
Bisindolyl Maleimides and Indolylmaleimide Derivatives—A Review of Their Synthesis and Bioactivity

 ,
, 
Abstract
1. Introduction
2. Synthesis of Bisindolylmaleimides and Derivatives
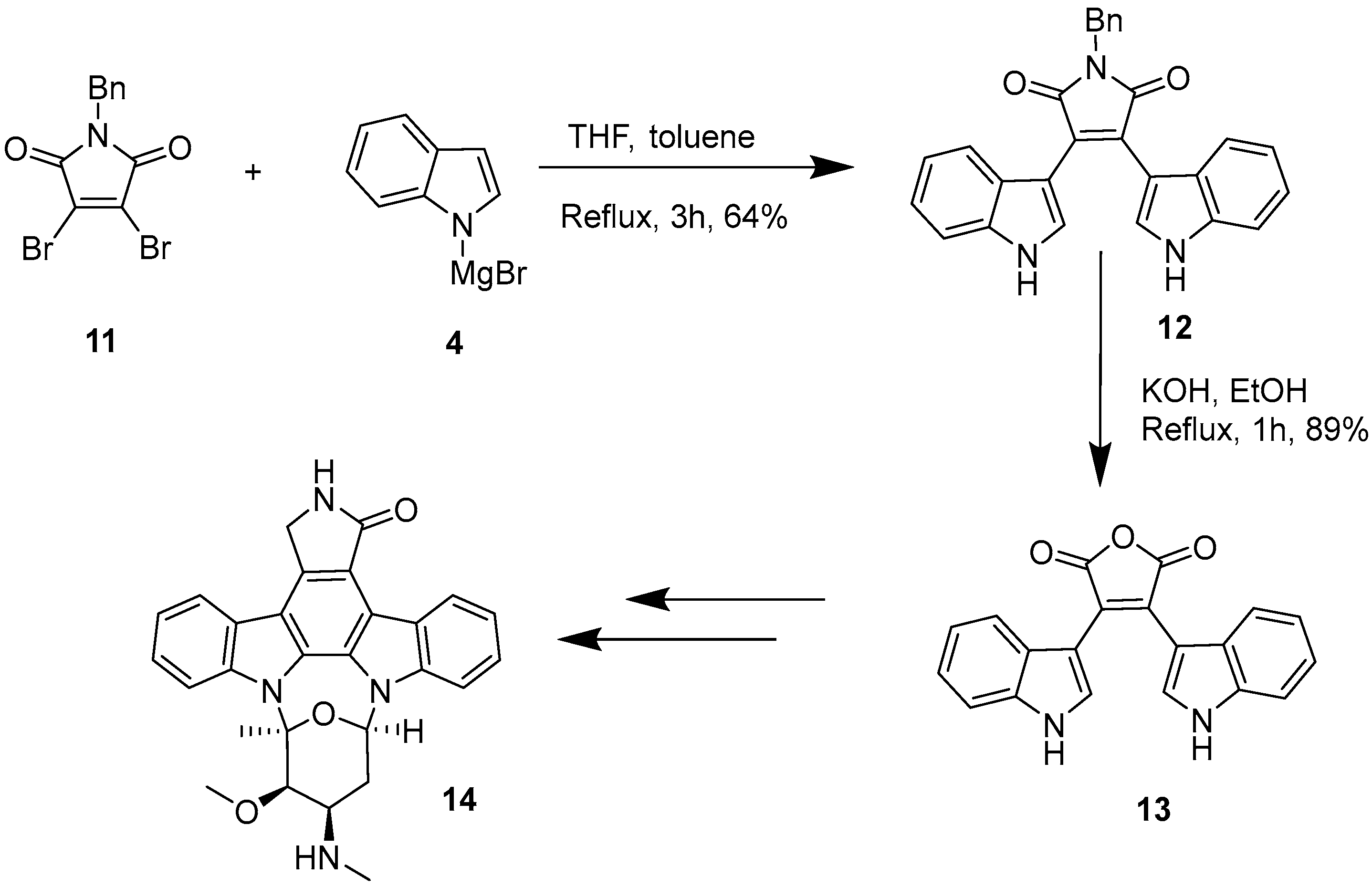
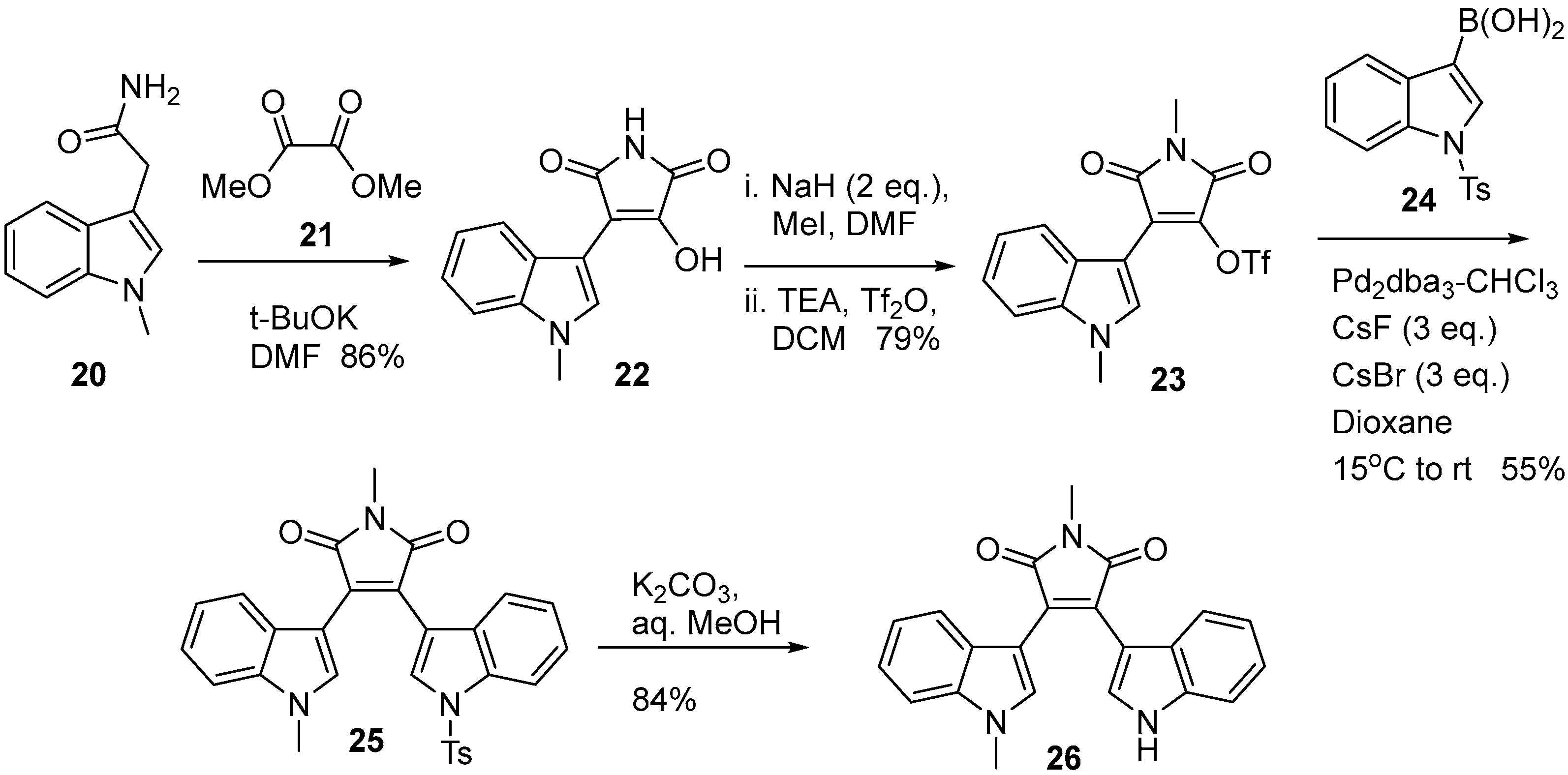
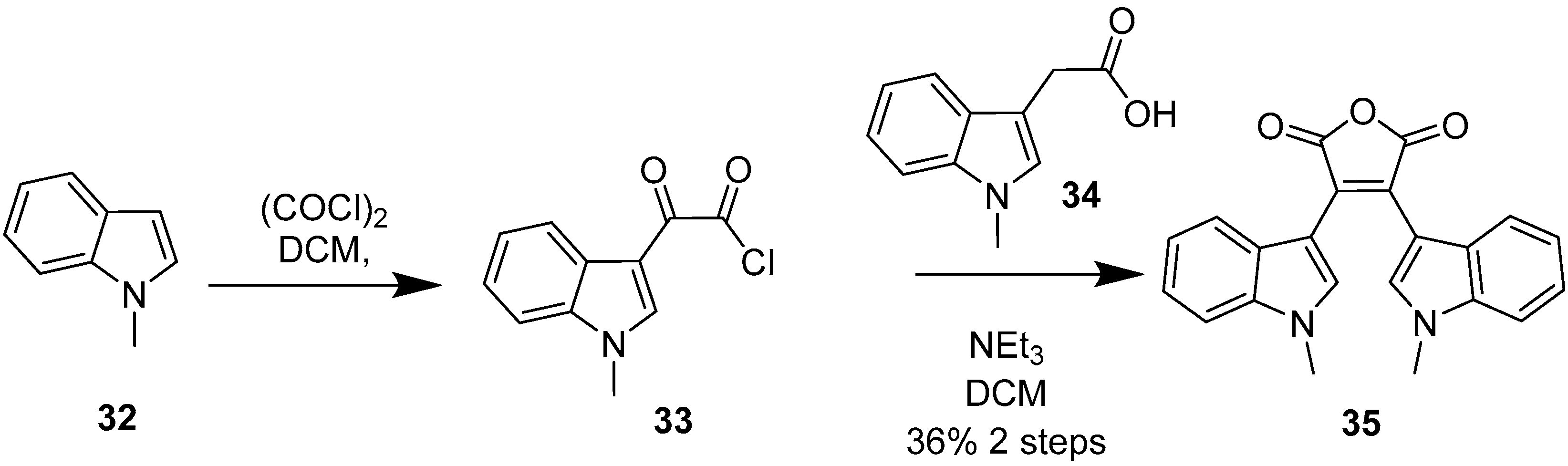
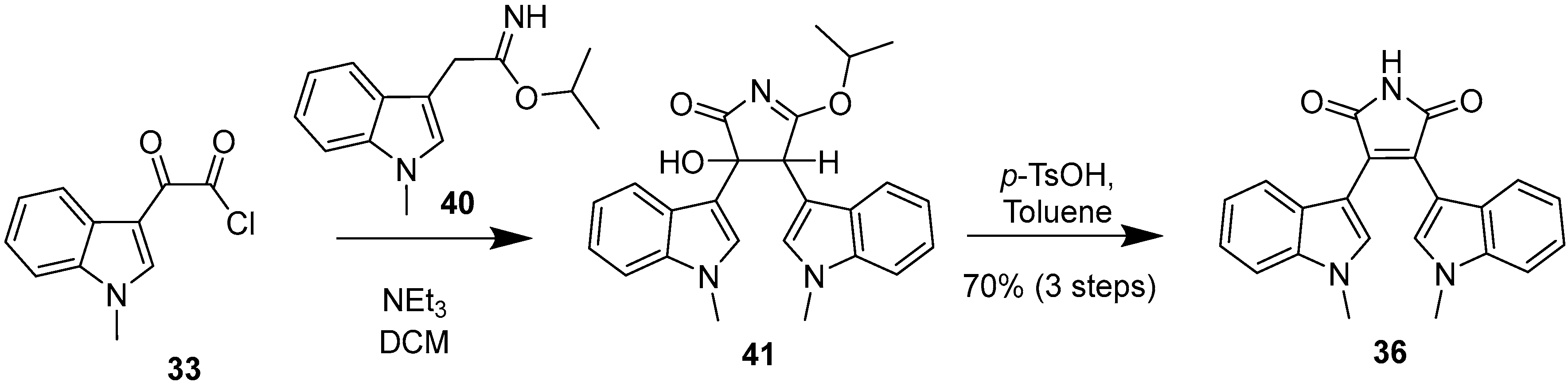
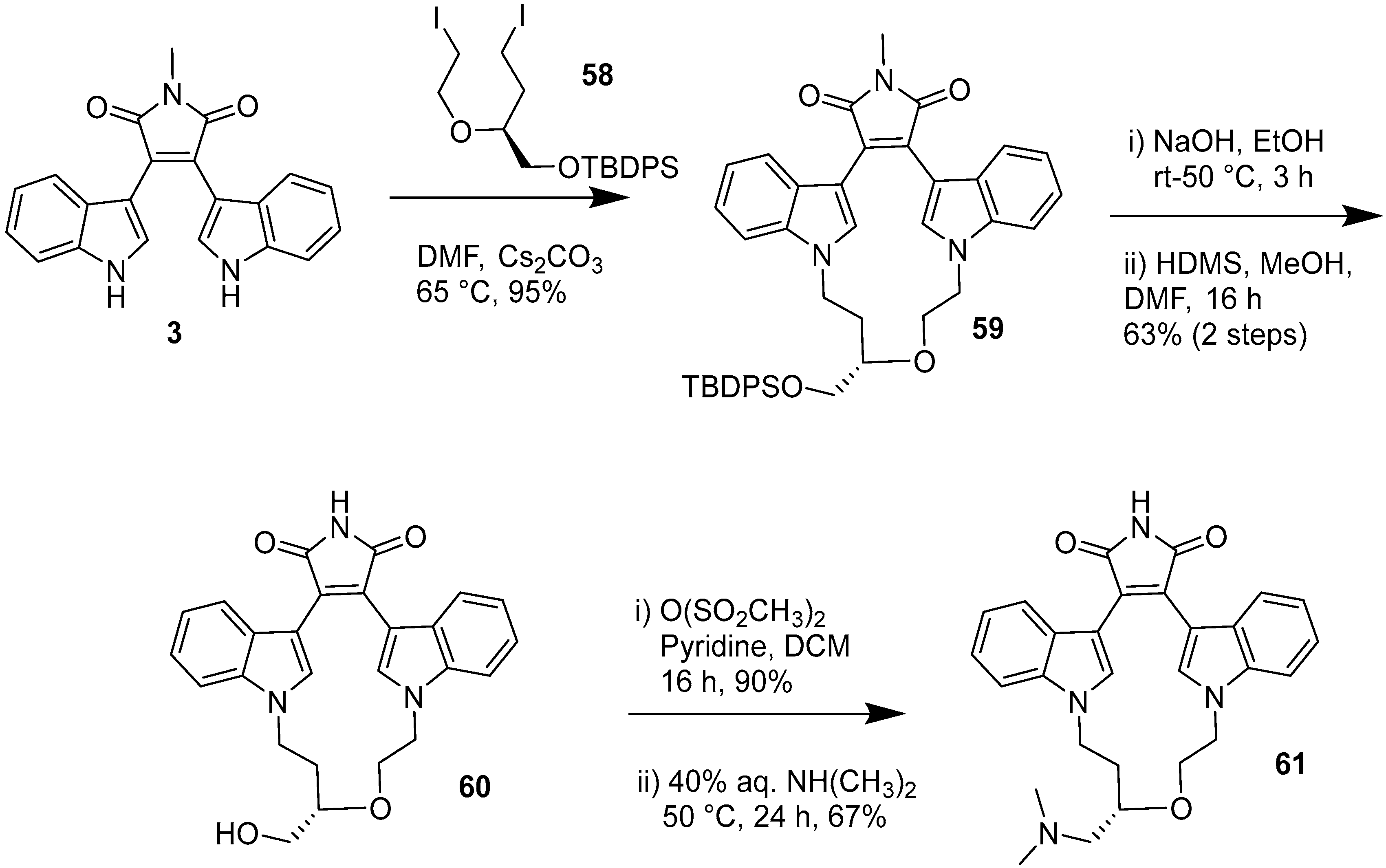
2.1. BIMs via the Use of Preformed Maleimides
2.2. BIMs via the Formation of the Maleimide Ring
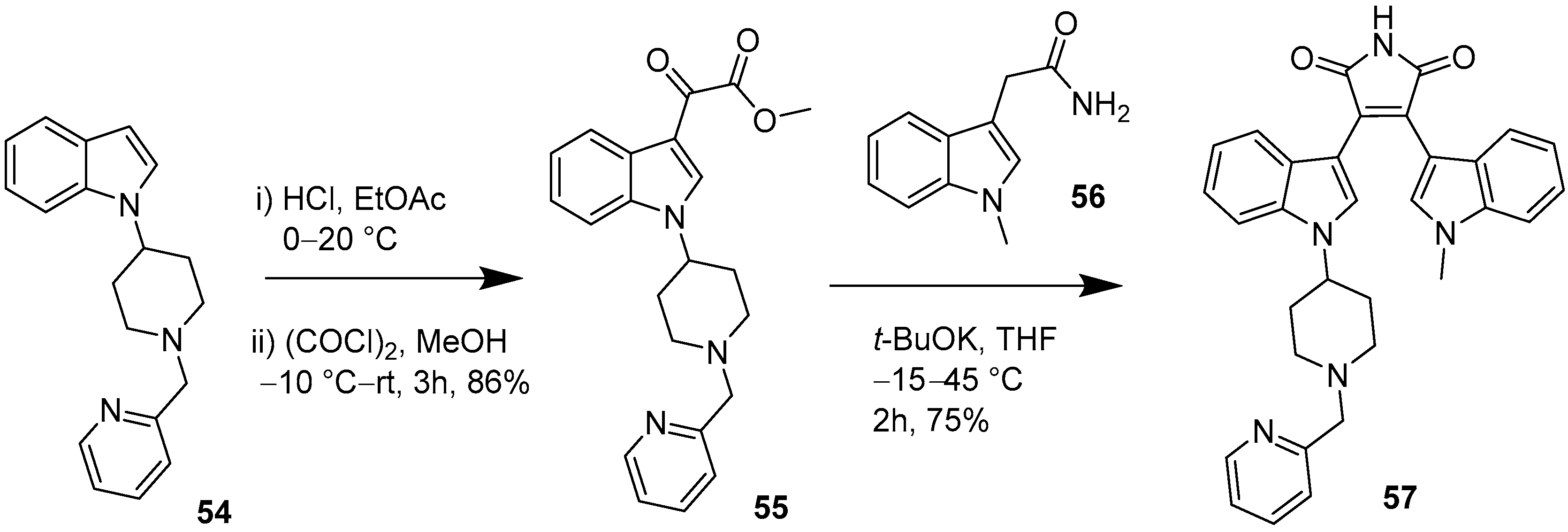
2.3. Synthesis of Clinically Relevant BIM Kinase Inhibitors
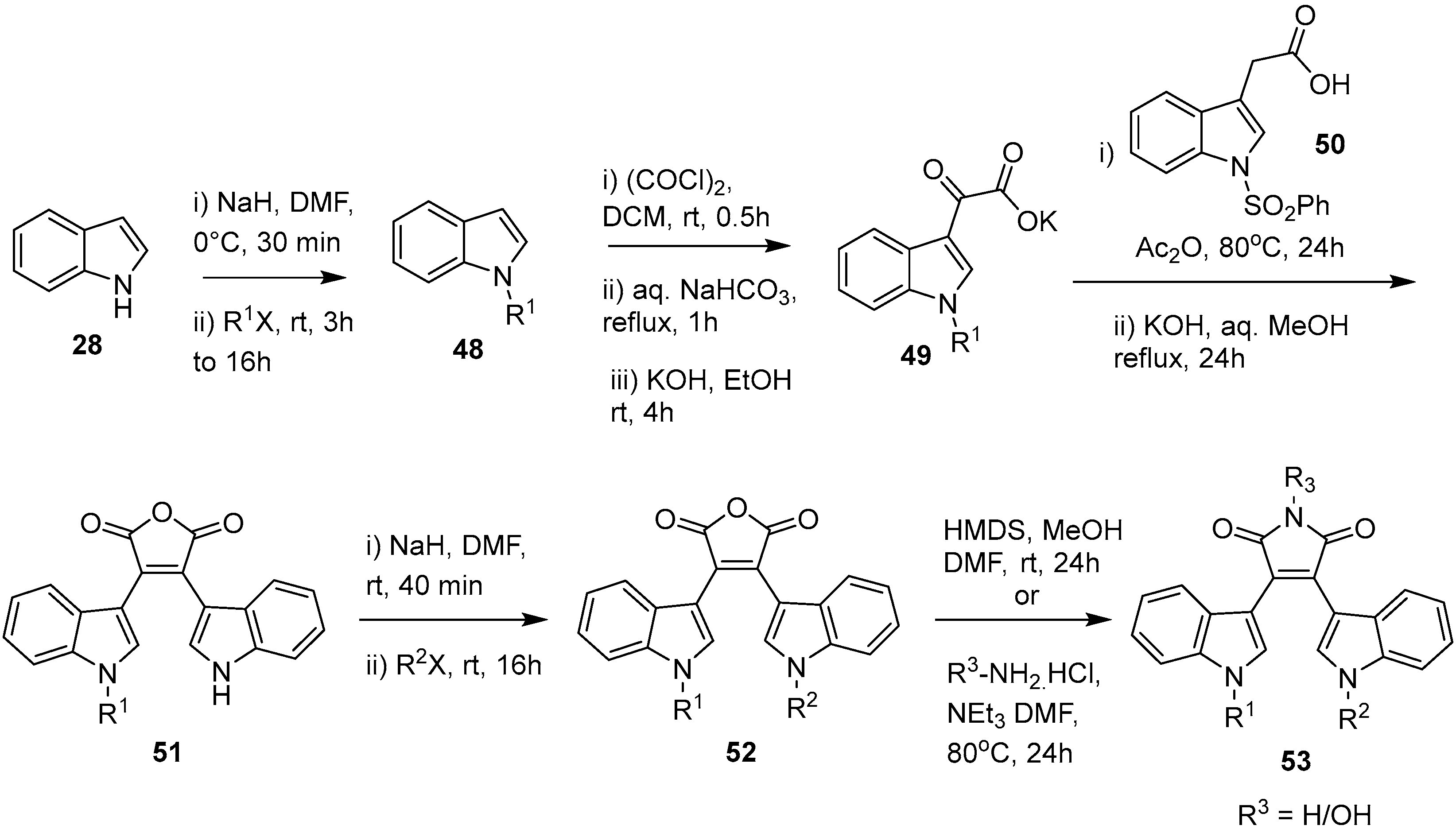
2.4. Preparation of Selected Modified Indolylmaleimides
3. Bioactivity of Bisindolylmaleimides and Derivatives
3.1. Bisindolylmaleimides (BIMs) and BIM-Type Inhibitors
3.2. Selected Modified Indolylmaleimides: Benzofuranylindolylmaleimides (BfIMs)
3.3. Selected Modified Indolylmaleimides: Napthylindolylmaleimides (NIMs)
3.4. Recent Applications of Bisindolylmaleimides and Derivatives
3.5. X-ray Crystal Structures of Bisindolylmaleimides and Kinases
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pajak, B.; Orzechowska, S.; Gajkowska, B.; Orzechowski, A. Bisindolylmaleimides in anti-cancer therapy–more than PKC inhibitors. Adv. Med. Sci. 2008, 53, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, C.; Mendez, C.; Salas, J.A. Indolocarbazole natural products: Occurrence, biosynthesis, and biological activity. Nat. Prod. Rep. 2006, 23, 1007–1045. [Google Scholar] [CrossRef] [PubMed]
