
Targeting SMAD-Dependent Signaling: Considerations in Epithelial and Mesenchymal Solid Tumors




Abstract
1. Introduction
2. Canonical TGF-β/SMAD Signaling
3. Activity and Nucleocytoplasmic Trafficking of SMADS
Mechanisms of SMAD Protein Nucleocytoplasmic Trafficking
4. Regulation of SMAD Activity by Post-Translational Modifications (PTMs)
4.1. Phosphorylation and Dephosphorylation
4.2. Ubiquitylation and Deubiquitylation
4.3. Acetylation, ADP-Ribosylation, and Sumoylation
5. Regulation of SMAD-Mediated Transcription
5.1. Histone Modification
5.2. Regulation of SMAD-Mediated Transcriptional Activity Post-Transcriptionally
6. Non-SMAD, Non-Canonical TGF-β Pathway Control
6.1. ERK/MAP Kinase Signaling
6.2. JNK and p38 MAP Kinase Signaling
6.3. JAK-STAT Signaling
6.4. PI3/AKT/mTOR Signaling
6.5. TGF-β Type I Receptor (TGF-βRI) Intracellular Domain Signaling
6.6. Rho-(like) GTPase Signaling
6.7. Crosstalk between SMAD and Other Signaling Pathway Molecules
7. TGF-β/SMAD Mediated Progression in Solid Tumors
7.1. Epithelial Cells
7.2. Cancer Cells with EMT, and Hybrid/Partial EMT
7.3. Cancer Cells with Mesenchymal Characteristics
7.4. Cancer Cells with Stemness Characteristics
7.5. Cancer Dissemination and Metastasis
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Shi, Y.; Massagué, J. Mechanisms of TGF-β Signaling from Cell Membrane to the Nucleus. Cell 2003, 113, 685–700. [Google Scholar] [CrossRef] [PubMed]
- Massagué, J. TGFβ signalling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, P.; Winter, S.L.; Alexandrow, M.G. Cell cycle arrest by transforming growth factor beta1 near G1/S is mediated by acute abrogation of prereplication complex activation involving an Rb-MCM interaction. Mol. Cell Biol. 2010, 30, 845–856. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.J.; Nieto, M.A. Epithelial-Mesenchymal Transitions in Development and Disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A.; Huang, R.Y.-J.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [PubMed]
- Pastushenko, I.; Brisebarre, A.; Sifrim, A.; Fioramonti, M.; Revenco, T.; Boumahdi, S.; Van Keymeulen, A.; Brown, D.; Moers, V.; Lemaire, S.; et al. Identification of the tumor transition states occurring during EMT. Nature 2018, 556, 463–468. [Google Scholar] [CrossRef]
- Varga, J.; Greten, F.R. Cell plasticity in epithelial homeostasis and tumorigenesis. Nat. Cell Biol. 2017, 19, 1133–1141. [Google Scholar] [CrossRef]
- Massagué, J. TGFbeta in Cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Lambert, A.W.; Pattabiraman, D.R.; Weinberg, R.A. Emerging Biological Principles of Metastasis. Cell 2017, 168, 670–691. [Google Scholar] [CrossRef]
- Huynh, L.K.; Hipolito, C.J.; Ten Dijke, P. A Perspective on the Development of TGF-β Inhibitors for Cancer Treatment. Biomolecules 2019, 9, 743. [Google Scholar] [CrossRef]
- Suzuki, H.I. MicroRNA Control of TGF-β Signaling. Int. J. Mol. Sci. 2018, 19, 1901. [Google Scholar] [CrossRef]
- Papoutsoglou, P.; Moustakas, A. Long non-coding RNAs and TGF-β signaling in cancer. Cancer Sci. 2020, 111, 2672–2681. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, X.; Xie, F.; Zhang, Z.; van Dam, H.; Zhang, L.; Zhou, F. The regulation of TGF-β/SMAD signaling by protein deubiquitination. Protein Cell 2014, 5, 503–517. [Google Scholar] [CrossRef]
- Robertson, I.B.; Rifkin, D.B. Regulation of the Bioavailability of TGF-β and TGF-β-Related Proteins. Cold Spring Harb. Perspect. Biol. 2016, 8, a021907. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Chen, M.; Sang, X.; You, X.; Wang, Y.; Paterson, I.C.; Hong, W.; Yang, X. Development of small molecule inhibitors targeting TGF-β ligand and receptor: Structures, mechanism, preclinical studies and clinical usage. Eur. J. Med. Chem. 2020, 191, 112154. [Google Scholar] [CrossRef]
- Kim, B.-G.; Malek, E.; Choi, S.H.; Ignatz-Hoover, J.J.; Driscoll, J.J. Novel therapies emerging in oncology to target the TGF-β pathway. J. Hematol. Oncol. 2021, 14, 55. [Google Scholar] [CrossRef]
- Huang, C.-Y.; Chung, C.-L.; Hu, T.-H.; Chen, J.-J.; Liu, P.-F.; Chen, C.-L. Recent progress in TGF-β inhibitors for cancer therapy. Biomed. Pharmacother. 2021, 134, 111046. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Li, Y.; Xiong, L.; Wang, W.; Wu, M.; Yuan, T.; Yang, W.; Tian, C.; Miao, Z.; Wang, T.; et al. Small molecules in targeted cancer therapy: Advances, challenges, and future perspectives. Signal Transduct. Target. Ther. 2021, 6, 201. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, A.F.; Ten Dijke, P.; Zhu, H.J. On-Target Anti-TGF-β Therapies Are Not Succeeding in Clinical Cancer Treatments: What Are Remaining Challenges? Front. Cell Dev. Biol. 2020, 8, 605. [Google Scholar] [CrossRef] [PubMed]
- Ciardiello, D.; Elez, E.; Tabernero, J.; Seoane, J. Clinical development of therapies targeting TGFβ: Current knowledge and future perspectives. Ann. Oncol. 2020, 31, 1336–1349. [Google Scholar] [CrossRef]
- Feng, X.H.; Derynck, R. Specificity and versatility in tgf-beta signaling through Smads. Annu. Rev. Cell Dev. Biol. 2005, 21, 659–693. [Google Scholar] [CrossRef]
- Wakefield, L.M.; Hill, C.S. Beyond TGFβ: Roles of other TGFβ superfamily members in cancer. Nat. Rev. Cancer 2013, 13, 328–341. [Google Scholar] [CrossRef]
- Hata, A.; Chen, Y.G. TGF-β Signaling from Receptors to Smads. Cold Spring Harb. Perspect. Biol. 2016, 8, a022061. [Google Scholar] [CrossRef]
- Hill, C.S. Transcriptional Control by the SMADs. Cold Spring Harb. Perspect. Biol. 2016, 8, a022079. [Google Scholar] [CrossRef]
- Heldin, C.-H.; Moustakas, A. Role of Smads in TGFβ signaling. Cell Tissue Res. 2012, 347, 21–36. [Google Scholar] [CrossRef]
- Miyazawa, K.; Shinozaki, M.; Hara, T.; Furuya, T.; Miyazono, K. Two major Smad pathways in TGF-β superfamily signalling. GenesCells 2002, 7, 1191–1204. [Google Scholar] [CrossRef]
- Locker, J. Book reveiw Review of Molecular Biology of Human Cancers (An Advanced Student’s Text, by Wolfgang Schulz, Department of Urology and Center for Biological and Medical Research, Heinrich Heine University, Düsseldorf) ISBN 1-4020-3185-8. Cancer Causes Control 2005, 16, 191–192. [Google Scholar] [CrossRef]
- Nicolás, F.J.; De Bosscher, K.; Schmierer, B.; Hill, C.S. Analysis of Smad nucleocytoplasmic shuttling in living cells. J. Cell Sci. 2004, 117 Pt 18, 4113–4125. [Google Scholar] [CrossRef] [PubMed]
- Huse, M.; Muir, T.W.; Xu, L.; Chen, Y.-G.; Kuriyan, J.; Massagué, J. The TGFβ Receptor Activation Process: An Inhibitor- to Substrate-Binding Switch. Mol. Cell 2001, 8, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Souchelnytskyi, S.; Tamaki, K.; Engström, U.; Wernstedt, C.; ten Dijke, P.; Heldin, C.-H. Phosphorylation of Ser465 and Ser467 in the C Terminus of Smad2 Mediates Interaction with Smad4 and Is Required for Transforming Growth Factor-β Signaling*. J. Biol. Chem. 1997, 272, 28107–28115. [Google Scholar] [CrossRef]
- Panopoulou, E.; Gillooly, D.J.; Wrana, J.L.; Zerial, M.; Stenmark, H.; Murphy, C.; Fotsis, T. Early endosomal regulation of Smad-dependent signaling in endothelial cells. J. Biol. Chem. 2002, 277, 18046–18052. [Google Scholar] [CrossRef] [PubMed]
- Tsukazaki, T.; Chiang, T.A.; Davison, A.F.; Attisano, L.; Wrana, J.L. SARA, a FYVE Domain Protein that Recruits Smad2 to the TGFβ Receptor. Cell 1998, 95, 779–791. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.K.; Bergmann, S.; Pandolfi, P.P. Cytoplasmic PML function in TGF-beta signalling. Nature 2004, 431, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Jin, G.; Gao, Y.; Lin, H.-K. Cytoplasmic PML: From Molecular Regulation to Biological Functions. J. Cell. Biochem. 2014, 115, 812–818. [Google Scholar] [CrossRef]
- Hocevar, B.A.; Smine, A.; Xu, X.-X.; Howe, P.H. The adaptor molecule Disabled-2 links the transforming growth factor β receptors to the Smad pathway. EMBO J. 2001, 20, 2789–2801. [Google Scholar] [CrossRef]
- Heldin, C.-H.; Miyazono, K.; ten Dijke, P. TGF-β signalling from cell membrane to nucleus through SMAD proteins. Nature 1997, 390, 465–471. [Google Scholar] [CrossRef]
- Wu, G.; Chen, Y.G.; Ozdamar, B.; Gyuricza, C.A.; Chong, P.A.; Wrana, J.L.; Massagué, J.; Shi, Y. Structural basis of Smad2 recognition by the Smad anchor for receptor activation. Science 2000, 287, 92–97. [Google Scholar] [CrossRef]
- Miura, S.; Takeshita, T.; Asao, H.; Kimura, Y.; Murata, K.; Sasaki, Y.; Hanai, J.I.; Beppu, H.; Tsukazaki, T.; Wrana, J.L.; et al. Hgs (Hrs), a FYVE domain protein, is involved in Smad signaling through cooperation with SARA. Mol. Cell Biol. 2000, 20, 9346–9355. [Google Scholar] [CrossRef] [PubMed]
- Miura, S.; Mishina, Y. Hepatocyte growth factor–regulated tyrosine kinase substrate (Hgs) is involved in BMP signaling through phosphorylation of smads and TAK1 in early mouse embryo. Dev. Dyn. 2011, 240, 2474–2481. [Google Scholar] [CrossRef] [PubMed]
- Chacko, B.M.; Qin, B.Y.; Tiwari, A.; Shi, G.; Lam, S.; Hayward, L.J.; De Caestecker, M.; Lin, K. Structural basis of heteromeric smad protein assembly in TGF-beta signaling. Mol. Cell 2004, 15, 813–823. [Google Scholar] [CrossRef]
- Kawabata, M.; Inoue, H.; Hanyu, A.; Imamura, T.; Miyazono, K. Smad proteins exist as monomers in vivo and undergo homo- and hetero-oligomerization upon activation by serine/threonine kinase receptors. EMBO J. 1998, 17, 4056–4065. [Google Scholar] [CrossRef] [PubMed]
- Tzavlaki, K.; Moustakas, A. TGF-β Signaling. Biomolecules 2020, 10, 487. [Google Scholar] [CrossRef] [PubMed]
- Lucarelli, P.; Schilling, M.; Kreutz, C.; Vlasov, A.; Boehm, M.E.; Iwamoto, N.; Steiert, B.; Lattermann, S.; Wäsch, M.; Stepath, M.; et al. Resolving the Combinatorial Complexity of Smad Protein Complex Formation and Its Link to Gene Expression. Cell Syst. 2018, 6, 75–89.e11. [Google Scholar] [CrossRef] [PubMed]
- Kurisaki, A.; Kose, S.; Yoneda, Y.; Heldin, C.H.; Moustakas, A. Transforming growth factor-beta induces nuclear import of Smad3 in an importin-beta1 and Ran-dependent manner. Mol. Biol. Cell 2001, 12, 1079–1091. [Google Scholar] [CrossRef] [PubMed]
- Kurisaki, A.; Kurisaki, K.; Kowanetz, M.; Sugino, H.; Yoneda, Y.; Heldin, C.H.; Moustakas, A. The mechanism of nuclear export of Smad3 involves exportin 4 and Ran. Mol. Cell Biol. 2006, 26, 1318–1332. [Google Scholar] [CrossRef]
- Dai, F.; Lin, X.; Chang, C.; Feng, X.H. Nuclear export of Smad2 and Smad3 by RanBP3 facilitates termination of TGF-beta signaling. Dev. Cell 2009, 16, 345–357. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.; Latek, R.; Lodish, H.F. An extended bipartite nuclear localization signal in Smad4 is required for its nuclear import and transcriptional activity. Oncogene 2003, 22, 1057–1069. [Google Scholar] [CrossRef]
- Xu, L.; Kang, Y.; Çöl, S.; Massagué, J. Smad2 Nucleocytoplasmic Shuttling by Nucleoporins CAN/Nup214 and Nup153 Feeds TGFβ Signaling Complexes in the Cytoplasm and Nucleus. Mol. Cell 2002, 10, 271–282. [Google Scholar] [CrossRef]
- Xu, L.; Alarcón, C.; Çöl, S.; Massaguè, J. Distinct Domain Utilization by Smad3 and Smad4 for Nucleoporin Interaction and Nuclear Import*. J. Biol. Chem. 2003, 278, 42569–42577. [Google Scholar] [CrossRef]
- Jin, Q.; Ding, W.; Mulder, K.M. Requirement for the Dynein Light Chain km23-1 in a Smad2-dependent Transforming Growth Factor-β Signaling Pathway*. J. Biol. Chem. 2007, 282, 19122–19132. [Google Scholar] [CrossRef] [PubMed]
- Jin, Q.; Ding, W.; Mulder, K.M. The TGFβ Receptor-interacting Protein km23-1/DYNLRB1 Plays an Adaptor Role in TGFβ1 Autoinduction via Its Association with Ras*. J. Biol. Chem. 2012, 287, 26453–26463. [Google Scholar] [CrossRef]
- Conery, A.R.; Cao, Y.; Thompson, E.A.; Townsend, C.M., Jr.; Ko, T.C.; Luo, K. Akt interacts directly with Smad3 to regulate the sensitivity to TGF-beta induced apoptosis. Nat. Cell Biol. 2004, 6, 366–372. [Google Scholar] [CrossRef] [PubMed]
- Remy, I.; Montmarquette, A.; Michnick, S.W. PKB/Akt modulates TGF-beta signalling through a direct interaction with Smad3. Nat. Cell Biol. 2004, 6, 358–365. [Google Scholar] [CrossRef]
- Kretzschmar, M.; Liu, F.; Hata, A.; Doody, J.; Massagué, J. The TGF-beta family mediator Smad1 is phosphorylated directly and activated functionally by the BMP receptor kinase. Genes Dev. 1997, 11, 984–995. [Google Scholar] [CrossRef]
- Matsuura, I.; Wang, G.; He, D.; Liu, F. Identification and Characterization of ERK MAP Kinase Phosphorylation Sites in Smad3. Biochemistry 2005, 44, 12546–12553. [Google Scholar] [CrossRef]
- Wang, G.; Matsuura, I.; He, D.; Liu, F. Transforming growth factor-{beta}-inducible phosphorylation of Smad3. J. Biol. Chem. 2009, 284, 9663–9673. [Google Scholar] [CrossRef]
- Matsuura, I.; Denissova, N.G.; Wang, G.; He, D.; Long, J.; Liu, F. Cyclin-dependent kinases regulate the antiproliferative function of Smads. Nature 2004, 430, 226–231. [Google Scholar] [CrossRef]
- Fuentealba, L.C.; Eivers, E.; Ikeda, A.; Hurtado, C.; Kuroda, H.; Pera, E.M.; De Robertis, E.M. Integrating patterning signals: Wnt/GSK3 regulates the duration of the BMP/Smad1 signal. Cell 2007, 131, 980–993. [Google Scholar] [CrossRef] [PubMed]
- Millet, C.; Yamashita, M.; Heller, M.; Yu, L.R.; Veenstra, T.D.; Zhang, Y.E. A negative feedback control of transforming growth factor-beta signaling by glycogen synthase kinase 3-mediated Smad3 linker phosphorylation at Ser-204. J. Biol. Chem. 2009, 284, 19808–19816. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Duan, X.; Liang, Y.-Y.; Su, Y.; Wrighton, K.H.; Long, J.; Hu, M.; Davis, C.M.; Wang, J.; Brunicardi, F.C.; et al. PPM1A Functions as a Smad Phosphatase to Terminate TGFβ Signaling. Cell 2006, 125, 915–928. [Google Scholar] [CrossRef] [PubMed]
- Dai, F.; Shen, T.; Li, Z.; Lin, X.; Feng, X.H. PPM1A dephosphorylates RanBP3 to enable efficient nuclear export of Smad2 and Smad3. EMBO Rep. 2011, 12, 1175–1181. [Google Scholar] [CrossRef]
- Yang, X.; Teng, Y.; Hou, N.; Fan, X.; Cheng, X.; Li, J.; Wang, L.; Wang, Y.; Wu, X.; Yang, X. Delayed Re-epithelialization in Ppm1a Gene-deficient Mice Is Mediated by Enhanced Activation of Smad2*. J. Biol. Chem. 2011, 286, 42267–42273. [Google Scholar] [CrossRef]
