
Niraparib and Advanced Ovarian Cancer: A Beacon in the Non-BRCA Mutated Setting

 ,
,  , and
, and
Abstract
:1. Introduction
2. Pharmacodynamics and Pharmacokinetics of Niraparib
Dosage and Administration Route
3. Niraparib Dosage and Use in Special Populations
3.1. Renal and Liver Impairment
3.2. Old Patients
4. Therapeutic Efficacy of Niraparib
4.1. Maintenance Treatment of Recurrent, Platinum-Sensitive, Advanced Ovarian Cancer
4.2. First-Line Monotherapy Maintenance Treatment of Advanced Platinum-Sensitive Ovarian Cancer
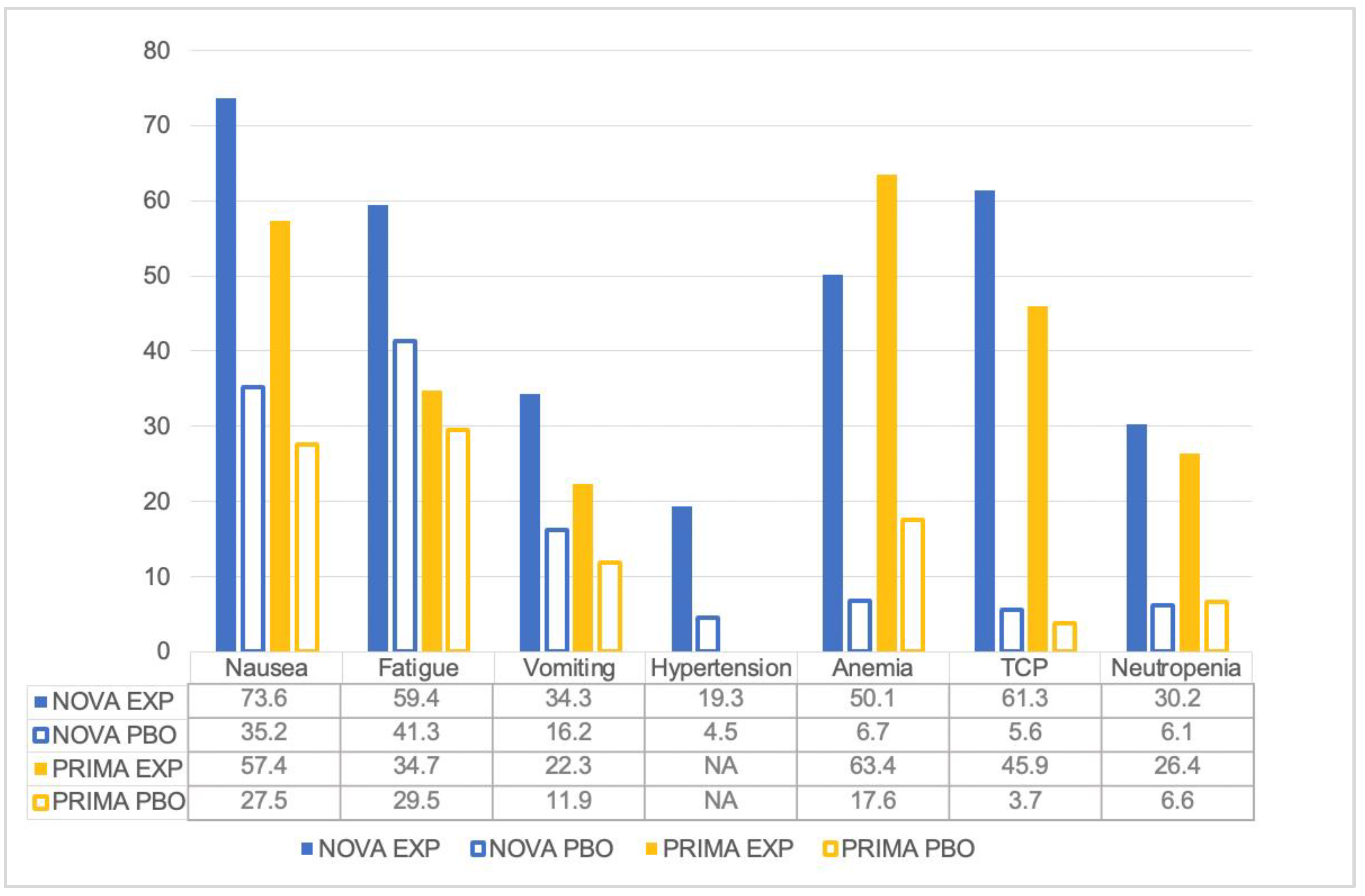
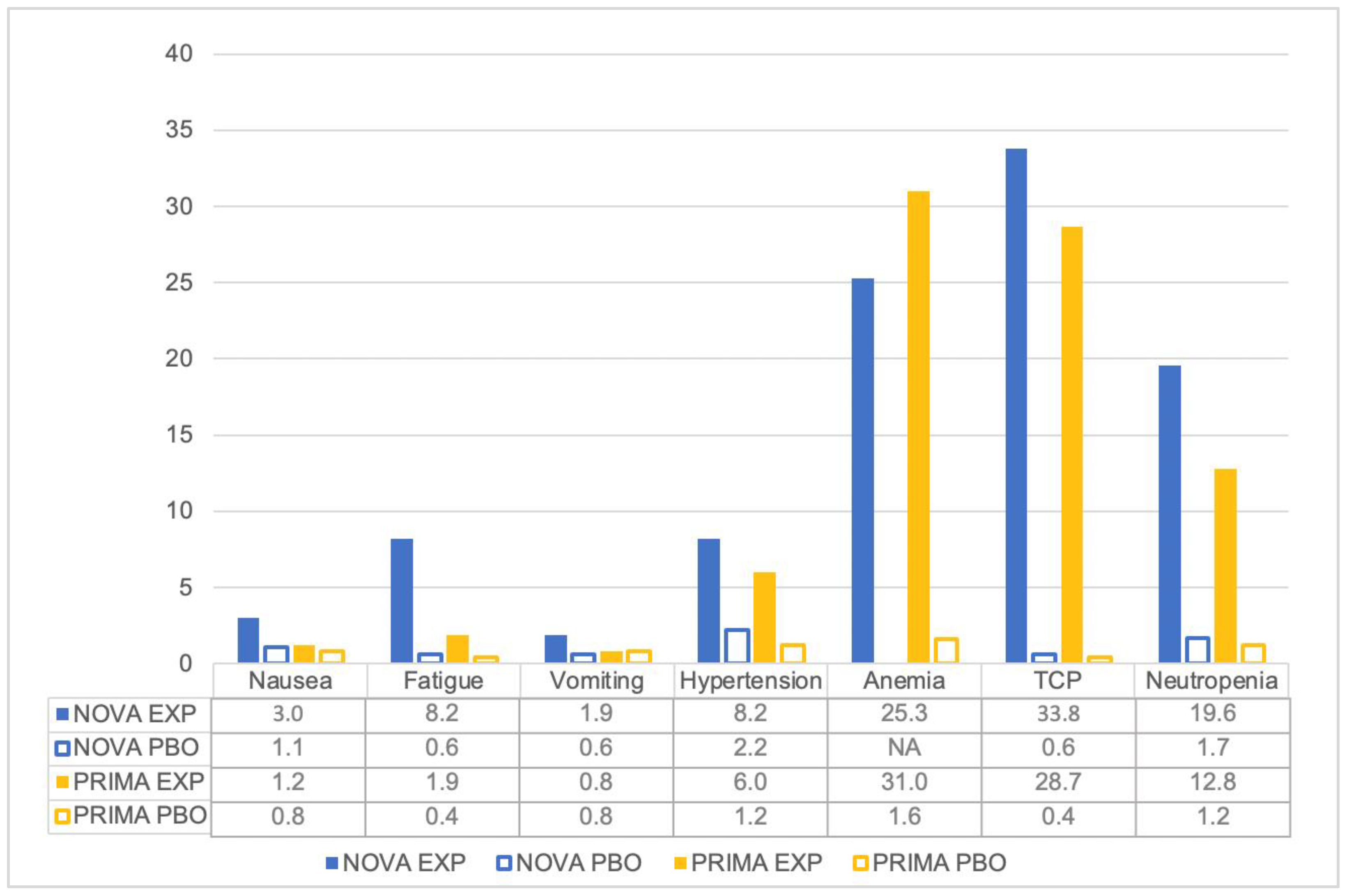
5. Tolerability of Niraparib
6. Future Perspectives and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics. CA A Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Cheung, A.; Shah, S.; Parker, J.; Soor, P.; Limbu, A.; Sheriff, M.; Boussios, S. Non-Epithelial Ovarian Cancers: How Much Do We Really Know? Int. J. Environ. Res. Public Health 2022, 19, 1106. [Google Scholar] [CrossRef] [PubMed]
- Pavlidis, N.; Rassy, E.; Vermorken, J.B.; Assi, T.; Kattan, J.; Boussios, S.; Smith-Gagen, J. The outcome of patients with serous papillary peritoneal cancer, fallopian tube cancer, and epithelial ovarian cancer by treatment eras: 27 years data from the SEER registry. Cancer Epidemiol. 2021, 75, 102045. [Google Scholar] [CrossRef] [PubMed]