- Nakano, H.; Ōmura, S. Chemical biology of natural indolocarbazole products: 30 years since the discovery of staurosporine. J. Antibiot. 2009, 62, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Janosik, T.; Rannug, A.; Rannug, U.; Wahlström, N.; Slätt, J.; Bergman, J. Chemistry and Properties of Indolocarbazoles. Chem. Rev. 2018, 118, 9058–9128. [Google Scholar] [CrossRef] [PubMed]
- Volvoikar, P.; Torney, P. An account of synthetic strategies towards indolocarbazole alkaloids: Arcyriaflavin A and staurosporinone. Tetrahedron 2021, 82, 131756. [Google Scholar] [CrossRef]
- Chambers, G.E.; Sayan, A.E.; Brown, R.C.D. The synthesis of biologically active indolocarbazole natural products. Nat. Prod. Rep. 2021, 38, 1794–1820. [Google Scholar] [CrossRef] [PubMed]
- Steglich, W. Slime moulds (Myxomycetes) as a source of new biologically active metabolites. Pure Appl. Chem. 1989, 61, 281–288. [Google Scholar] [CrossRef]
- Panov, A.A.; Simonov, A.Y.; Lavrenov, S.N.; Lakatosh, S.A.; Trenin, A.S. 3,4-Disubstituted maleimides: Synthesis and biological activity. Chem. Heterocycl. Comp. 2018, 54, 103–113. [Google Scholar] [CrossRef]
- Steglich, W.; Steffan, B.; Kopanski, L.; Eckhardt, G. Indole Pigments from the Fruiting Bodies of the Slime Mold Arcyria denudata. Angew. Chem. 1980, 19, 459–460. [Google Scholar] [CrossRef]
- Joyce, R.P.; Gainor, J.A.; Weinreb, S.M. Synthesis of the Aromatic and Monosaccharide Moieties of Staurosporine. J. Org. Chem. 1987, 52, 1177–1185. [Google Scholar] [CrossRef]
- Kaneko, T.; Wong, H.; Okamoto, K.T.; Clardy, J. Two synthetic approaches to rebeccamycin. Tetrahedron Lett. 1985, 26, 4015–4018. [Google Scholar] [CrossRef]