- Lo Sardo, F.; Strano, S.; Blandino, G. YAP and TAZ in Lung Cancer: Oncogenic Role and Clinical Targeting. Cancers 2018, 10, 137. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Zeng, C.; Ye, S.; Dai, X.; He, Q.; Yang, B.; Zhu, H. Yes-associated protein (YAP) and transcriptional coactivator with a PDZ-binding motif (TAZ): A nexus between hypoxia and cancer. Acta Pharm. Sin. B 2020, 10, 947–960. [Google Scholar] [CrossRef]
- Hiemer, S.E.; Szymaniak, A.D.; Varelas, X. The transcriptional regulators TAZ and YAP direct transforming growth factor β-induced tumorigenic phenotypes in breast cancer cells. J. Biol. Chem. 2014, 289, 13461–13474. [Google Scholar] [CrossRef] [PubMed]
- Varelas, X.; Sakuma, R.; Samavarchi-Tehrani, P.; Peerani, R.; Rao, B.M.; Dembowy, J.; Yaffe, M.B.; Zandstra, P.W.; Wrana, J.L. TAZ controls Smad nucleocytoplasmic shuttling and regulates human embryonic stem-cell self-renewal. Nat. Cell Biol. 2008, 10, 837–848. [Google Scholar] [CrossRef]
- Chen, H.B.; Rud, J.G.; Lin, K.; Xu, L. Nuclear targeting of transforming growth factor-beta-activated Smad complexes. J. Biol. Chem. 2005, 280, 21329–21336. [Google Scholar] [CrossRef]
- Chen, Y.L.; Law, P.Y.; Loh, H.H. Inhibition of Akt/Protein Kinase B Signaling by Naltrindole in Small Cell Lung Cancer Cells. Cancer Res. 2004, 64, 8723–8730. [Google Scholar] [CrossRef]
- DeFeo-Jones, D.; Barnett, S.F.; Fu, S.; Hancock, P.J.; Haskell, K.M.; Leander, K.R.; McAvoy, E.; Robinson, R.G.; Duggan, M.E.; Lindsley, C.W.; et al. Tumor cell sensitization to apoptotic stimuli by selective inhibition of specific Akt/PKB family members. Mol. Cancer Ther. 2005, 4, 271–279. [Google Scholar] [CrossRef]
- Lin, J.; Sampath, D.; Nannini, M.A.; Lee, B.B.; Degtyarev, M.; Oeh, J.; Savage, H.; Guan, Z.; Hong, R.; Kassees, R.; et al. Targeting Activated Akt with GDC-0068, a Novel Selective Akt Inhibitor That Is Efficacious in Multiple Tumor Models. Clin. Cancer Res. 2013, 19, 1760–1772. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Luo, G.; Qiu, Z. Akt inhibitor MK-2206 reduces pancreatic cancer cell viability and increases the efficacy of gemcitabine. Oncol. Lett. 2020, 19, 1999–2004. [Google Scholar] [CrossRef]
- Malkomes, P.; Lunger, I.; Luetticke, A.; Oppermann, E.; Haetscher, N.; Serve, H.; Holzer, K.; Bechstein, W.O.; Rieger, M.A. Selective AKT Inhibition by MK-2206 Represses Colorectal Cancer-Initiating Stem Cells. Ann. Surg. Oncol. 2016, 23, 2849–2857. [Google Scholar] [CrossRef]
- Huang, S.; Xiao, J.; Wu, J.; Liu, J.; Feng, X.; Yang, C.; Xiang, D.; Luo, S. Tizoxanide Promotes Apoptosis in Glioblastoma by Inhibiting CDK1 Activity. Front. Pharmacol. 2022, 13, 895573. [Google Scholar] [CrossRef] [PubMed]
- Nong, H.B.; Zhang, Y.N.; Bai, Y.G.; Zhang, Q.; Liu, M.F.; Zhou, Q.; Shi, Z.H.; Zeng, G.F.; Zong, S.H. Adapalene Inhibits Prostate Cancer Cell Proliferation In Vitro and In Vivo by Inducing DNA Damage, S-phase Cell Cycle Arrest, and Apoptosis. Front. Pharmacol. 2022, 13, 801624. [Google Scholar] [CrossRef] [PubMed]
- Mehraj, U.; Wani, N.A.; Hamid, A.; Alkhanani, M.; Almilaibary, A.; Mir, M.A. Adapalene inhibits the growth of triple-negative breast cancer cells by S-phase arrest and potentiates the antitumor efficacy of GDC-0941. Front. Pharmacol. 2022, 13, 958443. [Google Scholar] [CrossRef]
- Decker, J.T.; Ma, J.A.; Shea, L.D.; Jeruss, J.S. Implications of TGFβ Signaling and CDK Inhibition for the Treatment of Breast Cancer. Cancers 2021, 13, 5343. [Google Scholar] [CrossRef]
- Tarasewicz, E.; Rivas, L.; Hamdan, R.; Dokic, D.; Parimi, V.; Bernabe, B.P.; Thomas, A.; Shea, L.D.; Jeruss, J.S. Inhibition of CDK-mediated phosphorylation of Smad3 results in decreased oncogenesis in triple negative breast cancer cells. Cell Cycle 2014, 13, 3191–3201. [Google Scholar] [CrossRef]
- Zhu, Y.; Ke, K.-B.; Xia, Z.-K.; Li, H.-J.; Su, R.; Dong, C.; Zhou, F.-M.; Wang, L.; Chen, R.; Wu, S.-G.; et al. Discovery of vanoxerine dihydrochloride as a CDK2/4/6 triple-inhibitor for the treatment of human hepatocellular carcinoma. Mol. Med. 2021, 27, 15. [Google Scholar] [CrossRef]
- Duda, P.; Akula, S.M.; Abrams, S.L.; Steelman, L.S.; Martelli, A.M.; Cocco, L.; Ratti, S.; Candido, S.; Libra, M.; Montalto, G.; et al. Targeting GSK3 and Associated Signaling Pathways Involved in Cancer. Cells 2020, 9, 1110. [Google Scholar] [CrossRef]
- Kazi, A.; Xiang, S.; Yang, H.; Delitto, D.; Trevino, J.; Jiang, R.H.Y.; Ayaz, M.; Lawrence, H.R.; Kennedy, P.; Sebti, S.M. GSK3 suppression upregulates β-catenin and c-Myc to abrogate KRas-dependent tumors. Nat. Commun. 2018, 9, 5154. [Google Scholar] [CrossRef] [PubMed]
- Abushahba, W.; Olabisi, O.O.; Jeong, B.-S.; Boregowda, R.K.; Wen, Y.; Liu, F.; Goydos, J.S.; Lasfar, A.; Cohen-Solal, K.A. Non-Canonical Smads Phosphorylation Induced by the Glutamate Release Inhibitor, Riluzole, through GSK3 Activation in Melanoma. PLoS ONE 2012, 7, e47312. [Google Scholar] [CrossRef] [PubMed]
- Otręba, M.; Sjölander, J.J.; Grøtli, M.; Sunnerhagen, P. A Small Molecule Targeting Human MEK1/2 Enhances ERK and p38 Phosphorylation under Oxidative Stress or with Phenothiazines. Life 2021, 11, 297. [Google Scholar] [CrossRef] [PubMed]
- Mattmann, M.E.; Stoops, S.L.; Lindsley, C.W. Inhibition of Akt with small molecules and biologics: Historical perspective and current status of the patent landscape. Expert. Opin. Ther. Pat. 2011, 21, 1309–1338. [Google Scholar] [CrossRef]
- Xu, P.; Lin, X.; Feng, X.H. Posttranslational Regulation of Smads. Cold Spring Harb. Perspect. Biol. 2016, 8, a022087. [Google Scholar] [CrossRef]
- Funaba, M.; Zimmerman, C.M.; Mathews, L.S. Modulation of Smad2-mediated Signaling by Extracellular Signal-regulated Kinase*. J. Biol. Chem. 2002, 277, 41361–41368. [Google Scholar] [CrossRef]
- Yakymovych, I.; Ten Dijke, P.; Heldin, C.H.; Souchelnytskyi, S. Regulation of Smad signaling by protein kinase C. Faseb J. 2001, 15, 553–555. [Google Scholar] [CrossRef]
- Saura, M.; Zaragoza, C.; Herranz, B.; Griera, M.; Diez-Marqués, L.; Rodriguez-Puyol, D.; Rodriguez-Puyol, M. Nitric Oxide Regulates Transforming Growth Factor-β Signaling in Endothelial Cells. Circ. Res. 2005, 97, 1115–1123. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Iaria, J.; Simpson, R.J.; Zhu, H.-J. Ras enhances TGF-β signaling by decreasing cellular protein levels of its type II receptor negative regulator SPSB1. Cell Commun. Signal. 2018, 16, 10. [Google Scholar] [CrossRef]
- Guo, X.; Waddell, D.S.; Wang, W.; Wang, Z.; Liberati, N.T.; Yong, S.; Liu, X.; Wang, X.F. Ligand-dependent ubiquitination of Smad3 is regulated by casein kinase 1 gamma 2, an inhibitor of TGF-beta signaling. Oncogene 2008, 27, 7235–7247. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Li, Y.; Zhang, H.; Liu, F.; Cheng, Z.; Wang, D.; Wang, G.; Xu, H.; Zhao, Y.; Cao, L.; et al. Oncogenic PAK4 regulates Smad2/3 axis involving gastric tumorigenesis. Oncogene 2014, 33, 3473–3484. [Google Scholar] [CrossRef]
- Yan, X.; Zhang, J.; Sun, Q.; Tuazon, P.T.; Wu, X.; Traugh, J.A.; Chen, Y.-G. p21-activated Kinase 2 (PAK2) Inhibits TGF-β Signaling in Madin-Darby Canine Kidney (MDCK) Epithelial Cells by Interfering with the Receptor-Smad Interaction. J. Biol. Chem. 2012, 287, 13705–13712. [Google Scholar] [CrossRef] [PubMed]
- Nakao, A.; Imamura, T.; Souchelnytskyi, S.; Kawabata, M.; Ishisaki, A.; Oeda, E.; Tamaki, K.; Hanai, J.; Heldin, C.H.; Miyazono, K.; et al. TGF-beta receptor-mediated signalling through Smad2, Smad3 and Smad4. EMBO J. 1997, 16, 5353–5362. [Google Scholar] [CrossRef] [PubMed]
- Roelen, B.A.; Cohen, O.S.; Raychowdhury, M.K.; Chadee, D.N.; Zhang, Y.; Kyriakis, J.M.; Alessandrini, A.A.; Lin, H.Y. Phosphorylation of threonine 276 in Smad4 is involved in transforming growth factor-beta-induced nuclear accumulation. Am. J. Physiol. Cell Physiol. 2003, 285, C823–C830. [Google Scholar] [CrossRef] [PubMed]
- Morén, A.; Raja, E.; Heldin, C.H.; Moustakas, A. Negative regulation of TGFβ signaling by the kinase LKB1 and the scaffolding protein LIP1. J. Biol. Chem. 2011, 286, 341–353. [Google Scholar] [CrossRef] [PubMed]
- Seong, H.A.; Jung, H.; Ha, H. Murine protein serine/threonine kinase 38 stimulates TGF-beta signaling in a kinase-dependent manner via direct phosphorylation of Smad proteins. J. Biol. Chem. 2010, 285, 30959–30970. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Pan, L.; Qin, X.; Chen, H.; Xu, Y.; Chen, Y.; Tang, H. MTMR4 Attenuates Transforming Growth Factor β (TGFβ) Signaling by Dephosphorylating R-Smads in Endosomes*. J. Biol. Chem. 2010, 285, 8454–8462. [Google Scholar] [CrossRef] [PubMed]
- Wrighton, K.H.; Willis, D.; Long, J.; Liu, F.; Lin, X.; Feng, X.-H. Small C-terminal Domain Phosphatases Dephosphorylate the Regulatory Linker Regions of Smad2 and Smad3 to Enhance Transforming Growth Factor-β Signaling*. J. Biol. Chem. 2006, 281, 38365–38375. [Google Scholar] [CrossRef] [PubMed]
- Knockaert, M.; Sapkota, G.; Alarcón, C.; Massagué, J.; Brivanlou, A.H. Unique players in the BMP pathway: Small C-terminal domain phosphatases dephosphorylate Smad1 to attenuate BMP signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 11940–11945. [Google Scholar] [CrossRef]
- Lin, X.; Liang, M.; Feng, X.-H. Smurf2 Is a Ubiquitin E3 Ligase Mediating Proteasome-dependent Degradation of Smad2 in Transforming Growth Factor-β Signaling*210. J. Biol. Chem. 2000, 275, 36818–36822. [Google Scholar] [CrossRef]
- Zhang, Y.; Chang, C.; Gehling, D.J.; Hemmati-Brivanlou, A.; Derynck, R. Regulation of Smad degradation and activity by Smurf2, an E3 ubiquitin ligase. Proc. Natl. Acad. Sci. USA 2001, 98, 974–979. [Google Scholar] [CrossRef]
- Zhu, H.; Kavsak, P.; Abdollah, S.; Wrana, J.L.; Thomsen, G.H. A SMAD ubiquitin ligase targets the BMP pathway and affects embryonic pattern formation. Nature 1999, 400, 687–693. [Google Scholar] [CrossRef]
- Kuratomi, G.; Komuro, A.; Goto, K.; Shinozaki, M.; Miyazawa, K.; Miyazono, K.; Imamura, T. NEDD4-2 (neural precursor cell expressed, developmentally down-regulated 4-2) negatively regulates TGF-beta (transforming growth factor-beta) signalling by inducing ubiquitin-mediated degradation of Smad2 and TGF-beta type I receptor. Biochem. J. 2005, 386 Pt 3, 461–470. [Google Scholar] [CrossRef]
- Xin, H.; Xu, X.; Li, L.; Ning, H.; Rong, Y.; Shang, Y.; Wang, Y.; Fu, X.Y.; Chang, Z. CHIP controls the sensitivity of transforming growth factor-beta signaling by modulating the basal level of Smad3 through ubiquitin-mediated degradation. J. Biol. Chem. 2005, 280, 20842–20850. [Google Scholar] [CrossRef]
- Fukuchi, M.; Imamura, T.; Chiba, T.; Ebisawa, T.; Kawabata, M.; Tanaka, K.; Miyazono, K. Ligand-dependent Degradation of Smad3 by a Ubiquitin Ligase Complex of ROC1 and Associated Proteins. Mol. Biol. Cell 2001, 12, 1431–1443. [Google Scholar] [CrossRef]
- Guo, X.; Ramirez, A.; Waddell, D.S.; Li, Z.; Liu, X.; Wang, X.-F. Axin and GSK3-β control Smad3 protein stability and modulate TGF-β signaling. Genes Dev. 2008, 22, 106–120. [Google Scholar] [CrossRef] [PubMed]
- Levy, L.; Howell, M.; Das, D.; Harkin, S.; Episkopou, V.; Hill, C.S. Arkadia Activates Smad3/Smad4-Dependent Transcription by Triggering Signal-Induced SnoN Degradation. Mol. Cell. Biol. 2007, 27, 6068–6083. [Google Scholar] [CrossRef] [PubMed]
- Mavrakis, K.J.; Andrew, R.L.; Lee, K.L.; Petropoulou, C.; Dixon, J.E.; Navaratnam, N.; Norris, D.P.; Episkopou, V. Arkadia Enhances Nodal/TGF-β Signaling by Coupling Phospho-Smad2/3 Activity and Turnover. PLoS Biol. 2007, 5, e67. [Google Scholar] [CrossRef] [PubMed]
- Liang, M.; Liang, Y.-Y.; Wrighton, K.; Ungermannova, D.; Wang, X.-P.; Brunicardi, F.C.; Liu, X.; Feng, X.-H.; Lin, X. Ubiquitination and Proteolysis of Cancer-Derived Smad4 Mutants by SCFSkp2. Mol. Cell. Biol. 2004, 24, 7524–7537. [Google Scholar] [CrossRef]
- Agricola, E.; Randall, R.A.; Gaarenstroom, T.; Dupont, S.; Hill, C.S. Recruitment of TIF1γ to Chromatin via Its PHD Finger-Bromodomain Activates Its Ubiquitin Ligase and Transcriptional Repressor Activities. Mol. Cell 2011, 43, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Dupont, S.; Zacchigna, L.; Cordenonsi, M.; Soligo, S.; Adorno, M.; Rugge, M.; Piccolo, S. Germ-Layer Specification and Control of Cell Growth by Ectodermin, a Smad4 Ubiquitin Ligase. Cell 2005, 121, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Inui, M.; Manfrin, A.; Mamidi, A.; Martello, G.; Morsut, L.; Soligo, S.; Enzo, E.; Moro, S.; Polo, S.; Dupont, S.; et al. USP15 is a deubiquitylating enzyme for receptor-activated SMADs. Nat. Cell Biol. 2011, 13, 1368–1375. [Google Scholar] [CrossRef] [PubMed]
- Dupont, S.; Mamidi, A.; Cordenonsi, M.; Montagner, M.; Zacchigna, L.; Adorno, M.; Martello, G.; Stinchfield, M.J.; Soligo, S.; Morsut, L.; et al. FAM/USP9x, a deubiquitinating enzyme essential for TGFbeta signaling, controls Smad4 monoubiquitination. Cell 2009, 136, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Simonsson, M.; Kanduri, M.; Grönroos, E.; Heldin, C.-H.; Ericsson, J. The DNA Binding Activities of Smad2 and Smad3 Are Regulated by Coactivator-mediated Acetylation*. J. Biol. Chem. 2006, 281, 39870–39880. [Google Scholar] [CrossRef]
- Inoue, Y.; Itoh, Y.; Abe, K.; Okamoto, T.; Daitoku, H.; Fukamizu, A.; Onozaki, K.; Hayashi, H. Smad3 is acetylated by p300/CBP to regulate its transactivation activity. Oncogene 2007, 26, 500–508. [Google Scholar] [CrossRef]
- Tu, A.W.; Luo, K. Acetylation of Smad2 by the co-activator p300 regulates activin and transforming growth factor beta response. J. Biol. Chem. 2007, 282, 21187–21196. [Google Scholar] [CrossRef] [PubMed]
- Lönn, P.; van der Heide, L.P.; Dahl, M.; Hellman, U.; Heldin, C.H.; Moustakas, A. PARP-1 attenuates Smad-mediated transcription. Mol. Cell 2010, 40, 521–532. [Google Scholar] [CrossRef]
- Imoto, S.; Sugiyama, K.; Muromoto, R.; Sato, N.; Yamamoto, T.; Matsuda, T. Regulation of transforming growth factor-beta signaling by protein inhibitor of activated STAT, PIASy through Smad3. J. Biol. Chem. 2003, 278, 34253–34258. [Google Scholar] [CrossRef]
- Liang, M.; Melchior, F.; Feng, X.H.; Lin, X. Regulation of Smad4 sumoylation and transforming growth factor-beta signaling by protein inhibitor of activated STAT1. J. Biol. Chem. 2004, 279, 22857–22865. [Google Scholar] [CrossRef]
