


Mulberry Twig Alkaloids Improved the Progression of Metabolic-Associated Fatty Liver Disease in High-Fat Diet-Induced Obese Mice by Regulating the PGC1α/PPARα and KEAP1/NRF2 Pathways

 ,
,
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
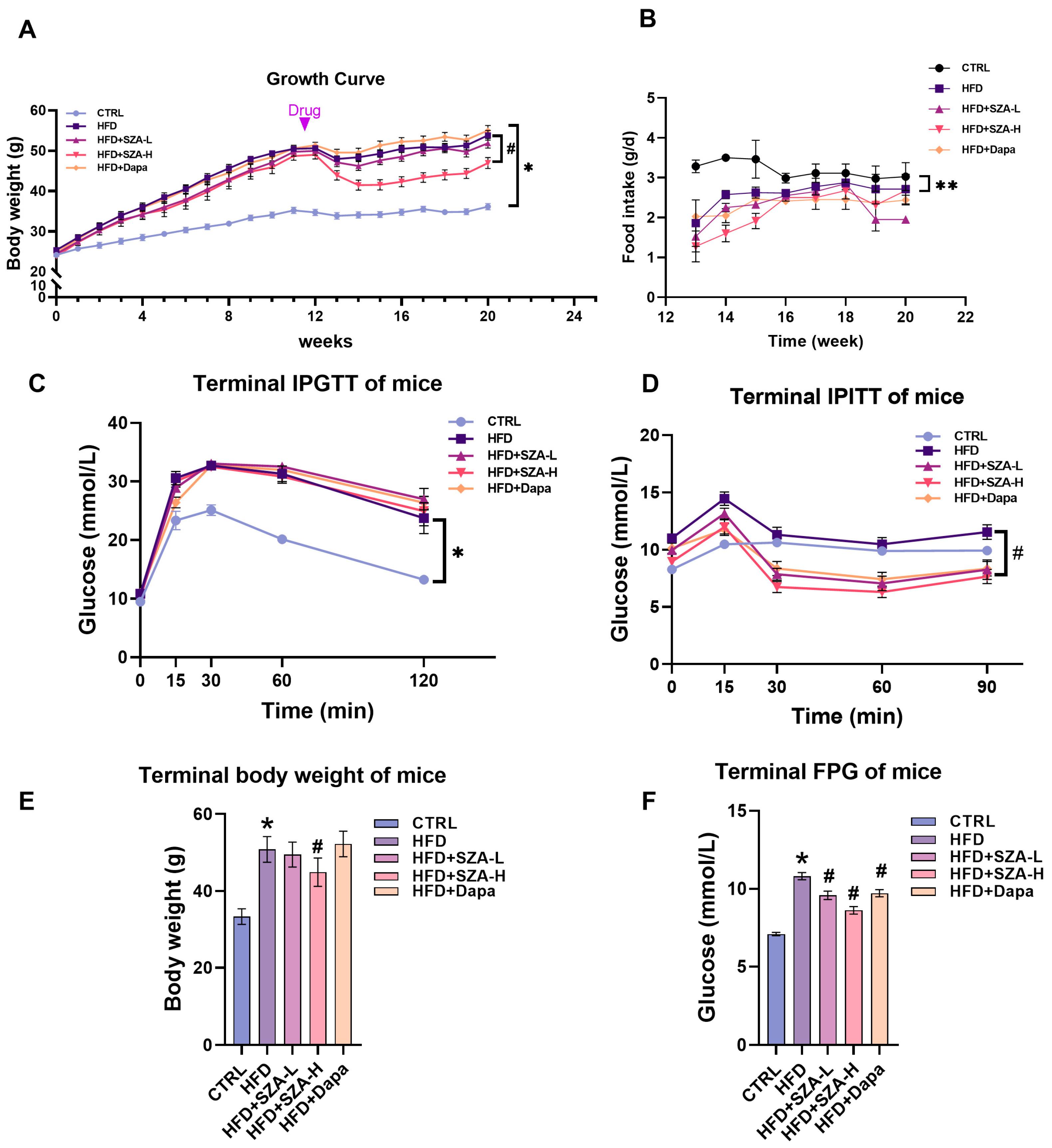
2.1. High-Dose SZ-A Decreased Body Weight and Improved Insulin Tolerance in Mice Fed with HFD
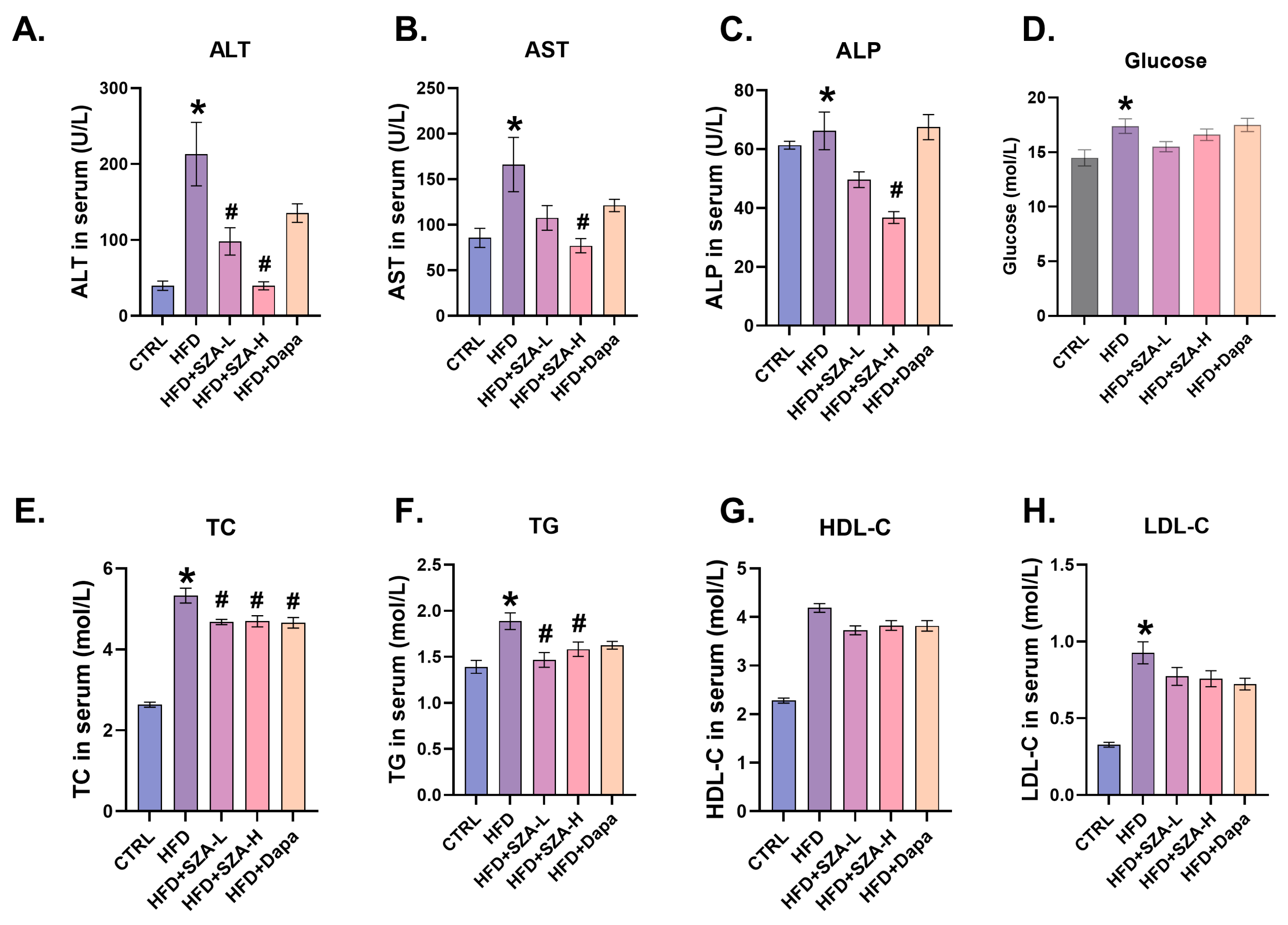
2.2. High-Dose SZ-A Significantly Reduced the Serum Liver Enzyme Concentrations and Improved Lipid Profiles in MAFLD Induced by HFD
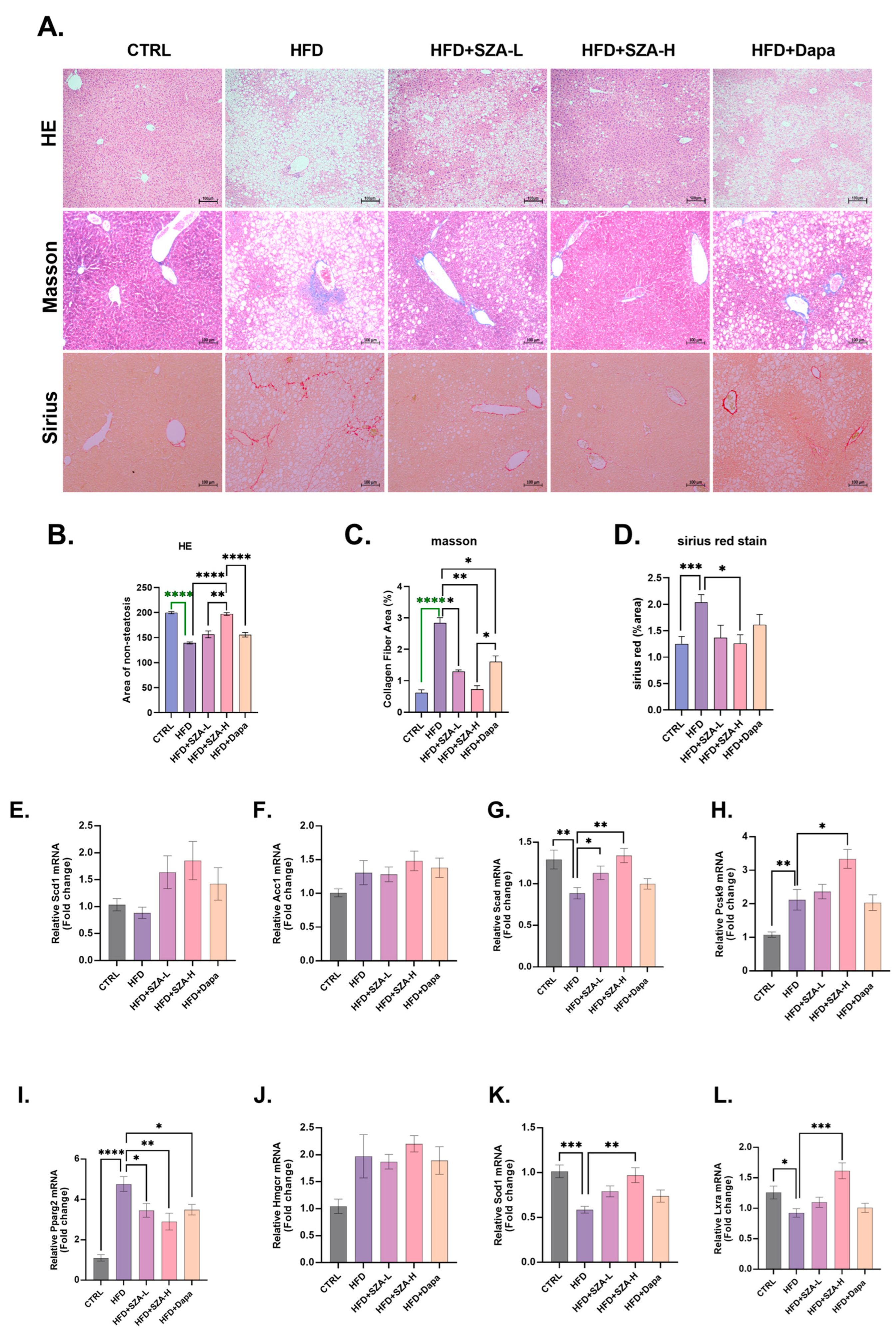
2.3. SZ-A Could Improve Liver Hepatic Lipids in MAFLD Induced by HFD
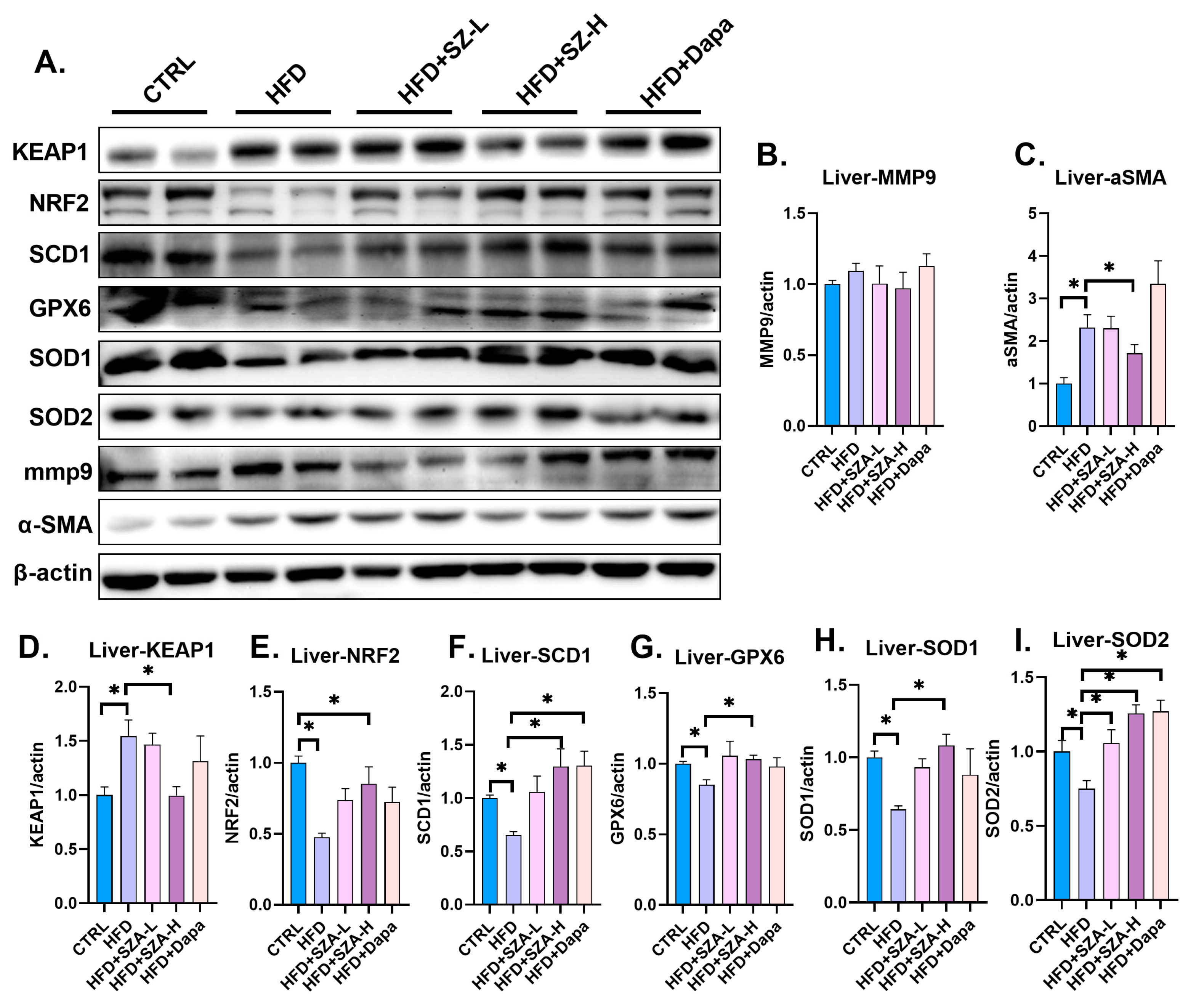
2.4. SZ-A Improved Liver Fibrosis in MAFLD Induced by HFD and Affected the Expression of Oxidative Stress Kinase
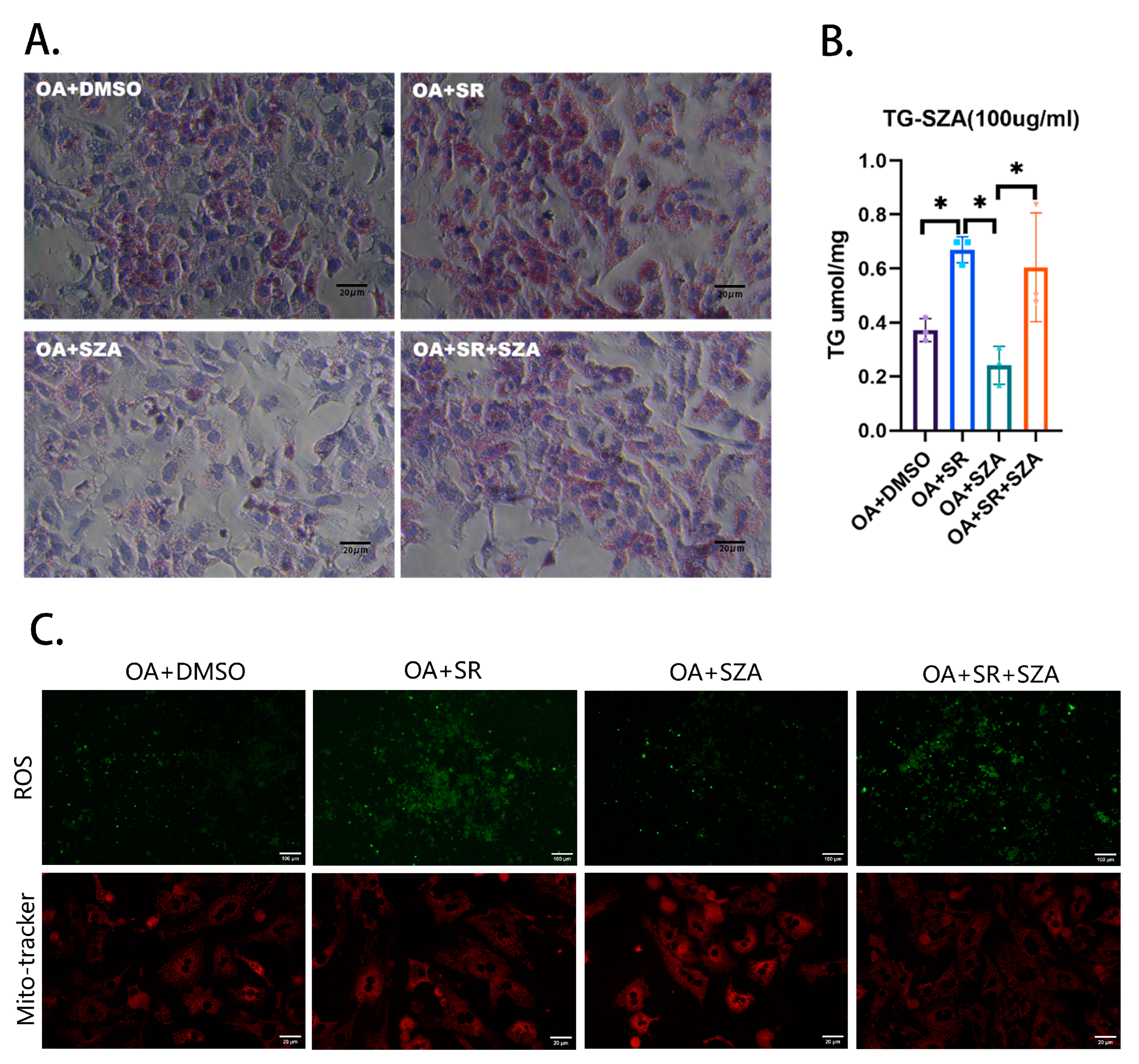
2.5. SZ-A Improved Liver Lipid Metabolism by Upregulating the Synthesis of PGC1α
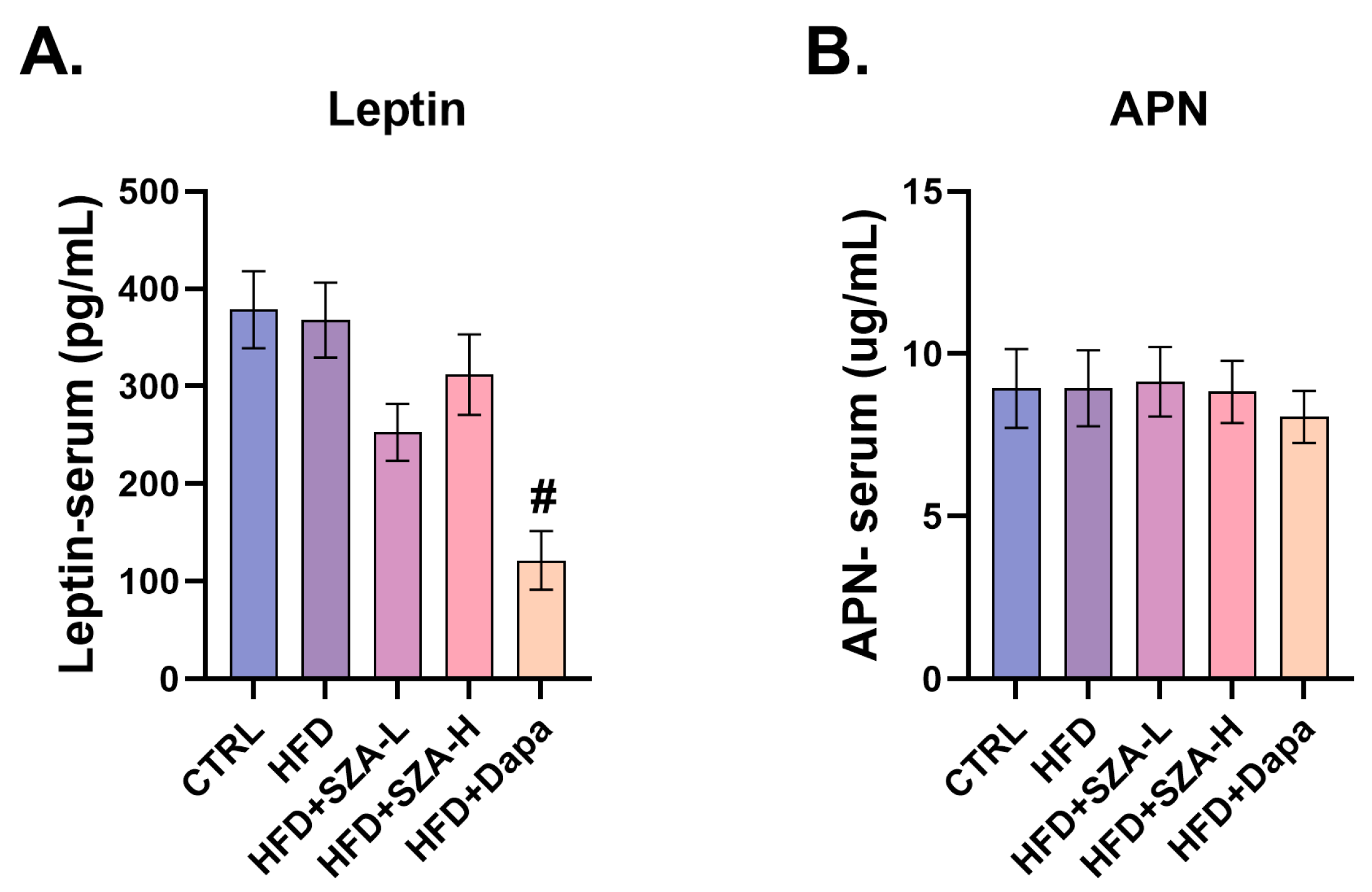
2.6. Adiponectin and Leptin Levels Were Not Significantly Affected by SZ-A
2.7. PGC1α Might Interact with NRF2 at the Molecular Level
3. Discussion
3.1. SZ-A Improved Liver Lipid Metabolism in MAFLD Mouse Models
3.2. SZ-A Improved Liver Fibrosis and Oxidative Stress in MAFLD Model Mice
3.3. Summary
4. Materials and Methods
4.1. Establishment of Animal Models
4.2. Serological Tests
