
Determination of the Cannabinoid CB1 Receptor’s Positive Allosteric Modulator Binding Site through Mutagenesis Studies

 , and
, and
Abstract
1. Introduction
2. Results
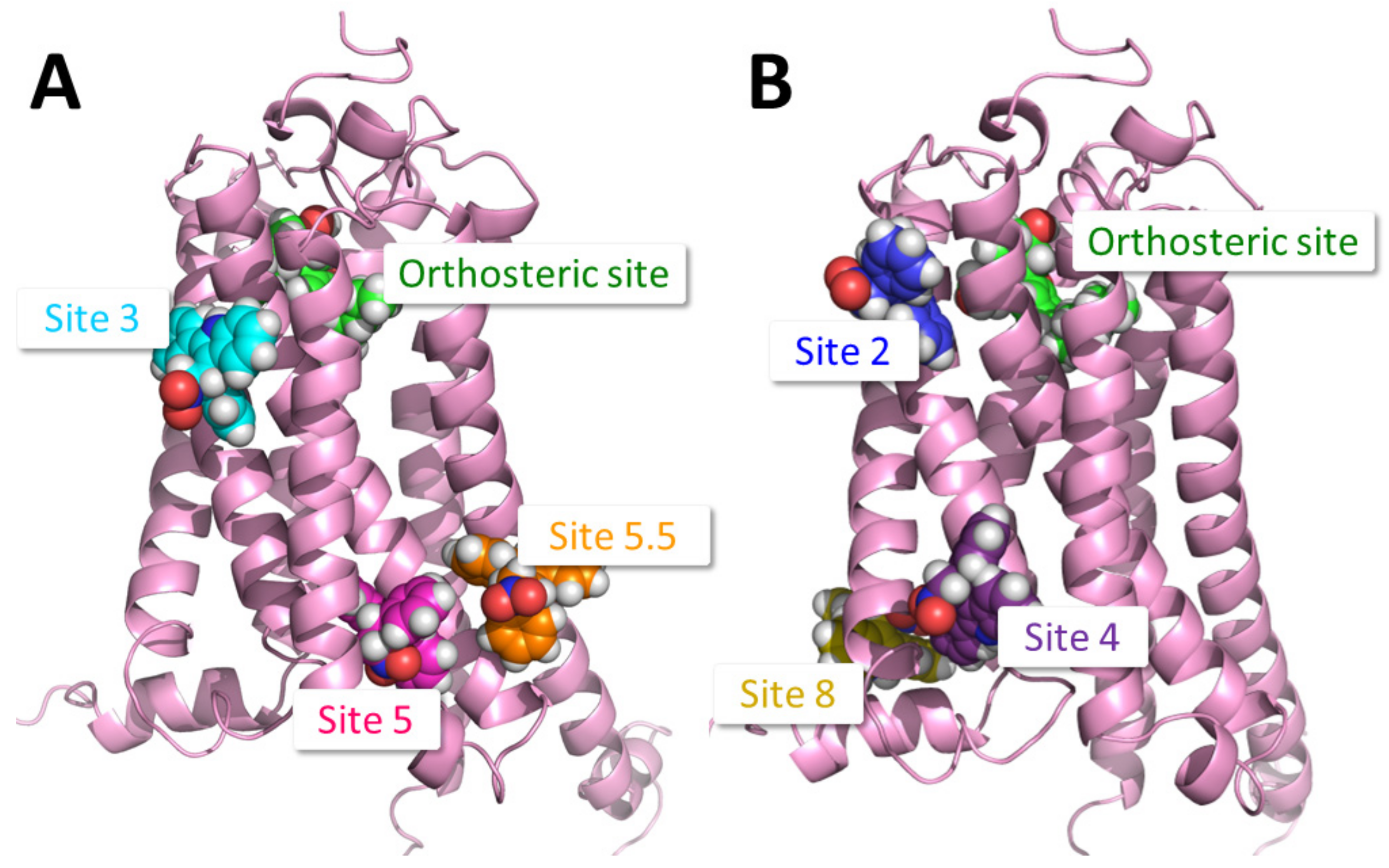
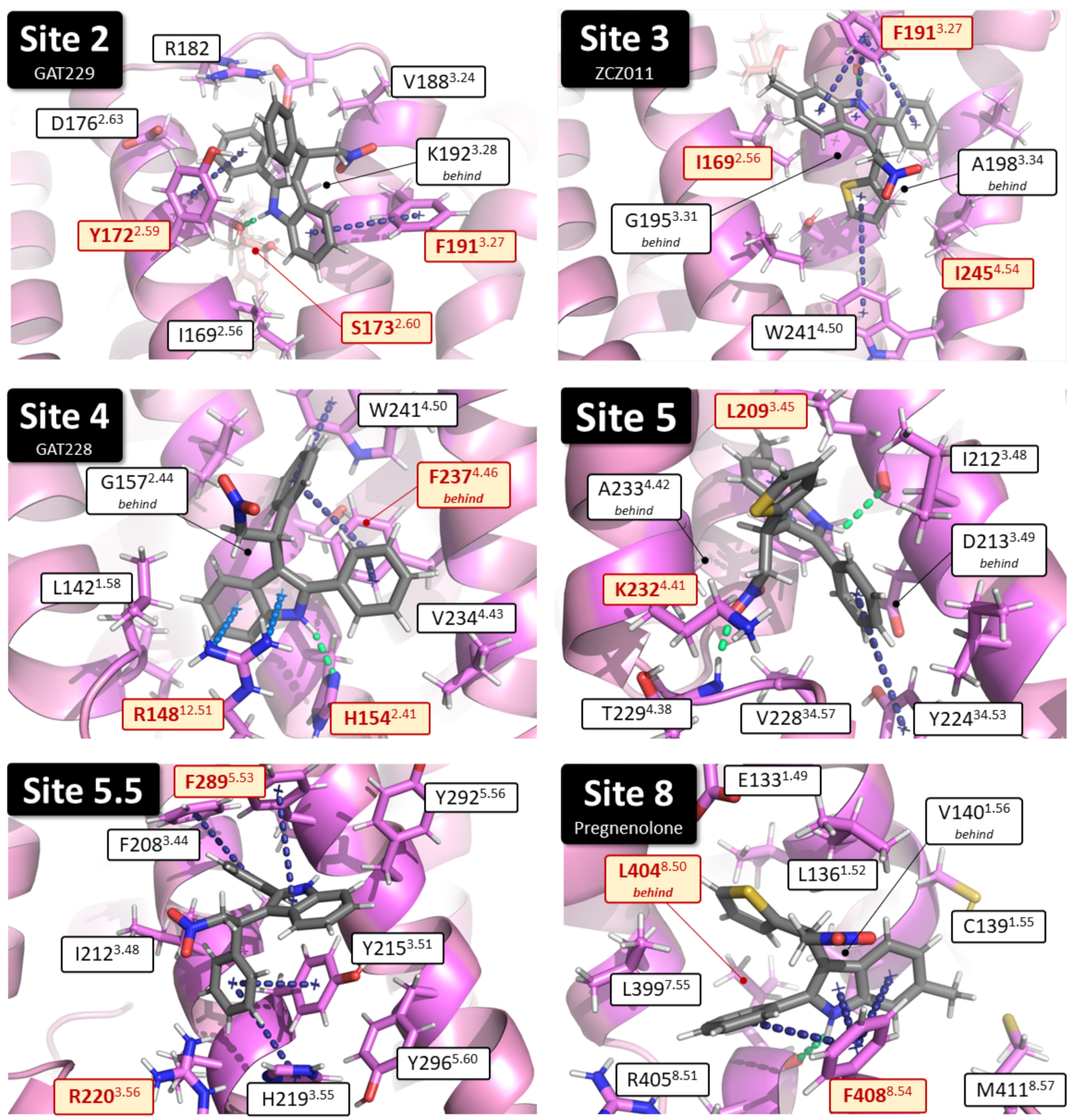
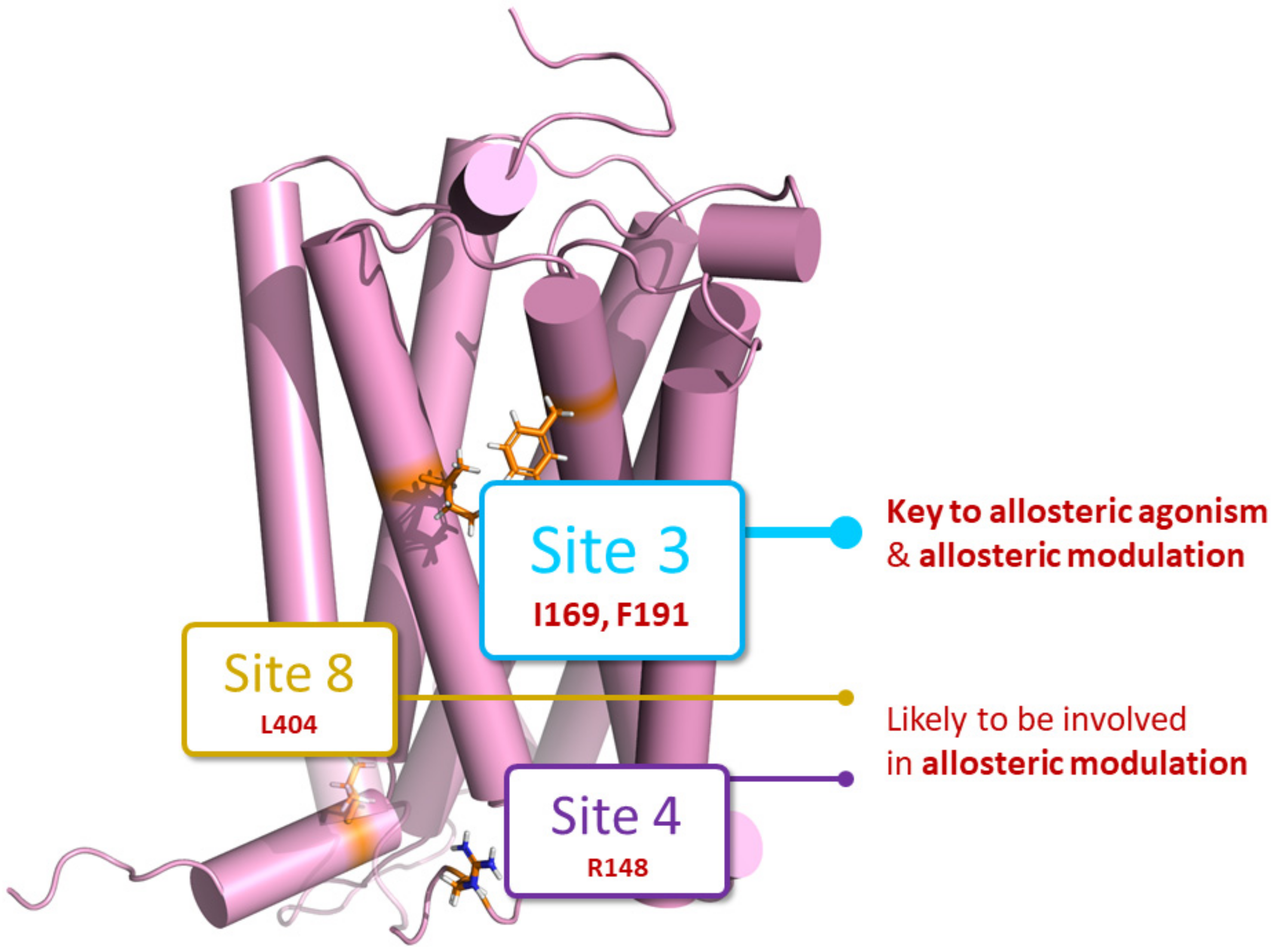
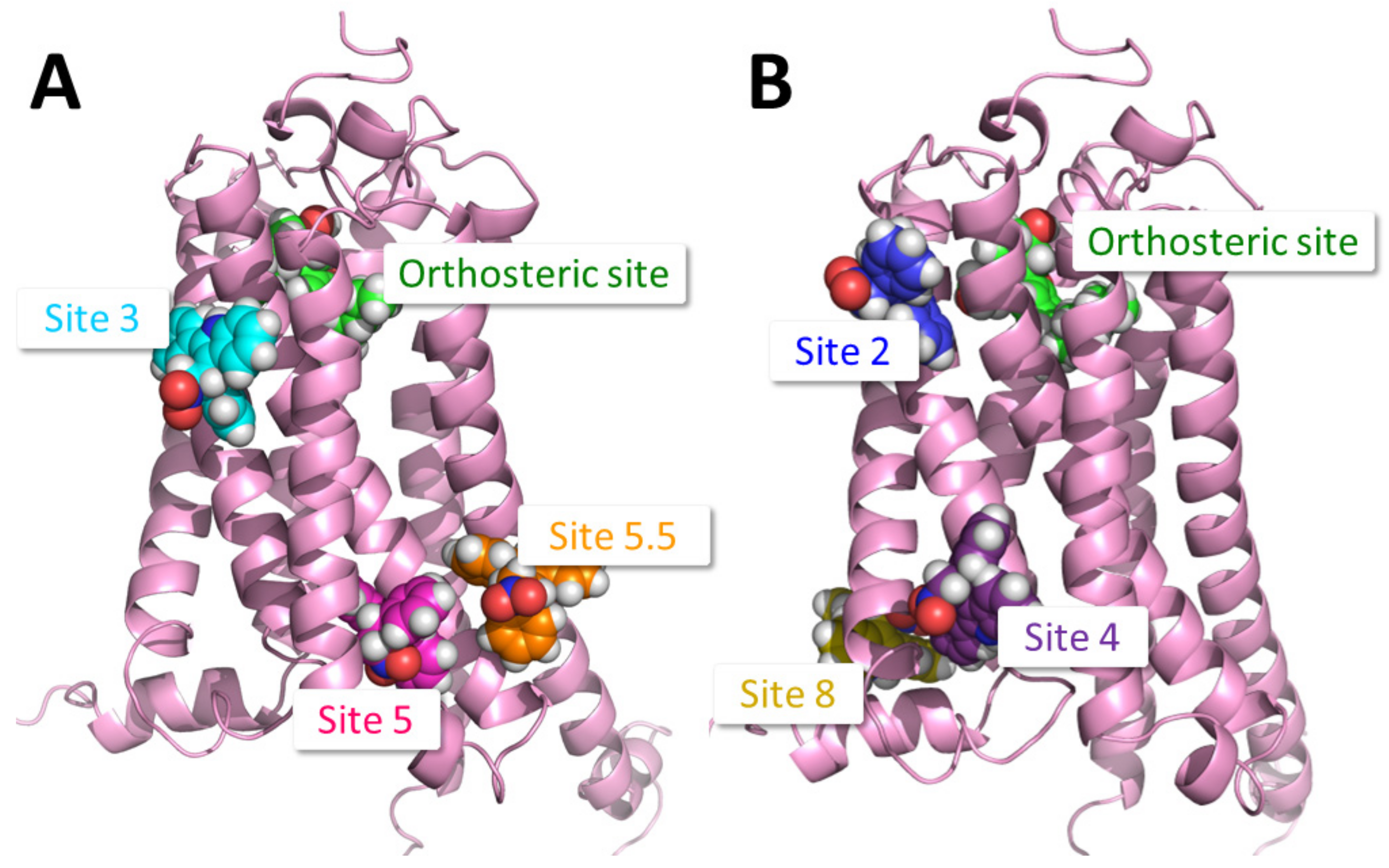
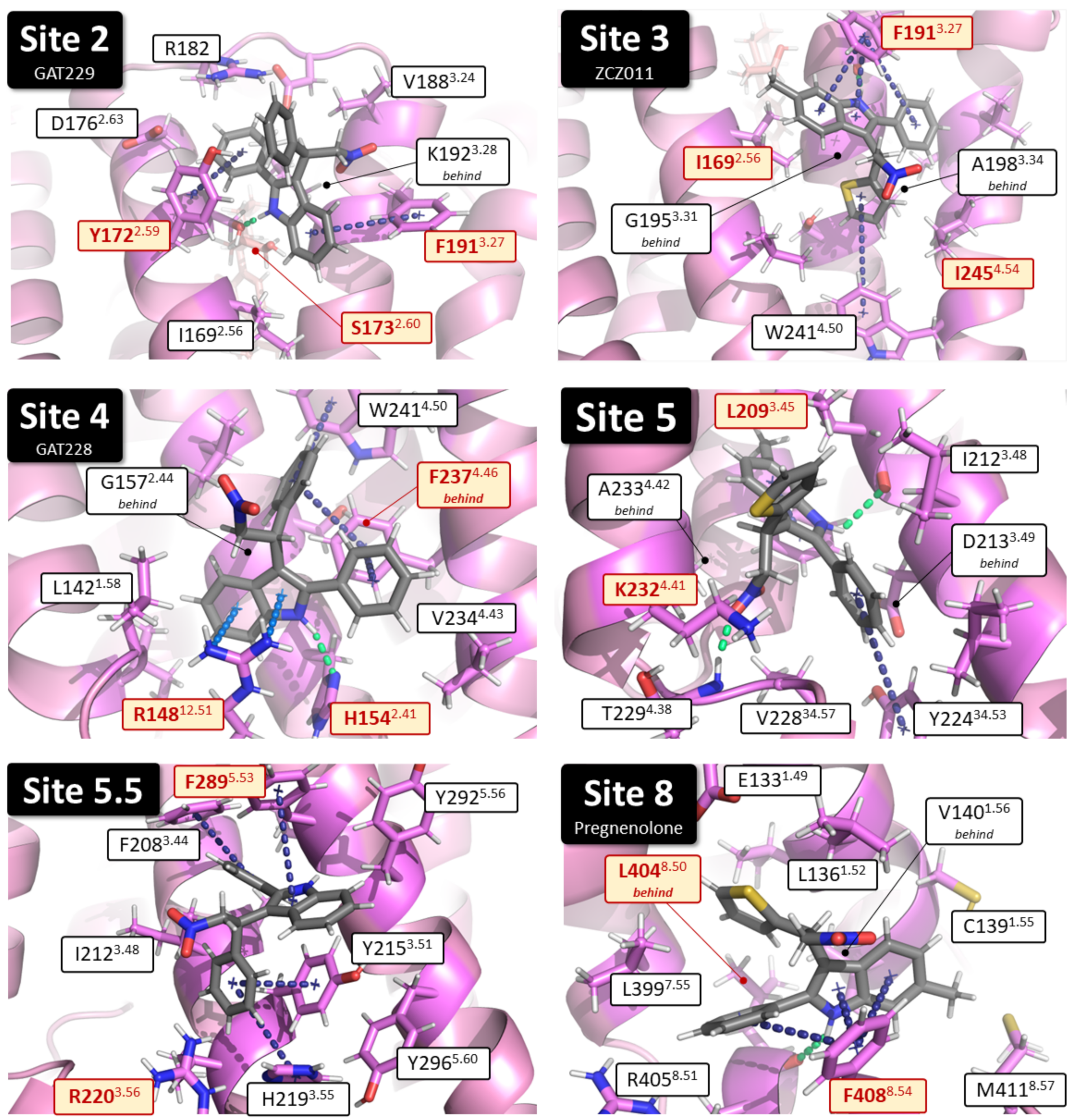
2.1. Identification of Putative PAM Binding Sites
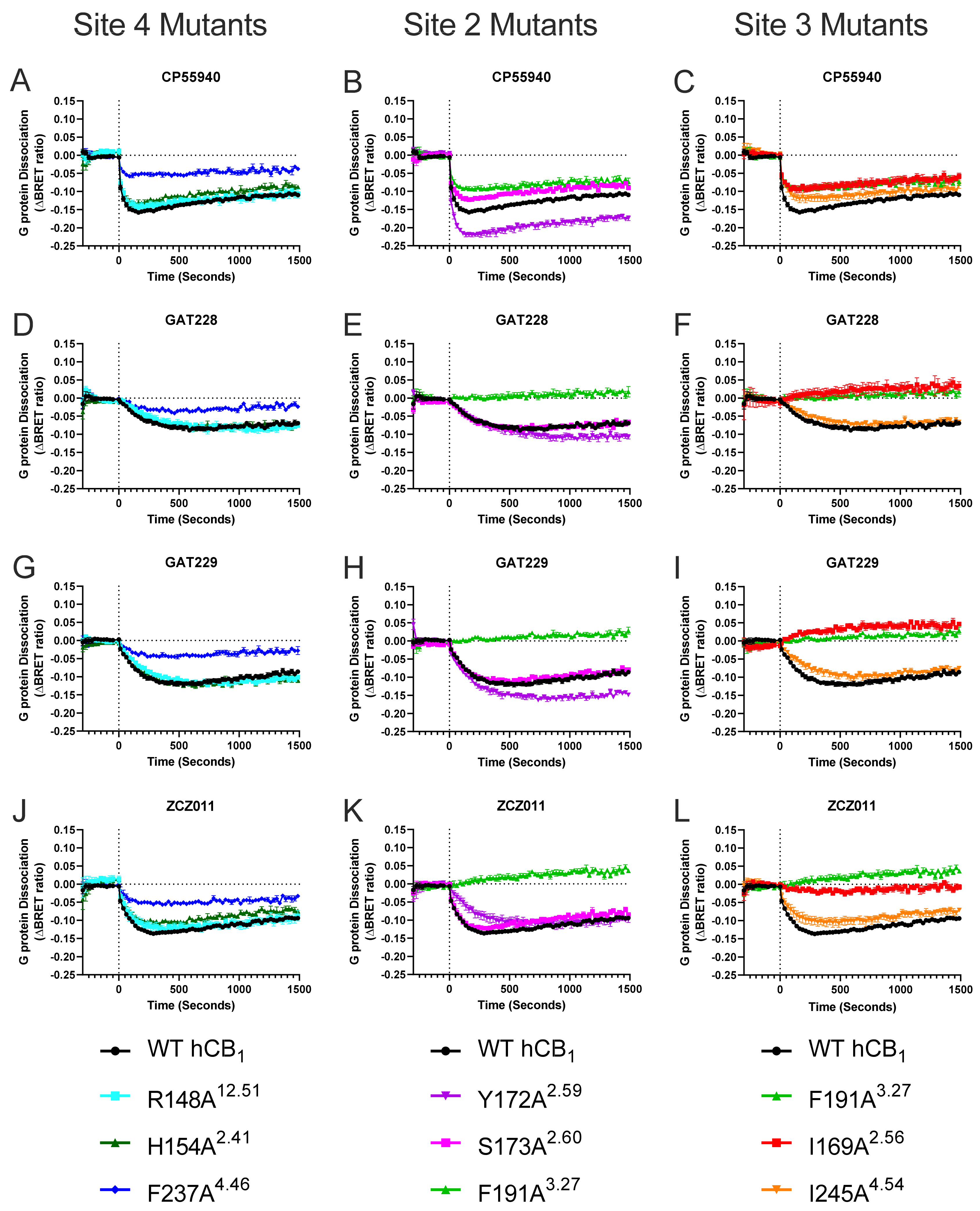
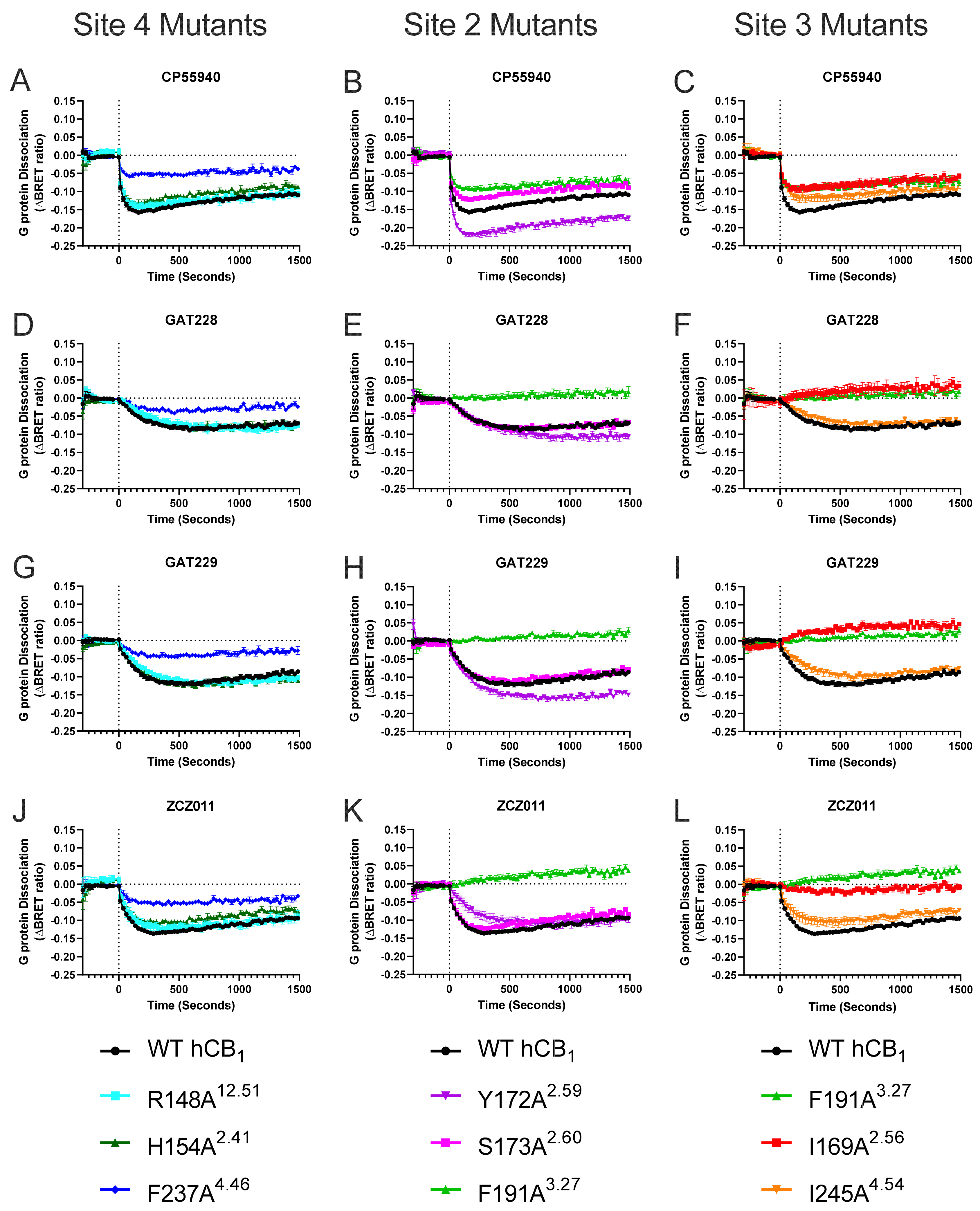
2.2. Effect of Mutating Key hCB1 Residues on Orthosteric and Allosteric Agonist/Inverse Agonist-Induced G Protein Dissociation
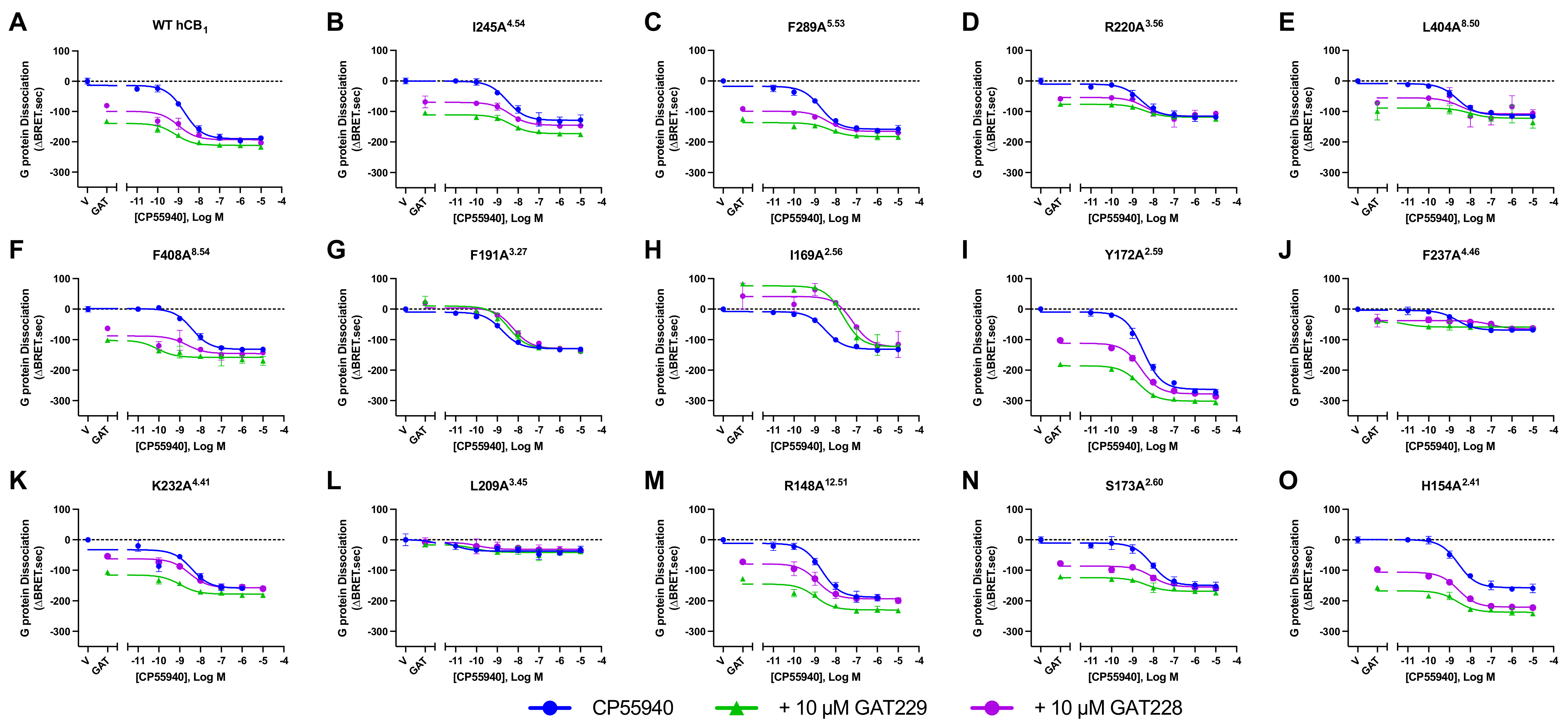
2.3. Effect of hCB1 Mutants on Orthosteric Agonist-Induced G Protein Dissociation in Combination with Allosteric Ligands
2.4. Receptor Expression
3. Discussion
4. Materials and Methods
4.1. Drugs
4.2. Cell Lines and Maintenance
4.3. Identification of Putative PAM Binding Sites
4.4. QuikChange® Mutagenesis—Development of hCB1 Mutants
4.5. pIRES G Protein Dissociation Assay
4.6. Immunocytochemistry for Receptor Expression
4.7. Data and Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Katona, I.; Freund, T.F. Endocannabinoid signaling as a synaptic circuit breaker in neurological disease. Nat. Med. 2008, 14, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Pertwee, R.G. The diverse CB1 and CB2 receptor pharmacology of three plant cannabinoids: Δ9-tetrahydrocannabinol, cannabidiol and Δ9-tetrahydrocannabivarin. Br. J. Pharmacol. 2008, 153, 199–215. [Google Scholar] [CrossRef] [PubMed]
- Buckner, J.D.; Heimberg, R.G.; Matthews, R.A.; Silgado, J. Marijuana-related problems and social anxiety: The role of marijuana behaviors in social situations. Psychol. Addict. Behav. 2012, 26, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Haspula, D.; Clark, M.A. Cannabinoid Receptors: An Update on Cell Signaling, Pathophysiological Roles and Therapeutic Opportunities in Neurological, Cardiovascular, and Inflammatory Diseases. Int. J. Mol. Sci. 2020, 21, 7693. [Google Scholar] [CrossRef] [PubMed]
