
Revolutionizing Medicinal Chemistry: The Application of Artificial Intelligence (AI) in Early Drug Discovery


Abstract
:1. Introduction
2. AI/ML Algorithms and Bio Big Data Utilized in Drug Discovery Research
2.1. Overview of ML Algorithms
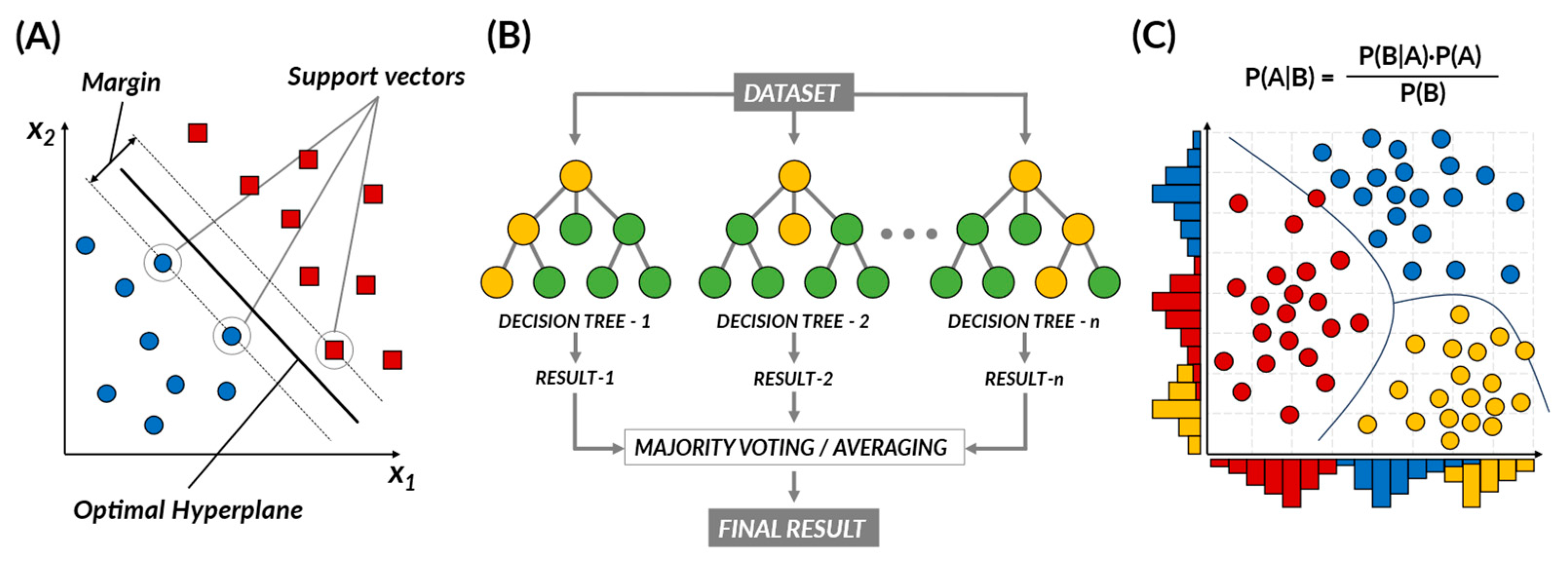
2.1.1. Supervised Learning
- (1)
- Support Vector Machine (SVM)
- (2)
- Naïve Bayes
- (3)
- Random Forest (RF)
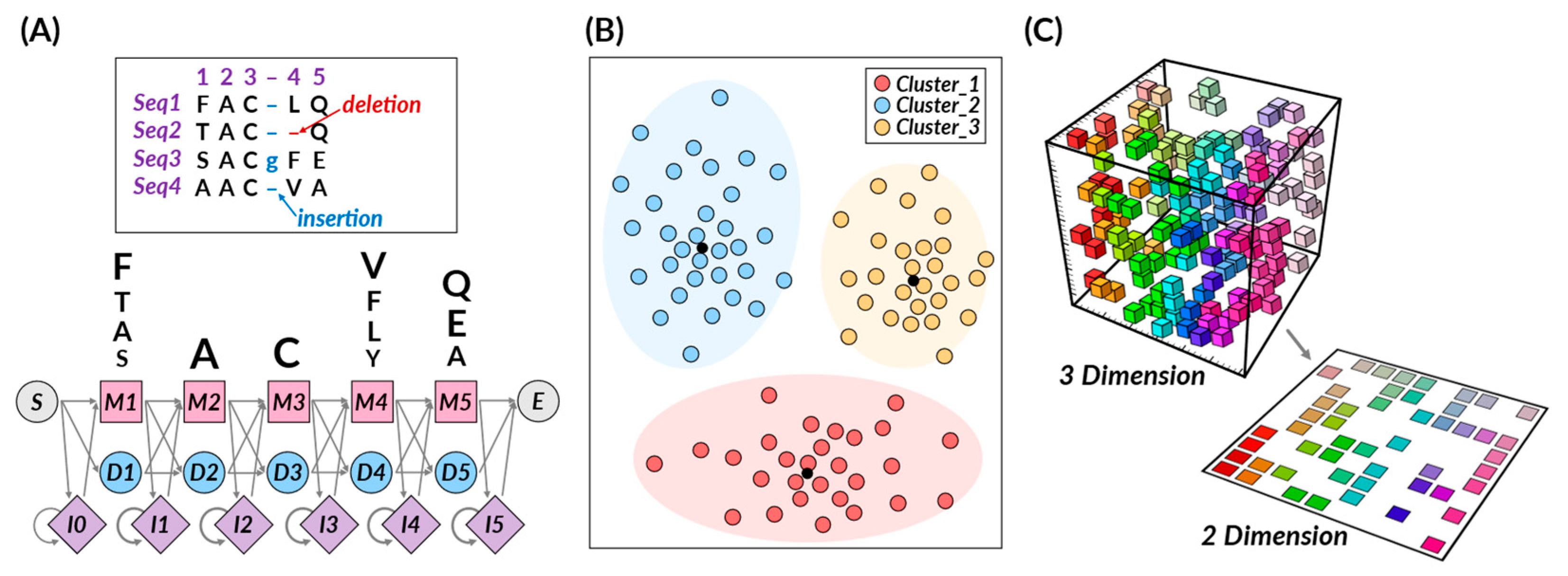
2.1.2. Unsupervised Learning
- (1)
- Hidden Markov Models (HMMs)
- (2)
- K-means Clustering
- (3)
- T-Distributed Stochastic Neighbor Embedding (t-SNE)
2.1.3. Reinforcement Learning
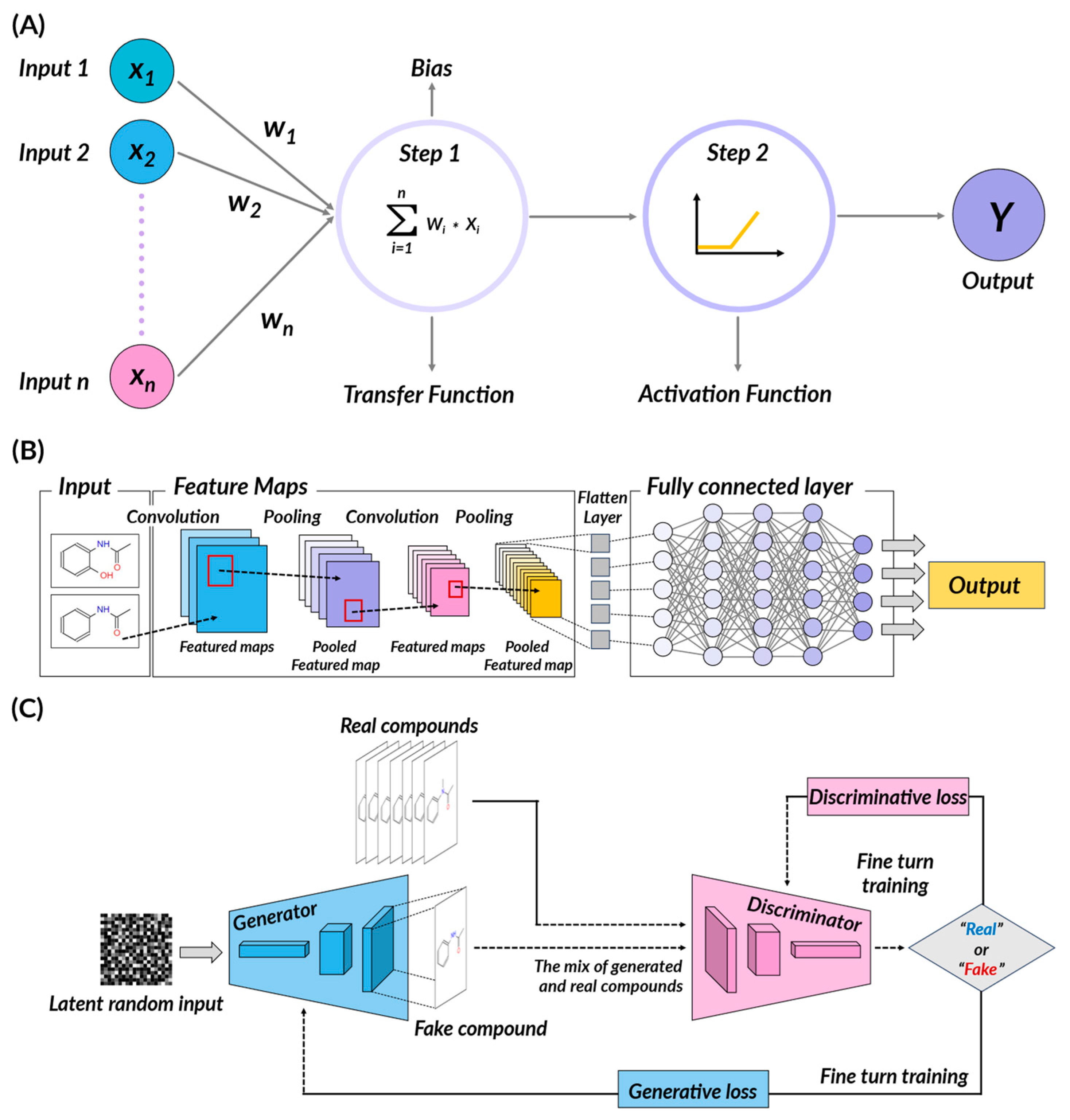
2.2. Deep Learning Method
- (1)
- Convolutional Neural Networks (CNNs)
- (2)
- RecurrentNeural Networks (RNNs)
- (3)
- Deep Belief Networks (DBNs)
- (4)
- Autoencoders
- (5)
- Generative Adversarial Networks (GANs)
2.3. Performance Metrics
- (1)
- Metrics for Classification Models
- (2)
- Metrics for Regression Models
2.4. Databases in Drug Research
3. AI in Structural Biology
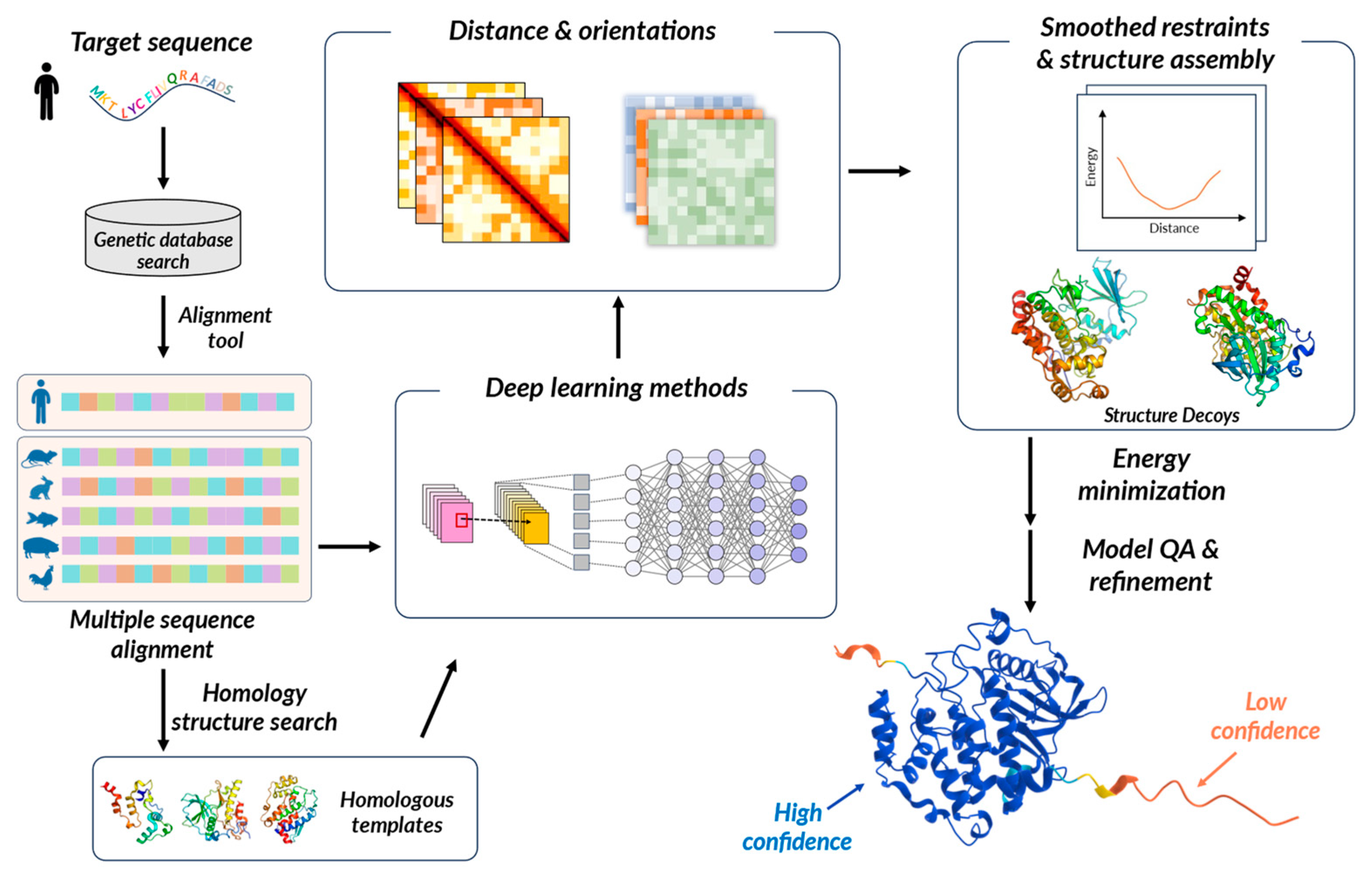
3.1. Protein Folding and Its Prediction
3.2. Biomolecular Structure Prediction by Computational Methods
3.3. Advancements in Protein Structural Research through AI
4. AI in Medicinal Chemistry or Cheminformatics
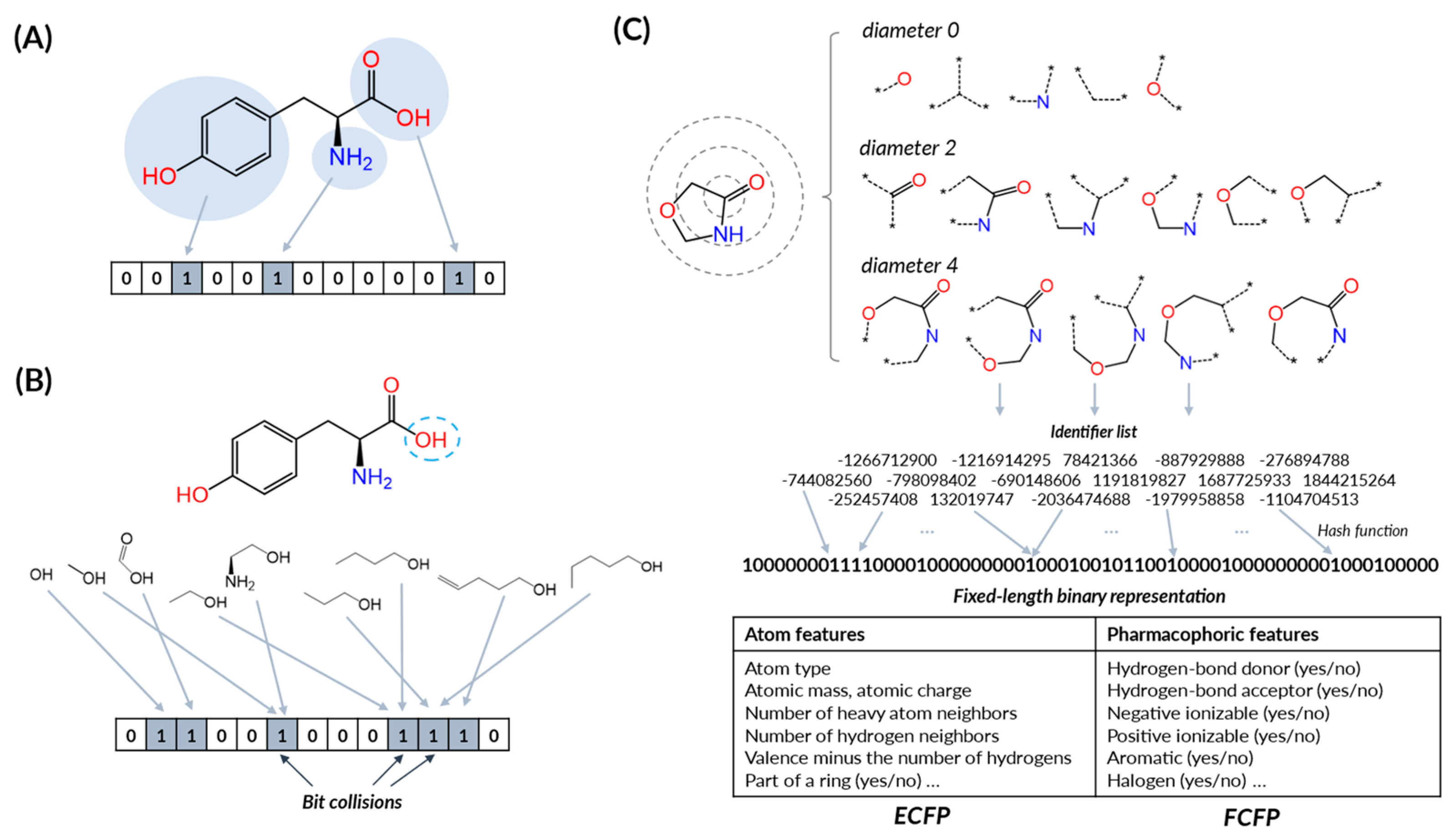
4.1. Molecular Fingerprints

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Molecular Fingerprint | Description | Ref. |
|---|---|---|---|
| Substructure key-based | Molecular ACCess system (MACCS) keys |
| [157] |
| PubChem fingerprint |
| [158] | |
| Topological | Daylight fingerprint |
| [150] |
| Atom pairs2D fingerprints (APFP) |
| [159] | |
| Circular | Extended-connectivity fingerprints (ECFP) |
| [152] |
| Molprint2D |
| [160] | |
| Pharmacophoric | Functional-class fingerprints (FCFP) |
| [152] |
| SMILES-based | SMIfp |
| [154] |
4.2. Deep Generative Model for Molecular Design
4.3. Prediction of Drug–Target Interaction (DTI)
4.4. Toxicity Prediction
- (1)
- Prediction of Hepatotoxicity
- (2)
- Prediction of Cardiotoxicity
- (3)
- Prediction of Mutagenicity and Carcinogenicity
5. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chen, W.; Liu, X.; Zhang, S.; Chen, S. Artificial intelligence for drug discovery: Resources, methods, and applications. Mol. Ther. Nucl. Acids 2023, 31, 691–702. [Google Scholar] [CrossRef] [PubMed]
- Cifci, M.A. A Deep Learning-Based Framework for Uncertainty Quantification in Medical Imaging Using the DropWeak Technique: An Empirical Study with Baresnet. Diagnostics 2023, 13, 800. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.H.; Zhang, Q.X. Deep Learning of Sparse Patterns in Medical IoT for Efficient Big Data Harnessing. IEEE Access 2023, 11, 25856–25864. [Google Scholar] [CrossRef]
- Alya, A.A. Artificial intelligence in drug design: Algorithms, applications, challenges and ethics. Future Drug Discov. 2021, 3, FDD59. [Google Scholar] [CrossRef]
- Gupta, R.; Srivastava, D.; Sahu, M.; Tiwari, S.; Ambasta, R.K.; Kumar, P. Artificial intelligence to deep learning: Machine intelligence approach for drug discovery. Mol. Divers 2021, 25, 1315–1360. [Google Scholar] [CrossRef]
- Zhu, H. Big Data and Artificial Intelligence Modeling for Drug Discovery. Annu. Rev. Pharmacol. 2020, 60, 573–589. [Google Scholar] [CrossRef]
- Hu, Y.; Lu, Y.; Wang, S.; Zhang, M.Y.; Qu, X.S.; Niu, B. Application of Machine Learning Approaches for the Design and Study of Anticancer Drugs. Curr. Drug Targets 2019, 20, 488–500. [Google Scholar] [CrossRef]
- Tong, X.C.; Liu, X.H.; Tan, X.Q.; Li, X.T.; Jiang, J.X.; Xiong, Z.P.; Xu, T.Y.; Jiang, H.L.; Qiao, N.; Zheng, M.Y. Generative Models for De Novo Drug Design. J. Med. Chem. 2021, 64, 14011–14027. [Google Scholar] [CrossRef]