- Piccart, M.J. Randomized Intergroup Trial of Cisplatin-Paclitaxel Versus Cisplatin-Cyclophosphamide in Women with Advanced Epithelial Ovarian Cancer: Three-Year Results. J. Natl. Cancer Inst. 2000, 92, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Ozols, R.F.; Bundy, B.N.; Greer, B.E.; Fowler, J.M.; Clarke-Pearson, D.; Burger, R.A.; Mannel, R.S.; DeGeest, K.; Hartenbach, E.M.; Baergen, R.; et al. Phase III Trial of Carboplatin and Paclitaxel Compared with Cisplatin and Paclitaxel in Patients with Optimally Resected Stage III Ovarian Cancer: A Gynecologic Oncology Group Study. JCO 2003, 21, 3194–3200. [Google Scholar] [CrossRef] [PubMed]
- Neijt, J.P.; Engelholm, S.A.; Tuxen, M.K.; Sørensen, P.G.; Hansen, M.; Sessa, C.; de Swart, C.A.M.; Hirsch, F.R.; Lund, B.; van Houwelingen, H.C. Exploratory Phase III Study of Paclitaxel and Cisplatin Versus Paclitaxel and Carboplatin in Advanced Ovarian Cancer. JCO 2000, 18, 3084–3092. [Google Scholar] [CrossRef]
- McGuire, W.P.; Hoskins, W.J.; Brady, M.F.; Kucera, P.R.; Partridge, E.E.; Look, K.Y.; Clarke-Pearson, D.L.; Davidson, M. Cyclophosphamide and Cisplatin Compared with Paclitaxel and Cisplatin in Patients with Stage III and Stage IV Ovarian Cancer. N. Engl. J. Med. 1996, 334, 1–6. [Google Scholar] [CrossRef]