- Kenakin, T. Allosteric Theory: Taking Therapeutic Advantage of the Malleable Nature of GPCRs. Curr. Neuropharmacol. 2007, 5, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Wootten, D.; Christopoulos, A.; Sexton, P.M. Emerging paradigms in GPCR allostery: Implications for drug discovery. Nat. Rev. Drug Discov. 2013, 12, 630–644. [Google Scholar] [CrossRef]
- Navarro, H.A.; Howard, J.L.; Pollard, G.T.; Carroll, F.I. Positive allosteric modulation of the human cannabinoid (CB1) receptor by RTI-371, a selective inhibitor of the dopamine transporter. Br. J. Pharmacol. 2009, 156, 1178–1184. [Google Scholar] [CrossRef]
- Ignatowska-Jankowska, B.M.; Baillie, G.L.; Kinsey, S.; Crowe, M.; Ghosh, S.; Owens, R.A.; Damaj, I.M.; Poklis, J.; Wiley, J.L.; Zanda, M.; et al. A Cannabinoid CB1 Receptor-Positive Allosteric Modulator Reduces Neuropathic Pain in the Mouse with No Psychoactive Effects. Neuropsychopharmacology 2015, 40, 2948–2959. [Google Scholar] [CrossRef]
- Laprairie, R.B.; Kulkarni, P.M.; Deschamps, J.R.; Kelly, M.E.M.; Janero, D.R.; Cascio, M.G.; Stevenson, L.A.; Pertwee, R.G.; Kenakin, T.P.; Denovan-Wright, E.M.; et al. Enantiospecific Allosteric Modulation of Cannabinoid 1 Receptor. ACS Chem. Neurosci. 2017, 8, 1188–1203. [Google Scholar] [CrossRef]
- Green, H.M.; Finlay, D.B.; Ross, R.A.; Greig, I.R.; Duffull, S.B.; Glass, M. In Vitro Characterization of 6-Methyl-3-(2-nitro-1-(thiophen-2-yl)ethyl)-2-phenyl-1 H-indole (ZCZ011) at the Type 1 Cannabinoid Receptor: Allosteric Agonist or Allosteric Modulator? ACS Pharmacol. Transl. Sci. 2022, 5, 1279–1291. [Google Scholar] [CrossRef]
- Cairns, E.A.; Szczesniak, A.-M.; Straiker, A.J.; Kulkarni, P.M.; Pertwee, R.G.; Thakur, G.A.; Baldridge, W.H.; Kelly, M.E. The In Vivo Effects of the CB1-Positive Allosteric Modulator GAT229 on Intraocular Pressure in Ocular Normotensive and Hypertensive Mice. J. Ocul. Pharmacol. Ther. 2017, 33, 582–590. [Google Scholar] [CrossRef]
- Slivicki, R.A.; Xu, Z.; Kulkarni, P.M.; Pertwee, R.G.; Mackie, K.; Thakur, G.A.; Hohmann, A.G. Positive Allosteric Modulation of Cannabinoid Receptor Type 1 Suppresses Pathological Pain Without Producing Tolerance or Dependence. Biol. Psychiatry 2018, 84, 722–733. [Google Scholar] [CrossRef]