- Cheng, Y.; Gong, Y.S.; Liu, Y.S.; Song, B.S.; Zou, Q. Molecular design in drug discovery: A comprehensive review of deep generative models. Brief Bioinform. 2021, 22, bbab344. [Google Scholar] [CrossRef]
- Xue, D.Y.; Gong, Y.K.; Yang, Z.Y.; Chuai, G.H.; Qu, S.; Shen, A.Z.; Yu, J.; Liu, Q. Advances and challenges in deep generative models for de novo molecule generation. Wires Comput. Mol. Sci. 2019, 9, e1395. [Google Scholar] [CrossRef]
- Vemula, D.; Jayasurya, P.; Sushmitha, V.; Kumar, Y.N.; Bhandari, V. CADD, AI and ML in drug discovery: A comprehensive review. Eur. J. Pharm. Sci. 2023, 181, 106324. [Google Scholar] [CrossRef] [PubMed]
- Cerchia, C.; Lavecchia, A. New avenues in artificial-intelligence-assisted drug discovery. Drug Discov. Today 2023, 28, 103516. [Google Scholar] [CrossRef] [PubMed]
- Dara, S.; Dhamercherla, S.; Jadav, S.S.; Babu, C.H.M.; Ahsan, M.J. Machine Learning in Drug Discovery: A Review. Artif. Intell. Rev. 2022, 55, 1947–1999. [Google Scholar] [CrossRef] [PubMed]
- Priya, S.; Tripathi, G.; Singh, D.B.; Jain, P.; Kumar, A. Machine learning approaches and their applications in drug discovery and design. Chem. Biol. Drug Des. 2022, 100, 136–153. [Google Scholar] [CrossRef]
- Guedes, I.A.; Barreto, A.M.S.; Marinho, D.; Krempser, E.; Kuenemann, M.A.; Sperandio, O.; Dardenne, L.E.; Miteva, M.A. New machine learning and physics-based scoring functions for drug discovery. Sci. Rep. 2021, 11, 3198. [Google Scholar] [CrossRef]
- Tamura, S.; Miyao, T.; Bajorath, J. Large-scale prediction of activity cliffs using machine and deep learning methods of increasing complexity. J. Cheminform. 2023, 15, 4. [Google Scholar] [CrossRef]
- Stumpfe, D.; Hu, H.B.; Bajorath, J. Advances in exploring activity cliffs. J. Comput. Aid. Mol. Des. 2020, 34, 929–942. [Google Scholar] [CrossRef]
- Heikamp, K.; Hu, X.Y.; Yan, A.X.; Bajorath, J. Prediction of Activity Cliffs Using Support Vector Machines. J. Chem. Inf. Model 2012, 52, 2354–2365. [Google Scholar] [CrossRef]
- Rodriguez-Perez, R.; Bajorath, J. Evolution of Support Vector Machine and Regression Modeling in Chemoinformatics and Drug Discovery. J. Comput. Aid. Mol. Des. 2022, 36, 355–362. [Google Scholar] [CrossRef]
- Warszycki, D.; Struski, L.; Smieja, M.; Kafel, R.; Kurczab, R. Pharmacoprint: A Combination of a Pharmacophore Fingerprint and Artificial Intelligence as a Tool for Computer-Aided Drug Design. J. Chem. Inf. Model 2021, 61, 5054–5065. [Google Scholar] [CrossRef]
- Jayaraj, P.B.; Jain, S. Ligand based virtual screening using SVM on GPU. Comput. Biol. Chem. 2019, 83, 107143. [Google Scholar] [CrossRef] [PubMed]
- Ogura, K.; Sato, T.; Tuki, H.; Honma, T. Support Vector Machine model for hERG inhibitory activities based on the integrated hERG database using descriptor selection by NSGA-II. Sci. Rep. 2019, 9, 12220. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Perez, R.; Vogt, M.; Bajorath, J. Support Vector Machine Classification and Regression Prioritize Different Structural Features for Binary Compound Activity and Potency Value Prediction. ACS Omega 2017, 2, 6371–6379. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Cruz, N.; Medina-Franco, J.L. Epigenetic Target Profiler: A Web Server to Predict Epigenetic Targets of Small Molecules. J. Chem. Inf. Model 2021, 61, 1550–1554. [Google Scholar] [CrossRef]
- Tong, Z.; Zhou, Y.; Wang, J. Identifying potential drug targets in hepatocellular carcinoma based on network analysis and one-class support vector machine. Sci. Rep. 2019, 9, 10442. [Google Scholar] [CrossRef]
