treatment-related AE = 34% pt. No grade 4 AE or treatment-related death. | ||||
| Phase III [444] | NCT03631706 | PD-L1-high advanced NSCLC (304 pts) received Bintrafusp Alfa or pembrolizumab | No significant difference in endpoints was observed between treatment groups. | About 3-fold more grade 3–4 AV in the Bintrafusp Alfa group than in the pembrolizumab group. The study was discontinued. | ||||
| SHR-1701 | TGF-β1, TGF-β3, PD-1 | Ligand trap-mAb bifunctional protein | Phase I | NCT03774979 | Recurrent or metastatic CC following platinum-based therapy (32 pt) | ORR = 15.6%, ongoing response in 80% of responders, DCR = 50%. | Treatment-related EA of grade 3 or 4 in 34% pts. No treatment-related deaths. | |
| Phase I | NCT03710265 | ASTS (171 pt) | 20% ORR 54.5% OSR | No DLT observed. | ||||
| YM101 | TGF-β1, TGF-β2, TGF-β3, PD-L1 | Bispecific mAb | [445] | None | BC, CRC, murine T cells in syngeneic mice | Counteract the biological effects of TGF-β and PD-1/PD-L1 pathways; superior antitumor activity compared to monotherapy by anti-TGF-β or anti-PD-1/PD-L1. | ||
| BiTP | TGF-β1, TGF-β2, TGF-β3, PD-L1 | Bispecific mAb | [446] | None | TNBC in syngeneic mice | Similarly effective compared to YM101; enhanced immune cell penetration by reducing collagen deposition. | ||
| Trx-SARA | SARA | Peptide Aptamer | [447] | None | NMuMG murine mammary epithelial cells | Binds to Smads 2 and 3, inhibits TGF-β responses. | Not assessed. | |
| APT-β1 | Active TGF-β1 | Nucleotide Aptamer | [448] | None | NSCLC xenografts in mice (±gefitinib) | Enhanced effectiveness of gefitinib on tumor regression. More potent than TGF-β1 mAb. | Not assessed. | |
| Aptamer S58 | TβRII extracellular domain | Nucleotide Aptamer | [449] | None | Human tendon fibroblasts | Inhibited aSMA expression and incorporation into stress fibers. | Not assessed. | |
| Trabedersen (AP12009) | TGF-β2 | ASO | [450] Phase I | NCT00844064 | CRC, PC, MM | |||
| [451] Phase II | NCT00431561, NCT00761280 | Refractory AA or secondary GBM (145 pts) | 19 pts CRR or PR, improved OS of responders. | Nervous disorders. | ||||
| Phase IIb | NCT05935774 + atezolizumab | Metastatic or recurrent NSCLC | Study withdrawn. | |||||
| ISTH0036 | TGF-β2 | ASO | [452] Phase I | NCT02406833 | Glaucoma patients, intravitreal injection | Likely effective. | Likely safe. | |
| AP11014 | TGF-β1 | ASO | [453] | PCa, CRC, NSCLC | Meeting abstract only, 2004. | Meeting abstract only, 2004. | ||
| ISTH0047 | TGF-β2 | ASO | [454] | Glioma | Inhibited TGF-β2 and growth and invasion of glioma cells, prolonged host survival. Not a well-controlled study. | Not adequately assessed. | ||
| ISTH10047 | TGF-β1 | ASO | [454] | Glioma | Inhibited TGF-β2 and growth and invasion of glioma cells, prolonged host survival. Not a well-controlled study. | Not adequately assessed. |
12.7. Monoclonal Antibodies
12.8. Ligand Traps
12.9. Bifunctional Fusion Proteins
12.10. Antisense Oligonucleotides (ASOs)
12.11. Aptamers
13. Summary and Future Prospects
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Roberts, A.B.; Sporn, M.B. Differential expression of the TGF-beta isoforms in embryogenesis suggests specific roles in developing and adult tissues. Mol. Reprod. Dev. 1992, 32, 91–98. [Google Scholar] [CrossRef]
- Massague, J.; Sheppard, D. TGF-beta signaling in health and disease. Cell 2023, 186, 4007–4037. [Google Scholar] [CrossRef] [PubMed]
- Batlle, E.; Massague, J. Transforming Growth Factor-beta Signaling in Immunity and Cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef] [PubMed]
- Tanguy, J.; Boutanquoi, P.M.; Burgy, O.; Dondaine, L.; Beltramo, G.; Uyanik, B.; Garrido, C.; Bonniaud, P.; Bellaye, P.S.; Goirand, F. HSPB5 Inhibition by NCI-41356 Reduces Experimental Lung Fibrosis by Blocking TGF-beta1 Signaling. Pharmaceuticals 2023, 16, 177. [Google Scholar] [CrossRef] [PubMed]
- David, C.J.; Massague, J. Contextual determinants of TGFbeta action in development, immunity and cancer. Nat. Rev. Mol. Cell Biol. 2018, 19, 419–435. [Google Scholar] [CrossRef] [PubMed]
- Lichtman, M.K.; Otero-Vinas, M.; Falanga, V. Transforming growth factor beta (TGF-beta) isoforms in wound healing and fibrosis. Wound Repair. Regen. 2016, 24, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Margadant, C.; Sonnenberg, A. Integrin-TGF-beta crosstalk in fibrosis, cancer and wound healing. EMBO Rep. 2010, 11, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Massague, J. TGFbeta in Cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef] [PubMed]
- David, C.J.; Huang, Y.H.; Chen, M.; Su, J.; Zou, Y.; Bardeesy, N.; Iacobuzio-Donahue, C.A.; Massague, J. TGF-beta Tumor Suppression through a Lethal EMT. Cell 2016, 164, 1015–1030. [Google Scholar] [CrossRef]
- Shi, Y.; Massague, J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 2003, 113, 685–700. [Google Scholar] [CrossRef]
- Massague, J.; Seoane, J.; Wotton, D. Smad transcription factors. Genes. Dev. 2005, 19, 2783–2810. [Google Scholar] [CrossRef] [PubMed]
- Parvani, J.G.; Taylor, M.A.; Schiemann, W.P. Noncanonical TGF-beta signaling during mammary tumorigenesis. J. Mammary Gland. Biol. Neoplasia 2011, 16, 127–146. [Google Scholar] [CrossRef]
- de Larco, J.E.; Todaro, G.J. Growth factors from murine sarcoma virus-transformed cells. Proc. Natl. Acad. Sci. USA 1978, 75, 4001–4005. [Google Scholar] [CrossRef] [PubMed]
- Anzano, M.A.; Roberts, A.B.; Smith, J.M.; Sporn, M.B.; De Larco, J.E. Sarcoma growth factor from conditioned medium of virally transformed cells is composed of both type alpha and type beta transforming growth factors. Proc. Natl. Acad. Sci. USA 1983, 80, 6264–6268. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.B.; Frolik, C.A.; Anzano, M.A.; Sporn, M.B. Transforming growth factors from neoplastic and nonneoplastic tissues. Fed. Proc. 1983, 42, 2621–2626. [Google Scholar] [PubMed]
- Faria, J.; de Andrade, C.; Goes, A.M.; Rodrigues, M.A.; Gomes, D.A. Effects of different ligands on epidermal growth factor receptor (EGFR) nuclear translocation. Biochem. Biophys. Res. Commun. 2016, 478, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Jarrett, J.A.; Chen, E.Y.; Eaton, D.H.; Bell, J.R.; Assoian, R.K.; Roberts, A.B.; Sporn, M.B.; Goeddel, D.V. Human transforming growth factor-beta complementary DNA sequence and expression in normal and transformed cells. Nature 1985, 316, 701–705. [Google Scholar] [CrossRef]
- Roberts, A.B.; Anzano, M.A.; Meyers, C.A.; Wideman, J.; Blacher, R.; Pan, Y.C.; Stein, S.; Lehrman, S.R.; Smith, J.M.; Lamb, L.C.; et al. Purification and properties of a type beta transforming growth factor from bovine kidney. Biochemistry 1983, 22, 5692–5698. [Google Scholar] [CrossRef] [PubMed]
- Coffey, R.J., Jr.; Derynck, R.; Wilcox, J.N.; Bringman, T.S.; Goustin, A.S.; Moses, H.L.; Pittelkow, M.R. Production and auto-induction of transforming growth factor-alpha in human keratinocytes. Nature 1987, 328, 817–820. [Google Scholar] [CrossRef]
- Frolik, C.A.; Dart, L.L.; Meyers, C.A.; Smith, D.M.; Sporn, M.B. Purification and initial characterization of a type beta transforming growth factor from human placenta. Proc. Natl. Acad. Sci. USA 1983, 80, 3676–3680. [Google Scholar] [CrossRef]
- Assoian, R.K.; Komoriya, A.; Meyers, C.A.; Miller, D.M.; Sporn, M.B. Transforming growth factor-beta in human platelets. Identification of a major storage site, purification, and characterization. J. Biol. Chem. 1983, 258, 7155–7160. [Google Scholar] [CrossRef]
- Childs, C.B.; Proper, J.A.; Tucker, R.F.; Moses, H.L. Serum contains a platelet-derived transforming growth factor. Proc. Natl. Acad. Sci. USA 1982, 79, 5312–5316. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.B.; Anzano, M.A.; Wakefield, L.M.; Roche, N.S.; Stern, D.F.; Sporn, M.B. Type beta transforming growth factor: A bifunctional regulator of cellular growth. Proc. Natl. Acad. Sci. USA 1985, 82, 119–123. [Google Scholar] [CrossRef]
- Tucker, R.F.; Shipley, G.D.; Moses, H.L.; Holley, R.W. Growth inhibitor from BSC-1 cells closely related to platelet type beta transforming growth factor. Science 1984, 226, 705–707. [Google Scholar] [CrossRef] [PubMed]
- Shipley, G.D.; Tucker, R.F.; Moses, H.L. Type beta transforming growth factor/growth inhibitor stimulates entry of monolayer cultures of AKR-2B cells into S phase after a prolonged prereplicative interval. Proc. Natl. Acad. Sci. USA 1985, 82, 4147–4151. [Google Scholar] [CrossRef] [PubMed]
- Moses, H.L.; Roberts, A.B.; Derynck, R. The Discovery and Early Days of TGF-beta: A Historical Perspective. Cold Spring Harb. Perspect. Biol. 2016, 8, a021865. [Google Scholar] [CrossRef] [PubMed]
- Silberstein, G.B.; Daniel, C.W. Reversible inhibition of mammary gland growth by transforming growth factor-beta. Science 1987, 237, 291–293. [Google Scholar] [CrossRef] [PubMed]
- Russell, W.E.; Coffey, R.J., Jr.; Ouellette, A.J.; Moses, H.L. Type beta transforming growth factor reversibly inhibits the early proliferative response to partial hepatectomy in the rat. Proc. Natl. Acad. Sci. USA 1988, 85, 5126–5130. [Google Scholar] [CrossRef] [PubMed]
- Jhappan, C.; Geiser, A.G.; Kordon, E.C.; Bagheri, D.; Hennighausen, L.; Roberts, A.B.; Smith, G.H.; Merlino, G. Targeting expression of a transforming growth factor beta 1 transgene to the pregnant mammary gland inhibits alveolar development and lactation. EMBO J. 1993, 12, 1835–1845. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.; Fowlis, D.J.; Bryson, S.; Duffie, E.; Ireland, H.; Balmain, A.; Akhurst, R.J. TGFbeta1 inhibits the formation of benign skin tumors, but enhances progression to invasive spindle carcinomas in transgenic mice. Cell 1996, 86, 531–542. [Google Scholar] [CrossRef]
- Markowitz, S.; Wang, J.; Myeroff, L.; Parsons, R.; Sun, L.; Lutterbaugh, J.; Fan, R.S.; Zborowska, E.; Kinzler, K.W.; Vogelstein, B.; et al. Inactivation of the type II TGF-beta receptor in colon cancer cells with microsatellite instability. Science 1995, 268, 1336–1338. [Google Scholar] [CrossRef]
- Pierce, D.F., Jr.; Gorska, A.E.; Chytil, A.; Meise, K.S.; Page, D.L.; Coffey, R.J., Jr.; Moses, H.L. Mammary tumor suppression by transforming growth factor beta 1 transgene expression. Proc. Natl. Acad. Sci. USA 1995, 92, 4254–4258. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wu, G.; Willson, J.K.; Zborowska, E.; Yang, J.; Rajkarunanayake, I.; Wang, J.; Gentry, L.E.; Wang, X.F.; Brattain, M.G. Expression of transforming growth factor beta type II receptor leads to reduced malignancy in human breast cancer MCF-7 cells. J. Biol. Chem. 1994, 269, 26449–26455. [Google Scholar] [CrossRef] [PubMed]
- Park, K.; Kim, S.J.; Bang, Y.J.; Park, J.G.; Kim, N.K.; Roberts, A.B.; Sporn, M.B. Genetic changes in the transforming growth factor beta (TGF-beta) type II receptor gene in human gastric cancer cells: Correlation with sensitivity to growth inhibition by TGF-beta. Proc. Natl. Acad. Sci. USA 1994, 91, 8772–8776. [Google Scholar] [CrossRef] [PubMed]
- Markowitz, S.D.; Roberts, A.B. Tumor suppressor activity of the TGF-beta pathway in human cancers. Cytokine Growth Factor. Rev. 1996, 7, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Amoroso, S.R.; Huang, N.; Roberts, A.B.; Potter, M.; Letterio, J.J. Consistent loss of functional transforming growth factor beta receptor expression in murine plasmacytomas. Proc. Natl. Acad. Sci. USA 1998, 95, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.; de Castro, K.; Barnes, H.E.; Parks, W.T.; Stewart, L.; Bottinger, E.P.; Danielpour, D.; Wakefield, L.M. Loss of responsiveness to transforming growth factor beta induces malignant transformation of nontumorigenic rat prostate epithelial cells. Cancer Res. 1999, 59, 4834–4842. [Google Scholar]
- Guo, Y.; Kyprianou, N. Restoration of transforming growth factor beta signaling pathway in human prostate cancer cells suppresses tumorigenicity via induction of caspase-1-mediated apoptosis. Cancer Res. 1999, 59, 1366–1371. [Google Scholar] [PubMed]
- Akhurst, R.J.; Derynck, R. TGF-beta signaling in cancer—A double-edged sword. Trends Cell Biol. 2001, 11, S44–S51. [Google Scholar] [CrossRef] [PubMed]
- Ikushima, H.; Miyazono, K. Cellular context-dependent “colors” of transforming growth factor-beta signaling. Cancer Sci. 2010, 101, 306–312. [Google Scholar] [CrossRef]
- Hanks, S.K.; Armour, R.; Baldwin, J.H.; Maldonado, F.; Spiess, J.; Holley, R.W. Amino acid sequence of the BSC-1 cell growth inhibitor (polyergin) deduced from the nucleotide sequence of the cDNA. Proc. Natl. Acad. Sci. USA 1988, 85, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Marquardt, H.; Lioubin, M.N.; Ikeda, T. Complete amino acid sequence of human transforming growth factor type beta 2. J. Biol. Chem. 1987, 262, 12127–12131. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, T.; Lioubin, M.N.; Marquardt, H. Human transforming growth factor type beta 2: Production by a prostatic adenocarcinoma cell line, purification, and initial characterization. Biochemistry 1987, 26, 2406–2410. [Google Scholar] [CrossRef] [PubMed]
- Cheifetz, S.; Weatherbee, J.A.; Tsang, M.L.; Anderson, J.K.; Mole, J.E.; Lucas, R.; Massague, J. The transforming growth factor-beta system, a complex pattern of cross-reactive ligands and receptors. Cell 1987, 48, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Flanders, K.C.; Yang, Y.A.; Herrmann, M.; Chen, J.; Mendoza, N.; Mirza, A.M.; Wakefield, L.M. Quantitation of TGF-beta proteins in mouse tissues shows reciprocal changes in TGF-beta1 and TGF-beta3 in normal vs neoplastic mammary epithelium. Oncotarget 2016, 7, 38164–38179. [Google Scholar] [CrossRef] [PubMed]
- Danielpour, D.; Kim, K.Y.; Dart, L.L.; Watanabe, S.; Roberts, A.B.; Sporn, M.B. Evidence for Differential Regulation of Tgf-Beta-1 and Tgf-Beta-2 Expression Invivo by Sandwich Enzyme-Linked Immunosorbent Assays. Ann. N. Y. Acad. Sci. 1990, 593, 300–302. [Google Scholar] [CrossRef]
- Derynck, R.; Lindquist, P.B.; Lee, A.; Wen, D.; Tamm, J.; Graycar, J.L.; Rhee, L.; Mason, A.J.; Miller, D.A.; Coffey, R.J.; et al. A new type of transforming growth factor-beta, TGF-beta 3. EMBO J. 1988, 7, 3737–3743. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.A.; Lee, A.; Pelton, R.W.; Chen, E.Y.; Moses, H.L.; Derynck, R. Murine transforming growth factor-beta 2 cDNA sequence and expression in adult tissues and embryos. Mol. Endocrinol. 1989, 3, 1108–1114. [Google Scholar] [CrossRef] [PubMed]
- Denhez, F.; Lafyatis, R.; Kondaiah, P.; Roberts, A.B.; Sporn, M.B. Cloning by polymerase chain reaction of a new mouse TGF-beta, mTGF-beta 3. Growth Factors 1990, 3, 139–146. [Google Scholar] [CrossRef]
- Danielpour, D.; Roberts, A.B. Specific and sensitive quantitation of transforming growth factor beta 3 by sandwich enzyme-linked immunosorbent assay. J. Immunol. Methods 1995, 180, 265–272. [Google Scholar] [CrossRef]
- ten Dijke, P.; Hansen, P.; Iwata, K.K.; Pieler, C.; Foulkes, J.G. Identification of another member of the transforming growth factor type beta gene family. Proc. Natl. Acad. Sci. USA 1988, 85, 4715–4719. [Google Scholar] [CrossRef] [PubMed]
- Piek, E.; Heldin, C.H.; Ten Dijke, P. Specificity, diversity, and regulation in TGF-beta superfamily signaling. FASEB J. 1999, 13, 2105–2124. [Google Scholar] [CrossRef] [PubMed]
- Levy, L.; Hill, C.S. Alterations in components of the TGF-beta superfamily signaling pathways in human cancer. Cytokine Growth Factor. Rev. 2006, 17, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.; Brown, C.W.; Matzuk, M.M. Genetic analysis of the mammalian transforming growth factor-beta superfamily. Endocr. Rev. 2002, 23, 787–823. [Google Scholar] [CrossRef] [PubMed]
- Macias, M.J.; Martin-Malpartida, P.; Massague, J. Structural determinants of Smad function in TGF-beta signaling. Trends Biochem. Sci. 2015, 40, 296–308. [Google Scholar] [CrossRef] [PubMed]
- Letterio, J.J.; Bottinger, E.P. TGF-beta knockout and dominant-negative receptor transgenic mice. Miner. Electrolyte Metab. 1998, 24, 161–167. [Google Scholar] [CrossRef]
- Kulkarni, A.B.; Huh, C.G.; Becker, D.; Geiser, A.; Lyght, M.; Flanders, K.C.; Roberts, A.B.; Sporn, M.B.; Ward, J.M.; Karlsson, S. Transforming growth factor beta 1 null mutation in mice causes excessive inflammatory response and early death. Proc. Natl. Acad. Sci. USA 1993, 90, 770–774. [Google Scholar] [CrossRef] [PubMed]