- Walker, J.L.; Brady, M.F.; Wenzel, L.; Fleming, G.F.; Huang, H.Q.; DiSilvestro, P.A.; Fujiwara, K.; Alberts, D.S.; Zheng, W.; Tewari, K.S.; et al. Randomized Trial of Intravenous Versus Intraperitoneal Chemotherapy Plus Bevacizumab in Advanced Ovarian Carcinoma: An NRG Oncology/Gynecologic Oncology Group Study. JCO 2019, 37, 1380–1390. [Google Scholar] [CrossRef]
- Kehoe, S.; Hook, J.; Nankivell, M.; Jayson, G.C.; Kitchener, H.; Lopes, T.; Luesley, D.; Perren, T.; Bannoo, S.; Mascarenhas, M.; et al. Primary chemotherapy versus primary surgery for newly diagnosed advanced ovarian cancer (CHORUS): An open-label, randomised, controlled, non-inferiority trial. Lancet 2015, 386, 249–257. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. The DNA damage response and cancer therapy. Nature 2012, 481, 287–294. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Boussios, S.; Rassy, E.; Moschetta, M.; Ghose, A.; Adeleke, S.; Sanchez, E.; Sheriff, M.; Chargari, C.; Pavlidis, N. BRCA Mutations in Ovarian and Prostate Cancer: Bench to Bedside. Cancers 2022, 14, 3888. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef] [PubMed]
- FDA Approved Olaparib. Available online: https://www.fda.gov/drugs/fda-approved-olaparib-lynparza-astrazeneca-pharmaceuticals-lp-maintenance-treatment-adult-patients#:~:text=On%20December%2019%2C%202018%2C%20the,ovarian%2C%20fallopian%20tube%20or%20primary (accessed on 21 April 2023).
