
Advancements in Phosphodiesterase 5 Inhibitors: Unveiling Present and Future Perspectives



Abstract
1. Introduction
2. Classification of Phosphodiesterases
3. Tissues and Organs of High Expression for PDE5
4. PDE5 Physiological Role
5. PDE5 as a Drug Target for Disease Treatment
5.1. Approved Clinical Uses of PDE5 Inhibitors
5.1.1. Erectile Dysfunction
5.1.2. Pulmonary Arterial Hypertension
5.1.3. Lower Urinary Tract Symptoms Secondary to Benign Prostatic Hyperplasia
5.2. Emerging and Future Uses of PDE5 Inhibitors
5.2.1. Cancer
- (1)
- Cell growth arrest and induction of apoptosis
- (2)
- Chemotherapy sensitization
- (3)
- Modulation of antitumor immune response
- (4)
- Chemopreventive mechanisms
5.2.2. CNS Diseases
5.2.3. Cardiovascular Diseases
5.2.4. Kidney Diseases
5.2.5. Cystic Fibrosis
5.2.6. Diabetes
5.2.7. Miscellaneous Indications
5.3. Side Effects and Contraindications of PDE5 Inhibitors
- (i)
- Although PDE5 is reported as a promising target for anti-cancer therapy, as explained earlier, the prolonged use of PDE5-Is has been linked to an increased risk of melanoma. Lie and co-workers reported an association between sildenafil use and an increased risk of melanoma in a prospective cohort study conducted on 25,848 men [208]. Several other cohorts and case-control studies have also reported a correlation between the use of sildenafil and tadalafil and the increased risk of melanoma [209,210]. However, this association between the prolonged use of PDE5-Is and the development of cancer was only reported for melanoma; even the risk of other types of skin cancer, such as squamous cell carcinoma and basal cell carcinoma, was not correlated to the use of PDE5-Is [211].
- (ii)
- Visual disturbances have been usually reported with the use of PDE5-Is because of PDE6 inhibition. However, several studies have reported more serious ophthalmologic side effects associated with the use of PDE5-Is, which include non-arteritic anterior ischemic optic neuropathy (NAION), which may eventually lead to vision loss [212]. Two case-crossover studies have shown a two-fold increase in the risk of NAION in men using PDE5-Is, and currently, all PDE5Is (Viagra®, Cialis®, Levitra® and Spedra®) mention NAION as a caution in their summary of product characteristics [213,214].
- (iii)
- Moreover, sensorineural hearing loss (SSHL) has been associated with the prolonged use of PDE5-Is. Two in vivo studies have shown that the prolonged use of sildenafil could lead to hearing loss in mice and rats [215,216]; in addition, published trials and pharmacovigilance agencies reported 47 cases of SSHL as a result of prolonged administration of sildenafil [217], and more specifically, Maddox et al. reported two cases of SSHL due to daily use of tadalafil 10 mg and sildenafil 50 mg + tadalafil 10 mg use where both patients did not recover after a follow-up [218]. Both NAION and SSHL are of unknown pathophysiology.
- (iv)
- Priapism (prolonged erection of the penis) is another less common side effect reported with the prolonged use of PDE5-Is, as only a few cases have been reported for priapism associated with the use of PDE5-Is [219]. The risk of priapism increases in the case of concomitant use of other ED medications along with the PDE5-Is.
6. PDE5 Inhibitors
6.1. PDE5 Inhibitors
6.1.1. Pyrimidinones

