
The Development and Evaluation of a Novel Highly Selective PET Radiotracer for Targeting BET BD1


Abstract
1. Introduction
2. Results
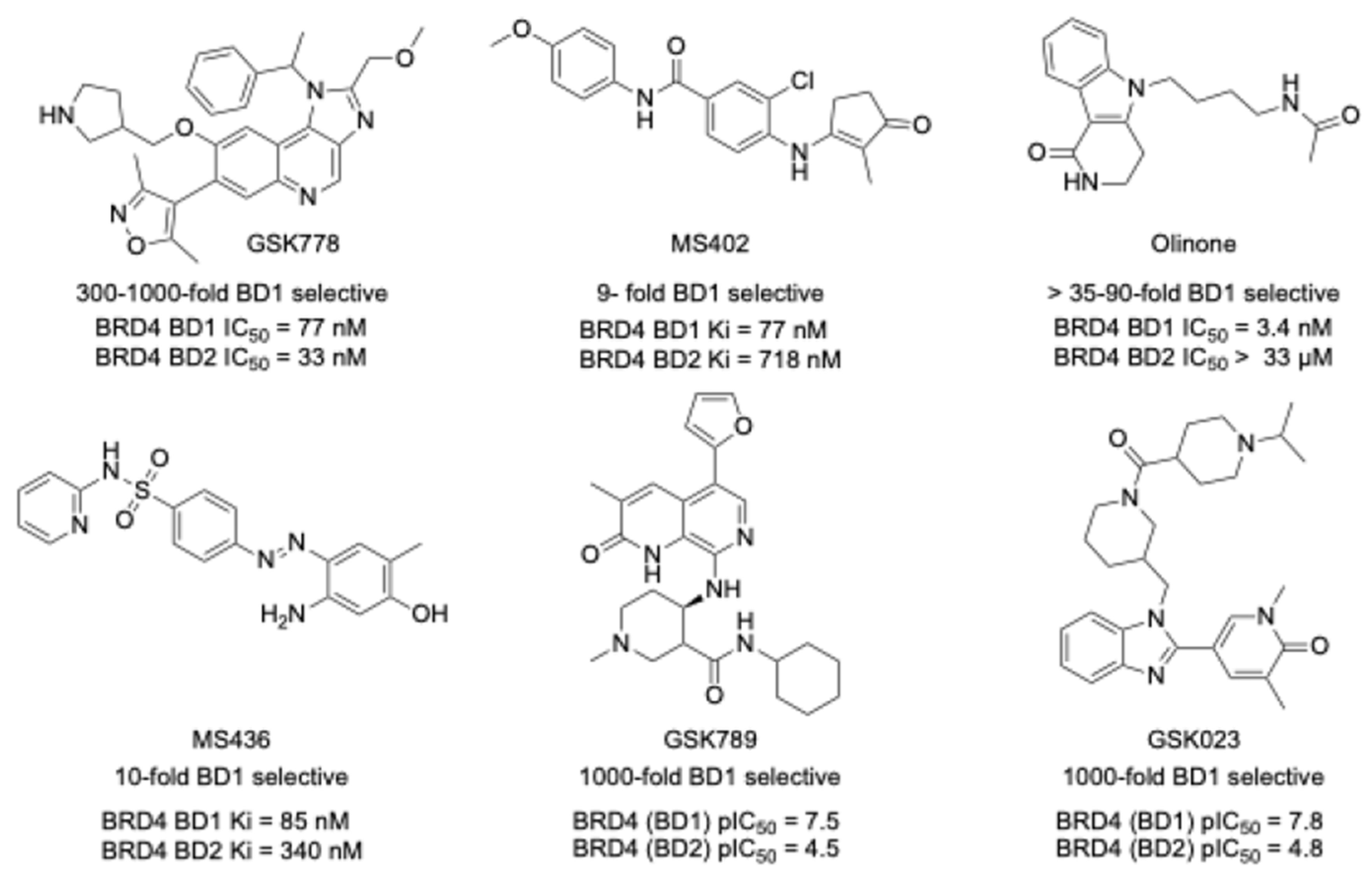
2.1. Optimizing GSK023 for PET Imaging: High Selectivity and Potency in BET BD1 Domain Targeting
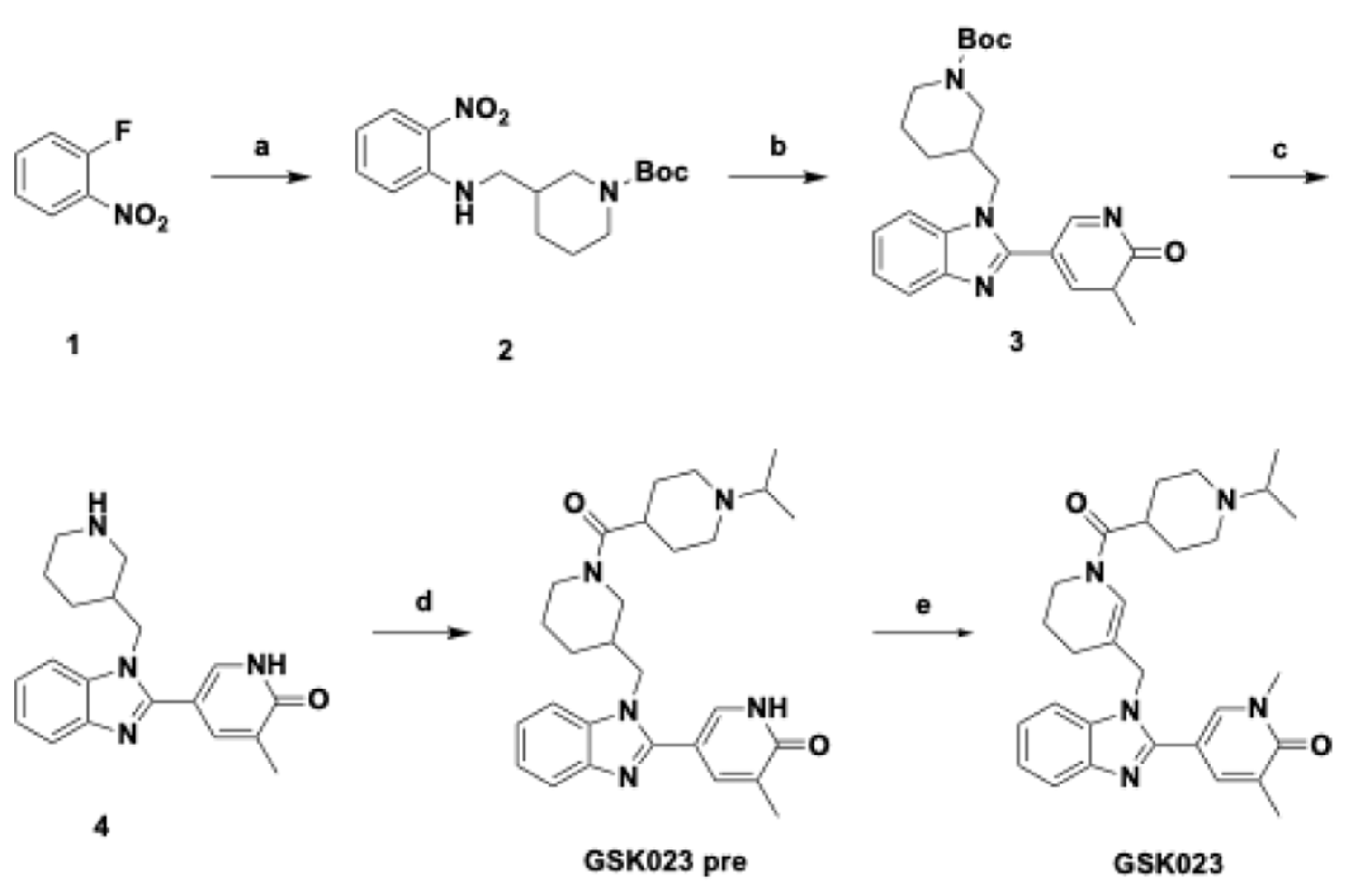
2.2. Chemical Synthesis for GSK023 and [11C]GSK023
2.2.1. Synthesis Summary of GSK023 and GSK023 Pre
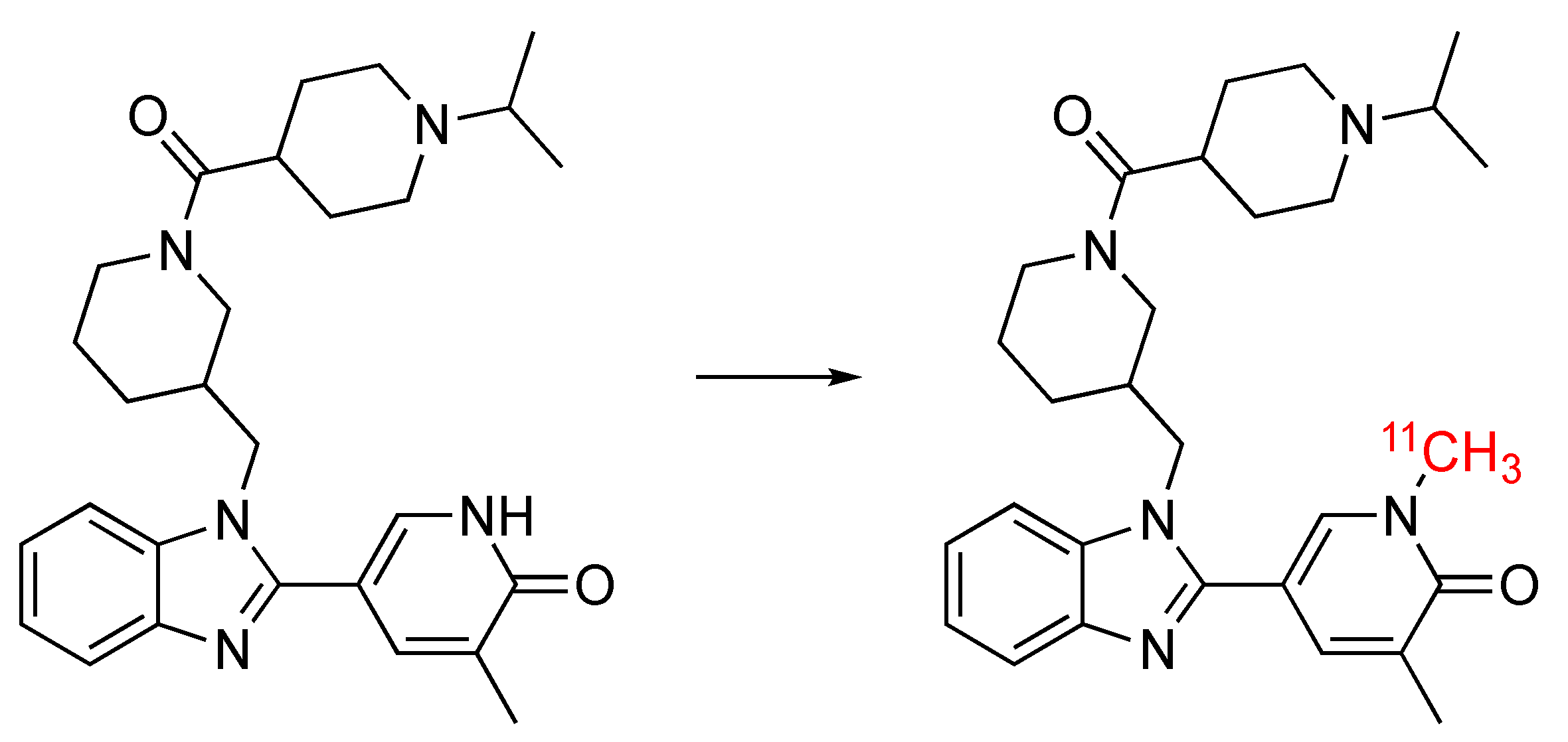
2.2.2. Radiolabeling of [11C]GSK023
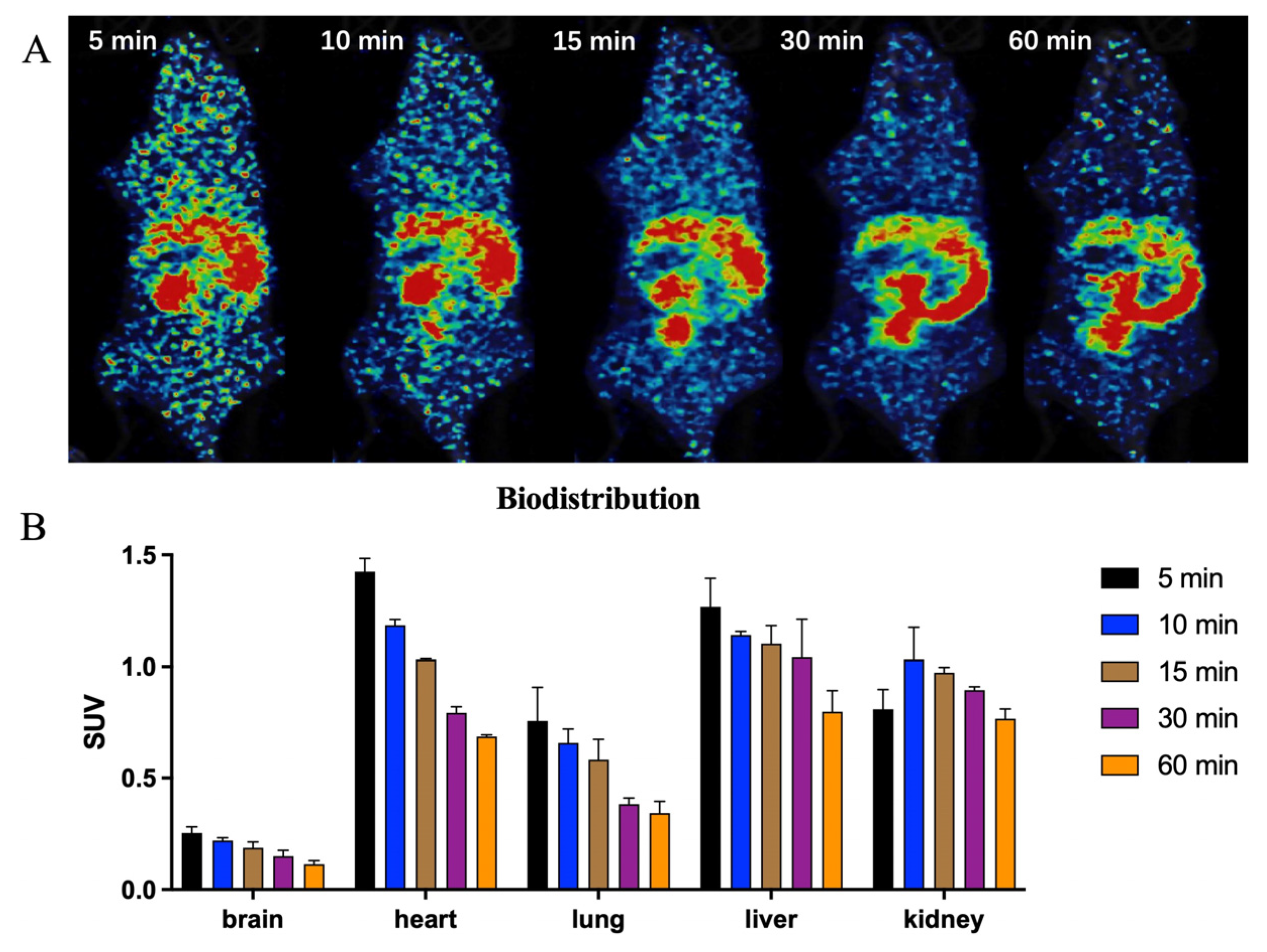
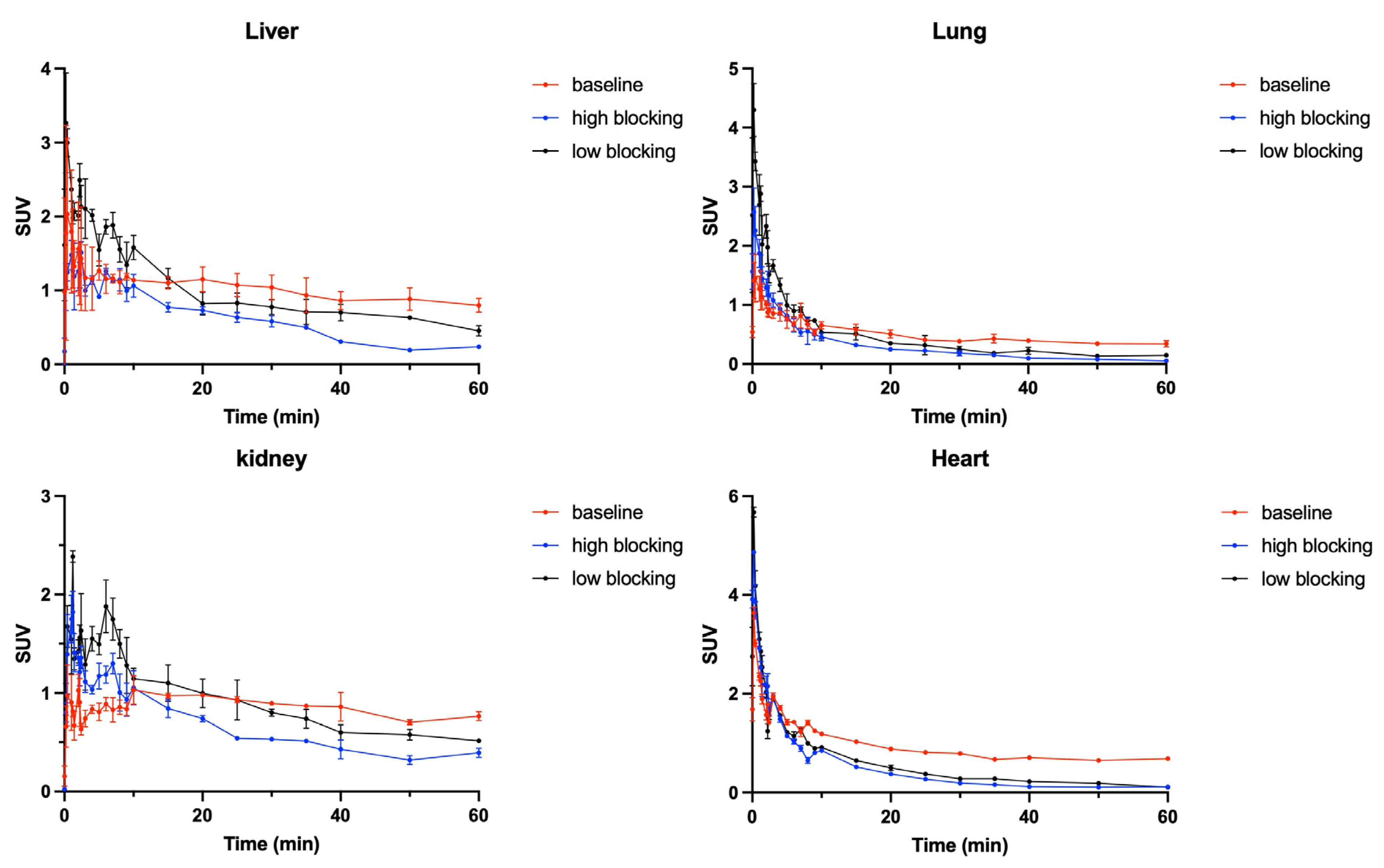
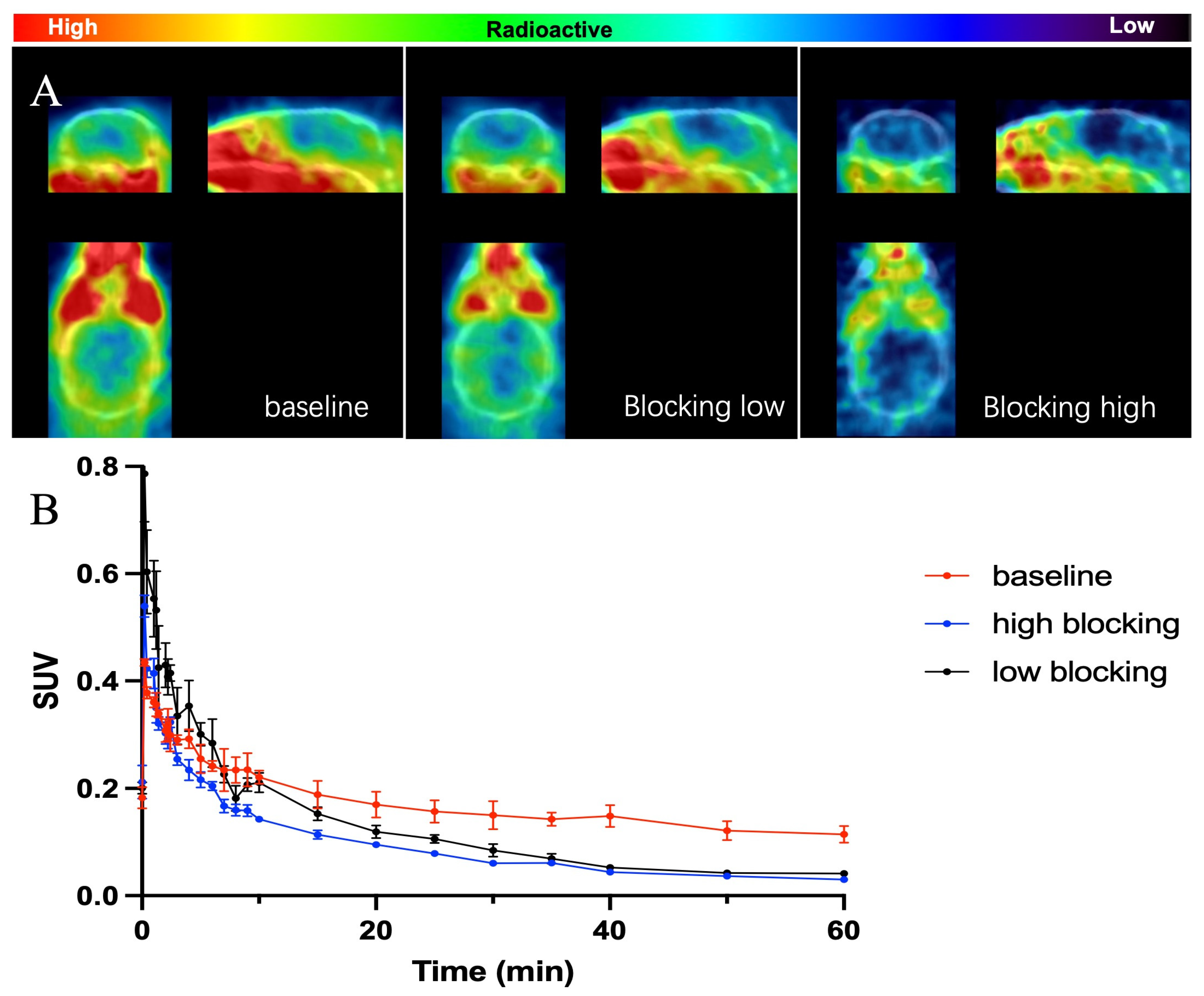
2.3. In Vivo PET-CT Imaging with [11C]GSK023 in Rodents
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Taniguchi, Y. The Bromodomain and Extra-Terminal Domain (BET) Family: Functional Anatomy of BET Paralogous Proteins. Int. J. Mol. Sci. 2016, 17, 1849. [Google Scholar] [CrossRef] [PubMed]
- Kulikowski, E.; Rakai, B.D.; Wong, N.C.W. Inhibitors of bromodomain and extra-terminal proteins for treating multiple human diseases. Med. Res. Rev. 2021, 41, 223–245. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hu, J.; Pan, D.; Li, G.; Chen, K.; Hu, X. Regulation of programmed cell death by Brd4. Cell Death Dis. 2022, 13, 1059. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wu, Y.; Wang, Y.; Diao, P.; Zhang, W.; Li, J.; Ge, H.; Song, Y.; Li, Z.; Wang, D.; Liu, L.; et al. Therapeutic Targeting of BRD4 in Head Neck Squamous Cell Carcinoma. Theranostics 2019, 9, 1777–1793. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Shorstova, T.; Foulkes, W.D.; Witcher, M. Achieving clinical success with BET inhibitors as anti-cancer agents. Br. J. Cancer 2021, 124, 1478–1490. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Bolinger, A.A.; Zhou, J.; Tian, B. Bromodomain-containing protein 4 (BRD4): A key player in inflammatory bowel disease and potential to inspire epigenetic therapeutics. Expert Opin. Ther. Targets 2023, 27, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Yongprayoon, V.; Wattanakul, N.; Khomate, W.; Apithanangsiri, N.; Kasitipradit, T.; Nantajit, D.; Tavassoli, M. Targeting BRD4: Potential therapeutic strategy for head and neck squamous cell carcinoma (Review). Oncol. Rep. 2024, 51, 74. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Padmanabhan, B.; Mathur, S.; Manjula, R.; Tripathi, S. Bromodomain and extra-terminal (BET) family proteins: New therapeutic targets in major diseases. J. Biosci. 2016, 41, 295–311. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Plotnikov, A.N.; Rusinova, E.; Shen, T.; Morohashi, K.; Joshua, J.; Zeng, L.; Mujtaba, S.; Ohlmeyer, M.; Zhou, M.M. Structure-guided design of potent diazobenzene inhibitors for the BET bromodomains. J. Med. Chem. 2013, 56, 9251–9264. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Cheung, K.; Lu, G.; Sharma, R.; Vincek, A.; Zhang, R.; Plotnikov, A.N.; Zhang, F.; Zhang, Q.; Ju, Y.; Hu, Y.; et al. BET N-terminal bromodomain inhibition selectively blocks Th17 cell differentiation and ameliorates colitis in mice. Proc. Natl. Acad. Sci. USA 2017, 114, 2952–2957. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gacias, M.; Gerona-Navarro, G.; Plotnikov, A.N.; Zhang, G.; Zeng, L.; Kaur, J.; Moy, G.; Rusinova, E.; Rodriguez, Y.; Matikainen, B.; et al. Selective chemical modulation of gene transcription favors oligodendrocyte lineage progression. Chem. Biol. 2014, 21, 841–854. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gilan, O.; Rioja, I.; Knezevic, K.; Bell, M.J.; Yeung, M.M.; Harker, N.R.; Lam, E.Y.N.; Chung, C.W.; Bamborough, P.; Petretich, M.; et al. Selective targeting of BD1 and BD2 of the BET proteins in cancer and immunoinflammation. Science 2020, 368, 387–394. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Watson, R.J.; Bamborough, P.; Barnett, H.; Chung, C.W.; Davis, R.; Gordon, L.; Grandi, P.; Petretich, M.; Phillipou, A.; Prinjha, R.K.; et al. GSK789: A Selective Inhibitor of the First Bromodomains (BD1) of the Bromo and Extra Terminal Domain (BET) Proteins. J. Med. Chem. 2020, 63, 9045–9069. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, Y.; Wang, Y.; Bai, P.; Hallisey, M.R.; Varela, B.L.; Siewko, A.; Wang, C.; Xu, Y. Development and Characterization of a Novel Carbon-11 Labeled Positron Emission Tomography Radiotracer for Neuroimaging of Sirtuin 1 with Benzoxazine-Based Compounds. Drug Des. Dev. Ther. 2024, 18, 819–827. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bradley, E.; Fusani, L.; Chung, C.W.; Craggs, P.D.; Demont, E.H.; Humphreys, P.G.; Mitchell, D.J.; Phillipou, A.; Rioja, I.; Shah, R.R.; et al. Structure-Guided Design of a Domain-Selective Bromodomain and Extra Terminal N-Terminal Bromodomain Chemical Probe. J. Med. Chem. 2023, 66, 15728–15749. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhu, A.; Lee, D.; Shim, H. Metabolic positron emission tomography imaging in cancer detection and therapy response. Semin. Oncol. 2011, 38, 55–69. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Pan, Z.; Zhao, Y.; Wang, X.; Xie, X.; Liu, M.; Zhang, K.; Wang, L.; Bai, D.; Foster, L.J.; Shu, R.; et al. Targeting bromodomain-containing proteins: Research advances of drug discovery. Mol. Biomed. 2023, 4, 13. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- AlRawashdeh, S.; Barakat, K.H. Applications of Molecular Dynamics Simulations in Drug Discovery. Methods Mol. Biol. 2024, 2714, 127–141. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, Y.; Liu, Y.; Cheng, H.; Dagnew, T.M.; Xu, Y.; Wang, C. Synthesis and Characterization of a New Carbon-11 Labeled Positron Emission Tomography Radiotracer for Orexin 2 Receptors Neuroimaging. Drug Des. Dev. Ther. 2024, 18, 215–222. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wu, Q.; Heidenreich, D.; Zhou, S.; Ackloo, S.; Krämer, A.; Nakka, K.; Lima-Fernandes, E.; Deblois, G.; Duan, S.; Vellanki, R.N.; et al. A chemical toolbox for the study of bromodomains and epigenetic signaling. Nat. Commun. 2019, 10, 1915. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Vaquero, J.J.; Kinahan, P. Positron Emission Tomography: Current Challenges and Opportunities for Technological Advances in Clinical and Preclinical Imaging Systems. Annu. Rev. Biomed. Eng. 2015, 17, 385–414. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bai, P.; Lu, X.; Liu, Y.; Lan, Y.; Wang, H.; Fiedler, S.; Striar, R.; Wang, C. Discovery of a Positron Emission Tomography Radiotracer Selectively Targeting the BD1 Bromodomains of BET Proteins. ACS Med. Chem. Lett. 2021, 12, 282–287. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Xu, Y.; Wang, Y.; Wang, H.; Wang, C. Synthesis and Characterization of Carbon-11 Labeled Iloperidone for Imaging of α(1)-Adrenoceptor in Brain. Front. Mol. Biosci. 2020, 7, 586327. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wang, C.; Schroeder, F.A.; Wey, H.Y.; Borra, R.; Wagner, F.F.; Reis, S.; Kim, S.W.; Holson, E.B.; Haggarty, S.J.; Hooker, J.M. In vivo imaging of histone deacetylases (HDACs) in the central nervous system and major peripheral organs. J. Med. Chem. 2014, 57, 7999–8009. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bai, P.; Yan, L.; Bagdasarian, F.A.; Wilks, M.Q.; Wey, H.Y.; Wang, C. A positron emission tomography imaging probe selectively targeting the BD1 bromodomain and extra-terminal domain. Chem. Commun. 2022, 58, 9654–9657. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Assay System | TR-FRET pIC50 | Fold Selectivity | BROMOscan pKd | Fold Selectivity | ||
|---|---|---|---|---|---|---|
| Bromodomain | BRD4 | BRD4 | ||||
| BD1 | BD2 | BD1 | BD2 | |||
| BRD4 | 7.8 | 4.8 | 1000-fold | 8.1 | 5.6 | 316-fold |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Wang, Y.; Xu, Y.; Kang, L.; Tocci, D.; Wang, C. The Development and Evaluation of a Novel Highly Selective PET Radiotracer for Targeting BET BD1. Pharmaceuticals 2024, 17, 1289. https://doi.org/10.3390/ph17101289
Wang Y, Wang Y, Xu Y, Kang L, Tocci D, Wang C. The Development and Evaluation of a Novel Highly Selective PET Radiotracer for Targeting BET BD1. Pharmaceuticals. 2024; 17(10):1289. https://doi.org/10.3390/ph17101289
Chicago/Turabian StyleWang, Yanli, Yongle Wang, Yulong Xu, Leyi Kang, Darcy Tocci, and Changning Wang. 2024. "The Development and Evaluation of a Novel Highly Selective PET Radiotracer for Targeting BET BD1" Pharmaceuticals 17, no. 10: 1289. https://doi.org/10.3390/ph17101289
APA StyleWang, Y., Wang, Y., Xu, Y., Kang, L., Tocci, D., & Wang, C. (2024). The Development and Evaluation of a Novel Highly Selective PET Radiotracer for Targeting BET BD1. Pharmaceuticals, 17(10), 1289. https://doi.org/10.3390/ph17101289