- Essel, K.G.; Moore, K.N. Niraparib for the treatment of ovarian cancer. Expert Rev. Anticancer Ther. 2018, 18, 727–733. [Google Scholar] [CrossRef]
- Longoria, T.C.; Tewari, K.S. Pharmacokinetic drug evaluation of niraparib for the treatment of ovarian cancer. Expert Opin. Drug Metab. Toxicol. 2018, 14, 543–550. [Google Scholar] [CrossRef]
- van Andel, L.; Zhang, Z.; Lu, S.; Kansra, V.; Agarwal, S.; Hughes, L.; Tibben, M.M.; Gebretensae, A.; Lucas, L.; Hillebrand, M.J.X.; et al. Human mass balance study and metabolite profiling of 14C-niraparib, a novel poly(ADP-Ribose) polymerase (PARP)-1 and PARP-2 inhibitor, in patients with advanced cancer. Investig. New Drugs 2017, 35, 751–765. [Google Scholar] [CrossRef]
- Zejula, Niraparib, European Medicines Agency. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/zejula (accessed on 21 April 2023).
- Lee, A. Niraparib: A Review in First-Line Maintenance Therapy in Advanced Ovarian Cancer. Targ. Oncol. 2021, 16, 839–845. [Google Scholar] [CrossRef]
- Sandhu, S.K.; Schelman, W.R.; Wilding, G.; Moreno, V.; Baird, R.D.; Miranda, S.; Hylands, L.; Riisnaes, R.; Forster, M.; Omlin, A.; et al. The poly(ADP-ribose) polymerase inhibitor niraparib (MK4827) in BRCA mutation carriers and patients with sporadic cancer: A phase 1 dose-escalation trial. Lancet Oncol. 2013, 14, 882–892. [Google Scholar] [CrossRef]
- Zejula, Niraparib, Food and Drug Administration. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/208447s015s017lbledt.pdf (accessed on 21 April 2023).
- Akce, M.; El-Khoueiry, A.; Piha-Paul, S.A.; Bacque, E.; Pan, P.; Zhang, Z.-Y.; Ewesuedo, R.; Gupta, D.; Tang, Y.; Milton, A.; et al. Pharmacokinetics and safety of niraparib in patients with moderate hepatic impairment. Cancer Chemother. Pharmacol. 2021, 88, 825–836. [Google Scholar] [CrossRef]
- Zhao, D.; Long, X.; Wang, J. Dose Adjustment of Poly (ADP Ribose) Polymerase Inhibitors in Patients with Hepatic or Renal Impairment. DDDT 2022, 16, 3947–3955. [Google Scholar] [CrossRef]
- González-Martín, A.; Pothuri, B.; Vergote, I.; DePont Christensen, R.; Graybill, W.; Mirza, M.R.; McCormick, C.; Lorusso, D.; Hoskins, P.; Freyer, G.; et al. Niraparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2391–2402. [Google Scholar] [CrossRef] [PubMed]
- Valabrega, G.; Pothuri, B.; Oaknin, A.; Graybill, W.; Sánchez, A.; Mccormick, C.; Baurain, J.-F.; Hoskins, P.; Denys, H.; O’cearbhaill, R.; et al. 819P Efficacy and safety of niraparib in older patients (pts) with advanced ovarian cancer (OC): Results from the PRIMA/ENGOT-OV26/GOG-3012 trial. Ann. Oncol. 2020, 31, S619. [Google Scholar] [CrossRef]
- Mirza, M.R.; Monk, B.J.; Herrstedt, J.; Oza, A.M.; Mahner, S.; Redondo, A.; Fabbro, M.; Ledermann, J.A.; Lorusso, D.; Vergote, I.; et al. Niraparib Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer. N. Engl. J. Med. 2016, 375, 2154–2164. [Google Scholar] [CrossRef]
