
Evaluating Fatty Acid Amide Hydrolase as a Suitable Target for Sleep Promotion in a Transgenic TauP301S Mouse Model of Neurodegeneration

 , ,
, ,
Abstract
1. Introduction
2. Results
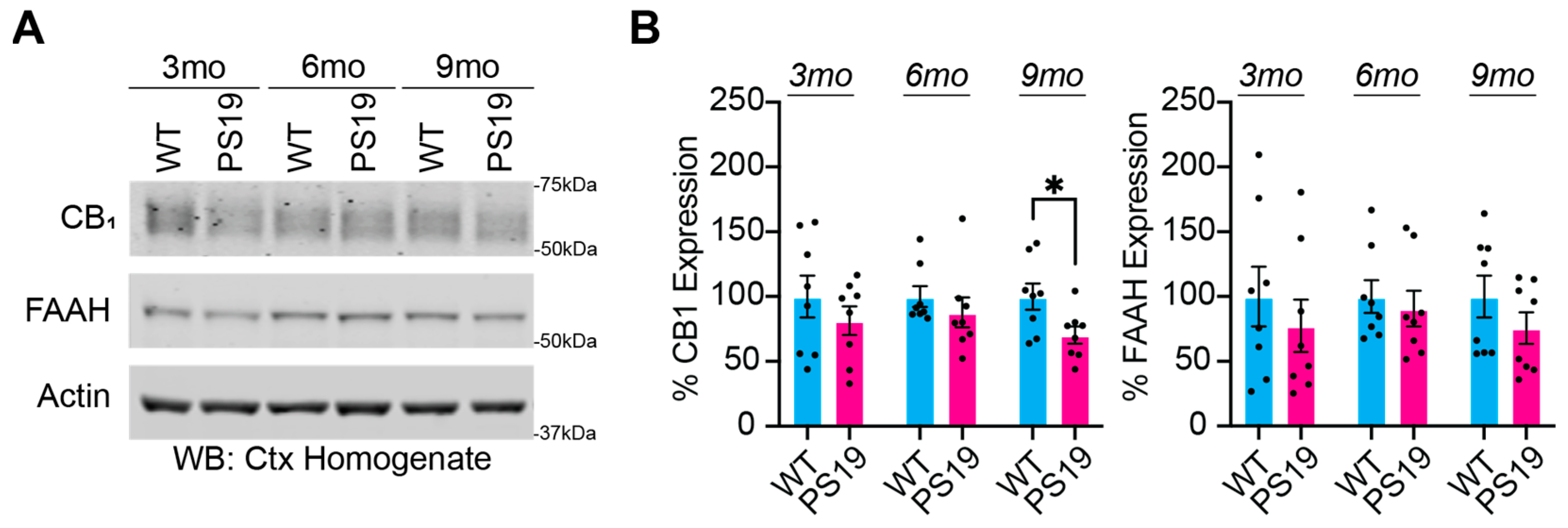
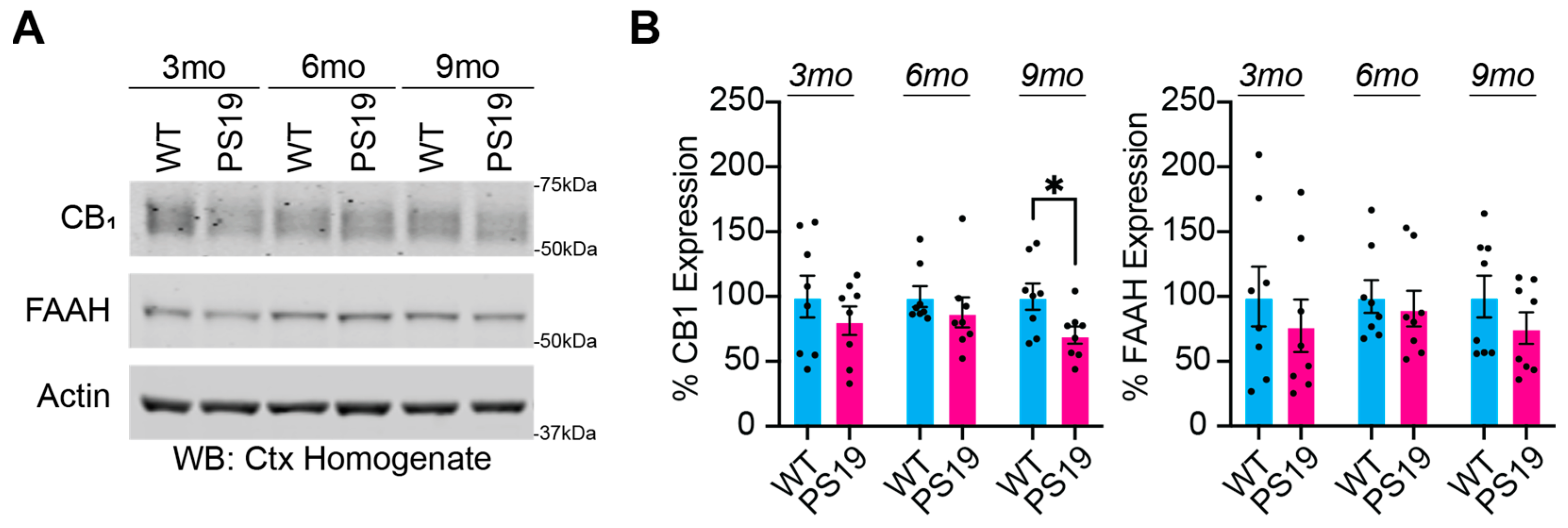
2.1. Dysregulation of the Endocannabinoid System in P301S Tau Mice
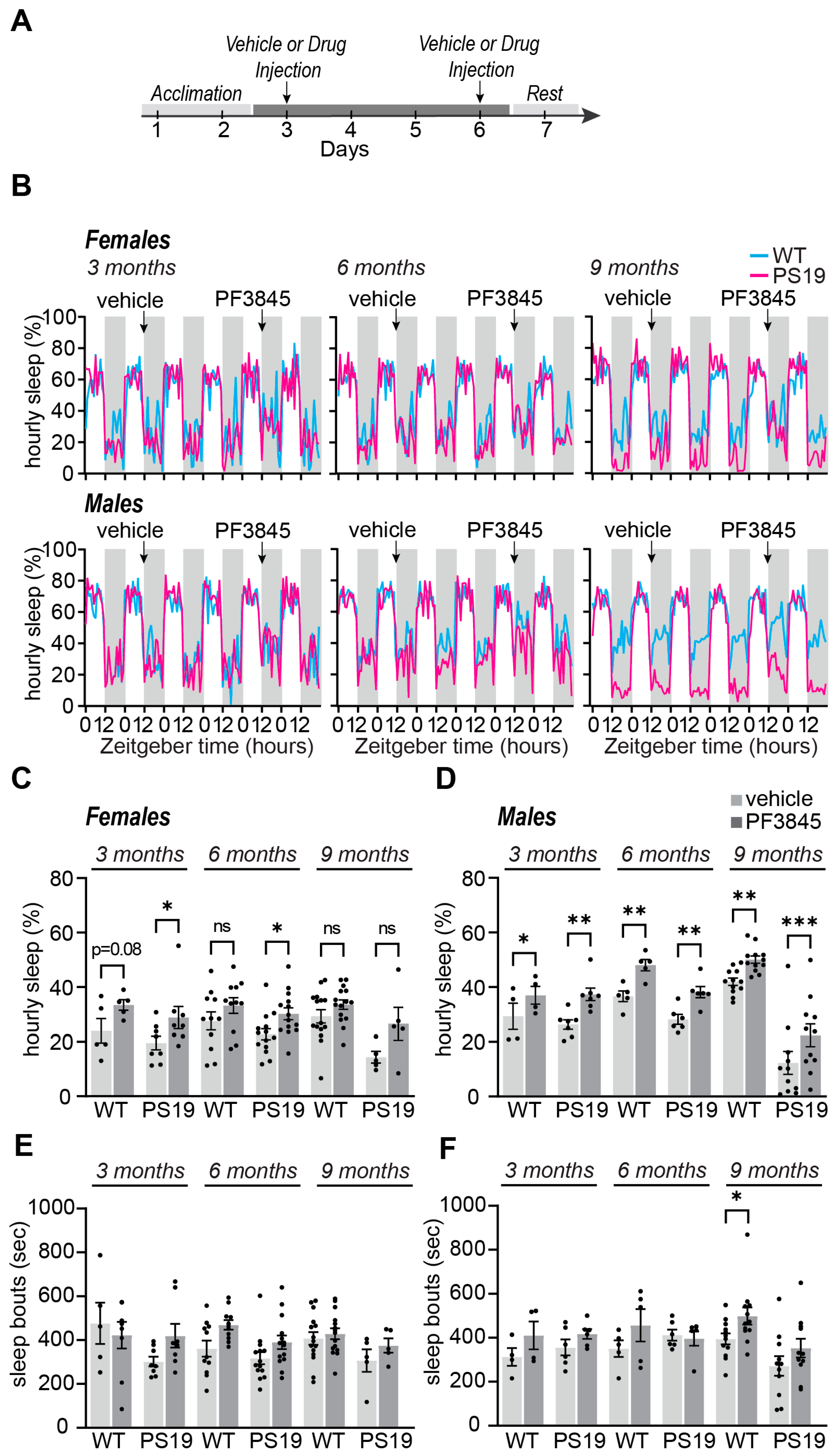
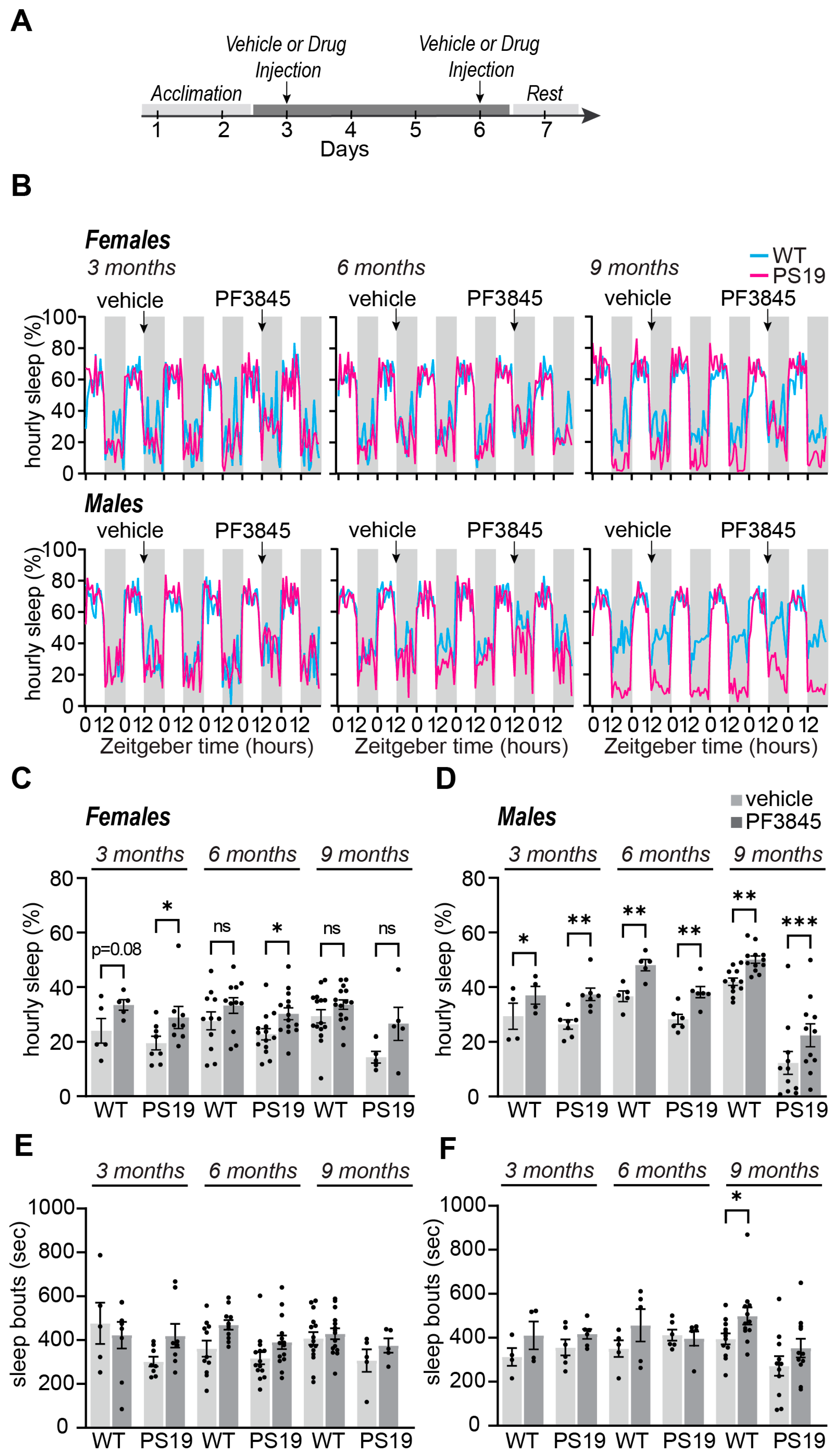
2.2. Selective Acute Inhibition of FAAH Promotes Sleep in P301S Tau Mice
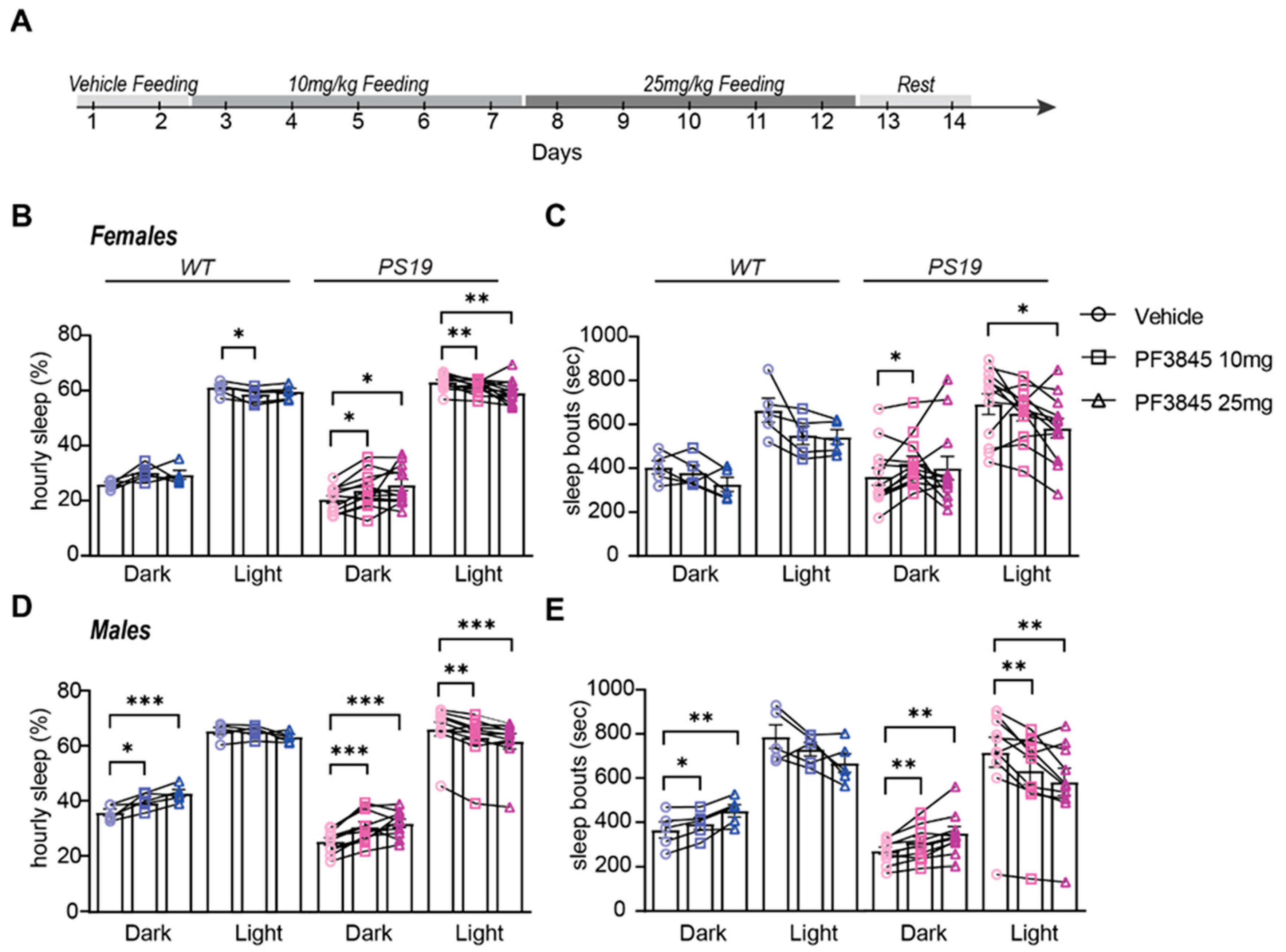
2.3. Selective Sustained Inhibition of FAAH Promotes Sleep in P301S Tau Mice
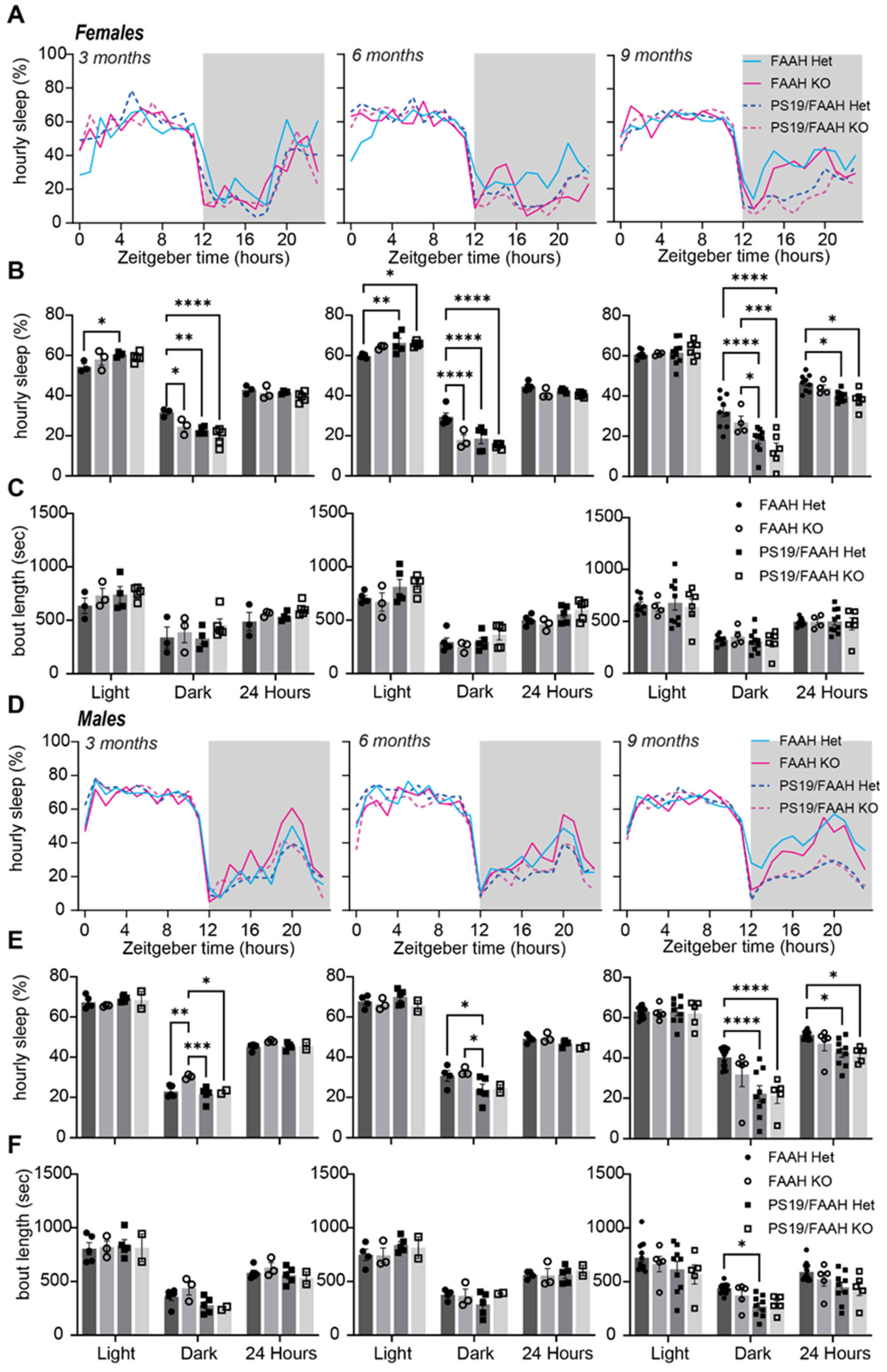
2.4. Lack of Sleep-Promoting Effects of FAAH Knockout in P301S Tau Mice
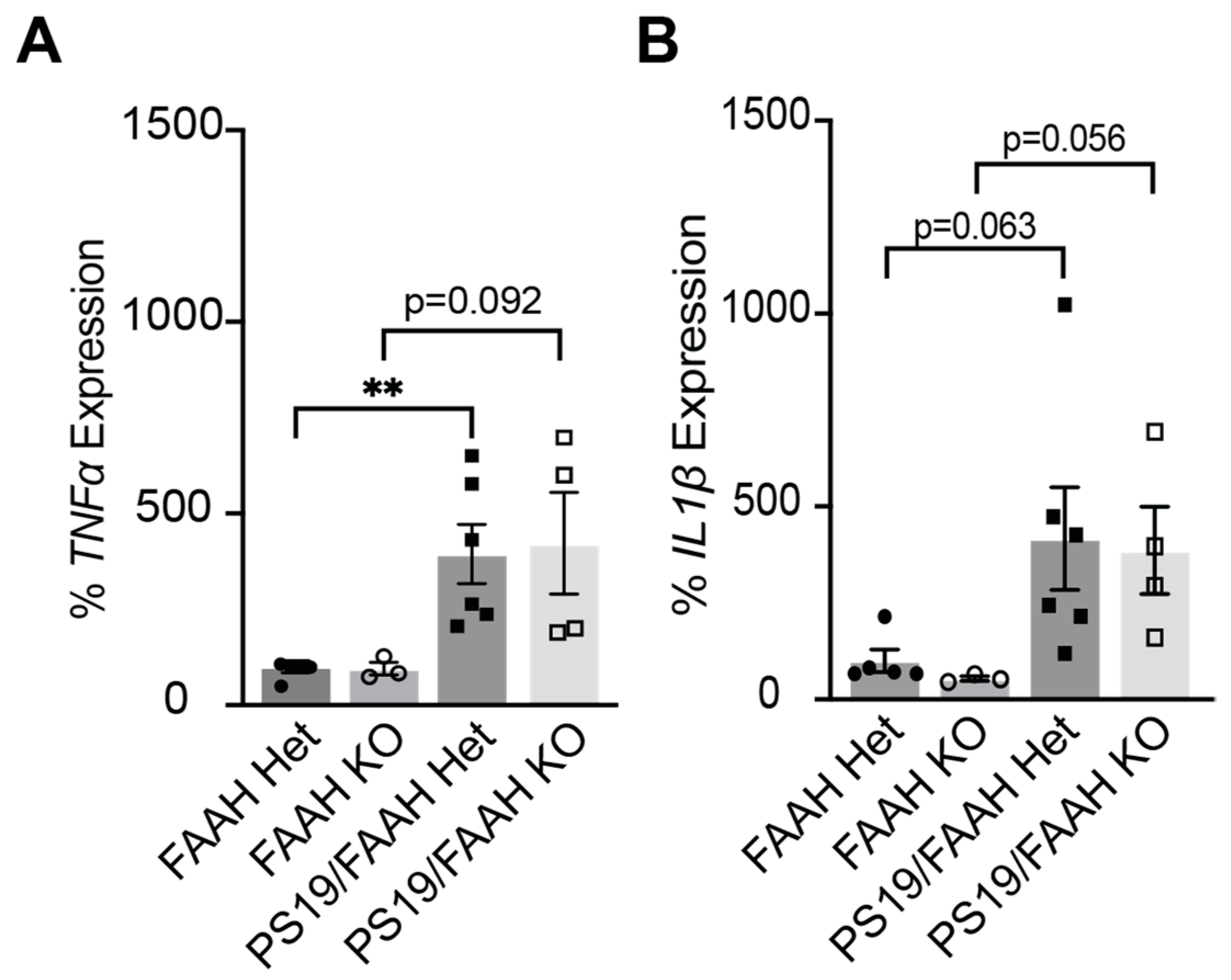
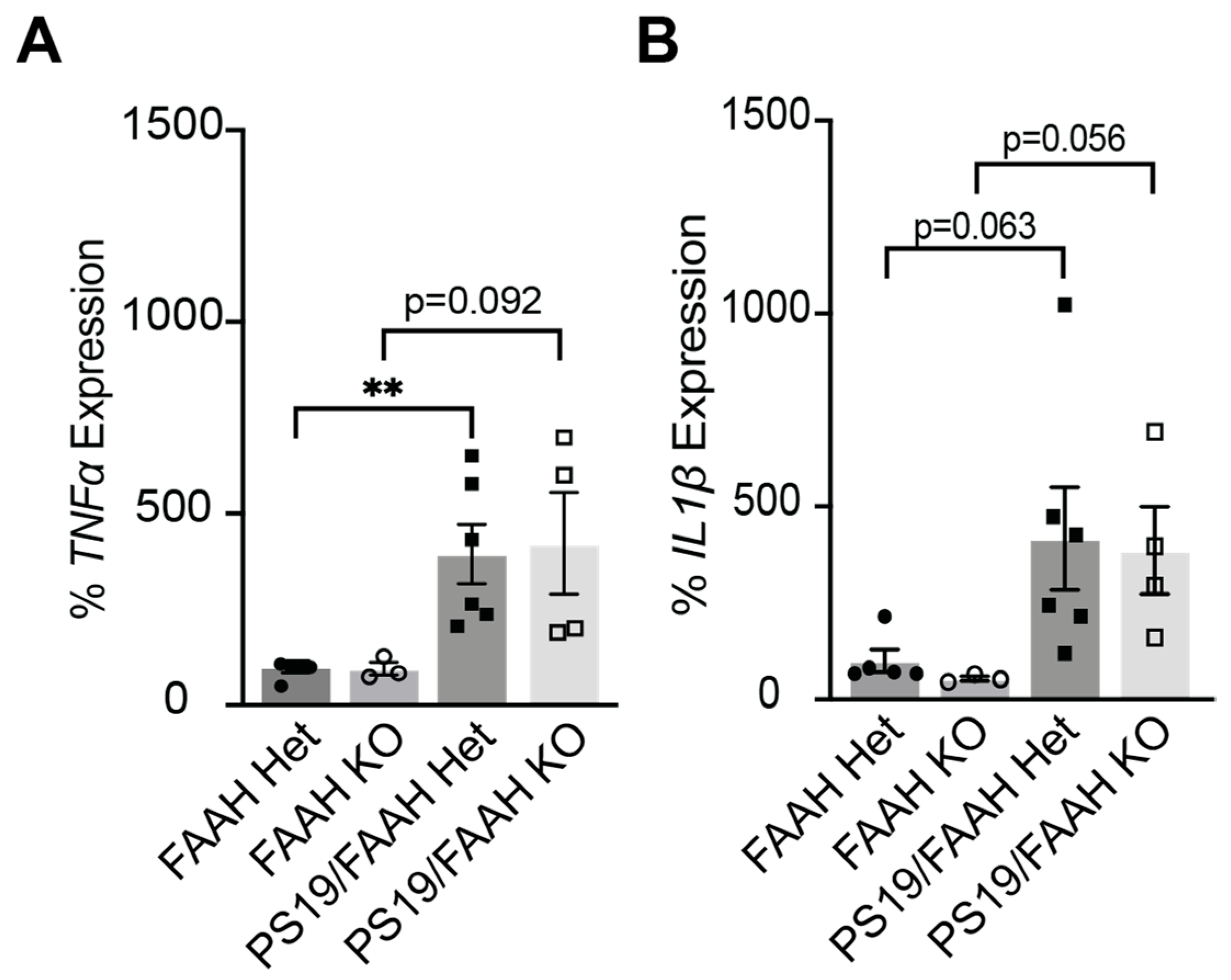
2.5. FAAH KO Does Not Protect against Inflammation in P301S Tau Mice
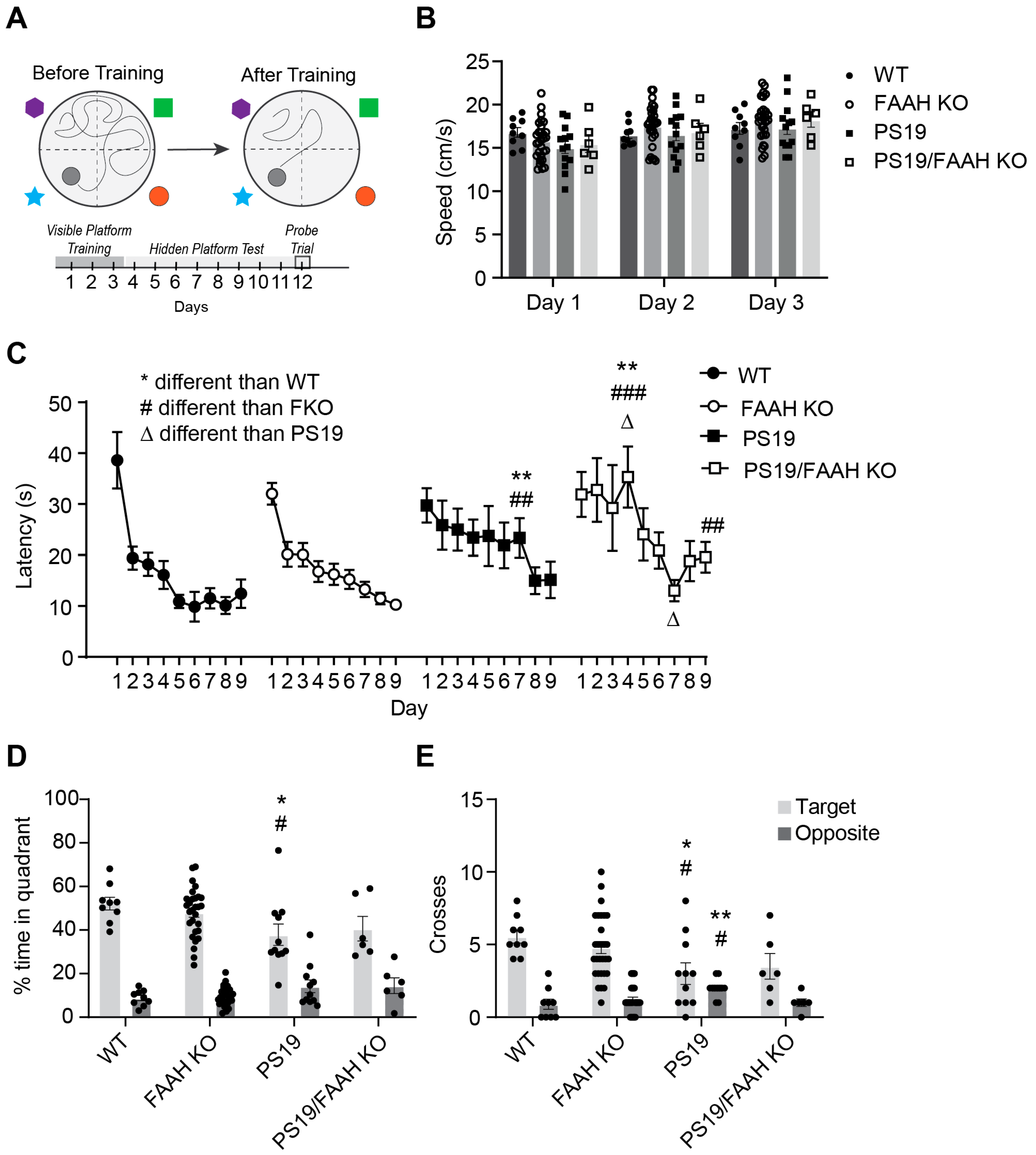
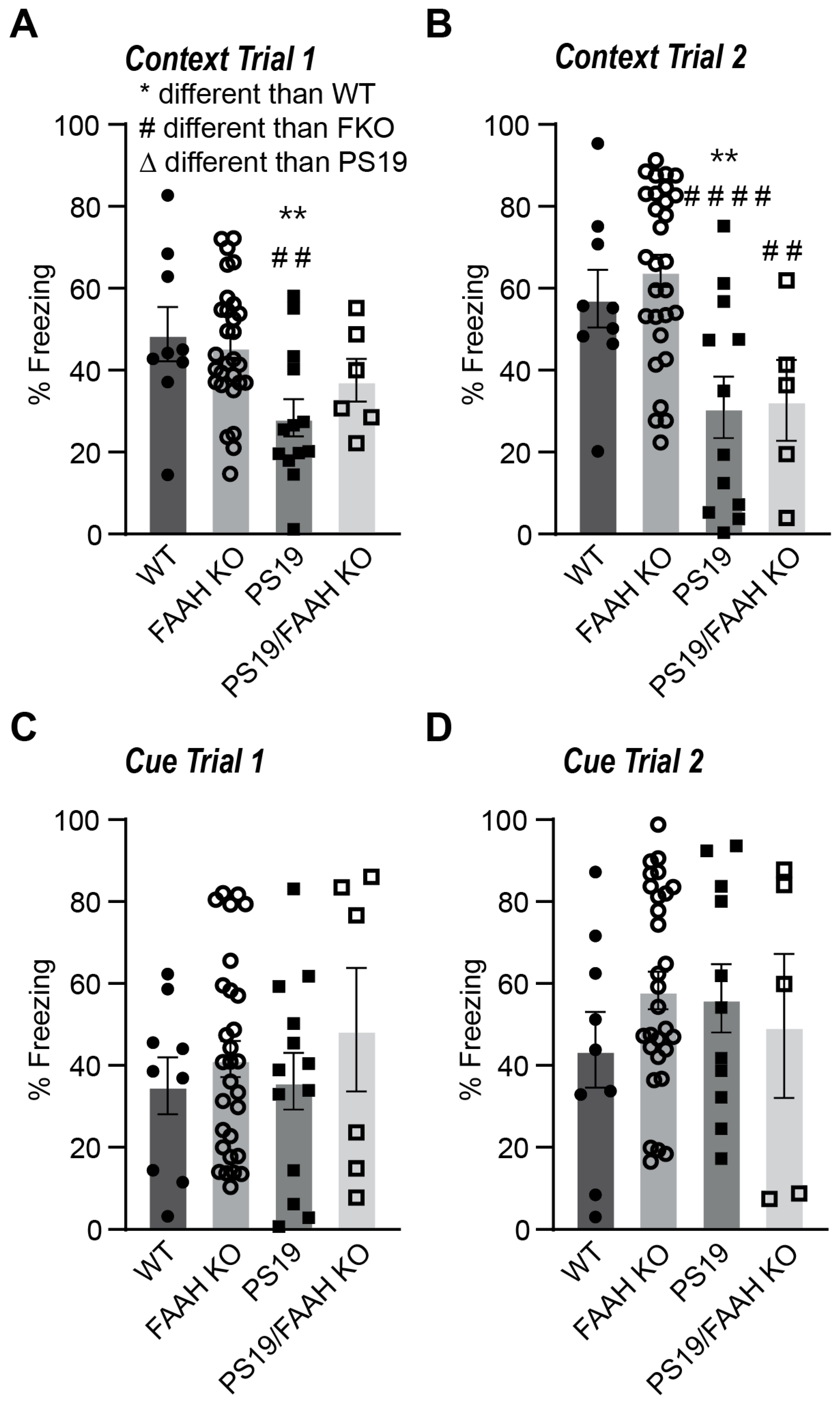
2.6. FAAH KO Does Not Protect P301S Tau Mice against Cognitive Decline
3. Discussion
3.1. Endocannabinoids and Relevance for Sleep
3.2. Sex-Specific Effects of Sleep Promotion
3.3. Effects of Acute vs. Sustained FAAH Inactivation
3.4. Evaluation of FAAH KO Model
4. Materials and Methods
4.1. Mice
4.2. Western Blot
4.3. Sleep Monitoring and Behavior Analysis
4.4. Drugs and Treatments
4.5. Quantitative Real-Time PCR
4.6. Behavioral Paradigm
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, C.; Holtzman, D.M. Bidirectional relationship between sleep and Alzheimer’s disease: Role of amyloid, tau, and other factors. Neuropsychopharmacology 2020, 45, 104–120. [Google Scholar] [CrossRef]
- Martin, S.C.; Joyce, K.K.; Harper, K.M.; Nikolova, V.D.; Cohen, T.J.; Moy, S.S.; Diering, G.H. Sleep disruption precedes forebrain synaptic Tau burden and contributes to cognitive decline in a sex-dependent manner in the P301S Tau transgenic mouse model. bioRxiv 2023. [Google Scholar] [CrossRef]
- Jack, C.R.; Holtzman, D.M. Biomarker modeling of Alzheimer’s disease. Neuron 2013, 80, 1347–1358. [Google Scholar] [CrossRef] [PubMed]
- Holth, J.; Patel, T.; Holtzman, D.M. Sleep in Alzheimer’s Disease—Beyond Amyloid. Neurobiol. Sleep Circadian Rhythm. 2017, 2, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Lim, M.M.; Gerstner, J.R.; Holtzman, D.M. The sleep-wake cycle and Alzheimer’s disease: What do we know? Neurodegener. Dis. Manag. 2014, 4, 351–362. [Google Scholar] [CrossRef] [PubMed]