- Laprairie, R.B.; Bagher, A.M.; Rourke, J.L.; Zrein, A.; Cairns, E.A.; Kelly, M.E.; Sinal, C.J.; Kulkarni, P.M.; Thakur, G.A.; Denovan-Wright, E.M. Positive allosteric modulation of the type 1 cannabinoid receptor reduces the signs and symptoms of Huntington’s disease in the R6/2 mouse model. Neuropharmacology 2019, 151, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Tseng, C.-C.; Baillie, G.; Donvito, G.; Mustafa, M.A.; Juola, S.E.; Zanato, C.; Massarenti, C.; Dall’angelo, S.; Harrison, W.T.A.; Lichtman, A.H.; et al. The Trifluoromethyl Group as a Bioisosteric Replacement of the Aliphatic Nitro Group in CB1 Receptor Positive Allosteric Modulators. J. Med. Chem. 2019, 62, 5049–5062. [Google Scholar] [CrossRef] [PubMed]
- Slivicki, R.A.; Iyer, V.; Mali, S.S.; Garai, S.; Thakur, G.A.; Crystal, J.D.; Hohmann, A.G. Positive Allosteric Modulation of CB1 Cannabinoid Receptor Signaling Enhances Morphine Antinociception and Attenuates Morphine Tolerance Without Enhancing Morphine- Induced Dependence or Reward. Front. Mol. Neurosci. 2020, 13, 54. [Google Scholar] [CrossRef] [PubMed]
- Dodu, J.C.; Moncayo, R.K.; Damaj, M.I.; Schlosburg, J.E.; Akbarali, H.I.; O’brien, L.D.; Kendall, D.A.; Wu, Z.; Lu, D.; Lichtman, A.H. The Cannabinoid Receptor Type 1 Positive Allosteric Modulator ZCZ011 Attenuates Naloxone-Precipitated Diarrhea and Weight Loss in Oxycodone-Dependent Mice. J. Pharmacol. Exp. Ther. 2022, 380, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Roebuck, A.J.; Greba, Q.; Smolyakova, A.-M.; Alaverdashvili, M.; Marks, W.N.; Garai, S.; Baglot, S.L.; Petrie, G.; Cain, S.M.; Snutch, T.P.; et al. Positive allosteric modulation of type 1 cannabinoid receptors reduces spike-and-wave discharges in Genetic Absence Epilepsy Rats from Strasbourg. Neuropharmacology 2021, 190, 108553. [Google Scholar] [CrossRef] [PubMed]
- McElroy, D.L.; Roebuck, A.J.; Greba, Q.; Garai, S.; Brandt, A.L.; Yilmaz, O.; Cain, S.M.; Snutch, T.P.; Thakur, G.A.; Laprairie, R.B.; et al. The type 1 cannabinoid receptor positive allosteric modulators GAT591 and GAT593 reduce spike-and-wave discharges in Genetic Absence Epilepsy Rats from Strasbourg. IBRO Neurosci. Rep. 2022, 12, 121–130. [Google Scholar] [CrossRef]