- Fabbro, M.; Moore, K.; Dørum, A.; Tinker, A.; Mahner, S.; Bover, I.; Banerjee, S.; Tognon, G.; Goffin, F.; Shapira-Frommer, R.; et al. Safety and Efficacy of Niraparib in Elderly Patients (Pts) with Recurrent Ovarian Cancer (OC). Ann. Oncol. 2017, 28, v332. [Google Scholar] [CrossRef]
- Wu, X.; Zhu, J.; Yin, R.; Yang, J.; Liu, J.; Wang, J.; Wu, L.; Liu, Z.; Gao, Y.; Wang, D.; et al. Niraparib maintenance therapy in patients with platinum-sensitive recurrent ovarian cancer using an individualized starting dose (NORA): A randomized, double-blind, placebo-controlled phase III trial☆. Ann. Oncol. 2021, 32, 512–521. [Google Scholar] [CrossRef]
- Maiorano, B.A.; Maiorano, M.F.P.; Lorusso, D.; Di Maio, M.; Maiello, E. Efficacy and safety of PARP inhibitors in elderly patients with advanced ovarian cancer: A systematic review and meta-analysis. Int. J. Gynecol. Cancer 2022, 32, 1410–1418. [Google Scholar] [CrossRef]
- del Campo, J.M.; Matulonis, U.A.; Malander, S.; Provencher, D.; Mahner, S.; Follana, P.; Waters, J.; Berek, J.S.; Woie, K.; Oza, A.M.; et al. Niraparib Maintenance Therapy in Patients With Recurrent Ovarian Cancer After a Partial Response to the Last Platinum-Based Chemotherapy in the ENGOT-OV16/NOVA Trial. JCO 2019, 37, 2968–2973. [Google Scholar] [CrossRef]
- Matulonis, U.; Herrstedt, J.; Oza, A.; Mahner, S.; Redondo, A.; Berton, D.; Berek, J.; Lund, B.; Marmé, F.; González-Martín, A.; et al. Long-term safety and secondary efficacy endpoints in the ENGOT-OV16/NOVA phase 3 trial of niraparib in recurrent ovarian cancer. In Proceedings of the Society of Gynecological Oncology 2021 Virtual Annual Meeting on Women’s Cancer, Virtual, 19–21 March 2021; Abstract 37. [Google Scholar]
- Matulonis, U.A.; Herrstedt, J.; Oza, A. Final overall survival and long-term safety in the ENGOT-OV16/NOVA phase III trial of niraparib in patients with recurrent ovarian cancer. In Proceedings of the SGO 2023, Tampa, FL, USA, 25–28 March 2023. [Google Scholar]
- Berek, J.S.; Matulonis, U.A.; Peen, U.; Ghatage, P.; Mahner, S.; Redondo, A.; Lesoin, A.; Colombo, N.; Vergote, I.; Rosengarten, O.; et al. Safety and dose modification for patients receiving niraparib. Ann. Oncol. 2018, 29, 1784–1792. [Google Scholar] [CrossRef]
- Wang, S.; Woodgate, S.; Potter, J.; Lawo, S.; Mikule, K. Evaluation of clinical-stage PARP inhibitors in cell-based assays to correlate PARP suppression with functional impact on DNA repair. Eur. J. Cancer 2016, 69, S123–S124. [Google Scholar] [CrossRef]
- Gonzalez, A.; Mirza, M.; Vergote, I.; Li, Y.; Hazard, S.; Clark, R.; Graybill, W.; Pothuri, B.; Monk, B. A prospective evaluation of tolerability of niraparib dosing based upon baseline body weight (wt) and platelet (blplt) count: Blinded pooled interim safety data from the PRIMA Study. Ann. Oncol. 2018, 29, viii335–viii336. [Google Scholar] [CrossRef]