- Kwon, S.; Bae, H.; Jo, J.; Yoon, S. Comprehensive ensemble in QSAR prediction for drug discovery. BMC Bioinform. 2019, 20, 521. [Google Scholar] [CrossRef]
- Hou, T.L.; Bian, Y.M.; McGuire, T.; Xie, X.Q. Integrated Multi-Class Classification and Prediction of GPCR Allosteric Modulators by Machine Learning Intelligence. Biomolecules 2021, 11, 870. [Google Scholar] [CrossRef]
- Kaiser, T.M.; Burger, P.B.; Butch, C.J.; Pelly, S.C.; Liotta, D.C. A Machine Learning Approach for Predicting HIV Reverse Transcriptase Mutation Susceptibility of Biologically Active Compounds. J. Chem. Inf. Model 2018, 58, 1544–1552. [Google Scholar] [CrossRef]
- Hu, J.; Zhou, L.W.; Li, B.; Zhang, X.L.; Chen, N.S. Improve hot region prediction by analyzing different machine learning algorithms. BMC Bioinform. 2021, 22, 522. [Google Scholar] [CrossRef]
- Celebi, R.; Uyar, H.; Yasar, E.; Gumus, O.; Dikenelli, O.; Dumontier, M. Evaluation of knowledge graph embedding approaches for drug-drug interaction prediction in realistic settings. BMC Bioinform. 2019, 20, 726. [Google Scholar] [CrossRef]
- Cai, C.P.; Guo, P.F.; Zhou, Y.D.; Zhou, J.W.; Wang, Q.; Zhang, F.X.; Fang, J.S.; Cheng, F.X. Deep Learning-Based Prediction of Drug-Induced Cardiotoxicity. J. Chem. Inf. Model 2019, 59, 1073–1084. [Google Scholar] [CrossRef] [PubMed]
- Madhukar, N.S.; Khade, P.K.; Huang, L.D.; Gayvert, K.; Galletti, G.; Stogniew, M.; Allen, J.E.; Giannakakou, P.; Elemento, O. A Bayesian machine learning approach for drug target identification using diverse data types. Nat. Commun. 2019, 10, 5221. [Google Scholar] [CrossRef]
- Saha, S.; Chatterjee, P.; Halder, A.K.; Nasipuri, M.; Basu, S.; Plewczynski, D. ML-DTD: Machine Learning-Based Drug Target Discovery for the Potential Treatment of COVID-19. Vaccines 2022, 10, 1643. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.K.A.; Malim, N.H.A.H. Comparative Studies on Resampling Techniques in Machine Learning and Deep Learning Models for Drug-Target Interaction Prediction. Molecules 2023, 28, 1663. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, K.A.; Huang, X.D. Machine Learning-based Virtual Screening and Its Applications to Alzheimer’s Drug Discovery: A Review. Curr. Pharm. Des. 2018, 24, 3347–3358. [Google Scholar] [CrossRef] [PubMed]
- Zakharov, A.V.; Varlamova, E.V.; Lagunin, A.A.; Dmitriev, A.V.; Muratov, E.N.; Fourches, D.; Kuz’min, V.E.; Poroikov, V.V.; Tropsha, A.; Nicklaus, M.C. QSAR Modeling and Prediction of Drug-Drug Interactions. Mol. Pharm. 2016, 13, 545–556. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.Y.; Lee, J.; Damjanovic, A.; Brooks, B.R. Protein pK(a) Prediction by Tree-Based Machine Learning. J. Chem. Theory Comput. 2022, 18, 2673–2686. [Google Scholar] [CrossRef]
- Cooper, K.; Baddeley, C.; French, B.; Gibson, K.; Golden, J.; Lee, T.; Pierre, S.; Weiss, B.; Yang, J. Novel Development of Predictive Feature Fingerprints to Identify Chemistry-Based Features for the Effective Drug Design of SARS-CoV-2 Target Antagonists and Inhibitors Using Machine Learning. ACS Omega 2021, 6, 4857–4877. [Google Scholar] [CrossRef]
- Brekkan, A.; Jonsson, S.; Karlsson, M.O.; Plan, E.L. Handling underlying discrete variables with bivariate mixed hidden Markov models in NONMEM. J. Pharmacokinet. Pharmacodyn. 2019, 46, 591–604. [Google Scholar] [CrossRef]
- Tamposis, I.A.; Tsirigos, K.D.; Theodoropoulou, M.C.; Kontou, P.I.; Bagos, P.G. Semi-supervised learning of Hidden Markov Models for biological sequence analysis. Bioinformatics 2019, 35, 2208–2215. [Google Scholar] [CrossRef]