- Garai, S.; Schaffer, P.C.; Laprairie, R.B.; Janero, D.R.; Pertwee, R.G.; Straiker, A.; Thakur, G.A. Design, synthesis, and pharmacological profiling of cannabinoid 1 receptor allosteric modulators: Preclinical efficacy of C2-group GAT211 congeners for reducing intraocular pressure. Bioorganic Med. Chem. 2021, 50, 116421. [Google Scholar] [CrossRef]
- Garai, S.; Kulkarni, P.M.; Schaffer, P.C.; Leo, L.M.; Brandt, A.L.; Zagzoog, A.; Black, T.; Lin, X.; Hurst, D.P.; Janero, D.R.; et al. Application of Fluorine- and Nitrogen-Walk Approaches: Defining the Structural and Functional Diversity of 2-Phenylindole Class of Cannabinoid 1 Receptor Positive Allosteric Modulators. J. Med. Chem. 2020, 63, 542–568. [Google Scholar] [CrossRef]
- Nepali, K.; Lee, H.-Y.; Liou, J.-P. Nitro-Group-Containing Drugs. J. Med. Chem. 2019, 62, 2851–2893. [Google Scholar] [CrossRef]
- Hurst, D.P.; Garai, S.; Kulkarni, P.M.; Schaffer, P.C.; Reggio, P.H.; Thakur, G.A. Identification of CB1 Receptor Allosteric Sites Using Force-Biased MMC Simulated Annealing and Validation by Structure–Activity Relationship Studies. ACS Med. Chem. Lett. 2019, 10, 1216–1221. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Wang, X.; Xu, Z.; Wu, C.; Zhou, Y.; Wang, Y.; Lin, G.; Li, K.; Wu, M.; Xia, A.; et al. Molecular mechanism of allosteric modulation for the cannabinoid receptor CB1. Nat. Chem. Biol. 2022, 18, 831–840. [Google Scholar] [CrossRef] [PubMed]
- Vallee, M.; Vitiello, S.; Bellocchio, L.; Hébert-Chatelain, E.; Monlezun, S.; Martín-García, E.; Kasanetz, F.; Baillie, G.L.; Panin, F.; Cathala, A.; et al. Pregnenolone Can Protect the Brain from Cannabis Intoxication. Science 2014, 343, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Ballesteros, J.A.; Weinstein, H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods Neurosci. 1995, 25, 366–428. [Google Scholar]
- Corey, R.A.; Stansfeld, P.J.; Sansom, M.S. The energetics of protein–lipid interactions as viewed by molecular simulations. Biochem. Soc. Trans. 2019, 48, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Shao, Z.; Yan, W.; Chapman, K.; Ramesh, K.; Ferrell, A.J.; Yin, J.; Wang, X.; Xu, Q.; Rosenbaum, D.M. Structure of an allosteric modulator bound to the CB1 cannabinoid receptor. Nat. Chem. Biol. 2019, 15, 1199–1205. [Google Scholar] [CrossRef] [PubMed]