4.3. Histological Examination
4.4. Gene and Protein Level Detection
4.5. Isolation of Murine Hepatocytes
4.6. Rescue Experiment of Liver Primary Cells
4.7. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sanyal, A.J.; Brunt, E.M.; Kleiner, D.E.; Kowdley, K.V.; Chalasani, N.; LaVine, J.E.; Ratziu, V.; McCullough, A. Endpoints and clinical trial design for nonalcoholic steatohepatitis. Hepatology 2011, 54, 344–353. [Google Scholar] [CrossRef]
- Chan, K.E.; Koh, T.J.L.; Tang, A.S.P.; Quek, J.; Yong, J.N.; Tay, P.; Tan, D.J.H.; Lim, W.H.; Lin, S.Y.; Huang, D.; et al. Global Prevalence and Clinical Characteristics of Metabolic Associated Fatty Liver Disease. A Meta-Analysis and Systematic Review of 10,739,607 Individuals. J. Clin. Endocrinol. Metab. 2022, 107, 2691–2700. [Google Scholar] [CrossRef]
- Abd El-Kader, S.M.; El-Den Ashmawy, E.M.S. Non-alcoholic fatty liver disease: The diagnosis and management. World J. Hepatol. 2015, 7, 846–858. [Google Scholar] [CrossRef] [PubMed]
- Riazi, K.; Azhari, H.; Charette, J.H.; Underwood, F.E.; King, J.A.; Afshar, E.E.; Swain, M.G.; Congly, S.E.; Kaplan, G.G.; Shaheen, A.-A. The prevalence and incidence of NAFLD worldwide: A systematic review and meta-analysis. Lancet Gastroenterol. Hepatol. 2022, 7, 851–861. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Mao, X.; Liu, Z.; Zhang, T.; Jin, L.; Chen, X. Global Burden of Nonalcoholic Fatty Liver Disease, 1990 to 2019: Findings from the Global Burden of Disease Study 2019. J. Clin. Gastroenterol. 2023, 57, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Garcia, D.; Hellberg, K.; Chaix, A.; Wallace, M.; Herzig, S.; Badur, M.G.; Lin, T.; Shokhirev, M.N.; Pinto, A.F.; Ross, D.S.; et al. Genetic Liver-Specific AMPK Activation Protects against Diet-Induced Obesity and NAFLD. Cell Rep. 2019, 26, 192–208.e6. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.-J.; Huang, W.-C.; Yu, M.-C.; Chen, Y.-L.; Shen, S.-C.; Yeh, K.-W.; Liou, C.-J. Tomatidine ameliorates obesity-induced nonalcoholic fatty liver disease in mice. J. Nutr. Biochem. 2021, 91, 108602. [Google Scholar] [CrossRef]
- Lee, G.-H.; Peng, C.; Park, S.-A.; Hoang, T.-H.; Lee, H.-Y.; Kim, J.; Kang, S.-I.; Lee, C.-H.; Lee, J.-S.; Chae, H.-J. Citrus Peel Extract Ameliorates High-Fat Diet-Induced NAFLD via Activation of AMPK Signaling. Nutrients 2020, 12, 673. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Fu, J.; Liu, D.; Sun, J.; Hou, Y.; Chen, C.; Shao, J.; Wang, L.; Wang, X.; Zhao, R.; et al. Hepatocyte-specific Nrf2 deficiency mitigates high-fat diet-induced hepatic steatosis: Involvement of reduced PPARγ expression. Redox Biol. 2020, 30, 101412. [Google Scholar] [CrossRef]