- LaFargue, C.J.; Dal Molin, G.Z.; Sood, A.K.; Coleman, R.L. Exploring and comparing adverse events between PARP inhibitors. Lancet Oncol. 2019, 20, e15–e28. [Google Scholar] [CrossRef] [PubMed]
- Perrotta, I.; Brunelli, E.; Sciangula, A.; Conforti, F.; Perrotta, E.; Tripepi, S.; Donato, G.; Cassese, M. iNOS induction and PARP-1 activation in human atherosclerotic lesions: An immunohistochemical and ultrastructural approach. Cardiovasc. Pathol. 2011, 20, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, L.; Zhang, F.; Zhang, C.; Deng, S.; Wang, R.; Zhang, Y.; Huang, D.; Huang, K. Inhibition of PARP prevents angiotensin II-induced aortic fibrosis in rats. Int. J. Cardiol. 2013, 167, 2285–2293. [Google Scholar] [CrossRef]
- Morice, P.M.; Ray-Coquard, I.; Moore, K.N.; Diéras, V.; Alexandre, J. PARP inhibitors and newly second primary malignancies in cancer patients: A systematic review and safety meta-analysis of placebo randomized controlled trials. Ann. Oncol. 2021, 32, 1048–1050. [Google Scholar] [CrossRef]
- Korenaga, T.K.; Tewari, K.S. Gynecologic cancer in pregnancy. Gynecol. Oncol. 2020, 157, 799–809. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Zhu, J.; Yin, R.; Wang, J.; Pan, L.; Kong, B.; Zheng, H.; Liu, J.; Wu, X.; Wang, L.; et al. Treatment With Niraparib Maintenance Therapy in Patients With Newly Diagnosed Advanced Ovarian Cancer: A Phase 3 Randomized Clinical Trial. JAMA Oncol. 2023. [Google Scholar] [CrossRef]
- Maiorano, B.A.; Lorusso, D.; Maiorano, M.F.P.; Ciardiello, D.; Parrella, P.; Petracca, A.; Cormio, G.; Maiello, E. The Interplay between PARP Inhibitors and Immunotherapy in Ovarian Cancer: The Rationale behind a New Combination Therapy. Int. J. Mol. Sci. 2022, 23, 3871. [Google Scholar] [CrossRef]
- Revythis, A.; Limbu, A.; Mikropoulos, C.; Ghose, A.; Sanchez, E.; Sheriff, M.; Boussios, S. Recent Insights into PARP and Immuno-Checkpoint Inhibitors in Epithelial Ovarian Cancer. Int. J. Environ. Res. Public Health 2022, 19, 8577. [Google Scholar] [CrossRef] [PubMed]
- Konstantinopoulos, P.A.; Waggoner, S.E.; Vidal, G.A.; Mita, M.M.; Fleming, G.F.; Holloway, R.W.; Van Le, L.; Sachdev, J.C.; Chapman-Davis, E.; Colon-Otero, G.; et al. TOPACIO/Keynote-162 (NCT02657889): A phase 1/2 study of niraparib + pembrolizumab in patients (pts) with advanced triple-negative breast cancer or recurrent ovarian cancer (ROC)—Results from ROC cohort. JCO 2018, 36 (Suppl. S15), 106. [Google Scholar] [CrossRef]
- Aliyuda, F.; Moschetta, M.; Ghose, A.; Sofia Rallis, K.; Sheriff, M.; Sanchez, E.; Rassy, E.; Boussios, S. Advances in Ovarian Cancer Treatment Beyond PARP Inhibitors. Curr. Cancer Drug Targets 2023, 23, 433–446. [Google Scholar] [CrossRef]
- Tentori, L.; Lacal, P.M.; Muzi, A.; Dorio, A.S.; Leonetti, C.; Scarsella, M.; Ruffini, F.; Xu, W.; Min, W.; Stoppacciaro, A.; et al. Poly(ADP-ribose) polymerase (PARP) inhibition or PARP-1 gene deletion reduces angiogenesis. Eur. J. Cancer 2007, 43, 2124–2133. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.; Yang, K.; Taylor-Harding, B.; Wiedemeyer, W.R.; Buckanovich, R.J. VEGFR3 Inhibition Chemosensitizes Ovarian Cancer Stemlike Cells through Down-Regulation of BRCA1 and BRCA2. Neoplasia 2014, 16, 343–353.e2. [Google Scholar] [CrossRef]