- Holth, J.K.; Mahan, T.E.; Robinson, G.O.; Rocha, A.; Holtzman, D.M. Altered sleep and EEG power in the P301S Tau transgenic mouse model. Ann. Clin. Transl. Neurol. 2017, 4, 180–190. [Google Scholar] [CrossRef]
- Roh, J.H.; Huang, Y.; Bero, A.W.; Kasten, T.; Stewart, F.R.; Bateman, R.J.; Holtzman, D.M. Disruption of the Sleep-Wake Cycle and Diurnal Fluctuation of -Amyloid in Mice with Alzheimer’s Disease Pathology. Sci. Transl. Med. 2012, 4, ra122–ra150. [Google Scholar] [CrossRef]
- Prospéro-García, O.; Amancio-Belmont, O.; Becerril Meléndez, A.L.; Ruiz-Contreras, A.E.; Méndez-Díaz, M. Endocannabinoids and sleep. Neurosci. Biobehav. Rev. 2016, 71, 671–679. [Google Scholar] [CrossRef]
- Kesner, A.J.; Lovinger, D.M. Cannabinoids, Endocannabinoids and Sleep. Front. Mol. Neurosci. 2020, 13, 125. [Google Scholar] [CrossRef]
- Kalifa, S.; Polston, E.K.; Allard, J.S.; Manaye, K.F. Distribution patterns of cannabinoid CB1 receptors in the hippocampus of APPswe/PS1ΔE9 double transgenic mice. Brain Res. 2011, 1376, 94–100. [Google Scholar] [CrossRef]
- Basavarajappa, B.S.; Shivakumar, M.; Joshi, V.; Subbanna, S. Endocannabinoid system in neurodegenerative disorders. J. Neurochem. 2017, 142, 624–648. [Google Scholar] [CrossRef]
- Pava, M.J.; Makriyannis, A.; Lovinger, D.M. Endocannabinoid signaling regulates sleep stability. PLoS ONE 2016, 11, e0152473. [Google Scholar] [CrossRef] [PubMed]
- Silvani, A.; Berteotti, C.; Bastianini, S.; Lo Martire, V.; Mazza, R.; Pagotto, U.; Quarta, C.; Zoccoli, G. Multiple sleep alterations in mice lacking cannabinoid type 1 receptors. PLoS ONE 2014, 9, e89432. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.D.S.C.; Gay, S.M.; Armstrong, M.L.; Pazhayam, N.M.; Reisdorph, N.; Diering, G.H. Tonic endocannabinoid signaling supports sleep through development in both sexes. Sleep 2022, 45, zsac083. [Google Scholar] [CrossRef] [PubMed]
- Diering, G.H.; Nirujogi, R.S.; Roth, R.H.; Worley, P.F.; Pandey, A.; Huganir, R.L. Homer1a drives homeostatic scaling-down of excitatory synapses during sleep. Science 2017, 355, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Ye, M.; Monroe, S.K.; Gay, S.M.; Armstrong, M.L.; Youngstrom, D.E.; Urbina, F.L.; Gupton, S.L.; Reisdorph, N.; Diering, G.H. Coordinated Regulation of CB1 Cannabinoid Receptors and Anandamide Metabolism Stabilizes Network Activity during Homeostatic Downscaling. eNeuro 2022, 9, ENEURO.0276-22. [Google Scholar] [CrossRef] [PubMed]