- Hayashida, T.; Decaestecker, M.; Schnaper, H.W. Cross-talk between ERK MAP kinase and Smad signaling pathways enhances TGF-beta-dependent responses in human mesangial cells. Faseb J. 2003, 17, 1576–1578. [Google Scholar] [CrossRef]
- Ragusa, M.; Statello, L.; Maugeri, M.; Majorana, A.; Barbagallo, D.; Salito, L.; Sammito, M.; Santonocito, M.; Angelica, R.; Cavallaro, A.; et al. Specific alterations of the microRNA transcriptome and global network structure in colorectal cancer after treatment with MAPK/ERK inhibitors. J. Mol. Med. 2012, 90, 1421–1438. [Google Scholar] [CrossRef] [PubMed]
- Samadani, R.; Zhang, J.; Brophy, A.; Oashi, T.; Priyakumar, U.D.; Raman, E.P.; St John, F.J.; Jung, K.Y.; Fletcher, S.; Pozharski, E.; et al. Small-molecule inhibitors of ERK-mediated immediate early gene expression and proliferation of melanoma cells expressing mutated BRaf. Biochem. J. 2015, 467, 425–438. [Google Scholar] [CrossRef] [PubMed]
- Kaoud, T.S.; Johnson, W.H.; Ebelt, N.D.; Piserchio, A.; Zamora-Olivares, D.; Van Ravenstein, S.X.; Pridgen, J.R.; Edupuganti, R.; Sammons, R.; Cano, M.; et al. Modulating multi-functional ERK complexes by covalent targeting of a recruitment site in vivo. Nat. Commun. 2019, 10, 5232. [Google Scholar] [CrossRef] [PubMed]
- Herrero, A.; Pinto, A.; Colón-Bolea, P.; Casar, B.; Jones, M.; Agudo-Ibáñez, L.; Vidal, R.; Tenbaum, S.P.; Nuciforo, P.; Valdizán, E.M.; et al. Small Molecule Inhibition of ERK Dimerization Prevents Tumorigenesis by RAS-ERK Pathway Oncogenes. Cancer Cell 2015, 28, 170–182. [Google Scholar] [CrossRef] [PubMed]
- Marques da Fonseca, L.; Jacques da Silva, L.R.; Santos dos Reis, J.; Rodrigues da Costa Santos, M.A.; de Sousa Chaves, V.; Monteiro da Costa, K.; Sa-Diniz, J.d.N.; Freire de Lima, C.G.; Morrot, A.; Nunes Franklim, T.; et al. Piperine Inhibits TGF-β Signaling Pathways and Disrupts EMT-Related Events in Human Lung Adenocarcinoma Cells. Medicines 2020, 7, 19. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Huang, J.; Yuan, Z.; Feng, S.; Xie, Y.; Ma, W. Protein kinase C inhibitor chelerythrine selectively inhibits proliferation of triple-negative breast cancer cells. Sci. Rep. 2017, 7, 2022. [Google Scholar] [CrossRef]
- Liu, S.; Zhang, Y.; Yang, Q.; Zhang, Y.; Liu, H.; Huang, M.H.; Wang, R.; Lu, F. PKC signal amplification suppresses non-small cell lung cancer growth by promoting p21 expression and phosphorylation. Heliyon 2022, 8, e10657. [Google Scholar] [CrossRef]
- Dükel, M.; Tavsan, Z.; Erdogan, D.; Erkan Gök, D.; Ayar Kayali, H. Protein kinase C Inhibitors selectively modulate dynamics of cell adhesion molecules and cell death in human colon cancer cells. Cell Adhes. Migr. 2019, 13, 83–97. [Google Scholar] [CrossRef]
- Whitt, J.D.; Li, N.; Tinsley, H.N.; Chen, X.; Zhang, W.; Li, Y.; Gary, B.D.; Keeton, A.B.; Xi, Y.; Abadi, A.H.; et al. A Novel Sulindac Derivative that Potently Suppresses Colon Tumor Cell Growth by Inhibiting cGMP Phosphodiesterase and β-Catenin Transcriptional Activity. Cancer Prev. Res. 2012, 5, 822–833. [Google Scholar] [CrossRef]
- Tinsley, H.N.; Mathew, B.; Chen, X.; Maxuitenko, Y.Y.; Li, N.; Lowe, W.M.; Whitt, J.D.; Zhang, W.; Gary, B.D.; Keeton, A.B.; et al. Novel Non-Cyclooxygenase Inhibitory Derivative of Sulindac Inhibits Breast Cancer Cell Growth In Vitro and Reduces Mammary Tumorigenesis in Rats. Cancers 2023, 15, 646. [Google Scholar] [CrossRef]
- Vena, F.; Bayle, S.; Nieto, A.; Quereda, V.; Aceti, M.; Frydman, S.M.; Sansil, S.S.; Grant, W.; Monastyrskyi, A.; McDonald, P.; et al. Targeting Casein Kinase 1 Delta Sensitizes Pancreatic and Bladder Cancer Cells to Gemcitabine Treatment by Upregulating Deoxycytidine Kinase. Mol. Cancer Ther. 2020, 19, 1623–1635. [Google Scholar] [CrossRef]
- Liu, M.; Hu, Y.; Lu, S.; Lu, M.; Li, J.; Chang, H.; Jia, H.; Zhou, M.; Ren, F.; Zhong, J. IC261, a specific inhibitor of CK1δ/ε, promotes aerobic glycolysis through p53-dependent mechanisms in colon cancer. Int. J. Biol. Sci. 2020, 16, 882–892. [Google Scholar] [CrossRef]
- Licciulli, S.; Maksimoska, J.; Zhou, C.; Troutman, S.; Kota, S.; Liu, Q.; Duron, S.; Campbell, D.; Chernoff, J.; Field, J.; et al. FRAX597, a small molecule inhibitor of the p21-activated kinases, inhibits tumorigenesis of neurofibromatosis type 2 (NF2)-associated Schwannomas. J. Biol. Chem. 2013, 288, 29105–29114. [Google Scholar] [CrossRef] [PubMed]
- Murray, B.W.; Guo, C.; Piraino, J.; Westwick, J.K.; Zhang, C.; Lamerdin, J.; Dagostino, E.; Knighton, D.; Loi, C.M.; Zager, M.; et al. Small-molecule p21-activated kinase inhibitor PF-3758309 is a potent inhibitor of oncogenic signaling and tumor growth. Proc. Natl. Acad. Sci. USA 2010, 107, 9446–9451. [Google Scholar] [CrossRef]
- Hao, C.; Zhao, F.; Song, H.; Guo, J.; Li, X.; Jiang, X.; Huan, R.; Song, S.; Zhang, Q.; Wang, R.; et al. Structure-Based Design of 6-Chloro-4-aminoquinazoline-2-carboxamide Derivatives as Potent and Selective p21-Activated Kinase 4 (PAK4) Inhibitors. J. Med. Chem. 2018, 61, 265–285. [Google Scholar] [CrossRef]
- Song, P.; Zhao, F.; Li, D.; Qu, J.; Yao, M.; Su, Y.; Wang, H.; Zhou, M.; Wang, Y.; Gao, Y.; et al. Synthesis of selective PAK4 inhibitors for lung metastasis of lung cancer and melanoma cells. Acta Pharm. Sin. B 2022, 12, 2905–2922. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, L.; McNamara, K.L.; Li, D.; Borgman, C.L.; McDermott, U.; Brandstetter, K.A.; Padera, R.F.; Chirieac, L.R.; Settleman, J.E.; Wong, K.K. Sunitinib prolongs survival in genetically engineered mouse models of multistep lung carcinogenesis. Cancer Prev. Res. 2009, 2, 330–337. [Google Scholar] [CrossRef]
- Andrade-Vieira, R.; Goguen, D.; Bentley, H.A.; Bowen, C.V.; Marignani, P.A. Pre-clinical study of drug combinations that reduce breast cancer burden due to aberrant mTOR and metabolism promoted by LKB1 loss. Oncotarget 2014, 5, 12738–12752. [Google Scholar] [CrossRef]
- Zheng, B.; Jeong, J.H.; Asara, J.M.; Yuan, Y.Y.; Granter, S.R.; Chin, L.; Cantley, L.C. Oncogenic B-RAF negatively regulates the tumor suppressor LKB1 to promote melanoma cell proliferation. Mol. Cell 2009, 33, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Farrington, C.C.; Yuan, E.; Mazhar, S.; Izadmehr, S.; Hurst, L.; Allen-Petersen, B.L.; Janghorban, M.; Chung, E.; Wolczanski, G.; Galsky, M.; et al. Protein phosphatase 2A activation as a therapeutic strategy for managing MYC-driven cancers. J. Biol. Chem. 2020, 295, 757–770. [Google Scholar] [CrossRef]
- McClinch, K.; Avelar, R.A.; Callejas, D.; Izadmehr, S.; Wiredja, D.; Perl, A.; Sangodkar, J.; Kastrinsky, D.B.; Schlatzer, D.; Cooper, M.; et al. Small-Molecule Activators of Protein Phosphatase 2A for the Treatment of Castration-Resistant Prostate Cancer. Cancer Res. 2018, 78, 2065–2080. [Google Scholar] [CrossRef]
- Wang, L.; Ge, S.; Zhou, F. MicroRNA-487a-3p inhibits the growth and invasiveness of oral squamous cell carcinoma by targeting PPM1A. Bioengineered 2021, 12, 937–947. [Google Scholar] [CrossRef] [PubMed]
- Kauko, O.; O’Connor, C.M.; Kulesskiy, E.; Sangodkar, J.; Aakula, A.; Izadmehr, S.; Yetukuri, L.; Yadav, B.; Padzik, A.; Laajala, T.D.; et al. PP2A inhibition is a druggable MEK inhibitor resistance mechanism in KRAS-mutant lung cancer cells. Sci. Transl. Med. 2018, 10, eaaq1093. [Google Scholar] [CrossRef] [PubMed]
- Sowa, N.; Horie, T.; Kuwabara, Y.; Baba, O.; Watanabe, S.; Nishi, H.; Kinoshita, M.; Takanabe-Mori, R.; Wada, H.; Shimatsu, A.; et al. MicroRNA 26b encoded by the intron of small CTD phosphatase (SCP) 1 has an antagonistic effect on its host gene. J. Cell Biochem. 2012, 113, 3455–3465. [Google Scholar] [CrossRef]
- Matsuoka, H.; Ando, K.; Swayze, E.J.; Unan, E.C.; Mathew, J.; Hu, Q.; Tsuda, Y.; Nakashima, Y.; Saeki, H.; Oki, E.; et al. CTDSP1 inhibitor rabeprazole regulates DNA-PKcs dependent topoisomerase I degradation and irinotecan drug resistance in colorectal cancer. PLoS ONE 2020, 15, e0228002. [Google Scholar] [CrossRef] [PubMed]
- Gu, M.; Zhang, Y.; Zhou, X.; Ma, H.; Yao, H.; Ji, F. Rabeprazole exhibits antiproliferative effects on human gastric cancer cell lines. Oncol. Lett. 2014, 8, 1739–1744. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Deng, J.; Yuan, J.; Tang, X.; Wang, Y.; Chen, H.; Liu, Y.; Zhou, L. Curcumin exerts its tumor suppressive function via inhibition of NEDD4 oncoprotein in glioma cancer cells. Int. J. Oncol. 2017, 51, 467–477. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Yu, Y.; Wang, W.; Jiang, Y.; Li, Y.; Jiang, X.; Qiao, Y.; Chen, L.; Zhao, X.; Liu, J.; et al. Targeting the E3 ligase NEDD4 as a novel therapeutic strategy for IGF1 signal pathway-driven gastric cancer. Oncogene 2023, 42, 1072–1087. [Google Scholar] [CrossRef]
- Zhang, J.; Xie, J.J.; Zhou, S.J.; Chen, J.; Hu, Q.; Pu, J.X.; Lu, J.L. Diosgenin inhibits the expression of NEDD4 in prostate cancer cells. Am. J. Transl. Res. 2019, 11, 3461–3471. [Google Scholar]
- Leu, J.I.; Pimkina, J.; Frank, A.; Murphy, M.E.; George, D.L. A small molecule inhibitor of inducible heat shock protein 70. Mol. Cell 2009, 36, 15–27. [Google Scholar] [CrossRef]
- Dublang, L.; Underhaug, J.; Flydal, M.I.; Velasco-Carneros, L.; Maréchal, J.D.; Moro, F.; Boyano, M.D.; Martinez, A.; Muga, A. Inhibition of the Human Hsc70 System by Small Ligands as a Potential Anticancer Approach. Cancers 2021, 13, 2936. [Google Scholar] [CrossRef]
- Liu, Y.Q.; Wang, X.L.; Cheng, X.; Lu, Y.Z.; Wang, G.Z.; Li, X.C.; Zhang, J.; Wen, Z.S.; Huang, Z.L.; Gao, Q.L.; et al. Skp1 in lung cancer: Clinical significance and therapeutic efficacy of its small molecule inhibitors. Oncotarget 2015, 6, 34953–34967. [Google Scholar] [CrossRef]
- Hussain, M.; Lu, Y.; Tariq, M.; Jiang, H.; Shu, Y.; Luo, S.; Zhu, Q.; Zhang, J.; Liu, J. A small-molecule Skp1 inhibitor elicits cell death by p53-dependent mechanism. iScience 2022, 25, 104591. [Google Scholar] [CrossRef]
- Wu, J.W.; Krawitz, A.R.; Chai, J.; Li, W.; Zhang, F.; Luo, K.; Shi, Y. Structural mechanism of Smad4 recognition by the nuclear oncoprotein Ski: Insights on Ski-mediated repression of TGF-beta signaling. Cell 2002, 111, 357–367. [Google Scholar] [CrossRef]
- Zhou, L.; Yu, X.; Li, M.; Gong, G.; Liu, W.; Li, T.; Zuo, H.; Li, W.; Gao, F.; Liu, H. Cdh1-mediated Skp2 degradation by dioscin reprogrammes aerobic glycolysis and inhibits colorectal cancer cells growth. EBioMedicine 2020, 51, 102570. [Google Scholar] [CrossRef]
- Chan, C.H.; Morrow, J.K.; Li, C.F.; Gao, Y.; Jin, G.; Moten, A.; Stagg, L.J.; Ladbury, J.E.; Cai, Z.; Xu, D.; et al. Pharmacological inactivation of Skp2 SCF ubiquitin ligase restricts cancer stem cell traits and cancer progression. Cell 2013, 154, 556–568. [Google Scholar] [CrossRef]
- Lasko, L.M.; Jakob, C.G.; Edalji, R.P.; Qiu, W.; Montgomery, D.; Digiammarino, E.L.; Hansen, T.M.; Risi, R.M.; Frey, R.; Manaves, V.; et al. Discovery of a selective catalytic p300/CBP inhibitor that targets lineage-specific tumours. Nature 2017, 550, 128–132. [Google Scholar] [CrossRef]
- Ansari, M.S.Z.; Stagni, V.; Iuzzolino, A.; Rotili, D.; Mai, A.; Del Bufalo, D.; Lavia, P.; Degrassi, F.; Trisciuoglio, D. Pharmacological targeting of CBP/p300 drives a redox/autophagy axis leading to senescence-induced growth arrest in non-small cell lung cancer cells. Cancer Gene Ther. 2023, 30, 124–136. [Google Scholar] [CrossRef]
- Cai, L.-Y.; Chen, S.-J.; Xiao, S.-H.; Sun, Q.-J.; Ding, C.-H.; Zheng, B.-N.; Zhu, X.-Y.; Liu, S.-Q.; Yang, F.; Yang, Y.-X.; et al. Targeting p300/CBP Attenuates Hepatocellular Carcinoma Progression through Epigenetic Regulation of Metabolism. Cancer Res. 2021, 81, 860–872. [Google Scholar] [CrossRef]
- Gajer, J.M.; Furdas, S.D.; Gründer, A.; Gothwal, M.; Heinicke, U.; Keller, K.; Colland, F.; Fulda, S.; Pahl, H.L.; Fichtner, I.; et al. Histone acetyltransferase inhibitors block neuroblastoma cell growth in vivo. Oncogenesis 2015, 4, e137. [Google Scholar] [CrossRef]
- Wang, Y.M.; Gu, M.L.; Meng, F.S.; Jiao, W.R.; Zhou, X.X.; Yao, H.P.; Ji, F. Histone acetyltransferase p300/CBP inhibitor C646 blocks the survival and invasion pathways of gastric cancer cell lines. Int. J. Oncol. 2017, 51, 1860–1868. [Google Scholar] [CrossRef]
- Illuzzi, G.; Staniszewska, A.D.; Gill, S.J.; Pike, A.; McWilliams, L.; Critchlow, S.E.; Cronin, A.; Fawell, S.; Hawthorne, G.; Jamal, K.; et al. Preclinical Characterization of AZD5305, A Next-Generation, Highly Selective PARP1 Inhibitor and Trapper. Clin. Cancer Res. 2022, 28, 4724–4736. [Google Scholar] [CrossRef]
- Sreekumar, S.; Zhou, D.; Mpoy, C.; Schenk, E.; Scott, J.; Arbeit, J.M.; Xu, J.; Rogers, B.E. Preclinical Efficacy of a PARP-1 Targeted Auger-Emitting Radionuclide in Prostate Cancer. Int. J. Mol. Sci. 2023, 24, 3083. [Google Scholar] [CrossRef]
- Shi, Y.; Wang, Y.-F.; Jayaraman, L.; Yang, H.; Massagué, J.; Pavletich, N.P. Crystal Structure of a Smad MH1 Domain Bound to DNA: Insights on DNA Binding in TGF-β Signaling. Cell 1998, 94, 585–594. [Google Scholar] [CrossRef]
- Zawel, L.; Le Dai, J.; Buckhaults, P.; Zhou, S.; Kinzler, K.W.; Vogelstein, B.; Kern, S.E. Human Smad3 and Smad4 Are Sequence-Specific Transcription Activators. Mol. Cell 1998, 1, 611–617. [Google Scholar] [CrossRef]
- Morikawa, M.; Koinuma, D.; Miyazono, K.; Heldin, C.H. Genome-wide mechanisms of Smad binding. Oncogene 2013, 32, 1609–1615. [Google Scholar] [CrossRef]
- Dennler, S.; Itoh, S.; Vivien, D.; ten Dijke, P.; Huet, S.; Gauthier, J.M. Direct binding of Smad3 and Smad4 to critical TGF beta-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. EMBO J 1998, 17, 3091–3100. [Google Scholar] [CrossRef]
- Liu, L.; Liu, X.; Ren, X.; Tian, Y.; Chen, Z.; Xu, X.; Du, Y.; Jiang, C.; Fang, Y.; Liu, Z.; et al. Smad2 and Smad3 have differential sensitivity in relaying TGFβ signaling and inversely regulate early lineage specification. Sci. Rep. 2016, 6, 21602. [Google Scholar] [CrossRef]
- Yagi, K.; Goto, D.; Hamamoto, T.; Takenoshita, S.; Kato, M.; Miyazono, K. Alternatively spliced variant of Smad2 lacking exon 3. Comparison with wild-type Smad2 and Smad3. J. Biol. Chem. 1999, 274, 703–709. [Google Scholar] [CrossRef]