- Brenner, M.; Rexhausen, H.; Steffan, B.; Steglich, W. Synthesis of arcyriarubin b and related bisindolylmaleimides. Tetrahedron 1988, 44, 2887–2892. [Google Scholar] [CrossRef]
- Harris, W.; Hill, C.H.; Keech, E.; Malsher, P. Oxidative cyclisations with palladium acetate. A short synthesis of staurosporine aglycone. Tetrahedron Lett. 1993, 34, 8361–8364. [Google Scholar] [CrossRef]
- Xie, G.; Lown, W.J. A facile synthesis of staurosporine aglycone. Tetrahedron Lett. 1994, 35, 5555–5558. [Google Scholar] [CrossRef]
- Faul, M.M.; Sullivan, K.A.; Winneroski, L.L. A General Approach to the Synthesis of Bisindolylmaleimides: Synthesis of Staurosporine Aglycone. Synthesis 1995, 1995, 1511–1516. [Google Scholar] [CrossRef]
- Ohkubo, M.; Nishimura, T.; Jona, H.; Honma, T.; Morishima, H. Practical synthesis of indolopyrrolocarbazoles. Tetrahedron 1996, 52, 8099–8112. [Google Scholar] [CrossRef]
- Neel, D.A.; Jirousek, M.R.; McDonald, J.H., III. Synthesis of bisindolylmaleimides using a palladium catalyzed cross-coupling reaction. Bioorg. Med. Chem. Lett. 1998, 8, 47–50. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Liu, Z. Synthesis of Arcyriarubin A and Arcyriaflavin A via Cross-Coupling of Indolylboronic Acid with Dibromomaleimides. Synth. Commun. 2010, 40, 144–150. [Google Scholar] [CrossRef]
- Shenyin, Z.; Yulong, A. A Kind of Preparation Method of Bisindole Maleimide Compound. Chinese Patent CN105153126(A), 10 September 2015. Available online: http://patents.google.com/patent/CN105153126B/ (accessed on 9 June 2023).