- Steinegger, M.; Meier, M.; Mirdita, M.; Vohringer, H.; Haunsberger, S.J.; Soding, J. HH-suite3 for fast remote homology detection and deep protein annotation. BMC Bioinform. 2019, 20, 473. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.P.; Nute, M.; Mirarab, S.; Warnow, T. HIPPI: Highly accurate protein family classification with ensembles of HMMs. BMC Genom. 2016, 17, 765. [Google Scholar] [CrossRef] [PubMed]
- Li, J.F.; Lee, J.Y.; Liao, L. A new algorithm to train hidden Markov models for biological sequences with partial labels. BMC Bioinform. 2021, 22, 162. [Google Scholar] [CrossRef] [PubMed]
- Tamposis, I.A.; Tsirigos, K.D.; Theodoropoulou, M.C.; Kontou, P.I.; Tsaousis, G.N.; Sarantopoulou, D.; Litou, Z.I.; Bagos, P.G. JUCHMME: A Java Utility for Class Hidden Markov Models and Extensions for biological sequence analysis. Bioinformatics 2019, 35, 5309–5312. [Google Scholar] [CrossRef]
- Kaur, H.; Lynn, A.M. Mapping the FtsQBL divisome components in bacterial NTD pathogens as potential drug targets. Front. Genet. 2023, 13, 1010870. [Google Scholar] [CrossRef]
- Gupta, A.; Zhou, H.X. Machine Learning-Enabled Pipeline for Large-Scale Virtual Drug Screening. J. Chem. Inf. Model 2021, 61, 4236–4244. [Google Scholar] [CrossRef]
- David, L.; Thakkar, A.; Mercado, R.; Engkvist, O. Molecular representations in AI-driven drug discovery: A review and practical guide. J. Cheminform. 2020, 12, 56. [Google Scholar] [CrossRef]
- Madugula, S.S.; John, L.; Nagamani, S.; Gaur, A.S.; Poroikov, V.V.; Sastry, G.N. Molecular descriptor analysis of approved drugs using unsupervised learning for drug repurposing. Comput. Biol. Med. 2021, 138, 104856. [Google Scholar] [CrossRef]
- Huang, L.; Luo, H.M.; Li, S.N.; Wu, F.X.; Wang, J.X. Drug-drug similarity measure and its applications. Brief Bioinform. 2021, 22, bbaa265. [Google Scholar] [CrossRef]
- Nedyalkova, M.; Simeonov, V. Partitioning Pattern of Natural Products Based on Molecular Properties Descriptors Representing Drug-Likeness. Symmetry 2021, 13, 546. [Google Scholar] [CrossRef]
- McKay, K.; Hamilton, N.B.; Remington, J.M.; Schneebeli, S.T.; Li, J.N. Essential Dynamics Ensemble Docking for Structure-Based GPCR Drug Discovery. Front. Mol. Biosci. 2022, 9, 879212. [Google Scholar] [CrossRef]
- Chandak, T.; Mayginnes, J.P.; Mayes, H.; Wong, C.F. Using machine learning to improve ensemble docking for drug discovery. Proteins 2020, 88, 1263–1270. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.L.; Li, Q.; Sun, J.; Tan, S.H.; Tang, Y.H.; Zhao, M.M.; Li, Y.Y.; Cao, X.; Zhao, J.C.; Yang, J.K. Potential drug discovery for COVID-19 treatment targeting Cathepsin L using a deep learning-based strategy. Comput. Struct. Biotechnol. J. 2022, 20, 2442–2454. [Google Scholar] [CrossRef] [PubMed]
- Andronov, M.; Fedorov, M.V.; Sosnin, S. Exploring Chemical Reaction Space with Reaction Difference Fingerprints and Parametric t-SNE. ACS Omega 2021, 6, 30743–30751. [Google Scholar] [CrossRef]
- Thomas, M.; Smith, R.T.; O’Boyle, N.M.; de Graaf, C.; Bender, A. Comparison of structure- and ligand-based scoring functions for deep generative models: A GPCR case study. J. Cheminform. 2021, 13, 39. [Google Scholar] [CrossRef]
- Barnard, T.; Hagan, H.; Tseng, S.; Sosso, G.C. Less may be more: An informed reflection on molecular descriptors for drug design and discovery. Mol. Syst. Des. Eng. 2020, 5, 317–329. [Google Scholar] [CrossRef]
- Liu, G.N.; Singha, M.; Pu, L.M.; Neupane, P.; Feinstein, J.; Wu, H.C.; Ramanujam, J.; Brylinski, M. GraphDTI: A robust deep learning predictor of drug-target interactions from multiple heterogeneous data. J. Cheminform. 2021, 13, 58. [Google Scholar] [CrossRef]
- Xu, X.L.; Xie, Z.M.; Yang, Z.Y.; Li, D.F.; Xu, X.M. A t-SNE Based Classification Approach to Compositional Microbiome Data. Front. Genet. 2020, 11, 620143. [Google Scholar] [CrossRef]