- Chen, Y.-M.; Lian, C.-F.; Sun, Q.-W.; Wang, T.-T.; Liu, Y.-Y.; Ye, J.; Gao, L.-L.; Yang, Y.-F.; Liu, S.-N.; Shen, Z.-F.; et al. Ramulus Mori (Sangzhi) Alkaloids Alleviate High-Fat Diet-Induced Obesity and Nonalcoholic Fatty Liver Disease in Mice. Antioxidants 2022, 11, 905. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Huang, X.; Ye, H.; Chen, Y.; Yu, J.; Yang, J.; Zhang, X. Randomized, Double-Blinded, Double-Dummy, Active-Controlled, and Multiple-Dose Clinical Study Comparing the Efficacy and Safety of Mulberry Twig (Ramulus Mori, Sangzhi) Alkaloid Tablet and Acarbose in Individuals with Type 2 Diabetes Mellitus. Evid. Based Complement. Altern. Med. 2016, 2016, 7121356. [Google Scholar] [CrossRef]
- Lei, L.; Huan, Y.; Liu, Q.; Li, C.; Cao, H.; Ji, W.; Gao, X.; Fu, Y.; Li, P.; Zhang, R.; et al. Morus alba L. (Sangzhi) Alkaloids Promote Insulin Secretion, Restore Diabetic β-Cell Function by Preventing Dedifferentiation and Apoptosis. Front. Pharmacol. 2022, 13, 841981. [Google Scholar] [CrossRef]
- Liu, Q.; Liu, S.; Cao, H.; Ji, W.; Li, C.; Huan, Y.; Lei, L.; Fu, Y.; Gao, X.; Liu, Y.; et al. Ramulus Mori (Sangzhi) Alkaloids (SZ-A) Ameliorate Glucose Metabolism Accompanied by the Modulation of Gut Microbiota and Ileal Inflammatory Damage in Type 2 Diabetic KKAy Mice. Front. Pharmacol. 2021, 12, 642400. [Google Scholar] [CrossRef]