- Maza, N.; Wang, D.; Kowalski, C.; Stoveken, H.M.; Dao, M.; Sial, O.K.; Giles, A.C.; Grill, B.; Martemyanov, K.A. Ptchd1 mediates opioid tolerance via cholesterol-dependent effects on μ-opioid receptor trafficking. Nat. Neurosci. 2022, 25, 1179–1190. [Google Scholar] [CrossRef] [PubMed]
- Díaz, O.; Renault, P.; Giraldo, J. Evaluating Allosteric Perturbations in Cannabinoid Receptor 1 by In Silico Single-Point Mutation. ACS Omega 2022, 7, 37873–37884. [Google Scholar] [CrossRef]
- Matti, C.; Salnikov, A.; Artinger, M.; D’Agostino, G.; Kindinger, I.; Uguccioni, M.; Thelen, M.; Legler, D.F. ACKR4 Recruits GRK3 Prior to β-Arrestins but Can Scavenge Chemokines in the Absence of β-Arrestins. Front. Immunol. 2020, 11, 720. [Google Scholar] [CrossRef]
- Finlay, D.B.; Cawston, E.E.; Grimsey, N.L.; Hunter, M.R.; Korde, A.; Vemuri, V.K.; Makriyannis, A.; Glass, M. Gαs signalling of the CB1 receptor and the influence of receptor number. Br. J. Pharmacol. 2017, 174, 2545–2562. [Google Scholar] [CrossRef]
- Halgren, T. New method for fast and accurate binding-site identification and analysis. Chem. Biol. Drug Des. 2007, 69, 146–148. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A. Identifying and characterizing binding sites and assessing druggability. J. Chem. Inf. Model. 2009, 49, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Hua, T.; Li, X.; Wu, L.; Iliopoulos-Tsoutsouvas, C.; Wang, Y.; Wu, M.; Shen, L.; Brust, C.A.; Nikas, S.P.; Song, F.; et al. Activation and Signaling Mechanism Revealed by Cannabinoid Receptor-Gi Complex Structures. Cell 2020, 180, 655–665.e18. [Google Scholar] [CrossRef] [PubMed]
- Manning, J.J.; Rawcliffe, G.; Finlay, D.B.; Glass, M. Cannabinoid 1 (CB1) receptor arrestin subtype-selectivity and phosphorylation dependence. Br. J. Pharmacol. 2023, 180, 369–382. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.; Matti, C.; Grimsey, N.L.; Legler, D.F.; Javitch, J.A.; Finlay, D.B.; Glass, M. Delineating the interactions between the cannabinoid CB2 receptor and its regulatory effectors; β-arrestins and GPCR kinases. Br. J. Pharmacol. 2022, 179, 2223–2239. [Google Scholar] [CrossRef] [PubMed]