- Pyrowolakis, G.; Hartmann, B.; Müller, B.; Basler, K.; Affolter, M. A Simple Molecular Complex Mediates Widespread BMP-Induced Repression during Drosophila Development. Dev. Cell 2004, 7, 229–240. [Google Scholar] [CrossRef]
- Weiss, A.; Charbonnier, E.; Ellertsdóttir, E.; Tsirigos, A.; Wolf, C.; Schuh, R.; Pyrowolakis, G.; Affolter, M. A conserved activation element in BMP signaling during Drosophila development. Nat. Struct. Mol. Biol. 2010, 17, 69–76. [Google Scholar] [CrossRef]
- Ito, T.; Ikehara, T.; Nakagawa, T.; Kraus, W.L.; Muramatsu, M. p300-Mediated acetylation facilitates the transfer of histone H2A–H2B dimers from nucleosomes to a histone chaperone. Genes Dev. 2000, 14, 1899–1907. [Google Scholar] [CrossRef] [PubMed]
- Kahata, K.; Hayashi, M.; Asaka, M.; Hellman, U.; Kitagawa, H.; Yanagisawa, J.; Kato, S.; Imamura, T.; Miyazono, K. Regulation of transforming growth factor-β and bone morphogenetic protein signalling by transcriptional coactivator GCN5. Genes Cells 2004, 9, 143–151. [Google Scholar] [CrossRef]
- Ross, S.; Cheung, E.; Petrakis, T.G.; Howell, M.; Kraus, W.L.; Hill, C.S. Smads orchestrate specific histone modifications and chromatin remodeling to activate transcription. EMBO J. 2006, 25, 4490–4502. [Google Scholar] [CrossRef]
- Du, D.; Katsuno, Y.; Meyer, D.; Budi, E.H.; Chen, S.-H.; Koeppen, H.; Wang, H.; Akhurst, R.J.; Derynck, R. Smad3-mediated recruitment of the methyltransferase SETDB1/ESET controls Snail1 expression and epithelial–mesenchymal transition. EMBO Rep. 2018, 19, 135–155. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.-H.; Zhang, Y.; Wu, R.-Y.; Derynck, R. The tumor suppressor Smad4/DPC4 and transcriptional adaptor CBP/p300 are coactivators for Smad3 in TGF-β-induced transcriptional activation. Genes Dev. 1998, 12, 2153–2163. [Google Scholar] [CrossRef]
- Song, C.; Zhou, C. HOXA10 mediates epithelial-mesenchymal transition to promote gastric cancer metastasis partly via modulation of TGFB2/Smad/METTL3 signaling axis. J. Exp. Clin. Cancer Res. 2021, 40, 62. [Google Scholar] [CrossRef]
- Aragón, E.; Wang, Q.; Zou, Y.; Morgani, S.M.; Ruiz, L.; Kaczmarska, Z.; Su, J.; Torner, C.; Tian, L.; Hu, J.; et al. Structural basis for distinct roles of SMAD2 and SMAD3 in FOXH1 pioneer-directed TGF-β signaling. Genes Dev. 2019, 33, 1506–1524. [Google Scholar] [CrossRef]
- Zhang, H.; Ma, Y.; Zhang, S.; Liu, H.; He, H.; Li, N.; Gong, Y.; Zhao, S.; Jiang, J.D.; Shao, R.G. Involvement of Ras GTPase-activating protein SH3 domain-binding protein 1 in the epithelial-to-mesenchymal transition-induced metastasis of breast cancer cells via the Smad signaling pathway. Oncotarget 2015, 6, 17039–17053. [Google Scholar] [CrossRef]
- Vervoort, S.J.; de Jong, O.G.; Roukens, M.G.; Frederiks, C.L.; Vermeulen, J.F.; Lourenço, A.R.; Bella, L.; Vidakovic, A.T.; Sandoval, J.L.; Moelans, C.; et al. Global transcriptional analysis identifies a novel role for SOX4 in tumor-induced angiogenesis. Elife 2018, 7, e27706. [Google Scholar] [CrossRef]
- Seoane, J.; Le, H.-V.; Shen, L.; Anderson, S.A.; Massagué, J. Integration of Smad and Forkhead Pathways in the Control of Neuroepithelial and Glioblastoma Cell Proliferation. Cell 2004, 117, 211–223. [Google Scholar] [CrossRef] [PubMed]
- González-González, A.; Muñoz-Muela, E.; Marchal, J.A.; Cara, F.E.; Molina, M.P.; Cruz-Lozano, M.; Jiménez, G.; Verma, A.; Ramírez, A.; Qian, W.; et al. Activating Transcription Factor 4 Modulates TGFβ-Induced Aggressiveness in Triple-Negative Breast Cancer via SMAD2/3/4 and mTORC2 Signaling. Clin. Cancer Res. 2018, 24, 5697–5709. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.W.; Hui, W.W.Y.; Wang, J.J.; Yung, M.M.H.; Hui, L.M.N.; Qin, Y.; Liang, R.R.; Leung, T.H.Y.; Xu, D.; Chan, K.K.L.; et al. DLX1 acts as a crucial target of FOXM1 to promote ovarian cancer aggressiveness by enhancing TGF-β/SMAD4 signaling. Oncogene 2017, 36, 1404–1416. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.R.; Kang, Y.; Siegel, P.M.; Massagué, J. E2F4/5 and p107 as Smad cofactors linking the TGFbeta receptor to c-myc repression. Cell 2002, 110, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Verrecchia, F.; Vindevoghel, L.; Lechleider, R.J.; Uitto, J.; Roberts, A.B.; Mauviel, A. Smad3/AP-1 interactions control transcriptional responses to TGF-β in a promoter-specific manner. Oncogene 2001, 20, 3332–3340. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Wu, J.; Jiang, Q.; Cheng, H.; Han, J.-D.J.; Chen, Y.-G. CXXC5 suppresses hepatocellular carcinoma by promoting TGF-β-induced cell cycle arrest and apoptosis. J. Mol. Cell Biol. 2017, 10, 48–59. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.; Tu, S.; Wang, J.; Luo, S.; Yan, X. CXXC5: A novel regulator and coordinator of TGF-β, BMP and Wnt signaling. J. Cell Mol. Med. 2019, 23, 740–749. [Google Scholar] [CrossRef] [PubMed]
- Adam, P.J.; Regan, C.P.; Hautmann, M.B.; Owens, G.K. Positive- and negative-acting Kruppel-like transcription factors bind a transforming growth factor beta control element required for expression of the smooth muscle cell differentiation marker SM22alpha in vivo. J. Biol. Chem. 2000, 275, 37798–37806. [Google Scholar] [CrossRef]
- Li, H.X.; Han, M.; Bernier, M.; Zheng, B.; Sun, S.G.; Su, M.; Zhang, R.; Fu, J.R.; Wen, J.K. Krüppel-like factor 4 promotes differentiation by transforming growth factor-beta receptor-mediated Smad and p38 MAPK signaling in vascular smooth muscle cells. J. Biol. Chem. 2010, 285, 17846–17856. [Google Scholar] [CrossRef]
- Spittau, B.; Krieglstein, K. Klf10 and Klf11 as mediators of TGF-beta superfamily signaling. Cell Tissue Res. 2012, 347, 65–72. [Google Scholar] [CrossRef]
- Buck, A.; Buchholz, M.; Wagner, M.; Adler, G.; Gress, T.; Ellenrieder, V. The tumor suppressor KLF11 mediates a novel mechanism in transforming growth factor beta-induced growth inhibition that is inactivated in pancreatic cancer. Mol. Cancer Res. 2006, 4, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Wu, H.; Xu, Y.; Zhang, Z.; Liu, T.; Lin, X.; Feng, X.H. Zinc finger protein 451 is a novel Smad corepressor in transforming growth factor-β signaling. J. Biol. Chem. 2014, 289, 2072–2083. [Google Scholar] [CrossRef]
- Xi, Q.; Wang, Z.; Zaromytidou, A.-I.; Zhang, X.H.F.; Chow-Tsang, L.-F.; Liu, J.X.; Kim, H.; Barlas, A.; Manova-Todorova, K.; Kaartinen, V.; et al. A Poised Chromatin Platform for TGF-β Access to Master Regulators. Cell 2011, 147, 1511–1524. [Google Scholar] [CrossRef]
- Tang, X.; Li, G.; Su, F.; Cai, Y.; Shi, L.; Meng, Y.; Liu, Z.; Sun, J.; Wang, M.; Qian, M.; et al. HDAC8 cooperates with SMAD3/4 complex to suppress SIRT7 and promote cell survival and migration. Nucleic Acids Res. 2020, 48, 2912–2923. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.S.; Hong, J.J.; Wu, J.F.; Yan, S.; Wu, D.; Liu, N.; Liu, Q.F.; Wu, Q.W.; Xie, Y.Y.; Liu, Y.J.; et al. OVOL2 antagonizes TGF-β signaling to regulate epithelial to mesenchymal transition during mammary tumor metastasis. Oncotarget 2017, 8, 39401–39416. [Google Scholar] [CrossRef]
- Stroschein, S.L.; Wang, W.; Zhou, S.; Zhou, Q.; Luo, K. Negative feedback regulation of TGF-beta signaling by the SnoN oncoprotein. Science 1999, 286, 771–774. [Google Scholar] [CrossRef] [PubMed]
- Luo, K.; Stroschein, S.L.; Wang, W.; Chen, D.; Martens, E.; Zhou, S.; Zhou, Q. The Ski oncoprotein interacts with the Smad proteins to repress TGFbeta signaling. Genes Dev. 1999, 13, 2196–2206. [Google Scholar] [CrossRef]
- Wotton, D.; Lo, R.S.; Swaby, L.-A.C.; Massagué, J. Multiple Modes of Repression by the Smad Transcriptional Corepressor TGIF*. J. Biol. Chem. 1999, 274, 37105–37110. [Google Scholar] [CrossRef]
- Guca, E.; Suñol, D.; Ruiz, L.; Konkol, A.; Cordero, J.; Torner, C.; Aragon, E.; Martin-Malpartida, P.; Riera, A.; Macias, M.J. TGIF1 homeodomain interacts with Smad MH1 domain and represses TGF-β signaling. Nucleic Acids Res. 2018, 46, 9220–9235. [Google Scholar] [CrossRef]
- Boon, R.A.; Fledderus, J.O.; Volger, O.L.; van Wanrooij, E.J.; Pardali, E.; Weesie, F.; Kuiper, J.; Pannekoek, H.; ten Dijke, P.; Horrevoets, A.J. KLF2 suppresses TGF-beta signaling in endothelium through induction of Smad7 and inhibition of AP-1. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 532–539. [Google Scholar] [CrossRef] [PubMed]
- Nakao, A.; Afrakhte, M.; Morén, A.; Nakayama, T.; Christian, J.L.; Heuchel, R.; Itoh, S.; Kawabata, M.; Heldin, N.E.; Heldin, C.H.; et al. Identification of Smad7, a TGFbeta-inducible antagonist of TGF-beta signalling. Nature 1997, 389, 631–635. [Google Scholar] [CrossRef]
- Itoh, S.; Landström, M.; Hermansson, A.; Itoh, F.; Heldin, C.-H.; Heldin, N.-E.; ten Dijke, P. Transforming Growth Factor β1 Induces Nuclear Export of Inhibitory Smad7*. J. Biol. Chem. 1998, 273, 29195–29201. [Google Scholar] [CrossRef] [PubMed]
- Elkouris, M.; Kontaki, H.; Stavropoulos, A.; Antonoglou, A.; Nikolaou, K.C.; Samiotaki, M.; Szantai, E.; Saviolaki, D.; Brown, P.J.; Sideras, P.; et al. SET9-Mediated Regulation of TGF-β Signaling Links Protein Methylation to Pulmonary Fibrosis. Cell Rep. 2016, 15, 2733–2744. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, V.; Sixt, K.M.; Gao, S.; Xu, X.; Huang, J.; Weigert, R.; Zhou, M.; Zhang, Y.E. Direct Regulation of Alternative Splicing by SMAD3 through PCBP1 Is Essential to the Tumor-Promoting Role of TGF-β. Mol. Cell 2016, 64, 549–564. [Google Scholar] [CrossRef] [PubMed]
- Davis, B.N.; Hilyard, A.C.; Lagna, G.; Hata, A. SMAD proteins control DROSHA-mediated microRNA maturation. Nature 2008, 454, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Davis, B.N.; Hilyard, A.C.; Nguyen, P.H.; Lagna, G.; Hata, A. Smad proteins bind a conserved RNA sequence to promote microRNA maturation by Drosha. Mol. Cell 2010, 39, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Xiong, X.; Chen, Y.-G. Feedback regulation of TGF-β signaling. Acta Biochim. Biophys. Sin. 2017, 50, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Adylova, A.; Mukhanbetzhanovna, A.A.; Attar, R.; Yulaevna, I.M.; Farooqi, A.A. Regulation of TGFβ/SMAD signaling by long non-coding RNAs in different cancers: Dark Knight in the Castle of molecular oncology. Noncoding RNA Res. 2021, 6, 23–28. [Google Scholar] [CrossRef]
- Bertero, A.; Brown, S.; Madrigal, P.; Osnato, A.; Ortmann, D.; Yiangou, L.; Kadiwala, J.; Hubner, N.C.; de Los Mozos, I.R.; Sadée, C.; et al. The SMAD2/3 interactome reveals that TGFβ controls m(6)A mRNA methylation in pluripotency. Nature 2018, 555, 256–259. [Google Scholar] [CrossRef]
- Bassi, Z.I.; Fillmore, M.C.; Miah, A.H.; Chapman, T.D.; Maller, C.; Roberts, E.J.; Davis, L.C.; Lewis, D.E.; Galwey, N.W.; Waddington, K.E.; et al. Modulating PCAF/GCN5 Immune Cell Function through a PROTAC Approach. ACS Chem. Biol. 2018, 13, 2862–2867. [Google Scholar] [CrossRef]
- Collins, H.M.; Abdelghany, M.K.; Messmer, M.; Yue, B.; Deeves, S.E.; Kindle, K.B.; Mantelingu, K.; Aslam, A.; Winkler, G.S.; Kundu, T.K.; et al. Differential effects of garcinol and curcumin on histone and p53 modifications in tumour cells. BMC Cancer 2013, 13, 37. [Google Scholar] [CrossRef]
- Mota, M.; Sweha, S.R.; Pun, M.; Natarajan, S.K.; Ding, Y.; Chung, C.; Hawes, D.; Yang, F.; Judkins, A.R.; Samajdar, S.; et al. Targeting SWI/SNF ATPases in H3.3K27M diffuse intrinsic pontine gliomas. Proc. Natl. Acad. Sci. USA 2023, 120, e2221175120. [Google Scholar] [CrossRef]
- Lee, D.; Lee, D.Y.; Hwang, Y.S.; Seo, H.R.; Lee, S.A.; Kwon, J. The Bromodomain Inhibitor PFI-3 Sensitizes Cancer Cells to DNA Damage by Targeting SWI/SNF. Mol. Cancer Res. 2021, 19, 900–912. [Google Scholar] [CrossRef]
- Papillon, J.P.N.; Nakajima, K.; Adair, C.D.; Hempel, J.; Jouk, A.O.; Karki, R.G.; Mathieu, S.; Möbitz, H.; Ntaganda, R.; Smith, T.; et al. Discovery of Orally Active Inhibitors of Brahma Homolog (BRM)/SMARCA2 ATPase Activity for the Treatment of Brahma Related Gene 1 (BRG1)/SMARCA4-Mutant Cancers. J. Med. Chem. 2018, 61, 10155–10172. [Google Scholar] [CrossRef]
- Federico, A.; Steinfass, T.; Larribère, L.; Novak, D.; Morís, F.; Núñez, L.E.; Umansky, V.; Utikal, J. Mithramycin A and Mithralog EC-8042 Inhibit SETDB1 Expression and Its Oncogenic Activity in Malignant Melanoma. Mol. Ther. Oncolytics 2020, 18, 83–99. [Google Scholar] [CrossRef]
- Lee, J.K.; Kim, K.C. DZNep, inhibitor of S-adenosylhomocysteine hydrolase, down-regulates expression of SETDB1 H3K9me3 HMTase in human lung cancer cells. Biochem. Biophys. Res. Commun. 2013, 438, 647–652. [Google Scholar] [CrossRef]
- Shao, Y.; Song, X.; Jiang, W.; Chen, Y.; Ning, Z.; Gu, W.; Jiang, J. MicroRNA-621 Acts as a Tumor Radiosensitizer by Directly Targeting SETDB1 in Hepatocellular Carcinoma. Mol. Ther. 2019, 27, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, M.H.; Gmachl, M.; Ramharter, J.; Savarese, F.; Gerlach, D.; Marszalek, J.R.; Sanderson, M.P.; Kessler, D.; Trapani, F.; Arnhof, H.; et al. BI-3406, a Potent and Selective SOS1-KRAS Interaction Inhibitor, Is Effective in KRAS-Driven Cancers through Combined MEK Inhibition. Cancer Discov. 2021, 11, 142–157. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Park, D.; Magis, A.T.; Zhang, J.; Zhou, W.; Sica, G.L.; Ramalingam, S.S.; Curran, W.J.; Deng, X. Small Molecule KRAS Agonist for Mutant KRAS Cancer Therapy. Mol. Cancer 2019, 18, 85. [Google Scholar] [CrossRef]
- Shima, F.; Yoshikawa, Y.; Ye, M.; Araki, M.; Matsumoto, S.; Liao, J.; Hu, L.; Sugimoto, T.; Ijiri, Y.; Takeda, A.; et al. In silico discovery of small-molecule Ras inhibitors that display antitumor activity by blocking the Ras–effector interaction. Proc. Natl. Acad. Sci. USA 2013, 110, 8182–8187. [Google Scholar] [CrossRef] [PubMed]
- Janes, M.R.; Zhang, J.; Li, L.-S.; Hansen, R.; Peters, U.; Guo, X.; Chen, Y.; Babbar, A.; Firdaus, S.J.; Darjania, L.; et al. Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor. Cell 2018, 172, 578–589.e17. [Google Scholar] [CrossRef] [PubMed]
- Afaloniati, H.; Angelopoulou, K.; Giakoustidis, A.; Hardas, A.; Pseftogas, A.; Makedou, K.; Gargavanis, A.; Goulopoulos, T.; Iliadis, S.; Papadopoulos, V.; et al. HDAC1/2 Inhibitor Romidepsin Suppresses DEN-Induced Hepatocellular Carcinogenesis in Mice. Onco Targets Ther. 2020, 13, 5575–5588. [Google Scholar] [CrossRef] [PubMed]