- Polyzos, S.A.; Kountouras, J.; Mantzoros, C.S. Obesity and nonalcoholic fatty liver disease: From pathophysiology to therapeutics. Metabolism 2019, 92, 82–97. [Google Scholar] [CrossRef]
- Leoni, S.; Tovoli, F.; Napoli, L.; Serio, I.; Ferri, S.; Bolondi, L. Current guidelines for the management of non-alcoholic fatty liver disease: A systematic review with comparative analysis. World J. Gastroenterol. 2018, 24, 3361–3373. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.-F.; Ku, H.-C.; Lin, H. PGC-1α as a Pivotal Factor in Lipid and Metabolic Regulation. Int. J. Mol. Sci. 2018, 19, 3447. [Google Scholar] [CrossRef] [PubMed]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Ramatchandirin, B.; Pearah, A.; He, L. Regulation of Liver Glucose and Lipid Metabolism by Transcriptional Factors and Coactivators. Life 2023, 13, 515. [Google Scholar] [CrossRef] [PubMed]
- Bougarne, N.; Weyers, B.; Desmet, S.J.; Deckers, J.; Ray, D.W.; Staels, B.; De Bosscher, K. Molecular Actions of PPARα in Lipid Metabolism and Inflammation. Endocr. Rev. 2018, 39, 760–802. [Google Scholar] [CrossRef] [PubMed]
- Szántó, M.; Gupte, R.; Kraus, W.L.; Pacher, P.; Bai, P. PARPs in lipid metabolism and related diseases. Prog. Lipid Res. 2021, 84, 101117. [Google Scholar] [CrossRef]
- Mohs, A.; Otto, T.; Schneider, K.M.; Peltzer, M.; Boekschoten, M.; Holland, C.H.; Kalveram, L.; Wiegand, S.; Saez-Rodriguez, J.; Longerich, T.; et al. Hepatocyte-specific NRF2 activation controls fibrogenesis and carcinogenesis in steatohepatitis. J. Hepatol. 2021, 74, 638–648. [Google Scholar] [CrossRef]