- Davis, P.D.; Bit, R.A.; Hurst, S.A. A convenient synthesis of bisindolyl- and indolylaryl maleic anhydrides. Tetrahedron Lett. 1990, 31, 2353–2356. [Google Scholar] [CrossRef]
- Davis, P.D.; Bit, R.A. A mild conversion of maleic anhydrides into maleimides. Tetrahedron Lett. 1990, 31, 5201–5204. [Google Scholar] [CrossRef]
- Sandler, S.R.; Karo, W. Organic Functional Group Preparations; Academic Press: London, UK, 1972; Volume 3, Chapter 7. [Google Scholar]
- Bit, R.A.; Crackett, P.H.; Harris, W.; Hill, C.H. A convenient synthesis of bisindolylmaleimides. Tetrahedron Lett. 1993, 34, 5623–5626. [Google Scholar] [CrossRef]
- Faul, M.M.; Winneroski, L.L.; Krumrich, C.A. A New, Efficient Method for the Synthesis of Bisindolylmaleimides. J. Org. Chem. 1998, 63, 6053–6058. [Google Scholar] [CrossRef] [PubMed]
- Bergman, J.; Koch, E.; Pelcman, B. Coupling reactions of indole-3-acetic acid derivatives. Synthesis of arcyriaflavin A. J. Chem. Soc. Perkin Trans. 1 2000, 16, 2609–2614. [Google Scholar] [CrossRef]
- Roy, S.; Roy, S.; Gribble, G.W. A Practical Method for the Synthesis of Indolylaryl- and Bisindolylmaleimides. Org. Lett. 2006, 8, 4975–4977. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.-C.; Jia, Y.-H.; Li, T.-T.; Hei, Z.-H.; Yang, F.-L.; Huang, M.-H.; Luo, Y.-J. Protecting-group-free synthesis of the bisindolylmaleimide GF109203X. Arkivoc 2015, 2015, 153–163. [Google Scholar] [CrossRef]
- Li, X.; Ma, H.; Li, L.; Chen, Y.; Sun, X.; Dong, Z.; Liu, J.-Y.; Zhu, W.; Zhang, J.-T. Novel synthetic bisindolylmaleimide alkaloids inhibit STAT3 activation by binding to the SH2 domain and suppress breast xenograft tumor growth. Oncogene 2018, 37, 2469–2480. [Google Scholar] [CrossRef] [PubMed]
- Winfield, H.J.; Cahill, M.M.; O’Shea, K.D.; Pierce, L.T.; Robert, T.; Ruchaud, S.; Bach, S.; Marchand, P.; McCarthy, F.O. Synthesis and anticancer activity of novel bisindolylhydroxymaleimide derivatives with potent GSK-3 kinase inhibition. Bioorg. Med. Chem. 2018, 26, 4209–4224. [Google Scholar] [CrossRef]
- Faul, M.M.; Grutsch, J.L.; Kobierski, M.E.; Kopach, M.E.; Krumrich, C.A.; Staszak, M.A.; Udodong, U.; Vicenzi, J.T.; Sullivan, K.A. Strategies for the synthesis of N-(azacycloalkyl)bisindolylmaleimides: Selective inhibitors of PKC-β. Tetrahedron 2003, 59, 7215–7229. [Google Scholar] [CrossRef]
- Jirousek, M.R.; Gillig, J.R.; Neel, D.A.; Rito, C.J.; O’Bannon, D.; Heath, W.F.; McDonald, J.H.; Faul, M.M.; Winneroski, L.L.; Melikian Badalian, A.; et al. Synthesis of bisindolylmaleimide macrocycles. Bioorg. Med. Chem. Lett. 1995, 5, 2093–2096. [Google Scholar] [CrossRef]
- Jirousek, M.R.; Gillig, J.R.; Gonzalez, C.M.; Heath, W.F.; McDonald, J.H.; Neel, D.A.; Rito, C.J.; Singh, U.; Stramm, L.E.; Melikian-Badalian, A.; et al. (S)-13-[(Dimethylamino)methyl]-10,11,14,15-tetrahydro-4,9:16,21-dimetheno-1H,13H-dibenzo[e,k]pyrrolo [3,4-h][1,4,13]oxadiazacyclohexadecene-1,3(2H)-dione (LY333531) and Related Analogues: Isozyme Selective Inhibitors of Protein Kinase Cβ. J. Med. Chem. 1996, 39, 2664–2671. [Google Scholar] [CrossRef]
- Kuo, G.-H.; Prouty, C.; DeAngelis, A.; Shen, L.; O’Neill, D.J.; Shah, C.; Connolly, P.J.; Murray, W.V.; Conway, B.R.; Cheung, P.; et al. Synthesis and Discovery of Macrocyclic Polyoxygenated Bis-7-azaindolylmaleimides as a Novel Series of Potent and Highly Selective Glycogen Synthase Kinase-3β Inhibitors. J. Med. Chem. 2003, 46, 4021–4031. [Google Scholar] [CrossRef] [PubMed]
- Kozikowski, A.P.; Gaisina, I.N.; Yuan, H.; Petukhov, P.A.; Blond, S.Y.; Fedolak, A.; Caldarone, B.; McGonigle, P. Structure-Based Design Leads to the Identification of Lithium Mimetics That Block Mania-like Effects in Rodents. Possible New GSK-3β Therapies for Bipolar Disorders. J. Am. Chem. Soc. 2007, 129, 8328–8332. [Google Scholar] [CrossRef] [PubMed]
- Gaisina, I.N.; Gallier, F.; Ougolkov, A.V.; Kim, K.H.; Kurome, T.; Guo, S.; Holzle, D.; Luchini, D.N.; Blond, S.Y.; Billadeau, D.D.; et al. From a Natural Product Lead to the Identification of Potent and Selective Benzofuran-3-yl-(indol-3-yl)maleimides as Glycogen Synthase Kinase 3 β Inhibitors That Suppress Proliferation and Survival of Pancreatic Cancer Cells. J. Med. Chem. 2009, 52, 1853–1863. [Google Scholar] [CrossRef] [PubMed]
- Faul, M.M.; Winneroski, L.L.; Krumrich, C.A. A new one step synthesis of maleimides by condensation of glyoxylate esters with acetamides. Tetrahedron Lett. 1999, 40, 1109–1112. [Google Scholar] [CrossRef]
- van Eis, M.J.; Evenou, J.; Schuler, W.; Zenke, G.; Vangrevelinghe, E.; Wagner, J.; von Matt, P. Indolyl-naphthyl-maleimides as potent and selective inhibitors of protein kinase C-α/β. Bioorg. Med. Chem. Lett. 2017, 27, 781–786. [Google Scholar] [CrossRef] [PubMed]
- Fabre, S.; Prudhomme, M. Protein Kinase C Inhibitors; Structure–Activity Relationships in K252c-Related Compounds. Bioorg. Med. Chem. 1993, 1, 193–196. [Google Scholar] [CrossRef] [PubMed]
- Shengyin, Z.; Zhiyu, S.; Weimin, Q.; Dengqing, Z. Research Progress of Indole Maleimide Protein Kinase C Inhibitors. Org. Chem. 2008, 28, 1676. [Google Scholar]
- Sancelme, M.; Fabre, S.; Prudhomme, M. Antimicrobial Activities of Indolocarbazole and Bis-indole Protein Kinase C Inhibitors. J. Antibiot. 1994, 47, 792–798. [Google Scholar] [CrossRef]
- Toullec, D.; Pianettis, P.; Coste, H.; Belleverguel, P.; Grand-Perret, T.; Ajakanee, M.; Baudet, V.; Boissin, P.; Boursier, E.; Loriolle, F.; et al. The Bisindolylmaleimide GF 109203X Is a Potent and Selective Inhibitor of Protein Kinase C. J. Biol. Chem. 1991, 266, 15771–15781. [Google Scholar] [CrossRef]
- Hu, H. Recent discovery and development of selective protein kinase C inhibitors. Drug Discov. Today 1996, 1, 438–447. [Google Scholar] [CrossRef]
- Zhou, F.; Huang, H.; Zhang, L. Bisindoylmaleimide I enhances osteogenic differentiation. Protein Cell 2012, 3, 311–320. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kosgodage, U.S.; Trindade, R.P.; Thompson, P.R.; Inal, J.M.; Lange, S. Chloramidine/Bisindolylmaleimide-I-Mediated Inhibition of Exosome and Microvesicle Release and Enhanced Efficacy of Cancer Chemotherapy. Int. J. Mol. Sci. 2017, 18, 1007. [Google Scholar] [CrossRef] [PubMed]
- Graff, J.R.; McNulty, A.M.; Hanna, K.R.; Konicek, B.W.; Lynch, R.L.; Bailey, S.N.; Banks, C.; Capen, A.; Goode, R.; Lewis, J.E.; et al. The Protein Kinase CB–Selective Inhibitor, Enzastaurin (LY317615.HCl), Suppresses Signaling through the AKT Pathway, Induces Apoptosis, and Suppresses Growth of Human Colon Cancer and Glioblastoma Xenografts. Cancer Res. 2005, 65, 7462–7469. [Google Scholar] [CrossRef] [PubMed]
- Keyes, K.A.; Mann, L.; Sherman, M.; Galbreath, E.; Schirtzinger, L.; Ballard, D.; Chen, Y.-F.; Iversen, P.; Teicher, B.A. LY317615 decreases plasma VEGF levels in human tumor xenograft-bearing mice. Cancer Chemother. Pharmacol. 2004, 53, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Wick, W.; Puduvalli, V.K.; Chamberlain, M.C.; van den Bent, M.J.; Carpentier, A.F.; Cher, L.M.; Mason, W.; Weller, M.; Hong, S.; Musib, L.; et al. Phase III study of enzastaurin compared with lomustine in the treatment of recurrent intracranial glioblastoma. J. Clin. Oncol. 2010, 28, 1168–1174. [Google Scholar] [CrossRef] [PubMed]
- Nowakowski, G.S.; Zhu, J.; Zhang, Q.; Brody, J.; Sun, X.; Maly, J.; Song, Y.; Rizvi, S.; Song, Y.; Lansigan, F.; et al. ENGINE: A Phase III randomized placebo controlled study of enzastaurin/R-CHOP as frontline therapy in high-risk diffuse large B-cell lymphoma patients with the genomic biomarker DGM1. Future Oncol. 2020, 16, 991–999. [Google Scholar] [CrossRef] [PubMed]
- A Trial of Enzastaurin Plus Temozolomide During and Following Radiation Therapy in Patients with Newly Diagnosed Glioblastoma with or without the Novel Genomic Biomarker, DGM1 2023. Available online: http://clinicaltrials.gov/study/NCT03776071 (accessed on 30 June 2023).