- De Vivo, L.; Bellesi, M.; Marshall, W.; Bushong, E.A.; Ellisman, M.H.; Tononi, G.; Cirelli, C. Ultrastructural evidence for synaptic scaling across the wake/sleep cycle. Science 2017, 355, 507–510. [Google Scholar] [CrossRef] [PubMed]
- Ahn, K.; Johnson, D.S.; Mileni, M.; Beidler, D.; Long, J.Z.; McKinney, M.K.; Weerapana, E.; Sadagopan, N.; Liimatta, M.; Smith, S.E.; et al. Discovery and characterization of a highly selective FAAH inhibitor that reduces inflammatory pain. Chem. Biol. 2009, 16, 411–420. [Google Scholar] [CrossRef]
- Holth, J.K.; Fritschi, S.K.; Wang, C.; Pedersen, N.P.; Cirrito, J.R.; Mahan, T.E.; Finn, M.B.; Manis, M.; Geerling, J.C.; Fuller, P.M.; et al. The sleep-wake cycle regulates brain interstitial fluid tau in mice and CSF tau in humans. Science 2019, 363, 880–884. [Google Scholar] [CrossRef]
- Yoshiyama, Y.; Higuchi, M.; Zhang, B.; Huang, S.M.; Iwata, N.; Saido, T.C.; Maeda, J.; Suhara, T.; Trojanowski, J.Q.; Lee, V.M. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 2007, 53, 337–351. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhan, G.; Fenik, P.; Brandes, M.; Bell, P.; Francois, N.; Shulman, K.; Veasey, S. Chronic Sleep Disruption Advances the Temporal Progression of Tauopathy in P301S Mutant Mice. J. Neurosci. 2018, 38, 10255–10270. [Google Scholar] [CrossRef]
- Chalermpalanupap, T.; Schroeder, J.P.; Rorabaugh, J.M.; Liles, L.C.; Lah, J.J.; Levey, A.I.; Weinshenker, D. Locus Coeruleus Ablation Exacerbates Cognitive Deficits, Neuropathology, and Lethality in P301S Tau Transgenic Mice. J. Neurosci. 2018, 38, 74–92. [Google Scholar] [CrossRef] [PubMed]
- Niphakis, M.J.; Cognetta, I.I.I.A.B.; Chang, J.W.; Buczynski, M.W.; Parsons, L.H.; Byrne, F.; Burston, J.J.; Chapman, V.; Cravatt, B.F. Evaluation of NHS carbamates as a potent and selective class of endocannabinoid hydrolase inhibitors. ACS Chem. Neurosci. 2013, 4, 1322–1332. [Google Scholar] [CrossRef] [PubMed]
- Mang, G.M.; Nicod, J.; Emmenegger, Y.; Donohue, K.D.; O’Hara, B.F.; Franken, P. Evaluation of a piezoelectric system as an alternative to electroencephalogram/electromyogram recordings in mouse sleep studies. Sleep 2014, 37, 1383–1392. [Google Scholar] [CrossRef]