- Kaartinen, V.; Voncken, J.W.; Shuler, C.; Warburton, D.; Bu, D.; Heisterkamp, N.; Groffen, J. Abnormal lung development and cleft palate in mice lacking TGF-beta 3 indicates defects of epithelial-mesenchymal interaction. Nat. Genet. 1995, 11, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Gaussin, V.; Van de Putte, T.; Mishina, Y.; Hanks, M.C.; Zwijsen, A.; Huylebroeck, D.; Behringer, R.R.; Schneider, M.D. Endocardial cushion and myocardial defects after cardiac myocyte-specific conditional deletion of the bone morphogenetic protein receptor ALK3. Proc. Natl. Acad. Sci. USA 2002, 99, 2878–2883. [Google Scholar] [CrossRef]
- Proetzel, G.; Pawlowski, S.A.; Wiles, M.V.; Yin, M.; Boivin, G.P.; Howles, P.N.; Ding, J.; Ferguson, M.W.; Doetschman, T. Transforming growth factor-beta 3 is required for secondary palate fusion. Nat. Genet. 1995, 11, 409–414. [Google Scholar] [CrossRef]
- Roberts, A.B.; Kim, S.J.; Noma, T.; Glick, A.B.; Lafyatis, R.; Lechleider, R.; Jakowlew, S.B.; Geiser, A.; O’Reilly, M.A.; Danielpour, D.; et al. Multiple forms of TGF-beta: Distinct promoters and differential expression. Ciba Found. Symp. 1991, 157, 7–15; discussion 15–28. [Google Scholar] [CrossRef] [PubMed]
- Komai, T.; Okamura, T.; Inoue, M.; Yamamoto, K.; Fujio, K. Reevaluation of Pluripotent Cytokine TGF-beta3 in Immunity. Int. J. Mol. Sci. 2018, 19, 2261. [Google Scholar] [CrossRef] [PubMed]
- Rubtsov, Y.P.; Rudensky, A.Y. TGFbeta signalling in control of T-cell-mediated self-reactivity. Nat. Rev. Immunol. 2007, 7, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.J.; Datta, A.; Littlefield, C.; Kalra, A.; Chapron, C.; Wawersik, S.; Dagbay, K.B.; Brueckner, C.T.; Nikiforov, A.; Danehy, F.T., Jr.; et al. Selective inhibition of TGFbeta1 activation overcomes primary resistance to checkpoint blockade therapy by altering tumor immune landscape. Sci. Transl. Med. 2020, 12, eaay8456. [Google Scholar] [CrossRef] [PubMed]
- Bang, Y.J.; Kim, S.J.; Danielpour, D.; O’Reilly, M.A.; Kim, K.Y.; Myers, C.E.; Trepel, J.B. Cyclic AMP induces transforming growth factor beta 2 gene expression and growth arrest in the human androgen-independent prostate carcinoma cell line PC-3. Proc. Natl. Acad. Sci. USA 1992, 89, 3556–3560. [Google Scholar] [CrossRef] [PubMed]
- Geiser, A.G.; Kim, S.J.; Roberts, A.B.; Sporn, M.B. Characterization of the mouse transforming growth factor-beta 1 promoter and activation by the Ha-ras oncogene. Mol. Cell. Biol. 1991, 11, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Glick, A.B.; Sporn, M.B.; Yuspa, S.H. Altered regulation of TGF-beta 1 and TGF-alpha in primary keratinocytes and papillomas expressing v-Ha-ras. Mol. Carcinog. 1991, 4, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Danielpour, D.; Kim, K.Y.; Winokur, T.S.; Sporn, M.B. Differential regulation of the expression of transforming growth factor-beta s 1 and 2 by retinoic acid, epidermal growth factor, and dexamethasone in NRK-49F and A549 cells. J. Cell. Physiol. 1991, 148, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Glick, A.B.; Danielpour, D.; Morgan, D.; Sporn, M.B.; Yuspa, S.H. Induction and autocrine receptor binding of transforming growth factor-beta 2 during terminal differentiation of primary mouse keratinocytes. Mol. Endocrinol. 1990, 4, 46–52. [Google Scholar] [CrossRef]
- Glick, A.B.; Flanders, K.C.; Danielpour, D.; Yuspa, S.H.; Sporn, M.B. Retinoic acid induces transforming growth factor-beta 2 in cultured keratinocytes and mouse epidermis. Cell Regul. 1989, 1, 87–97. [Google Scholar] [CrossRef]
- Danielpour, D. Induction of transforming growth factor-beta autocrine activity by all-trans-retinoic acid and 1 alpha,25-dihydroxyvitamin D3 in NRP-152 rat prostatic epithelial cells. J. Cell. Physiol. 1996, 166, 231–239. [Google Scholar] [CrossRef]
- Danielpour, D. Transdifferentiation of NRP-152 rat prostatic basal epithelial cells toward a luminal phenotype: Regulation by glucocorticoid, insulin-like growth factor-I and transforming growth factor-beta. J. Cell Sci. 1999, 112 Pt 2, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Pierce, G.F.; Mustoe, T.A.; Lingelbach, J.; Masakowski, V.R.; Griffin, G.L.; Senior, R.M.; Deuel, T.F. Platelet-derived growth factor and transforming growth factor-beta enhance tissue repair activities by unique mechanisms. J. Cell Biol. 1989, 109, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Fong, K.D.; Trindade, M.C.; Wang, Z.; Nacamuli, R.P.; Pham, H.; Fang, T.D.; Song, H.M.; Smith, R.L.; Longaker, M.T.; Chang, J. Microarray analysis of mechanical shear effects on flexor tendon cells. Plast. Reconstr. Surg. 2005, 116, 1393–1404; discussion 1405–1406. [Google Scholar] [CrossRef] [PubMed]
- Colletta, A.A.; Wakefield, L.M.; Howell, F.V.; van Roozendaal, K.E.; Danielpour, D.; Ebbs, S.R.; Sporn, M.B.; Baum, M. Anti-oestrogens induce the secretion of active transforming growth factor beta from human fetal fibroblasts. Br. J. Cancer 1990, 62, 405–409. [Google Scholar] [CrossRef] [PubMed]
- Lucia, M.S.; Sporn, M.B.; Roberts, A.B.; Stewart, L.V.; Danielpour, D. The role of transforming growth factor-beta1, -beta2, and -beta3 in androgen-responsive growth of NRP-152 rat prostatic epithelial cells. J. Cell. Physiol. 1998, 175, 184–192. [Google Scholar] [CrossRef]
- Kim, S.J.; Wagner, S.; Liu, F.; O’Reilly, M.A.; Robbins, P.D.; Green, M.R. Retinoblastoma gene product activates expression of the human TGF-beta 2 gene through transcription factor ATF-2. Nature 1992, 358, 331–334. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Minigh, J.; Miles, S.; Niles, R.M. Retinoic acid decreases ATF-2 phosphorylation and sensitizes melanoma cells to taxol-mediated growth inhibition. J. Mol. Signal. 2008, 3, 3. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Namachivayam, K.; MohanKumar, K.; Arbach, D.; Jagadeeswaran, R.; Jain, S.K.; Natarajan, V.; Mehta, D.; Jankov, R.P.; Maheshwari, A. All-Trans Retinoic Acid Induces TGF-beta2 in Intestinal Epithelial Cells via RhoA- and p38alpha MAPK-Mediated Activation of the Transcription Factor ATF2. PLoS ONE 2015, 10, e0134003. [Google Scholar] [CrossRef] [PubMed]
- Feng, C.; Zuo, Z. Regulatory factor X1-induced down-regulation of transforming growth factor beta2 transcription in human neuroblastoma cells. J. Biol. Chem. 2012, 287, 22730–22739. [Google Scholar] [CrossRef]
- Samatar, A.A.; Wang, L.; Mirza, A.; Koseoglu, S.; Liu, S.; Kumar, C.C. Transforming growth factor-beta 2 is a transcriptional target for Akt/protein kinase B via forkhead transcription factor. J. Biol. Chem. 2002, 277, 28118–28126. [Google Scholar] [CrossRef] [PubMed]
- Murvai, M.; Borbely, A.A.; Konya, J.; Gergely, L.; Veress, G. Effect of human papillomavirus type 16 E6 and E7 oncogenes on the activity of the transforming growth factor-beta2 (TGF-beta2) promoter. Arch. Virol. 2004, 149, 2379–2392. [Google Scholar] [CrossRef]
- Peralta-Zaragoza, O.; Bermudez-Morales, V.; Gutierrez-Xicotencatl, L.; Alcocer-Gonzalez, J.; Recillas-Targa, F.; Madrid-Marina, V. E6 and E7 oncoproteins from human papillomavirus type 16 induce activation of human transforming growth factor beta1 promoter throughout Sp1 recognition sequence. Viral Immunol. 2006, 19, 468–480. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, M.A.; Danielpour, D.; Roberts, A.B.; Sporn, M.B. Regulation of expression of transforming growth factor-beta 2 by transforming growth factor-beta isoforms is dependent upon cell type. Growth Factors 1992, 6, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia 2015, 3, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Akman, H.O.; Zhang, H.; Siddiqui, M.A.; Solomon, W.; Smith, E.L.; Batuman, O.A. Response to hypoxia involves transforming growth factor-beta2 and Smad proteins in human endothelial cells. Blood 2001, 98, 3324–3331. [Google Scholar] [CrossRef] [PubMed]
- Shields, M.A.; Ebine, K.; Sahai, V.; Kumar, K.; Siddiqui, K.; Hwang, R.F.; Grippo, P.J.; Munshi, H.G. Snail cooperates with KrasG12D to promote pancreatic fibrosis. Mol. Cancer Res. 2013, 11, 1078–1087. [Google Scholar] [CrossRef] [PubMed]
- Hong, L.; Li, F.; Tang, C.; Li, L.; Sun, L.; Li, X.; Zhu, L. Semaphorin 7A promotes endothelial to mesenchymal transition through ATF3 mediated TGF-beta2/Smad signaling. Cell Death Dis. 2020, 11, 695. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.H.; Wang, F.; Wang, F.L.; Liu, Q.; Zhou, J. Regulation of transforming growth factor beta-mediated epithelial-mesenchymal transition of lens epithelial cells by c-Src kinase under high glucose conditions. Exp. Ther. Med. 2018, 16, 1520–1528. [Google Scholar] [CrossRef]
- Shah, C.A.; Wang, H.; Bei, L.; Platanias, L.C.; Eklund, E.A. HoxA10 regulates transcription of the gene encoding transforming growth factor beta2 (TGFbeta2) in myeloid cells. J. Biol. Chem. 2011, 286, 3161–3176. [Google Scholar] [CrossRef]
- Chida, T.; Ito, M.; Nakashima, K.; Kanegae, Y.; Aoshima, T.; Takabayashi, S.; Kawata, K.; Nakagawa, Y.; Yamamoto, M.; Shimano, H.; et al. Critical role of CREBH-mediated induction of transforming growth factor beta2 by hepatitis C virus infection in fibrogenic responses in hepatic stellate cells. Hepatology 2017, 66, 1430–1443. [Google Scholar] [CrossRef]
- Deng, L.; Li, Y.; Huang, J.M.; Zhou, G.; Qian, W.; Xu, K. Effects of p-CREB-1 on transforming growth factor-beta3 auto-regulation in hepatic stellate cells. J. Cell. Biochem. 2011, 112, 1046–1054. [Google Scholar] [CrossRef] [PubMed]
- Laverty, H.G.; Wakefield, L.M.; Occleston, N.L.; O’Kane, S.; Ferguson, M.W. TGF-beta3 and cancer: A review. Cytokine Growth Factor. Rev. 2009, 20, 305–317. [Google Scholar] [CrossRef] [PubMed]
- Chang, Z.; Kishimoto, Y.; Hasan, A.; Welham, N.V. TGF-beta3 modulates the inflammatory environment and reduces scar formation following vocal fold mucosal injury in rats. Dis. Model. Mech. 2014, 7, 83–91. [Google Scholar] [CrossRef]
- Gato, A.; Martinez, M.L.; Tudela, C.; Alonso, I.; Moro, J.A.; Formoso, M.A.; Ferguson, M.W.; Martinez-Alvarez, C. TGF-beta(3)-induced chondroitin sulphate proteoglycan mediates palatal shelf adhesion. Dev. Biol. 2002, 250, 393–405. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Taya, Y.; O’Kane, S.; Ferguson, M.W. Pathogenesis of cleft palate in TGF-beta3 knockout mice. Development 1999, 126, 3869–3879. [Google Scholar] [CrossRef] [PubMed]
- Tudela, C.; Formoso, M.A.; Martinez, T.; Perez, R.; Aparicio, M.; Maestro, C.; Del Rio, A.; Martinez, E.; Ferguson, M.; Martinez-Alvarez, C. TGF-beta3 is required for the adhesion and intercalation of medial edge epithelial cells during palate fusion. Int. J. Dev. Biol. 2002, 46, 333–336. [Google Scholar] [PubMed]
- Yang, L.T.; Kaartinen, V. Tgfb1 expressed in the Tgfb3 locus partially rescues the cleft palate phenotype of Tgfb3 null mutants. Dev. Biol. 2007, 312, 384–395. [Google Scholar] [CrossRef] [PubMed]
- Hall, B.E.; Wankhade, U.D.; Konkel, J.E.; Cherukuri, K.; Nagineni, C.N.; Flanders, K.C.; Arany, P.R.; Chen, W.; Rane, S.G.; Kulkarni, A.B. Transforming growth factor-beta3 (TGF-beta3) knock-in ameliorates inflammation due to TGF-beta1 deficiency while promoting glucose tolerance. J. Biol. Chem. 2013, 288, 32074–32092. [Google Scholar] [CrossRef] [PubMed]
- Danielpour, D.; Sporn, M.B. Differential inhibition of transforming growth factor beta 1 and beta 2 activity by alpha 2-macroglobulin. J. Biol. Chem. 1990, 265, 6973–6977. [Google Scholar] [CrossRef]
- Anderton, M.J.; Mellor, H.R.; Bell, A.; Sadler, C.; Pass, M.; Powell, S.; Steele, S.J.; Roberts, R.R.; Heier, A. Induction of heart valve lesions by small-molecule ALK5 inhibitors. Toxicol. Pathol. 2011, 39, 916–924. [Google Scholar] [CrossRef] [PubMed]
- Mitra, M.S.; Lancaster, K.; Adedeji, A.O.; Palanisamy, G.S.; Dave, R.A.; Zhong, F.; Holdren, M.S.; Turley, S.J.; Liang, W.C.; Wu, Y.; et al. A Potent Pan-TGFbeta Neutralizing Monoclonal Antibody Elicits Cardiovascular Toxicity in Mice and Cynomolgus Monkeys. Toxicol. Sci. 2020, 175, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Sanford, L.P.; Ormsby, I.; Gittenberger-de Groot, A.C.; Sariola, H.; Friedman, R.; Boivin, G.P.; Cardell, E.L.; Doetschman, T. TGFbeta2 knockout mice have multiple developmental defects that are non-overlapping with other TGFbeta knockout phenotypes. Development 1997, 124, 2659–2670. [Google Scholar] [CrossRef] [PubMed]
- Shull, M.M.; Ormsby, I.; Kier, A.B.; Pawlowski, S.; Diebold, R.J.; Yin, M.; Allen, R.; Sidman, C.; Proetzel, G.; Calvin, D.; et al. Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature 1992, 359, 693–699. [Google Scholar] [CrossRef] [PubMed]
- Lindsay, M.E.; Schepers, D.; Bolar, N.A.; Doyle, J.J.; Gallo, E.; Fert-Bober, J.; Kempers, M.J.; Fishman, E.K.; Chen, Y.; Myers, L.; et al. Loss-of-function mutations in TGFB2 cause a syndromic presentation of thoracic aortic aneurysm. Nat. Genet. 2012, 44, 922–927. [Google Scholar] [CrossRef] [PubMed]
- Boileau, C.; Guo, D.C.; Hanna, N.; Regalado, E.S.; Detaint, D.; Gong, L.; Varret, M.; Prakash, S.K.; Li, A.H.; d’Indy, H.; et al. TGFB2 mutations cause familial thoracic aortic aneurysms and dissections associated with mild systemic features of Marfan syndrome. Nat. Genet. 2012, 44, 916–921. [Google Scholar] [CrossRef] [PubMed]
- Renard, M.; Callewaert, B.; Malfait, F.; Campens, L.; Sharif, S.; del Campo, M.; Valenzuela, I.; McWilliam, C.; Coucke, P.; De Paepe, A.; et al. Thoracic aortic-aneurysm and dissection in association with significant mitral valve disease caused by mutations in TGFB2. Int. J. Cardiol. 2013, 165, 584–587. [Google Scholar] [CrossRef] [PubMed]
- Disha, K.; Schulz, S.; Kuntze, T.; Girdauskas, E. Transforming Growth Factor Beta-2 Mutations in Barlow’s Disease and Aortic Dilatation. Ann. Thorac. Surg. 2017, 104, e19–e21. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bertoli-Avella, A.M.; Gillis, E.; Morisaki, H.; Verhagen, J.M.A.; de Graaf, B.M.; van de Beek, G.; Gallo, E.; Kruithof, B.P.T.; Venselaar, H.; Myers, L.A.; et al. Mutations in a TGF-beta ligand, TGFB3, cause syndromic aortic aneurysms and dissections. J. Am. Coll. Cardiol. 2015, 65, 1324–1336. [Google Scholar] [CrossRef]
- Beffagna, G.; Occhi, G.; Nava, A.; Vitiello, L.; Ditadi, A.; Basso, C.; Bauce, B.; Carraro, G.; Thiene, G.; Towbin, J.A.; et al. Regulatory mutations in transforming growth factor-beta3 gene cause arrhythmogenic right ventricular cardiomyopathy type 1. Cardiovasc. Res. 2005, 65, 366–373. [Google Scholar] [CrossRef]
- Gentry, L.E.; Lioubin, M.N.; Purchio, A.F.; Marquardt, H. Molecular events in the processing of recombinant type 1 pre-pro-transforming growth factor beta to the mature polypeptide. Mol. Cell. Biol. 1988, 8, 4162–4168. [Google Scholar] [CrossRef] [PubMed]
- Gentry, L.E.; Twardzik, D.R.; Lim, G.J.; Ranchalis, J.E.; Lee, D.C. Expression and characterization of transforming growth factor alpha precursor protein in transfected mammalian cells. Mol. Cell. Biol. 1987, 7, 1585–1591. [Google Scholar] [CrossRef] [PubMed]
- Miyazono, K.; Ichijo, H.; Heldin, C.H. Transforming growth factor-beta: Latent forms, binding proteins and receptors. Growth Factors 1993, 8, 11–22. [Google Scholar] [CrossRef]
- Olofsson, A.; Ichijo, H.; Moren, A.; ten Dijke, P.; Miyazono, K.; Heldin, C.H. Efficient association of an amino-terminally extended form of human latent transforming growth factor-beta binding protein with the extracellular matrix. J. Biol. Chem. 1995, 270, 31294–31297. [Google Scholar] [CrossRef] [PubMed]
- Moren, A.; Olofsson, A.; Stenman, G.; Sahlin, P.; Kanzaki, T.; Claesson-Welsh, L.; ten Dijke, P.; Miyazono, K.; Heldin, C.H. Identification and characterization of LTBP-2, a novel latent transforming growth factor-beta-binding protein. J. Biol. Chem. 1994, 269, 32469–32478. [Google Scholar] [CrossRef] [PubMed]
- Annes, J.P.; Munger, J.S.; Rifkin, D.B. Making sense of latent TGFbeta activation. J. Cell Sci. 2003, 116, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Wipff, P.J.; Rifkin, D.B.; Meister, J.J.; Hinz, B. Myofibroblast contraction activates latent TGF-beta1 from the extracellular matrix. J. Cell Biol. 2007, 179, 1311–1323. [Google Scholar] [CrossRef] [PubMed]