- Bindra, R.S.; Gibson, S.L.; Meng, A.; Westermark, U.; Jasin, M.; Pierce, A.J.; Bristow, R.G.; Classon, M.K.; Glazer, P.M. Hypoxia-Induced Down-regulation of BRCA1 Expression by E2Fs. Cancer Res. 2005, 65, 11597–11604. [Google Scholar] [CrossRef] [PubMed]
- Mirza, M.R.; Lundqvist, E.; Birrer, M.J.; Christensen, R.D.; Nyvang, G.-B.; Malander, S.; Anttila, M.; Werner, T.L.; Lund, B.; Lindahl, G.; et al. Niraparib plus bevacizumab versus niraparib alone for platinum-sensitive recurrent ovarian cancer (NSGO-AVANOVA2/ENGOT-ov24): A randomised, phase 2, superiority trial. Lancet Oncol. 2019, 20, 1409–1419. [Google Scholar] [CrossRef] [PubMed]
- Hardesty, M.M.; Krivak, T.C.; Wright, G.S.; Hamilton, E.; Fleming, E.L.; Belotte, J.; Keeton, E.K.; Wang, P.; Gupta, D.; Clements, A.; et al. OVARIO phase II trial of combination niraparib plus bevacizumab maintenance therapy in advanced ovarian cancer following first-line platinum-based chemotherapy with bevacizumab. Gynecol. Oncol. 2022, 166, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Lim, M.C.; Lee, J.-K.; Jeong, D.H.; Kim, S.I.; Choi, M.C.; Kim, B.-G.; Lee, J.-Y. A single-arm, phase II study of niraparib and bevacizumab maintenance therapy in platinum-sensitive, recurrent ovarian cancer patients previously treated with a PARP inhibitor: Korean Gynecologic Oncology Group (KGOG 3056)/NIRVANA-R trial. J. Gynecol. Oncol. 2022, 33, e12. [Google Scholar] [CrossRef]
- Liu, G.; Feng, Y.; Li, J.; Deng, T.; Yin, A.; Yan, L.; Zheng, M.; Xiong, Y.; Li, J.; Huang, Y.; et al. A novel combination of niraparib and anlotinib in platinum-resistant ovarian cancer: Efficacy and safety results from the phase II, multi-center ANNIE study. eClinicalMedicine 2022, 54, 101767. [Google Scholar] [CrossRef]
- Maiorano, B.A.; Maiorano, M.F.P.; Maiello, E. Olaparib and advanced ovarian cancer: Summary of the past and looking into the future. Front. Pharmacol. 2023, 14, 1162665. [Google Scholar] [CrossRef]
- Pujade-Lauraine, E.; Selle, F.; Scambia, G.; Asselain, B.; Marmé, F.; Lindemann, K.; Colombo, N.; Madry, R.; Glasspool, R.; Dubot, C.; et al. LBA33 maintenance olaparib rechallenge in patients (pts) with ovarian carcinoma (OC) previously treated with a PARP inhibitor (PARPi): Phase IIIb OReO/ENGOT Ov-38 trial. Ann. Oncol. 2021, 32, S1308–S1309. [Google Scholar] [CrossRef]
- Frenel, J.S.; Kim, J.W.; Aryal, N.; Asher, R.; Berton, D.; Vidal, L.; Pautier, P.; Ledermann, J.A.; Penson, R.T.; Oza, A.M.; et al. Efficacy of subsequent chemotherapy for patients with BRCA1/2-mutated recurrent epithelial ovarian cancer progressing on olaparib versus placebo maintenance: Post-hoc analyses of the SOLO2/ENGOT Ov-21 trial. Ann. Oncol. 2022, 33, 1021–1028. [Google Scholar] [CrossRef]