- Mayr, C.; Kiesslich, T.; Erber, S.; Bekric, D.; Dobias, H.; Beyreis, M.; Ritter, M.; Jäger, T.; Neumayer, B.; Winkelmann, P.; et al. HDAC Screening Identifies the HDAC Class I Inhibitor Romidepsin as a Promising Epigenetic Drug for Biliary Tract Cancer. Cancers 2021, 13, 3862. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.-H.; Zhang, X.-B.; Han, X.-Q.; Feng, C.-R.; Wang, F.S.; Wang, P.G.; Shen, J.; Shi, Y.-K. Antitumor effects of a novel histone deacetylase inhibitor NK-HDAC-1 on breast cancer. Oncol. Rep. 2013, 30, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S.; Park, S.B.; Kim, S.A.; Kwon, S.K.; Cha, H.; Lee, D.Y.; Ro, S.; Cho, J.M.; Song, S.Y. A novel HDAC inhibitor, CG200745, inhibits pancreatic cancer cell growth and overcomes gemcitabine resistance. Sci. Rep. 2017, 7, 41615. [Google Scholar] [CrossRef]
- Chun, S.-M.; Lee, J.-Y.; Choi, J.; Lee, J.-H.; Hwang, J.J.; Kim, C.-S.; Suh, Y.-A.; Jang, S.J. Epigenetic Modulation with HDAC Inhibitor CG200745 Induces Anti-Proliferation in Non-Small Cell Lung Cancer Cells. PLoS ONE 2015, 10, e0119379. [Google Scholar] [CrossRef] [PubMed]
- Sandor, V.; Bakke, S.; Robey, R.W.; Kang, M.H.; Blagosklonny, M.V.; Bender, J.; Brooks, R.; Piekarz, R.L.; Tucker, E.; Figg, W.D.; et al. Phase I trial of the histone deacetylase inhibitor, depsipeptide (FR901228, NSC 630176), in patients with refractory neoplasms. Clin. Cancer Res. 2002, 8, 718–728. [Google Scholar] [PubMed]
- Sandor, V.; Senderowicz, A.; Mertins, S.; Sackett, D.; Sausville, E.; Blagosklonny, M.V.; Bates, S.E. P21-dependent G1arrest with downregulation of cyclin D1 and upregulation of cyclin E by the histone deacetylase inhibitor FR901228. Br. J. Cancer 2000, 83, 817–825. [Google Scholar] [CrossRef]
- Nian, H.; Bisson, W.H.; Dashwood, W.-M.; Pinto, J.T.; Dashwood, R.H. α-Keto acid metabolites of organoselenium compounds inhibit histone deacetylase activity in human colon cancer cells. Carcinogenesis 2009, 30, 1416–1423. [Google Scholar] [CrossRef]
- Suzuki, T.; Muto, N.; Bando, M.; Itoh, Y.; Masaki, A.; Ri, M.; Ota, Y.; Nakagawa, H.; Iida, S.; Shirahige, K.; et al. Design, Synthesis, and Biological Activity of NCC149 Derivatives as Histone Deacetylase 8-Selective Inhibitors. ChemMedChem 2014, 9, 657–664. [Google Scholar] [CrossRef]
- Rettig, I.; Koeneke, E.; Trippel, F.; Mueller, W.C.; Burhenne, J.; Kopp-Schneider, A.; Fabian, J.; Schober, A.; Fernekorn, U.; von Deimling, A.; et al. Selective inhibition of HDAC8 decreases neuroblastoma growth in vitro and in vivo and enhances retinoic acid-mediated differentiation. Cell Death Dis. 2015, 6, e1657. [Google Scholar] [CrossRef]
- Du, Y.; Yuan, Y.; Xu, L.; Zhao, F.; Wang, W.; Xu, Y.; Tian, X. Discovery of METTL3 Small Molecule Inhibitors by Virtual Screening of Natural Products. Front Pharmacol 2022, 13, 878135. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Zhao, R.; Meng, W.; Liao, Y. Effects and translatomics characteristics of a small-molecule inhibitor of METTL3 against non-small cell lung cancer. J. Pharm. Anal. 2023, 13, 625–639. [Google Scholar] [CrossRef] [PubMed]
- Barsyte-Lovejoy, D.; Li, F.; Oudhoff, M.J.; Tatlock, J.H.; Dong, A.; Zeng, H.; Wu, H.; Freeman, S.A.; Schapira, M.; Senisterra, G.A.; et al. (R)-PFI-2 is a potent and selective inhibitor of SETD7 methyltransferase activity in cells. Proc. Natl. Acad. Sci. USA 2014, 111, 12853–12858. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.K.; Pardoux, C.; Hall, M.C.; Lee, P.S.; Warburton, D.; Qing, J.; Smith, S.M.; Derynck, R. TGF-beta activates Erk MAP kinase signalling through direct phosphorylation of ShcA. Embo J. 2007, 26, 3957–3967. [Google Scholar] [CrossRef] [PubMed]
- Zuo, W.; Chen, Y.G. Specific activation of mitogen-activated protein kinase by transforming growth factor-beta receptors in lipid rafts is required for epithelial cell plasticity. Mol. Biol. Cell 2009, 20, 1020–1029. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, A.; Thakur, N.; Grimsby, S.; Marcusson, A.; von Bulow, V.; Schuster, N.; Zhang, S.; Heldin, C.-H.; Landström, M. The type I TGF-β receptor engages TRAF6 to activate TAK1 in a receptor kinase-independent manner. Nat. Cell Biol. 2008, 10, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, M.; Fatyol, K.; Jin, C.; Wang, X.; Liu, Z.; Zhang, Y.E. TRAF6 mediates Smad-independent activation of JNK and p38 by TGF-beta. Mol. Cell 2008, 31, 918–924. [Google Scholar] [CrossRef] [PubMed]
- Yumoto, K.; Thomas, P.S.; Lane, J.; Matsuzaki, K.; Inagaki, M.; Ninomiya-Tsuji, J.; Scott, G.J.; Ray, M.K.; Ishii, M.; Maxson, R.; et al. TGF-β-activated kinase 1 (Tak1) mediates agonist-induced Smad activation and linker region phosphorylation in embryonic craniofacial neural crest-derived cells. J. Biol. Chem. 2013, 288, 13467–13480. [Google Scholar] [CrossRef]
- Edlund, S.; Bu, S.; Schuster, N.; Aspenström, P.; Heuchel, R.; Heldin, N.E.; ten Dijke, P.; Heldin, C.H.; Landström, M. Transforming growth factor-beta1 (TGF-beta)-induced apoptosis of prostate cancer cells involves Smad7-dependent activation of p38 by TGF-beta-activated kinase 1 and mitogen-activated protein kinase kinase 3. Mol. Biol. Cell 2003, 14, 529–544. [Google Scholar] [CrossRef]
- Jung, S.M.; Lee, J.H.; Park, J.; Oh, Y.S.; Lee, S.K.; Park, J.S.; Lee, Y.S.; Kim, J.H.; Lee, J.Y.; Bae, Y.S.; et al. Smad6 inhibits non-canonical TGF-β1 signaling by recruiting the deubiquitinase A20 to TRAF6. Nat. Commun. 2013, 4, 2562. [Google Scholar] [CrossRef]
- Zhang, L.; Zhou, F.; García de Vinuesa, A.; de Kruijf, E.M.; Mesker, W.E.; Hui, L.; Drabsch, Y.; Li, Y.; Bauer, A.; Rousseau, A.; et al. TRAF4 Promotes TGF-β Receptor Signaling and Drives Breast Cancer Metastasis. Mol. Cell 2013, 51, 559–572. [Google Scholar] [CrossRef] [PubMed]
- Owen, K.L.; Brockwell, N.K.; Parker, B.S. JAK-STAT Signaling: A Double-Edged Sword of Immune Regulation and Cancer Progression. Cancers 2019, 11, 2002. [Google Scholar] [CrossRef]
- Tang, L.Y.; Heller, M.; Meng, Z.; Yu, L.R.; Tang, Y.; Zhou, M.; Zhang, Y.E. Transforming Growth Factor-β (TGF-β) Directly Activates the JAK1-STAT3 Axis to Induce Hepatic Fibrosis in Coordination with the SMAD Pathway. J. Biol. Chem. 2017, 292, 4302–4312. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.Y.; Shin, I.; Arteaga, C.L. Type I Transforming Growth Factor β Receptor Binds to and Activates Phosphatidylinositol 3-Kinase*. J. Biol. Chem. 2005, 280, 10870–10876. [Google Scholar] [CrossRef] [PubMed]
- Hamidi, A.; Song, J.; Thakur, N.; Itoh, S.; Marcusson, A.; Bergh, A.; Heldin, C.H.; Landström, M. TGF-β promotes PI3K-AKT signaling and prostate cancer cell migration through the TRAF6-mediated ubiquitylation of p85α. Sci. Signal 2017, 10, aal4186. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Connolly, E.; Smyth, J.W.; Akhurst, R.J.; Derynck, R. TGF-β-induced activation of mTOR complex 2 drives epithelial-mesenchymal transition and cell invasion. J. Cell Sci. 2012, 125 Pt 5, 1259–1273. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Derynck, R. Cell size and invasion in TGF-beta-induced epithelial to mesenchymal transition is regulated by activation of the mTOR pathway. J. Cell Biol. 2007, 178, 437–451. [Google Scholar] [CrossRef]
- Mu, Y.; Sundar, R.; Thakur, N.; Ekman, M.; Gudey, S.K.; Yakymovych, M.; Hermansson, A.; Dimitriou, H.; Bengoechea-Alonso, M.T.; Ericsson, J.; et al. TRAF6 ubiquitinates TGFβ type I receptor to promote its cleavage and nuclear translocation in cancer. Nat. Commun. 2011, 2, 330. [Google Scholar] [CrossRef]
- Gudey, S.K.; Sundar, R.; Mu, Y.; Wallenius, A.; Zang, G.; Bergh, A.; Heldin, C.H.; Landström, M. TRAF6 stimulates the tumor-promoting effects of TGFβ type I receptor through polyubiquitination and activation of presenilin 1. Sci. Signal 2014, 7, ra2. [Google Scholar] [CrossRef]
- Sundar, R.; Gudey, S.K.; Heldin, C.-H.; Landström, M. TRAF6 promotes TGFβ-induced invasion and cell-cycle regulation via Lys63-linked polyubiquitination of Lys178 in TGFβ type I receptor. Cell Cycle 2015, 14, 554–565. [Google Scholar] [CrossRef]
- Bhowmick, N.A.; Ghiassi, M.; Bakin, A.; Aakre, M.; Lundquist, C.A.; Engel, M.E.; Arteaga, C.L.; Moses, H.L. Transforming growth factor-beta1 mediates epithelial to mesenchymal transdifferentiation through a RhoA-dependent mechanism. Mol. Biol. Cell 2001, 12, 27–36. [Google Scholar] [CrossRef]
- Edlund, S.; Landström, M.; Heldin, C.-H.; Aspenström, P. Transforming Growth Factor-β–induced Mobilization of Actin Cytoskeleton Requires Signaling by Small GTPases Cdc42 and RhoA. Mol. Biol. Cell 2002, 13, 902–914. [Google Scholar] [CrossRef] [PubMed]
- Ozdamar, B.; Bose, R.; Barrios-Rodiles, M.; Wang, H.-R.; Zhang, Y.; Wrana, J.L. Regulation of the Polarity Protein Par6 by TGFß Receptors Controls Epithelial Cell Plasticity. Science 2005, 307, 1603–1609. [Google Scholar] [CrossRef] [PubMed]
- Wilkes, M.C.; Murphy, S.J.; Garamszegi, N.; Leof, E.B. Cell-Type-Specific Activation of PAK2 by Transforming Growth Factor β Independent of Smad2 and Smad3. Mol. Cell. Biol. 2003, 23, 8878–8889. [Google Scholar] [CrossRef] [PubMed]
- Ferrigno, O.; Lallemand, F.; Verrecchia, F.; L’Hoste, S.; Camonis, J.; Atfi, A.; Mauviel, A. Yes-associated protein (YAP65) interacts with Smad7 and potentiates its inhibitory activity against TGF-β/Smad signaling. Oncogene 2002, 21, 4879–4884. [Google Scholar] [CrossRef] [PubMed]
- Alarcón, C.; Zaromytidou, A.-I.; Xi, Q.; Gao, S.; Yu, J.; Fujisawa, S.; Barlas, A.; Miller, A.N.; Manova-Todorova, K.; Macias, M.J.; et al. Nuclear CDKs Drive Smad Transcriptional Activation and Turnover in BMP and TGF-β Pathways. Cell 2009, 139, 757–769. [Google Scholar] [CrossRef] [PubMed]
- Nallet-Staub, F.; Yin, X.; Gilbert, C.; Marsaud, V.; Ben Mimoun, S.; Javelaud, D.; Leof, E.B.; Mauviel, A. Cell Density Sensing Alters TGF-β Signaling in a Cell-Type-Specific Manner, Independent from Hippo Pathway Activation. Dev. Cell 2015, 32, 640–651. [Google Scholar] [CrossRef] [PubMed]
- Szeto, S.G.; Narimatsu, M.; Lu, M.; He, X.; Sidiqi, A.M.; Tolosa, M.F.; Chan, L.; De Freitas, K.; Bialik, J.F.; Majumder, S.; et al. YAP/TAZ Are Mechanoregulators of TGF-β-Smad Signaling and Renal Fibrogenesis. J. Am. Soc. Nephrol. 2016, 27, 3117–3128. [Google Scholar] [CrossRef]
- Lee, D.-H.; Park, J.O.; Kim, T.-S.; Kim, S.-K.; Kim, T.-h.; Kim, M.-c.; Park, G.S.; Kim, J.-H.; Kuninaka, S.; Olson, E.N.; et al. LATS-YAP/TAZ controls lineage specification by regulating TGFβ signaling and Hnf4α expression during liver development. Nat. Commun. 2016, 7, 11961. [Google Scholar] [CrossRef]
- Wang, L.; Zhu, Y.; Sharma, K. Transforming Growth Factor-β1 Stimulates Protein Kinase A in Mesangial Cells*. J. Biol. Chem. 1998, 273, 8522–8527. [Google Scholar] [CrossRef]
- Yang, H.; Li, G.; Wu, J.J.; Wang, L.; Uhler, M.; Simeone, D.M. Protein kinase A modulates transforming growth factor-β signaling through a direct interaction with Smad4 protein. J. Biol. Chem. 2013, 288, 8737–8749. [Google Scholar] [CrossRef]
- Felici, A.; Wurthner, J.U.; Parks, W.T.; Ruh-yu Giam, L.; Reiss, M.; Karpova, T.S.; McNally, J.G.; Roberts, A.B. TLP, a novel modulator of TGF-β signaling, has opposite effects on Smad2- and Smad3-dependent signaling. EMBO J. 2003, 22, 4465–4477. [Google Scholar] [CrossRef]
- Lin, Y.; Zhang, B.; Liang, H.; Lu, Y.; Ai, X.; Zhang, B.; Chen, X. JNK inhibitor SP600125 enhances TGF-β-induced apoptosis of RBE human cholangiocarcinoma cells in a Smad-dependent manner. Mol. Med. Rep. 2013, 8, 1623–1629. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.Y.; Chen, T.S.; Wang, X.P.; Qu, J.L.; Chen, M. The JNK inhibitor SP600125 enhances dihydroartemisinin-induced apoptosis by accelerating Bax translocation into mitochondria in human lung adenocarcinoma cells. FEBS Lett. 2010, 584, 4019–4026. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, Y.; Chen, L.; Yu, F.; Li, X.; Dan, T.; Zhao, J.; Zhou, S. Polyphyllin I induces G2/M phase arrest and apoptosis in U251 human glioma cells via mitochondrial dysfunction and the JNK signaling pathway. Acta Biochim. Biophys. Sin. 2017, 49, 479–486. [Google Scholar] [CrossRef]
- Cerbone, A.; Toaldo, C.; Pizzimenti, S.; Pettazzoni, P.; Dianzani, C.; Minelli, R.; Ciamporcero, E.; Roma, G.; Dianzani, M.U.; Canaparo, R.; et al. AS601245, an Anti-Inflammatory JNK Inhibitor, and Clofibrate Have a Synergistic Effect in Inducing Cell Responses and in Affecting the Gene Expression Profile in CaCo-2 Colon Cancer Cells. PPAR Res. 2012, 2012, 269751. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Yang, X.; Kang, Q.; Lu, J.; Denzinger, M.; Kornmann, M.; Traub, B. JNK inhibitor IX restrains pancreatic cancer through p53 and p21. Front. Oncol. 2022, 12, 1006131. [Google Scholar] [CrossRef]
- Ebelt, N.D.; Kaoud, T.S.; Edupuganti, R.; Van Ravenstein, S.; Dalby, K.N.; Van, C.L. A c-Jun N-terminal kinase inhibitor, JNK-IN-8, sensitizes triple negative breast cancer cells to lapatinib. Oncotarget 2017, 8, 104894. [Google Scholar] [CrossRef] [PubMed]
- Campbell, R.M.; Anderson, B.D.; Brooks, N.A.; Brooks, H.B.; Chan, E.M.; De Dios, A.; Gilmour, R.; Graff, J.R.; Jambrina, E.; Mader, M.; et al. Characterization of LY2228820 Dimesylate, a Potent and Selective Inhibitor of p38 MAPK with Antitumor Activity. Mol. Cancer Ther. 2014, 13, 364–374. [Google Scholar] [CrossRef]
- Jin, X.; Mo, Q.; Zhang, Y.; Gao, Y.; Wu, Y.; Li, J.; Hao, X.; Ma, D.; Gao, Q.; Chen, P. The p38 MAPK inhibitor BIRB796 enhances the antitumor effects of VX680 in cervical cancer. Cancer Biol. Ther. 2016, 17, 566–576. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Wang, Y.; Xu, Y.; Sun, Q.; Liu, H.; Chen, Q.; Liu, B. BIRB796, an Inhibitor of p38 Mitogen-Activated Protein Kinase, Inhibits Proliferation and Invasion in Glioblastoma Cells. ACS Omega 2021, 6, 11466–11473. [Google Scholar] [CrossRef]
- Kim, M.N.; Lee, S.M.; Kim, J.S.; Hwang, S.G. Preclinical efficacy of a novel dual PI3K/mTOR inhibitor, CMG002, alone and in combination with sorafenib in hepatocellular carcinoma. Cancer Chemother. Pharmacol. 2019, 84, 809–817. [Google Scholar] [CrossRef] [PubMed]
- Liao, G.; Gao, B.; Gao, Y.; Yang, X.; Cheng, X.; Ou, Y. Phycocyanin Inhibits Tumorigenic Potential of Pancreatic Cancer Cells: Role of Apoptosis and Autophagy. Sci. Rep. 2016, 6, 34564. [Google Scholar] [CrossRef] [PubMed]