- Huitron-Resendiz, S.; Sanchez-Alavez, M.; Wills, D.N.; Cravatt, B.F.; Henriksen, S.J. Characterization of the sleep-wake patterns in mice lacking fatty acid amide hydrolase. Sleep 2004, 27, 857–865. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef]
- Lue, L.F.; Rydel, R.; Brigham, E.F.; Yang, L.B.; Hampel, H.; Murphy, G.M., Jr.; Brachova, L.; Yan, S.D.; Walker, D.G.; Shen, Y.; et al. Inflammatory repertoire of Alzheimer’s disease and nondemented elderly microglia in vitro. Glia 2001, 35, 72–79. [Google Scholar] [CrossRef]
- Tarkowski, E.; Andreasen, N.; Tarkowski, A.; Blennow, K. Intrathecal inflammation precedes development of Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 2003, 74, 1200–1205. [Google Scholar] [CrossRef]
- Takeuchi, H.; Iba, M.; Inoue, H.; Higuchi, M.; Takao, K.; Tsukita, K.; Karatsu, Y.; Iwamoto, Y.; Miyakawa, T.; Suhara, T.; et al. P301S mutant human tau transgenic mice manifest early symptoms of human tauopathies with dementia and altered sensorimotor gating. PLoS ONE 2011, 6, e21050. [Google Scholar] [CrossRef]
- Lasagna-Reeves, C.A.; de Haro, M.; Hao, S.; Park, J.; Rousseaux, M.W.; Al-Ramahi, I.; Jafar-Nejad, P.; Vilanova-Velez, L.; See, L.; De Maio, A.; et al. Reduction of Nuak1 Decreases Tau and Reverses Phenotypes in a Tauopathy Mouse Model. Neuron 2016, 92, 407–418. [Google Scholar] [CrossRef]
- Scheltens, P.; De Strooper, B.; Kivipelto, M.; Holstege, H.; Chételat, G.; Teunissen, C.E.; Cummings, J.; van der Flier, W.M. Alzheimer’s disease. Lancet 2021, 397, 1577–1590. [Google Scholar] [CrossRef] [PubMed]
- Owen, J.E.; Zhu, Y.; Fenik, P.; Zhan, G.; Bell, P.; Liu, C.; Veasey, S. Late-in-life neurodegeneration after chronic sleep loss in young adult mice. Sleep 2021, 44, zsab057. [Google Scholar] [CrossRef] [PubMed]
- Bellesi, M.; Vivo LDe Chini, M.; Gilli, F.; Tononi, G.; Cirelli, C. Sleep Loss Promotes Astrocytic Phagocytosis and Microglial Activation in Mouse Cerebral Cortex. J. Neurosci. 2017, 37, 5263–5273. [Google Scholar] [CrossRef] [PubMed]
- Hurtado-Alvarado, G.; Pavón, L.; Castillo-García, S.A.; Hernández, M.E.; Domínguez-Salazar, E.; Velázquez-Moctezuma, J.; Gómez-González, B. Sleep loss as a factor to induce cellular and molecular inflammatory variations. Clin. Dev. Immunol. 2013, 2013, 801341. [Google Scholar] [CrossRef] [PubMed]
- Pava, M.J.; Den Hartog, C.R.; Blanco-Centurion, C.; Shiromani, P.J.; Woodward, J.J. Endocannabinoid modulation of cortical up-states and NREM sleep. PLoS ONE 2014, 9, e88672. [Google Scholar] [CrossRef] [PubMed]