6.1.2. Aminopyrimidines

6.1.3. Pyrido-Pyrimidines
6.1.4. Pyrazolopyrimidinones

6.1.5. Tetrahydro-β-Carbolines (THβCs)

6.1.6. Quinazolines

6.1.7. Quinazolinedihydro-β-Carbolines

6.1.8. Pyrroloquinolones

6.1.9. Quinolines

6.1.10. Tetrahydrobenzo[b][1,6]Naphthyridine
6.1.11. Chromenopyrrolones

6.1.12. Azepinoindolones

6.1.13. Pyridopyrazinones
6.1.14. Thienopyrimidines

6.1.15. PDE5 Allosteric Inhibitors
Evodiamine Derivatives

Trisubstituted Pyrazolines

6.2. PDE5 in the Context of Dual Inhibitors
6.2.1. Compounds with Dual PDE5 and HDAC Inhibitory Activities



6.2.2. Compounds with Dual PDE5 and AchE Inhibitory Activities

6.2.3. Dual PDE5 Inhibitor and NO Donor

6.2.4. Compounds with Dual PDE5 and Topoisomerase 2 Inhibitory Activities

6.3. PDE5 Inhibitors for Radiodiagnosis

7. Binding Modes of PDE5 Inhibitors
7.1. The PDE5 Active Site
- A metal-binding site (M site) that contains Zn+2 and Mg+2 ions, together with several aspartate and histidine residues.
- A core pocket (Q pocket) lined by Gln817, Phe820, Val782, and Tyr612.
- A hydrophobic pocket (H pocket).
- The Q2-pocket, which is lined by Phe786, Phe787, Leu804, Ile813, Met816.
- The Lid region consists of Tyr664, Met816, Ala823 and Gly819.
7.2. The Evo Pocket
8. Recent Update on Clinical Trials Involving PDE5 Inhibitors
9. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Francis, S.H.; Blount, M.A.; Corbin, J.D. Mammalian cyclic nucleotide phosphodiesterases: Molecular mechanisms and physiological functions. Physiol. Rev. 2011, 91, 651–690. [Google Scholar] [CrossRef]
- Lin, C.-S.; Lin, G.; Xin, Z.-C.; Lue, T.F. Expression, distribution and regulation of phosphodiesterase 5. Curr. Pharm. Des. 2006, 12, 3439–3457. [Google Scholar] [CrossRef] [PubMed]
- Wood, E.R.; Bledsoe, R.; Chai, J.; Daka, P.; Deng, H.; Ding, Y.; Harris-Gurley, S.; Kryn, L.H.; Nartey, E.; Nichols, J. The role of phosphodiesterase 12 (PDE12) as a negative regulator of the innate immune response and the discovery of antiviral inhibitors. J. Biol. Chem. 2015, 290, 19681–19696. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Cruz, J.; Cabrera-Leon, A.; Martın-Morales, A.; Fernandez, A.; Burgos, R.; Rejas, J. Male erectile dysfunction and health-related quality of life. Eur. Urol. 2003, 44, 245–253. [Google Scholar] [CrossRef]
- Corinaldesi, C.; Di Luigi, L.; Lenzi, A.; Crescioli, C. Phosphodiesterase type 5 inhibitors: Back and forward from cardiac indications. J. Endocrinol. Investig. 2016, 39, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Loughney, K.; Hill, T.R.; Florio, V.A.; Uher, L.; Rosman, G.J.; Wolda, S.L.; Jones, B.A.; Howard, M.L.; McAllister-Lucas, L.M.; Sonnenburg, W.K. Isolation and characterization of cDNAs encoding PDE5A, a human cGMP-binding, cGMP-specific 3′, 5′-cyclic nucleotide phosphodiesterase. Gene 1998, 216, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Dimitriadis, F.; Tsounapi, P.; Saito, M.; Watanabe, T.; Sylakos, A.; Tsabalas, S.; Miyagawa, I.; Sofikitis, N. Is there a role for PDE5 inhibitors in the management of male infertility due to defects in testicular or epididymal function? Curr. Pharm. Des. 2009, 15, 3506–3520. [Google Scholar] [CrossRef]
- Ückert, S.; Kuczyk, M.A. Cyclic nucleotide metabolism including nitric oxide and phosphodiesterase-related targets in the lower urinary tract. Urin. Tract 2011, 2011, 527–542. [Google Scholar]
- Bers, D.M. Cardiac excitation–contraction coupling. Nature 2002, 415, 198–205. [Google Scholar] [CrossRef]
- Denninger, J.W.; Marletta, M.A. Guanylate cyclase and the⋅ NO/cGMP signaling pathway. Biochim. Biophys. Acta (BBA)-Bioenerg. 1999, 1411, 334–350. [Google Scholar] [CrossRef]
- Murad, F. Nitric oxide and cyclic GMP in cell signaling and drug development. N. Engl. J. Med. 2006, 355, 2003–2011. [Google Scholar] [CrossRef] [PubMed]
- Schlossmann, J.; Ammendola, A.; Ashman, K.; Zong, X.; Huber, A.; Neubauer, G.; Wang, G.X.; Allescher, H.D.; Korth, M.; Wilm, M.; et al. Regulation of intracellular calcium by a signalling complex of IRAG, IP3 receptor and cGMP kinase Iβ. Nature 2000, 404, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, F.; Ammendola, A.; Schlossmann, J. Rising behind NO: cGMP-dependent protein kinases. J. Cell Sci. 2000, 113, 1671–1676. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Strada, S.J. The novel functions of cGMP-specific phosphodiesterase 5 and its inhibitors in carcinoma cells and pulmonary/cardiovascular vessels. Curr. Top. Med. Chem. 2007, 7, 437–454. [Google Scholar] [CrossRef]
- Lincoln, T.M.; Dey, N.; Sellak, H. Invited review: cGMP-dependent protein kinase signaling mechanisms in smooth muscle: From the regulation of tone to gene expression. J. Appl. Physiol. 2001, 91, 1421–1430. [Google Scholar] [CrossRef]
- Rondina, M.T.; Weyrich, A.S. Targeting phosphodiesterases in anti-platelet therapy. Antiplatelet Agents 2012, 210, 225–238. [Google Scholar]
- Coskuner, E.R.; Ozkan, B. Reno-protective effects of Phosphodiesterase 5 inhibitors. Clin. Exp. Nephrol. 2021, 25, 585–597. [Google Scholar] [CrossRef]
- Blount, M.A.; Beasley, A.; Zoraghi, R.; Sekhar, K.R.; Bessay, E.P.; Francis, S.H.; Corbin, J.D. Binding of tritiated sildenafil, tadalafil, or vardenafil to the phosphodiesterase-5 catalytic site displays potency, specificity, heterogeneity, and cGMP stimulation. Mol. Pharmacol. 2004, 66, 144–152. [Google Scholar] [CrossRef]
- Bruzziches, R.; Francomano, D.; Gareri, P.; Lenzi, A.; Aversa, A. An update on pharmacological treatment of erectile dysfunction with phosphodiesterase type 5 inhibitors. Expert Opin. Pharmacother. 2013, 14, 1333–1344. [Google Scholar] [CrossRef]
- Kedia, G.T.; Ückert, S.; Assadi-Pour, F.; Kuczyk, M.A.; Albrecht, K. Avanafil for the treatment of erectile dysfunction: Initial data and clinical key properties. Ther. Adv. Urol. 2013, 5, 35–41. [Google Scholar] [CrossRef]
- Schellack, N.; Agoro, A. A review of phosphodiesterase type 5 inhibitors. S. Afr. Fam. Pract. 2014, 56, 96–101. [Google Scholar] [CrossRef]
- Gupta, M.; Kovar, A.; Meibohm, B. The clinical pharmacokinetics of phosphodiesterase-5 inhibitors for erectile dysfunction. J. Clin. Pharmacol. 2005, 45, 987–1003. [Google Scholar] [CrossRef] [PubMed]
- Ückert, S.; Kuczyk, M.A.; Oelke, M. Phosphodiesterase inhibitors in clinical urology. Expert Rev. Clin. Pharmacol. 2013, 6, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Francis, S.H.; Corbin, J.D. Phosphodiesterase-5 inhibition: The molecular biology of erectile function and dysfunction. Urol. Clin. 2005, 32, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Corbin, J.D.; Francis, S.H. Molecular Biology and Pharmacology of PDE-5—Inhibitor Therapy for Erectile Dysfunction. J. Androl. 2003, 24, S38–S41. [Google Scholar] [CrossRef]
- Carson, C.C.; Burnett, A.L.; Levine, L.A.; Nehra, A. The efficacy of sildenafil citrate (Viagra®) in clinical populations: An update. Urology 2002, 60, 12–27. [Google Scholar] [CrossRef]
- Hellstrom, W.J.; Gittelman, M.; Karlin, G.; Segerson, T.; Thibonnier, M.; Taylor, T.; Padma-Nathan, H.; Group, V.S. Sustained efficacy and tolerability of vardenafil, a highly potent selective phosphodiesterase type 5 inhibitor, in men with erectile dysfunction: Results of a randomized, double-blind, 26-week placebo-controlled pivotal trial. Urology 2003, 61, 8–14. [Google Scholar] [CrossRef]
- Brock, G.B.; McMahon, C.G.; Chen, K.; Costigan, T.; Shen, W.; Watkins, V.; Anglin, G.; Whitaker, S. Efficacy and safety of tadalafil for the treatment of erectile dysfunction: Results of integrated analyses. J. Urol. 2002, 168, 1332–1336. [Google Scholar] [CrossRef]
- Goldstein, I.; McCullough, A.R.; Jones, L.A.; Hellstrom, W.J.; Bowden, C.H.; DiDonato, K.; Trask, B.; Day, W.W. A randomized, double-blind, placebo-controlled evaluation of the safety and efficacy of avanafil in subjects with erectile dysfunction. J. Sex. Med. 2012, 9, 1122–1133. [Google Scholar] [CrossRef]
- Cui, Y.-S.; Li, N.; Zong, H.-T.; Yan, H.-L.; Zhang, Y. Avanafil for male erectile dysfunction: A systematic review and meta-analysis. Asian J. Androl. 2014, 16, 472. [Google Scholar]
- Jannini, E.A.; DeRogatis, L.R.; Chung, E.; Brock, G.B. How to evaluate the efficacy of the phosphodiesterase type 5 inhibitors. J. Sex. Med. 2012, 9, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Mirone, V.; Fusco, F.; Rossi, A.; Sicuteri, R.; Montorsi, F. Tadalafil and vardenafil vs sildenafil: A review of patient-preference studies. BJU Int. 2009, 103, 1212–1217. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; Morrell, N.W.; Archer, S.L.; Stenmark, K.R.; MacLean, M.R.; Lang, I.M.; Christman, B.W.; Weir, E.K.; Eickelberg, O.; Voelkel, N.F. Cellular and molecular pathobiology of pulmonary arterial hypertension. J. Am. Coll. Cardiol. 2004, 43, S13–S24. [Google Scholar] [CrossRef] [PubMed]
- Corbin, J.D.; Beasley, A.; Blount, M.A.; Francis, S.H. High lung PDE5: A strong basis for treating pulmonary hypertension with PDE5 inhibitors. Biochem. Biophys. Res. Commun. 2005, 334, 930–938. [Google Scholar] [CrossRef]
- Giaid, A.; Saleh, D. Reduced expression of endothelial nitric oxide synthase in the lungs of patients with pulmonary hypertension. N. Engl. J. Med. 1995, 333, 214–221. [Google Scholar] [CrossRef]
- Shenoy, P.; Agarwal, V. Phosphodiesterase inhibitors in the management of autoimmune disease. Autoimmun. Rev. 2010, 9, 511–515. [Google Scholar] [CrossRef]
- Nguyen, H.; Amanullah, A.M. Therapeutic potentials of phosphodiesterase-5 inhibitors in cardiovascular disease. Rev. Cardiovasc. Med. 2014, 15, 158–167. [Google Scholar] [CrossRef]
- Yamamura, A.; Fujitomi, E.; Ohara, N.; Tsukamoto, K.; Sato, M.; Yamamura, H. Tadalafil induces antiproliferation, apoptosis, and phosphodiesterase type 5 downregulation in idiopathic pulmonary arterial hypertension in vitro. Eur. J. Pharmacol. 2017, 810, 44–50. [Google Scholar] [CrossRef]
- Foresta, C.; De Toni, L.; Di Mambro, A.; Garolla, A.; Ferlin, A.; Zuccarello, D. BASIC SCIENCE: The PDE5 Inhibitor Sildenafil Increases Circulating Endothelial Progenitor Cells and CXCR4 Expression. J. Sex. Med. 2009, 6, 369–372. [Google Scholar] [CrossRef]
- Rubin, L.J.; Badesch, D.B.; Barst, R.J.; Galiè, N.; Black, C.M.; Keogh, A.; Pulido, T.; Frost, A.; Roux, S.; Leconte, I. Bosentan therapy for pulmonary arterial hypertension. N. Engl. J. Med. 2002, 346, 896–903. [Google Scholar] [CrossRef]
- Pepke-Zaba, J.; Gilbert, C.; Collings, L.; Brown, M.C. Sildenafil improves health-related quality of life in patients with pulmonary arterial hypertension. Chest 2008, 133, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Oudiz, R.J.; Brundage, B.H.; Galiè, N.; Ghofrani, H.A.; Simonneau, G.; Botros, F.T.; Chan, M.; Beardsworth, A.; Barst, R.J.; PHIRST Study Group. Tadalafil for the treatment of pulmonary arterial hypertension: A double-blind 52-week uncontrolled extension study. J. Am. Coll. Cardiol. 2012, 60, 768–774. [Google Scholar] [CrossRef] [PubMed]
- Pepke-Zaba, J.; Beardsworth, A.; Chan, M.; Angalakuditi, M. Tadalafil therapy and health-related quality of life in pulmonary arterial hypertension. Curr. Med. Res. Opin. 2009, 25, 2479–2485. [Google Scholar] [CrossRef] [PubMed]
- Kukreja, R.C.; Salloum, F.N.; Das, A.; Koka, S.; Ockaili, R.A.; Xi, L. Emerging new uses of phosphodiesterase-5 inhibitors in cardiovascular diseases. Exp. Clin. Cardiol. 2011, 16, e30. [Google Scholar] [PubMed]
- Silvera, F.; Blasina, M.; Vaamonde, L.; Tellechea, S.; Godoy, C.; Zabala, S.; Mañana, G.; Martell, M.; Olivera, W. Sildenafil prevents the increase of extravascular lung water and pulmonary hypertension after meconium aspiration in newborn piglets. Braz. J. Med. Biol. Res. 2011, 44, 778–785. [Google Scholar] [CrossRef]
- Ghofrani, H.A.; Voswinckel, R.; Reichenberger, F.; Olschewski, H.; Haredza, P.; Karadaş, B.; Schermuly, R.T.; Weissmann, N.; Seeger, W.; Grimminger, F. Differences in hemodynamic and oxygenation responses to three different phosphodiesterase-5 inhibitors in patients with pulmonary arterial hypertension: A randomized prospective study. J. Am. Coll. Cardiol. 2004, 44, 1488–1496. [Google Scholar]
- Schwartz, B.G.; Levine, L.A.; Comstock, G.; Stecher, V.J.; Kloner, R.A. Cardiac uses of phosphodiesterase-5 inhibitors. J. Am. Coll. Cardiol. 2012, 59, 9–15. [Google Scholar] [CrossRef][Green Version]
- Simonneau, G.; Rubin, L.J.; Galie, N.; Barst, R.J.; Fleming, T.R.; Frost, A.E.; Engel, P.J.; Kramer, M.R.; Burgess, G.; Collings, L. Addition of sildenafil to long-term intravenous epoprostenol therapy in patients with pulmonary arterial hypertension: A randomized trial. Ann. Intern. Med. 2008, 149, 521–530. [Google Scholar] [CrossRef]
- Stehlik, J.; Movsesian, M.A. Combined use of PDE5 inhibitors and nitrates in the treatment of pulmonary arterial hypertension in patients with heart failure. J. Card. Fail. 2009, 15, 31–34. [Google Scholar] [CrossRef]
- Bowles, E.A.; Moody, G.N.; Yeragunta, Y.; Stephenson, A.H.; Ellsworth, M.L.; Sprague, R.S. Phosphodiesterase 5 inhibitors augment UT-15C-stimulated ATP release from erythrocytes of humans with pulmonary arterial hypertension. Exp. Biol. Med. 2015, 240, 121–127. [Google Scholar] [CrossRef]
- Zhao, L.; Mason, N.A.; Morrell, N.W.; Kojonazarov, B.; Sadykov, A.; Maripov, A.; Mirrakhimov, M.M.; Aldashev, A.; Wilkins, M.R. Sildenafil inhibits hypoxia-induced pulmonary hypertension. Circulation 2001, 104, 424–428. [Google Scholar] [CrossRef]
- Richalet, J.-P.; Gratadour, P.; Robach, P.; Pham, I.; Déchaux, M.; Joncquiert-Latarjet, A.; Mollard, P.; Brugniaux, J.; Cornolo, J. Sildenafil inhibits altitude-induced hypoxemia and pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2005, 171, 275–281. [Google Scholar] [CrossRef] [PubMed]
- McVary, K.T.; McKenna, K.E. The relationship between erectile dysfunction and lower urinary tract symptoms: Epidemiological, clinical, and basic science evidence. Curr. Prostate Rep. 2004, 2, 71–77. [Google Scholar] [CrossRef]
- Kerschan-Schindl, K.; Uher, E.; Wiesinger, G.; Kaider, A.; Ebenbichler, G.; Nicolakis, P.; Kollmitzer, J.; Preisinger, E.; Fialka-Moser, V. Reliability of pelvic floor muscle strength measurement in elderly incontinent women. Neurourol. Urodyn. Off. J. Int. Cont. Soc. 2002, 21, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Wong, P.; Lawrentschuk, N.; Bolton, D.M. Phosphodiesterase 5 inhibitors in the management of benign prostatic hyperplasia and erectile dysfunction: The best of both worlds. Curr. Opin. Urol. 2009, 19, 7–12. [Google Scholar] [CrossRef]
- Fusco, F.; di Villa Bianca, R.D.E.; Mitidieri, E.; Cirino, G.; Sorrentino, R.; Mirone, V. Sildenafil effect on the human bladder involves the L-cysteine/hydrogen sulfide pathway: A novel mechanism of action of phosphodiesterase type 5 inhibitors. Eur. Urol. 2012, 62, 1174–1180. [Google Scholar] [CrossRef] [PubMed]
- Peixoto, C.A.; dos Gomes, F.O.S. The role of phosphodiesterase-5 inhibitors in prostatic inflammation: A review. J. Inflamm. 2015, 12, 54. [Google Scholar] [CrossRef] [PubMed]
- Mónica, F.Z.; De Nucci, G. Tadalafil for the treatment of benign prostatic hyperplasia. Expert Opin. Pharmacother. 2019, 20, 929–937. [Google Scholar] [CrossRef]
- Nomiya, M.; Burmeister, D.M.; Sawada, N.; Campeau, L.; Zarifpour, M.; Keys, T.; Peyton, C.; Yamaguchi, O.; Andersson, K.-E. Prophylactic effect of tadalafil on bladder function in a rat model of chronic bladder ischemia. J. Urol. 2013, 189, 754–761. [Google Scholar] [CrossRef]
- Sairam, K.; Kulinskaya, E.; McNicholas, T.; Boustead, G.; Hanbury, D. Sildenafil influences lower urinary tract symptoms. BJU Int. 2002, 90, 836–839. [Google Scholar] [CrossRef]
- McVary, K.T.; Roehrborn, C.G.; Kaminetsky, J.C.; Auerbach, S.M.; Wachs, B.; Young, J.M.; Esler, A.; Sides, G.D.; Denes, B.S. Tadalafil relieves lower urinary tract symptoms secondary to benign prostatic hyperplasia. J. Urol. 2007, 177, 1401–1407. [Google Scholar] [CrossRef] [PubMed]
- Roehrborn, C.G.; Kaminetsky, J.C.; Auerbach, S.M.; Montelongo, R.M.; Elion-Mboussa, A.; Viktrup, L. Changes in peak urinary flow and voiding efficiency in men with signs and symptoms of benign prostatic hyperplasia during once daily tadalafil treatment. BJU Int. 2010, 105, 502–507. [Google Scholar] [CrossRef] [PubMed]
- Dmochowski, R.; Roehrborn, C.; Klise, S.; Xu, L.; Kaminetsky, J.; Kraus, S. Urodynamic effects of once daily tadalafil in men with lower urinary tract symptoms secondary to clinical benign prostatic hyperplasia: A randomized, placebo controlled 12-week clinical trial. J. Urol. 2010, 183, 1092–1097. [Google Scholar] [CrossRef] [PubMed]
- Stief, C.G.; Porst, H.; Neuser, D.; Beneke, M.; Ulbrich, E. A randomised, placebo-controlled study to assess the efficacy of twice-daily vardenafil in the treatment of lower urinary tract symptoms secondary to benign prostatic hyperplasia. Eur. Urol. 2008, 53, 1236–1244. [Google Scholar] [CrossRef]