- Rafiq, R.A.; Quadri, A.; Nazir, L.A.; Peerzada, K.; Ganai, B.A.; Tasduq, S.A. A Potent Inhibitor of Phosphoinositide 3-Kinase (PI3K) and Mitogen Activated Protein (MAP) Kinase Signalling, Quercetin (3, 3′, 4′, 5, 7-Pentahydroxyflavone) Promotes Cell Death in Ultraviolet (UV)-B-Irradiated B16F10 Melanoma Cells. PLoS ONE 2015, 10, e0131253. [Google Scholar] [CrossRef] [PubMed]
- Raina, K.; Agarwal, C.; Wadhwa, R.; Serkova, N.J.; Agarwal, R. Energy deprivation by silibinin in colorectal cancer cells. Autophagy 2013, 9, 697–713. [Google Scholar] [CrossRef] [PubMed]
- Nanta, R.; Shrivastava, A.; Sharma, J.; Shankar, S.; Srivastava, R.K. Inhibition of sonic hedgehog and PI3K/Akt/mTOR pathways cooperate in suppressing survival, self-renewal and tumorigenic potential of glioblastoma-initiating cells. Mol. Cell. Biochem. 2019, 454, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Li, C.F.; Chen, J.Y.; Ho, Y.H.; Hsu, W.H.; Wu, L.C.; Lan, H.Y.; Hsu, D.S.; Tai, S.K.; Chang, Y.C.; Yang, M.H. Snail-induced claudin-11 prompts collective migration for tumour progression. Nat. Cell Biol. 2019, 21, 251–262. [Google Scholar] [CrossRef]
- Qi, Y.; Zhao, X.; Chen, J.; Pradipta, A.R.; Wei, J.; Ruan, H.; Zhou, L.; Hsung, R.P.; Tanaka, K. In vitro and in vivo cancer cell apoptosis triggered by competitive binding of Cinchona alkaloids to the RING domain of TRAF6. Biosci. Biotechnol. Biochem. 2019, 83, 1011–1026. [Google Scholar] [CrossRef]
- Shang, X.; Marchioni, F.; Sipes, N.; Evelyn, C.R.; Jerabek-Willemsen, M.; Duhr, S.; Seibel, W.; Wortman, M.; Zheng, Y. Rational Design of Small Molecule Inhibitors Targeting RhoA Subfamily Rho GTPases. Chem. Biol. 2012, 19, 699–710. [Google Scholar] [CrossRef]
- Shang, X.; Marchioni, F.; Evelyn, C.R.; Sipes, N.; Zhou, X.; Seibel, W.; Wortman, M.; Zheng, Y. Small-molecule inhibitors targeting G-protein-coupled Rho guanine nucleotide exchange factors. Proc. Natl. Acad. Sci. USA 2013, 110, 3155–3160. [Google Scholar] [CrossRef]
- Evelyn, C.R.; Wade, S.M.; Wang, Q.; Wu, M.; Iñiguez-Lluhí, J.A.; Merajver, S.D.; Neubig, R.R. CCG-1423: A small-molecule inhibitor of RhoA transcriptional signaling. Mol. Cancer Ther. 2007, 6, 2249–2260. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, K.I.; Watanabe, B.; Nakagawa, Y.; Minami, S.; Morita, T. RPEL Proteins Are the Molecular Targets for CCG-1423, an Inhibitor of Rho Signaling. PLoS ONE 2014, 9, e89016. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.C.; Huang, T.H.; Chang, C.S.; Tsai, Y.R.; Lin, R.H.; Lee, P.W.; Hsu, M.F.; Huang, L.J.; Wang, J.P. Signaling mechanisms of inhibition of phospholipase D activation by CHS-111 in formyl peptide-stimulated neutrophils. Biochem. Pharmacol. 2011, 81, 269–278. [Google Scholar] [CrossRef]
- Hoy, J.J.; Salinas Parra, N.; Park, J.; Kuhn, S.; Iglesias-Bartolome, R. Protein kinase A inhibitor proteins (PKIs) divert GPCR-Gαs-cAMP signaling toward EPAC and ERK activation and are involved in tumor growth. Faseb J. 2020, 34, 13900–13917. [Google Scholar] [CrossRef] [PubMed]
- Zynda, E.R.; Matveev, V.; Makhanov, M.; Chenchik, A.; Kandel, E.S. Protein kinase A type II-α regulatory subunit regulates the response of prostate cancer cells to taxane treatment. Cell Cycle 2014, 13, 3292–3301. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cassoni, P.; Sapino, A.; Fortunati, N.; Munaron, L.; Chini, B.; Bussolati, G. Oxytocin inhibits the proliferation of MDA-MB231 human breast-cancer cells via cyclic adenosine monophosphate and protein kinase A. Int. J. Cancer 1997, 72, 340–344. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, W.E.; Zhang, X.; Li, Y.; Chen, B.; Liu, C.; Ai, X.; Zhang, X.; Tian, Y.; Zhang, C.; et al. Cardiomyocyte PKA Ablation Enhances Basal Contractility While Eliminates Cardiac β-Adrenergic Response Without Adverse Effects on the Heart. Circ. Res. 2019, 124, 1760–1777. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Hong, W.; Wei, X. The molecular mechanisms and therapeutic strategies of EMT in tumor progression and metastasis. J. Hematol. Oncol. 2022, 15, 129. [Google Scholar] [CrossRef]
- Jan, R.; Chaudhry, G.E. Understanding Apoptosis and Apoptotic Pathways Targeted Cancer Therapeutics. Adv. Pharm. Bull. 2019, 9, 205–218. [Google Scholar] [CrossRef]
- Ramesh, S.; Wildey, G.M.; Howe, P.H. Transforming growth factor beta (TGFbeta)-induced apoptosis: The rise & fall of Bim. Cell Cycle 2009, 8, 11–17. [Google Scholar] [CrossRef]
- Zhang, Y.; Alexander, P.B.; Wang, X.F. TGF-β Family Signaling in the Control of Cell Proliferation and Survival. Cold Spring Harb. Perspect. Biol. 2017, 9, a022145. [Google Scholar] [CrossRef]
- Meulmeester, E.; ten Dijke, P. The dynamic roles of TGF-β in cancer. J. Pathol. 2011, 223, 206–219. [Google Scholar] [CrossRef]
- Yu, L.; Hébert, M.C.; Zhang, Y.E. TGF-beta receptor-activated p38 MAP kinase mediates Smad-independent TGF-beta responses. EMBO J. 2002, 21, 3749–3759. [Google Scholar] [CrossRef]
- Chen, B.; Mu, C.; Zhang, Z.; He, X.; Liu, X. The Love-Hate Relationship Between TGF-β Signaling and the Immune System During Development and Tumorigenesis. Front. Immunol. 2022, 13, 891268. [Google Scholar] [CrossRef]
- Schrantz, N.; Auffredou, M.T.; Bourgeade, M.F.; Besnault, L.; Leca, G.; Vazquez, A. Zinc-mediated regulation of caspases activity: Dose-dependent inhibition or activation of caspase-3 in the human Burkitt lymphoma B cells (Ramos). Cell Death Differ. 2001, 8, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Hu, Y.; Fan, X.; Wu, X.; Mao, Y.; Hu, B.; Guo, H.; Wen, L.; Tang, F. Single-cell RNA-seq analysis unveils a prevalent epithelial/mesenchymal hybrid state during mouse organogenesis. Genome Biol. 2018, 19, 31. [Google Scholar] [CrossRef]
- Puram, S.V.; Tirosh, I.; Parikh, A.S.; Patel, A.P.; Yizhak, K.; Gillespie, S.; Rodman, C.; Luo, C.L.; Mroz, E.A.; Emerick, K.S.; et al. Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell 2017, 171, 1611–1624.e1624. [Google Scholar] [CrossRef]
- Aiello, N.M.; Maddipati, R.; Norgard, R.J.; Balli, D.; Li, J.; Yuan, S.; Yamazoe, T.; Black, T.; Sahmoud, A.; Furth, E.E.; et al. EMT Subtype Influences Epithelial Plasticity and Mode of Cell Migration. Dev. Cell 2018, 45, 681–695.e684. [Google Scholar] [CrossRef] [PubMed]
- Medici, D.; Kalluri, R. Endothelial–mesenchymal transition and its contribution to the emergence of stem cell phenotype. Semin. Cancer Biol. 2012, 22, 379–384. [Google Scholar] [CrossRef] [PubMed]
- Piera-Velazquez, S.; Jimenez, S.A. Endothelial to Mesenchymal Transition: Role in Physiology and in the Pathogenesis of Human Diseases. Physiol. Rev. 2019, 99, 1281–1324. [Google Scholar] [CrossRef]
- Derynck, R.; Weinberg, R.A. EMT and Cancer: More Than Meets the Eye. Dev. Cell 2019, 49, 313–316. [Google Scholar] [CrossRef]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef]
- Kajita, M.; McClinic, K.N.; Wade, P.A. Aberrant expression of the transcription factors snail and slug alters the response to genotoxic stress. Mol. Cell Biol. 2004, 24, 7559–7566. [Google Scholar] [CrossRef] [PubMed]
- Qiao, B.; Johnson, N.W.; Gao, J. Epithelial-mesenchymal transition in oral squamous cell carcinoma triggered by transforming growth factor-beta1 is Snail family-dependent and correlates with matrix metalloproteinase-2 and -9 expressions. Int. J. Oncol. 2010, 37, 663–668. [Google Scholar] [CrossRef]
- Li, W.; Shen, M.; Jiang, Y.Z.; Zhang, R.; Zheng, H.; Wei, Y.; Shao, Z.M.; Kang, Y. Deubiquitinase USP20 promotes breast cancer metastasis by stabilizing SNAI2. Genes Dev. 2020, 34, 1310–1315. [Google Scholar] [CrossRef]
- Fan, L.; Lei, H.; Zhang, S.; Peng, Y.; Fu, C.; Shu, G.; Yin, G. Non-canonical signaling pathway of SNAI2 induces EMT in ovarian cancer cells by suppressing miR-222-3p transcription and upregulating PDCD10. Theranostics 2020, 10, 5895–5913. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Gross, K.M.; Kuperwasser, C. Molecular regulation of Snail2 in development and disease. J. Cell Sci. 2019, 132, 235127. [Google Scholar] [CrossRef]
- Chaffer, C.L.; Marjanovic, N.D.; Lee, T.; Bell, G.; Kleer, C.G.; Reinhardt, F.; D’Alessio, A.C.; Young, R.A.; Weinberg, R.A. Poised Chromatin at the ZEB1 Promoter Enables Breast Cancer Cell Plasticity and Enhances Tumorigenicity. Cell 2013, 154, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Krebs, A.M.; Mitschke, J.; Lasierra Losada, M.; Schmalhofer, O.; Boerries, M.; Busch, H.; Boettcher, M.; Mougiakakos, D.; Reichardt, W.; Bronsert, P.; et al. The EMT-activator Zeb1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer. Nat. Cell Biol. 2017, 19, 518–529. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Chen, B.; Zhu, Z.; Ye, W.; Zeng, J.; Liu, G.; Wang, S.; Gao, J.; Xu, G.; Huang, Z. Prognostic value of ZEB-1 in solid tumors: A meta-analysis. BMC Cancer 2019, 19, 635. [Google Scholar] [CrossRef]
- Zhang, G.J.; Zhou, T.; Tian, H.P.; Liu, Z.L.; Xia, S.S. High expression of ZEB1 correlates with liver metastasis and poor prognosis in colorectal cancer. Oncol. Lett. 2013, 5, 564–568. [Google Scholar] [CrossRef] [PubMed]
- Jørgensen, C.L.T.; Forsare, C.; Bendahl, P.O.; Falck, A.K.; Fernö, M.; Lövgren, K.; Aaltonen, K.; Rydén, L. Expression of epithelial-mesenchymal transition-related markers and phenotypes during breast cancer progression. Breast Cancer Res. Treat. 2020, 181, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Kurahara, H.; Takao, S.; Maemura, K.; Mataki, Y.; Kuwahata, T.; Maeda, K.; Ding, Q.; Sakoda, M.; Iino, S.; Ishigami, S.; et al. Epithelial–mesenchymal transition and mesenchymal–epithelial transition via regulation of ZEB-1 and ZEB-2 expression in pancreatic cancer. J. Surg. Oncol. 2012, 105, 655–661. [Google Scholar] [CrossRef]
- Yang, M.H.; Wu, M.Z.; Chiou, S.H.; Chen, P.M.; Chang, S.Y.; Liu, C.J.; Teng, S.C.; Wu, K.J. Direct regulation of TWIST by HIF-1alpha promotes metastasis. Nat. Cell Biol. 2008, 10, 295–305. [Google Scholar] [CrossRef]
- Meng, J.; Chen, S.; Han, J.X.; Qian, B.; Wang, X.R.; Zhong, W.L.; Qin, Y.; Zhang, H.; Gao, W.F.; Lei, Y.Y.; et al. Twist1 Regulates Vimentin through Cul2 Circular RNA to Promote EMT in Hepatocellular Carcinoma. Cancer Res. 2018, 78, 4150–4162. [Google Scholar] [CrossRef]
- Yang, J.; Mani, S.A.; Donaher, J.L.; Ramaswamy, S.; Itzykson, R.A.; Come, C.; Savagner, P.; Gitelman, I.; Richardson, A.; Weinberg, R.A. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 2004, 117, 927–939. [Google Scholar] [CrossRef]
- Lee, T.K.; Poon, R.T.; Yuen, A.P.; Ling, M.T.; Kwok, W.K.; Wang, X.H.; Wong, Y.C.; Guan, X.Y.; Man, K.; Chau, K.L.; et al. Twist overexpression correlates with hepatocellular carcinoma metastasis through induction of epithelial-mesenchymal transition. Clin. Cancer Res. 2006, 12, 5369–5376. [Google Scholar] [CrossRef]
- Pattabiraman, D.R.; Weinberg, R.A. Tackling the cancer stem cells—What challenges do they pose? Nat. Rev. Drug Discov. 2014, 13, 497–512. [Google Scholar] [CrossRef]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Scheel, C.; Eaton, E.N.; Li, S.H.; Chaffer, C.L.; Reinhardt, F.; Kah, K.J.; Bell, G.; Guo, W.; Rubin, J.; Richardson, A.L.; et al. Paracrine and autocrine signals induce and maintain mesenchymal and stem cell states in the breast. Cell 2011, 145, 926–940. [Google Scholar] [CrossRef]
- McCormack, N.; O’Dea, S. Regulation of epithelial to mesenchymal transition by bone morphogenetic proteins. Cell Signal. 2013, 25, 2856–2862. [Google Scholar] [CrossRef] [PubMed]
- Giampieri, S.; Manning, C.; Hooper, S.; Jones, L.; Hill, C.S.; Sahai, E. Localized and reversible TGFbeta signalling switches breast cancer cells from cohesive to single cell motility. Nat. Cell Biol. 2009, 11, 1287–1296. [Google Scholar] [CrossRef] [PubMed]
- Katsuno, Y.; Derynck, R. Epithelial plasticity, epithelial-mesenchymal transition, and the TGF-β family. Dev. Cell 2021, 56, 726–746. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Turley, S.J.; Akhurst, R.J. TGFβ biology in cancer progression and immunotherapy. Nat. Rev. Clin. Oncol. 2021, 18, 9–34. [Google Scholar] [CrossRef] [PubMed]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef]
- Yu, M.; Bardia, A.; Wittner, B.S.; Stott, S.L.; Smas, M.E.; Ting, D.T.; Isakoff, S.J.; Ciciliano, J.C.; Wells, M.N.; Shah, A.M.; et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 2013, 339, 580–584. [Google Scholar] [CrossRef]
- Cheung, K.J.; Gabrielson, E.; Werb, Z.; Ewald, A.J. Collective invasion in breast cancer requires a conserved basal epithelial program. Cell 2013, 155, 1639–1651. [Google Scholar] [CrossRef]
- Katsuno, Y.; Meyer, D.S.; Zhang, Z.; Shokat, K.M.; Akhurst, R.J.; Miyazono, K.; Derynck, R. Chronic TGF-β exposure drives stabilized EMT, tumor stemness, and cancer drug resistance with vulnerability to bitopic mTOR inhibition. Sci. Signal 2019, 12, aau8544. [Google Scholar] [CrossRef] [PubMed]
- Ruscetti, M.; Quach, B.; Dadashian, E.L.; Mulholland, D.J.; Wu, H. Tracking and Functional Characterization of Epithelial-Mesenchymal Transition and Mesenchymal Tumor Cells during Prostate Cancer Metastasis. Cancer Res. 2015, 75, 2749–2759. [Google Scholar] [CrossRef]
- Fares, J.; Fares, M.Y.; Khachfe, H.H.; Salhab, H.A.; Fares, Y. Molecular principles of metastasis: A hallmark of cancer revisited. Signal Transduct. Target. Ther. 2020, 5, 28. [Google Scholar] [CrossRef]
- Gomis, R.R.; Gawrzak, S. Tumor cell dormancy. Mol. Oncol. 2017, 11, 62–78. [Google Scholar] [CrossRef]
- Yeh, A.C.; Ramaswamy, S. Mechanisms of Cancer Cell Dormancy--Another Hallmark of Cancer? Cancer Res. 2015, 75, 5014–5022. [Google Scholar] [CrossRef] [PubMed]
- Harper, K.L.; Sosa, M.S.; Entenberg, D.; Hosseini, H.; Cheung, J.F.; Nobre, R.; Avivar-Valderas, A.; Nagi, C.; Girnius, N.; Davis, R.J.; et al. Mechanism of early dissemination and metastasis in Her2+ mammary cancer. Nature 2016, 540, 588–592. [Google Scholar] [CrossRef] [PubMed]
- Rhim, A.D.; Mirek, E.T.; Aiello, N.M.; Maitra, A.; Bailey, J.M.; McAllister, F.; Reichert, M.; Beatty, G.L.; Rustgi, A.K.; Vonderheide, R.H.; et al. EMT and dissemination precede pancreatic tumor formation. Cell 2012, 148, 349–361. [Google Scholar] [CrossRef] [PubMed]