- Ge, C.; Tan, J.; Lou, D.; Zhu, L.; Zhong, Z.; Dai, X.; Sun, Y.; Kuang, Q.; Zhao, J.; Wang, L.; et al. Mulberrin confers protection against hepatic fibrosis by Trim31/Nrf2 signaling. Redox Biol. 2022, 51, 102274. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimnezhad, N.; Nayebifar, S.; Soltani, Z.; Khoramipour, K. High-intensity interval training reduced oxidative stress and apoptosis in the hippocampus of male rats with type 2 diabetes: The role of the PGC1α-Keap1-Nrf2 signaling pathway. Iran. J. Basic Med. Sci. 2023, 26, 1313–1319. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Pi, J.; Zhang, Q. Signal amplification in the KEAP1-NRF2-ARE antioxidant response pathway. Redox Biol. 2022, 54, 102389. [Google Scholar] [CrossRef]
- Ulasov, A.V.; Rosenkranz, A.A.; Georgiev, G.P.; Sobolev, A.S. Nrf2/Keap1/ARE signaling: Towards specific regulation. Life Sci. 2022, 291, 120111. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 System: A Thiol-Based Sensor-Effector Apparatus for Maintaining Redox Homeostasis. Physiol. Rev. 2018, 98, 1169–1203. [Google Scholar] [CrossRef] [PubMed]