- Gacci, M.; Andersson, K.-E.; Chapple, C.; Maggi, M.; Mirone, V.; Oelke, M.; Porst, H.; Roehrborn, C.; Stief, C.; Giuliano, F. Latest evidence on the use of phosphodiesterase type 5 inhibitors for the treatment of lower urinary tract symptoms secondary to benign prostatic hyperplasia. Eur. Urol. 2016, 70, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Liguori, G.; Trombetta, C.; De Giorgi, G.; Pomara, G.; Maio, G.; Vecchio, D.; Ocello, G.; Ollandini, G.; Bucci, S.; Belgrano, E. Efficacy and Safety of Combined Oral Therapy with Tadalafil andAlfuzosin: An Integrated Approach to the Management of Patientswith Lower Urinary Tract Symptoms and Erectile Dysfunction. Preliminary Report. J. Sex. Med. 2009, 6, 544–552. [Google Scholar] [CrossRef]
- Kaplan, S.A.; Gonzalez, R.R.; Te, A.E. Combination of alfuzosin and sildenafil is superior to monotherapy in treating lower urinary tract symptoms and erectile dysfunction. Eur. Urol. 2007, 51, 1717–1723. [Google Scholar] [CrossRef]
- Gacci, M.; Corona, G.; Salvi, M.; Vignozzi, L.; McVary, K.T.; Kaplan, S.A.; Roehrborn, C.G.; Serni, S.; Mirone, V.; Carini, M.; et al. A systematic review and meta-analysis on the use of phosphodiesterase 5 inhibitors alone or in combination with alpha-blockers for lower urinary tract symptoms due to benign prostatic hyperplasia. Eur. Urol. 2012, 61, 994–1003. [Google Scholar] [CrossRef]
- Tuncel, A.; Nalcacioglu, V.; Ener, K.; Aslan, Y.; Aydin, O.; Atan, A. Sildenafil citrate and tamsulosin combination is not superior to monotherapy in treating lower urinary tract symptoms and erectile dysfunction. World J. Urol. 2010, 28, 17–22. [Google Scholar] [CrossRef]
- Bechara, A.; Romano, S.; Casabé, A.; Haime, S.; Dedola, P.; Hernández, C.; Rey, H. Comparative efficacy assessment of tamsulosin vs. tamsulosin plus tadalafil in the treatment of LUTS/BPH. Pilot study. J. Sex. Med. 2008, 5, 2170–2178. [Google Scholar] [CrossRef]
- Guo, B.; Chen, X.; Wang, M.; Hou, H.; Zhang, Z.; Liu, M. Comparative Effectiveness of Tadalafil versus Tamsulosin in Treating Lower Urinary Tract Symptoms Suggestive of Benign Prostate Hyperplasia: A Meta-Analysis of Randomized Controlled Trials. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2020, 26, e923179-1–e923179-8. [Google Scholar] [CrossRef] [PubMed]
- Barone, I.; Giordano, C.; Bonofiglio, D.; Andò, S.; Catalano, S. Phosphodiesterase type 5 and cancers: Progress and challenges. Oncotarget 2017, 8, 99179. [Google Scholar] [CrossRef] [PubMed]
- Catalano, S.; Campana, A.; Giordano, C.; Győrffy, B.; Tarallo, R.; Rinaldi, A.; Bruno, G.; Ferraro, A.; Romeo, F.; Lanzino, M. Expression and Function of Phosphodiesterase Type 5 in Human Breast Cancer Cell Lines and Tissues: Implications for Targeted TherapyPDE5 Enhances Breast Cancer Cell Invasive Potential. Clin. Cancer Res. 2016, 22, 2271–2282. [Google Scholar] [CrossRef]
- Sponziello, M.; Verrienti, A.; Rosignolo, F.; De Rose, R.F.; Pecce, V.; Maggisano, V.; Durante, C.; Bulotta, S.; Damante, G.; Giacomelli, L. PDE5 expression in human thyroid tumors and effects of PDE5 inhibitors on growth and migration of cancer cells. Endocrine 2015, 50, 434–441. [Google Scholar] [CrossRef]
- Peak, T.C.; Richman, A.; Gur, S.; Yafi, F.A.; Hellstrom, W.J. The role of PDE5 inhibitors and the NO/cGMP pathway in cancer. Sex. Med. Rev. 2016, 4, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Pantziarka, P.; Sukhatme, V.; Crispino, S.; Bouche, G.; Meheus, L.; Sukhatme, V.P. Repurposing drugs in oncology (ReDO)—Selective PDE5 inhibitors as anti-cancer agents. Ecancermedicalscience 2018, 12, 824. [Google Scholar] [CrossRef] [PubMed]
- Sarfati, M.; Mateo, V.; Baudet, S.; Rubio, M.; Fernandez, C.; Davi, F.; Binet, J.L.; Delic, J.; Merle-Beral, H. Sildenafil and vardenafil, types 5 and 6 phosphodiesterase inhibitors, induce caspase-dependent apoptosis of B-chronic lymphocytic leukemia cells. Blood 2003, 101, 265–269. [Google Scholar] [CrossRef]
- Mei, X.-L.; Yang, Y.; Zhang, Y.-J.; Li, Y.; Zhao, J.-M.; Qiu, J.-G.; Zhang, W.-J.; Jiang, Q.-W.; Xue, Y.-Q.; Zheng, D.-W. Sildenafil inhibits the growth of human colorectal cancer in vitro and in vivo. Am. J. Cancer Res. 2015, 5, 3311. [Google Scholar]
- Li, H.; Liu, L.; David, M.L.; Whitehead, C.M.; Chen, M.; Fetter, J.R.; Sperl, G.J.; Pamukcu, R.; Thompson, W.J. Pro-apoptotic actions of exisulind and CP461 in SW480 colon tumor cells involve β-catenin and cyclin D1 down-regulation. Biochem. Pharmacol. 2002, 64, 1325–1336. [Google Scholar] [CrossRef]
- Tinsley, H.N.; Gary, B.D.; Keeton, A.B.; Lu, W.; Li, Y.; Piazza, G.A. Inhibition of PDE5 by sulindac sulfide selectively induces apoptosis and attenuates oncogenic Wnt/β-catenin–mediated transcription in human breast tumor cells. Cancer Prev. Res. 2011, 4, 1275–1284. [Google Scholar] [CrossRef]
- Rice, P.L.; Beard, K.S.; Driggers, L.J.; Ahnen, D.J. Inhibition of extracellular-signal regulated kinases 1/2 is required for apoptosis of human colon cancer cells in vitro by sulindac metabolites. Cancer Res. 2004, 64, 8148–8151. [Google Scholar] [CrossRef]
- Aono, Y.; Horinaka, M.; Iizumi, Y.; Watanabe, M.; Taniguchi, T.; Yasuda, S.; Sakai, T. Sulindac sulfone inhibits the mTORC1 pathway in colon cancer cells by directly targeting voltage-dependent anion channel 1 and 2. Biochem. Biophys. Res. Commun. 2018, 505, 1203–1210. [Google Scholar] [CrossRef] [PubMed]
- Thompson, W.J.; Piazza, G.A.; Li, H.; Liu, L.; Fetter, J.; Zhu, B.; Sperl, G.; Ahnen, D.; Pamukcu, R. Exisulind induction of apoptosis involves guanosine 3′, 5′-cyclic monophosphate phosphodiesterase inhibition, protein kinase G activation, and attenuated β-catenin. Cancer Res. 2000, 60, 3338–3342. [Google Scholar]
- Soh, J.-W.; Mao, Y.; Kim, M.-G.; Pamukcu, R.; Li, H.; Piazza, G.A.; Thompson, W.J.; Weinstein, I.B. Cyclic GMP mediates apoptosis induced by sulindac derivatives via activation of c-Jun NH2-terminal kinase. Clin. Cancer Res. 2000, 6, 4136–4141. [Google Scholar] [PubMed]
- Li, Q.; Shu, Y. Pharmacological modulation of cytotoxicity and cellular uptake of anti-cancer drugs by PDE5 inhibitors in lung cancer cells. Pharm. Res. 2014, 31, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Durrant, D.; Salloum, F.N.; Xi, L.; Kukreja, R.C. PDE5 inhibitors as therapeutics for heart disease, diabetes and cancer. Pharmacol. Ther. 2015, 147, 12–21. [Google Scholar] [CrossRef]
- Booth, L.; Roberts, J.; Cruickshanks, N.; Conley, A.; Durrant, D.; Das, A.; Fisher, P.; Kukreja, R.; Grant, S.; Poklepovic, A. PDE5 inhibitors enhance chemotherapy killing in gastrointestinal/genitourinary cancer cells. Mol. Pharmacol. 2014, 85, 408–419. [Google Scholar] [CrossRef]
- Ahn, K.-S.; Sim, W.-S.; Lee, I.-K.; Seu, Y.-B.; Kim, I.-H. Decursinol angelate: A cytotoxic and protein kinase C activating agent from the root of Angelica gigas. Planta Med. 1997, 63, 360–361. [Google Scholar] [CrossRef]
- Ding, P.-R.; Tiwari, A.K.; Ohnuma, S.; Lee, J.W.; An, X.; Dai, C.-L.; Lu, Q.-S.; Singh, S.; Yang, D.-H.; Talele, T.T. The phosphodiesterase-5 inhibitor vardenafil is a potent inhibitor of ABCB1/P-glycoprotein transporter. PLoS ONE 2011, 6, e19329. [Google Scholar] [CrossRef]
- Shi, Z.; Tiwari, A.K.; Shukla, S.; Robey, R.W.; Singh, S.; Kim, I.-W.; Bates, S.E.; Peng, X.; Abraham, I.; Ambudkar, S.V. Sildenafil reverses ABCB1-and ABCG2-mediated chemotherapeutic drug resistance. Cancer Res. 2011, 71, 3029–3041. [Google Scholar] [CrossRef]
- Black, K.L.; Yin, D.; Ong, J.M.; Hu, J.; Konda, B.M.; Wang, X.; Ko, M.K.; Bayan, J.-A.; Sacapano, M.R.; Espinoza, A. PDE5 inhibitors enhance tumor permeability and efficacy of chemotherapy in a rat brain tumor model. Brain Res. 2008, 1230, 290–302. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Ljubimova, J.Y.; Inoue, S.; Konda, B.; Patil, R.; Ding, H.; Espinoza, A.; Wawrowsky, K.A.; Patil, C.; Ljubimov, A.V. Phosphodiesterase type 5 inhibitors increase Herceptin transport and treatment efficacy in mouse metastatic brain tumor models. PLoS ONE 2010, 5, e10108. [Google Scholar] [CrossRef] [PubMed]
- Booth, L.; Roberts, J.L.; Cruickshanks, N.; Tavallai, S.; Webb, T.; Samuel, P.; Conley, A.; Binion, B.; Young, H.F.; Poklepovic, A. PDE5 inhibitors enhance celecoxib killing in multiple tumor types. J. Cell. Physiol. 2015, 230, 1115–1127. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Durrant, D.; Mitchell, C.; Mayton, E.; Hoke, N.N.; Salloum, F.N.; Park, M.A.; Qureshi, I.; Lee, R.; Dent, P. Sildenafil increases chemotherapeutic efficacy of doxorubicin in prostate cancer and ameliorates cardiac dysfunction. Proc. Natl. Acad. Sci. USA 2010, 107, 18202–18207. [Google Scholar] [CrossRef]
- Chang, J.-F.; Hsu, J.-L.; Sheng, Y.-H.; Leu, W.-J.; Yu, C.-C.; Chan, S.-H.; Chan, M.-L.; Hsu, L.-C.; Liu, S.-P.; Guh, J.-H. Phosphodiesterase type 5 (PDE5) inhibitors sensitize topoisomerase II inhibitors in killing prostate cancer through PDE5-independent impairment of HR and NHEJ DNA repair systems. Front. Oncol. 2019, 8, 681. [Google Scholar] [CrossRef]
- Hsu, J.-L.; Leu, W.-J.; Hsu, L.-C.; Ho, C.-H.; Liu, S.-P.; Guh, J.-H. Phosphodiesterase Type 5 Inhibitors Synergize Vincristine in Killing Castration-Resistant Prostate Cancer Through Amplifying Mitotic Arrest Signaling. Front. Oncol. 2020, 10, 1274. [Google Scholar] [CrossRef]
- Tavallai, M.; Hamed, H.A.; Roberts, J.L.; Cruickshanks, N.; Chuckalovcak, J.; Poklepovic, A.; Booth, L.; Dent, P. Nexavar/Stivarga and viagra interact to kill tumor cells. J. Cell. Physiol. 2015, 230, 2281–2298. [Google Scholar] [CrossRef]
- Booth, L.; Roberts, J.L.; Poklepovic, A.; Dent, P. PDE5 inhibitors enhance the lethality of [pemetrexed+ sorafenib]. Oncotarget 2017, 8, 13464. [Google Scholar] [CrossRef]
- Chuckalovcak, J.; Carter, J.; Poklepovic, A.; Dent, P. OSU-03012 and Viagra treatment inhibits the activity of multiple chaperone proteins and disrupts the blood brain barrier: Implications for anti-cancer therapies. J. Cell. Physiol. 2015, 230, 1982–1998. [Google Scholar]
- Chen, L.; Liu, Y.; Becher, A.; Diepold, K.; Schmid, E.; Fehn, A.; Brunner, C.; Rouhi, A.; Chiosis, G.; Cronauer, M. Sildenafil triggers tumor lethality through altered expression of HSP90 and degradation of PKD2. Carcinogenesis 2020, 41, 1421–1431. [Google Scholar] [CrossRef]
- Serafini, P.; Meckel, K.; Kelso, M.; Noonan, K.; Califano, J.; Koch, W.; Dolcetti, L.; Bronte, V.; Borrello, I. Phosphodiesterase-5 inhibition augments endogenous antitumor immunity by reducing myeloid-derived suppressor cell function. J. Exp. Med. 2006, 203, 2691–2702. [Google Scholar] [CrossRef] [PubMed]
- Califano, J.A.; Khan, Z.; Noonan, K.A.; Rudraraju, L.; Zhang, Z.; Wang, H.; Goodman, S.; Gourin, C.G.; Ha, P.K.; Fakhry, C. Tadalafil augments tumor specific immunity in patients with head and neck squamous cell carcinoma. Clin. Cancer Res. 2015, 21, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Sundquist, J.; Sundquist, K.; Ji, J. Use of phosphodiesterase 5 inhibitors is associated with lower risk of colorectal cancer in men with benign colorectal neoplasms. Gastroenterology 2019, 157, 672–681. [Google Scholar] [CrossRef] [PubMed]
- Chhonker, S.K.; Rawat, D.; Koiri, R.K. Protective and therapeutic effects of sildenafil and tadalafil on aflatoxin B1-induced hepatocellular carcinoma. Mol. Cell. Biochem. 2021, 476, 1195–1209. [Google Scholar] [CrossRef] [PubMed]
- Chhonker, S.K.; Rawat, D.; Koiri, R.K. Repurposing PDE5 inhibitor tadalafil and sildenafil as anticancer agent against hepatocellular carcinoma via targeting key events of glucose metabolism and multidrug resistance. J. Biochem. Mol. Toxicol. 2022, 36, e23100. [Google Scholar] [CrossRef]
- Liu, N.; Mei, L.; Fan, X.; Tang, C.; Ji, X.; Hu, X.; Shi, W.; Qian, Y.; Hussain, M.; Wu, J. Phosphodiesterase 5/protein kinase G signal governs stemness of prostate cancer stem cells through Hippo pathway. Cancer Lett. 2016, 378, 38–50. [Google Scholar] [CrossRef]
- Puzzo, D.; Sapienza, S.; Arancio, O.; Palmeri, A. Role of phosphodiesterase 5 in synaptic plasticity and memory. Neuropsychiatr. Dis. Treat. 2008, 4, 371–387. [Google Scholar] [CrossRef]
- Lu, Y.-F.; Kandel, E.R.; Hawkins, R.D. Nitric oxide signaling contributes to late-phase LTP and CREB phosphorylation in the hippocampus. J. Neurosci. 1999, 19, 10250–10261. [Google Scholar] [CrossRef]
- Ben Aissa, M.; Lee, S.H.; Bennett, B.M.; Thatcher, G.R.J. Targeting NO/cGMP signaling in the CNS for neurodegeneration and Alzheimer’s disease. Curr. Med. Chem. 2016, 23, 2770–2788. [Google Scholar] [CrossRef]
- Kawasaki, K.; Smith Jr, R.S.; Hsieh, C.-M.; Sun, J.; Chao, J.; Liao, J.K. Activation of the phosphatidylinositol 3-kinase/protein kinase Akt pathway mediates nitric oxide-induced endothelial cell migration and angiogenesis. Mol. Cell. Biol. 2003, 23, 5726–5737. [Google Scholar] [CrossRef]
- García-Osta, A.; Cuadrado-Tejedor, M.; García-Barroso, C.; Oyarzabal, J.; Franco, R. Phosphodiesterases as therapeutic targets for Alzheimer’s disease. ACS Chem. Neurosci. 2012, 3, 832–844. [Google Scholar] [CrossRef] [PubMed]
- Sabayan, B.; Zamiri, N.; Farshchizarabi, S.; Sabayan, B. Phoshphodiesterase-5 inhibitors: Novel weapons against Alzheimer’s disease? Int. J. Neurosci. 2010, 120, 746–751. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado-Tejedor, M.; Hervias, I.; Ricobaraza, A.; Puerta, E.; Pérez-Roldán, J.; García-Barroso, C.; Franco, R.; Aguirre, N.; García-Osta, A. Sildenafil restores cognitive function without affecting β-amyloid burden in a mouse model of Alzheimer’s disease. Br. J. Pharmacol. 2011, 164, 2029–2041. [Google Scholar] [CrossRef] [PubMed]
- Puzzo, D.; Staniszewski, A.; Deng, S.X.; Privitera, L.; Leznik, E.; Liu, S.; Zhang, H.; Feng, Y.; Palmeri, A.; Landry, D.W. Phosphodiesterase 5 inhibition improves synaptic function, memory, and amyloid-β load in an Alzheimer’s disease mouse model. J. Neurosci. 2009, 29, 8075–8086. [Google Scholar] [CrossRef]
- Zhang, J.; Guo, J.; Zhao, X.; Chen, Z.; Wang, G.; Liu, A.; Wang, Q.; Zhou, W.; Xu, Y.; Wang, C. Phosphodiesterase-5 inhibitor sildenafil prevents neuroinflammation, lowers beta-amyloid levels and improves cognitive performance in APP/PS1 transgenic mice. Behav. Brain Res. 2013, 250, 230–237. [Google Scholar] [CrossRef]
- García-Barroso, C.; Ricobaraza, A.; Pascual-Lucas, M.; Unceta, N.; Rico, A.J.; Goicolea, M.A.; Sallés, J.; Lanciego, J.L.; Oyarzabal, J.; Franco, R. Tadalafil crosses the blood–brain barrier and reverses cognitive dysfunction in a mouse model of AD. Neuropharmacology 2013, 64, 114–123. [Google Scholar] [CrossRef]
- Devan, B.D.; Sierra-Mercado, D., Jr.; Jimenez, M.; Bowker, J.L.; Duffy, K.B.; Spangler, E.L.; Ingram, D.K. Phosphodiesterase inhibition by sildenafil citrate attenuates the learning impairment induced by blockade of cholinergic muscarinic receptors in rats. Pharmacol. Biochem. Behav. 2004, 79, 691–699. [Google Scholar] [CrossRef] [PubMed]
- Reneerkens, O.A.; Rutten, K.; Akkerman, S.; Blokland, A.; Shaffer, C.L.; Menniti, F.S.; Steinbusch, H.W.; Prickaerts, J. Phosphodiesterase type 5 (PDE5) inhibition improves object recognition memory: Indications for central and peripheral mechanisms. Neurobiol. Learn. Mem. 2012, 97, 370–379. [Google Scholar] [CrossRef]
- Orejana, L.; Barros-Miñones, L.; Jordán, J.; Puerta, E.; Aguirre, N. Sildenafil ameliorates cognitive deficits and tau pathology in a senescence-accelerated mouse model. Neurobiol. Aging 2012, 33, 625.e611–625.e620. [Google Scholar] [CrossRef]
- Sikandaner, H.E.; Park, S.Y.; Kim, M.J.; Park, S.N.; Yang, D.W. Neuroprotective effects of sildenafil against oxidative stress and memory dysfunction in mice exposed to noise stress. Behav. Brain Res. 2017, 319, 37–47. [Google Scholar] [CrossRef]
- Al-Amin, M.M.; Hasan, S.N.; Alam, T.; Hasan, A.T.; Hossain, I.; Didar, R.R.; Alam, M.A.; Rahman, M.M. Tadalafil enhances working memory, and reduces hippocampal oxidative stress in both young and aged mice. Eur. J. Pharmacol. 2014, 745, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Xu, H.; Ding, S.; Wang, D.; Song, G.; Huang, X. Phosphodiesterase 5 inhibitors as novel agents for the treatment of Alzheimer’s disease. Brain Res. Bull. 2019, 153, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Zuccarello, E.; Acquarone, E.; Calcagno, E.; Argyrousi, E.K.; Deng, S.-X.; Landry, D.W.; Arancio, O.; Fiorito, J. Development of novel phosphodiesterase 5 inhibitors for the therapy of Alzheimer’s disease. Biochem. Pharmacol. 2020, 176, 113818. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, Z.; Zhang, R.L.; Cui, Y.; LaPointe, M.C.; Silver, B.; Chopp, M. Tadalafil, a long-acting type 5 phosphodiesterase isoenzyme inhibitor, improves neurological functional recovery in a rat model of embolic stroke. Brain Res. 2006, 1118, 192–198. [Google Scholar] [CrossRef]
- Mendes Soares, L.; Prickaerts, J.; Milani, H.; Del Bel, E.; Wilhelm Maria Steinbusch, H.; Maria Weffort de Oliveira, R. Phosphodiesterase inhibition as a therapeutic target for brain ischemia. CNS Neurol. Disord.-Drug Targets (Former. Curr. Drug Targets-CNS Neurol. Disord.) 2015, 14, 1012–1023. [Google Scholar]