- Murillo-Rodríguez, E.; Sánchez-Alavez, M.; Navarro, L.; Martínez-González, D.; Drucker-Colín, R.; Prospéro-García, O. Anandamide modulates sleep and memory in rats. Brain Res. 1998, 812, 270–274. [Google Scholar] [CrossRef]
- Pérez-Morales, M.; De La Herrán-Arita, A.K.; Méndez-Díaz, M.; Ruiz-Contreras, A.E.; Drucker-Colín, R.; Prospéro-García, O. 2-AG into the lateral hypothalamus increases REM sleep and cFos expression in melanin concentrating hormone neurons in rats. Pharmacol. Biochem. Behav. 2013, 108, 1–7. [Google Scholar] [CrossRef]
- Rueda-Orozco, P.E.; Soria-Gómez, E.; Montes-Rodríguez, C.J.; Pérez-Morales, M.; Prospéro-García, O. Intrahippocampal administration of anandamide increases REM sleep. Neurosci. Lett. 2010, 473, 158–162. [Google Scholar] [CrossRef]
- Herrera-Solís, A.; Vásquez, K.G.; Prospéro-García, O. Acute and subchronic administration of anandamide or oleamide increases REM sleep in rats. Pharmacol. Biochem. Behav. 2010, 95, 106–112. [Google Scholar] [CrossRef]
- Santucci, V.; Storme, J.J.; Soubrié, P.; Le Fur, G. Arousal-enhancing properties of the CB1 cannabinoid receptor antagonist SR 141716A in rats as assessed by electroencephalographic spectral and sleep-waking cycle analysis. Life Sci. 1996, 58, PL103–PL110. [Google Scholar] [CrossRef]
- Bogáthy, E.; Papp, N.; Vas, S.; Bagdy, G.; Tóthfalusi, L. AM-251, A Cannabinoid Antagonist, Modifies the Dynamics of Sleep-Wake Cycles in Rats. Front. Pharmacol. 2019, 10, 831. [Google Scholar] [CrossRef]
- Reich, C.G.; Taylor, M.E.; McCarthy, M.M. Differential effects of chronic unpredictable stress on hippocampal CB1 receptors in male and female rats. Behav. Brain Res. 2009, 203, 264–269. [Google Scholar] [CrossRef]
- Riebe, C.J.N.; Hill, M.N.; Lee, T.T.Y.; Hillard, C.J.; Gorzalka, B.B. Estrogenic regulation of limbic cannabinoid receptor binding. Psychoneuroendocrinology 2010, 35, 1265–1269. [Google Scholar] [CrossRef]
- Liu, X.; Li, X.; Zhao, G.; Wang, F.; Wang, L. Sexual dimorphic distribution of cannabinoid 1 receptor mRNA in adult C57BL/6J mice. J. Comp. Neurol. 2020, 528, 1986–1999. [Google Scholar] [CrossRef]
- Bedse, G.; Romano, A.; Cianci, S.; Lavecchia, A.M.; Lorenzo, P.; Elphick, M.R.; LaFerla, F.M.; Vendemiale, G.; Grillo, C.; Altieri, F.; et al. Altered expression of the CB1 cannabinoid receptor in the triple transgenic mouse model of Alzheimer’s disease. J. Alzheimers Dis. 2014, 40, 701–712. [Google Scholar] [CrossRef]