- Hall, D.A.; Langmead, C.J. Matching models to data: A receptor pharmacologist’s guide. Br. J. Pharmacol. 2010, 161, 1276–1290. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Binding Site | Proposed Ligand | Mutation | BW Number a | % Conserved b | Consensus Residue b | % CB1 c |
|---|---|---|---|---|---|---|
| Site 2 | GAT229 | Y172A | 2.59 × 59 | 36 | F | 1 |
| S173A | 2.60 × 60 | 12 | V | 1 | ||
| Site 2/3 | F191A | 3.27 × 27 | 29 | L | 13 | |
| Site 3 | ZCZ011 | F191A | 3.27 × 27 | 29 | L | 13 |
| I169A | 2.56 × 56 | 24 | T | 6 | ||
| I245A | 4.54 × 54 | 28 | L | 10 | ||
| Site 4 | GAT228 | R148A | 12.51 × 51 | 28 | Gap d | - |
| H154A | 2.41 × 41 | 22 | I | 1 | ||
| F237A | 4.46 × 46 | 29 | I | 1 | ||
| Site 5 | Cholesterol | K232A | 4.41 × 41 | 25 | R | 8 |
| L209A | 3.45 × 45 | 38 | A | 13 | ||
| Site 5.5 | Cholesterol | F289A | 5.53 × 53 | 30 | I | 5 |
| R220A | 3.56 × 56 | 32 | H | 13 | ||
| Site 8 | Pregnenolone | L404A | 8.50 × 50 | 60 | F | 9 |
| F408A | 8.54 × 54 | 33 | F | 35 |
| CP55940 Alone | CP55940 + 10 μM GAT229 | CP55940 + 10 μM GAT228 | |||||
|---|---|---|---|---|---|---|---|
| Site | Mutation | pEC50 | EMAX | pEC50 | EMAX | pEC50 | EMAX |
| - | WT b | 8.59 ± 0.06 | −184.1 ± 3.8 | 8.65 ± 0.13 | −205.1 ± 7.8 † | 8.42 ± 0.08 † | −194.2 ± 7.3 |
| Site 2 | Y172A2.59 | 8.46 ± 0.02 | −273.2 ± 6.9 * | 8.62 ± 0.03 † | −309.0 ± 3.5 † | 8.60 ± 0.04 † | −298.9 ± 5.4 † |
| S173A2.60 | 8.00 ± 0.08 * | −143.0 ± 5.7 * | 8.20 ± 0.08 † | −186.4 ± 7.5 † | 7.91 ± 0.10 | −176.1 ± 6.9 † | |
| Site 2/3 | F191A3.27 | 8.87 ± 0.06 | −117.3 ± 6.1 * | 8.46 ± 0.07 † | −119.1 ± 7.8 | 8.33 ± 0.06 † | −123.9 ± 7.4 |
| Site 3 | I169A2.56 | 8.20 ± 0.08 * | −110.7 ± 10.3 * | 7.65 ± 0.03 † | −109.7 ± 8.3 | 7.76 ± 0.16 | −117.0 ± 7.4 |
| I245A4.54 | 8.51 ± 0.07 | −150.1 ± 9.8 | 8.58 ± 0.18 | −182.9 ± 7.0 † | 8.26 ± 0.16 | −163.7 ± 8.2 | |
| Site 4 | R148A12.51 | 8.46 ± 0.10 | −175.6 ± 11.4 | 8.93 ± 0.06 † | −243.9 ± 5.4 † | 8.67 ± 0.09 † | −218.2 ± 8.5 |
| H154A2.41 | 8.60 ± 0.10 | −151.8 ± 5.9 * | 9.06 ± 0.40 | −210.6 ± 10.1 † | 8.53 ± 0.13 | −200.5 ± 11.6 † | |