- Buscemi, L.; Ramonet, D.; Klingberg, F.; Formey, A.; Smith-Clerc, J.; Meister, J.J.; Hinz, B. The single-molecule mechanics of the latent TGF-beta1 complex. Curr. Biol. 2011, 21, 2046–2054. [Google Scholar] [CrossRef] [PubMed]
- Barcellos-Hoff, M.H.; Dix, T.A. Redox-mediated activation of latent transforming growth factor-beta 1. Mol. Endocrinol. 1996, 10, 1077–1083. [Google Scholar] [CrossRef]
- Koli, K.; Saharinen, J.; Hyytiainen, M.; Penttinen, C.; Keski-Oja, J. Latency, activation, and binding proteins of TGF-beta. Microsc. Res. Tech. 2001, 52, 354–362. [Google Scholar] [CrossRef]
- Keski-Oja, J.; Koli, K.; von Melchner, H. TGF-beta activation by traction? Trends Cell Biol. 2004, 14, 657–659. [Google Scholar] [CrossRef] [PubMed]
- Bizik, J.; Felnerova, D.; Grofova, M.; Vaheri, A. Active transforming growth factor-beta in human melanoma cell lines: No evidence for plasmin-related activation of latent TGF-beta. J. Cell. Biochem. 1996, 62, 113–122. [Google Scholar] [CrossRef]
- Schultz-Cherry, S.; Ribeiro, S.; Gentry, L.; Murphy-Ullrich, J.E. Thrombospondin binds and activates the small and large forms of latent transforming growth factor-beta in a chemically defined system. J. Biol. Chem. 1994, 269, 26775–26782. [Google Scholar] [CrossRef] [PubMed]
- Barcellos-Hoff, M.H. Radiation-induced transforming growth factor beta and subsequent extracellular matrix reorganization in murine mammary gland. Cancer Res. 1993, 53, 3880–3886. [Google Scholar] [PubMed]
- Stockis, J.; Colau, D.; Coulie, P.G.; Lucas, S. Membrane protein GARP is a receptor for latent TGF-beta on the surface of activated human Treg. Eur. J. Immunol. 2009, 39, 3315–3322. [Google Scholar] [CrossRef] [PubMed]
- Tran, D.Q.; Andersson, J.; Wang, R.; Ramsey, H.; Unutmaz, D.; Shevach, E.M. GARP (LRRC32) is essential for the surface expression of latent TGF-β on platelets and activated FOXP3 regulatory T cells. Proc. Natl. Acad. Sci. USA 2009, 106, 13445–13450. [Google Scholar] [CrossRef] [PubMed]
- Lahimchi, M.R.; Eslami, M.; Yousefi, B. New insight into GARP striking role in cancer progression: Application for cancer therapy. Med. Oncol. 2022, 40, 33. [Google Scholar] [CrossRef] [PubMed]
- Metelli, A.; Wu, B.X.; Riesenberg, B.; Guglietta, S.; Huck, J.D.; Mills, C.; Li, A.; Rachidi, S.; Krieg, C.; Rubinstein, M.P.; et al. Thrombin contributes to cancer immune evasion via proteolysis of platelet-bound GARP to activate LTGF-beta. Sci. Transl. Med. 2020, 12, eaay4860. [Google Scholar] [CrossRef]
- Koivisto, L.; Bi, J.; Hakkinen, L.; Larjava, H. Integrin alphavbeta6: Structure, function and role in health and disease. Int. J. Biochem. Cell Biol. 2018, 99, 186–196. [Google Scholar] [CrossRef]
- Dallas, S.L.; Zhao, S.; Cramer, S.D.; Chen, Z.; Peehl, D.M.; Bonewald, L.F. Preferential production of latent transforming growth factor beta-2 by primary prostatic epithelial cells and its activation by prostate-specific antigen. J. Cell. Physiol. 2005, 202, 361–370. [Google Scholar] [CrossRef]
- Jobling, M.F.; Mott, J.D.; Finnegan, M.T.; Jurukovski, V.; Erickson, A.C.; Walian, P.J.; Taylor, S.E.; Ledbetter, S.; Lawrence, C.M.; Rifkin, D.B.; et al. Isoform-specific activation of latent transforming growth factor beta (LTGF-beta) by reactive oxygen species. Radiat. Res. 2006, 166, 839–848. [Google Scholar] [CrossRef] [PubMed]
- Walsh, L.A.; Damjanovski, S. IGF-1 increases invasive potential of MCF 7 breast cancer cells and induces activation of latent TGF-beta1 resulting in epithelial to mesenchymal transition. Cell Commun. Signal 2011, 9, 10. [Google Scholar] [CrossRef] [PubMed]
- Breuss, J.M.; Gallo, J.; DeLisser, H.M.; Klimanskaya, I.V.; Folkesson, H.G.; Pittet, J.F.; Nishimura, S.L.; Aldape, K.; Landers, D.V.; Carpenter, W.; et al. Expression of the beta 6 integrin subunit in development, neoplasia and tissue repair suggests a role in epithelial remodeling. J. Cell Sci. 1995, 108 Pt 6, 2241–2251. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Zhu, J.; Wang, R.; Chen, X.; Mi, L.; Walz, T.; Springer, T.A. Latent TGF-beta structure and activation. Nature 2011, 474, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Huang, Z.; Liang, W.C.; Yin, J.; Lin, W.Y.; Wu, J.; Vernes, J.M.; Lutman, J.; Caplazi, P.; Jeet, S.; et al. TGFbeta2 and TGFbeta3 isoforms drive fibrotic disease pathogenesis. Sci. Transl. Med. 2021, 13, eabe0407. [Google Scholar] [CrossRef]
- O’Connor-McCourt, M.D.; Wakefield, L.M. Latent transforming growth factor-beta in serum. A specific complex with alpha 2-macroglobulin. J. Biol. Chem. 1987, 262, 14090–14099. [Google Scholar] [CrossRef] [PubMed]
- Wakefield, L.M.; Smith, D.M.; Flanders, K.C.; Sporn, M.B. Latent transforming growth factor-beta from human platelets. A high molecular weight complex containing precursor sequences. J. Biol. Chem. 1988, 263, 7646–7654. [Google Scholar] [CrossRef] [PubMed]
- LaMarre, J.; Wollenberg, G.K.; Gauldie, J.; Hayes, M.A. Alpha 2-macroglobulin and serum preferentially counteract the mitoinhibitory effect of transforming growth factor-beta 2 in rat hepatocytes. Lab. Investig. 1990, 62, 545–551. [Google Scholar] [PubMed]
- Baghy, K.; Reszegi, A.; Tatrai, P.; Kovalszky, I. Decorin in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1272, 17–38. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Mann, D.M.; Ruoslahti, E. Negative regulation of transforming growth factor-beta by the proteoglycan decorin. Nature 1990, 346, 281–284. [Google Scholar] [CrossRef]
- Hildebrand, A.; Romaris, M.; Rasmussen, L.M.; Heinegard, D.; Twardzik, D.R.; Border, W.A.; Ruoslahti, E. Interaction of the small interstitial proteoglycans biglycan, decorin and fibromodulin with transforming growth factor beta. Biochem. J. 1994, 302 Pt 2, 527–534. [Google Scholar] [CrossRef] [PubMed]
- Wrana, J.L.; Attisano, L.; Carcamo, J.; Zentella, A.; Doody, J.; Laiho, M.; Wang, X.F.; Massague, J. TGF beta signals through a heteromeric protein kinase receptor complex. Cell 1992, 71, 1003–1014. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, M.; Moustakas, A.; Lin, H.Y.; Lodish, H.F.; Carr, B.I. Growth inhibition by transforming growth factor beta (TGF-beta) type I is restored in TGF-beta-resistant hepatoma cells after expression of TGF-beta receptor type II cDNA. Proc. Natl. Acad. Sci. USA 1993, 90, 5359–5363. [Google Scholar] [CrossRef] [PubMed]
- Wrana, J.L.; Attisano, L.; Wieser, R.; Ventura, F.; Massague, J. Mechanism of activation of the TGF-beta receptor. Nature 1994, 370, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Ebner, R.; Chen, R.H.; Shum, L.; Lawler, S.; Zioncheck, T.F.; Lee, A.; Lopez, A.R.; Derynck, R. Cloning of a type I TGF-beta receptor and its effect on TGF-beta binding to the type II receptor. Science 1993, 260, 1344–1348. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.F.; Lin, H.Y.; Ng-Eaton, E.; Downward, J.; Lodish, H.F.; Weinberg, R.A. Expression cloning and characterization of the TGF-beta type III receptor. Cell 1991, 67, 797–805. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.Y.; Wang, X.F.; Ng-Eaton, E.; Weinberg, R.A.; Lodish, H.F. Expression cloning of the TGF-beta type II receptor, a functional transmembrane serine/threonine kinase. Cell 1992, 68, 775–785. [Google Scholar] [CrossRef] [PubMed]
- Attisano, L.; Wrana, J.L.; Lopez-Casillas, F.; Massague, J. TGF-beta receptors and actions. Biochim. Biophys. Acta 1994, 1222, 71–80. [Google Scholar] [CrossRef]
- MacKay, K.; Danielpour, D. Novel 150- and 180-kDa glycoproteins that bind transforming growth factor (TGF)-beta 1 but not TGF-beta 2 are present in several cell lines. J. Biol. Chem. 1991, 266, 9907–9911. [Google Scholar] [CrossRef]
- MacKay, K.; Danielpour, D.; Miller, D.; Border, W.A.; Robbins, A.R. The 260-kDa transforming growth factor (TGF)-beta binding protein in rat glomeruli is a complex comprised of 170- and 85-kDa TGF-beta binding proteins. J. Biol. Chem. 1992, 267, 11449–11454. [Google Scholar] [CrossRef]
- MacKay, K.; Kondaiah, P.; Danielpour, D.; Austin, H.A., 3rd; Brown, P.D. Expression of transforming growth factor-beta 1 and beta 2 in rat glomeruli. Kidney Int. 1990, 38, 1095–1100. [Google Scholar] [CrossRef] [PubMed]
- Henis, Y.I.; Moustakas, A.; Lin, H.Y.; Lodish, H.F. The types II and III transforming growth factor-beta receptors form homo-oligomers. J. Cell Biol. 1994, 126, 139–154. [Google Scholar] [CrossRef] [PubMed]
- Luo, K.; Lodish, H.F. Signaling by chimeric erythropoietin-TGF-beta receptors: Homodimerization of the cytoplasmic domain of the type I TGF-beta receptor and heterodimerization with the type II receptor are both required for intracellular signal transduction. EMBO J. 1996, 15, 4485–4496. [Google Scholar] [CrossRef] [PubMed]
- Luo, K.; Lodish, H.F. Positive and negative regulation of type II TGF-beta receptor signal transduction by autophosphorylation on multiple serine residues. EMBO J. 1997, 16, 1970–1981. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Casillas, F.; Wrana, J.L.; Massague, J. Betaglycan presents ligand to the TGF beta signaling receptor. Cell 1993, 73, 1435–1444. [Google Scholar] [CrossRef] [PubMed]
- Massague, J. TGF-beta signal transduction. Annu. Rev. Biochem. 1998, 67, 753–791. [Google Scholar] [CrossRef] [PubMed]
- Andres, J.L.; Stanley, K.; Cheifetz, S.; Massague, J. Membrane-anchored and soluble forms of betaglycan, a polymorphic proteoglycan that binds transforming growth factor-beta. J. Cell Biol. 1989, 109, 3137–3145. [Google Scholar] [CrossRef] [PubMed]
- Tazat, K.; Hector-Greene, M.; Blobe, G.C.; Henis, Y.I. TbetaRIII independently binds type I and type II TGF-beta receptors to inhibit TGF-beta signaling. Mol. Biol. Cell 2015, 26, 3535–3545. [Google Scholar] [CrossRef] [PubMed]
- Charng, M.J.; Kinnunen, P.; Hawker, J.; Brand, T.; Schneider, M.D. FKBP-12 recognition is dispensable for signal generation by type I transforming growth factor-beta receptors. J. Biol. Chem. 1996, 271, 22941–22944. [Google Scholar] [CrossRef]
- Charng, M.J.; Zhang, D.; Kinnunen, P.; Schneider, M.D. A novel protein distinguishes between quiescent and activated forms of the type I transforming growth factor beta receptor. J. Biol. Chem. 1998, 273, 9365–9368. [Google Scholar] [CrossRef]
- Chen, Y.G.; Liu, F.; Massague, J. Mechanism of TGFbeta receptor inhibition by FKBP12. EMBO J. 1997, 16, 3866–3876. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.S.; Olson, E.N. Regulation of differentiation of the BC3H1 muscle cell line through cAMP-dependent and -independent pathways. J. Biol. Chem. 1988, 263, 19670–19677. [Google Scholar] [CrossRef] [PubMed]
- McKay, M.M.; Morrison, D.K. Integrating signals from RTKs to ERK/MAPK. Oncogene 2007, 26, 3113–3121. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.K.; Pardoux, C.; Hall, M.C.; Lee, P.S.; Warburton, D.; Qing, J.; Smith, S.M.; Derynck, R. TGF-beta activates Erk MAP kinase signalling through direct phosphorylation of ShcA. EMBO J. 2007, 26, 3957–3967. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Yan, W.; Wells, R.G.; Rimm, D.L.; McNiff, J.; Leffell, D.; Reiss, M. Novel inactivating mutations of transforming growth factor-beta type I receptor gene in head-and-neck cancer metastases. Int. J. Cancer 2001, 93, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Carter, D.; Garrigue-Antar, L.; Reiss, M. Transforming growth factor beta type I receptor kinase mutant associated with metastatic breast cancer. Cancer Res. 1998, 58, 4805–4810. [Google Scholar] [PubMed]
- Grady, W.M.; Myeroff, L.L.; Swinler, S.E.; Rajput, A.; Thiagalingam, S.; Lutterbaugh, J.D.; Neumann, A.; Brattain, M.G.; Chang, J.; Kim, S.J.; et al. Mutational inactivation of transforming growth factor beta receptor type II in microsatellite stable colon cancers. Cancer Res. 1999, 59, 320–324. [Google Scholar] [PubMed]
- Knaus, P.I.; Lindemann, D.; DeCoteau, J.F.; Perlman, R.; Yankelev, H.; Hille, M.; Kadin, M.E.; Lodish, H.F. A dominant inhibitory mutant of the type II transforming growth factor beta receptor in the malignant progression of a cutaneous T-cell lymphoma. Mol. Cell. Biol. 1996, 16, 3480–3489. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ma, Y.; He, S.; Gao, A.; Zhang, Y.; Zhu, Q.; Wang, P.; Yang, B.; Yin, H.; Li, Y.; Song, J.; et al. Methylation silencing of TGF-beta receptor type II is involved in malignant transformation of esophageal squamous cell carcinoma. Clin. Epigenet. 2020, 12, 25. [Google Scholar] [CrossRef]
- Guo, W.; Dong, Z.; Guo, Y.; Kuang, G.; Yang, Z.; Shan, B. Concordant repression and aberrant methylation of transforming growth factor-beta signaling pathway genes occurs early in gastric cardia adenocarcinoma. Mol. Biol. Rep. 2012, 39, 9453–9462. [Google Scholar] [CrossRef]
- Bebek, G.; Bennett, K.L.; Funchain, P.; Campbell, R.; Seth, R.; Scharpf, J.; Burkey, B.; Eng, C. Microbiomic subprofiles and MDR1 promoter methylation in head and neck squamous cell carcinoma. Hum. Mol. Genet. 2012, 21, 1557–1565. [Google Scholar] [CrossRef]
- Munoz-Antonia, T.; Torrellas-Ruiz, M.; Clavell, J.; Mathews, L.A.; Muro-Cacho, C.A.; Baez, A. Aberrant methylation inactivates transforming growth factor Beta receptor I in head and neck squamous cell carcinoma. Int. J. Otolaryngol. 2009, 2009, 848695. [Google Scholar] [CrossRef] [PubMed]
- Shima, K.; Morikawa, T.; Yamauchi, M.; Kuchiba, A.; Imamura, Y.; Liao, X.; Meyerhardt, J.A.; Fuchs, C.S.; Ogino, S. TGFBR2 and BAX mononucleotide tract mutations, microsatellite instability, and prognosis in 1072 colorectal cancers. PLoS ONE 2011, 6, e25062. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.M.; Boyd, K.; Davies, F.E. The potential role of epigenetic therapy in multiple myeloma. Br. J. Haematol. 2010, 148, 702–713. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Ghosh, P.; Osawa, H.; Sasaki, C.Y.; Rezanka, L.; Yang, J.; O’Farrell, T.J.; Longo, D.L. Resistance to TGF-beta 1 correlates with aberrant expression of TGF-beta receptor II in human B-cell lymphoma cell lines. Blood 2007, 109, 5301–5307. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhang, Q.; Rubenstein, J.N.; Liu, V.C.; Park, I.; Jang, T.; Lee, C. Restoration of expression of transforming growth factor-beta type II receptor in murine renal cell carcinoma (renca) cells by 5-Aza-2’-deoxycytidine. Life Sci. 2005, 76, 1159–1166. [Google Scholar] [CrossRef]
- Ammanamanchi, S.; Brattain, M.G. Restoration of transforming growth factor-beta signaling through receptor RI induction by histone deacetylase activity inhibition in breast cancer cells. J. Biol. Chem. 2004, 279, 32620–32625. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.T.; Chen, X.F.; Wang, M.H.; Wang, J.C.; Qi, Q.Y.; Zhang, R.M.; Xu, W.Q.; Fei, Q.Y.; Wang, F.; Cheng, Q.Q.; et al. Defective expression of transforming growth factor beta receptor type II is associated with CpG methylated promoter in primary non-small cell lung cancer. Clin. Cancer Res. 2004, 10, 2359–2367. [Google Scholar] [CrossRef] [PubMed]
- de Jonge, R.R.; Garrigue-Antar, L.; Vellucci, V.F.; Reiss, M. Frequent inactivation of the transforming growth factor beta type II receptor in small-cell lung carcinoma cells. Oncol. Res. 1997, 9, 89–98. [Google Scholar] [PubMed]
- Song, K.; Wang, H.; Krebs, T.L.; Kim, S.J.; Danielpour, D. Androgenic control of transforming growth factor-beta signaling in prostate epithelial cells through transcriptional suppression of transforming growth factor-beta receptor II. Cancer Res. 2008, 68, 8173–8182. [Google Scholar] [CrossRef]
- Engel, M.E.; Datta, P.K.; Moses, H.L. Signal transduction by transforming growth factor-beta: A cooperative paradigm with extensive negative regulation. J. Cell. Biochem. 1998, 72, 111–122. [Google Scholar] [CrossRef]
- Massague, J. TGFbeta signaling: Receptors, transducers, and Mad proteins. Cell 1996, 85, 947–950. [Google Scholar] [CrossRef]
- Wrana, J.; Pawson, T. Signal transduction. Mad about SMADs. Nature 1997, 388, 28–29. [Google Scholar] [CrossRef] [PubMed]
- Kretzschmar, M.; Massague, J. SMADs: Mediators and regulators of TGF-beta signaling. Curr. Opin. Genet. Dev. 1998, 8, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Yagi, K.; Goto, D.; Hamamoto, T.; Takenoshita, S.; Kato, M.; Miyazono, K. Alternatively spliced variant of Smad2 lacking exon 3. Comparison with wild-type Smad2 and Smad3. J. Biol. Chem. 1999, 274, 703–709. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.H.; Derynck, R. A kinase subdomain of transforming growth factor-beta (TGF-beta) type I receptor determines the TGF-beta intracellular signaling specificity. EMBO J. 1997, 16, 3912–3923. [Google Scholar] [CrossRef] [PubMed]