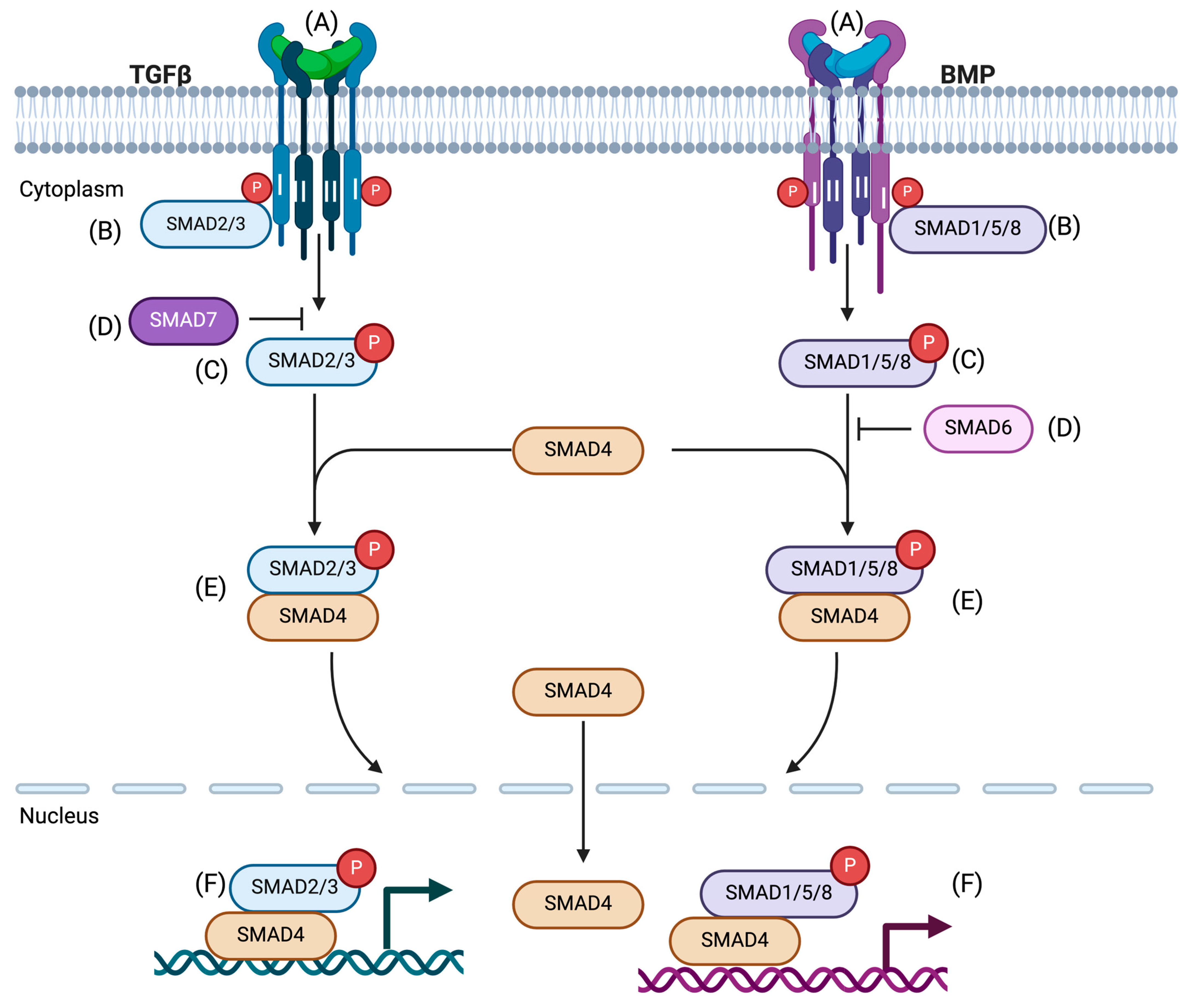{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Name of Inhibitors | Characteristics/Functions | Preclinical Study In Vitro (Cell Type) and In Vivo Models | Cancer Type | Reference |
|---|---|---|---|---|---|
| AKT/PKB (Se/Thr kinase/protein kinase B) | Naltrindole | A classic δ opioid antagonist that reduces cell growth and prompts apoptosis. | NCI-H69, NCI-H345, and NCI-H510 | Lung cancer | [69] |
| Small-molecule inhibitors of AKT (Aktis): Akti-1/2a, Akti-1, Akti-2, Akti1/2 | The inhibitors have pleckstrin homology domain-dependent, isozyme-specific activity. They sensitize tumor cells to apoptotic stimuli. | NCaP, MDA-MB468 A2780, BT474, HT29 | Breast cancer | [70] | |
| GDC-0068 | ATP-competitive AKT inhibitor. GDC-0068 inhibits AKT functions, resulting in inhibition of cell cycle progress and cancer cell viability. | MCF7-neo/Her2, BT47M1, PC-3 In vivo broad-spectrum human cancer xenograft models | Multiple solid tumors | [71] | |
| MK2206 | An allosteric AKT inhibitor. MK2206 and gemcitabine inhibit AKT phosphorylation and reduce viability of pancreatic cancer cells. | PANC1, Mia PaCa-2, BxPC-3, AsPC-1, SW1990 | Pancreatic cancer | [72] | |
| MK2206 | MK2206 decreases cell proliferation and stemness capacity to form colon spheres and initiate tumor formation. | Human CRC cell line HCT-116 In vivo mouse xenograft | Colorectal cancer | [73] | |
| CDKS (cycline-dependent kinase) | Nitazoxanide (NTZ) | TIZ constrains CDK1 phosphorylation at Thr161 and decreases CDK1/cyclin B1 complex function. TIZ induces the cell cycle arrest at the G2/M phase. In vivo, TIZ reduces the growth of subcutaneous and intracranial orthotopic xenograft models of glioblastoma. | U87, U118, and A172 human glioblastoma cells In vivo mouse xenograft model | Glioblastoma | [74] |
| Adapalene (ADA) synthetic retinoid | ADA inhibits cancer cell proliferation, migration, and invasion. ADA reduces tumor growth and bone damage. | RM-1 prostate cancer cell line | Prostate cancer | [75] | |
| Adapalene (ADA) and combination of PI3K inhibitor (GDC-0941) | The combination of inhibitors showed synergistic effect. Reactive oxygen species accumulation from ADA and GDC caused apoptosis and enhanced sensitivity to GDC in TNBC. | Breast cancer cell lines: MDA-MB-231, MDA-MB-468 MCF-7 | Triple-negative breast cancer (TNBC) | [76] | |
| CDKs (2/4) | CDK2i, CDK4i, | Phosphorylation and gene reporter functions of SMAD2 and SMAD3 are reduced by CDK inhibitors with TGF-β stimulation. | MDA-MB-231, MDA-MB-436, and Hs578T cell lines | Breast cancer | [77,78] |
| CDKs(2.4/6) | Vanoxerine dihydrochloride (CDK2/4/6 triple inhibitor) | Vanoxerine dihydrochloride arrests the cell cycle, induces apoptosis, and produces a synergistic cytotoxic effect in HCC cells. In vivo, tumor growth can be reduced in mouse models. | The human HCC cell lines QGY7703 and Huh7 In vivo mouse model | Hepatocellular carcinome | [79] |
| GSK3b axin/glycogen synthase kinase 3 beta | Tideglusib, AZD1080, BIO | The inhibitors target GSK3 and associated signaling pathways and modify the phosphorylation of GSK-3 substrates, such as T53 on c-MYC and S33/S37/T41 on β-catenin. Modulation of KRAS-dependent tumor growth is initiated by the inhibitors. | Human lung, colon pancreatic, and prostate cancer cell lines. Calu-6, A549, H460, PC9, and H4006 SW620, DLD-1, and HCT-8 MiaPaCa2 L3.6pl DU145 HEK293 Female athymic nude mice (human tumor xenograft) | Lung cancer Pancreatic cancer Prostate cancer | [80,81] |
| The glutamate release inhibitor, Riluzole | Pro-oncogenic function of SMADs is modulated by Riluzole. It increases linker region phosphorylation of SMAD2 and SMAD3 at serine clusters through GSK3. | Melanoma cell lines WM793, WM278, and 1205LU SiRNA GSK3α/β knock-down | Melanoma Cancer | [82] | |
| ERK (extracellular signal-regulated kinase) | INR119 | INR119 inhibits MEK1/2 in cancer cells. It can induce ROS by ERK signaling with increased kinase activity. As a result, the proapoptotic genes (TP53, BAX) are highly expressed, resulting in apoptosis. | Human breast cancer cell MCF-7 | Breast cancer | [83] |
| Target | Name of Inhibitors | Characteristics/Functions | Preclinical Study In Vitro (Cell Type) and In Vivo Models | Cancer Type | Reference |
|---|---|---|---|---|---|
| Erk Mapk | MEK/ERK inhibitors | The inhibitor targets MEK/ERK by decreasing serine phosphorylation of SMAD2/3, except phosphorylation of the C-terminal motif. | Human mesangial cells Mouse mammary gland epithelial cells (NMuMGs) | [120] | |
| ERK1/2 | FR180204 | FR180204 affects cell proliferation, apoptosis, and migration in CRC cells with inhibition of MEK/ERK signaling. | HCT116, Caco-2 | Colorectal | [121] |
| Thienyl benzenesulfonate scaffold | Thienyl benzenesulfonate scaffold selectively inhibits ERK1/2 substrates that specifically have an F-site or docking site with DEF motif for ERK. It induces apoptosis in melanoma cells containing mutated Braf. | HeLa cervical cancer cell, leukemia cell; Jurkat T cell, melanoma cell; A375 and RPMI7951 | Melanoma | [122] | |
| BI-78D3 | BI-78D3 binds DRS (D-recruitment site) of ERK2 by making a covalent bond with a cysteine residue (C15.9) and disrupts TGF-β/SMAD signaling. It induces apoptosis in melanoma cells. | HEK293T, CRL-3216 melanoma cell line: A357, CRL-1619 | Melanoma | [123] | |
| DEL-22379 | DEL-22379 inhibits ERK dimerization without affecting its phosphorylation by RAS-ERK pathway, leading to apoptosis and preventing tumor progression. | Human cell lines Mutant BRAF RAS In vivo mouse tumor model | [124] | ||
| Piperine | Piperine reduces the phosphorylation of SMAD2 and ERK1/2 and shows an anti-EMT effect. | A549, MDA-MB-231, and HepG2 | Lung cancer | [125] | |
| PKC (protein kinase C) | Chelerythrine | A natural benzophenanthridine alkaloid. Targeting PKC shows a selective antiproliferative effect on TNBC cells. | MDA-MB-231, BT-549, HCC1937, MDA-MB-453, MDA-MB-468 MCF7, ZR-75, and SK-BR3 In vivo xenograft mouse model | Breast cancer | [126] |
| BD-15 | BD-15 enhances PKC signal and upregulates p21 expression and phosphorylation. | Lung cancer cell lines | Lung cancer | [127] | |
| Bisindolylmaleimide I, Gö6976, and Rottlerin | The inhibitors induce ROS-induced apoptosis in cancer cells in human colon cancer cells. | CCD18Co and Caco-2 colon adenocarcinoma cells, CCD18Co, normal colon cell | Colon cancer | [128] | |
| PKG1 (Protein kinase G) | SBA (sulindac benzylamine)—a novel sulindac derivative lacking cyclooxygenase (COX)-inhibitory activity) | SBA is known as cyclic guanosine 3′,5′, -monophosphate phosphodiesterase (cGMP PDE). SBA inhibits cGMP hydrolysis in colon tumor cells and activates cGMP-mediated PKG, which suppresses tumor cell growth. | HT-29, SW480, HCT116, and FHC (human fetal colonocytes) | Colon cancer | [129] |
| SSA (sulindac sulfide amide—lacks COX-inhibitory activity) | cGMP PDE inhibitor. SSA targets cGMP/PKG and inhibits b-catenin/Tcf transcriptional activity, resulting in apoptosis of breast cancer cells and mammary tumorigenesis in rats. | Hs578t, MCF-7, ZR-75, SKBr3, MDA-MB3-231 In vivo rat model | Breast cancer | [130] | |
| CK1 (Casein kinase 1) | SR3029 | SR3029 targets CK1d and upregulates deoxycytidine kinase (dCK). A combination of SR3029 with gemcitabine-induced synergistic antiproliferative and enhanced apoptosis. | Human PDA cell lines: BxPC-3, MIAPaCa-2, PANC1 Bladder cancer cell lines: UM-UC3, TCCSUP, 5637, HT-1376, J82, T24, Orthotopic pancreatic and bladder cancer model in mice | Pancreatic cancer Bladder cancer | [131] |
| IC261 | IC261 targets CK1(d/e) isoforms and influences colon cancer cell growth and apoptosis by increasing aerobic glycolysis through p53-dependent mechanism. | HCT116, RKO, LOVO, SW480 | Colon cancer | [132] | |
| PAK2 (p21 Activated Kinase | FRAX597 | FRAX597 is an ATP-competitive, which significantly reduces NF2-deficient Schwann cell growth in vitro and tumor in a xenograft model. | SC4 cells, Nf2−/−SC4 Schwann cells In vivo tumor model in mouse | Neurofibromatosis type 2 (NF2)-associated schwannomas | [133] |
| PAK4 | PF3758309 Small-molecule P21-activated kinase inhibitor | ATP-competitive pyrrolopyrazole inhibitor of PAK4-dependent pathway blocked multiple tumor xenografts. | 92 tumor cell lines Human xenograft tumor model | Breast cancer Pancreatic cancer Colorectal cancer Non-small-cell lung cancer | [134] |
| Compound 31 Compound55 | Compound 31 inhibits cell proliferation, migration, and invasion of tumor cells by modulating the PAK4-mediated signaling pathways. Potential in antitumor metastatic efficacy and mitigation of TGF-β1-induced epithelial–mesenchymal transition (EMT). | Lung cancer cell A549 and pharmacokinetic assessment in rats Lung cancer cell A549 and melanoma line B16 In vivo zebrafish embryo and mouse model | Lung cancer Lung cancer melanoma | [135,136] | |
| LKB1 | Sunitinib | Sunitinib is multitarget angiogenesis. It reduces tumor size and necrosis. Metastatic and nonmetastatic mouse models show an increase in median survival. | KW-634 KW-857 In vivo mouse model | Non-small-cell lung cancer | [137] |
| AZD8055/2-DG | A combined treatment of AZD8055/2-DG reduced mammary gland tumorigenesis by inhibiting mTOR pathways and glycolytic metabolism. | Primary, mammary epithelial cells, LKB1−/−NIC mice, and wild-type mice | Breast cancer Metastatic lung tumor | [138] | |
| B-RAF-V600E | Targets LBK-AMK RAF-MEK-ERK signaling, allows activation of AMK, and inhibits melanoma cell proliferation. | K-Mel-28, UACC62, UACC257, SK-Mel-31, and MeWo Cell | Melanoma cancer | [139] | |
| PPM1A/PP2CA Phosphatase | SAMP (small-molecule activators of SAMPs) SMAP-2 | SAMPs persistently inhibit MYC expression and MYC transcriptional activity. Cancer cell proliferation is inhibited in vitro. Tumor growth inhibition was observed in vivo. SMAP-2 decreases cellular viability, induces apoptosis, and reduces tumor growth. | Lung cancer cell line H441 Breast cancer cell line: BT-549, MDA-MB-453, MDA-MB-231, SUM149, and HCC1143 In vivo mouse xenograft LNCaP, 22Rv1 in vitro and in vivo mouse model | Non-small-cell lung cancer Breast cancer Castration-resistant prostate cancer | [140,141] |
| MicroRNA-487a-3p | MicroRNA-487a-3p binds directly with the 3′UTR of PPMIA phosphatase. It can effectively inhibit the expression of the phosphatase enzyme. | CAL-27, CA8113 | Oral squamous cell carcinoma | [142] | |
| PP2A DT-061 | A combination of PP2A DT-061 and MEK inhibitor AZD6224 suppresses p-AKT and MYC. Tumor growth in mouse mode was reduced by the action of the inhibitor. | Cell lines: A549, H460, H358, H441, and H2122 In vivo mouse model | Lung cancer | [143] | |
| SCP(1,2,3) | miRNA-26b | miRNA-26 b has antagonist effect of host gene SCP1. | Rat cardiomyocytes | Cardiac hypertrophy | [144] |
| Rabeprazole | Rabeprazole is Scp/TFIIF-interacting CTD phosphatase (Fcp/SCP) family. It binds to the hydrophobic binding pocket of SCPs, a proton pump inhibitor that specifically inhibits SCP1. It regulates irinotecan drug resistance topoisomerase 1 degradation. | Cell lines: HCT116, HT29, DLD1, LoVo. Patient study | Colorectal cancer Gastric cancer | [145,146] | |
| NEDD4-2/NEDD4L | Curcumin | Curcumin promotes glioma cell growth inhibition and induces apoptosis. Glioma cell proliferation, migration, and invasion were reduced with reduced expression of NEED4, Notch1, and pAKT. | SNB19 and A1207 | Glioma cancer | [147] |
| OSI906 | OSI906 targets NEDD4, leading to inhibition of gastric cancer cell proliferation dependent on IGF1/IGF1R signaling pathway. | Human GC cell line: BGC803, MKN45, SGC7901, MKN28 Xenograft nude mouse model Patient data | Gastric cancer | [148] | |
| Diosgenin | Diosgenin inhibits the expression of NEDD4, resulting in anti-tumor effects (inhibition of cell growth, cell cycle arrest, apoptosis, inhibition of cell migration and invasion) in prostate cancer. | PC-3 | Prostate cancer | [149] | |
| HSC70-interacting protein (CHIP) | PES(2-Phenylethylenesulfonamide) | PES selectively interacts with HSP70 and disrupts the interaction of many co-chaperons, substrate proteins, and multiple signaling pathways. This suppresses tumor growth in mouse models. | Transgenic Eμ-Myc mouse model of lymphomagenesis | Lymphogenesis | [150] |
| Pinaverium bromide | Pinaverium bromide inhibits the intracellular chaperon activity of HSP70 system and elicits cytotoxic activity by activating apoptosis in melanoma cells. | Tumorigenic melanoma cell lines: A2058 and MeWo | Melanoma | [151] | |
| SCF (Skp1, Cullin1 and Fbw1a)/ROC | 6-OAP | Binding of SKP1 and 6-OAP regulates the interaction of SKPI-SKP2, resulting in prometaphase arrest. | 16HBE, HLF, 293T, and a panel of lung cancer cell lines In vivo murine model | Lung cancer | [152] |
| Z0933M | Binds C-terminal of SKP1 and inhibits the association of F-box protein to make stable SCF E3 ligase. It disrupts SCF and induces cell death by p53-dependent mechanism. | A panel of different cancer cells. MDA-231, MCF-7, Hela, BTC6, HEK293, HepG2, HCT and A-431 | Breast cancer | [153] | |
| SCFskp2 E3 ligase | C-series compound (C1, C2, C16, C20) | C-series compounds inhibit Cks1 activity to destabilize SKP2-p27 interaction, enhance p27 accumulation, and promote cell type-specific blocks in G1 or G2/M phase. | MCF-7, T47D, 501Mel, SK-MEL-173, SK-MEL-147 | Melanoma | [154] |
| Dioscin | Dioscin promotes SKP2-CDH1 interaction to induce CDH1-mediated degradation of SKP2 and delays tumor growth. | Colorectal cell line: DLD, HCT116, SW480, HT29, HCT8, SW620 In vivo mouse model | Colorectal cancer | [155] | |
| Compound 25 (C25) | C25 inhibits interaction of SKP2 with adaptor SKP1 and the ligase activity of SKP2, resulting in cancer progression. | 293T, PC3,A549, H460, H1299, Hep3B, U2OS | Liver, lung, prostate, and osteosarcoma | [156] | |
| P300 CREB protein and P300 Histone acetylase) | A-485 | A-485 arrested p300/pCBP-mediated histone acetylation marks of cell senescence in NSCLC. It regulates antitumor activity in many solid tumors. | Non-small-cell lung cancer (NSCLC) cell lines: NCI H460, NCI 1650, H1299 PC-3 In vivo mouse xenograft | Non-small-cell lung cancer Prostate cancer Melanoma | [157,158] |