- Karagiannaki, I.; Gourlia, K.; Lagani, V.; Pantazis, Y.; Tsamardinos, I. Learning biologically-interpretable latent representations for gene expression data. Mach. Learn. 2022, 1–31. [Google Scholar] [CrossRef]
- Zhang, L.P.; Tang, L.; Zhang, S.L.; Wang, Z.Z.; Shen, X.H.; Zhang, Z.Q. A Self-Adaptive Reinforcement-Exploration Q-Learning Algorithm. Symmetry 2021, 13, 1057. [Google Scholar] [CrossRef]
- Tang, B.W.; He, F.M.; Liu, D.P.; He, F.; Wu, T.; Fang, M.J.; Niu, Z.M.; Wu, Z.; Xu, D. AI-Aided Design of Novel Targeted Covalent Inhibitors against SARS-CoV-2. Biomolecules 2022, 12, 746. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.X.; Qian, Y.J.; Gao, H.Y.; Coley, C.W.; Mo, Y.M.; Barzilay, R.; Jensen, K.F. Towards efficient discovery of green synthetic pathways with Monte Carlo tree search and reinforcement learning. Chem. Sci. 2020, 11, 10959–10972. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.; Jang, G.H.; Kang, H.Y.; Song, G. Predicting aptamer sequences that interact with target proteins using an aptamer-protein interaction classifier and a Monte Carlo tree search approach. PLoS ONE 2021, 16, e0253760. [Google Scholar] [CrossRef] [PubMed]
- Yoshizawa, T.; Ishida, S.; Sato, T.; Ohta, M.; Honma, T.; Terayama, K. Selective Inhibitor Design for Kinase Homologs Using Multiobjective Monte Carlo Tree Search. J. Chem. Inf. Model 2022, 62, 5351–5360. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.B.; Pei, J.F.; Lai, L.H. Structure-based de novo drug design using 3D deep generative models. Chem. Sci. 2021, 12, 13664–13675. [Google Scholar] [CrossRef] [PubMed]
- Genheden, S.; Thakkar, A.; Chadimova, V.; Reymond, J.L.; Engkvist, O.; Bjerrum, E. AiZynthFinder: A fast, robust and flexible open-source software for retrosynthetic planning. J. Cheminform. 2020, 12, 70. [Google Scholar] [CrossRef]
- Skalic, M.; Martinez-Rosell, G.; Jimenez, J.; De Fabritiis, G. PlayMolecule BindScope: Large scale CNN-based virtual screening on the web. Bioinformatics 2019, 35, 1237–1238. [Google Scholar] [CrossRef]
- Haneczok, J.; Delijewski, M. Machine learning enabled identification of potential SARS-CoV-2 3CLpro inhibitors based on fixed molecular fingerprints and Graph-CNN neural representations. J. Biomed. Inform. 2021, 119, 103821. [Google Scholar] [CrossRef]
- Huo, X.; Xu, J.; Xu, M.; Chen, H. An improved 3D quantitative structure-activity relationships (QSAR) of molecules with CNN-based partial least squares model. Artif. Intell. Life Sci. 2023, 3, 100065. [Google Scholar] [CrossRef]
- Qian, Y.; Wu, J.; Zhang, Q. CAT-CPI: Combining CNN and transformer to learn compound image features for predicting compound-protein interactions. Front. Mol. Biosci. 2022, 9, 963912. [Google Scholar] [CrossRef]
- Jiang, M.J.; Wei, Z.Q.; Zhang, S.G.; Wang, S.; Wang, X.F.; Li, Z. FRSite: Protein drug binding site prediction based on faster R-CNN. J. Mol. Graph. Model. 2019, 93, 107454. [Google Scholar] [CrossRef] [PubMed]
- Hirohara, M.; Saito, Y.; Koda, Y.; Sato, K.; Sakakibara, Y. Convolutional neural network based on SMILES representation of compounds for detecting chemical motif. BMC Bioinform. 2018, 19, 526. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.W.; Zou, J.P.; Yu, J.L.; Shi, S.P. De novo drug design based on Stack-RNN with multi-objective reward-weighted sum and reinforcement learning. J. Mol. Model. 2023, 29, 121. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.N.; Yang, L.J.; Ding, N.; Li, G.W.; Cai, J.J.; An, X.L.; Wang, Z.J.; Qin, J.; Niu, Y.Z. Recurrent neural network (RNN) model accelerates the development of antibacterial metronidazole derivatives. RSC Adv. 2022, 12, 22893–22901. [Google Scholar] [CrossRef]