- Ölmestig, J.N.; Marlet, I.R.; Hainsworth, A.H.; Kruuse, C. Phosphodiesterase 5 inhibition as a therapeutic target for ischemic stroke: A systematic review of preclinical studies. Cell. Signal. 2017, 38, 39–48. [Google Scholar] [CrossRef]
- Silver, B.; McCarthy, S.; Lu, M.; Mitsias, P.; Russman, A.N.; Katramados, A.; Morris, D.C.; Lewandowski, C.A.; Chopp, M. Sildenafil treatment of subacute ischemic stroke: A safety study at 25-mg daily for 2 weeks. J. Stroke Cerebrovasc. Dis. 2009, 18, 381–383. [Google Scholar] [CrossRef]
- Ozdegirmenci, O.; Kucukozkan, T.; Akdag, E.; Topal, T.; Haberal, A.; Kayir, H.; Oter, S.; Akyol, M.; Uzbay, T. Effects of sildenafil and tadalafil on ischemia/reperfusion injury in fetal rat brain. J. Matern.-Fetal Neonatal Med. 2011, 24, 317–323. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, Z.G.; Zhang, R.L.; Chopp, M. Activation of the PI3-K/Akt pathway mediates cGMP enhanced-neurogenesis in the adult progenitor cells derived from the subventricular zone. J. Cereb. Blood Flow Metab. 2005, 25, 1150–1158. [Google Scholar] [CrossRef]
- Santos, A.I.; Carreira, B.P.; Nobre, R.J.; Carvalho, C.M.; Araújo, I.M. Stimulation of neural stem cell proliferation by inhibition of phosphodiesterase 5. Stem Cells Int. 2014, 2014, 878397. [Google Scholar] [CrossRef]
- Son, Y.; Kim, K.; Cho, H.-R. Sildenafil protects neuronal cells from mitochondrial toxicity induced by β-amyloid peptide via ATP-sensitive K+ channels. Biochem. Biophys. Res. Commun. 2018, 500, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Thakur, T.; Sharma, S.; Kumar, K.; Deshmukh, R.; Sharma, P.L. Neuroprotective role of PDE4 and PDE5 inhibitors in 3-nitropropionic acid induced behavioral and biochemical toxicities in rats. Eur. J. Pharmacol. 2013, 714, 515–521. [Google Scholar] [CrossRef] [PubMed]
- Kenney, K.; Amyot, F.; Moore, C.; Haber, M.; Turtzo, L.C.; Shenouda, C.; Silverman, E.; Gong, Y.; Qu, B.X.; Harburg, L. Phosphodiesterase-5 inhibition potentiates cerebrovascular reactivity in chronic traumatic brain injury. Ann. Clin. Transl. Neurol. 2018, 5, 418–428. [Google Scholar] [CrossRef]
- de Santana Nunes, A.K.; Rapôso, C.; de Oliveira, W.H.; Thomé, R.; Verinaud, L.; Tovar-Moll, F.; Peixoto, C.A. Phosphodiesterase-5 inhibition promotes remyelination by MCP-1/CCR-2 and MMP-9 regulation in a cuprizone-induced demyelination model. Exp. Neurol. 2016, 275, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Pifarre, P.; Prado, J.; Baltrons, M.A.; Giralt, M.; Gabarro, P.; Feinstein, D.L.; Hidalgo, J.; Garcia, A. Sildenafil (Viagra) ameliorates clinical symptoms and neuropathology in a mouse model of multiple sclerosis. Acta Neuropathol. 2011, 121, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Liebenberg, N.; Harvey, B.H.; Brand, L.; Wegener, G.; Brink, C.B. Chronic treatment with the phosphodiesterase type 5 inhibitors sildenafil and tadalafil display anxiolytic effects in Flinders Sensitive Line rats. Metab. Brain Dis. 2012, 27, 337–340. [Google Scholar] [CrossRef]
- Otari, K.; Upasani, C. Antidepressant-like effect of tadalafil, a phosphodiesterase type 5 inhibitor, in the forced swim test: Dose and duration of treatment dependence. Neurochem. J. 2015, 9, 306–310. [Google Scholar] [CrossRef]
- Matsushita, H.; Matsuzaki, M.; Han, X.-J.; Nishiki, T.-I.; Ohmori, I.; Michiue, H.; Matsui, H.; Tomizawa, K. Antidepressant-like effect of sildenafil through oxytocin-dependent cyclic AMP response element-binding protein phosphorylation. Neuroscience 2012, 200, 13–18. [Google Scholar] [CrossRef]
- Jaumann, M.; Dettling, J.; Gubelt, M.; Zimmermann, U.; Gerling, A.; Paquet-Durand, F.; Feil, S.; Wolpert, S.; Franz, C.; Varakina, K. cGMP-Prkg1 signaling and Pde5 inhibition shelter cochlear hair cells and hearing function. Nat. Med. 2012, 18, 252–259. [Google Scholar] [CrossRef]
- Huang, L.J.; Yoon, M.H.; Choi, J.I.; Kim, W.M.; Lee, H.G.; Kim, Y.O. Effect of sildenafil on neuropathic pain and hemodynamics in rats. Yonsei Med. J. 2010, 51, 82–87. [Google Scholar] [CrossRef]
- Vieira, M.C.; de Monte, F.B.M.; Eduardo Dematte, B.; Montagnoli, T.L.; Montes, G.C.; da Silva, J.S.; Mendez-Otero, R.; Trachez, M.M.; Sudo, R.T.; Zapata-Sudo, G. Antinociceptive Effect of Lodenafil Carbonate in Rodent Models of Inflammatory Pain and Spinal Nerve Ligation-Induced Neuropathic Pain. J. Pain Res. 2021, 14, 857–866. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Chopp, M.; Szalad, A.; Liu, Z.; Bolz, M.; Ãlvarez, F.M.; Lu, M.; Zhang, L.; Cui, Y.; Zhang, R.L. Phosphodiesterase-5 is a therapeutic target for peripheral neuropathy in diabetic mice. Neuroscience 2011, 193, 399–410. [Google Scholar] [CrossRef]
- Hackett, G. PDE5 inhibitors in diabetic peripheral neuropathy. Int. J. Clin. Pract. 2006, 60, 1123–1126. [Google Scholar] [CrossRef] [PubMed]
- Finnerup, N.B.; Sindrup, S.H.; Jensen, T.S. Chronic neuropathic pain: Mechanisms, drug targets and measurement. Fundam. Clin. Pharmacol. 2007, 21, 129–136. [Google Scholar] [CrossRef]
- Wallis, R.M.; Corbin, J.D.; Francis, S.H.; Ellis, P. Tissue distribution of phosphodiesterase families and the effects of sildenafil on tissue cyclic nucleotides, platelet function, and the contractile responses of trabeculae carneae and aortic rings in vitro. Am. J. Cardiol. 1999, 83, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Korkmaz-Icöz, S.; Radovits, T.; Szabó, G. Targeting phosphodiesterase 5 as a therapeutic option against myocardial ischaemia/reperfusion injury and for treating heart failure. Br. J. Pharmacol. 2018, 175, 223–231. [Google Scholar] [CrossRef]
- du Toit, E.F.; Rossouw, E.; Salie, R.; Opie, L.H.; Lochner, A. Effect of sildenafil on reperfusion function, infarct size, and cyclic nucleotide levels in the isolated rat heart model. Cardiovasc. Drugs Ther. 2005, 19, 23–31. [Google Scholar] [CrossRef]
- Sesti, C.; Florio, V.; Johnson, E.; Kloner, R. The phosphodiesterase-5 inhibitor tadalafil reduces myocardial infarct size. Int. J. Impot. Res. 2007, 19, 55–61. [Google Scholar] [CrossRef]
- Ahmad, N.; Wang, Y.; Ali, A.K.; Ashraf, M. Long-acting phosphodiesterase-5 inhibitor, tadalafil, induces sustained cardioprotection against lethal ischemic injury. Am. J. Physiol.-Heart Circ. Physiol. 2009, 297, H387–H391. [Google Scholar] [CrossRef]
- Maas, O.; Donat, U.; Frenzel, M.; Rütz, T.; Kroemer, H.; Felix, S.; Krieg, T. Vardenafil protects isolated rat hearts at reperfusion dependent on GC and PKG. Br. J. Pharmacol. 2008, 154, 25–31. [Google Scholar] [CrossRef]
- Das, A.; Xi, L.; Kukreja, R.C. Phosphodiesterase-5 inhibitor sildenafil preconditions adult cardiac myocytes against necrosis and apoptosis: Essential role of nitric oxide signaling. J. Biol. Chem. 2005, 280, 12944–12955. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, H.; Nagayama, T.; Blanton, R.M.; Seo, K.; Zhang, M.; Zhu, G.; Lee, D.I.; Bedja, D.; Hsu, S.; Tsukamoto, O. PDE5 inhibitor efficacy is estrogen dependent in female heart disease. J. Clin. Investig. 2014, 124, 2464–2471. [Google Scholar] [CrossRef] [PubMed]
- Hoke, N.N.; Salloum, F.N.; Kass, D.A.; Das, A.; Kukreja, R.C. Preconditioning by phosphodiesterase-5 inhibition improves therapeutic efficacy of adipose-derived stem cells following myocardial infarction in mice. Stem Cells 2012, 30, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Salloum, F.N.; Chau, V.Q.; Hoke, N.N.; Abbate, A.; Varma, A.; Ockaili, R.A.; Toldo, S.; Kukreja, R.C. Phosphodiesterase-5 inhibitor, tadalafil, protects against myocardial ischemia/reperfusion through protein-kinase G–dependent generation of hydrogen sulfide. Circulation 2009, 120, S31–S36. [Google Scholar] [CrossRef]
- Koka, S.; Xi, L.; Kukreja, R.C. Chronic inhibition of phosphodiesterase 5 with tadalafil affords cardioprotection in a mouse model of metabolic syndrome: Role of nitric oxide. Mol. Cell. Biochem. 2020, 468, 47–58. [Google Scholar] [CrossRef]
- Hutchings, D.C.; Anderson, S.G.; Caldwell, J.L.; Trafford, A.W. Phosphodiesterase-5 inhibitors and the heart: Compound cardioprotection? Heart 2018, 104, 1244–1250. [Google Scholar] [CrossRef] [PubMed]
- Nagendran, J.; Archer, S.L.; Soliman, D.; Gurtu, V.; Moudgil, R.; Haromy, A.; St. Aubin, C.; Webster, L.; Rebeyka, I.M.; Ross, D.B. Phosphodiesterase type 5 is highly expressed in the hypertrophied human right ventricle, and acute inhibition of phosphodiesterase type 5 improves contractility. Circulation 2007, 116, 238–248. [Google Scholar] [CrossRef]
- Lewis, G.D.; Lachmann, J.; Camuso, J.; Lepore, J.J.; Shin, J.; Martinovic, M.E.; Systrom, D.M.; Bloch, K.D.; Semigran, M.J. Sildenafil improves exercise hemodynamics and oxygen uptake in patients with systolic heart failure. Circulation 2007, 115, 59–66. [Google Scholar] [CrossRef]
- Guazzi, M.; Casali, M.; Berti, F.; Rossoni, G.; D’Gennaro Colonna, V.; Guazzi, M. Endothelium-mediated modulation of ergoreflex and improvement in exercise ventilation by acute sildenafil in heart failure patients. Clin. Pharmacol. Ther. 2008, 83, 336–341. [Google Scholar] [CrossRef]
- De Vecchis, R.; Cesaro, A.; Ariano, C.; Giasi, A.; Cioppa, C. Phosphodiesterase-5 inhibitors improve clinical outcomes, exercise capacity and pulmonary hemodynamics in patients with heart failure with reduced left ventricular ejection fraction: A meta-analysis. J. Clin. Med. Res. 2017, 9, 488. [Google Scholar] [CrossRef]
- Fisher, P.W.; Salloum, F.; Das, A.; Hyder, H.; Kukreja, R.C. Phosphodiesterase-5 inhibition with sildenafil attenuates cardiomyocyte apoptosis and left ventricular dysfunction in a chronic model of doxorubicin cardiotoxicity. Circulation 2005, 111, 1601–1610. [Google Scholar] [CrossRef] [PubMed]
- Koka, S.; Das, A.; Zhu, S.-G.; Durrant, D.; Xi, L.; Kukreja, R.C. Long-acting phosphodiesterase-5 inhibitor tadalafil attenuates doxorubicin-induced cardiomyopathy without interfering with chemotherapeutic effect. J. Pharmacol. Exp. Ther. 2010, 334, 1023–1030. [Google Scholar] [CrossRef] [PubMed]
- Yaguas, K.; Bautista, R.; Quiroz, Y.; Ferrebuz, A.; Pons, H.; Franco, M.; Vaziri, N.D.; Rodriguez-Iturbe, B. Chronic sildenafil treatment corrects endothelial dysfunction and improves hypertension. Am. J. Nephrol. 2010, 31, 283–291. [Google Scholar] [CrossRef]
- Lee, T.-M.; Chen, C.-C.; Chung, T.-H.; Chang, N.-C. Effect of sildenafil on ventricular arrhythmias in post-infarcted rat hearts. Eur. J. Pharmacol. 2012, 690, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Nagy, O.; Hajnal, Á.; Parratt, J.R.; Végh, Á. Sildenafil (Viagra) reduces arrhythmia severity during ischaemia 24 h after oral administration in dogs. Br. J. Pharmacol. 2004, 141, 549–551. [Google Scholar] [CrossRef]
- De Bon, E.; Bonanni, G.; Saggiorato, G.; Bassi, P.; Cella, G. Effects of tadalafil on platelets and endothelium in patients with erectile dysfunction and cardiovascular risk factors: A pilot study. Angiology 2010, 61, 602–606. [Google Scholar] [CrossRef]
- Toque, H.; Teixeira, C.; Priviero, F.; Morganti, R.; Antunes, E.; De Nucci, G. Vardenafil, but not sildenafil or tadalafil, has calcium-channel blocking activity in rabbit isolated pulmonary artery and human washed platelets. Br. J. Pharmacol. 2008, 154, 787–796. [Google Scholar] [CrossRef]
- Lewis, G.D.; Witzke, C.; Colon-Hernandez, P.; Guerrero, J.L.; Bloch, K.D.; Semigran, M.J. Sildenafil improves coronary artery patency in a canine model of platelet-mediated cyclic coronary occlusion after thrombolysis. J. Am. Coll. Cardiol. 2006, 47, 1471–1477. [Google Scholar] [CrossRef]
- Saeed, O.; Rangasamy, S.; Selevany, I.; Madan, S.; Fertel, J.; Eisenberg, R.; Aljoudi, M.; Patel, S.R.; Shin, J.; Sims, D.B. Sildenafil is associated with reduced device thrombosis and ischemic stroke despite low-level hemolysis on Heart Mate II support. Circ. Heart Fail. 2017, 10, e004222. [Google Scholar] [CrossRef]
- Gudmundsdóttir, I.J.; McRobbie, S.J.; Robinson, S.D.; Newby, D.E.; Megson, I.L. Sildenafil potentiates nitric oxide mediated inhibition of human platelet aggregation. Biochem. Biophys. Res. Commun. 2005, 337, 382–385. [Google Scholar] [CrossRef]
- Shenoy, P.D.; Kumar, S.; Jha, L.K.; Choudhary, S.K.; Singh, U.; Misra, R.; Agarwal, V. Efficacy of tadalafil in secondary Raynaud’s phenomenon resistant to vasodilator therapy: A double-blind randomized cross-over trial. Rheumatology 2010, 49, 2420–2428. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Griffiths, B.; Allen, J. Thermographic and symptomatic effect of a single dose of sildenafil citrate on Raynaud’s phenomenon in patients with systemic sclerosis: A potential treatment. J. Rheumatol. 2006, 33, 1918–1919. [Google Scholar] [PubMed]
- Kloner, R.A.; Goldstein, I.; Kirby, M.G.; Parker, J.D.; Sadovsky, R. Cardiovascular safety of phosphodiesterase type 5 inhibitors after nearly 2 decades on the market. Sex. Med. Rev. 2018, 6, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Kukreja, R. Cardiovascular protection with sildenafil following chronic inhibition of nitric oxide synthase. Br. J. Pharmacol. 2007, 150, 538–540. [Google Scholar] [CrossRef]
- Salloum, F.N.; Ockaili, R.A.; Wittkamp, M.; Marwaha, V.R.; Kukreja, R.C. Vardenafil: A novel type 5 phosphodiesterase inhibitor reduces myocardial infarct size following ischemia/reperfusion injury via opening of mitochondrial KATP channels in rabbits. J. Mol. Cell. Cardiol. 2006, 40, 405–411. [Google Scholar] [CrossRef]
- Wang, X.; Fisher, P.W.; Xi, L.; Kukreja, R.C. Essential role of mitochondrial Ca2+-activated and ATP-sensitive K+ channels in sildenafil-induced late cardioprotection. J. Mol. Cell. Cardiol. 2008, 44, 105–113. [Google Scholar] [CrossRef]
- Madhani, M.; Hall, A.R.; Cuello, F.; Charles, R.L.; Burgoyne, J.R.; Fuller, W.; Hobbs, A.J.; Shattock, M.J.; Eaton, P. Phospholemman Ser69 phosphorylation contributes to sildenafil-induced cardioprotection against reperfusion injury. Am. J. Physiol.-Heart Circ. Physiol. 2010, 299, H827–H836. [Google Scholar] [CrossRef]
- Inserte, J.; Barba, I.; Poncelas-Nozal, M.; Hernando, V.; Agulló, L.; Ruiz-Meana, M.; Garcia-Dorado, D. cGMP/PKG pathway mediates myocardial postconditioning protection in rat hearts by delaying normalization of intracellular acidosis during reperfusion. J. Mol. Cell. Cardiol. 2011, 50, 903–909. [Google Scholar] [CrossRef]
- Nagayama, T.; Hsu, S.; Zhang, M.; Koitabashi, N.; Bedja, D.; Gabrielson, K.L.; Takimoto, E.; Kass, D.A. Sildenafil stops progressive chamber, cellular, and molecular remodeling and improves calcium handling and function in hearts with pre-existing advanced hypertrophy caused by pressure overload. J. Am. Coll. Cardiol. 2009, 53, 207–215. [Google Scholar] [CrossRef]
- Li, N.; Yuan, Y.; Li, S.; Zeng, C.; Yu, W.; Shen, M.; Zhang, R.; Li, C.; Zhang, Y.; Wang, H. PDE5 inhibitors protect against post-infarction heart failure. Front. Biosci.-Landmark 2016, 21, 1194–1210. [Google Scholar]
- Chau, V.Q.; Salloum, F.N.; Hoke, N.N.; Abbate, A.; Kukreja, R.C. Mitigation of the progression of heart failure with sildenafil involves inhibition of RhoA/Rho-kinase pathway. Am. J. Physiol.-Heart Circ. Physiol. 2011, 300, H2272–H2279. [Google Scholar] [CrossRef]
- Afsar, B.; Ortiz, A.; Covic, A.; Gaipov, A.; Esen, T.; Goldsmith, D.; Kanbay, M. Phosphodiesterase type 5 inhibitors and kidney disease. Int. Urol. Nephrol. 2015, 47, 1521–1528. [Google Scholar] [CrossRef] [PubMed]
- Köktürk, S.; Benli, E.; Ayyıldız, A.; Cırrık, S.; Çetinkol, Y.; Ayyıldız, S.N.; Noyan, T. Positive outcomes of phosphodiesterase type 5 inhibitor on histopathologic and biochemical changes induced by ureteral obstruction. Rev. Da Assoc. Médica Bras. 2019, 65, 388–393. [Google Scholar] [CrossRef] [PubMed]
- Noel, S.; Dhooghe, B.; Leal, T. PDE5 inhibitors as potential tools in the treatment of cystic fibrosis. Front. Pharmacol. 2012, 3, 167. [Google Scholar] [CrossRef] [PubMed]
- Lubamba, B.; Lebacq, J.; Reychler, G.; Marbaix, E.; Wallemacq, P.; Lebecque, P.; Leal, T. Inhaled phosphodiesterase type 5 inhibitors restore chloride transport in cystic fibrosis mice. Eur. Respir. J. 2011, 37, 72–78. [Google Scholar] [CrossRef]
- Rodriguez-Miguelez, P.; Ishii, H.; Seigler, N.; Crandall, R.; Thomas, J.; Forseen, C.; McKie, K.T.; Harris, R.A. Sildenafil improves exercise capacity in patients with cystic fibrosis: A proof-of-concept clinical trial. Ther. Adv. Chronic Dis. 2019, 10, 2040622319887879. [Google Scholar] [CrossRef]
- Dormer, R.L.; Harris, C.M.; Clark, Z.; Pereira, M.M.C.; Doull, I.J.M.; Norez, C.; Becq, F.; McPherson, M.A. Sildenafil (Viagra) corrects ΔF508-CFTR location in nasal epithelial cells from patients with cystic fibrosis. Thorax 2005, 60, 55–59. [Google Scholar] [CrossRef][Green Version]
- Lubamba, B.; Lecourt, H.; Lebacq, J.; Lebecque, P.; De Jonge, H.; Wallemacq, P.; Leal, T. Preclinical evidence that sildenafil and vardenafil activate chloride transport in cystic fibrosis. Am. J. Respir. Crit. Care Med. 2008, 177, 506–515. [Google Scholar] [CrossRef]