| B029-2 | B029-2 inhibits glycolysis and induces tumor cell cycle arrest by reducing through modulation of histone acetylation. | Huh7, Hep3B In vivo mouse xenograft | Hepatocellular carcinoma | [159] | |
| PU141 | PU141 is a selective inhibitor to p300/pCBP that reduces tumor growth in vivo through the reduction of histone lysine acetylation. | SK-N-SH neuroblastoma cells In vivo mouse xenograft | Neuroblastoma | [160] | |
| C646 | C646 selectively inhibits p300 and CBP functions. It inhibited cell proliferation and induced apoptosis in vitro. | Human gastric epithelial cells, GES-1 and gastric cancer cell line SGC 7901, MKN45,BGC823, KATOIII | Gastric cancer | [161] | |
| PRAP1 | AZD5305 | AZD5305 shows anti-proliferative effects in vitro. It potentially and selectively inhibits PRAP1 functions. | In vitro and in vivo mouse xenograft and PDX model. Rat preclinical model cell; MDA-MB-436, DLD-1, DLD BRACA2−/− | Breast cancer | [162] |
| [77Br]Br-WC-DZ | A radio-brominated Auger emitting inhibitor targeting PARP-1. The inhibitor shows cytotoxicity in prostate cancer cells and promotes DNA damage and cell cycle arrest at G2/M phase. | Prostate cancer cell lines: PC-3, IGR-CaP1 In vivo, prostate cancer xenograft model | Prostate cancer | [163] |
| Target | Name of Inhibitors | Characteristics/Functions | Preclinical Study In Vitro (Cell Type) and In Vivo Models | Cancer Type | Reference |
|---|---|---|---|---|---|
| HAT Histone Acetyltransferase GNC5/PCAF | PU139 Pan-inhibitor | PU139 inhibits GNC5/PCAF function and triggers caspase-independent cell death. It blocks growth of SK-N-SH neuroblastoma xenografts in mice. | SK-N-SH neuroblastoma cell In vivo mouse xenograft | Neuroblastoma cell | [160] |
| GSK983 PROTAC (proteolysis-targeting chimeras) | GSK983 targets GNC5/PCAF and modulates immunity through mediators released by LPS-induced immune cells. | Immune cells | [210] | ||
| Garcinol and curcumin (garcinol derivative LTK4) | Garcinol blocks PCAF by modulating the acetylation of the C-terminal domain of p53 in tumor cells. | MCF7 and osteosarcoma cell lines U2OS and SaOS2 | Breast cancer | [211] | |
| SWI/SNF | PROTAC Tool Compound AU5330 | The inhibitor targets SWI/SNF complex, simultaneously degrades ATPases SMARC4, SMAR2, and PBRMI, and selectively kills H3.3K27M. | BT245, DIPG-007, DIPG-X*IIIp, H3.3 K27M | Lethal pediatric brain cancer | [212] |
| The bromodomain inhibitor-PFI3 | SWI/SNF‘s chromatin binding is directly blocked by PF13, resulting in DBS repair defects and alternations in damage checkpoints. As a result, necrosis and senescence increase cell death. | A549, HT29, H460, H1299, and U2OS | Several cancer types | [213] | |
| BRM and BRG1 inhibitors | The inhibitors target ATPase activity of SWI/SNF complex, downregulate BRM-dependent gene expression, and show antiproliferative activity in a BRG1-mutant lung tumor xenograft model. | In vivo mouse xenograft model | Lung tumor | [214] | |
| SETDB1/ESET | Mithramycin A and mithramycin analog (mithralog) EC8042 | The inhibitors suppress the expression of SETDB1 and induce changes at transcriptomic, morphological, and functional levels, leading to antitumor effects. | SK-HI SETDB1 melanoma cell line | Malignant melanoma | [215] |
| DZNep (deazaneplanocin)A | DZNep inhibits histone methylations, including H3K27me3 and HCK9me3. The reduced levels of H3K27me3 and H3K9me3 decrease the EZH2 and SETDB1 protein levels in lung cancer cells. | Lung epithelial carcinoma cell A549, H1299, H460 | Lung cancer | [216] | |
| miRNA-621 | miRNA-621 could directly target the 3′ UTR of SETDB1. Direct inhibition of SETDB1 further boosts the radiosensitivity of HCC cells. | LO2.HepG2, Smmc-7721, Bel 7404 HCC mouse model | Hepatocellular carcinoma | [217] | |
| RAS Small GTPase | BI-3406 | BI-3406 is a SOS1/MEK inhibitor that enables tumor growth in different KRAS-driven tumor models. | NCI-H23, FLAG-SOS1, SOS1, and SOS2-negative cells Patient-derived xenograft study Cell-derived efficacy study in mouse model | KRAS-driven cancer | [218] |
| KRAS agonist 533 | KRAS-533 binds the GTP/GDP-binding pocket of KRAS. KRA-533 increases KRAS activity and suppresses cell growth in lung cancer patients. | A459 Lung cncer xenograft | Lung cancer | [219] | |
| Kobe0065 Kobe2602 | In vitro and in vivo, the inhibitors show inhibitory effect binding with H-Raf-GTP-c-Raf-1. They induce apoptosis and inhibit cell growth. | NIH 3T3 cells transformed with H-rasG12V SW480 Tumor xenograft | Colorectal cancer | [220] | |
| ARS-1620 | The inhibitor dissects oncogenic KRAS decency. | Subcutaneous xenograft models bearing KRAS p.G12C. | NSCLC | [221] | |
| HDAC1 Histone Deacetylase | Romidepsin | HDAC1/2 inhibitor, which suppresses diethylnitrosamine (DEN)-induced hepatocellular carcinoma (HCC). HDAC1 inhibitor. Cells treated with romidepsin showed apoptotic cell death and reduced HDAC activity. | C56BL/6 mice A panel of 8 BTC cell lines | Hepatocellular carcinoma (HCC) Biliary tract cancer (BTC) | [222,223] |
| NK-HDAC-1 | Cell cycle arrest, apoptosis effects, and inhibition of cell growth were observed. | MDA-MB-231, MCF-7 In vivo mice | Breast cancer | [224] | |
| CG200745 | Inhibition of pancreatic cancer cell growth by overcoming gemcitabine resistance. | BxPC3, Cfpac-1, HPAC Xenograft mouse model | Pancreatic cancer | [225] | |
| CG200745 causes epigenetic reactivation of critical genes and induces antiproliferation in NSCLC cancer. | Lung cancer cell lines; NSCLC and Beas-2B (Beas-2B) | Lung cancer | [226] | ||
| FR901228 | Targets CDKA1A/p21 to induce cell cycle arrest. | In vitro MCF-10A, PC-3, DU145, SW620, IGROV, MCF-7, A549 | Many cancer types: breast cancer, prostate cancer, ovarian cancer, colon cancer, and lung cancer | [227,228] | |
| HDAC8 | Organoselenium compounds MSC—methyselenocysteine SM—selenomethionine | MSC and SM are HDAC inhibitors (HDAC1 and HDAC8) that generate metabolites from α-keto acid and potentially affect histone and chromatin remodeling. | Human HT29 and HCT116, HCT116(53−/−), HCT116(53+/+) | Colon cancer | [229] |
| NCC149 derivatives | A selective inhibitor of HDAC8 that increases a-tubulin acetylation and suppresses T-cell lymphoma cells. | HeLa cells | Cervical cancer | [230] | |
| HDAC8 selective inhibitors (cpd2, PCI-04051, PCI-48000, PCI-48012) with retinoic acid | In vitro and in vivo, the inhibitors reduce neuroblastoma growth by selective inhibition of HDAC8 with retinoic acid. | Human neuroblastoma cell lines; BE(2)-2,IMR-32, SH-SY5Y, SK-N-AS and SH-EP Mouse xenograft study with HDAC8 knockdown | Neuroblastoma | [231] | |
| m6A Methyltransferase | Quercetin (derived from natural products) | Quercetin can inhibit METTL3(methyltransferase complex), decrease m6A level, and inhibit tumor cell proliferation. | MIA PaCa-2 | Pancreatic cancer | [232] |
| STM2457 | A small-molecule inhibitor of METTL3. STM2457 affects the inhibition of catalytic activity and upregulation of METTL3, resulting in upregulation of PD-L1 and reduction of tumor progression. | A panel of lung cancer and lung epithelial cell lines A549, H1975 HBE135, BEAS-2B In vivo mouse model | Non-small-cell lung cancer Oral | [233] | |
| BI-78D3 | BI-78D3 binds DRS (D-recruitment site) of ERK2 by making covalent bond at C159. Apoptosis was induced in different melanoma cell lines, including BRAF inhibitor-naive and resistant melanoma cells. | HEK293T, CRL-3216 Melanoma cell line: A357, CRL-1619 | Melanoma | [123] | |
| SET (7/9) Methyltransferase | (R)-PFI-2 | (R)-PFI-2 is a selective inhibitor of SET 7. It can cause modulation of Hippo pathway by increasing nuclear YAP and YAP-mediated gene transcription. | Murine embryonic fibroblasts (MEFs) MCF7 cells | Breast cancer | [234] |
| Target | Name of Inhibitors | Characteristics/Functions | Preclinical Study In Vitro (Cell Type) and In Vivo Models | Cancer Type | Reference |
|---|---|---|---|---|---|
| c-Jun N-terminal kinase (JNK) | SP0016125 | Inhibition of JNK and induction of apoptosis by SMAD-mediated caspase activation. Targeting JNK and activating BAX and dihydroartemisinin (DHA)-induced human lung adenocarcinoma cell apoptosis. | The human cholangiocarcinoma cell RBE and PT67 ASTC-a-1, A549 | Cholangiocarcinoma Lung adenocarcinoma | [264,265] |
| Polyphylin I (PPI) | JNK pathway is targeted by PPI. In glioblastoma cells, G2/M phase arrest and apoptosis are observed. Although the expression of Bax, p-JNK, and cytochromes are upregulated, anti-apoptotic Bcl-2 protein is downregulated. | U251 glioblastoma cell | Glioblastoma | [266] | |
| AS601245 and clofibrate (PPARa agonist) | Inhibition of JNK pathway. STAT3 signal is reduced. | CoCo-2, HepG2 | Colon cancer | [267] | |
| JNK-in-IX | JNK-in-IX is specific inhibitor against JNK2. It causes DNA damage through G2 arrest mediated by p53 and p21. | ASpC-1, BxPC-3 MIA Paca-2 Human pancreatic organoid | Pancreatic cancer | [268] | |
| JNK-IN-8 | Lapatinib and JNK-IN-8 synergistically inhibit transcriptional activity of AP-1, Nrf2, and NFӄ to promote apoptosis. | MDA-MB-436, HCC1569, MDA-MB-231 In vivo mice | Breast cancer | [269] | |
| p38 MAP kinase | LY2228820 | LY2228820 is an ATP-competitive inhibitor of the α- and β-isoforms of p38 MAPK. It inhibited tumor growth in various in vivo cancer models. | A549, U-87MG, HeLa, MDA-MB-468, 786-O, OPM-2, A2780 In vivo mice xenograft model | Melanoma, non-smal—cell lung cancer, ovarian, glioma, myeloma, breast | [270] |
| BIRB-796 with VX680 | Dual blocking of Aurora kinase and p38 MAPK. Reduced cell proliferation of cervical cancer. | HeLa, Caski, and SiHa Human tumor xenograft in nude mice | Cervical cancer | [271] | |
| BRIB796 | RIB796 blocks G1 phase cycle and inhibits cell proliferation, migration, and migration in GMB cell lines. | U87, U251 | Glioblastoma | [272] | |
| AKT | CMG002 and sorafenib | HCC cell proliferation and tumor growth were reduced by inhibition of MAPK and PI3K/AKT/mTOR pathways. | Human HCC cell line, Huh-7, and HepG2 | Hepatocellular carcinoma | [273] |
| Phycocyanin | Phycocyanin modulates MAPK, Akt/mTOR, and NF-ӄBA pathways to induce apoptosis and autophagic cancer cell death. Complex regulation of the MAPK, Akt/mTOR, and NF-κB signaling pathways. | Pancreatic cancer cell lines: PANC-1, BxPC-3 Other cells: MCF-7, HepG2,HK-2, BGC-823 | Pancreatic cancer | [274] | |
| Quercetin | Inhibition of PI3/AKT and MEK/ERK pathways and induction of apoptosis. | Melanoma B6-F10 | Melanoma | [275] | |
| Silibinin | Triggering the MAP2K1/2-MAPK1/3 pathway but blocking the PI3/AKT/mTOR pathway to induce autophagy and apoptosis. | Colorectal cancer SW 480, HT29, and LoVo cells In vivo mice | Colorectal cancer | [276] | |
| NVD-LD-225 NVP-BEZ-235 | Targets sonic hedgehog and PI3/AKT/mTOR pathways and suppresses tumorigenic potential of glioblastoma initiating cells. | Glioblastoma-initiating cells from patients In vivo mice | Glioblastoma | [277] | |
| TRAF6 | TMBPS [bis (4-hydroxy-3,5-dimethylphenyl) sulfone] | TMBPS directly targets TRAF6 to reduce its level. Thereby, it controls multiple pathways like protein kinase B, AKT, and ERK1/2, resulting in cell cycle arrest, apoptosis, and inhibition of tumor growth. | In vitro In vivo mouse xenograft model | Hepatocellular carcinoma | [278] |
| Cinchona alkaloids (small-molecule inhibitor, competitive inhibitor of ring domain of TRAF6) | Cinchona alkaloids are potential anti-tumor inhibitors that induce apoptosis both in vitro and in vivo. Ubiquitination and phosphorylation of AKT and TAK1 are inhibited, and Bax/Bcl-2 is upregulated. In vivo, study shows an increase in cytokine production like TNF-α, IFN-γ, and IgG. | HeLa cells | Human cancer | [279] | |
| RhoA Selected small-molecule inhibitor | Rhosin | Inhibition of RhoA activation; blocks GEF binding. | NIH3T3, HME, MCF-7 | Breast cancer | [280] |
| Y16 | Inhibition of RhoA activation by targeting LARG; blocks RhoA binding. | MCF-7, MSF10A | Breast cancer | [281] | |
| CCG-1423 | Inhibition of RhoA activation by targeting MKL1; blocks RhoA-dependent gene transcription. | HK293T, PC-3 NIH3T3 | Prostate cancer | [282,283] | |
| CHS-111 | Inhibition of RhoA activation by targeting PLD; blocks RhoA membrane recruitment. | Rat neutrophile | [284] | ||
| PKA | PKI (PK inhibitors): PKIA, PKAIB, and PKIG Synthetic peptide analogs of PKI | Alteration of PKA activation, which drives GPCR-Gαs-cAMP signaling toward EPAC-RAP1 and MAPK. | HEK293 Prostate epithelial cell line: RWPE Prostrate adenocarcinoma cell line: LNCaP, VCaP, DU145, and PC3 In vivo mouse | Prostate cancer | [285] |
| PKI (6–22) amide | PKI modulates the responses of cancer cells treated by Taxol and Taxane therapeutics. It inhibits the cAMP-PKA pathway in breast cancer cells and reverts the proliferative effect of oxytocin-treated tumors. | Tet-activator expressing LNCaP (LNGK9) and DU145 cells MDA-MB231 | Prostate cancer Breast cancer | [286,287] | |
| PKI (1–25) amide | Cardiac protection through cAMP-dependent EPAC/Rac1/ERK signaling pathway. | Transgenic mouse cardiomyocytes in mice | Cardiomyocytes | [288] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Runa, F.; Ortiz-Soto, G.; de Barros, N.R.; Kelber, J.A. Targeting SMAD-Dependent Signaling: Considerations in Epithelial and Mesenchymal Solid Tumors. Pharmaceuticals 2024, 17, 326. https://doi.org/10.3390/ph17030326
Runa F, Ortiz-Soto G, de Barros NR, Kelber JA. Targeting SMAD-Dependent Signaling: Considerations in Epithelial and Mesenchymal Solid Tumors. Pharmaceuticals. 2024; 17(3):326. https://doi.org/10.3390/ph17030326
Chicago/Turabian StyleRuna, Farhana, Gabriela Ortiz-Soto, Natan Roberto de Barros, and Jonathan A. Kelber. 2024. "Targeting SMAD-Dependent Signaling: Considerations in Epithelial and Mesenchymal Solid Tumors" Pharmaceuticals 17, no. 3: 326. https://doi.org/10.3390/ph17030326
APA StyleRuna, F., Ortiz-Soto, G., de Barros, N. R., & Kelber, J. A. (2024). Targeting SMAD-Dependent Signaling: Considerations in Epithelial and Mesenchymal Solid Tumors. Pharmaceuticals, 17(3), 326. https://doi.org/10.3390/ph17030326