- Ju, Y.E.S.; Lucey, B.P.; Holtzman, D.M. Sleep and Alzheimer disease pathology—A bidirectional relationship. Nat. Rev. Neurol. 2014, 10, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.E.; Lim, M.M.; Bateman, R.J.; Lee, J.J.; Smyth, L.P.; Cirrito, J.R.; Fujiki, N.; Nishino, S.; Holtzman, D.M. Amyloid-beta dynamics are regulated by orexin and the sleep-wake cycle. Science 2009, 326, 1005–1007. [Google Scholar] [CrossRef] [PubMed]
- Saarelainen, L.; Taipale, H.; Koponen, M.; Tanskanen, A.; Tolppanen, A.M.; Tiihonen, J.; Hartikainen, S. The Incidence of Benzodiazepine and Related Drug Use in Persons with and without Alzheimer’s Disease. J. Alzheimers Dis. 2016, 49, 809–818. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.T.; Lin, F.J.; Erickson, S.R.; Hong, J.L.; Wu, C.H. The Association Between the Use of Zolpidem and the Risk of Alzheimer’s Disease Among Older People. J. Am. Geriatr. Soc. 2017, 65, 2488–2495. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Yagyu, K.; Sackett, S.; Zhang, Y. Anti-Inflammatory Effects by Pharmacological Inhibition or Knockdown of Fatty Acid Amide Hydrolase in BV2 Microglial Cells. Cells 2019, 8, 491. [Google Scholar] [CrossRef] [PubMed]
- de Ceglia, M.; Micioni Di Bonaventura, M.V.; Romano, A.; Friuli, M.; Micioni Di Bonaventura, E.; Gavito, A.L.; Botticelli, L.; Gaetani, S.; de Fonseca, F.R.; Cifani, C. Anxiety associated with palatable food withdrawal is reversed by the selective FAAH inhibitor PF-3845: A regional analysis of the contribution of endocannabinoid signaling machinery. Int. J. Eat. Disord. 2023, 56, 1098–1113. [Google Scholar] [CrossRef] [PubMed]
- Schlosburg, J.E.; Blankman, J.L.; Long, J.Z.; Nomura, D.K.; Pan, B.; Kinsey, S.G.; Nguyen, P.T.; Ramesh, D.; Booker, L.; Burston, J.J.; et al. Chronic monoacylglycerol lipase blockade causes functional antagonism of the endocannabinoid system. Nat. Neurosci. 2010, 13, 1113–1119. [Google Scholar] [CrossRef] [PubMed]
- Ahn, K.; Johnson, D.S.; Cravatt, B.F. Fatty acid amide hydrolase as a potential therapeutic target for the treatment of pain and CNS disorders. Expert Opin. Drug Discov. 2009, 4, 763–784. [Google Scholar] [CrossRef] [PubMed]