- Lo, R.S.; Chen, Y.G.; Shi, Y.; Pavletich, N.P.; Massague, J. The L3 loop: A structural motif determining specific interactions between SMAD proteins and TGF-beta receptors. EMBO J. 1998, 17, 996–1005. [Google Scholar] [CrossRef]
- Miura, S.; Takeshita, T.; Asao, H.; Kimura, Y.; Murata, K.; Sasaki, Y.; Hanai, J.I.; Beppu, H.; Tsukazaki, T.; Wrana, J.L.; et al. Hgs (Hrs), a FYVE domain protein, is involved in Smad signaling through cooperation with SARA. Mol. Cell. Biol. 2000, 20, 9346–9355. [Google Scholar] [CrossRef] [PubMed]
- Tsukazaki, T.; Chiang, T.A.; Davison, A.F.; Attisano, L.; Wrana, J.L. SARA, a FYVE domain protein that recruits Smad2 to the TGFbeta receptor. Cell 1998, 95, 779–791. [Google Scholar] [CrossRef]
- Wu, R.Y.; Zhang, Y.; Feng, X.H.; Derynck, R. Heteromeric and homomeric interactions correlate with signaling activity and functional cooperativity of Smad3 and Smad4/DPC4. Mol. Cell. Biol. 1997, 17, 2521–2528. [Google Scholar] [CrossRef]
- Xiao, Z.; Liu, X.; Lodish, H.F. Importin beta mediates nuclear translocation of Smad 3. J. Biol. Chem. 2000, 275, 23425–23428. [Google Scholar] [CrossRef]
- Hocevar, B.A.; Smine, A.; Xu, X.X.; Howe, P.H. The adaptor molecule Disabled-2 links the transforming growth factor beta receptors to the Smad pathway. EMBO J. 2001, 20, 2789–2801. [Google Scholar] [CrossRef]
- Liu, F. PCTA: A new player in TGF-beta signaling. Sci. Signal 2008, 1, pe49. [Google Scholar] [CrossRef] [PubMed]
- Ten Dijke, P.; Goumans, M.J.; Itoh, F.; Itoh, S. Regulation of cell proliferation by Smad proteins. J. Cell. Physiol. 2002, 191, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Sun, Y.; Constantinescu, S.N.; Karam, E.; Weinberg, R.A.; Lodish, H.F. Transforming growth factor beta-induced phosphorylation of Smad3 is required for growth inhibition and transcriptional induction in epithelial cells. Proc. Natl. Acad. Sci. USA 1997, 94, 10669–10674. [Google Scholar] [CrossRef] [PubMed]
- Yamamura, Y.; Hua, X.; Bergelson, S.; Lodish, H.F. Critical role of Smads and AP-1 complex in transforming growth factor-beta -dependent apoptosis. J. Biol. Chem. 2000, 275, 36295–36302. [Google Scholar] [CrossRef] [PubMed]
- Jonk, L.J.; Itoh, S.; Heldin, C.H.; ten Dijke, P.; Kruijer, W. Identification and functional characterization of a Smad binding element (SBE) in the JunB promoter that acts as a transforming growth factor-beta, activin, and bone morphogenetic protein-inducible enhancer. J. Biol. Chem. 1998, 273, 21145–21152. [Google Scholar] [CrossRef] [PubMed]
- Chipuk, J.E.; Cornelius, S.C.; Pultz, N.J.; Jorgensen, J.S.; Bonham, M.J.; Kim, S.J.; Danielpour, D. The androgen receptor represses transforming growth factor-beta signaling through interaction with Smad3. J. Biol. Chem. 2002, 277, 1240–1248. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, K. Smad phosphoisoform signaling specificity: The right place at the right time. Carcinogenesis 2011, 32, 1578–1588. [Google Scholar] [CrossRef] [PubMed]
- Wendt, M.K.; Tian, M.; Schiemann, W.P. Deconstructing the mechanisms and consequences of TGF-beta-induced EMT during cancer progression. Cell Tissue Res. 2012, 347, 85–101. [Google Scholar] [CrossRef] [PubMed]
- Nomura, M.; Li, E. Smad2 role in mesoderm formation, left-right patterning and craniofacial development. Nature 1998, 393, 786–790. [Google Scholar] [CrossRef]
- Waldrip, W.R.; Bikoff, E.K.; Hoodless, P.A.; Wrana, J.L.; Robertson, E.J. Smad2 signaling in extraembryonic tissues determines anterior-posterior polarity of the early mouse embryo. Cell 1998, 92, 797–808. [Google Scholar] [CrossRef]
- Weinstein, M.; Yang, X.; Li, C.; Xu, X.; Gotay, J.; Deng, C.X. Failure of egg cylinder elongation and mesoderm induction in mouse embryos lacking the tumor suppressor smad2. Proc. Natl. Acad. Sci. USA 1998, 95, 9378–9383. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Richardson, J.A.; Parada, L.F.; Graff, J.M. Smad3 mutant mice develop metastatic colorectal cancer. Cell 1998, 94, 703–714. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Letterio, J.J.; Lechleider, R.J.; Chen, L.; Hayman, R.; Gu, H.; Roberts, A.B.; Deng, C. Targeted disruption of SMAD3 results in impaired mucosal immunity and diminished T cell responsiveness to TGF-beta. EMBO J. 1999, 18, 1280–1291. [Google Scholar] [CrossRef]
- Datto, M.B.; Frederick, J.P.; Pan, L.; Borton, A.J.; Zhuang, Y.; Wang, X.F. Targeted disruption of Smad3 reveals an essential role in transforming growth factor beta-mediated signal transduction. Mol. Cell. Biol. 1999, 19, 2495–2504. [Google Scholar] [CrossRef] [PubMed]
- Ashcroft, G.S.; Yang, X.; Glick, A.B.; Weinstein, M.; Letterio, J.L.; Mizel, D.E.; Anzano, M.; Greenwell-Wild, T.; Wahl, S.M.; Deng, C.; et al. Mice lacking Smad3 show accelerated wound healing and an impaired local inflammatory response. Nat. Cell Biol. 1999, 1, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Sirard, C.; de la Pompa, J.L.; Elia, A.; Itie, A.; Mirtsos, C.; Cheung, A.; Hahn, S.; Wakeham, A.; Schwartz, L.; Kern, S.E.; et al. The tumor suppressor gene Smad4/Dpc4 is required for gastrulation and later for anterior development of the mouse embryo. Genes. Dev. 1998, 12, 107–119. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Li, C.; Xu, X.; Deng, C. The tumor suppressor SMAD4/DPC4 is essential for epiblast proliferation and mesoderm induction in mice. Proc. Natl. Acad. Sci. USA 1998, 95, 3667–3672. [Google Scholar] [CrossRef]
- Millet, C.; Zhang, Y.E. Roles of Smad3 in TGF-beta signaling during carcinogenesis. Crit. Rev. Eukaryot. Gene Expr. 2007, 17, 281–293. [Google Scholar] [CrossRef]
- Song, K.; Wang, H.; Krebs, T.L.; Wang, B.; Kelley, T.J.; Danielpour, D. DHT selectively reverses Smad3-mediated/TGF-beta-induced responses through transcriptional down-regulation of Smad3 in prostate epithelial cells. Mol. Endocrinol. 2010, 24, 2019–2029. [Google Scholar] [CrossRef]
- Huang, C.; Hu, F.; Song, D.; Sun, X.; Liu, A.; Wu, Q.; She, X.; Chen, Y.; Chen, L.; Hu, F.; et al. EZH2-triggered methylation of SMAD3 promotes its activation and tumor metastasis. J. Clin. Investig. 2022, 132, e152394. [Google Scholar] [CrossRef] [PubMed]
- Inoue, Y.; Imamura, T. Regulation of TGF-beta family signaling by E3 ubiquitin ligases. Cancer Sci. 2008, 99, 2107–2112. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Liang, M.; Feng, X.H. Smurf2 is a ubiquitin E3 ligase mediating proteasome-dependent degradation of Smad2 in transforming growth factor-beta signaling. J. Biol. Chem. 2000, 275, 36818–36822. [Google Scholar] [CrossRef] [PubMed]
- Lo, R.S.; Massague, J. Ubiquitin-dependent degradation of TGF-beta-activated smad2. Nat. Cell Biol. 1999, 1, 472–478. [Google Scholar] [CrossRef] [PubMed]
- Izzi, L.; Attisano, L. Regulation of the TGFbeta signalling pathway by ubiquitin-mediated degradation. Oncogene 2004, 23, 2071–2078. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Duan, X.; Liang, Y.Y.; Su, Y.; Wrighton, K.H.; Long, J.; Hu, M.; Davis, C.M.; Wang, J.; Brunicardi, F.C.; et al. PPM1A functions as a Smad phosphatase to terminate TGFbeta signaling. Cell 2006, 125, 915–928. [Google Scholar] [CrossRef] [PubMed]
- Runa, F.; Ortiz-Soto, G.; de Barros, N.R.; Kelber, J.A. Targeting SMAD-Dependent Signaling: Considerations in Epithelial and Mesenchymal Solid Tumors. Pharmaceuticals 2024, 17, 326. [Google Scholar] [CrossRef] [PubMed]
- Heldin, C.H.; Moustakas, A. Signaling Receptors for TGF-beta Family Members. Cold Spring Harb. Perspect. Biol. 2016, 8, a022053. [Google Scholar] [CrossRef] [PubMed]
- Moustakas, A.; Heldin, C.H. Non-Smad TGF-beta signals. J. Cell Sci. 2005, 118, 3573–3584. [Google Scholar] [CrossRef]
- Sorrentino, A.; Thakur, N.; Grimsby, S.; Marcusson, A.; von Bulow, V.; Schuster, N.; Zhang, S.; Heldin, C.H.; Landstrom, M. The type I TGF-beta receptor engages TRAF6 to activate TAK1 in a receptor kinase-independent manner. Nat. Cell Biol. 2008, 10, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Akhurst, R.J.; Balmain, A. TGF-beta signaling in tumor suppression and cancer progression. Nat. Genet. 2001, 29, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Wahdan-Alaswad, R.; Danielpour, D. Critical role of Smad2 in tumor suppression and transforming growth factor-beta-induced apoptosis of prostate epithelial cells. Cancer Res. 2009, 69, 2185–2190. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, M.; Fatyol, K.; Jin, C.; Wang, X.; Liu, Z.; Zhang, Y.E. TRAF6 mediates Smad-independent activation of JNK and p38 by TGF-beta. Mol. Cell 2008, 31, 918–924. [Google Scholar] [CrossRef] [PubMed]
- Ozdamar, B.; Bose, R.; Barrios-Rodiles, M.; Wang, H.R.; Zhang, Y.; Wrana, J.L. Regulation of the polarity protein Par6 by TGFbeta receptors controls epithelial cell plasticity. Science 2005, 307, 1603–1609. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.J.; Barnes, A.P.; Hand, R.; Polleux, F.; Ehlers, M.D. TGF-beta signaling specifies axons during brain development. Cell 2010, 142, 144–157. [Google Scholar] [CrossRef] [PubMed]
- Conery, A.R.; Cao, Y.; Thompson, E.A.; Townsend, C.M., Jr.; Ko, T.C.; Luo, K. Akt interacts directly with Smad3 to regulate the sensitivity to TGF-beta induced apoptosis. Nat. Cell Biol. 2004, 6, 366–372. [Google Scholar] [CrossRef] [PubMed]
- Song, K.; Wang, H.; Krebs, T.L.; Danielpour, D. Novel roles of Akt and mTOR in suppressing TGF-beta/ALK5-mediated Smad3 activation. EMBO J. 2006, 25, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Katsuno, Y.; Derynck, R. Epithelial plasticity, epithelial-mesenchymal transition, and the TGF-beta family. Dev. Cell 2021, 56, 726–746. [Google Scholar] [CrossRef]
- Derynck, R.; Turley, S.J.; Akhurst, R.J. TGFbeta biology in cancer progression and immunotherapy. Nat. Rev. Clin. Oncol. 2021, 18, 9–34. [Google Scholar] [CrossRef]
- Wu, M.; Chen, G.; Li, Y.P. TGF-beta and BMP signaling in osteoblast, skeletal development, and bone formation, homeostasis and disease. Bone Res. 2016, 4, 16009. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, G.; Cook, B.D.; Terushkin, V.; Pintucci, G.; Mignatti, P. Transforming growth factor-beta 1 (TGF-beta1) induces angiogenesis through vascular endothelial growth factor (VEGF)-mediated apoptosis. J. Cell. Physiol. 2009, 219, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Goumans, M.J.; Ten Dijke, P. TGF-beta Signaling in Control of Cardiovascular Function. Cold Spring Harb. Perspect. Biol. 2018, 10, a022210. [Google Scholar] [CrossRef] [PubMed]
- Meyers, E.A.; Kessler, J.A. TGF-beta Family Signaling in Neural and Neuronal Differentiation, Development, and Function. Cold Spring Harb. Perspect. Biol. 2017, 9, a022244. [Google Scholar] [CrossRef]
- Robson, C.N.; Gnanapragasam, V.; Byrne, R.L.; Collins, A.T.; Neal, D.E. Transforming growth factor-beta1 up-regulates p15, p21 and p27 and blocks cell cycling in G1 in human prostate epithelium. J. Endocrinol. 1999, 160, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Iavarone, A.; Massague, J. E2F and histone deacetylase mediate transforming growth factor beta repression of cdc25A during keratinocyte cell cycle arrest. Mol. Cell. Biol. 1999, 19, 916–922. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Seoane, J.; Pouponnot, C.; Staller, P.; Schader, M.; Eilers, M.; Massague, J. TGFbeta influences Myc, Miz-1 and Smad to control the CDK inhibitor p15INK4b. Nat. Cell Biol. 2001, 3, 400–408. [Google Scholar] [CrossRef] [PubMed]
- Staller, P.; Peukert, K.; Kiermaier, A.; Seoane, J.; Lukas, J.; Karsunky, H.; Moroy, T.; Bartek, J.; Massague, J.; Hanel, F.; et al. Repression of p15INK4b expression by Myc through association with Miz-1. Nat. Cell Biol. 2001, 3, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Shima, Y.; Nakao, K.; Nakashima, T.; Kawakami, A.; Nakata, K.; Hamasaki, K.; Kato, Y.; Eguchi, K.; Ishii, N. Activation of caspase-8 in transforming growth factor-beta-induced apoptosis of human hepatoma cells. Hepatology 1999, 30, 1215–1222. [Google Scholar] [CrossRef]
- Chipuk, J.E.; Bhat, M.; Hsing, A.Y.; Ma, J.; Danielpour, D. Bcl-xL blocks transforming growth factor-beta 1-induced apoptosis by inhibiting cytochrome c release and not by directly antagonizing Apaf-1-dependent caspase activation in prostate epithelial cells. J. Biol. Chem. 2001, 276, 26614–26621. [Google Scholar] [CrossRef]
- Ahmed, M.M.; Alcock, R.A.; Chendil, D.; Dey, S.; Das, A.; Venkatasubbarao, K.; Mohiuddin, M.; Sun, L.; Strodel, W.E.; Freeman, J.W. Restoration of transforming growth factor-beta signaling enhances radiosensitivity by altering the Bcl-2/Bax ratio in the p53 mutant pancreatic cancer cell line MIA PaCa-2. J. Biol. Chem. 2002, 277, 2234–2246. [Google Scholar] [CrossRef]
- Jang, C.W.; Chen, C.H.; Chen, C.C.; Chen, J.Y.; Su, Y.H.; Chen, R.H. TGF-beta induces apoptosis through Smad-mediated expression of DAP-kinase. Nat. Cell Biol. 2002, 4, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, K.; Nagai, S.; Ninomiya-Tsuji, J.; Nishita, M.; Tamai, K.; Irie, K.; Ueno, N.; Nishida, E.; Shibuya, H.; Matsumoto, K. XIAP, a cellular member of the inhibitor of apoptosis protein family, links the receptors to TAB1-TAK1 in the BMP signaling pathway. EMBO J. 1999, 18, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Perlman, R.; Schiemann, W.P.; Brooks, M.W.; Lodish, H.F.; Weinberg, R.A. TGF-beta-induced apoptosis is mediated by the adapter protein Daxx that facilitates JNK activation. Nat. Cell Biol. 2001, 3, 708–714. [Google Scholar] [CrossRef] [PubMed]
- Saile, B.; Matthes, N.; El Armouche, H.; Neubauer, K.; Ramadori, G. The bcl, NFkappaB and p53/p21WAF1 systems are involved in spontaneous apoptosis and in the anti-apoptotic effect of TGF-beta or TNF-alpha on activated hepatic stellate cells. Eur. J. Cell Biol. 2001, 80, 554–561. [Google Scholar] [CrossRef] [PubMed]
- Edlund, S.; Bu, S.; Schuster, N.; Aspenstrom, P.; Heuchel, R.; Heldin, N.E.; ten Dijke, P.; Heldin, C.H.; Landstrom, M. Transforming growth factor-beta1 (TGF-beta)-induced apoptosis of prostate cancer cells involves Smad7-dependent activation of p38 by TGF-beta-activated kinase 1 and mitogen-activated protein kinase kinase 3. Mol. Biol. Cell 2003, 14, 529–544. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, S.; Wildey, G.M.; Howe, P.H. Transforming growth factor beta (TGFbeta)-induced apoptosis: The rise & fall of Bim. Cell Cycle 2009, 8, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.; Ghiassi, M.; Jirmanova, L.; Balliet, A.G.; Hoffman, B.; Fornace, A.J., Jr.; Liebermann, D.A.; Bottinger, E.P.; Roberts, A.B. Transforming growth factor-beta-induced apoptosis is mediated by Smad-dependent expression of GADD45b through p38 activation. J. Biol. Chem. 2003, 278, 43001–43007. [Google Scholar] [CrossRef]
- Yang, J.; Song, K.; Krebs, T.L.; Jackson, M.W.; Danielpour, D. Rb/E2F4 and Smad2/3 link survivin to TGF-beta-induced apoptosis and tumor progression. Oncogene 2008, 27, 5326–5338. [Google Scholar] [CrossRef]
- Wang, J.; Yang, L.; Yang, J.; Kuropatwinski, K.; Wang, W.; Liu, X.Q.; Hauser, J.; Brattain, M.G. Transforming growth factor beta induces apoptosis through repressing the phosphoinositide 3-kinase/AKT/survivin pathway in colon cancer cells. Cancer Res. 2008, 68, 3152–3160. [Google Scholar] [CrossRef]
- Nastiuk, K.L.; Yoo, K.; Lo, K.; Su, K.; Yeung, P.; Kutaka, J.; Danielpour, D.; Krolewski, J.J. FLICE-like inhibitory protein blocks transforming growth factor beta 1-induced caspase activation and apoptosis in prostate epithelial cells. Mol. Cancer Res. 2008, 6, 231–242. [Google Scholar] [CrossRef]
- Letterio, J.J.; Geiser, A.G.; Kulkarni, A.B.; Dang, H.; Kong, L.; Nakabayashi, T.; Mackall, C.L.; Gress, R.E.; Roberts, A.B. Autoimmunity associated with TGF-beta1-deficiency in mice is dependent on MHC class II antigen expression. J. Clin. Investig. 1996, 98, 2109–2119. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Yoshida, K.; Ward, J.M.; Letterio, J.J.; Longenecker, G.; Yaswen, L.; Mittleman, B.; Mozes, E.; Roberts, A.B.; Karlsson, S.; et al. Beta 2-microglobulin-deficient background ameliorates lethal phenotype of the TGF-beta 1 null mouse. J. Immunol. 1999, 163, 4013–4019. [Google Scholar] [CrossRef] [PubMed]