- Aljohani, A.M.; Syed, D.N.; Ntambi, J.M. Insights into Stearoyl-CoA Desaturase-1 Regulation of Systemic Metabolism. Trends Endocrinol. Metab. 2017, 28, 831–842. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, M.; Flowers, M.T.; Sampath, H.; Chu, K.; Otzelberger, C.; Liu, X.; Ntambi, J.M. Hepatic stearoyl-CoA desaturase-1 deficiency protects mice from carbohydrate-induced adiposity and hepatic steatosis. Cell Metab. 2007, 6, 484–496. [Google Scholar] [CrossRef] [PubMed]
- Xuan, Y.; Wang, H.; Yung, M.M.H.; Chen, F.; Chan, W.-S.; Chan, Y.-S.; Tsui, S.K.W.; Ngan, H.Y.S.; Chan, K.K.L.; Chan, D.W. SCD1/FADS2 fatty acid desaturases equipoise lipid metabolic activity and redox-driven ferroptosis in ascites-derived ovarian cancer cells. Theranostics 2022, 12, 3534–3552. [Google Scholar] [CrossRef]
- Morieri, M.L.; Vitturi, N.; Avogaro, A.; Targher, G.; Fadini, G.P. Prevalence of hepatic steatosis in patients with type 2 diabetes and response to glucose-lowering treatments. A multicenter retrospective study in Italian specialist care. J. Endocrinol. Investig. 2021, 44, 1879–1889. [Google Scholar] [CrossRef]
- National Research Council. Guide for the Care and Use of Laboratory Animals, 8th ed.; The National Academies Press: Washington, DC, USA, 2011. [Google Scholar] [CrossRef]