| F237A4.46 | 8.60 ± 0.11 | −69.8 ± 6.7 * | 8.60 ± 0.70 | −67.4 ± 8.3 | 7.52 ± 0.30 † | −69.4 ± 6.4 | |
| Site 5 | K232A4.41 | 8.61 ± 0.05 | −160.6 ± 7.6 | 8.58 ± 0.20 | −194.9 ± 9.0 † | 8.45 ± 0.11 | −181.9 ± 8.6 |
| L209A3.45 | ND | −39.8 ± 1.1 * | ND | −51.9 ± 2.8 † | ND | −43.2 ± 4.5 | |
| Site 5.5 | F289A5.53 | 8.62 ± 0.11 | −145.4 ± 5.7 | 7.97 ± 0.22 | −161.1 ± 6.5 † | 8.17 ± 0.10 † | −156.7 ± 5.3 |
| R220A3.56 | 8.58 ± 0.08 | −110.8 ± 2.8 * | 8.76 ± 0.34 | −144.6 ± 10.4 | 8.41 ± 0.14 | −140.1 ± 13.5 | |
| Site 8 | L404A8.50 | 8.66 ± 0.05 | −103.1 ± 3.6 * | 8.36 ± 0.45 | −122.3 ± 7.0 | 8.39 ± 0.12 † | −115.1 ± 8.3 |
| F408A8.54 | 8.56 ± 0.07 | −128.6 ± 12.1 * | 8.98 ± 0.38 | −154.0 ± 14.5 † | 8.11 ± 0.20 | −148.4 ± 11.4 † | |
| Site | Mutation | 10 μM CP55940 | 10 μM GAT229 | 10 μM GAT228 | 10 μM ZCZ011 | 10 μM CP55940 + 10 μM ZCZ011 |
|---|---|---|---|---|---|---|
| - | WT b | −188.7 ± 3.6 | −148.6 ± 7.2 | −105.7 ± 7.2 | −167.3 ± 3.2 | −215.9 ± 2.9 † |
| Site 2 | Y172A2.59 | −284.7 ± 8.4 * | −207.0 ± 7.1 * | −133.4 ± 9.8 * | −142.1 ± 14.4 | −305.2 ± 5.7 † |
| S173A2.60 | −146.2 ± 5.5 * | −137.3 ± 4.8 | −105.5 ± 7.6 | −144.2 ± 6.8 | −186.8 ± 5.5 † | |
| Site 2/3 | F191A3.27 | −117.8 ± 7.5 * | 22.1 ± 6.4 * | 19.5 ± 11.9 * | 35.0 ± 8.4 * | −134.7 ± 12.4 † |
| Site 3 | I169A2.56 | −113.1 ± 11.7 * | 54.8 ± 7.5 * | 30.0 ± 5.9 * | −20.0 ± 9.0 * | −120.0 ± 10.9 |
| I245A4.54 | −154.9 ± 10.6 | −121.6 ± 8.0 | −90.3 ± 8.5 | −130.4 ± 12.9 | −183.2 ± 7.2 † | |
| Site 4 | R148A12.51 | −179.5 ± 12.2 | −148.5 ± 7.7 | −102.7 ± 11.8 | −150.1 ± 10.2 | −237.3 ± 11.6 † |
| H154A2.41 | −155.3 ± 6.5 * | −154.6 ± 7.8 | −101.6 ± 6.9 | −129.4 ± 5.1 * | −189.5 ± 8.5 † | |
| F237A4.46 | −69.8 ± 6.5 * | −52.4 ± 5.2 * | −40.8 ± 2.8 * | −64.2 ± 4.1 * | −85.2 ± 9.9 † | |
| Site 5 | K232A4.41 | −167.7 ± 9.6 | −134.5 ± 7.9 | −94.0 ± 10.5 | −140.0 ± 7.3 | −194.9 ± 7.2 † |
| L209A3.45 | −42.7 ± 2.8 * | −25.1 ± 3.5 * | −19.0 ± 3.3 * | −34.4 ± 2.9 * | −71.3 ± 4.2 † | |
| Site 5.5 | F289A5.53 | −148.1 ± 5.2 | −123.6 ± 5.9 | −97.4 ± 7.2 | −135.5 ± 6.3 | −167.8 ± 6.9 † |
| R220A3.56 | −112.1 ± 2.9 * | −102.9 ± 9.9 | −85.2 ± 13.6 | −95.7 ± 7.7 * | −137.8 ± 5.0 † | |