- Coleman, R.L.; Oza, A.; Lorusso, D.; Aghajanian, C.; Oaknin, A.; Dean, A.; Colombo, N.; Weberpals, J.; Clamp, A.; Scambia, G.; et al. Overall survival results from ARIEL3: A phase 3 randomized, double-blind study of rucaparib vs placebo following response to platinum-based chemotherapy for recurrent ovarian carcinoma. In Proceedings of the IGCS 2022 Annual Global Meeting, New York, NY, USA, 29 September–1 October 2022. [Google Scholar]
- Ghose, A.; Gullapalli, S.V.N.; Chohan, N.; Bolina, A.; Moschetta, M.; Rassy, E.; Boussios, S. Applications of Proteomics in Ovarian Cancer: Dawn of a New Era. Proteomes 2022, 10, 16. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Study Name (NCT)—Year | Phase | Target Population (Number of pts) | Niraparib Administration Protocol | Primary EP | Results | |
|---|---|---|---|---|---|---|
| mPFS | mOS | |||||
| Maintenance treatment of PS-ROC | ||||||
| ENGOT-OV16/NOVA (NCT01847274)—2017 | III | PS-ROC (n = 553) niraparib arm (n = 367) PBO arm (n = 179) gBRCAm (n = 203) Non-gBRCA (n = 350) | 300 mg OD | mPFS | gBRCAm subgroup: 21.0 mos vs. 5.5 mos (HR 0.27; 95% CI 0.2–0.4; p < 0.0001) Non-gBRCA subgroup: 9.3 mos vs. 3.9 mos (HR 0.45; 95% CI, 0.34 to 0.61; p < 0.0001) | gBRCAm subgroup: 40.9 vs. 38.1 mos (HR 0.85, 95% CI 0.61–1.20) Non-gBRCA subgroup: 31.0 vs. 34.8 mos (HR 1.06, 95% CI 0.81–1.37) |
| First-line maintenance in newly diagnosed platinum-sensitive OC | ||||||
| PRIMA/ENGOT-OV26/GOG-3012 (NCT02655016)—2020 | III | Overall (n = 733) HRD+ (n = 373) niraparib arm (n = 487) PBO (n = 246) | 300 mg OD (initial protocol) 200 mg OD (from Nov 2017) | mPFS | Overall: mPFS 13.8 mos vs. 8.2 mos (HR 0.62; 95% CI, 0.50–0.76, p < 0.001) HRD+: mPFS 21.9 mos vs. 10.4 mos (HR 0.43; 95% CI, 0.31–0.59, p < 0.001) | Overall: 24-mos OS rate 84% vs. 77% (HR 0.70; 95% CI, 0.44–1.11). |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maiorano, M.F.P.; Maiorano, B.A.; Biancofiore, A.; Cormio, G.; Maiello, E. Niraparib and Advanced Ovarian Cancer: A Beacon in the Non-BRCA Mutated Setting. Pharmaceuticals 2023, 16, 1261. https://doi.org/10.3390/ph16091261
Maiorano MFP, Maiorano BA, Biancofiore A, Cormio G, Maiello E. Niraparib and Advanced Ovarian Cancer: A Beacon in the Non-BRCA Mutated Setting. Pharmaceuticals. 2023; 16(9):1261. https://doi.org/10.3390/ph16091261
Chicago/Turabian StyleMaiorano, Mauro Francesco Pio, Brigida Anna Maiorano, Annalucia Biancofiore, Gennaro Cormio, and Evaristo Maiello. 2023. "Niraparib and Advanced Ovarian Cancer: A Beacon in the Non-BRCA Mutated Setting" Pharmaceuticals 16, no. 9: 1261. https://doi.org/10.3390/ph16091261
APA StyleMaiorano, M. F. P., Maiorano, B. A., Biancofiore, A., Cormio, G., & Maiello, E. (2023). Niraparib and Advanced Ovarian Cancer: A Beacon in the Non-BRCA Mutated Setting. Pharmaceuticals, 16(9), 1261. https://doi.org/10.3390/ph16091261