- Lim, J.; Ryu, S.; Park, K.; Choe, Y.J.; Ham, J.; Kim, W.Y. Predicting Drug-Target Interaction Using a Novel Graph Neural Network with 3D Structure-Embedded Graph Representation. J. Chem. Inf. Model. 2019, 59, 3981–3988. [Google Scholar] [CrossRef]
- Saldivar-Gonzalez, F.I.; Aldas-Bulos, V.D.; Medina-Franco, J.L.; Plisson, F. Natural product drug discovery in the artificial intelligence era. Chem. Sci. 2022, 13, 1526–1546. [Google Scholar] [CrossRef]
- Yang, H.; Hu, B.; Pan, X.; Yan, S.; Feng, Y.; Zhang, X.; Yin, L.; Hu, C. Deep belief network-based drug identification using near infrared spectroscopy. J. Innov. Opt. Health Sci. 2017, 10, 1630011. [Google Scholar] [CrossRef]
- Griffiths, R.R.; Hernandez-Lobato, J.M. Constrained Bayesian optimization for automatic chemical design using variational autoencoders. Chem. Sci. 2020, 11, 577–586. [Google Scholar] [CrossRef]
- Sajadi, S.Z.; Chahooki, M.A.Z.; Gharaghani, S.; Abbasi, K. AutoDTI plus plus: Deep unsupervised learning for DTI prediction by autoencoders. BMC Bioinform. 2021, 22, 204. [Google Scholar] [CrossRef]
- Zhang, Y.; Hu, Y.Q.; Li, H.H.; Liu, X.Y. Drug-protein interaction prediction via variational autoencoders and attention mechanisms. Front. Genet. 2022, 13, 1032779. [Google Scholar] [CrossRef]
- Song, T.; Ren, Y.Q.; Wang, S.; Han, P.F.; Wang, L.L.; Li, X.; Rodriguez-Paton, A. DNMG: Deep molecular generative model by fusion of 3D information for de novo drug design. Methods 2023, 211, 10–22. [Google Scholar] [CrossRef]
- Hussain, S.; Anees, A.; Das, A.; Nguyen, B.P.; Marzuki, M.; Lin, S.P.; Wright, G.; Singhal, A. High-content image generation for drug discovery using generative adversarial networks. Neural Netw. 2020, 132, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Li, K.; Shi, J. DGANDDI: Double Generative Adversarial Networks for Drug-Drug Interaction Prediction. IEEE/ACM Trans. Comput. Biol. Bioinform. 2023, 20, 1854–1863. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.L.; Wang, J.J.; Pang, L.; Liu, Y.; Zhang, J. GANsDTA: Predicting Drug-Target Binding Affinity Using GANs. Front. Genet. 2020, 10, 1243. [Google Scholar] [CrossRef] [PubMed]
- Hicks, S.A.; Strumke, I.; Thambawita, V.; Hammou, M.; Riegler, M.A.; Halvorsen, P.; Parasa, S. On evaluation metrics for medical applications of artificial intelligence. Sci. Rep. 2022, 12, 5979. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.J.; Gindulyte, A.; He, J.; He, S.Q.; Li, Q.L.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2023 update. Nucleic Acids Res. 2022, 51, D1373–D1380. [Google Scholar] [CrossRef]
- Mendez, D.; Gaulton, A.; Bento, A.P.; Chambers, J.; De Veij, M.; Felix, E.; Magarinos, M.P.; Mosquera, J.F.; Mutowo, P.; Nowotka, M.; et al. ChEMBL: Towards direct deposition of bioassay data. Nucleic Acids Res. 2019, 47, D930–D940. [Google Scholar] [CrossRef]
- Irwin, J.J.; Tang, K.G.; Young, J.; Dandarchuluun, C.; Wong, B.R.; Khurelbaatar, M.; Moroz, Y.S.; Mayfield, J.; Sayle, R.A. ZINC20-A Free Ultralarge-Scale Chemical Database for Ligand Discovery. J. Chem. Inf. Model. 2020, 60, 6065–6073. [Google Scholar] [CrossRef]
- Pence, H.E.; Williams, A. ChemSpider: An Online Chemical Information Resource. J. Chem. Educ. 2010, 87, 1123–1124. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Avram, S.; Wilson, T.B.; Curpan, R.; Halip, L.; Borota, A.; Bora, A.; Bologa, C.G.; Holmes, J.; Knockel, J.; Yang, J.J.; et al. DrugCentral 2023 extends human clinical data and integrates veterinary drugs. Nucleic Acids Res. 2022, 51, D1276–D1287. [Google Scholar] [CrossRef] [PubMed]
- Drugs@FDA: FDA-Approved Drugs. Available online: https://www.accessdata.fda.gov/scripts/cder/daf/ (accessed on 22 July 2023).