- Bota, D.; Butowski, N.; Piccioni, D.; De la Fuente, M.; Mao, Y.; Li, W.-B.; Trusheim, J.; Fink, K.; Campian, J.; Lobbous, M.; et al. CTNI-08. DB102-01 ENGAGE study: A biomarker-guided, randomized, double-blind, placebo-controlled, multi-center Phase 3 clinical trial of DB102 in patients with newly diagnosed glioblastoma (GBM). Neuro-Oncology 2021, 23, 60. [Google Scholar] [CrossRef]
- Investigate Efficacy, Safety, and Pharmacokinetics of Enzastaurin for the Prevention of Arterial Events in Patients with Vascular Ehlers-Danlos Syndrome (PREVEnt). 2022. Available online: http://clinicaltrials.gov/ct2/show/NCT05463679 (accessed on 30 June 2023).
- Sobhia, M.E.; Grewal, B.K.; ML, S.P.; Patel, J.; Kaur, A.; Haokip, T.; Kokkula, A. Protein kinase C inhibitors: A patent review (2008–2009). Expert Opin. Therap. Pat. 2013, 23, 1–19. [Google Scholar] [CrossRef]
- Teicher, B.A.; Alvarez, E.; Mendelsohn, L.G. Enzymatic rational and preclinical support for a potent protein kinase Cβ inhibitor in cancer therapy. Adv. Enzyme Regul. 1999, 39, 313–327. [Google Scholar] [CrossRef]
- Shiying, L.; Yuyang, J.; Jian, C.; Feng, L.; Li, M.; Yufen, Z. Inhibitors of protein kinase C. Chin. Sci. Bull. 2005, 50, 1293–1304. [Google Scholar]
- McGill, J.B.; King, G.L.; Berg, P.H.; Price, K.L.; Kles, K.A.; Bastyr, E.J.; Hyslop, D.L. Clinical safety of the selective PKC-β inhibitor, ruboxistaurin. Expert Opin. Drug Saf. 2006, 5, 835–845. [Google Scholar] [CrossRef] [PubMed]
- Javey, G.; Schwartz, S.G.; Flynn, H.W.; Aiello, L.P.; Sheetz, M.J. Ruboxistaurin: Review of Safety and Efficacy in the Treatment of Diabetic Retinopathy. Clin. Med. Insights Ther. 2010, 2, CMT-S5046. [Google Scholar] [CrossRef]
- Eli Lilly Nederland, B.V. ARxxant Withdrawal Assessment Report European Medicines Agency (EMEA/H/C/000753). Available online: http://www.ema.europa.eu/en/medicines/human/withdrawn-applications/arxxant (accessed on 9 June 2023).
- Randomized and Open Label Study for Safety and Efficacy of DBI-102 vs. Vehicle vs. Hydroquinone on Skin Pigmentation and Lentigos. Available online: https://clinicaltrials.gov/study/NCT05511948 (accessed on 9 June 2023).
- Davis, P.D.; Hill, C.H.; Lawton, G.; Nixon, J.S.; Wilkinson, S.E.; Hurst, S.A.; Keech, E.; Turner, S.E. Inhibitors of Protein Kinase, C. I. 1. 2,3-Bisarylmaleimides. J. Med. Chem. 1992, 35, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Ugolkov, A.V.; Bondarenko, G.I.; Dubrovskyi, O.; Berbegall, A.P.; Navarro, S.; Noguera, R.; O’Halloran, T.V.; Hendrix, M.J.; Giles, F.J.; Mazar, A.P. 9-ING-41, a small molecule Glycogen Synthase Kinase-3 inhibitor, is active in neuroblastoma. Anticancer Drugs 2018, 29, 717–724. [Google Scholar] [CrossRef] [PubMed]
- 9-ING-41 in Patients with Advanced Cancers. 2019. Available online: http://www.clinicaltrials.gov/study/NCT03678883 (accessed on 30 June 2023).
- Jeffers, A.; Qin, W.; Owens, S.; Koenig, K.B.; Komatsu, S.; Giles, F.J.; Schmitt, D.M.; Idell, S.; Tucker, T.A. Glycogen Synthase Kinase-3β Inhibition with 9-ING-41 Attenuates the Progression of Pulmonary Fibrosis. Sci. Rep. 2019, 9, 18925. [Google Scholar] [CrossRef] [PubMed]
- Anraku, T.; Kuroki, H.; Kazama, A.; Bilim, V.; Tasaki, M.; Schmitt, D.; Mazar, A.; Giles, F.J.; Ugolkov, A.V.; Tomita, Y. Clinically relevant GSK-3β inhibitor 9-ING-41 is active as a single agent and in combination with other antitumour therapies in human renal cancer. Int. J. Mol. Med. 2020, 45, 315–323. [Google Scholar] [PubMed]
- Peifer, C.; Stoiber, T.; Unger, E.; Totzke, F.; Schächtele, C.; Marme, D.; Brenk, R.; Klebe, G.; Schollmeyer, D.; Dannhardt, G. Design, Synthesis, and Biological Evaluation of 3,4-Diarylmaleimides as Angiogenesis Inhibitors. J. Med. Chem. 2006, 49, 1271–1281. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Jia, D.; Ao, J.; Liu, H.; Zang, Y.; Azam, M.; Habib, S.L.; Li, J.; Ruan, X.; Jia, H.; et al. Identification of Bisindolylmaleimide IX as a potential agent to treat drug-resistant BCR-ABL positive leukemia. Oncotarget 2016, 7, 69945–69960. [Google Scholar] [CrossRef] [PubMed]
- Mayati, A.; Bruyere, A.; Moreau, A.; Jouan, E.; Denizot, C.; Parmentier, Y.; Fardel, O. Protein Kinase C-Independent inhibition of Organic Cation Transporter 1 activity by the bisindolylmaleimide Ro 31-8220. PLoS ONE 2015, 10, e0144667. [Google Scholar] [CrossRef]
- Sosa-Peinado, A.; Fructuoso-García, K.; Vásquez-Bochm, L.X.; González-Andrade, M. Bisindolylmaleimides new ligands of CaM Protein. Molecules 2022, 27, 7161. [Google Scholar] [CrossRef]
- Hers, I.; Tavare, J.M.; Denton, R.M. The protein kinase C inhibitors bisindolylmaleimide I (GF 109203x) and IX (Ro 31-8220) are potent inhibitors of glycogen synthase kinase-3 activity. FEBS Lett. 1999, 460, 433–436. [Google Scholar] [CrossRef] [PubMed]
- Coultrap, S.J.; Sun, H.; Tenner, T.E., Jr.; Machu, T.K. Competitive antagonism of the mouse 5-hydroxytryptamine3 receptor by bisindolylmaleimide I, a “selective” protein kinase C inhibitor. J. Pharmacol. Exp. Ther. 1999, 290, 76–82. [Google Scholar] [PubMed]
- Robey, R.W.; Shukla, S.; Steadman, K.; Obrzut, T.; Finley, E.M.; Ambudkar, S.V.; Bates, S.E. Inhibition of ABCG2-mediated transport by protein kinase inhibitors with a bisindolylmaleimide or indolocarbazole structure. Mol. Cancer Ther. 2007, 6, 1877–1885. [Google Scholar]
- Deane, F.M.; Lin, A.J.S.; Hains, P.G.; Pilgrim, S.L.; Robinson, P.J.; McCluskey, A. FD5180, a Novel Protein Kinase Affinity Probe, and the Effect of Bead Loading on Protein Kinase Identification. ACS Omega 2017, 2, 3828–3838. [Google Scholar] [CrossRef] [PubMed]