- Li, M.O.; Flavell, R.A. Contextual regulation of inflammation: A duet by transforming growth factor-beta and interleukin-10. Immunity 2008, 28, 468–476. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhang, P.; Li, J.; Kulkarni, A.B.; Perruche, S.; Chen, W. A critical function for TGF-beta signaling in the development of natural CD4+CD25+Foxp3+ regulatory T cells. Nat. Immunol. 2008, 9, 632–640. [Google Scholar] [CrossRef] [PubMed]
- Li, M.O.; Wan, Y.Y.; Flavell, R.A. T cell-produced transforming growth factor-beta1 controls T cell tolerance and regulates Th1- and Th17-cell differentiation. Immunity 2007, 26, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Robinson, R.T.; Gorham, J.D. TGF-beta 1 regulates antigen-specific CD4+ T cell responses in the periphery. J. Immunol. 2007, 179, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Wahl, S.M.; Swisher, J.; McCartney-Francis, N.; Chen, W. TGF-beta: The perpetrator of immune suppression by regulatory T cells and suicidal T cells. J. Leukoc. Biol. 2004, 76, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Tone, Y.; Furuuchi, K.; Kojima, Y.; Tykocinski, M.L.; Greene, M.I.; Tone, M. Smad3 and NFAT cooperate to induce Foxp3 expression through its enhancer. Nat. Immunol. 2008, 9, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Martinez, G.J.; Zhang, Z.; Reynolds, J.M.; Tanaka, S.; Chung, Y.; Liu, T.; Robertson, E.; Lin, X.; Feng, X.H.; Dong, C. Smad2 positively regulates the generation of Th17 cells. J. Biol. Chem. 2010, 285, 29039–29043. [Google Scholar] [CrossRef]
- Zhou, L.; Lopes, J.E.; Chong, M.M.; Ivanov, I.I.; Min, R.; Victora, G.D.; Shen, Y.; Du, J.; Rubtsov, Y.P.; Rudensky, A.Y.; et al. TGF-beta-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORgammat function. Nature 2008, 453, 236–240. [Google Scholar] [CrossRef] [PubMed]
- Sanjabi, S.; Oh, S.A.; Li, M.O. Regulation of the Immune Response by TGF-beta: From Conception to Autoimmunity and Infection. Cold Spring Harb. Perspect. Biol. 2017, 9, a022236. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.A.; Massague, J. TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell 2005, 8, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Brabletz, T.; Pfeuffer, I.; Schorr, E.; Siebelt, F.; Wirth, T.; Serfling, E. Transforming growth factor beta and cyclosporin A inhibit the inducible activity of the interleukin-2 gene in T cells through a noncanonical octamer-binding site. Mol. Cell. Biol. 1993, 13, 1155–1162. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.; Ellingsworth, L.R.; Gillis, S.; Wall, R.; Kincade, P.W. Beta transforming growth factors are potential regulators of B lymphopoiesis. J. Exp. Med. 1987, 166, 1290–1299. [Google Scholar] [CrossRef] [PubMed]
- Kehrl, J.H.; Roberts, A.B.; Wakefield, L.M.; Jakowlew, S.; Sporn, M.B.; Fauci, A.S. Transforming growth factor beta is an important immunomodulatory protein for human B lymphocytes. J. Immunol. 1986, 137, 3855–3860. [Google Scholar] [CrossRef] [PubMed]
- Park, S.R.; Seo, G.Y.; Choi, A.J.; Stavnezer, J.; Kim, P.H. Analysis of transforming growth factor-beta1-induced Ig germ-line gamma2b transcription and its implication for IgA isotype switching. Eur. J. Immunol. 2005, 35, 946–956. [Google Scholar] [CrossRef] [PubMed]
- Assoian, R.K.; Sporn, M.B. Type beta transforming growth factor in human platelets: Release during platelet degranulation and action on vascular smooth muscle cells. J. Cell Biol. 1986, 102, 1217–1223. [Google Scholar] [CrossRef] [PubMed]
- Hosgood, G. Wound healing. The role of platelet-derived growth factor and transforming growth factor beta. Vet. Surg. 1993, 22, 490–495. [Google Scholar] [CrossRef]
- Bissell, D.M. Chronic liver injury, TGF-beta, and cancer. Exp. Mol. Med. 2001, 33, 179–190. [Google Scholar] [CrossRef]
- Crowe, M.J.; Doetschman, T.; Greenhalgh, D.G. Delayed wound healing in immunodeficient TGF-beta 1 knockout mice. J. Investig. Dermatol. 2000, 115, 3–11. [Google Scholar] [CrossRef]
- Roberts, A.B.; Tian, F.; Byfield, S.D.; Stuelten, C.; Ooshima, A.; Saika, S.; Flanders, K.C. Smad3 is key to TGF-beta-mediated epithelial-to-mesenchymal transition, fibrosis, tumor suppression and metastasis. Cytokine Growth Factor. Rev. 2006, 17, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Shi, W.; Wang, Y.L.; Chen, H.; Bringas, P., Jr.; Datto, M.B.; Frederick, J.P.; Wang, X.F.; Warburton, D. Smad3 deficiency attenuates bleomycin-induced pulmonary fibrosis in mice. Am. J. Physiol. Lung Cell Mol. Physiol. 2002, 282, L585–L593. [Google Scholar] [CrossRef] [PubMed]
- Bonniaud, P.; Kolb, M.; Galt, T.; Robertson, J.; Robbins, C.; Stampfli, M.; Lavery, C.; Margetts, P.J.; Roberts, A.B.; Gauldie, J. Smad3 null mice develop airspace enlargement and are resistant to TGF-beta-mediated pulmonary fibrosis. J. Immunol. 2004, 173, 2099–2108. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Huang, X.R.; Canlas, E.; Oka, K.; Truong, L.D.; Deng, C.; Bhowmick, N.A.; Ju, W.; Bottinger, E.P.; Lan, H.Y. Essential role of Smad3 in angiotensin II-induced vascular fibrosis. Circ. Res. 2006, 98, 1032–1039. [Google Scholar] [CrossRef] [PubMed]
- You, Y.K.; Wu, W.F.; Huang, X.R.; Li, H.D.; Ren, Y.P.; Zeng, J.C.; Chen, H.; Lan, H.Y. Deletion of Smad3 protects against C-reactive protein-induced renal fibrosis and inflammation in obstructive nephropathy. Int. J. Biol. Sci. 2021, 17, 3911–3922. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wang, G.; Hu, M.; Dai, R.; Li, C.; Cao, Y. Specific inhibitor of Smad3 (SIS3) alleviated submandibular gland fibrosis and dysfunction after dominant duct ligation in mice. J. Dent. Sci. 2023, 18, 865–871. [Google Scholar] [CrossRef] [PubMed]
- Thuault, S.; Valcourt, U.; Petersen, M.; Manfioletti, G.; Heldin, C.H.; Moustakas, A. Transforming growth factor-beta employs HMGA2 to elicit epithelial-mesenchymal transition. J. Cell Biol. 2006, 174, 175–183. [Google Scholar] [CrossRef]
- Derynck, R.; Weinberg, R.A. EMT and Cancer: More Than Meets the Eye. Dev. Cell 2019, 49, 313–316. [Google Scholar] [CrossRef]
- Galliher, A.J.; Schiemann, W.P. Beta3 integrin and Src facilitate transforming growth factor-beta mediated induction of epithelial-mesenchymal transition in mammary epithelial cells. Breast Cancer Res. 2006, 8, R42. [Google Scholar] [CrossRef]
- Galliher, A.J.; Schiemann, W.P. Src phosphorylates Tyr284 in TGF-beta type II receptor and regulates TGF-beta stimulation of p38 MAPK during breast cancer cell proliferation and invasion. Cancer Res. 2007, 67, 3752–3758. [Google Scholar] [CrossRef]
- Galliher-Beckley, A.J.; Schiemann, W.P. Grb2 binding to Tyr284 in TbetaR-II is essential for mammary tumor growth and metastasis stimulated by TGF-beta. Carcinogenesis 2008, 29, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Shipitsin, M.; Campbell, L.L.; Argani, P.; Weremowicz, S.; Bloushtain-Qimron, N.; Yao, J.; Nikolskaya, T.; Serebryiskaya, T.; Beroukhim, R.; Hu, M.; et al. Molecular definition of breast tumor heterogeneity. Cancer Cell 2007, 11, 259–273. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Ghiaur, G.; Gerber, J.; Jones, R.J. Concise review: Cancer stem cells and minimal residual disease. Stem Cells 2012, 30, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Jolly, M.K.; Jia, D.; Boareto, M.; Mani, S.A.; Pienta, K.J.; Ben-Jacob, E.; Levine, H. Coupling the modules of EMT and stemness: A tunable ‘stemness window’ model. Oncotarget 2015, 6, 25161–25174. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Settleman, J. EMT, cancer stem cells and drug resistance: An emerging axis of evil in the war on cancer. Oncogene 2010, 29, 4741–4751. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Holzel, M.; Knijnenburg, T.; Schlicker, A.; Roepman, P.; McDermott, U.; Garnett, M.; Grernrum, W.; Sun, C.; Prahallad, A.; et al. MED12 controls the response to multiple cancer drugs through regulation of TGF-beta receptor signaling. Cell 2012, 151, 937–950. [Google Scholar] [CrossRef] [PubMed]
- Anido, J.; Saez-Borderias, A.; Gonzalez-Junca, A.; Rodon, L.; Folch, G.; Carmona, M.A.; Prieto-Sanchez, R.M.; Barba, I.; Martinez-Saez, E.; Prudkin, L.; et al. TGF-beta Receptor Inhibitors Target the CD44(high)/Id1(high) Glioma-Initiating Cell Population in Human Glioblastoma. Cancer Cell 2010, 18, 655–668. [Google Scholar] [CrossRef]
- Papageorgis, P.; Lambert, A.W.; Ozturk, S.; Gao, F.; Pan, H.; Manne, U.; Alekseyev, Y.O.; Thiagalingam, A.; Abdolmaleky, H.M.; Lenburg, M.; et al. Smad signaling is required to maintain epigenetic silencing during breast cancer progression. Cancer Res. 2010, 70, 968–978. [Google Scholar] [CrossRef] [PubMed]
- Naka, K.; Hoshii, T.; Muraguchi, T.; Tadokoro, Y.; Ooshio, T.; Kondo, Y.; Nakao, S.; Motoyama, N.; Hirao, A. TGF-beta-FOXO signalling maintains leukaemia-initiating cells in chronic myeloid leukaemia. Nature 2010, 463, 676–680. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Wang, L.; Zhang, B.; Li, J.; Dou, X.; Zhao, R.C. TGF-beta1-induced PI3K/Akt/NF-kappaB/MMP9 signalling pathway is activated in Philadelphia chromosome-positive chronic myeloid leukaemia hemangioblasts. J. Biochem. 2011, 149, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Hannigan, A.; Smith, P.; Kalna, G.; Lo Nigro, C.; Orange, C.; O’Brien, D.I.; Shah, R.; Syed, N.; Spender, L.C.; Herrera, B.; et al. Epigenetic downregulation of human disabled homolog 2 switches TGF-beta from a tumor suppressor to a tumor promoter. J. Clin. Investig. 2010, 120, 2842–2857. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Seok Kang, Y.; Soo Kim, J.; Shin, N.Y.; Hanks, S.K.; Song, W.K. The integrin-coupled signaling adaptor p130Cas suppresses Smad3 function in transforming growth factor-beta signaling. Mol. Biol. Cell 2008, 19, 2135–2146. [Google Scholar] [CrossRef] [PubMed]
- Wendt, M.K.; Smith, J.A.; Schiemann, W.P. p130Cas is required for mammary tumor growth and transforming growth factor-beta-mediated metastasis through regulation of Smad2/3 activity. J. Biol. Chem. 2009, 284, 34145–34156. [Google Scholar] [CrossRef] [PubMed]
- Neil, J.R.; Schiemann, W.P. Altered TAB1:I kappaB kinase interaction promotes transforming growth factor beta-mediated nuclear factor-kappaB activation during breast cancer progression. Cancer Res. 2008, 68, 1462–1470. [Google Scholar] [CrossRef] [PubMed]
- Neil, J.R.; Tian, M.; Schiemann, W.P. X-linked inhibitor of apoptosis protein and its E3 ligase activity promote transforming growth factor-beta-mediated nuclear factor-kappaB activation during breast cancer progression. J. Biol. Chem. 2009, 284, 21209–21217. [Google Scholar] [CrossRef] [PubMed]
- Neil, J.R.; Johnson, K.M.; Nemenoff, R.A.; Schiemann, W.P. Cox-2 inactivates Smad signaling and enhances EMT stimulated by TGF-beta through a PGE2-dependent mechanisms. Carcinogenesis 2008, 29, 2227–2235. [Google Scholar] [CrossRef]
- Kretzschmar, M.; Doody, J.; Timokhina, I.; Massague, J. A mechanism of repression of TGFbeta/ Smad signaling by oncogenic Ras. Genes. Dev. 1999, 13, 804–816. [Google Scholar] [CrossRef]
- Singha, P.K.; Yeh, I.T.; Venkatachalam, M.A.; Saikumar, P. Transforming growth factor-beta (TGF-beta)-inducible gene TMEPAI converts TGF-beta from a tumor suppressor to a tumor promoter in breast cancer. Cancer Res. 2010, 70, 6377–6383. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Itoh, S.; Goto, T.; Ohnishi, E.; Inamitsu, M.; Itoh, F.; Satoh, K.; Wiercinska, E.; Yang, W.; Shi, L.; et al. TMEPAI, a transmembrane TGF-beta-inducible protein, sequesters Smad proteins from active participation in TGF-beta signaling. Mol. Cell 2010, 37, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Haque, M.A.; Abdelaziz, M.; Puteri, M.U.; Vo Nguyen, T.T.; Kudo, K.; Watanabe, Y.; Kato, M. PMEPA1/TMEPAI Is a Unique Tumorigenic Activator of AKT Promoting Proteasomal Degradation of PHLPP1 in Triple-Negative Breast Cancer Cells. Cancers 2021, 13, 4934. [Google Scholar] [CrossRef] [PubMed]
- Ettahar, A.; Ferrigno, O.; Zhang, M.Z.; Ohnishi, M.; Ferrand, N.; Prunier, C.; Levy, L.; Bourgeade, M.F.; Bieche, I.; Romero, D.G.; et al. Identification of PHRF1 as a tumor suppressor that promotes the TGF-beta cytostatic program through selective release of TGIF-driven PML inactivation. Cell Rep. 2013, 4, 530–541. [Google Scholar] [CrossRef] [PubMed]
- Kong, B.; Michalski, C.W.; Hong, X.; Valkovskaya, N.; Rieder, S.; Abiatari, I.; Streit, S.; Erkan, M.; Esposito, I.; Friess, H.; et al. AZGP1 is a tumor suppressor in pancreatic cancer inducing mesenchymal-to-epithelial transdifferentiation by inhibiting TGF-beta-mediated ERK signaling. Oncogene 2010, 29, 5146–5158. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Bao, W.; Zhang, B.; Zhang, S.; Chen, Z.; Zhu, X.; He, B.; Wu, L.; Chen, X.; Deng, T.; et al. AZGP1 activation by lenvatinib suppresses intrahepatic cholangiocarcinoma epithelial-mesenchymal transition through the TGF-beta1/Smad3 pathway. Cell Death Dis. 2023, 14, 590. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.Y.; Chen, R.; Yu, J.X.; Liu, T.; Qu, Y.; Lu, L.G. AZGP1 suppresses epithelial-to-mesenchymal transition and hepatic carcinogenesis by blocking TGFbeta1-ERK2 pathways. Cancer Lett. 2016, 374, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Sorensen-Zender, I.; Rong, S.; Haller, H.; Schmitt, R. The Therapeutic Potential of Zinc-Alpha2-Glycoprotein (AZGP1) in Fibrotic Kidney Disease. Int. J. Mol. Sci. 2022, 23, 646. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Qu, X.; Ma, Y.; Zheng, J.; Chu, D.; Liu, B.; Li, X.; Wang, M.; Xu, C.; Liu, N.; et al. Tumor suppressor NDRG2 tips the balance of oncogenic TGF-beta via EMT inhibition in colorectal cancer. Oncogenesis 2014, 3, e86. [Google Scholar] [CrossRef]
- Lee, K.W.; Lim, S.; Kim, K.D. The Function of N-Myc Downstream-Regulated Gene 2 (NDRG2) as a Negative Regulator in Tumor Cell Metastasis. Int. J. Mol. Sci. 2022, 23, 9365. [Google Scholar] [CrossRef]
- Moradi Monfared, M.; Alizadeh Zarei, M.; Rafiei Dehbidi, G.; Behzad Behbahani, A.; Arabsolghar, R.; Takhshid, M.A. NDRG2 Regulates the Expression of Genes Involved in Epithelial Mesenchymal Transition of Prostate Cancer Cells. Iran. J. Med. Sci. 2019, 44, 118–126. [Google Scholar] [PubMed]
- Jin, Z.; Gu, C.; Tian, F.; Jia, Z.; Yang, J. NDRG2 knockdown promotes fibrosis in renal tubular epithelial cells through TGF-beta1/Smad3 pathway. Cell Tissue Res. 2017, 369, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, W.; Chen, S.; Cai, J.; Ban, Y.; Peng, Q.; Zhou, Y.; Zeng, Z.; Li, X.; Xiong, W.; et al. FOXA1 reprograms the TGF-beta-stimulated transcriptional program from a metastasis promoter to a tumor suppressor in nasopharyngeal carcinoma. Cancer Lett. 2019, 442, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Jin, W. TrkB-Induced Inhibition of R-SMAD/SMAD4 Activation is Essential for TGF-beta-Mediated Tumor Suppressor Activity. Cancers 2020, 12, 1048. [Google Scholar] [CrossRef] [PubMed]
- Gorelik, L.; Flavell, R.A. Immune-mediated eradication of tumors through the blockade of transforming growth factor-beta signaling in T cells. Nat. Med. 2001, 7, 1118–1122. [Google Scholar] [CrossRef] [PubMed]
- Donkor, M.K.; Sarkar, A.; Savage, P.A.; Franklin, R.A.; Johnson, L.K.; Jungbluth, A.A.; Allison, J.P.; Li, M.O. T cell surveillance of oncogene-induced prostate cancer is impeded by T cell-derived TGF-beta1 cytokine. Immunity 2011, 35, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Ahmadzadeh, M.; Rosenberg, S.A. TGF-beta 1 attenuates the acquisition and expression of effector function by tumor antigen-specific human memory CD8 T cells. J. Immunol. 2005, 174, 5215–5223. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Jin, W.; Hardegen, N.; Lei, K.J.; Li, L.; Marinos, N.; McGrady, G.; Wahl, S.M. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J. Exp. Med. 2003, 198, 1875–1886. [Google Scholar] [CrossRef] [PubMed]
- Ravi, R.; Noonan, K.A.; Pham, V.; Bedi, R.; Zhavoronkov, A.; Ozerov, I.V.; Makarev, E.; Artemov, V.A.; Wysocki, P.T.; Mehra, R.; et al. Bifunctional immune checkpoint-targeted antibody-ligand traps that simultaneously disable TGFbeta enhance the efficacy of cancer immunotherapy. Nat. Commun. 2018, 9, 741. [Google Scholar] [CrossRef]