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Karp, P.D.; Billington, R.; Caspi, R.; Fulcher, C.A.; Latendresse, M.; Kothari, A.; Keseler, I.M.; Krummenacker, M.; Midford, P.E.; Ong, Q.; et al. The BioCyc collection of microbial genomes and metabolic pathways. Brief Bioinform. 2019, 20, 1085–1093. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, M.; Jassal, B.; Stephan, R.; Milacic, M.; Rothfels, K.; Senff-Ribeiro, A.; Griss, J.; Sevilla, C.; Matthews, L.; Gong, C.Q.; et al. The reactome pathway knowledgebase 2022. Nucleic Acids Res. 2022, 50, D687–D692. [Google Scholar] [CrossRef]
- Wishart, D.S.; Guo, A.C.; Oler, E.; Wang, F.; Anjum, A.; Peters, H.; Dizon, R.; Sayeeda, Z.; Tian, S.Y.; Lee, B.L.; et al. HMDB 5.0: The Human Metabolome Database for 2022. Nucleic Acids Res. 2022, 50, D622–D631. [Google Scholar] [CrossRef]
- Orchard, S.; Ammari, M.; Aranda, B.; Breuza, L.; Briganti, L.; Broackes-Carter, F.; Campbell, N.H.; Chavali, G.; Chen, C.; del-Toro, N.; et al. The MIntAct project-IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res. 2014, 42, D358–D363. [Google Scholar] [CrossRef]
- Oughtred, R.; Rust, J.; Chang, C.; Breitkreutz, B.J.; Stark, C.; Willems, A.; Boucher, L.; Leung, G.; Kolas, N.; Zhang, F.; et al. The BioGRID database: A comprehensive biomedical resource of curated protein, genetic, and chemical interactions. Protein Sci. 2021, 30, 187–200. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING database in 2023: Protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2022, 51, D638–D646. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Santos, A.; von Mering, C.; Jensen, L.J.; Bork, P.; Kuhn, M. STITCH 5: Augmenting protein-chemical interaction networks with tissue and affinity data. Nucleic Acids Res. 2016, 44, D380–D384. [Google Scholar] [CrossRef]
- Gilson, M.K.; Liu, T.Q.; Baitaluk, M.; Nicola, G.; Hwang, L.; Chong, J. BindingDB in 2015: A public database for medicinal chemistry, computational chemistry and systems pharmacology. Nucleic Acids Res. 2016, 44, D1045–D1053. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhang, Y.T.; Lian, X.C.; Li, F.C.; Wang, C.X.; Zhu, F.; Qiu, Y.Q.; Chen, Y.Z. Therapeutic target database update 2022: Facilitating drug discovery with enriched comparative data of targeted agents. Nucleic Acids Res. 2022, 50, D1398–D1407. [Google Scholar] [CrossRef]
- Harding, S.D.; Armstrong, J.F.; Faccenda, E.; Southan, C.; Alexander, S.P.H.; Davenport, A.P.; Pawson, A.J.; Spedding, M.; Davies, J.A.; NC-IUPHAR. The IUPHAR/BPS guide to PHARMACOLOGY in 2022: Curating pharmacology for COVID-19, malaria and antibacterials. Nucleic Acids Res. 2022, 50, D1282–D1294. [Google Scholar] [CrossRef] [PubMed]
- Freshour, S.L.; Kiwala, S.; Cotto, K.C.; Coffman, A.C.; McMichael, J.F.; Song, J.J.; Griffith, M.; Griffith, O.L.; Wagner, A.H. Integration of the Drug-Gene Interaction Database (DGIdb 4.0) with open crowdsource efforts. Nucleic Acids Res. 2021, 49, D1144–D1151. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.P.; Wiegers, T.C.; Johnson, R.J.; Sciaky, D.; Wiegers, J.; Mattingly, C.J. Comparative Toxicogenomics Database (CTD): Update 2023. Nucleic Acids Res. 2022, 51, D1257–D1262. [Google Scholar] [CrossRef] [PubMed]
- Ganter, B.; Snyder, R.D.; Halbert, D.N.; Lee, M.D. Toxicogenomics in drug discovery and development: Mechanistic analysis of compound/class-dependent effects using the DrugMatrix® database. Pharmacogenomics 2006, 7, 1025–1044. [Google Scholar] [CrossRef] [PubMed]
- OECD eChemPortal. Available online: https://www.echemportal.org/echemportal/ (accessed on 25 July 2023).