- Birchall, A.M.; Bishop, J.; Bradshaw, D.; Cline, A.; Coffey, J.; Elliott, L.H.; Gibson, V.M.; Greenham, A.; Hallam, T.J.; Harris, W.; et al. Ro 32-0432, a selective and orally active inhibitor of protein kinase C prevents T-cell activation. J. Pharmacol. Exp. Ther. 1994, 268, 922–929. [Google Scholar] [PubMed]
- Bit, R.A.; Davis, P.D.; Elliott, L.H.; Harris, W.; Hill, C.H.; Keech, E.; Kumar, H.; Lawton, G.; Maw, A.; Nixon, J.S.; et al. Inhibitors of protein kinase C. 3. Potent and highly selective bisindolylmaleimides by conformational restriction. J. Med. Chem. 1993, 36, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Komander, D.; Kular, G.S.; Schüttelkopf, A.W.; Deak, M.; Prakash, K.R.C.; Bain, J.; Elliott, M.; Garrido-Franco, M.; Kozikowski, A.P.; Alessi, D.R.; et al. Interactions of LY333531 and Other Bisindolyl Maleimide Inhibitors with PDK1. Structure 2004, 12, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Davies, S.P.; Reddy, H.; Caivano, M.; Chen, P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem. J. 2000, 351, 95–105. [Google Scholar] [CrossRef]
- Gupta, Y.; Maciorowski, D.; Zak, S.E.; Jones, K.A.; Kathayat, R.S.; Azizi, S.-A.; Mathur, R.; Pearce, C.M.; Ilc, D.J.; Husein, H.; et al. Bisindolylmaleimide IX: A novel anti-SARS-CoV2 agent targeting viral main protease 3CLpro demonstrated by virtual screening pipeline and in-vitro validation assays. Methods 2021, 195, 57–71. [Google Scholar] [CrossRef]
- Huang, C.; Feng, F.; Shi, Y.; Li, W.; Wang, Z.; Zhu, Y.; Yuan, S.; Hu, D.; Dai, J.; Jiang, Q.; et al. Protein Kinase C inhibitors reduce SARS-CoV-2 replication in cultured cells. Microbiol. Spectr. 2022, 10, e01056-22. [Google Scholar] [CrossRef]
- Sun, X.; Li, L.; Ma, H.-G.; Sun, P.; Wang, Q.-L.; Zhang, T.-T.; Shen, Y.-M.; Zhu, W.-M.; Li, X. Bisindolylmaleimide alkaloid BMA-155Cl induces autophagy and apoptosis in human hepatocarcinoma HepG-2 cells through the NF-κB p65 pathway. Acta Pharmacol. Sin. 2017, 38, 524–538. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Sannapureddi, R.K.R.; Todankar, C.S.; Ramanathan, R.; Biswas, A.; Sathyamoorthy, B.; Pradeepkumar, P.I. Bisindolylmaleimide ligands stabilize c-MYC G-Quadruplex DNA structure and downregulate gene expression. Biochemistry 2022, 61, 1064–1076. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Wang, J.; Chen, H.; Mei, X.; Su, Z.; Wu, J.; Lin, Z.; Ling, Q. Solvent-dependent and highly selective anion sensing and molecular logic application of bisindolylmaleimide derivatives. RSC Adv. 2017, 7, 12161–12169. [Google Scholar] [CrossRef]
- Knapp, S.; Debreczeni, J.; Bullock, A.; von Delft, F.; Sundstrom, M.; Arrowsmith, C.; Edwards, A.; Guo, K. PDB Entry–1XWS. Crystal Structure of the Human PIM1 Kinase Domain. Available online: https://doi.org/10.2210/pdb1xws/pdb (accessed on 18 July 2023).
- Knapp, S.; Debreczeni, J.; Bullock, A.; von Delft, F.; Sundstrom, M.; Arrowsmith, C.; Edwards, A.; Guo, K. PDB Entry–2BIK. Human Pim1 Phosphorylated on Ser261. Available online: https://doi.org/10.2210/pdb2bik/pdb (accessed on 18 July 2023).
- Knapp, S.; Debreczeni, J.; Bullock, A.; von Delft, F.; Sundstrom, M.; Arrowsmith, C.; Edwards, A.; Guo, K. PDB Entry–2BIL. The Human Protein Kinase Pim1 in Complex with Its Consensus Peptide Pimtide. Available online: https://doi.org/10.2210/pdb2bil/pdb (accessed on 18 July 2023).
- Messerschmidt, A.; Macieira, S.; Velarde, M.; Bädeker, M.; Benda, C.; Jestel, A.; Brandstetter, H.; Neuefeind, T.; Blaesse, M. Crystal structure of the catalytic domain of human atypical protein kinase C-iota reveals interaction mode of phosphorylation site in turn motif. J. Mol. Biol. 2005, 352, 918–931. [Google Scholar] [CrossRef] [PubMed]
- Sadowsky, J.D.; Burlingame, M.A.; Wolan, D.W.; McClendon, C.L.; Jacobson, M.P.; Wells, J.A. Turning a protein kinase on or off from a single allosteric site via disulfide trapping. Proc. Natl. Acad. Sci. USA 2011, 108, 6056–6061. [Google Scholar] [CrossRef] [PubMed]
- Elkins, J.M.; Amos, A.; Niesen, F.H.; Pike, A.C.W.; Fedorov, O.; Knapp, S. Structure of dystrophia myotonica protein kinase. Protein Sci. 2009, 18, 782–791. [Google Scholar] [CrossRef] [PubMed]
- Fedorov, O.; Marsden, B.; Pogacic, V.; Rellos, P.; Müller, S.; Bullock, A.N.; Schwaller, J.; Sundström, M.; Knapp, S. A systematic interaction map of validated kinase inhibitors with Ser/Thr kinases. Proc. Natl. Acad. Sci. USA 2007, 104, 20523–20528. [Google Scholar] [CrossRef]
- Mesecar, A.M.; Walters, R.L. PDB Entry–3SD0. Identification of a Glycogen Synthase Kinase-3b Inhibitor that Attenuates Hyperactivity in CLOCK Mutant Mice. Available online: https://doi.org/10.2210/pdb3sd0/pdb (accessed on 18 July 2023).
- Grodsky, N.; Li, Y.; Bouzida, D.; Love, R.; Jensen, J.; Nodes, B.; Nonomiya, J.; Grant, S. Structure of the catalytic domain of human protein kinase C beta II complexed with a bisindolylmaleimide inhibitor. Biochemistry 2006, 45, 13970–13981. [Google Scholar] [CrossRef]
- Zhang, H.-C.; Boñaga, L.V.R.; Ye, H.; Derian, C.K.; Damiano, B.P.; Maryanoff, B.E. Novel bis(indolyl)maleimide pyridinophanes that are potent, selective inhibitors of glycogen synthase kinase-3. Bioorg. Med. Chem. Lett. 2007, 17, 2863–2868. [Google Scholar] [CrossRef]
- Allard, J.; Nikolcheva, T.; Gong, L.; Wang, J.; Dunten, P.; Avnur, Z.; Waters, R.; Sun, Q.; Skinner, B. PDB Entry–1R0E. Glycogen Synthase Kinase-3 Beta in Complex with 3-Indolyl-4-Arylmaleimide Inhibitor. Available online: https://doi.org/10.2210/pdb1r0e/pdb (accessed on 18 July 2023).