- Gonçalves, L.A.; Vigário, A.M.; Penha-Gonçalves, C. Improved isolation of murine hepatocytes for in vitro malaria liver stage studies. Malar. J. 2007, 6, 169. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, M.; Guo, C.; Li, Z.; Cai, X.; Wen, X.; Lv, F.; Lin, C.; Ji, L. Mulberry Twig Alkaloids Improved the Progression of Metabolic-Associated Fatty Liver Disease in High-Fat Diet-Induced Obese Mice by Regulating the PGC1α/PPARα and KEAP1/NRF2 Pathways. Pharmaceuticals 2024, 17, 1287. https://doi.org/10.3390/ph17101287
Zhang M, Guo C, Li Z, Cai X, Wen X, Lv F, Lin C, Ji L. Mulberry Twig Alkaloids Improved the Progression of Metabolic-Associated Fatty Liver Disease in High-Fat Diet-Induced Obese Mice by Regulating the PGC1α/PPARα and KEAP1/NRF2 Pathways. Pharmaceuticals. 2024; 17(10):1287. https://doi.org/10.3390/ph17101287
Chicago/Turabian StyleZhang, Mengqing, Chengcheng Guo, Zonglin Li, Xiaoling Cai, Xin Wen, Fang Lv, Chu Lin, and Linong Ji. 2024. "Mulberry Twig Alkaloids Improved the Progression of Metabolic-Associated Fatty Liver Disease in High-Fat Diet-Induced Obese Mice by Regulating the PGC1α/PPARα and KEAP1/NRF2 Pathways" Pharmaceuticals 17, no. 10: 1287. https://doi.org/10.3390/ph17101287
APA StyleZhang, M., Guo, C., Li, Z., Cai, X., Wen, X., Lv, F., Lin, C., & Ji, L. (2024). Mulberry Twig Alkaloids Improved the Progression of Metabolic-Associated Fatty Liver Disease in High-Fat Diet-Induced Obese Mice by Regulating the PGC1α/PPARα and KEAP1/NRF2 Pathways. Pharmaceuticals, 17(10), 1287. https://doi.org/10.3390/ph17101287