- Kathuria, S.; Gaetani, S.; Fegley, D.; Valiño, F.; Duranti, A.; Tontini, A.; Mor, M.; Tarzia, G.; Rana, G.L.; Calignano, A.; et al. Modulation of anxiety through blockade of anandamide hydrolysis. Nat. Med. 2003, 9, 76–81. [Google Scholar] [CrossRef]
- Moreira, F.A.; Kaiser, N.; Monory, K.; Lutz, B. Reduced anxiety-like behaviour induced by genetic and pharmacological inhibition of the endocannabinoid-degrading enzyme fatty acid amide hydrolase (FAAH) is mediated by CB1 receptors. Neuropharmacology 2008, 54, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Naidu, P.S.; Varvel, S.A.; Ahn, K.; Cravatt, B.F.; Martin, B.R.; Lichtman, A.H. Evaluation of fatty acid amide hydrolase inhibition in murine models of emotionality. Psychopharmacology 2007, 192, 61–70. [Google Scholar] [CrossRef]
- Schmidt, M.E.; Liebowitz, M.R.; Stein, M.B.; Grunfeld, J.; Van Hove, I.; Simmons, W.K.; Van Der Ark, P.; Palmer, J.A.; Saad, Z.S.; Pemberton, D.J.; et al. ARTICLE The effects of inhibition of fatty acid amide hydrolase (FAAH) by JNJ-42165279 in social anxiety disorder: A double-blind, randomized, placebo-controlled proof-of-concept study. Neuropsychopharmacology 2021, 46, 1004–1010. [Google Scholar] [CrossRef]
- Cravatt, B.F.; Saghatelian, A.; Hawkins, E.G.; Clement, A.B.; Bracey, M.H.; Lichtman, A.H. Functional disassociation of the central and peripheral fatty acid amide signaling systems. Proc. Natl. Acad. Sci. USA 2004, 101, 10821–10826. [Google Scholar] [CrossRef]
- Holt, S.; Comelli, F.; Costa, B.; Fowler, C.J. Inhibitors of fatty acid amide hydrolase reduce carrageenan-induced hind paw inflammation in pentobarbital-treated mice: Comparison with indomethacin and possible involvement of cannabinoid receptors. Br. J. Pharmacol. 2005, 146, 467–476. [Google Scholar] [CrossRef]
- Wang, H.; Xie, H.; Guo, Y.; Zhang, H.; Takahashi, T.; Kingsley, P.J.; Marnett, L.J.; Das, S.K.; Cravatt, B.F.; Dey, S.K. Fatty acid amide hydrolase deficiency limits early pregnancy events. J. Clin. Investig. 2006, 116, 2122–2131. [Google Scholar] [CrossRef]
- Sun, X.; Deng, W.; Li, Y.; Tang, S.; Leishman, E.; Bradshaw, H.B.; Dey, S.K. Sustained Endocannabinoid Signaling Compromises Decidual Function and Promotes Inflammation-induced Preterm Birth. J. Biol. Chem. 2016, 291, 8231–8240. [Google Scholar] [CrossRef] [PubMed]
- Lord, J.S.; Gay, S.M.; Harper, K.M.; Nikolova, V.D.; Smith, K.M.; Moy, S.S.; Diering, G.H. Early life sleep disruption potentiates lasting sex-specific changes in behavior in genetically vulnerable Shank3 heterozygous autism model mice. Mol. Autism. 2022, 13, 35. [Google Scholar] [CrossRef] [PubMed]