- Nandan, D.; Reiner, N.E. TGF-beta attenuates the class II transactivator and reveals an accessory pathway of IFN-gamma action. J. Immunol. 1997, 158, 1095–1101. [Google Scholar] [CrossRef]
- Porta, C.; Rimoldi, M.; Raes, G.; Brys, L.; Ghezzi, P.; Di Liberto, D.; Dieli, F.; Ghisletti, S.; Natoli, G.; De Baetselier, P.; et al. Tolerance and M2 (alternative) macrophage polarization are related processes orchestrated by p50 nuclear factor kappaB. Proc. Natl. Acad. Sci. USA 2009, 106, 14978–14983. [Google Scholar] [CrossRef] [PubMed]
- Comito, G.; Giannoni, E.; Segura, C.P.; Barcellos-de-Souza, P.; Raspollini, M.R.; Baroni, G.; Lanciotti, M.; Serni, S.; Chiarugi, P. Cancer-associated fibroblasts and M2-polarized macrophages synergize during prostate carcinoma progression. Oncogene 2014, 33, 2423–2431. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chen, S.; Wang, W.; Ning, B.F.; Chen, F.; Shen, W.; Ding, J.; Chen, W.; Xie, W.F.; Zhang, X. Cancer-associated fibroblasts promote hepatocellular carcinoma metastasis through chemokine-activated hedgehog and TGF-beta pathways. Cancer Lett. 2016, 379, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Barcellos-de-Souza, P.; Comito, G.; Pons-Segura, C.; Taddei, M.L.; Gori, V.; Becherucci, V.; Bambi, F.; Margheri, F.; Laurenzana, A.; Del Rosso, M.; et al. Mesenchymal Stem Cells are Recruited and Activated into Carcinoma-Associated Fibroblasts by Prostate Cancer Microenvironment-Derived TGF-beta1. Stem Cells 2016, 34, 2536–2547. [Google Scholar] [CrossRef] [PubMed]
- Koochekpour, S.; Merzak, A.; Pilkington, G.J. Vascular endothelial growth factor production is stimulated by gangliosides and TGF-beta isoforms in human glioma cells in vitro. Cancer Lett. 1996, 102, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Budi, E.H.; Mamai, O.; Hoffman, S.; Akhurst, R.J.; Derynck, R. Enhanced TGF-beta Signaling Contributes to the Insulin-Induced Angiogenic Responses of Endothelial Cells. iScience 2019, 11, 474–491. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.; Meng, Q.; Xu, J.; Wang, W.; Zhang, B.; Liu, J.; Liang, C.; Hua, J.; Zhao, Y.; Yu, X.; et al. Signaling pathways in cancer-associated fibroblasts: Recent advances and future perspectives. Cancer Commun. 2023, 43, 3–41. [Google Scholar] [CrossRef] [PubMed]
- Akinbo, D.B.; Ajayi, O.I. Thrombotic Pathogenesis and Laboratory Diagnosis in Cancer Patients, An Update. Int. J. Gen. Med. 2023, 16, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Kamp, D.W. Idiopathic pulmonary fibrosis: The inflammation hypothesis revisited. Chest 2003, 124, 1187–1190. [Google Scholar] [CrossRef]
- Sheppard, D. Transforming growth factor beta: A central modulator of pulmonary and airway inflammation and fibrosis. Proc. Am. Thorac. Soc. 2006, 3, 413–417. [Google Scholar] [CrossRef] [PubMed]
- Moretti, L.; Stalfort, J.; Barker, T.H.; Abebayehu, D. The interplay of fibroblasts, the extracellular matrix, and inflammation in scar formation. J. Biol. Chem. 2022, 298, 101530. [Google Scholar] [CrossRef] [PubMed]
- Ignotz, R.A.; Massague, J. Transforming growth factor-beta stimulates the expression of fibronectin and collagen and their incorporation into the extracellular matrix. J. Biol. Chem. 1986, 261, 4337–4345. [Google Scholar] [CrossRef] [PubMed]
- Border, W.A.; Ruoslahti, E. Transforming growth factor-beta in disease: The dark side of tissue repair. J. Clin. Investig. 1992, 90, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Stampfer, M.R.; Yaswen, P.; Alhadeff, M.; Hosoda, J. TGF beta induction of extracellular matrix associated proteins in normal and transformed human mammary epithelial cells in culture is independent of growth effects. J. Cell. Physiol. 1993, 155, 210–221. [Google Scholar] [CrossRef] [PubMed]
- Hocevar, B.A.; Brown, T.L.; Howe, P.H. TGF-beta induces fibronectin synthesis through a c-Jun N-terminal kinase-dependent, Smad4-independent pathway. EMBO J. 1999, 18, 1345–1356. [Google Scholar] [CrossRef] [PubMed]
- Piek, E.; Ju, W.J.; Heyer, J.; Escalante-Alcalde, D.; Stewart, C.L.; Weinstein, M.; Deng, C.; Kucherlapati, R.; Bottinger, E.P.; Roberts, A.B. Functional characterization of transforming growth factor beta signaling in Smad2- and Smad3-deficient fibroblasts. J. Biol. Chem. 2001, 276, 19945–19953. [Google Scholar] [CrossRef] [PubMed]
- Eickelberg, O.; Kohler, E.; Reichenberger, F.; Bertschin, S.; Woodtli, T.; Erne, P.; Perruchoud, A.P.; Roth, M. Extracellular matrix deposition by primary human lung fibroblasts in response to TGF-beta1 and TGF-beta3. Am. J. Physiol. 1999, 276, L814–L824. [Google Scholar] [CrossRef]
- Murphy, A.M.; Wong, A.L.; Bezuhly, M. Modulation of angiotensin II signaling in the prevention of fibrosis. Fibrogenesis Tissue Repair. 2015, 8, 7. [Google Scholar] [CrossRef]
- Kano, M.R.; Bae, Y.; Iwata, C.; Morishita, Y.; Yashiro, M.; Oka, M.; Fujii, T.; Komuro, A.; Kiyono, K.; Kaminishi, M.; et al. Improvement of cancer-targeting therapy, using nanocarriers for intractable solid tumors by inhibition of TGF-beta signaling. Proc. Natl. Acad. Sci. USA 2007, 104, 3460–3465. [Google Scholar] [CrossRef]
- Fujii, M.; Nakanishi, H.; Toyoda, T.; Tanaka, I.; Kondo, Y.; Osada, H.; Sekido, Y. Convergent signaling in the regulation of connective tissue growth factor in malignant mesothelioma: TGFbeta signaling and defects in the Hippo signaling cascade. Cell Cycle 2012, 11, 3373–3379. [Google Scholar] [CrossRef] [PubMed]
- Ng, M.R.; Brugge, J.S. A stiff blow from the stroma: Collagen crosslinking drives tumor progression. Cancer Cell 2009, 16, 455–457. [Google Scholar] [CrossRef] [PubMed]
- Levental, K.R.; Yu, H.; Kass, L.; Lakins, J.N.; Egeblad, M.; Erler, J.T.; Fong, S.F.; Csiszar, K.; Giaccia, A.; Weninger, W.; et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 2009, 139, 891–906. [Google Scholar] [CrossRef] [PubMed]
- Paszek, M.J.; Zahir, N.; Johnson, K.R.; Lakins, J.N.; Rozenberg, G.I.; Gefen, A.; Reinhart-King, C.A.; Margulies, S.S.; Dembo, M.; Boettiger, D.; et al. Tensional homeostasis and the malignant phenotype. Cancer Cell 2005, 8, 241–254. [Google Scholar] [CrossRef] [PubMed]
- Erler, J.T.; Bennewith, K.L.; Cox, T.R.; Lang, G.; Bird, D.; Koong, A.; Le, Q.T.; Giaccia, A.J. Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer Cell 2009, 15, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Erler, J.T.; Bennewith, K.L.; Nicolau, M.; Dornhofer, N.; Kong, C.; Le, Q.T.; Chi, J.T.; Jeffrey, S.S.; Giaccia, A.J. Lysyl oxidase is essential for hypoxia-induced metastasis. Nature 2006, 440, 1222–1226. [Google Scholar] [CrossRef] [PubMed]
- Payne, S.L.; Hendrix, M.J.; Kirschmann, D.A. Lysyl oxidase regulates actin filament formation through the p130(Cas)/Crk/DOCK180 signaling complex. J. Cell. Biochem. 2006, 98, 827–837. [Google Scholar] [CrossRef] [PubMed]
- Ying, M.; Chen, Y.; Yuan, B. Knockdown of lysyl oxidase like 1 inhibits the proliferation and pro-fibrotic effects of transforming growth factor-beta1-induced hypertrophic scar fibroblasts. Can. J. Physiol. Pharmacol. 2021, 99, 1272–1279. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhu, X.; Cao, X.; Lu, Y.; Zhou, J.; Zhang, X. Single-cell analysis reveals lysyl oxidase (Lox)(+) fibroblast subset involved in cardiac fibrosis of diabetic mice. J. Adv. Res. 2023, 54, 223–237. [Google Scholar] [CrossRef]
- Stark, K.; Massberg, S. Interplay between inflammation and thrombosis in cardiovascular pathology. Nat. Rev. Cardiol. 2021, 18, 666–682. [Google Scholar] [CrossRef]
- Wang, Y.; Golden, J.B.; Fritz, Y.; Zhang, X.; Diaconu, D.; Camhi, M.I.; Gao, H.; Dawes, S.M.; Xing, X.; Ganesh, S.K.; et al. Interleukin 6 regulates psoriasiform inflammation-associated thrombosis. JCI Insight 2016, 1, e89384. [Google Scholar] [CrossRef] [PubMed]
- Wakefield, T.W.; Strieter, R.M.; Downing, L.J.; Kadell, A.M.; Wilke, C.A.; Burdick, M.D.; Wrobleski, S.K.; Phillips, M.L.; Paulson, J.C.; Anderson, D.C.; et al. P-selectin and TNF inhibition reduce venous thrombosis inflammation. J. Surg. Res. 1996, 64, 26–31. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, G.; Villa, C.; Tamborini, A.; Villa, S. Evaluation of the main coagulation tests in the presence of hemolysis in healthy subjects and patients on oral anticoagulant therapy. Int. J. Lab. Hematol. 2015, 37, 819–833. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, A.; Gerosa, S.; D’Angelo, S.V.; Mailhac, A.; Colombo, A.; Agazzi, A.; Mazzola, G.; Chierchia, S. Protein S and protein C anticoagulant activity in acute and chronic cardiac ischemic syndromes. Relationship to inflammation, complement activation and in vivo thrombin activity. Thromb. Res. 1994, 75, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Falanga, V.; Qian, S.W.; Danielpour, D.; Katz, M.H.; Roberts, A.B.; Sporn, M.B. Hypoxia upregulates the synthesis of TGF-beta 1 by human dermal fibroblasts. J. Investig. Dermatol. 1991, 97, 634–637. [Google Scholar] [CrossRef] [PubMed]
- Mingyuan, X.; Qianqian, P.; Shengquan, X.; Chenyi, Y.; Rui, L.; Yichen, S.; Jinghong, X. Hypoxia-inducible factor-1alpha activates transforming growth factor-beta1/Smad signaling and increases collagen deposition in dermal fibroblasts. Oncotarget 2018, 9, 3188–3197. [Google Scholar] [CrossRef] [PubMed]
- Morello, S.; Caiazzo, E.; Turiello, R.; Cicala, C. Thrombo-Inflammation: A Focus on NTPDase1/CD39. Cells 2021, 10, 2223. [Google Scholar] [CrossRef] [PubMed]
- Hockel, M.; Vaupel, P. Tumor hypoxia: Definitions and current clinical, biologic, and molecular aspects. J. Natl. Cancer Inst. 2001, 93, 266–276. [Google Scholar] [CrossRef] [PubMed]
- Kim, L.C.; Simon, M.C. Hypoxia-Inducible Factors in Cancer. Cancer Res. 2022, 82, 195–196. [Google Scholar] [CrossRef]
- Rosell-Garcia, T.; Palomo-Alvarez, O.; Rodriguez-Pascual, F. A hierarchical network of hypoxia-inducible factor and SMAD proteins governs procollagen lysyl hydroxylase 2 induction by hypoxia and transforming growth factor beta1. J. Biol. Chem. 2019, 294, 14308–14318. [Google Scholar] [CrossRef]
- Donovan, D.; Harmey, J.H.; Toomey, D.; Osborne, D.H.; Redmond, H.P.; Bouchier-Hayes, D.J. TGF beta-1 regulation of VEGF production by breast cancer cells. Ann. Surg. Oncol. 1997, 4, 621–627. [Google Scholar] [CrossRef] [PubMed]
- Shih, S.C.; Claffey, K.P. Role of AP-1 and HIF-1 transcription factors in TGF-beta activation of VEGF expression. Growth Factors 2001, 19, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Jung, K. Normalization of the tumor microenvironment by harnessing vascular and immune modulation to achieve enhanced cancer therapy. Exp. Mol. Med. 2023, 55, 2308–2319. [Google Scholar] [CrossRef] [PubMed]
- Kyrle, P.A.; Eichinger, S. Deep vein thrombosis. Lancet 2005, 365, 1163–1174. [Google Scholar] [CrossRef] [PubMed]
- Mallikarjuna, P.; Zhou, Y.; Landstrom, M. The Synergistic Cooperation between TGF-beta and Hypoxia in Cancer and Fibrosis. Biomolecules 2022, 12, 635. [Google Scholar] [CrossRef] [PubMed]
- Ejaz, U.; Akhtar, F.; Xue, J.; Wan, X.; Zhang, T.; He, S. Review: Inhibitory potential of low molecular weight Heparin in cell adhesion; emphasis on tumor metastasis. Eur. J. Pharmacol. 2021, 892, 173778. [Google Scholar] [CrossRef] [PubMed]
- Mousavi, S.; Moradi, M.; Khorshidahmad, T.; Motamedi, M. Anti-Inflammatory Effects of Heparin and Its Derivatives: A Systematic Review. Adv. Pharmacol. Sci. 2015, 2015, 507151. [Google Scholar] [CrossRef]
- Obi, A.T.; Diaz, J.A.; Ballard-Lipka, N.L.; Roelofs, K.J.; Farris, D.M.; Lawrence, D.A.; Henke, P.K.; Wakefield, T.W. Low-molecular-weight heparin modulates vein wall fibrotic response in a plasminogen activator inhibitor 1-dependent manner. J. Vasc. Surg. Venous Lymphat. Disord. 2014, 2, 441–450.e441. [Google Scholar] [CrossRef] [PubMed]
- Sanford, D.; Lazo-Langner, A. The effect of low molecular weight heparin on survival in cancer patients: An updated systematic review and meta-analysis of randomized trials: Reply. J. Thromb. Haemost. 2014, 12, 1574–1575. [Google Scholar] [CrossRef]
- Park, C.H.; Yoo, T.H. TGF-beta Inhibitors for Therapeutic Management of Kidney Fibrosis. Pharmaceuticals 2022, 15, 1485. [Google Scholar] [CrossRef]
- Vistnes, M. Hitting the Target! Challenges and Opportunities for TGF-beta Inhibition for the Treatment of Cardiac fibrosis. Pharmaceuticals 2024, 17, 267. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Stappenbeck, F.; Parhami, F. Oxy210, a Semi-Synthetic Oxysterol, Inhibits Profibrotic Signaling in Cellular Models of Lung and Kidney Fibrosis. Pharmaceuticals 2023, 16, 114. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, O.A.; Abdel-Reheim, M.A.; Saleh, L.A.; Alamri, M.M.S.; Alfaifi, J.; Adam, M.I.E.; Farrag, A.A.; AlQahtani, A.A.J.; BinAfif, W.F.; Hashish, A.A.; et al. Alvespimycin Exhibits Potential Anti-TGF-beta Signaling in the Setting of a Proteasome Activator in Rats with Bleomycin-Induced Pulmonary Fibrosis: A Promising Novel Approach. Pharmaceuticals 2023, 16, 1123. [Google Scholar] [CrossRef] [PubMed]
- Akhurst, R.J. Targeting TGF-beta Signaling for Therapeutic Gain. Cold Spring Harb. Perspect. Biol. 2017, 9, a022301. [Google Scholar] [CrossRef] [PubMed]
- Dickson, M.C.; Martin, J.S.; Cousins, F.M.; Kulkarni, A.B.; Karlsson, S.; Akhurst, R.J. Defective haematopoiesis and vasculogenesis in transforming growth factor-beta 1 knock out mice. Development 1995, 121, 1845–1854. [Google Scholar] [CrossRef] [PubMed]
- Frazier, K.; Thomas, R.; Scicchitano, M.; Mirabile, R.; Boyce, R.; Zimmerman, D.; Grygielko, E.; Nold, J.; DeGouville, A.C.; Huet, S.; et al. Inhibition of ALK5 signaling induces physeal dysplasia in rats. Toxicol. Pathol. 2007, 35, 284–295. [Google Scholar] [CrossRef] [PubMed]
- Gueorguieva, I.; Cleverly, A.L.; Stauber, A.; Sada Pillay, N.; Rodon, J.A.; Miles, C.P.; Yingling, J.M.; Lahn, M.M. Defining a therapeutic window for the novel TGF-beta inhibitor LY2157299 monohydrate based on a pharmacokinetic/pharmacodynamic model. Br. J. Clin. Pharmacol. 2014, 77, 796–807. [Google Scholar] [CrossRef] [PubMed]
- Allison, R.S.; Mumy, M.L.; Wakefield, L.M. Translational control elements in the major human transforming growth factor-beta 1 mRNA. Growth Factors 1998, 16, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Duan, F.F.; Barron, G.; Meliton, A.; Mutlu, G.M.; Dulin, N.O.; Schuger, L. P311 Promotes Lung Fibrosis via Stimulation of Transforming Growth Factor-beta1, -beta2, and -beta3 Translation. Am. J. Respir. Cell Mol. Biol. 2019, 60, 221–231. [Google Scholar] [CrossRef]
- Anscher, M.S.; Crocker, I.R.; Jirtle, R.L. Transforming growth factor-beta 1 expression in irradiated liver. Radiat. Res. 1990, 122, 77–85. [Google Scholar] [CrossRef]
- Khalil, N.; Whitman, C.; Zuo, L.; Danielpour, D.; Greenberg, A. Regulation of alveolar macrophage transforming growth factor-beta secretion by corticosteroids in bleomycin-induced pulmonary inflammation in the rat. J. Clin. Investig. 1993, 92, 1812–1818. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Gu, X.; Xia, L.; Zhou, Y.; Bouamar, H.; Yang, J.; Ding, X.; Zwieb, C.; Zhang, J.; Hinck, A.P.; et al. A Novel TGFbeta Trap Blocks Chemotherapeutics-Induced TGFbeta1 Signaling and Enhances Their Anticancer Activity in Gynecologic Cancers. Clin. Cancer Res. 2018, 24, 2780–2793. [Google Scholar] [CrossRef] [PubMed]
- Bhola, N.E.; Balko, J.M.; Dugger, T.C.; Kuba, M.G.; Sanchez, V.; Sanders, M.; Stanford, J.; Cook, R.S.; Arteaga, C.L. TGF-beta inhibition enhances chemotherapy action against triple-negative breast cancer. J. Clin. Investig. 2013, 123, 1348–1358. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhao, Y.; Zhang, H.; Zheng, W.; Wang, R.; Gu, X. Predictive value of tumor mutational burden for PD-1/PD-L1 inhibitors in NSCLC: A meta-analysis. Medicine 2023, 102, e34990. [Google Scholar] [CrossRef] [PubMed]
- Cercek, A.; Lumish, M.; Sinopoli, J.; Weiss, J.; Shia, J.; Lamendola-Essel, M.; El Dika, I.H.; Segal, N.; Shcherba, M.; Sugarman, R.; et al. PD-1 Blockade in Mismatch Repair-Deficient, Locally Advanced Rectal Cancer. N. Engl. J. Med. 2022, 386, 2363–2376. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.; Yan, X.; Sheng, X.; Si, L.; Cui, C.; Kong, Y.; Mao, L.; Lian, B.; Bai, X.; Wang, X.; et al. Safety and clinical activity with an anti-PD-1 antibody JS001 in advanced melanoma or urologic cancer patients. J. Hematol. Oncol. 2019, 12, 7. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zhang, S.; Guo, X.; Li, C.; Yang, B.; Qu, X.; Wang, S. PD-1 inhibitors combined with paclitaxel and cisplatin in first-line treatment of esophageal squamous cell carcinoma (ESCC): A network meta-analysis. BMC Cancer 2023, 23, 1221. [Google Scholar] [CrossRef] [PubMed]