- Dhooghe, B.; Noël, S.; Bouzin, C.; Behets-Wydemans, G.; Leal, T. Correction of chloride transport and mislocalization of CFTR protein by vardenafil in the gastrointestinal tract of cystic fibrosis mice. PLoS ONE 2013, 8, e77314. [Google Scholar] [CrossRef]
- Poschet, J.F.; Timmins, G.S.; Taylor-Cousar, J.L.; Ornatowski, W.; Fazio, J.; Perkett, E.; Wilson, K.R.; Yu, H.D.; de Jonge, H.R.; Deretic, V. Pharmacological modulation of cGMP levels by phosphodiesterase 5 inhibitors as a therapeutic strategy for treatment of respiratory pathology in cystic fibrosis. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2007, 293, L712–L719. [Google Scholar] [CrossRef]
- Noel, S.; Panin, N.; Beka, M.; Dhooghe, B.; Huaux, F.; Leal, T. Vardenafil reduces macrophage pro-inflammatory overresponses in cystic fibrosis through PDE5-and CFTR-dependent mechanisms. Clin. Sci. 2017, 131, 1107–1121. [Google Scholar] [CrossRef] [PubMed]
- Poschet, J.F.; Fazio, J.A.; Timmins, G.S.; Ornatowski, W.; Perkett, E.; Delgado, M.; Deretic, V. Endosomal hyperacidification in cystic fibrosis is due to defective nitric oxide–cylic GMP signalling cascade. EMBO Rep. 2006, 7, 553–559. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Miguelez, P.; Lee, N.; Tucker, M.A.; Csányi, G.; McKie, K.T.; Forseen, C.; Harris, R.A. Sildenafil improves vascular endothelial function in patients with cystic fibrosis. Am. J. Physiol.-Heart Circ. Physiol. 2018, 315, H1486–H1494. [Google Scholar] [CrossRef] [PubMed]
- Corbin, J. Mechanisms of action of PDE5 inhibition in erectile dysfunction. Int. J. Impot. Res. 2004, 16, S4–S7. [Google Scholar] [CrossRef] [PubMed]
- Kukreja, R.C.; Wang, R.; Koka, S.; Das, A.; Samidurai, A.; Xi, L. Treating diabetes with combination of phosphodiesterase 5 inhibitors and hydroxychloroquine—A possible prevention strategy for COVID-19? Mol. Cell. Biochem. 2023, 479, 679–696. [Google Scholar] [CrossRef] [PubMed]
- Salonia, A.; Maga, T.; Colombo, R.; Scattoni, V.; Briganti, A.; Cestari, A.; Guazzoni, G.; Rigatti, P.; Montorsi, F. A prospective study comparing paroxetine alone versus paroxetine plus sildenafil in patients with premature ejaculation. J. Urol. 2002, 168, 2486–2489. [Google Scholar] [CrossRef]
- Gökçe, A.; Halis, F.; Demirtas, A.; Ekmekcioglu, O. The effects of three phosphodiesterase type 5 inhibitors on ejaculation latency time in lifelong premature ejaculators: A double-blind laboratory setting study. BJU Int. 2011, 107, 1274–1277. [Google Scholar] [CrossRef]
- Mcmahon, C.G.; Mcmahon, C.N.; Leow, L.J.; Winestock, C.G. Efficacy of type-5 phosphodiesterase inhibitors in the drug treatment of premature ejaculation: A systematic review. BJU Int. 2006, 98, 259–272. [Google Scholar] [CrossRef]
- Ferrini, M.G.; Kovanecz, I.; Nolazco, G.; Rajfer, J.; Gonzalez-Cadavid, N.F. Effects of long-term vardenafil treatment on the development of fibrotic plaques in a rat model of Peyronie’s disease. BJU Int. 2006, 97, 625–633. [Google Scholar] [CrossRef]
- Gonzalez-Cadavid, N.F.; Rajfer, J. Treatment of Peyronie’s disease with PDE5 inhibitors: An antifibrotic strategy. Nat. Rev. Urol. 2010, 7, 215–221. [Google Scholar] [CrossRef]
- Dimitriadis, F.; Giannakis, D.; Pardalidis, N.; Zikopoulos, K.; Paraskevaidis, E.; Giotitsas, N.; Kalaboki, V.; Tsounapi, P.; Baltogiannis, D.; Georgiou, I. Effects of phosphodiesterase 5 inhibitors on sperm parameters and fertilizing capacity. Asian J. Androl. 2008, 10, 115–133. [Google Scholar] [CrossRef] [PubMed]
- Jannini, E.A.; Lombardo, F.; Salacone, P.; Gandini, L.; Lenzi, A. Treatment of sexual dysfunctions secondary to male infertility with sildenafil citrate. Fertil. Steril. 2004, 81, 705–707. [Google Scholar] [CrossRef] [PubMed]
- Andric, S.A.; Janjic, M.M.; Stojkov, N.J.; Kostic, T.S. Sildenafil treatment in vivo stimulates Leydig cell steroidogenesis via the cAMP/cGMP signaling pathway. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E544–E550. [Google Scholar] [CrossRef] [PubMed]
- Janjic, M.M.; Stojkov, N.J.; Bjelic, M.M.; Mihajlovic, A.I.; Andric, S.A.; Kostic, T.S. Transient rise of serum testosterone level after single sildenafil treatment of adult male rats. J. Sex. Med. 2012, 9, 2534–2543. [Google Scholar] [CrossRef]
- Magawa, S.; Nii, M.; Tanaka, H.; Furuhashi, F.; Maki, S.; Kubo, M.; Tanaka, K.; Kondo, E.; Ikeda, T. Phase-1 clinical study of tadalafil administered for selective fetal growth restriction in twin pregnancy. J. Matern.-Fetal Neonatal Med. 2021, 34, 1075–1082. [Google Scholar] [CrossRef]
- Isidori, A.M.; Giannetta, E.; Pofi, R.; Venneri, M.A.; Gianfrilli, D.; Campolo, F.; Mastroianni, C.M.; Lenzi, A.; d’Ettorre, G. Targeting the NO-cGMP-PDE5 pathway in COVID-19 infection. The DEDALO project. Andrology 2021, 9, 33–38. [Google Scholar] [CrossRef]
- Zurawin, J.L.; Stewart, C.A.; Anaissie, J.E.; Yafi, F.A.; Hellstrom, W.J. Avanafil for the treatment of erectile dysfunction. Expert Rev. Clin. Pharmacol. 2016, 9, 1163–1170. [Google Scholar] [CrossRef]
- Li, W.-Q.; Qureshi, A.A.; Robinson, K.C.; Han, J. Sildenafil use and increased risk of incident melanoma in US men: A prospective cohort study. JAMA Intern. Med. 2014, 174, 964–970. [Google Scholar] [CrossRef]
- Loeb, S.; Folkvaljon, Y.; Lambe, M.; Robinson, D.; Garmo, H.; Ingvar, C.; Stattin, P. Use of phosphodiesterase type 5 inhibitors for erectile dysfunction and risk of malignant melanoma. JAMA 2015, 313, 2449–2455. [Google Scholar] [CrossRef]
- Pottegård, A.; Schmidt, S.A.J.; Olesen, A.B.; Achacoso, N.; Van Den Eeden, S.K.; Hallas, J.; Sørensen, H.T.; Friis, S.; Habel, L.A. Use of sildenafil or other phosphodiesterase inhibitors and risk of melanoma. Br. J. Cancer 2016, 115, 895–900. [Google Scholar] [CrossRef]
- Gul, M.; Serefoglu, E.C. An update on the drug safety of treating erectile dysfunction. Expert Opin. Drug Saf. 2019, 18, 965–975. [Google Scholar] [CrossRef] [PubMed]
- Kerr, N.M.; Danesh-Meyer, H.V. Phosphodiesterase inhibitors and the eye. Clin. Exp. Ophthalmol. 2009, 37, 514–523. [Google Scholar] [CrossRef]
- Campbell, U.B.; Walker, A.M.; Gaffney, M.; Petronis, K.R.; Creanga, D.; Quinn, S.; Klein, B.E.; Laties, A.M.; Lewis, M.; Sharlip, I.D. Acute nonarteritic anterior ischemic optic neuropathy and exposure to phosphodiesterase type 5 inhibitors. J. Sex. Med. 2015, 12, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Flahavan, E.M.; Li, H.; Gupte-Singh, K.; Rizk, R.T.; Ruff, D.D.; Francis, J.L.; Kinchen, K.S. Prospective case-crossover study investigating the possible association between nonarteritic anterior ischemic optic neuropathy and phosphodiesterase type 5 inhibitor exposure. Urology 2017, 105, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Hong, B.N.; Yi, T.H.; Kim, S.Y.; Kang, T.H. High dosage sildenafil induces hearing impairment in mice. Biol. Pharm. Bull. 2008, 31, 1981–1984. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bakir, S.; Firat, U.; Gün, R.; Bozkurt, Y.; Yorgancilar, E.; Kiniş, V.; Penbegül, N.; Gökalp, O.; Topçu, İ. Histopathologic results of long-term sildenafil administration on rat inner ear. Am. J. Otolaryngol. 2012, 33, 667–672. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.S.; Sheikh, Z.; Khan, S.; Dwivedi, R.; Benjamin, E. Viagra deafness—Sensorineural hearing loss and phosphodiesterase-5 inhibitors. Laryngoscope 2011, 121, 1049–1054. [Google Scholar] [CrossRef]
- Maddox, P.T.; Saunders, J.; Chandrasekhar, S.S. Sudden hearing loss from PDE-5 inhibitors: A possible cellular stress etiology. Laryngoscope 2009, 119, 1586–1589. [Google Scholar] [CrossRef]
- Broderick, G.A.; Kadioglu, A.; Bivalacqua, T.J.; Ghanem, H.; Nehra, A.; Shamloul, R. Priapism: Pathogenesis, epidemiology, and management. J. Sex. Med. 2010, 7, 476–500. [Google Scholar] [CrossRef]
- Nehra, A.; Jackson, G.; Miner, M.; Billups, K.L.; Burnett, A.L.; Buvat, J.; Carson, C.C.; Cunningham, G.R.; Ganz, P.; Goldstein, I. The Princeton III Consensus recommendations for the management of erectile dysfunction and cardiovascular disease. Mayo Clin. Proc. 2012, 87, 766–778. [Google Scholar] [CrossRef]
- Kayık, G.; Tüzün, N.Ş.; Durdagi, S. Investigation of PDE5/PDE6 and PDE5/PDE11 selective potent tadalafil-like PDE5 inhibitors using combination of molecular modeling approaches, molecular fingerprint-based virtual screening protocols and structure-based pharmacophore development. J. Enzym. Inhib. Med. Chem. 2017, 32, 311–330. [Google Scholar] [CrossRef]
- Padma-Nathan, H.; Giuliano, F. Oral drug therapy for erectile dysfunction. Urol. Clin. N. Am. 2001, 28, 321–334. [Google Scholar] [CrossRef] [PubMed]
- Terrett, N.K.; Bell, A.S.; Brown, D.; Ellis, P. Sildenafil (VIAGRATM), a potent and selective inhibitor of type 5 cGMP phosphodiesterase with utility for the treatment of male erectile dysfunction. Bioorg. Med. Chem. Lett. 1996, 6, 1819–1824. [Google Scholar] [CrossRef]
- Haning, H.; Niewöhner, U.; Schenke, T.; Es-Sayed, M.; Schmidt, G.; Lampe, T.; Bischoff, E. Imidazo [5, 1-f], triazin-4 (3H)-ones, a new class of potent PDE 5 inhibitors. Bioorg. Med. Chem. Lett. 2002, 12, 865–868. [Google Scholar] [CrossRef] [PubMed]
- Carson, C.C., III. Phosphodiesterase type 5 inhibitors: State of the therapeutic class. Urol. Clin. N. Am. 2007, 34, 507–515. [Google Scholar] [CrossRef]
- Briganti, A.; Salonia, A.; Gallina, A.; Saccà, A.; Montorsi, P.; Rigatti, P.; Montorsi, F. Drug insight: Oral phosphodiesterase type 5 inhibitors for erectile dysfunction. Nat. Clin. Pract. Urol. 2005, 2, 239–247. [Google Scholar] [CrossRef]
- Daugan, A.; Grondin, P.; Ruault, C.; Le Monnier de Gouville, A.-C.; Coste, H.; Kirilovsky, J.; Hyafil, F.; Labaudinière, R. The discovery of Tadalafil: A novel and highly selective PDE5 inhibitor. 1: 5, 6, 11, 11a-Tetrahydro-1 H-imidazo [1′, 5′: 1, 6] pyrido [3, 4-b] indole-1, 3 (2 H)-dione analogues. J. Med. Chem. 2003, 46, 4525–4532. [Google Scholar] [CrossRef]
- Daugan, A.; Grondin, P.; Ruault, C.; Le Monnier de Gouville, A.-C.; Coste, H.; Linget, J.M.; Kirilovsky, J.; Hyafil, F.; Labaudinière, R. The discovery of tadalafil: A novel and highly selective PDE5 inhibitor. 2: 2, 3, 6, 7, 12, 12a-hexahydropyrazino [1′, 2′: 1, 6] pyrido [3, 4-b] indole-1, 4-dione analogues. J. Med. Chem. 2003, 46, 4533–4542. [Google Scholar] [CrossRef]
- Wang, G.; Liu, Z.; Chen, T.; Wang, Z.; Yang, H.; Zheng, M.; Ren, J.; Tian, G.; Yang, X.; Li, L. Design, synthesis, and pharmacological evaluation of monocyclic pyrimidinones as novel inhibitors of PDE5. J. Med. Chem. 2012, 55, 10540–10550. [Google Scholar] [CrossRef]
- Gong, X.; Wang, G.; Ren, J.; Liu, Z.; Wang, Z.; Chen, T.; Yang, X.; Jiang, X.; Shen, J.; Jiang, H. Exploration of the 5-bromopyrimidin-4 (3H)-ones as potent inhibitors of PDE5. Bioorg. Med. Chem. Lett. 2013, 23, 4944–4947. [Google Scholar] [CrossRef]
- Sakamoto, T.; Koga, Y.; Hikota, M.; Matsuki, K.; Murakami, M.; Kikkawa, K.; Fujishige, K.; Kotera, J.; Omori, K.; Morimoto, H. Design and synthesis of novel 5-(3, 4, 5-trimethoxybenzoyl)-4-aminopyrimidine derivatives as potent and selective phosphodiesterase 5 inhibitors: Scaffold hopping using a pseudo-ring by intramolecular hydrogen bond formation. Bioorg. Med. Chem. Lett. 2014, 24, 5175–5180. [Google Scholar] [CrossRef]
- Sakamoto, T.; Koga, Y.; Hikota, M.; Matsuki, K.; Murakami, M.; Kikkawa, K.; Fujishige, K.; Kotera, J.; Omori, K.; Morimoto, H. The discovery of avanafil for the treatment of erectile dysfunction: A novel pyrimidine-5-carboxamide derivative as a potent and highly selective phosphodiesterase 5 inhibitor. Bioorg. Med. Chem. Lett. 2014, 24, 5460–5465. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, T.; Koga, Y.; Hikota, M.; Matsuki, K.; Mochida, H.; Kikkawa, K.; Fujishige, K.; Kotera, J.; Omori, K.; Morimoto, H. 8-(3-Chloro-4-methoxybenzyl)-8H-pyrido [2, 3-d] pyrimidin-7-one derivatives as potent and selective phosphodiesterase 5 inhibitors. Bioorg. Med. Chem. Lett. 2015, 25, 1431–1435. [Google Scholar] [CrossRef] [PubMed]
- Sawant, S.D.; Reddy, G.L.; Dar, M.I.; Srinivas, M.; Gupta, G.; Sahu, P.K.; Mahajan, P.; Nargotra, A.; Singh, S.; Sharma, S.C. Discovery of novel pyrazolopyrimidinone analogs as potent inhibitors of phosphodiesterase type-5. Bioorg. Med. Chem. 2015, 23, 2121–2128. [Google Scholar] [CrossRef]
- Reddy, G.L.; Dar, M.I.; Hudwekar, A.D.; Mahajan, P.; Nargotra, A.; Baba, A.M.; Nandi, U.; Wazir, P.; Singh, G.; Vishwakarma, R.A. Design, synthesis and biological evaluation of pyrazolopyrimidinone based potent and selective PDE5 inhibitors for treatment of erectile dysfunction. Bioorg. Chem. 2019, 89, 103022. [Google Scholar] [CrossRef]
- Rawson, D.J.; Ballard, S.; Barber, C.; Barker, L.; Beaumont, K.; Bunnage, M.; Cole, S.; Corless, M.; Denton, S.; Ellis, D. The discovery of UK-369003, a novel PDE5 inhibitor with the potential for oral bioavailability and dose-proportional pharmacokinetics. Bioorg. Med. Chem. 2012, 20, 498–509. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.S. Tadalafil: 15 years’ journey in male erectile dysfunction and beyond. Drug Dev. Res. 2019, 80, 683–701. [Google Scholar] [CrossRef]
- El-Gamil, D.S.; Ahmed, N.S.; Gary, B.D.; Piazza, G.A.; Engel, M.; Hartmann, R.W.; Abadi, A.H. Design of novel β-carboline derivatives with pendant 5-bromothienyl and their evaluation as phosphodiesterase-5 inhibitors. Arch. Der Pharm. 2013, 346, 23–33. [Google Scholar] [CrossRef]
- Elhady, A.K.; Sigler, S.C.; Noureldin, N.; Canzoneri, J.C.; Ahmed, N.S.; Piazza, G.A.; Abadi, A.H. Structure-based design of novel tetrahydro-beta-carboline derivatives with a hydrophilic side chain as potential phosphodiesterase inhibitors. Sci. Pharm. 2016, 84, 428–446. [Google Scholar] [CrossRef]
- Zheng, H.; Wu, Y.; Sun, B.; Cheng, C.; Qiao, Y.; Jiang, Y.; Zhao, S.; Xie, Z.; Tan, J.; Lou, H. Discovery of furyl/thienyl β-carboline derivatives as potent and selective PDE5 inhibitors with excellent vasorelaxant effect. Eur. J. Med. Chem. 2018, 158, 767–780. [Google Scholar] [CrossRef]
- Ahmed, N.S.; Ali, A.H.; El-Nashar, S.M.; Gary, B.D.; Fajardo, A.M.; Tinsley, H.N.; Piazza, G.A.; Negri, M.; Abadi, A.H. Exploring the PDE5 H-pocket by ensemble docking and structure-based design and synthesis of novel β-carboline derivatives. Eur. J. Med. Chem. 2012, 57, 329–343. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.S.; Gary, B.D.; Tinsley, H.N.; Piazza, G.A.; Laufer, S.; Abadi, A.H. Design, Synthesis and Structure–Activity Relationship of Functionalized Tetrahydro-β-carboline Derivatives as Novel PDE5 Inhibitors. Arch. Der Pharm. 2011, 344, 149–157. [Google Scholar] [CrossRef]
- Takase, Y.; Saeki, T.; Watanabe, N.; Adachi, H.; Souda, S.; Saito, I. Cyclic GMP phosphodiesterase inhibitors. 2. Requirement of 6-substitution of quinazoline derivatives for potent and selective inhibitory activity. J. Med. Chem. 1994, 37, 2106–2111. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Konishi, Y.; Yu, D.T.; Miskowski, T.A.; Riviello, C.M.; Macina, O.T.; Frierson, M.R.; Kondo, K.; Sugitani, M. Discovery of potent cyclic GMP phosphodiesterase inhibitors. 2-Pyridyl-and 2-imidazolylquinazolines possessing cyclic GMP phosphodiesterase and thromboxane synthesis inhibitory activities. J. Med. Chem. 1995, 38, 3547–3557. [Google Scholar] [CrossRef]
- Somnarin, T.; Pobsuk, N.; Chantakul, R.; Panklai, T.; Temkitthawon, P.; Hannongbua, S.; Chootip, K.; Ingkaninan, K.; Boonyarattanakalin, K.; Gleeson, D. Computational design, synthesis and biological evaluation of PDE5 inhibitors based on N2, N4-diaminoquinazoline and N2, N6-diaminopurine scaffolds. Bioorg. Med. Chem. 2022, 76, 117092. [Google Scholar] [CrossRef] [PubMed]
- Pobsuk, N.; Paracha, T.U.; Chaichamnong, N.; Salaloy, N.; Suphakun, P.; Hannongbua, S.; Choowongkomon, K.; Pekthong, D.; Chootip, K.; Ingkaninan, K. Design, synthesis and evaluation of N2, N4-diaminoquinazoline based inhibitors of phosphodiesterase type 5. Bioorg. Med. Chem. Lett. 2019, 29, 267–270. [Google Scholar] [CrossRef] [PubMed]
- Chatturong, U.; Martin, H.; Totoson, P.; Ingkaninan, K.; Temkitthawon, P.; Sermsenaphorn, S.; Somarin, T.; Konsue, A.; Gleeson, M.P.; Demougeot, C. Quinazoline-based human phosphodiesterase 5 inhibitors exhibited a selective vasorelaxant effect on rat isolated pulmonary arteries involving NO-sGC-cGMP pathway and calcium inhibitory effects. Vasc. Pharmacol. 2022, 147, 107111. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.-F.; Dong, Y.-H.; Wang, J.-H.; Ke, H.-M.; Song, G.-Q.; Xu, D.-F. Novel PDE5 inhibitors derived from rutaecarpine for the treatment of Alzheimer’s disease. Bioorg. Med. Chem. Lett. 2020, 30, 127097. [Google Scholar] [CrossRef]
- Zheng, H.; Li, L.; Sun, B.; Gao, Y.; Song, W.; Zhao, X.; Gao, Y.; Xie, Z.; Zhang, N.; Ji, J. Design and synthesis of furyl/thineyl pyrroloquinolones based on natural alkaloid perlolyrine, lead to the discovery of potent and selective PDE5 inhibitors. Eur. J. Med. Chem. 2018, 150, 30–38. [Google Scholar] [CrossRef]