- Thoma, G.; Nuninger, F.; Falchetto, R.; Hermes, E.; Tavares, G.A.; Vangrevelinghe, E.; Zerwes, H.-G. Identification of a potent Janus kinase 3 inhibitor with high selectivity within the Janus kinase family. J. Med. Chem. 2011, 54, 284–288. [Google Scholar] [CrossRef]
- Eathiraj, S.; Palma, R.; Volckova, E.; Hirschi, M.; France, D.S.; Ashwell, M.A.; Chan, T.C.K. Discovery of a novel mode of protein kinase inhibition characterized by the mechanism of inhibition of human mesenchymal-epithelial transition factor (c-Met) protein autophosphorylation by ARQ 197. J. Biol. Chem. 2011, 286, 20666–20676. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.; von Matt, P.; Sedrani, R.; Albert, R.; Cooke, N.; Ehrhardt, C.; Geiser, M.; Rummel, G.; Stark, W.; Strauss, A.; et al. Discovery of 3-(1H-indol-3-yl)-4-[2-(4-methylpiperazin-1-yl)quinazolin-4-yl]pyrrole-2,5-dione (AEB071), a potent and selective inhibitor of protein kinase C isotypes. J. Med. Chem. 2009, 52, 6193–6196. [Google Scholar] [CrossRef] [PubMed]
- Cahill, M.M.; O’Shea, K.D.; Pierce, L.T.; Winfield, H.J.; Eccles, K.S.; Lawrence, S.E.; McCarthy, F.O. Synthesis and Antiproliferative Activity of Novel Heterocyclic Indole-Trimethoxyphenyl Conjugates. Pharmaceuticals 2017, 10, 62. [Google Scholar] [CrossRef] [PubMed]
- Pierce, L.T.; Cahill, M.M.; McCarthy, F.O. Design and synthesis of novel 5,6-bisindolylpyrimidin-4-ones structurally related to ruboxistaurin (LY333531). Tetrahedron 2010, 66, 9754–9761. [Google Scholar] [CrossRef]
- Pierce, L.T.; Cahill, M.M.; Winfield, H.J.; McCarthy, F.O. Synthesis and identification of novel indolo [2,3-a]pyrimido[5,4-c]carbazoles as a new class of anti-cancer agents. Eur. J. Med. Chem. 2012, 56, 292–300. [Google Scholar] [CrossRef] [PubMed]
- Pierce, L.T.; Cahill, M.M.; McCarthy, F.O. Synthesis of novel 3,4-diaryl-5-aminopyrazoles as potential kinase inhibitors. Tetrahedron 2011, 67, 4601–4611. [Google Scholar] [CrossRef]



































{kind=link}
{kind=link}
{kind=link}
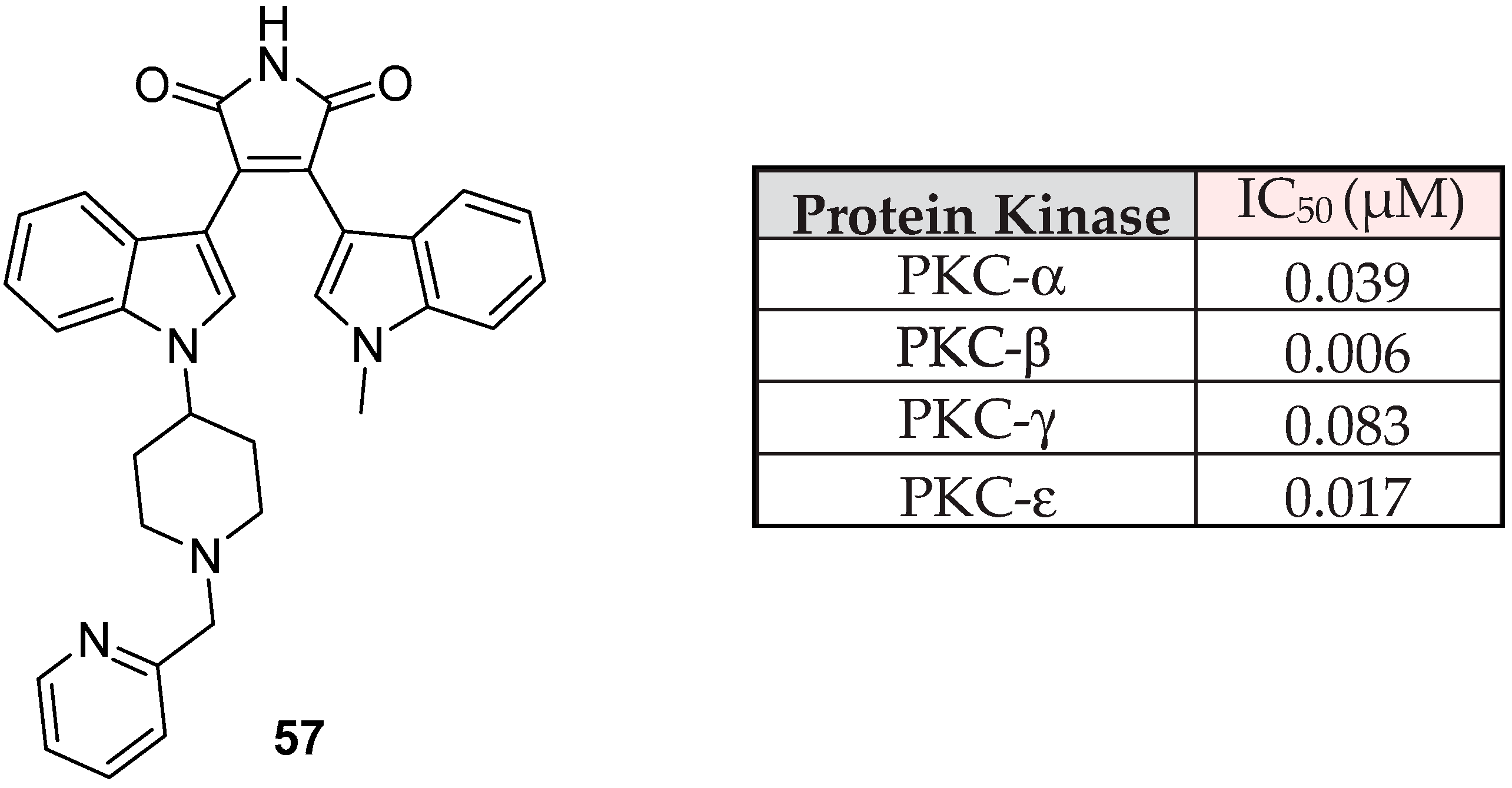{kind=link}
{kind=link}
{kind=link}
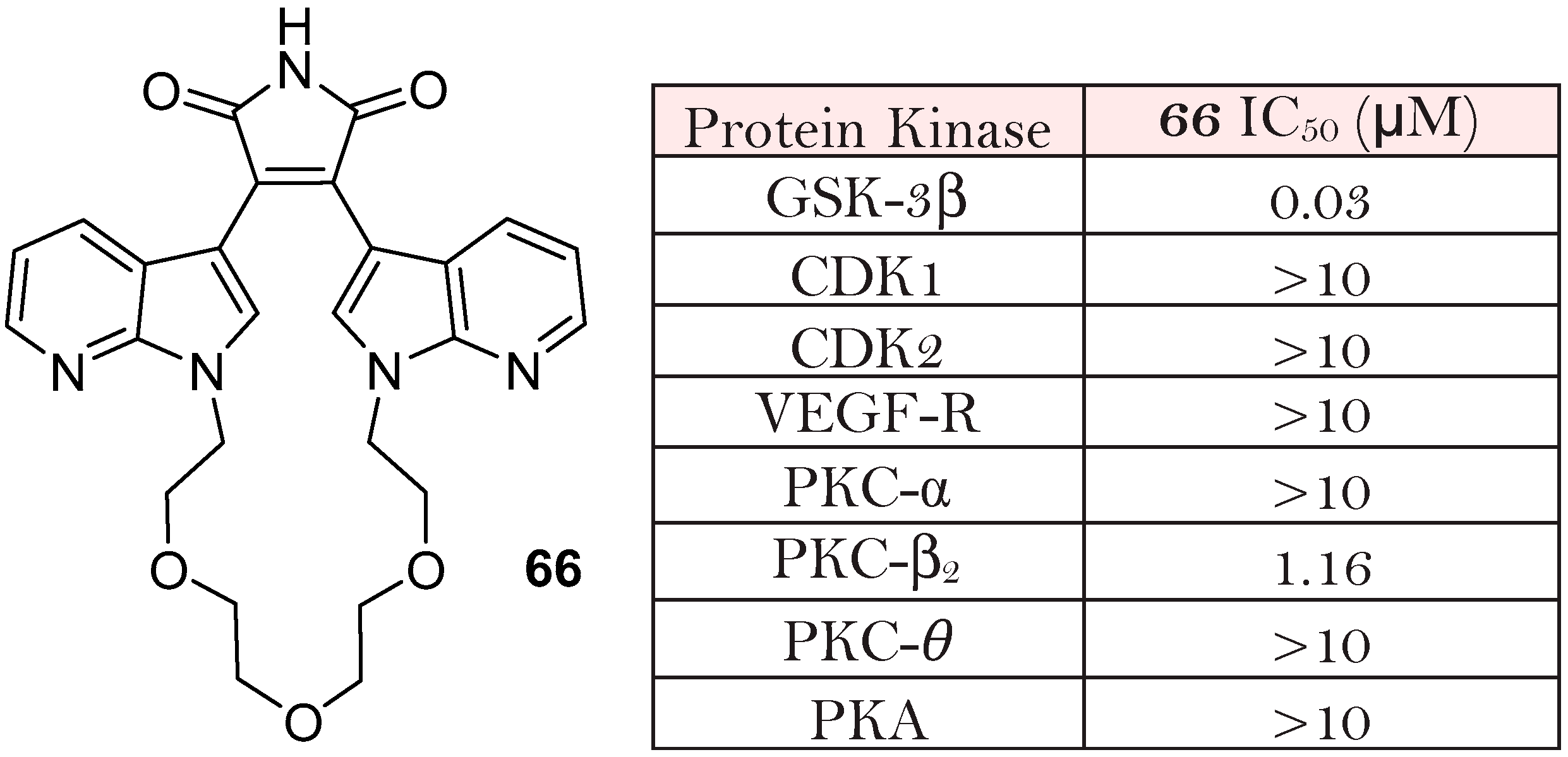{kind=link}
{kind=link}
{kind=link}
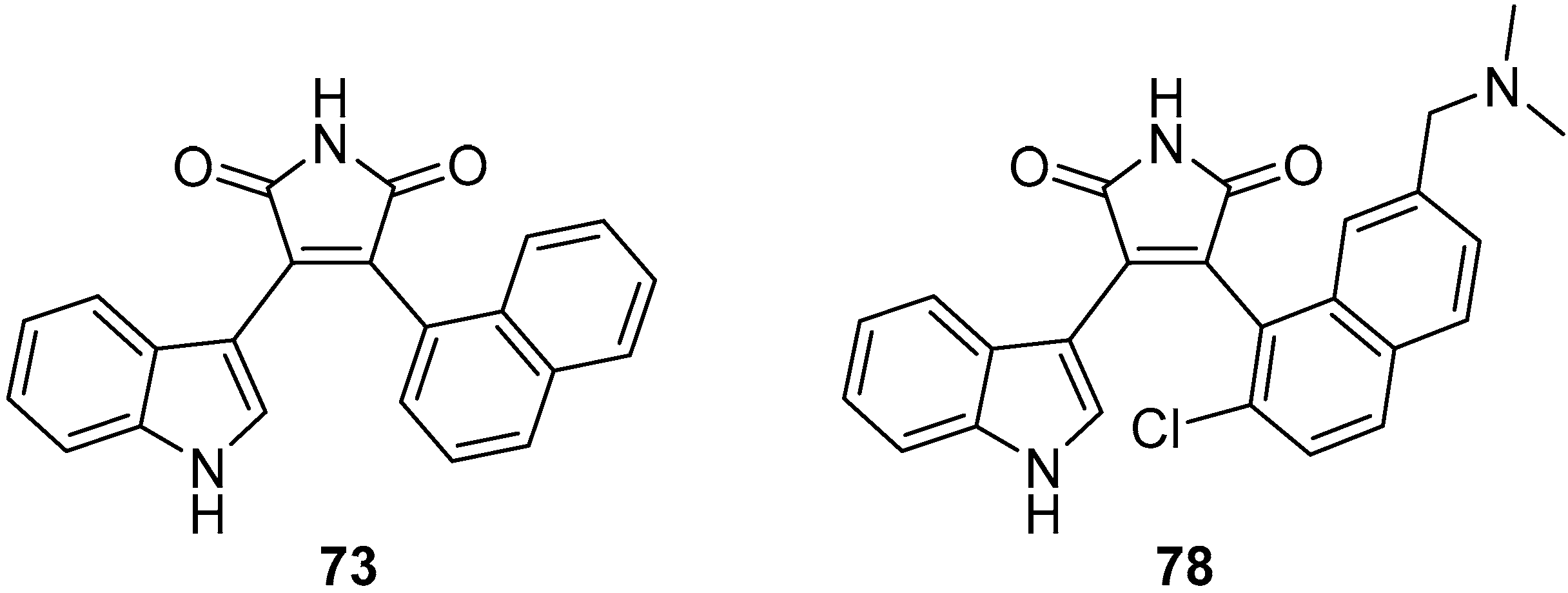{kind=link}
{kind=link}
{kind=link}
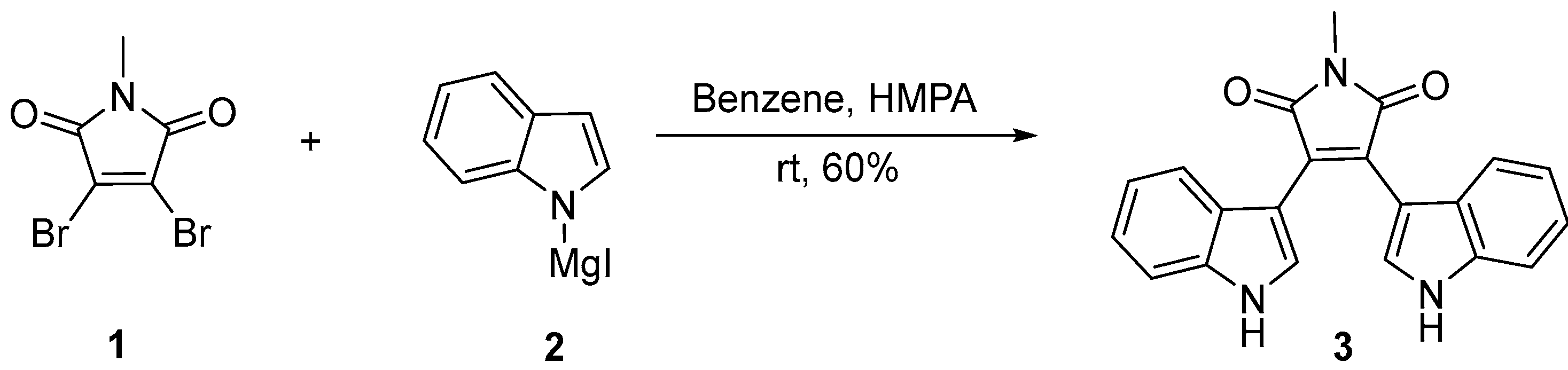{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| BIM | Proposed Targets |
|---|---|
| BIM-I | PKC, GSK-3β, 5-HT3, ABCG2, OCT-1, PDK1, MSK1, MAPKAP-K1, S6K1, Chk1, PKA, DYRK1 |