- Yi, M.; Jiao, D.; Xu, H.; Liu, Q.; Zhao, W.; Han, X.; Wu, K. Biomarkers for predicting efficacy of PD-1/PD-L1 inhibitors. Mol. Cancer 2018, 17, 129. [Google Scholar] [CrossRef] [PubMed]
- Yi, M.; Zheng, X.; Niu, M.; Zhu, S.; Ge, H.; Wu, K. Combination strategies with PD-1/PD-L1 blockade: Current advances and future directions. Mol. Cancer 2022, 21, 28. [Google Scholar] [CrossRef]
- Darvin, P.; Toor, S.M.; Sasidharan Nair, V.; Elkord, E. Immune checkpoint inhibitors: Recent progress and potential biomarkers. Exp. Mol. Med. 2018, 50, 1–11. [Google Scholar] [CrossRef]
- Bai, X.; Yi, M.; Jiao, Y.; Chu, Q.; Wu, K. Blocking TGF-beta Signaling To Enhance The Efficacy Of Immune Checkpoint Inhibitor. Onco Targets Ther. 2019, 12, 9527–9538. [Google Scholar] [CrossRef]
- Huang, C.Y.; Chung, C.L.; Hu, T.H.; Chen, J.J.; Liu, P.F.; Chen, C.L. Recent progress in TGF-beta inhibitors for cancer therapy. Biomed. Pharmacother. 2021, 134, 111046. [Google Scholar] [CrossRef] [PubMed]
- Powles, T.; Kockx, M.; Rodriguez-Vida, A.; Duran, I.; Crabb, S.J.; Van Der Heijden, M.S.; Szabados, B.; Pous, A.F.; Gravis, G.; Herranz, U.A.; et al. Clinical efficacy and biomarker analysis of neoadjuvant atezolizumab in operable urothelial carcinoma in the ABACUS trial. Nat. Med. 2019, 25, 1706–1714. [Google Scholar] [CrossRef] [PubMed]
- Inman, G.J.; Nicolas, F.J.; Callahan, J.F.; Harling, J.D.; Gaster, L.M.; Reith, A.D.; Laping, N.J.; Hill, C.S. SB-431542 is a potent and specific inhibitor of transforming growth factor-beta superfamily type I activin receptor-like kinase (ALK) receptors ALK4, ALK5, and ALK7. Mol. Pharmacol. 2002, 62, 65–74. [Google Scholar] [CrossRef] [PubMed]
- DaCosta Byfield, S.; Major, C.; Laping, N.J.; Roberts, A.B. SB-505124 is a selective inhibitor of transforming growth factor-beta type I receptors ALK4, ALK5, and ALK7. Mol. Pharmacol. 2004, 65, 744–752. [Google Scholar] [CrossRef] [PubMed]
- Gellibert, F.; Woolven, J.; Fouchet, M.H.; Mathews, N.; Goodland, H.; Lovegrove, V.; Laroze, A.; Nguyen, V.L.; Sautet, S.; Wang, R.; et al. Identification of 1,5-naphthyridine derivatives as a novel series of potent and selective TGF-beta type I receptor inhibitors. J. Med. Chem. 2004, 47, 4494–4506. [Google Scholar] [CrossRef] [PubMed]
- Grygielko, E.T.; Martin, W.M.; Tweed, C.; Thornton, P.; Harling, J.; Brooks, D.P.; Laping, N.J. Inhibition of gene markers of fibrosis with a novel inhibitor of transforming growth factor-beta type I receptor kinase in puromycin-induced nephritis. J. Pharmacol. Exp. Ther. 2005, 313, 943–951. [Google Scholar] [CrossRef] [PubMed]
- Ehata, S.; Hanyu, A.; Fujime, M.; Katsuno, Y.; Fukunaga, E.; Goto, K.; Ishikawa, Y.; Nomura, K.; Yokoo, H.; Shimizu, T.; et al. Ki26894, a novel transforming growth factor-beta type I receptor kinase inhibitor, inhibits in vitro invasion and in vivo bone metastasis of a human breast cancer cell line. Cancer Sci. 2007, 98, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Tojo, M.; Hamashima, Y.; Hanyu, A.; Kajimoto, T.; Saitoh, M.; Miyazono, K.; Node, M.; Imamura, T. The ALK-5 inhibitor A-83-01 inhibits Smad signaling and epithelial-to-mesenchymal transition by transforming growth factor-beta. Cancer Sci. 2005, 96, 791–800. [Google Scholar] [CrossRef]
- Uhl, M.; Aulwurm, S.; Wischhusen, J.; Weiler, M.; Ma, J.Y.; Almirez, R.; Mangadu, R.; Liu, Y.W.; Platten, M.; Herrlinger, U.; et al. SD-208, a novel transforming growth factor beta receptor I kinase inhibitor, inhibits growth and invasiveness and enhances immunogenicity of murine and human glioma cells in vitro and in vivo. Cancer Res. 2004, 64, 7954–7961. [Google Scholar] [CrossRef]
- Melisi, D.; Ishiyama, S.; Sclabas, G.M.; Fleming, J.B.; Xia, Q.; Tortora, G.; Abbruzzese, J.L.; Chiao, P.J. LY2109761, a novel transforming growth factor beta receptor type I and type II dual inhibitor, as a therapeutic approach to suppressing pancreatic cancer metastasis. Mol. Cancer Ther. 2008, 7, 829–840. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Zhang, Y.; Wang, H.; Guo, Z.; Wang, X.; Li, X.; Chang, S.; Sun, T.; Yu, Z.; Xu, T.; et al. Synthesis and biological evaluation of 4-(pyridin-4-oxy)-3-(3,3-difluorocyclobutyl)-pyrazole derivatives as novel potent transforming growth factor-beta type 1 receptor inhibitors. Eur. J. Med. Chem. 2020, 198, 112354. [Google Scholar] [CrossRef]
- Yap, T.A.; Vieito, M.; Baldini, C.; Sepulveda-Sanchez, J.M.; Kondo, S.; Simonelli, M.; Cosman, R.; van der Westhuizen, A.; Atkinson, V.; Carpentier, A.F.; et al. First-In-Human Phase I Study of a Next-Generation, Oral, TGFbeta Receptor 1 Inhibitor, LY3200882, in Patients with Advanced Cancer. Clin. Cancer Res. 2021, 27, 6666–6676. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.H.; Krishnaiah, M.; Sreenu, D.; Subrahmanyam, V.B.; Rao, K.S.; Lee, H.J.; Park, S.J.; Park, H.J.; Lee, K.; Sheen, Y.Y.; et al. Discovery of N-((4-([1,2,4]triazolo [1,5-a]pyridin-6-yl)-5-(6-methylpyridin-2-yl)-1H-imidazol-2-yl)methyl)-2-fluoroaniline (EW-7197): A highly potent, selective, and orally bioavailable inhibitor of TGF-beta type I receptor kinase as cancer immunotherapeutic/antifibrotic agent. J. Med. Chem. 2014, 57, 4213–4238. [Google Scholar] [CrossRef] [PubMed]
- Bueno, L.; de Alwis, D.P.; Pitou, C.; Yingling, J.; Lahn, M.; Glatt, S.; Troconiz, I.F. Semi-mechanistic modelling of the tumour growth inhibitory effects of LY2157299, a new type I receptor TGF-beta kinase antagonist, in mice. Eur. J. Cancer 2008, 44, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Wang, Z.S.; Zhou, H.; Pan, H.M.; Han, W.D.; Deng, Y.H.; Zhang, Y.Q.; Zhang, Y.; Wang, S.; Wang, J.; et al. GFH018, a small molecular inhibitor targeting TGF-βRI kinase, in patients with advanced solid tumors: Final results of the phase I study. J. Clin. Oncol. 2023, 41. [Google Scholar] [CrossRef]
- Li, J.; Liu, T.S.; Bao, H.Y.; Xu, Z.S.; Shu, Y.; Zheng, B.H. A phase I study of a TGF-β receptor I kinase inhibitor YL-13027 in patients with advanced solid tumors. J. Clin. Oncol. 2021, 39. [Google Scholar] [CrossRef]
- Callahan, J.F.; Burgess, J.L.; Fornwald, J.A.; Gaster, L.M.; Harling, J.D.; Harrington, F.P.; Heer, J.; Kwon, C.; Lehr, R.; Mathur, A.; et al. Identification of novel inhibitors of the transforming growth factor beta1 (TGF-beta1) type 1 receptor (ALK5). J. Med. Chem. 2002, 45, 999–1001. [Google Scholar] [CrossRef] [PubMed]
- Ceglia, I.; Dueck, A.C.; Masiello, F.; Martelli, F.; He, W.; Federici, G.; Petricoin, E.F., 3rd; Zeuner, A.; Iancu-Rubin, C.; Weinberg, R.; et al. Preclinical rationale for TGF-beta inhibition as a therapeutic target for the treatment of myelofibrosis. Exp. Hematol. 2016, 44, 1138–1155.e1134. [Google Scholar] [CrossRef]
- Heo, J.Y.; Do, J.Y.; Lho, Y.; Kim, A.Y.; Kim, S.W.; Kang, S.H. TGF-beta1 Receptor Inhibitor SB525334 Attenuates the Epithelial to Mesenchymal Transition of Peritoneal Mesothelial Cells via the TGF-beta1 Signaling Pathway. Biomedicines 2021, 9, 839. [Google Scholar] [CrossRef]
- Son, J.Y.; Park, S.Y.; Kim, S.J.; Lee, S.J.; Park, S.A.; Kim, M.J.; Kim, S.W.; Kim, D.K.; Nam, J.S.; Sheen, Y.Y. EW-7197, a novel ALK-5 kinase inhibitor, potently inhibits breast to lung metastasis. Mol. Cancer Ther. 2014, 13, 1704–1716. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.H.; Myers, J.; Tomchuck, S.; Bonner, M.; Eid, S.; Kingsley, D.; VanHeyst, K.; Kim, S.J.; Kim, B.G.; Huang, A.Y. Oral TGF-betaR1 inhibitor Vactosertib promotes osteosarcoma regression by targeting tumor proliferation and enhancing anti-tumor immunity. Res. Sq. 2023. [Google Scholar] [CrossRef] [PubMed]
- Hong, E.; Barczak, W.; Park, S.; Heo, J.S.; Ooshima, A.; Munro, S.; Hong, C.P.; Park, J.; An, H.; Park, J.O.; et al. Combination treatment of T1-44, a PRMT5 inhibitor with Vactosertib, an inhibitor of TGF-beta signaling, inhibits invasion and prolongs survival in a mouse model of pancreatic tumors. Cell Death Dis. 2023, 14, 93. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.Y.; Hwang, S.; Clarke, J.M.; Bauer, T.M.; Keedy, V.L.; Lee, H.; Park, N.; Kim, S.J.; Lee, J.I. Pharmacokinetic characteristics of vactosertib, a new activin receptor-like kinase 5 inhibitor, in patients with advanced solid tumors in a first-in-human phase 1 study. Investig. New Drugs 2020, 38, 812–820. [Google Scholar] [CrossRef] [PubMed]
- Kelley, R.K.; Gane, E.; Assenat, E.; Siebler, J.; Galle, P.R.; Merle, P.; Hourmand, I.O.; Cleverly, A.; Zhao, Y.; Gueorguieva, I.; et al. A Phase 2 Study of Galunisertib (TGF-beta1 Receptor Type I Inhibitor) and Sorafenib in Patients with Advanced Hepatocellular Carcinoma. Clin. Transl. Gastroenterol. 2019, 10, e00056. [Google Scholar] [CrossRef] [PubMed]
- Santini, V.; Valcarcel, D.; Platzbecker, U.; Komrokji, R.S.; Cleverly, A.L.; Lahn, M.M.; Janssen, J.; Zhao, Y.; Chiang, A.; Giagounidis, A.; et al. Phase II Study of the ALK5 Inhibitor Galunisertib in Very Low-, Low-, and Intermediate-Risk Myelodysplastic Syndromes. Clin. Cancer Res. 2019, 25, 6976–6985. [Google Scholar] [CrossRef]
- Wick, A.; Desjardins, A.; Suarez, C.; Forsyth, P.; Gueorguieva, I.; Burkholder, T.; Cleverly, A.L.; Estrem, S.T.; Wang, S.; Lahn, M.M.; et al. Phase 1b/2a study of galunisertib, a small molecule inhibitor of transforming growth factor-beta receptor I, in combination with standard temozolomide-based radiochemotherapy in patients with newly diagnosed malignant glioma. Investig. New Drugs 2020, 38, 1570–1579. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, T.; Gunderson, A.J.; Gilchrist, M.; Whiteford, M.; Kiely, M.X.; Hayman, A.; O’Brien, D.; Ahmad, R.; Manchio, J.V.; Fox, N.; et al. Galunisertib plus neoadjuvant chemoradiotherapy in patients with locally advanced rectal cancer: A single-arm, phase 2 trial. Lancet Oncol. 2022, 23, 1189–1200. [Google Scholar] [CrossRef] [PubMed]
- Melisi, D.; Oh, D.Y.; Hollebecque, A.; Calvo, E.; Varghese, A.; Borazanci, E.; Macarulla, T.; Merz, V.; Zecchetto, C.; Zhao, Y.; et al. Safety and activity of the TGFbeta receptor I kinase inhibitor galunisertib plus the anti-PD-L1 antibody durvalumab in metastatic pancreatic cancer. J. Immunother. Cancer 2021, 9, e002068. [Google Scholar] [CrossRef]
- Harding, J.J.; Do, R.K.; Yaqubie, A.; Cleverly, A.; Zhao, Y.; Gueorguieva, I.; Lahn, M.; Benhadji, K.A.; Kelley, R.K.; Abou-Alfa, G.K. Phase 1b study of galunisertib and ramucirumab in patients with advanced hepatocellular carcinoma. Cancer Med. 2021, 10, 3059–3067. [Google Scholar] [CrossRef]
- Zhang, Y.; Parrish, K.E.; Tortolani, D.R.; Poss, M.A.; Huang, A.; Wan, H.; Purandare, A.V.; Donnell, A.F.; Kempson, J.; Hou, X.; et al. Long-Acting Tumor-Activated Prodrug of a TGFbetaR Inhibitor. J. Med. Chem. 2021, 64, 15787–15798. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.C.; Tan, A.R.; Olencki, T.E.; Shapiro, G.I.; Dezube, B.J.; Reiss, M.; Hsu, F.J.; Berzofsky, J.A.; Lawrence, D.P. Phase I study of GC1008 (fresolimumab): A human anti-transforming growth factor-beta (TGFbeta) monoclonal antibody in patients with advanced malignant melanoma or renal cell carcinoma. PLoS ONE 2014, 9, e90353. [Google Scholar] [CrossRef] [PubMed]
- Formenti, S.C.; Lee, P.; Adams, S.; Goldberg, J.D.; Li, X.; Xie, M.W.; Ratikan, J.A.; Felix, C.; Hwang, L.; Faull, K.F.; et al. Focal Irradiation and Systemic TGFbeta Blockade in Metastatic Breast Cancer. Clin. Cancer Res. 2018, 24, 2493–2504. [Google Scholar] [CrossRef] [PubMed]
- Cohn, A.; Lahn, M.M.; Williams, K.E.; Cleverly, A.L.; Pitou, C.; Kadam, S.K.; Farmen, M.W.; Desaiah, D.; Raju, R.; Conkling, P.; et al. A phase I dose-escalation study to a predefined dose of a transforming growth factor-beta1 monoclonal antibody (TbetaM1) in patients with metastatic cancer. Int. J. Oncol. 2014, 45, 2221–2231. [Google Scholar] [CrossRef] [PubMed]
- Bauer, T.M.; Santoro, A.; Lin, C.C.; Garrido-Laguna, I.; Joerger, M.; Greil, R.; Spreafico, A.; Yau, T.; Goebeler, M.E.; Hutter-Kronke, M.L.; et al. Phase I/Ib, open-label, multicenter, dose-escalation study of the anti-TGF-beta monoclonal antibody, NIS793, in combination with spartalizumab in adult patients with advanced tumors. J. Immunother. Cancer 2023, 11, e007353. [Google Scholar] [CrossRef] [PubMed]
- Greco, R.; Qu, H.; Qu, H.; Theilhaber, J.; Shapiro, G.; Gregory, R.; Winter, C.; Malkova, N.; Sun, F.; Jaworski, J.; et al. Pan-TGFbeta inhibition by SAR439459 relieves immunosuppression and improves antitumor efficacy of PD-1 blockade. Oncoimmunology 2020, 9, 1811605. [Google Scholar] [CrossRef] [PubMed]
- Williamson, S.K.; Hodi, F.S.; Johnson, M.L.; Barve, M.A.; Juric, D.; Baranda, J.C.; Schneider, R.E.; Bauer, T.M.; Lin, T.T.; Wang, R.; et al. Safety, pharmacokinetic and pharmacodynamic results from dose escalation of SAR439459, a TGFβ inhibitor, as monotherapy or in combination with cemiplimab in a phase 1/1b study. J. Clin. Oncol. 2021, 39, 2510. [Google Scholar] [CrossRef]
- Dodagatta-Marri, E.; Meyer, D.S.; Reeves, M.Q.; Paniagua, R.; To, M.D.; Binnewies, M.; Broz, M.L.; Mori, H.; Wu, D.; Adoumie, M.; et al. alpha-PD-1 therapy elevates Treg/Th balance and increases tumor cell pSmad3 that are both targeted by alpha-TGFbeta antibody to promote durable rejection and immunity in squamous cell carcinomas. J. Immunother. Cancer 2019, 7, 62. [Google Scholar] [CrossRef] [PubMed]
- Tolcher, A.W.; Berlin, J.D.; Cosaert, J.; Kauh, J.; Chan, E.; Piha-Paul, S.A.; Amaya, A.; Tang, S.; Driscoll, K.; Kimbung, R.; et al. A phase 1 study of anti-TGFbeta receptor type-II monoclonal antibody LY3022859 in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2017, 79, 673–680. [Google Scholar] [CrossRef]
- Gabriely, G.; da Cunha, A.P.; Rezende, R.M.; Kenyon, B.; Madi, A.; Vandeventer, T.; Skillin, N.; Rubino, S.; Garo, L.; Mazzola, M.A.; et al. Targeting latency-associated peptide promotes antitumor immunity. Sci. Immunol. 2017, 2, eaaj1738. [Google Scholar] [CrossRef]
- Welsh, B.T.; Faucette, R.; Bilic, S.; Martin, C.J.; Schurpf, T.; Chen, D.; Nicholls, S.; Lansita, J.; Kalra, A. Nonclinical Development of SRK-181: An Anti-Latent TGFbeta1 Monoclonal Antibody for the Treatment of Locally Advanced or Metastatic Solid Tumors. Int. J. Toxicol. 2021, 40, 226–241. [Google Scholar] [CrossRef]
- Powderly, J.; Shimizu, T.; Lorusso, P.; Razak, A.; Miller, K.D.; Balar, A.V.; Bruix, J.; Michel, L.S.; Blaney, M.; Guan, X.; et al. Phase I first-in-human study of ABBV-151 as monotherapy or in combination with budigalimab in patients with locally advanced or metastatic solid tumours. Ann. Oncol. 2020, 31, S499. [Google Scholar] [CrossRef]
- Satoh, K.; Kobayashi, Y.; Fujimaki, K.; Hayashi, S.; Ishida, S.; Sugiyama, D.; Sato, T.; Lim, K.; Miyamoto, M.; Kozuma, S.; et al. Novel anti-GARP antibody DS-1055a augments anti-tumor immunity by depleting highly suppressive GARP+ regulatory T cells. Int. Immunol. 2021, 33, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Takasaka, N.; Seed, R.I.; Cormier, A.; Bondesson, A.J.; Lou, J.; Elattma, A.; Ito, S.; Yanagisawa, H.; Hashimoto, M.; Ma, R.; et al. Integrin alphavbeta8-expressing tumor cells evade host immunity by regulating TGF-beta activation in immune cells. JCI Insight 2018, 3, e122591. [Google Scholar] [CrossRef] [PubMed]
- Dodagatta-Marri, E.; Ma, H.Y.; Liang, B.; Li, J.; Meyer, D.S.; Chen, S.Y.; Sun, K.H.; Ren, X.; Zivak, B.; Rosenblum, M.D.; et al. Integrin alphavbeta8 on T cells suppresses anti-tumor immunity in multiple models and is a promising target for tumor immunotherapy. Cell Rep. 2021, 36, 109309. [Google Scholar] [CrossRef] [PubMed]
- Mascarenhas, J.; Migliaccio, A.R.; Kosiorek, H.; Bhave, R.; Palmer, J.; Kuykendall, A.; Mesa, R.; Rampal, R.K.; Gerds, A.T.; Yacoub, A.; et al. A Phase Ib Trial of AVID200, a TGFbeta 1/3 Trap, in Patients with Myelofibrosis. Clin. Cancer Res. 2023, 29, 3622–3632. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Liu, M.; Do, M.H.; Chou, C.; Stamatiades, E.G.; Nixon, B.G.; Shi, W.; Zhang, X.; Li, P.; Gao, S.; et al. Cancer immunotherapy via targeted TGF-beta signalling blockade in T(H) cells. Nature 2020, 587, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Jochems, C.; Tritsch, S.R.; Pellom, S.T.; Su, Z.; Soon-Shiong, P.; Wong, H.C.; Gulley, J.L.; Schlom, J. Analyses of functions of an anti-PD-L1/TGFbetaR2 bispecific fusion protein (M7824). Oncotarget 2017, 8, 75217–75231. [Google Scholar] [CrossRef] [PubMed]