- Fiorito, J.; Saeed, F.; Zhang, H.; Staniszewski, A.; Feng, Y.; Francis, Y.I.; Rao, S.; Thakkar, D.M.; Deng, S.-X.; Landry, D.W. Synthesis of quinoline derivatives: Discovery of a potent and selective phosphodiesterase 5 inhibitor for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2013, 60, 285–294. [Google Scholar] [CrossRef]
- Fiorito, J.; Vendome, J.; Saeed, F.; Staniszewski, A.; Zhang, H.; Yan, S.; Deng, S.-X.; Arancio, O.; Landry, D.W. Identification of a novel 1, 2, 3, 4-tetrahydrobenzo [b][1, 6] naphthyridine analogue as a potent phosphodiesterase 5 inhibitor with improved aqueous solubility for the treatment of Alzheimer’s disease. J. Med. Chem. 2017, 60, 8858–8875. [Google Scholar] [CrossRef] [PubMed]
- Shang, N.-N.; Shao, Y.-X.; Cai, Y.-H.; Guan, M.; Huang, M.; Cui, W.; He, L.; Yu, Y.-J.; Huang, L.; Li, Z. Discovery of 3-(4-hydroxybenzyl)-1-(thiophen-2-yl) chromeno [2, 3-c] pyrrol-9 (2H)-one as a phosphodiesterase-5 inhibitor and its complex crystal structure. Biochem. Pharmacol. 2014, 89, 86–98. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Zhang, T.; Chen, Y.; Huang, Y.; Geng, H.; Yu, Y.; Zhang, C.; Lai, Z.; Wu, Y.; Guo, X. Discovery and optimization of chromeno [2, 3-c] pyrrol-9 (2 H)-ones as novel selective and orally bioavailable phosphodiesterase 5 inhibitors for the treatment of pulmonary arterial hypertension. J. Med. Chem. 2017, 60, 6622–6637. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Huang, Y.; Chen, Y.; Huang, Y.-Y.; Geng, H.; Zhang, T.; Zhang, C.; Li, Z.; Guo, L.; Chen, J. Optimization of Chromeno [2, 3-c] pyrrol-9 (2 H)-ones as Highly Potent, Selective, and Orally Bioavailable PDE5 Inhibitors: Structure–Activity Relationship, X-ray Crystal Structure, and Pharmacodynamic Effect on Pulmonary Arterial Hypertension. J. Med. Chem. 2018, 61, 8468–8473. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Zheng, X.; Liu, R.; Li, Z.; Jiang, Z.; Zhou, Q.; Huang, Y.; Wu, X.-N.; Zhang, C.; Huang, Y.-Y. Free energy perturbation (FEP)-guided scaffold hopping. Acta Pharm. Sin. B 2022, 12, 1351–1362. [Google Scholar] [CrossRef] [PubMed]
- Amin, K.M.; El-Badry, O.M.; Abdel Rahman, D.E.; Abdellattif, M.H.; Abourehab, M.A.; El-Maghrabey, M.H.; Elsaid, F.G.; El Hamd, M.A.; Elkamhawy, A.; Ammar, U.M. Scaffold repurposing reveals new nanomolar phosphodiesterase type 5 (PDE5) inhibitors based on pyridopyrazinone scaffold: Investigation of in vitro and in silico properties. Pharmaceutics 2022, 14, 1954. [Google Scholar] [CrossRef] [PubMed]
- Owen, D.R.; Walker, J.K.; Jacobsen, E.J.; Freskos, J.N.; Hughes, R.O.; Brown, D.L.; Bell, A.S.; Brown, D.G.; Phillips, C.; Mischke, B.V. Identification, synthesis and SAR of amino substituted pyrido [3, 2b] pyrazinones as potent and selective PDE5 inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 4088–4091. [Google Scholar] [CrossRef][Green Version]
- Hughes, R.O.; Walker, J.K.; Cubbage, J.W.; Fobian, Y.M.; Rogier, D.J.; Heasley, S.E.; Blevis-Bal, R.M.; Benson, A.G.; Owen, D.R.; Jacobsen, E.J. Investigation of aminopyridiopyrazinones as PDE5 inhibitors: Evaluation of modifications to the central ring system. Bioorg. Med. Chem. Lett. 2009, 19, 4092–4096. [Google Scholar] [CrossRef]
- El-Sharkawy, L.Y.; El-Sakhawy, R.A.; Abdel-Halim, M.; Lee, K.; Piazza, G.A.; Ducho, C.; Hartmann, R.W.; Abadi, A.H. Design and synthesis of novel annulated thienopyrimidines as phosphodiesterase 5 (PDE5) inhibitors. Arch. Der Pharm. 2018, 351, 1800018. [Google Scholar] [CrossRef]
- Zhang, T.; Lai, Z.; Yuan, S.; Huang, Y.-Y.; Dong, G.; Sheng, C.; Ke, H.; Luo, H.-B. Discovery of evodiamine derivatives as highly selective PDE5 inhibitors targeting a unique allosteric pocket. J. Med. Chem. 2020, 63, 9828–9837. [Google Scholar] [CrossRef]
- Abdel-Halim, M.; Sigler, S.; Racheed, N.A.; Hefnawy, A.; Fathalla, R.K.; Hammam, M.A.; Maher, A.; Maxuitenko, Y.; Keeton, A.B.; Hartmann, R.W. From celecoxib to a novel class of phosphodiesterase 5 inhibitors: Trisubstituted pyrazolines as novel phosphodiesterase 5 inhibitors with extremely high potency and phosphodiesterase isozyme selectivity. J. Med. Chem. 2021, 64, 4462–4477. [Google Scholar] [CrossRef] [PubMed]
- Rabal, O.; Sánchez-Arias, J.A.; Cuadrado-Tejedor, M.; de Miguel, I.; Perez-Gonzalez, M.; García-Barroso, C.; Ugarte, A.; Estella-Hermoso de Mendoza, A.; Sáez, E.; Espelosin, M. Design, synthesis, and biological evaluation of first-in-class dual acting histone deacetylases (HDACs) and phosphodiesterase 5 (PDE5) inhibitors for the treatment of Alzheimer’s disease. J. Med. Chem. 2016, 59, 8967–9004. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Arias, J.A.; Rabal, O.; Cuadrado-Tejedor, M.; de Miguel, I.; Pérez-González, M.; Ugarte, A.; Saez, E.; Espelosin, M.; Ursua, S.; Haizhong, T. Impact of scaffold exploration on novel dual-acting histone deacetylases and phosphodiesterase 5 inhibitors for the treatment of Alzheimer’s disease. ACS Chem. Neurosci. 2017, 8, 638–661. [Google Scholar] [CrossRef]
- Rabal, O.; Sánchez-Arias, J.A.; Cuadrado-Tejedor, M.; de Miguel, I.; Pérez-González, M.; García-Barroso, C.; Ugarte, A.; Estella-Hermoso de Mendoza, A.; Sáez, E.; Espelosin, M. Discovery of in vivo chemical probes for treating Alzheimer’s disease: Dual phosphodiesterase 5 (PDE5) and class I histone deacetylase selective inhibitors. ACS Chem. Neurosci. 2018, 10, 1765–1782. [Google Scholar] [CrossRef] [PubMed]
- Rabal, O.; Sánchez-Arias, J.A.; Cuadrado-Tejedor, M.; de Miguel, I.; Pérez-González, M.; García-Barroso, C.; Ugarte, A.; de Mendoza, A.E.-H.; Sáez, E.; Espelosin, M. Design, synthesis, biological evaluation and in vivo testing of dual phosphodiesterase 5 (PDE5) and histone deacetylase 6 (HDAC6)-selective inhibitors for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2018, 150, 506–524. [Google Scholar] [CrossRef]
- Cuadrado-Tejedor, M.; Garcia-Barroso, C.; Sánchez-Arias, J.A.; Rabal, O.; Pérez-González, M.; Mederos, S.; Ugarte, A.; Franco, R.; Segura, V.; Perea, G. A first-in-class small-molecule that acts as a dual inhibitor of HDAC and PDE5 and that rescues hippocampal synaptic impairment in Alzheimer’s disease mice. Neuropsychopharmacology 2017, 42, 524–539. [Google Scholar] [CrossRef]
- Claveria-Cabello, A.; Colyn, L.; Uriarte, I.; Latasa, M.U.; Arechederra, M.; Herranz, J.M.; Alvarez, L.; Urman, J.M.; Martinez-Chantar, M.L.; Banales, J.M. Dual Pharmacological Targeting of HDACs and PDE5 Inhibits Liver Disease Progression in a Mouse Model of Biliary Inflammation and Fibrosis. Cancers 2020, 12, 3748. [Google Scholar] [CrossRef]
- Ma, N.; Luo, Y.; Wang, Y.; Liao, C.; Ye, W.-C.; Jiang, S. Selective histone deacetylase inhibitors with anticancer activity. Curr. Top. Med. Chem. 2016, 16, 415–426. [Google Scholar] [CrossRef]
- ElHady, A.K.; Shih, S.-P.; Chen, Y.-C.; Liu, Y.-C.; Ahmed, N.S.; Keeton, A.B.; Piazza, G.A.; Engel, M.; Abadi, A.H.; Abdel-Halim, M. Extending the use of tadalafil scaffold: Development of novel selective phosphodiesterase 5 inhibitors and histone deacetylase inhibitors. Bioorg. Chem. 2020, 98, 103742. [Google Scholar] [CrossRef]
- Mao, F.; Wang, H.; Ni, W.; Zheng, X.; Wang, M.; Bao, K.; Ling, D.; Li, X.; Xu, Y.; Zhang, H. Design, synthesis, and biological evaluation of orally available first-generation dual-target selective inhibitors of acetylcholinesterase (AChE) and phosphodiesterase 5 (PDE5) for the treatment of Alzheimer’s disease. ACS Chem. Neurosci. 2018, 9, 328–345. [Google Scholar] [CrossRef]
- Greenwald, M.B.-Y.; Tacconi, C.; Jukic, M.; Joshi, N.; Hiebert, P.; Brinckmann, J.; Tenor, H.; Naef, R.; Werner, S. A dual-acting nitric oxide donor and phosphodiesterase 5 inhibitor promotes wound healing in normal mice and mice with diabetes. J. Investig. Dermatol. 2021, 141, 415–426. [Google Scholar] [CrossRef]
- Ammar, L.; Lin, H.-Y.; Shih, S.-P.; Tsai, T.-N.; Syu, Y.-T.; Abdel-Halim, M.; Hwang, T.-L.; Abadi, A.H. Novel 9-Benzylaminoacridine Derivatives as Dual Inhibitors of Phosphodiesterase 5 and Topoisomerase II for the Treatment of Colon Cancer. Molecules 2023, 28, 840. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wenzel, B.; Dukic-Stefanovic, S.; Teodoro, R.; Ludwig, F.-A.; Deuther-Conrad, W.; Schröder, S.; Chezal, J.-M.; Moreau, E.; Brust, P. Development of a new radiofluorinated quinoline analog for PET imaging of phosphodiesterase 5 (PDE5) in brain. Pharmaceuticals 2016, 9, 22. [Google Scholar] [CrossRef] [PubMed]
- Chekol, R.; Gheysens, O.; Cleynhens, J.; Pokreisz, P.; Vanhoof, G.; Ahamed, M.; Janssens, S.; Verbruggen, A.; Bormans, G. Evaluation of PET radioligands for in vivo visualization of phosphodiesterase 5 (PDE5). Nucl. Med. Biol. 2014, 41, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Chekol, R.; Gheysens, O.; Ahamed, M.; Cleynhens, J.; Pokreisz, P.; Vanhoof, G.; Janssens, S.; Verbruggen, A.; Bormans, G. Carbon-11 and fluorine-18 radiolabeled pyridopyrazinone derivatives for positron emission tomography (PET) imaging of phosphodiesterase-5 (PDE5). J. Med. Chem. 2017, 60, 486–496. [Google Scholar] [CrossRef]
- Liu, J.; Maisonial-Besset, A.; Wenzel, B.; Canitrot, D.; Baufond, A.; Chezal, J.-M.; Brust, P.; Moreau, E. Synthesis and in vitro evaluation of new fluorinated quinoline derivatives with high affinity for PDE5: Towards the development of new PET neuroimaging probes. Eur. J. Med. Chem. 2017, 136, 548–560. [Google Scholar] [CrossRef]
- Xu, Z.; Jia, L.; Liu, W.; Li, W.; Song, Y.; Zheng, Q.-H. Radiosynthesis of a carbon-11 labeled PDE5 inhibitor [11C] TPN171 as a new potential PET heart imaging agent. Appl. Radiat. Isot. 2020, 162, 109190. [Google Scholar] [CrossRef]
- He, Y.-F.; Liu, Y.; Yu, J.-H.; Cheng, H.; Odilov, A.; Yang, F.-P.; Tian, G.-H.; Yao, X.-M.; Duan, H.-Q.; Yu, C.-Y. Pharmacokinetics, mass balance, and metabolism of [14C] TPN171, a novel PDE5 inhibitor, in humans for the treatment of pulmonary arterial hypertension. Acta Pharmacol. Sin. 2023, 44, 221–233. [Google Scholar] [CrossRef]
- Wenzel, B.; Liu, J.; Dukic-Stefanovic, S.; Deuther-Conrad, W.; Teodoro, R.; Ludwig, F.-A.; Chezal, J.-M.; Moreau, E.; Brust, P.; Maisonial-Besset, A. Targeting cyclic nucleotide phosphodiesterase 5 (PDE5) in brain: Toward the development of a PET radioligand labeled with fluorine-18. Bioorg. Chem. 2019, 86, 346–362. [Google Scholar] [CrossRef]
- Dong, F.; Du, J.; Miao, C.; Jia, L.; Li, W.; Wang, M.; Zheng, Q.-H.; Xu, Z. Radiosynthesis of carbon-11 labeled PDE5 inhibitors as new potential PET radiotracers for imaging of Alzheimer’s disease. Appl. Radiat. Isot. 2019, 154, 108873. [Google Scholar] [CrossRef]
- El-Kawy, O.; Ibrahim, I.; Shewatah, H.; El-Azony, K. Preparation and evaluation of radioiodinated avanafil: A novel potential radiopharmaceutical for the diagnostic evaluation of erectile dysfunction. Appl. Radiat. Isot. 2022, 183, 110160. [Google Scholar] [CrossRef] [PubMed]
- Zoraghi, R.; Francis, S.H.; Corbin, J.D. Critical amino acids in phosphodiesterase-5 catalytic site that provide for high-affinity interaction with cyclic guanosine monophosphate and inhibitors. Biochemistry 2007, 46, 13554–13563. [Google Scholar] [CrossRef] [PubMed]
- Falk, J.A.; Philip, K.J.; Schwarz, E.R. The emergence of oral tadalafil as a once-daily treatment for pulmonary arterial hypertension. Vasc. Health Risk Manag. 2010, 6, 273–280. [Google Scholar]
- Miller, M.S. Role of phosphodiesterase type 5 inhibitors for lower urinary tract symptoms. Ann. Pharmacother. 2013, 47, 278–283. [Google Scholar] [CrossRef]
- Gur, S.; Kadowitz, P.J.; Can Serefoglu, E.; Hellstrom, W.J.G. PDE5 inhibitor treatment options for urologic and non-urologic indications: 2012 update. Curr. Pharm. Des. 2012, 18, 5590–5606. [Google Scholar] [CrossRef]
- Sanati, M.; Aminyavari, S.; Mollazadeh, H.; Bibak, B.; Mohtashami, E.; Afshari, A.R. How do phosphodiesterase-5 inhibitors affect cancer? A focus on glioblastoma multiforme. Pharmacol. Rep. 2022, 74, 323–339. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Kloner, R.A.; Salloum, F.N.; Jovin, I.S. Cardiac effects of phosphodiesterase-5 inhibitors: Efficacy and safety. Cardiovasc. Drugs Ther. 2021, 37, 793–806. [Google Scholar] [CrossRef]
- Pels, A.; Derks, J.; Elvan-Taspinar, A.; van Drongelen, J.; de Boer, M.; Duvekot, H.; van Laar, J.; van Eyck, J.; Al-Nasiry, S.; Sueters, M. Maternal sildenafil vs placebo in pregnant women with severe early-onset fetal growth restriction: A randomized clinical trial. JAMA Netw. Open 2020, 3, e205323. [Google Scholar] [CrossRef]
- Behr, J.; Nathan, S.D.; Wuyts, W.A.; Bishop, N.M.; Bouros, D.E.; Antoniou, K.; Guiot, J.; Kramer, M.R.; Kirchgaessler, K.-U.; Bengus, M. Efficacy and safety of sildenafil added to pirfenidone in patients with advanced idiopathic pulmonary fibrosis and risk of pulmonary hypertension: A double-blind, randomised, placebo-controlled, phase 2b trial. Lancet Respir. Med. 2021, 9, 85–95. [Google Scholar] [CrossRef]
- Kuwana, M.; Blair, C.; Takahashi, T.; Langley, J.; Coghlan, J.G. Initial combination therapy of ambrisentan and tadalafil in connective tissue disease-associated pulmonary arterial hypertension (CTD-PAH) in the modified intention-to-treat population of the AMBITION study: Post hoc analysis. Ann. Rheum. Dis. 2020, 79, 626–634. [Google Scholar] [CrossRef]
- Luginbuhl, A.J.; Johnson, J.M.; Harshyne, L.A.; Linnenbach, A.J.; Shukla, S.K.; Alnemri, A.; Kumar, G.; Cognetti, D.M.; Curry, J.M.; Kotlov, N. Tadalafil enhances immune signatures in response to neoadjuvant nivolumab in resectable head and neck squamous cell carcinoma. Clin. Cancer Res. 2022, 28, 915–927. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.-J.; Song, S.; Chang, S.-A.; Kim, H.-K.; Jung, H.-O.; Choi, J.-H.; Lee, J.S.; Kim, K.-H.; Jeong, J.-O.; Lee, J.H. Efficacy and safety of udenafil for the treatment of pulmonary arterial hypertension: A placebo-controlled, double-blind, phase IIb clinical trial. Clin. Ther. 2019, 41, 1499–1507. [Google Scholar] [CrossRef] [PubMed]
- Santamarina, M.G.; Beddings, I.; Lomakin, F.M.; Boisier Riscal, D.; Gutierrez Claveria, M.; Vidal Marambio, J.; Retamal Baez, N.; Pavez Novoa, C.; Reyes Allende, C.; Ferreira Perey, P. Sildenafil for treating patients with COVID-19 and perfusion mismatch: A pilot randomized trial. Crit. Care 2022, 26, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Hubers, S.A.; Wan, S.H.; Adel, F.W.; Benike, S.L.; Burnett, J.C., Jr.; Scott, C.; Chen, H.H. Cardiorenal Effects of Long-Term Phosphodiesterase V Inhibition in Pre–Heart Failure. J. Am. Heart Assoc. 2022, 11, e022126. [Google Scholar] [CrossRef]
- Ferguson-Sells, L.; Velez de Mendizabal, N.; Li, B.; Small, D. Population pharmacokinetics of tadalafil in pediatric patients with pulmonary arterial hypertension: A combined adult/pediatric model. Clin. Pharmacokinet. 2022, 61, 249–262. [Google Scholar] [CrossRef]
- Omarjee, L.; Le Pabic, E.; Custaud, M.-A.; Fontaine, C.; Locher, C.; Renault, A.; Jaquinandi, V.; Azzola, V.; Barbeau-Terrier, C.; Laporte, I. Effects of sildenafil on maximum walking time in patients with arterial claudication: The ARTERIOFIL study. Vasc. Pharmacol. 2019, 118, 106563. [Google Scholar] [CrossRef] [PubMed]
- Greutmann, M.; Tobler, D.; Engel, R.; Heg, D.; Mueller, C.; Frenk, A.; Gabriel, H.; Rutz, T.; Buechel, R.R.; Willhelm, M. Effect of phosphodiesterase-5 inhibition on SystEmic Right VEntricular size and function—A multi-center, double-blind, randomized, placebo-controlled trial–SERVE. Eur. J. Heart Fail. 2023, 25, 1105–1114. [Google Scholar] [CrossRef]
- Jagdish, R.K.; Kamaal, A.; Shasthry, S.M.; Benjamin, J.; Maiwall, R.; Jindal, A.; Choudhary, A.; Rajan, V.; Arora, V.; Bhardwaj, A. Tadalafil improves erectile dysfunction and quality of life in men with cirrhosis: A randomized double blind placebo controlled trial. Hepatol. Int. 2021, 17, 434–451. [Google Scholar] [CrossRef]
- Di Maria, M.V.; Goldberg, D.J.; Zak, V.; Hu, C.; Lubert, A.M.; Dragulescu, A.; Mackie, A.S.; McCrary, A.; Weingarten, A.; Parthiban, A. Impact of Udenafil on Echocardiographic Indices of Single Ventricle Size and Function in FUEL Study Participants. Circ. Cardiovasc. Imaging 2022, 15, e013676. [Google Scholar] [CrossRef]
- Andersen, A.; Waziri, F.; Schultz, J.G.; Holmboe, S.; Becker, S.W.; Jensen, T.; Søndergaard, H.M.; Dodt, K.K.; May, O.; Mortensen, U.M. Pulmonary vasodilation by sildenafil in acute intermediate-high risk pulmonary embolism: A randomized explorative trial. BMC Pulm. Med. 2021, 21, 72. [Google Scholar] [CrossRef]
- Pierce, C.M.; Zhang, M.H.; Jonsson, B.; Iorga, D.; Cheruvu, N.; Balagtas, C.C.; Steinhorn, R.H. Efficacy and safety of IV sildenafil in the treatment of newborn infants with, or at risk of, persistent pulmonary hypertension of the newborn (PPHN): A multicenter, randomized, placebo-controlled trial. J. Pediatr. 2021, 237, 154–161.e153. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Miguelez, P.; Seigler, N.; Ishii, H.; Crandall, R.; McKie, K.T.; Forseen, C.; Harris, R.A. Exercise intolerance in cystic fibrosis: Importance of skeletal muscle. Med. Sci. Sports Exerc. 2021, 53, 684. [Google Scholar] [CrossRef] [PubMed]