| BIM-II | PKC, ABCG2, OCT-1, PDK1, MSK1, MAPKAP-K1, PKA, DYRK1 |
| BIM-III | PKC, ABCG2, OCT-1, S6K1, MAPKAPK1, RSK2, MSK1, PDK1 |
| BIM-IV | PKC, PKA, MAPKAPK1, MSK1, ABCG2 |
| BIM-V | ABCG2, SK6 |
| BIM-VI | OCT-1 |
| BIM-VII | OCT-1 |
| BIM-VIII | PKC, GSK-3β, carbachol-evoked noradrenaline release, OCT-1, PDK1, MSK1, S6K1, PKA, DYRK1, MAPKAPK1 |
| BIM-IX | PKC, GSK-3β, MAPKAPK1, MSK1, OCT-1, S6K1 |
| BIM-X | Multiple Protein Kinases |
| BIM-XI | PKC, T-cell activation |
| BIM-Type Ligand | Kinase | Family | PDB | Reference |
|---|---|---|---|---|
| BIM-I | PDK1 | AGC | 1UU8 | [74] |
| PIM1 | CAMK | 1XWS | [81] | |
| PIM1 | CAMK | 2BIK | [82] | |
| PIM1 | CAMK | 2BIL | [83] | |
| PKC-1 | AGC | 1ZRZ | [84] | |
| BIM-II | PDK1 | AGC | 1UU7 | [74] |
| PDK1 | AGC | 3ORZ | [85] | |
| PDK1 | AGC | 3OTU | [85] | |
| BIM-III | PDK1 | AGC | 1UU9 | [74] |
| BIM-VII | DMPK1 | AGC | 2VD5 | [86] |
| BIM-VIII | PDK1 | AGC | 1UVR | [74] |
| Ruboxistaurin 61 | PDK1 | AGC | 1UU3 | [74] |
| PIM1 | CAMK | 2J2I | [87] | |
| BfIM 71 | GSK-3β | CMGC | 3SD0 | [88] |
 | PKC-β | AGC | 2I0E | [89] |
 | GSK-3β | CMGC | 2OW3 | [90] |
| Indolyl phenyl maleimide 94 | GSK-3β | CMGC | 1R0E | [91] |
| Indolyl pyrimidinyl maleimide 95 | JAK3 | TK | 3PJC | [92] |
| Indolyl pyrroloquinolinyl maleimide 96 | MET | TK | 3RHK | [93] |
| Indolyl quinazolinyl maleimide 97 | PKC-α | AGC | 3IW4 | [94] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cooney, L.N.; O’Shea, K.D.; Winfield, H.J.; Cahill, M.M.; Pierce, L.T.; McCarthy, F.O. Bisindolyl Maleimides and Indolylmaleimide Derivatives—A Review of Their Synthesis and Bioactivity. Pharmaceuticals 2023, 16, 1191. https://doi.org/10.3390/ph16091191
Cooney LN, O’Shea KD, Winfield HJ, Cahill MM, Pierce LT, McCarthy FO. Bisindolyl Maleimides and Indolylmaleimide Derivatives—A Review of Their Synthesis and Bioactivity. Pharmaceuticals. 2023; 16(9):1191. https://doi.org/10.3390/ph16091191
Chicago/Turabian StyleCooney, Louise N., Kevin D. O’Shea, Hannah J. Winfield, Michael M. Cahill, Larry T. Pierce, and Florence O. McCarthy. 2023. "Bisindolyl Maleimides and Indolylmaleimide Derivatives—A Review of Their Synthesis and Bioactivity" Pharmaceuticals 16, no. 9: 1191. https://doi.org/10.3390/ph16091191
APA StyleCooney, L. N., O’Shea, K. D., Winfield, H. J., Cahill, M. M., Pierce, L. T., & McCarthy, F. O. (2023). Bisindolyl Maleimides and Indolylmaleimide Derivatives—A Review of Their Synthesis and Bioactivity. Pharmaceuticals, 16(9), 1191. https://doi.org/10.3390/ph16091191