- Burvenich, I.; Goh, Y.W.; Guo, N.; Gan, H.; Rigopoulos, A.; Liu, Z.Q.; Ackermann, U.; Wichmann, C.; McDonald, A.; O’Keefe, G.; et al. Preclinical evaluation of Zr-Df-radiolabeled bispecific anti-PD-L1/TGF-βRII fusion protein bintrafusp alfa. J. Nucl. Med. 2021, 62, 66. [Google Scholar]
- Paz-Ares, L.; Kim, T.M.; Vicente, D.; Felip, E.; Lee, D.H.; Lee, K.H.; Lin, C.C.; Flor, M.J.; Di Nicola, M.; Alvarez, R.M.; et al. Bintrafusp Alfa, a Bifunctional Fusion Protein Targeting TGF-beta and PD-L1, in Second-Line Treatment of Patients With NSCLC: Results From an Expansion Cohort of a Phase 1 Trial. J. Thorac. Oncol. 2020, 15, 1210–1222. [Google Scholar] [CrossRef]
- Kang, Y.K.; Bang, Y.J.; Kondo, S.; Chung, H.C.; Muro, K.; Dussault, I.; Helwig, C.; Osada, M.; Doi, T. Safety and Tolerability of Bintrafusp Alfa, a Bifunctional Fusion Protein Targeting TGFbeta and PD-L1, in Asian Patients with Pretreated Recurrent or Refractory Gastric Cancer. Clin. Cancer Res. 2020, 26, 3202–3210. [Google Scholar] [CrossRef] [PubMed]
- Doi, T.; Fujiwara, Y.; Koyama, T.; Ikeda, M.; Helwig, C.; Watanabe, M.; Vugmeyster, Y.; Kudo, M. Phase I Study of the Bifunctional Fusion Protein Bintrafusp Alfa in Asian Patients with Advanced Solid Tumors, Including a Hepatocellular Carcinoma Safety-Assessment Cohort. Oncologist 2020, 25, e1292–e1302. [Google Scholar] [CrossRef] [PubMed]
- Cho, B.C.; Daste, A.; Ravaud, A.; Salas, S.; Isambert, N.; McClay, E.; Awada, A.; Borel, C.; Ojalvo, L.S.; Helwig, C.; et al. Bintrafusp alfa, a bifunctional fusion protein targeting TGF-beta and PD-L1, in advanced squamous cell carcinoma of the head and neck: Results from a phase I cohort. J. Immunother. Cancer 2020, 8, e000664. [Google Scholar] [CrossRef] [PubMed]
- Cho, B.C.; Lee, J.S.; Wu, Y.L.; Cicin, I.; Dols, M.C.; Ahn, M.J.; Cuppens, K.; Veillon, R.; Nadal, E.; Dias, J.M.; et al. Bintrafusp Alfa Versus Pembrolizumab in Patients With Treatment-Naive, Programmed Death-Ligand 1-High Advanced NSCLC: A Randomized, Open-Label, Phase 3 Trial. J. Thorac. Oncol. 2023, 18, 1731–1742. [Google Scholar] [CrossRef] [PubMed]
- Yi, M.; Zhang, J.; Li, A.; Niu, M.; Yan, Y.; Jiao, Y.; Luo, S.; Zhou, P.; Wu, K. The construction, expression, and enhanced anti-tumor activity of YM101: A bispecific antibody simultaneously targeting TGF-beta and PD-L1. J. Hematol. Oncol. 2021, 14, 27. [Google Scholar] [CrossRef] [PubMed]
- Yi, M.; Wu, Y.; Niu, M.; Zhu, S.; Zhang, J.; Yan, Y.; Zhou, P.; Dai, Z.; Wu, K. Anti-TGF-beta/PD-L1 bispecific antibody promotes T cell infiltration and exhibits enhanced antitumor activity in triple-negative breast cancer. J. Immunother. Cancer 2022, 10, e005543. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Rossi, J. Aptamers as targeted therapeutics: Current potential and challenges. Nat. Rev. Drug Discov. 2017, 16, 181–202. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Hashimoto, Y.; Nakamura, Y. Anti-TGF-beta1 aptamer enhances therapeutic effect of tyrosine kinase inhibitor, gefitinib, on non-small cell lung cancer in xenograft model. Mol. Ther. Nucleic Acids 2022, 29, 969–978. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Li, L.; Zou, L.; Zhu, X.; Xian, G.; Li, H.; Tan, Y.; Xie, L. A novel aptamer targeting TGF-beta receptor II inhibits transdifferentiation of human tenon’s fibroblasts into myofibroblast. Investig. Ophthalmol. Vis. Sci. 2012, 53, 6897–6903. [Google Scholar] [CrossRef]
- Bogdahn, U.; Hau, P.; Stockhammer, G.; Venkataramana, N.K.; Mahapatra, A.K.; Suri, A.; Balasubramaniam, A.; Nair, S.; Oliushine, V.; Parfenov, V.; et al. Targeted therapy for high-grade glioma with the TGF-beta2 inhibitor trabedersen: Results of a randomized and controlled phase IIb study. Neuro Oncol. 2011, 13, 132–142. [Google Scholar] [CrossRef]
- Uckun, F.M.; Qazi, S.; Hwang, L.; Trieu, V.N. Recurrent or Refractory High-Grade Gliomas Treated by Convection-Enhanced Delivery of a TGFbeta2-Targeting RNA Therapeutic: A Post-Hoc Analysis with Long-Term Follow-Up. Cancers 2019, 11, 1892. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, N.; Voykov, B.; Renieri, G.; Bell, K.; Richter, P.; Weigel, M.; Thieme, H.; Wilhelm, B.; Lorenz, K.; Feindor, M.; et al. First-in-human phase I study of ISTH0036, an antisense oligonucleotide selectively targeting transforming growth factor beta 2 (TGF-beta2), in subjects with open-angle glaucoma undergoing glaucoma filtration surgery. PLoS ONE 2017, 12, e0188899. [Google Scholar] [CrossRef] [PubMed]
- Schlingensiepen, K.H.; Bischof, A.; Egger, T.; Hafner, M.; Herrmuth, H.; Jachimczak, P.; Kielmanowicz, M.; Niewel, M.; Zavadova, E.; Stauder, G. The TGF-beta1 antisense oligonucleotide AP 11014 for the treatment of non-small cell lung, colorectal and prostate cancer: Preclinical studies. J. Clin. Oncol. 2004, 22, 227s. [Google Scholar] [CrossRef]
- Papachristodoulou, A.; Silginer, M.; Weller, M.; Schneider, H.; Hasenbach, K.; Janicot, M.; Roth, P. Therapeutic Targeting of TGFbeta Ligands in Glioblastoma Using Novel Antisense Oligonucleotides Reduces the Growth of Experimental Gliomas. Clin. Cancer Res. 2019, 25, 7189–7201. [Google Scholar] [CrossRef] [PubMed]
- Ge, R.; Rajeev, V.; Ray, P.; Lattime, E.; Rittling, S.; Medicherla, S.; Protter, A.; Murphy, A.; Chakravarty, J.; Dugar, S.; et al. Inhibition of growth and metastasis of mouse mammary carcinoma by selective inhibitor of transforming growth factor-beta type I receptor kinase in vivo. Clin. Cancer Res. 2006, 12, 4315–4330. [Google Scholar] [CrossRef] [PubMed]
- Green, A.C.; Lath, D.; Hudson, K.; Walkley, B.; Down, J.M.; Owen, R.; Evans, H.R.; Paton-Hough, J.; Reilly, G.C.; Lawson, M.A.; et al. TGFbeta Inhibition Stimulates Collagen Maturation to Enhance Bone Repair and Fracture Resistance in a Murine Myeloma Model. J. Bone Miner. Res. 2019, 34, 2311–2326. [Google Scholar] [CrossRef] [PubMed]
- Fournier, P.G.; Juarez, P.; Jiang, G.; Clines, G.A.; Niewolna, M.; Kim, H.S.; Walton, H.W.; Peng, X.H.; Liu, Y.; Mohammad, K.S.; et al. The TGF-beta Signaling Regulator PMEPA1 Suppresses Prostate Cancer Metastases to Bone. Cancer Cell 2015, 27, 809–821. [Google Scholar] [CrossRef] [PubMed]
- Geyh, S.; Rodriguez-Paredes, M.; Jager, P.; Koch, A.; Bormann, F.; Gutekunst, J.; Zilkens, C.; Germing, U.; Kobbe, G.; Lyko, F.; et al. Transforming growth factor beta1-mediated functional inhibition of mesenchymal stromal cells in myelodysplastic syndromes and acute myeloid leukemia. Haematologica 2018, 103, 1462–1471. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, S.D.; Wu, X.; He, Y.; Chen, S.; Yang, H.; Staser, K.W.; Wang, J.; Zhang, P.; Jiang, C.; Yokota, H.; et al. Hyperactive transforming growth factor-beta1 signaling potentiates skeletal defects in a neurofibromatosis type 1 mouse model. J. Bone Miner. Res. 2013, 28, 2476–2489. [Google Scholar] [CrossRef]
- Medicherla, S.; Li, L.; Ma, J.Y.; Kapoun, A.M.; Gaspar, N.J.; Liu, Y.W.; Mangadu, R.; O’Young, G.; Protter, A.A.; Schreiner, G.F.; et al. Antitumor activity of TGF-beta inhibitor is dependent on the microenvironment. Anticancer. Res. 2007, 27, 4149–4157. [Google Scholar]
- Gaspar, N.J.; Li, L.; Kapoun, A.M.; Medicherla, S.; Reddy, M.; Li, G.; O’Young, G.; Quon, D.; Henson, M.; Damm, D.L.; et al. Inhibition of transforming growth factor beta signaling reduces pancreatic adenocarcinoma growth and invasiveness. Mol. Pharmacol. 2007, 72, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, K.S.; Javelaud, D.; Fournier, P.G.; Niewolna, M.; McKenna, C.R.; Peng, X.H.; Duong, V.; Dunn, L.K.; Mauviel, A.; Guise, T.A. TGF-beta-RI kinase inhibitor SD-208 reduces the development and progression of melanoma bone metastases. Cancer Res. 2011, 71, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Akbari, A.; Amanpour, S.; Muhammadnejad, S.; Ghahremani, M.H.; Ghaffari, S.H.; Dehpour, A.R.; Mobini, G.R.; Shidfar, F.; Abastabar, M.; Khoshzaban, A.; et al. Evaluation of antitumor activity of a TGF-beta receptor I inhibitor (SD-208) on human colon adenocarcinoma. Daru 2014, 22, 47. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Zhang, Y.; Zhang, Q.; Li, Y.; Gong, T.; Zhang, Z.; Ding, R.; Sun, X. Development and validation of an LC-MS/MS method for the determination of SB-505124 in rat plasma: Application to pharmacokinetic study. J. Pharm. Biomed. Anal. 2016, 117, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Blum, A.E.; Venkitachalam, S.; Ravillah, D.; Chelluboyina, A.K.; Kieber-Emmons, A.M.; Ravi, L.; Kresak, A.; Chandar, A.K.; Markowitz, S.D.; Canto, M.I.; et al. Systems Biology Analyses Show Hyperactivation of Transforming Growth Factor-beta and JNK Signaling Pathways in Esophageal Cancer. Gastroenterology 2019, 156, 1761–1774. [Google Scholar] [CrossRef] [PubMed]
- Park, S.A.; Kim, M.J.; Park, S.Y.; Kim, J.S.; Lee, S.J.; Woo, H.A.; Kim, D.K.; Nam, J.S.; Sheen, Y.Y. EW-7197 inhibits hepatic, renal, and pulmonary fibrosis by blocking TGF-beta/Smad and ROS signaling. Cell. Mol. Life Sci. 2015, 72, 2023–2039. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.M.; Hwang, Y.C.; Liu, I.J.; Lee, C.C.; Tsai, H.Z.; Li, H.J.; Wu, H.C. Development of therapeutic antibodies for the treatment of diseases. J. Biomed. Sci. 2020, 27, 1. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef] [PubMed]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016, 165, 35–44. [Google Scholar] [CrossRef]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel, E.E., III; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef]
- Tauriello, D.V.F.; Palomo-Ponce, S.; Stork, D.; Berenguer-Llergo, A.; Badia-Ramentol, J.; Iglesias, M.; Sevillano, M.; Ibiza, S.; Canellas, A.; Hernando-Momblona, X.; et al. TGFbeta drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018, 554, 538–543. [Google Scholar] [CrossRef]
- Lan, Y.; Zhang, D.; Xu, C.; Hance, K.W.; Marelli, B.; Qi, J.; Yu, H.; Qin, G.; Sircar, A.; Hernandez, V.M.; et al. Enhanced preclinical antitumor activity of M7824, a bifunctional fusion protein simultaneously targeting PD-L1 and TGF-beta. Sci. Transl. Med. 2018, 10, eaan5488. [Google Scholar] [CrossRef] [PubMed]
- Knudson, K.M.; Hicks, K.C.; Luo, X.; Chen, J.Q.; Schlom, J.; Gameiro, S.R. M7824, a novel bifunctional anti-PD-L1/TGFbeta Trap fusion protein, promotes anti-tumor efficacy as monotherapy and in combination with vaccine. Oncoimmunology 2018, 7, e1426519. [Google Scholar] [CrossRef] [PubMed]
- Strauss, J.; Gatti-Mays, M.E.; Cho, B.C.; Hill, A.; Salas, S.; McClay, E.; Redman, J.M.; Sater, H.A.; Donahue, R.N.; Jochems, C.; et al. Bintrafusp alfa, a bifunctional fusion protein targeting TGF-beta and PD-L1, in patients with human papillomavirus-associated malignancies. J. Immunother. Cancer 2020, 8, e001395. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.C.; Doi, T.; Muro, K.; Hou, M.M.; Esaki, T.; Hara, H.; Chung, H.C.; Helwig, C.; Dussault, I.; Osada, M.; et al. Bintrafusp Alfa, a Bifunctional Fusion Protein Targeting TGFbeta and PD-L1, in Patients with Esophageal Squamous Cell Carcinoma: Results from a Phase 1 Cohort in Asia. Target. Oncol. 2021, 16, 447–459. [Google Scholar] [CrossRef] [PubMed]
- Tan, B.; Khattak, A.; Felip, E.; Kelly, K.; Rich, P.; Wang, D.; Helwig, C.; Dussault, I.; Ojalvo, L.S.; Isambert, N. Bintrafusp Alfa, a Bifunctional Fusion Protein Targeting TGF-beta and PD-L1, in Patients with Esophageal Adenocarcinoma: Results from a Phase 1 Cohort. Target. Oncol. 2021, 16, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Spira, A.; Wertheim, M.S.; Kim, E.J.; Tan, B.; Lenz, H.J.; Nikolinakos, P.; Rich, P.L.; Jehl, G.; Machl, A.; Ito, R.; et al. Bintrafusp Alfa: A Bifunctional Fusion Protein Targeting PD-L1 and TGF-beta, in Patients with Pretreated Colorectal Cancer: Results from a Phase I Trial. Oncologist 2023, 28, e124–e127. [Google Scholar] [CrossRef] [PubMed]
- Yoo, C.; Oh, D.Y.; Choi, H.J.; Kudo, M.; Ueno, M.; Kondo, S.; Chen, L.T.; Osada, M.; Helwig, C.; Dussault, I.; et al. Phase I study of bintrafusp alfa, a bifunctional fusion protein targeting TGF-beta and PD-L1, in patients with pretreated biliary tract cancer. J. Immunother. Cancer 2020, 8, e000564. [Google Scholar] [CrossRef]
- Wilkins, J.J.; Vugmeyster, Y.; Dussault, I.; Girard, P.; Khandelwal, A. Population Pharmacokinetic Analysis of Bintrafusp Alfa in Different Cancer Types. Adv. Ther. 2019, 36, 2414–2433. [Google Scholar] [CrossRef]
- Longoria, T.C.; Tewari, K.S. Evaluation of the pharmacokinetics and metabolism of pembrolizumab in the treatment of melanoma. Expert. Opin. Drug Metab. Toxicol. 2016, 12, 1247–1253. [Google Scholar] [CrossRef]
- Kok, I.C.; Hooiveld, J.S.; van de Donk, P.P.; Giesen, D.; van der Veen, E.L.; Lub-de Hooge, M.N.; Brouwers, A.H.; Hiltermann, T.J.N.; van der Wekken, A.J.; Hijmering-Kappelle, L.B.M.; et al. (89)Zr-pembrolizumab imaging as a non-invasive approach to assess clinical response to PD-1 blockade in cancer. Ann. Oncol. 2022, 33, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Tang, D.; Wang, J.; Zhou, Q.; Peng, J.; Lou, H.; Sun, Y.; Cai, Y.; Chen, H.; Yang, J.; et al. SHR-1701, a Bifunctional Fusion Protein Targeting PD-L1 and TGFbeta, for Recurrent or Metastatic Cervical Cancer: A Clinical Expansion Cohort of a Phase I Study. Clin. Cancer Res. 2022, 28, 5297–5305. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Zhou, J.; Wang, Y.; Li, M.; Jiang, H.; Liu, Y.; Yin, X.; Ge, M.; Xiang, X.; Ying, J.; et al. Bifunctional anti-PD-L1/TGF-betaRII agent SHR-1701 in advanced solid tumors: A dose-escalation, dose-expansion, and clinical-expansion phase 1 trial. BMC Med. 2022, 20, 408. [Google Scholar] [CrossRef] [PubMed]
- Cheng, B.; Ding, K.; Chen, P.; Ji, J.; Luo, T.; Guo, X.; Qiu, W.; Ma, C.; Meng, X.; Wang, J.; et al. Anti-PD-L1/TGF-betaR fusion protein (SHR-1701) overcomes disrupted lymphocyte recovery-induced resistance to PD-1/PD-L1 inhibitors in lung cancer. Cancer Commun. 2022, 42, 17–36. [Google Scholar] [CrossRef] [PubMed]
- Xiong, H.; Veedu, R.N.; Diermeier, S.D. Recent Advances in Oligonucleotide Therapeutics in Oncology. Int. J. Mol. Sci. 2021, 22, 3295. [Google Scholar] [CrossRef]
- Wick, W.; Weller, M. Trabedersen to target transforming growth factor-beta: When the journey is not the reward, in reference to Bogdahn et al. (Neuro-Oncology 2011;13:132-142). Neuro-Oncology 2011, 13, 559–560; author reply 561–562. [Google Scholar] [CrossRef] [PubMed]
- Roy, L.O.; Poirier, M.B.; Fortin, D. Differential Expression and Clinical Significance of Transforming Growth Factor-Beta Isoforms in GBM Tumors. Int. J. Mol. Sci. 2018, 19, 1113. [Google Scholar] [CrossRef]
- Schlingensiepen, K.H.; Jaschinski, F.; Lang, S.A.; Moser, C.; Geissler, E.K.; Schlitt, H.J.; Kielmanowicz, M.; Schneider, A. Transforming growth factor-beta 2 gene silencing with trabedersen (AP 12009) in pancreatic cancer. Cancer Sci. 2011, 102, 1193–1200. [Google Scholar] [CrossRef]
- Reverdatto, S.; Burz, D.S.; Shekhtman, A. Peptide aptamers: Development and applications. Curr. Top. Med. Chem. 2015, 15, 1082–1101. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Danielpour, D. Advances and Challenges in Targeting TGF-β Isoforms for Therapeutic Intervention of Cancer: A Mechanism-Based Perspective. Pharmaceuticals 2024, 17, 533. https://doi.org/10.3390/ph17040533
Danielpour D. Advances and Challenges in Targeting TGF-β Isoforms for Therapeutic Intervention of Cancer: A Mechanism-Based Perspective. Pharmaceuticals. 2024; 17(4):533. https://doi.org/10.3390/ph17040533
Chicago/Turabian StyleDanielpour, David. 2024. "Advances and Challenges in Targeting TGF-β Isoforms for Therapeutic Intervention of Cancer: A Mechanism-Based Perspective" Pharmaceuticals 17, no. 4: 533. https://doi.org/10.3390/ph17040533
APA StyleDanielpour, D. (2024). Advances and Challenges in Targeting TGF-β Isoforms for Therapeutic Intervention of Cancer: A Mechanism-Based Perspective. Pharmaceuticals, 17(4), 533. https://doi.org/10.3390/ph17040533