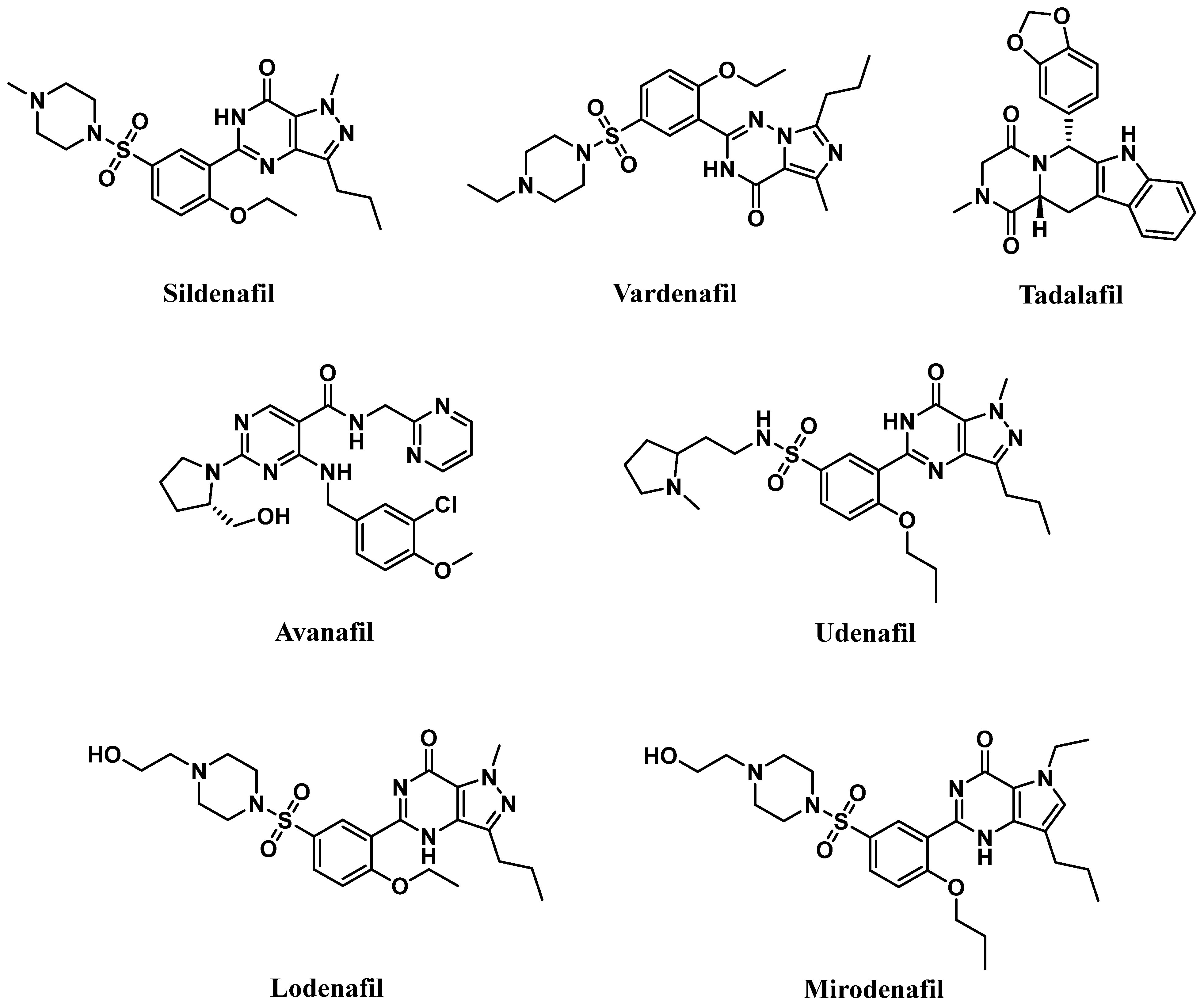{kind=link}
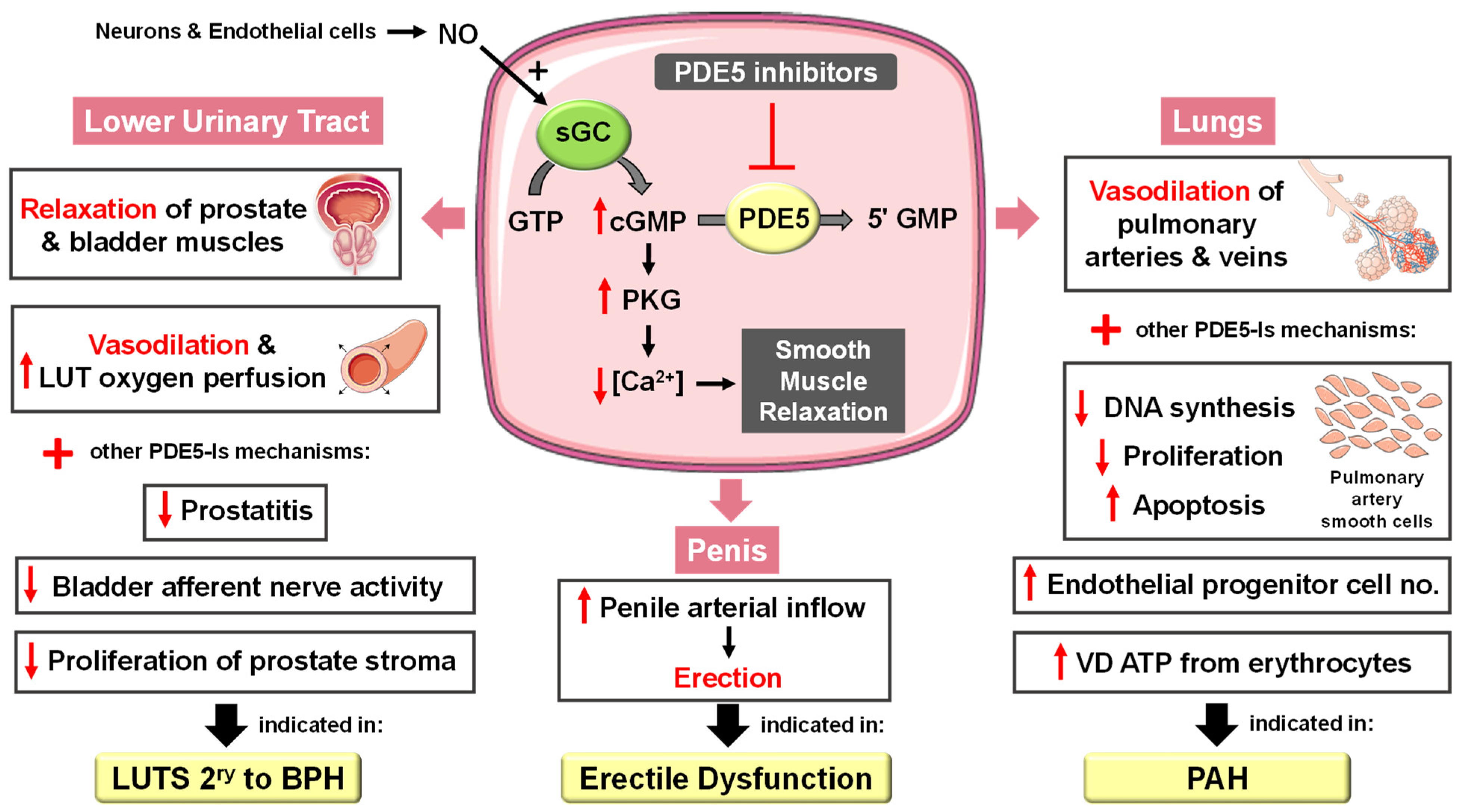{kind=link}
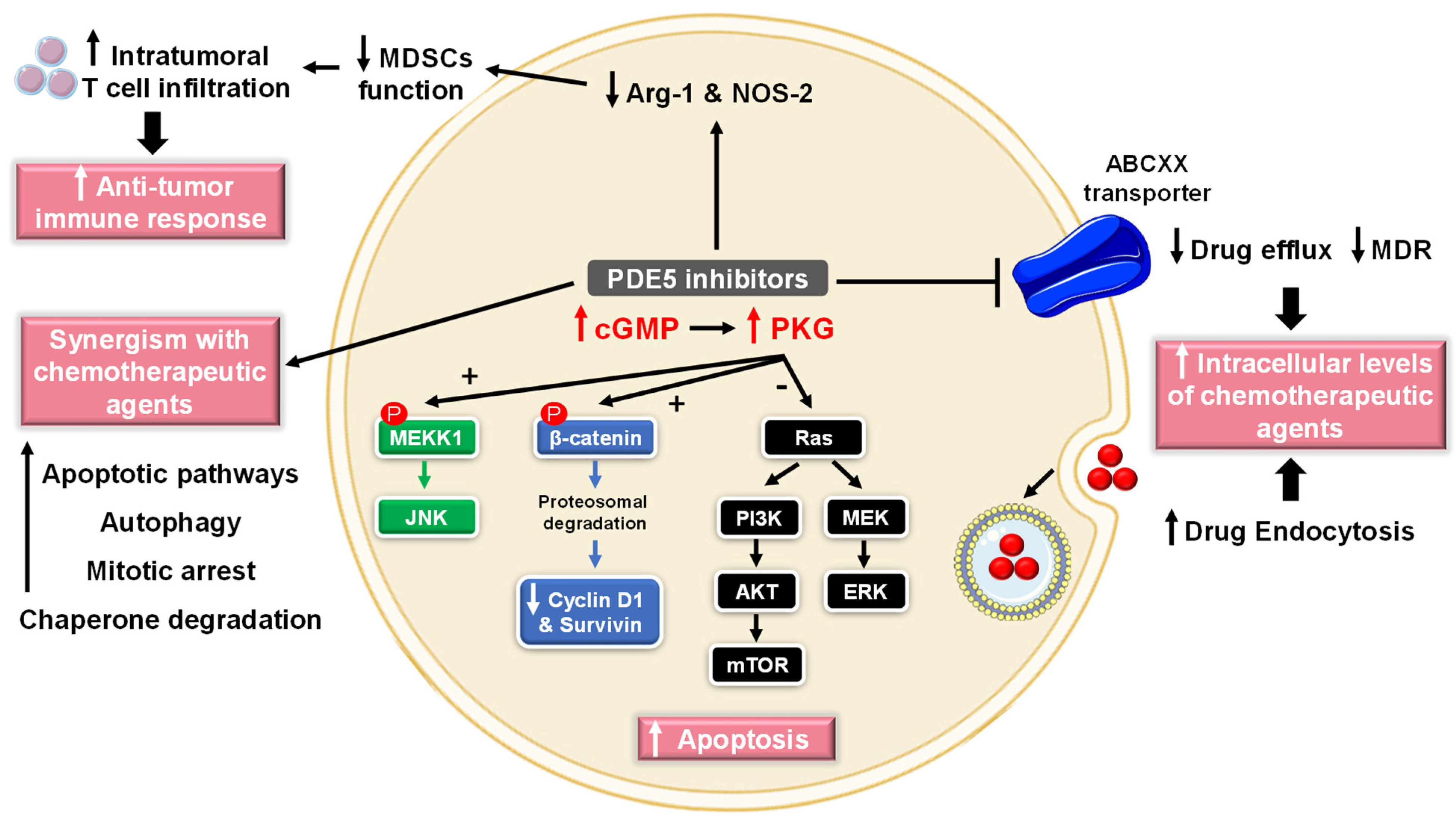{kind=link}
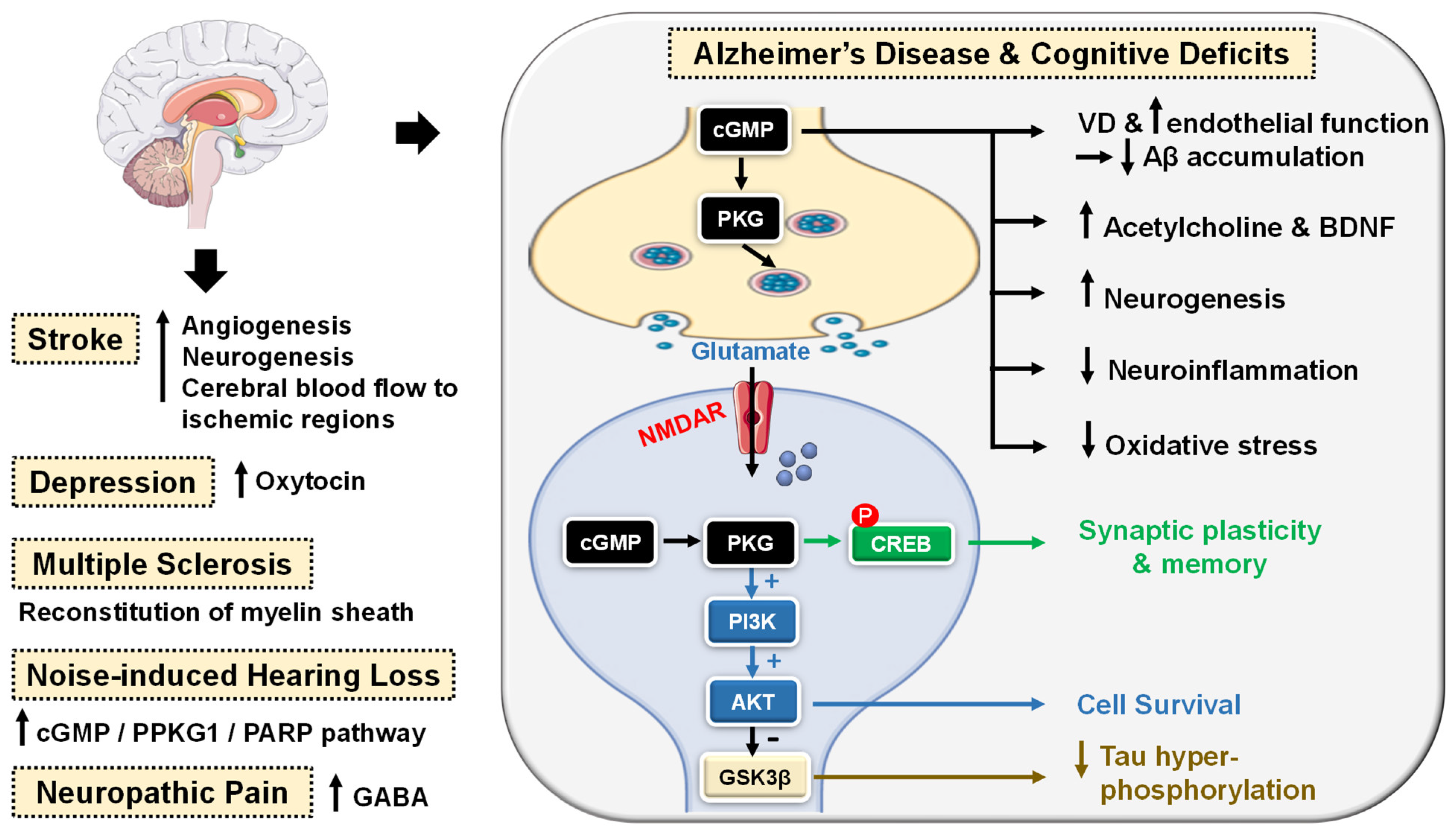{kind=link}
{kind=link}
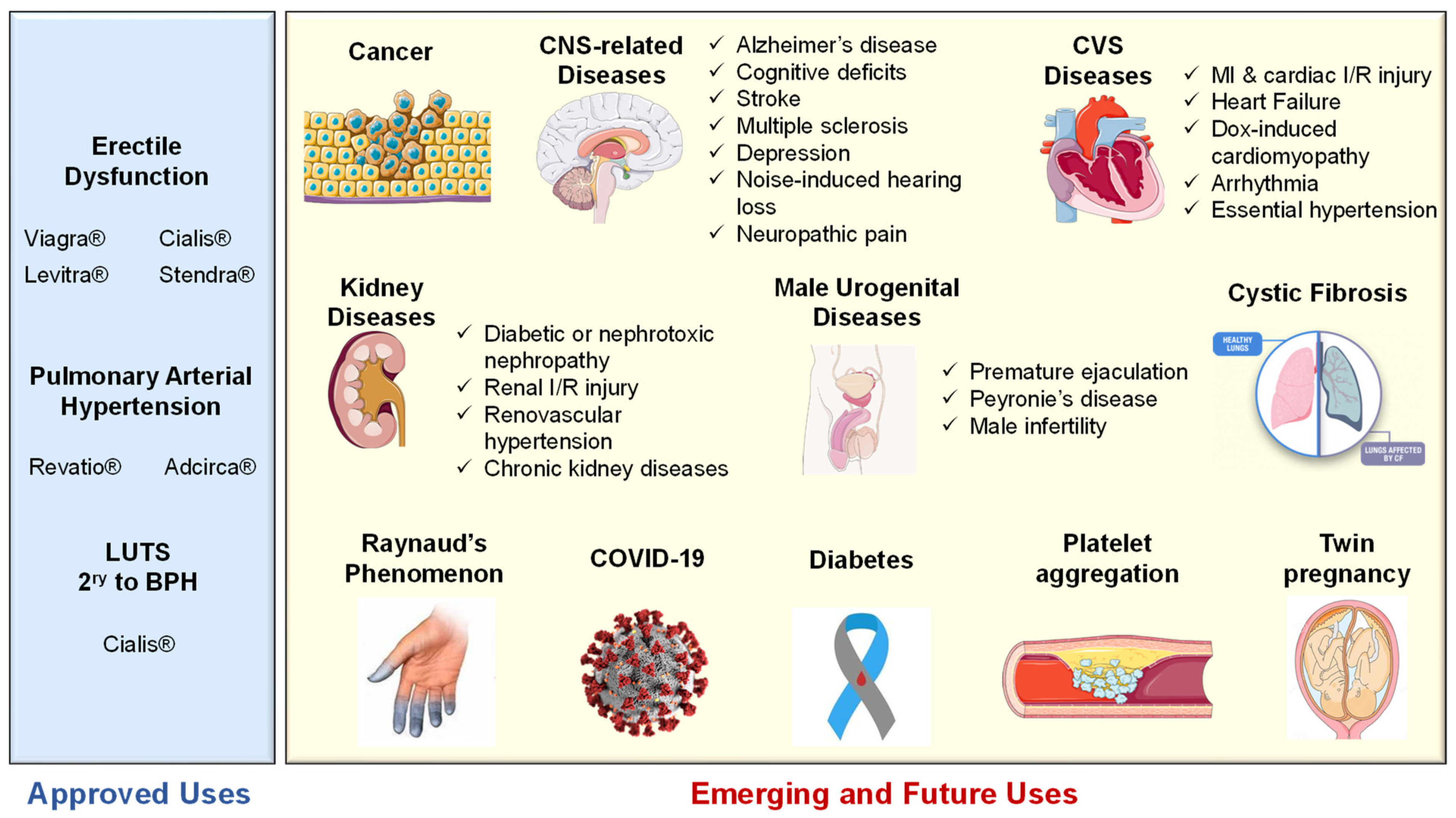{kind=link}
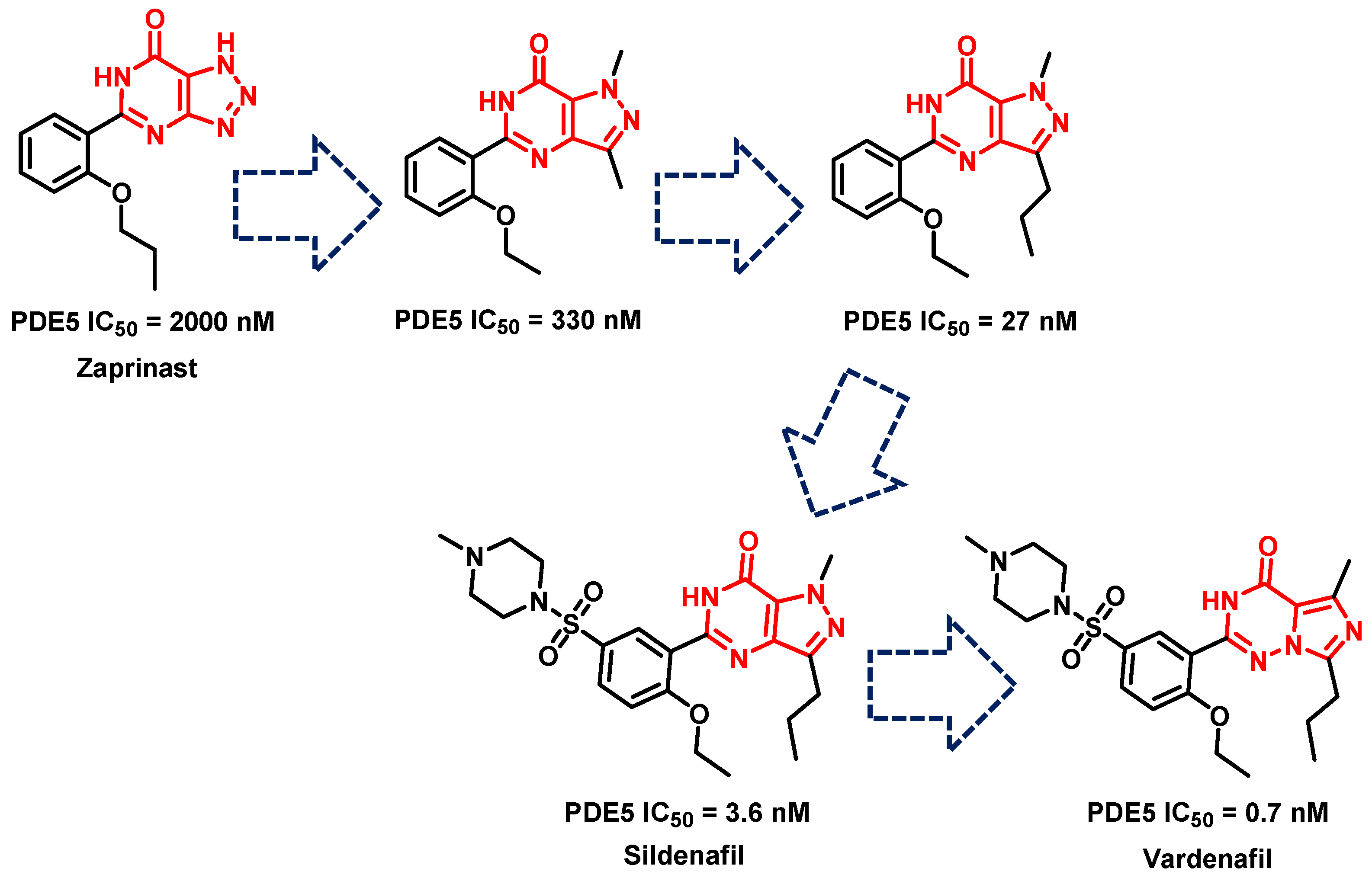{kind=link}
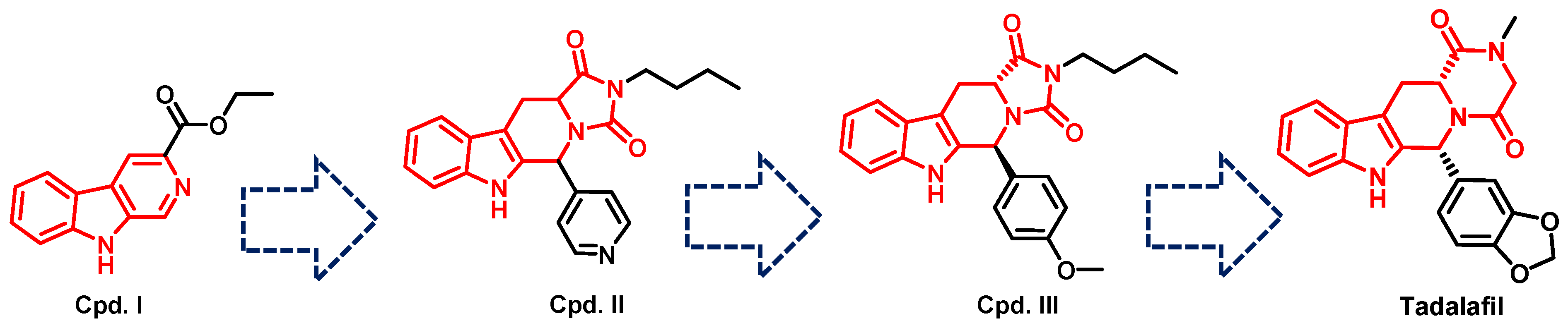{kind=link}
| Scaffold and Compound No. | PDB Code | Cocrystal Structure | Key Ligand Interactions with the PDE5 Active Site |
|---|---|---|---|
| Pyrimidinone, Cpd. 2 | 4G2W |  Sildenafil-like | The pyrimidinone core is anchored through; a bidentate hydrogen bond with Gln817, CH-pi stacking with Phe820, Val782 and Phe786, in addition to a hydrogen bond with Met816 |
| Pyrimidinone, Cpd. 4 | 4I9Z |  Sildenafil-like | The pyrimidinone core is anchored through; a bidentate hydrogen bond with Gln817, π-π stacking with Phe820, a CH-pi interaction with Val782, in addition to a hydrogen bond with Met816 |
| Pyrimidinone, Cpd. 5 | 4IAO |  Tadalafil-like | The pyrimidinone core is anchored through; a monodentate hydrogen bond with Gln817, π-π stacking with Phe820, a CH-pi interaction with Val782, in addition to a hydrogen bond with Met816 |
| Pyrimidine, Cpd. 9 (Avanafil) | 6L6E |  Sildenafil-like | The pyrimidine core is anchored through; a bidentate hydrogen bond between the terminal hydroxy group on the pyrrolidine ring and Gln817, π-π stacking with Phe820, in addition to a CH-pi interaction with Leu725, a hydrogen bond with Gln775 and Leu804, as well as a halogen bond with Ala779 |
| Chromenopyrrolones Cpd. 32 | 4MD6 |  Tadalafil-like | The chromenopyrrolone core is anchored through; a monodentate hydrogen bond between the pyrrole ring and Gln817, π-π stacking with Phe820 and three CH-pi interactions with Val782 |
| Chromenopyrrolones, Cpd. 36 | 5ZZ2 |  Sildenafil-like | The chromenopyrrolone core is anchored through; a bidentate hydrogen bond with Gln817, π-π stacking with Phe820, three CH-pi interactions with Val782, in addition to; a CH-pi interaction with Phe786, and a hydrogen bond between the benzodioxole moiety and Gln817 |
| Azepinoindolone, Cpd. 39 | 7FAQ |  Tadalafil-like | The tetrahydro-1H-azepinoindolo-1-one core is anchored through: a monodentate hydrogen bond with Gln817, π-π stacking with Phe820, and two CH-pi interactions with Val782 |
| Azepinoindolone, Cpd. 40 | 7FAR |  Tadalafil-like | The tetrahydro-1H-azepinoindolo-1-one core is anchored through: a monodentate hydrogen bond with Gln817, π-π stacking with Phe820, three CH-pi interactions with Val782, a CH-pi interaction with Phe786 and a hydrogen bond with His613 |
| Indolopyrazinoquinazolinone, Cpd. 46 | 6VIB |  Allosteric Inhibitor | The compound is anchored in the allosteric site through; four hydrogen bonds with Asp563, a hydrogen bond with His617 and two CH-pi interactions with Arg616 and Ala767 |
| Identifier | Phase | Intervention | No. of Subjects | Targeted Disease | Main Finding(s) | Ref. |
|---|---|---|---|---|---|---|
| NCT02277132 | II/III | Sildenafil 25 mg TID vs. placebo | 216 (108 sildenafil and 108 placebo) | Severe early-onset fetal growth restriction in pregnant women | Antenatal maternal sildenafil administration did not reduce the risk of perinatal mortality or major neonatal morbidity. The trial was terminated due to increased risk of neonatal pulmonary hypertension. | [288] |
| NCT02951429 | II | Pirfenidone 801 mg TID + 20 mg TID sildenafil or placebo | 177 (88 sildenafil and 89 placebo) | Advanced idiopathic pulmonary fibrosis and risk of pulmonary hypertension | No difference in the proportion of patients with disease progression over 52 weeks between the sildenafil and placebo groups. Similar treatment-emergent adverse events and/or mortality were reported in patients in both groups. | [289] |
| NCT01178073 | III | Tadalafil + ambrisentan (COMB) vs. monotherapy of either agent (MONO) | 216 (117 COMB and 99 MONO) | Connective tissue disease-associated pulmonary arterial hypertension and systemic sclerosis-pulmonary arterial hypertension | The risk of clinical failure was lower with COMB vs. MONO (risk reduction: CTD-PAH 51.7%, SSc-PAH 53.7%), particularly in patients with haemodynamic parameters characteristic of typical PAH without signs of left heart disease and/or restrictive lung disease at baseline. | [290] |
| NCT03238365 | Early I | Nivolumab 240 mg IV on days 1 and 15 followed by surgery on day 28 + tadalafil 10 mg orally once daily for 4 weeks or placebo | 50 | Resectable Head and Neck Squamous Cell Carcinoma | After 4 weeks of treatment, preoperative nivolumab and tadalafil combination was found safe and more than 50% of the patients showed at least 20% treatment response. Posttreatment specimens showed augmentation of the immune microenvironment (B- and natural killer cell gene sets in the tumor and effector T cells in the periphery) with the addition of tadalafil. | [291] |
| NCT01553721 | II | Udenafil 50 mg BID vs. placebo | 63 | Pulmonary Arterial Hypertension | Udenafil improves exercise capacity among the patients with a history of endothelin receptor antagonist therapy. There were no significant differences in the Borg dyspnea score and time to clinical worsening between groups. | [292] |