- Trzeciakiewicz, H.; Ajit, D.; Tseng, J.H.; Chen, Y.; Ajit, A.; Tabassum, Z.; Lobrovich, R.; Peterson, C.; Riddick, N.V.; Itano, M.S.; et al. An HDAC6-dependent surveillance mechanism suppresses tau-mediated neurodegeneration and cognitive decline. Nat. Commun. 2020, 11, 5522. [Google Scholar] [CrossRef] [PubMed]
- Necarsulmer, J.C.; Simon, J.M.; Evangelista, B.A.; Chen, Y.; Tian, X.; Nafees, S.; Marquez, A.B.; Jiang, H.; Wang, P.; Ajit, D.; et al. RNA-binding deficient TDP-43 drives cognitive decline in a mouse model of TDP-43 proteinopathy. eLife 2023, 12, RP85921. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Species | Source | Concentration |
|---|---|---|---|
| Anti-AT8 | Mouse | Thermo Fisher UJ2859471 | 1:1000 |
| Anti-CB1 | Rabbit | Cell Signaling CST 93815 | 1:1000 |
| Anti-FAAH | Mouse | Abcam AB54615 | 1:1000 |
| Anti-Actin | Mouse | EMD Millipore MAB1501 | 1:10,000 |
| Anti-Hsc70 | Mouse | EMD Millipore MABE1120 | 1:1000 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martin, S.C.; Joyce, K.K.; Harper, K.M.; Harp, S.J.; Cohen, T.J.; Moy, S.S.; Diering, G.H. Evaluating Fatty Acid Amide Hydrolase as a Suitable Target for Sleep Promotion in a Transgenic TauP301S Mouse Model of Neurodegeneration. Pharmaceuticals 2024, 17, 319. https://doi.org/10.3390/ph17030319
Martin SC, Joyce KK, Harper KM, Harp SJ, Cohen TJ, Moy SS, Diering GH. Evaluating Fatty Acid Amide Hydrolase as a Suitable Target for Sleep Promotion in a Transgenic TauP301S Mouse Model of Neurodegeneration. Pharmaceuticals. 2024; 17(3):319. https://doi.org/10.3390/ph17030319
Chicago/Turabian StyleMartin, Shenée C., Kathryn K. Joyce, Kathryn M. Harper, Samuel J. Harp, Todd J. Cohen, Sheryl S. Moy, and Graham H. Diering. 2024. "Evaluating Fatty Acid Amide Hydrolase as a Suitable Target for Sleep Promotion in a Transgenic TauP301S Mouse Model of Neurodegeneration" Pharmaceuticals 17, no. 3: 319. https://doi.org/10.3390/ph17030319
APA StyleMartin, S. C., Joyce, K. K., Harper, K. M., Harp, S. J., Cohen, T. J., Moy, S. S., & Diering, G. H. (2024). Evaluating Fatty Acid Amide Hydrolase as a Suitable Target for Sleep Promotion in a Transgenic TauP301S Mouse Model of Neurodegeneration. Pharmaceuticals, 17(3), 319. https://doi.org/10.3390/ph17030319