| Site 8 | L404A8.50 | −110.3 ± 3.7 * | −88.9 ± 6.2 | −63.2 ± 5.6 | −85.2 ± 7.3 * | −133.5 ± 5.6 † |
| F408A8.54 | −131.7 ± 10.4 * | −110.0 ± 8.6 | −90.3 ± 8.3 | −116.9 ± 10.1 * | −159.7 ± 13.4 † |
| Site | Mutant | Surface Receptor Expression | Total Receptor Expression |
|---|---|---|---|
| - | WT b | 224.5 ± 37.7 | 895.1 ± 141.3 |
| Site 2 | Y172A2.59 | 482.7 ± 104.8 | 1568 ± 448.5 |
| S173A2.60 | 313.2 ± 62.7 | 1477 ± 425.9 | |
| Site 2/3 | F191A3.27 | 82.9 ± 43.6 | 620.2 ± 214.6 |
| Site 3 | I169A2.56 | 88.9 ± 33.8 | 525.5 ± 231.3 |
| I245A4.54 | 117.9 ± 60.1 | 631.2 ± 226.6 | |
| Site 4 | R148A12.51 | 125.7 ± 34.4 * | 966.5 ± 351.3 |
| H154A2.41 | 217.8 ± 70.1 | 1047 ± 394.8 | |
| F237A4.46 | 71.5 ± 22.9 * | 1117 ± 354.8 | |
| Site 5 | K232A4.41 | 207.4 ± 50.5 | 1154 ± 351.2 |
| L209A3.45 | 18.7 ± 17.4 * | 532.2 ± 173.6 * | |
| Site 5.5 | F289A5.53 | 217.4 ± 93.5 | 1043 ± 340.5 |
| R220A3.56 | 160.2 ± 56.3 | 813.6 ± 192.8 | |
| Site 8 | L404A8.50 | 164.1 ± 126.2 | 389.7.1 ± 154.2 * |
| F408A8.54 | 110.9 ± 55.6 | 634.9 ± 257.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Green, H.M.; Fellner, D.M.J.; Finlay, D.B.; Furkert, D.P.; Glass, M. Determination of the Cannabinoid CB1 Receptor’s Positive Allosteric Modulator Binding Site through Mutagenesis Studies. Pharmaceuticals 2024, 17, 154. https://doi.org/10.3390/ph17020154
Green HM, Fellner DMJ, Finlay DB, Furkert DP, Glass M. Determination of the Cannabinoid CB1 Receptor’s Positive Allosteric Modulator Binding Site through Mutagenesis Studies. Pharmaceuticals. 2024; 17(2):154. https://doi.org/10.3390/ph17020154
Chicago/Turabian StyleGreen, Hayley M., Daniel M. J. Fellner, David B. Finlay, Daniel P. Furkert, and Michelle Glass. 2024. "Determination of the Cannabinoid CB1 Receptor’s Positive Allosteric Modulator Binding Site through Mutagenesis Studies" Pharmaceuticals 17, no. 2: 154. https://doi.org/10.3390/ph17020154
APA StyleGreen, H. M., Fellner, D. M. J., Finlay, D. B., Furkert, D. P., & Glass, M. (2024). Determination of the Cannabinoid CB1 Receptor’s Positive Allosteric Modulator Binding Site through Mutagenesis Studies. Pharmaceuticals, 17(2), 154. https://doi.org/10.3390/ph17020154