| NCT04489446 | I/II | Sildenafil 25 mg orally TID for seven days vs. placebo | 40 (20 sildenafil and 20 placebo) | COVID-19 in patients showing perfusion abnormalities | Sildenafil led to significantly shorter median length of hospital stay than the placebo group (9 IQR 7–12 days vs. 12 IQR 9–21 days, p = 0.04). No statistically significant differences were found in the oxygenation parameters (PaO2/FiO2 ratios and A-a gradients). | [293] |
| NCT01970176 | I/II | 20 mg tadalafil vs. placebo, repeated after 12 weeks | 20 (13 tadalafil and 7 placebo) | Pre-Heart Failure | Long-term tadalafil did not improve glomerular filtration rate (median increase of 2.0 mL/min in the tadalafil group versus 13.5 mL/min in the placebo group; p = 0.54). There was no difference in urinary sodium or cGMP excretion with tadalafil following short-term saline loading. | [294] |
| NCT01484431 | I/II | Tadalafil 2.5, 10, 20, or 40 mg orally, once daily | 19 | Pediatric Patients with Pulmonary Arterial Hypertension | Plasma tadalafil concentrations in pediatric patients aged 2 to <18 years were similar to those in adults at similar doses in a previous trial and confirmed that dosing of 40 mg once daily in pediatric patients with a bodyweight ≥ 40 kg, and a dose of 20 mg once daily in patients with a body weight < 40 kg and aged ≥ 2 years are suitable for phase III evaluation. | [295] |
| NCT02832570 | III | Sildenafil 100 mg oral single dose | 14 | Arterial claudication | Maximal walking time was significantly improved during the sildenafil period compared with the placebo period (300 s [95% CI 172 s–428 s] vs. 402 s [95% CI 274 s–529 s] p < 0.01). Sildenafil had no significant effect on pain-free walking time or skin tissue oxygenation during exercise. | [296] |
| NCT03049540 | III | Tadalafil 20 mg once daily vs. placebo | 100 | Congenital heart disease with systemic right ventricles | Right ventricular systolic function, exercise capacity and neuro-hormonal activation remained stable over a 3-year follow-up period where no significant treatment effect of tadalafil was observed. | [297] |
| NCT03566914 | II | Tadalafil 10 mg daily vs. placebo for 12 weeks | 140 (70 tadalafil and 70 placebo) | Erectile dysfunction in patients with cirrhosis | More patients in tadalafil group achieved > 5 points increase in the erectile function domain of the International Index of Erectile Function when compared with the placebo group [44(62.9%) vs. 21(30%), p < 0.001]. Patients receiving tadalafil had significantly more decline in the scores of GAD-7 (assessing anxiety) and PHQ-9 (assessing depression). | [298] |
| NCT02741115 | III | Udenafil 87.5 mg oral BID | 386 (191 udenafil and 195 placebo) | Single ventricle heart disease | Udenafil group had significantly improved between baseline and 26 weeks visits compared to placebo group in myocardial performance index (p = 0.03), atrioventricular valve inflow peak E (p = 0.009), and A velocities (p = 0.034), and annular Doppler tissue imaging-derived peak e’ velocity (p = 0.008). There were no significant differences in change in single ventricle size, systolic function, atrioventricular valve regurgitation severity, or mean fenestration gradient. | [299] |
| NCT04283240 | Early I | Sildenafil 20 mg single oral dose vs. placebo as add-on to conventional therapy | 20 (10 sildenafil and 10 placebo) | Acute intermediate-high risk pulmonary embolism | Sildenafil did not improve cardiac index compared to baseline (0.02 ± 0.36 l/min/m2, p = 0.89) and neither did placebo (0.00 ± 0.34 l/min/m2, p = 0.97). Sildenafil lowered mean arterial blood pressure (−19 ± 10 mmHg, p < 0.001) which was not observed in the placebo group (0 ± 9 mmHg, p = 0.97). | [300] |
| NCT01720524 | III | Sildenafil IV (loading: 0.1 mg/kg, over 30 min; maintenance: 0.03 mg/kg/h) vs. placebo, for 14 days | 59 (29 sildenafil and 30 placebo) | Persistent pulmonary hypertension of newborn (receiving iNO) | Treatment failure rates did not differ with sildenafil (27.6%) vs. placebo (20.0%; p = 0.4935). Mean time on iNO was not different with sildenafil (4.1 days) vs. placebo (4.1 days; p = 0.9850). | [301] |
| NCT02057458 | II | Sildenafil 20 mg TID for 4 weeks | 19 | Cystic fibrosis | 4 weeks of sildenafil improved skeletal muscle O2 utilization during exercise to similar values observed in healthy individuals. | [302] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
ElHady, A.K.; El-Gamil, D.S.; Abdel-Halim, M.; Abadi, A.H. Advancements in Phosphodiesterase 5 Inhibitors: Unveiling Present and Future Perspectives. Pharmaceuticals 2023, 16, 1266. https://doi.org/10.3390/ph16091266
ElHady AK, El-Gamil DS, Abdel-Halim M, Abadi AH. Advancements in Phosphodiesterase 5 Inhibitors: Unveiling Present and Future Perspectives. Pharmaceuticals. 2023; 16(9):1266. https://doi.org/10.3390/ph16091266
Chicago/Turabian StyleElHady, Ahmed K., Dalia S. El-Gamil, Mohammad Abdel-Halim, and Ashraf H. Abadi. 2023. "Advancements in Phosphodiesterase 5 Inhibitors: Unveiling Present and Future Perspectives" Pharmaceuticals 16, no. 9: 1266. https://doi.org/10.3390/ph16091266
APA StyleElHady, A. K., El-Gamil, D. S., Abdel-Halim, M., & Abadi, A. H. (2023). Advancements in Phosphodiesterase 5 Inhibitors: Unveiling Present and Future Perspectives. Pharmaceuticals, 16(9), 1266. https://doi.org/10.3390/ph16091266