- Kuhn, M.; Letunic, I.; Jensen, L.J.; Bork, P. The SIDER database of drugs and side effects. Nucleic Acids Res. 2016, 44, D1075–D1079. [Google Scholar] [CrossRef] [PubMed]
- Bateman, A.; Martin, M.J.; Orchard, S.; Magrane, M.; Ahmad, S.; Alpi, E.; Bowler-Barnett, E.H.; Britto, R.; Cukura, A.; Denny, P.; et al. UniProt: The Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 2022, 51, D523–D531. [Google Scholar] [CrossRef]
- Paysan-Lafosse, T.; Blum, M.; Chuguransky, S.; Grego, T.; Pinto, B.L.; Salazar, G.A.; Bileschi, M.L.; Bork, P.; Bridge, A.; Colwell, L.; et al. InterPro in 2022. Nucleic Acids Res. 2022, 51, D418–D427. [Google Scholar] [CrossRef]
- Benson, D.A.; Cavanaugh, M.; Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2012, 41, D36–D42. [Google Scholar] [CrossRef]
- Burley, S.K.; Bhikadiya, C.; Bi, C.X.; Bittrich, S.; Chao, H.Y.; Chen, L.; Craig, P.A.; Crichlow, G.V.; Dalenberg, K.; Duarte, J.M.; et al. RCSB Protein Data Bank (RCSB.org): Delivery of experimentally-determined PDB structures alongside one million computed structure models of proteins from artificial intelligence/machine learning. Nucleic Acids Res. 2022, 51, D488–D508. [Google Scholar] [CrossRef]
- Feng, Z.K.; Chen, L.; Maddula, H.; Akcan, O.; Oughtred, R.; Berman, H.M.; Westbrook, J. Ligand Depot: A data warehouse for ligands bound to macromolecules. Bioinformatics 2004, 20, 2153–2155. [Google Scholar] [CrossRef] [PubMed]
- Keenan, A.B.; Jenkins, S.L.; Jagodnik, K.M.; Koplev, S.; He, E.; Torre, D.; Wang, Z.C.; Dohlman, A.B.; Silverstein, M.C.; Lachmann, A.; et al. The Library of Integrated Network-Based Cellular Signatures NIH Program: System-Level Cataloging of Human Cells Response to Perturbations. Cell Syst. 2018, 6, 13–24. [Google Scholar] [CrossRef]
- Chang, A.; Jeske, L.; Ulbrich, S.; Hofmann, J.; Koblitz, J.; Schomburg, I.; Neumann-Schaal, M.; Jahn, D.; Schomburg, D. BRENDA, the ELIXIR core data resource in 2021: New developments and updates. Nucleic Acids Res. 2021, 49, D498–D508. [Google Scholar] [CrossRef] [PubMed]
- Sorokina, M.; Merseburger, P.; Rajan, K.; Yirik, M.A.; Steinbeck, C. COCONUT online: Collection of Open Natural Products database. J. Cheminform. 2021, 13, 2. [Google Scholar] [CrossRef]
- Landaburu, L.U.; Berenstein, A.J.; Videla, S.; Maru, P.; Shanmugam, D.; Chernomoretz, A.; Aguero, F. TDR Targets 6: Driving drug discovery for human pathogens through intensive chemogenomic data integration. Nucleic Acids Res. 2020, 48, D992–D1005. [Google Scholar] [CrossRef]
- Bryant, P. Deep learning for protein complex structure prediction. Curr. Opin. Struct. Biol. 2023, 79, 102529. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Ivankov, D.N.; Finkelstein, A.V. Solution of Levinthal’s Paradox and a Physical Theory of Protein Folding Times. Biomolecules 2020, 10, 250. [Google Scholar] [CrossRef]
- Rose, G.D. Protein folding—Seeing is deceiving. Protein Sci. 2021, 30, 1606–1616. [Google Scholar] [CrossRef]
- Sorokina, I.; Mushegian, A.R.; Koonin, E.V. Is Protein Folding a Thermodynamically Unfavorable, Active, Energy-Dependent Process? Int. J. Mol. Sci. 2022, 23, 521. [Google Scholar] [CrossRef]
- Muhammed, M.T.; Aki-Yalcin, E. Homology modeling in drug discovery: Overview, current applications, and future perspectives. Chem. Biol. Drug Des. 2019, 93, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Burley, S.K.; Berman, H.M.; Duarte, J.M.; Feng, Z.K.; Flatt, J.W.; Hudson, B.P.; Lowe, R.; Peisach, E.; Piehl, D.W.; Rose, Y.; et al. Protein Data Bank: A Comprehensive Review of 3D Structure Holdings and Worldwide Utilization by Researchers, Educators, and Students. Biomolecules 2022, 12, 1425. [Google Scholar] [CrossRef] [PubMed]
- Burley, S.K.; Bhikadiya, C.; Bi, C.X.; Bittrich, S.; Chen, L.; Crichlow, G.V.; Duarte, J.M.; Dutta, S.; Fayazi, M.; Feng, Z.K.; et al. RCSB Protein Data Bank: Celebrating 50 years of the PDB with new tools for understanding and visualizing biological macromolecules in 3D. Protein Sci. 2022, 31, 187–208. [Google Scholar] [CrossRef]
- Sali, A.; Blundell, T.L. Comparative Protein Modeling by Satisfaction of Spatial Restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef]
- Webb, B.; Sali, A. Protein Structure Modeling with MODELLER. Methods Mol. Biol. 2021, 2199, 239–255. [Google Scholar] [CrossRef]
- Studer, G.; Tauriello, G.; Bienert, S.; Biasini, M.; Johner, N.; Schwede, T. ProMod3—A versatile homology modelling toolbox. PLoS Comput. Biol. 2021, 17, e1008667. [Google Scholar] [CrossRef]
- Studer, G.; Rempfer, C.; Waterhouse, A.M.; Gumienny, R.; Haas, J.; Schwede, T. QMEANDisCo-distance constraints applied on model quality estimation. Bioinformatics 2020, 36, 1765–1771. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinform. 2008, 9, 40. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef]
- Kryshtafovych, A.; Monastyrskyy, B.; Fidelis, K.; Moult, J.; Schwede, T.; Tramontano, A. Evaluation of the template-based modeling in CASP12. Proteins 2018, 86, 321–334. [Google Scholar] [CrossRef]
- Zheng, W.; Li, Y.; Zhang, C.X.; Zhou, X.G.; Pearce, R.; Bell, E.W.; Huang, X.Q.; Zhang, Y. Protein structure prediction using deep learning distance and hydrogen-bonding restraints in CASP14. Proteins 2021, 89, 1734–1751. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Applying and improving AlphaFold at CASP14. Proteins 2021, 89, 1711–1721. [Google Scholar] [CrossRef]
- Senior, A.W.; Evans, R.; Jumper, J.; Kirkpatrick, J.; Sifre, L.; Green, T.; Qin, C.L.; Zidek, A.; Nelson, A.W.R.; Bridgland, A.; et al. Improved protein structure prediction using potentials from deep learning. Nature 2020, 577, 706–710. [Google Scholar] [CrossRef] [PubMed]
- Anishchenko, I.; Baek, M.; Park, H.; Hiranuma, N.; Kim, D.E.; Dauparas, J.; Mansoor, S.; Humphreys, I.R.; Baker, D. Protein tertiary structure prediction and refinement using deep learning and Rosetta in CASP14. Proteins 2021, 89, 1722–1733. [Google Scholar] [CrossRef] [PubMed]
- Evans, R.; O’Neill, M.; Pritzel, A.; Antropova, N.; Senior, A.; Green, T.; Žídek, A.; Bates, R.; Blackwell, S.; Yim, J.; et al. Protein complex prediction with AlphaFold-Multimer. bioRxiv 2022. bioRxiv:10.1101/2021.10.04.463034. [Google Scholar]
- Gao, M.; An, D.N.; Parks, J.M.; Skolnick, J. AF2Complex predicts direct physical interactions in multimeric proteins with deep learning. Nat. Commun. 2022, 13, 1744. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.M.; Akin, H.; Rao, R.S.; Hie, B.; Zhu, Z.K.; Lu, W.T.; Smetanin, N.; Verkuil, R.; Kabeli, O.; Shmueli, Y.; et al. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 2023, 379, 1123–1130. [Google Scholar] [CrossRef]
- Azzaz, F.; Yahi, N.; Chahinian, H.; Fantini, J. The Epigenetic Dimension of Protein Structure Is an Intrinsic Weakness of the AlphaFold Program. Biomolecules 2022, 12, 1527. [Google Scholar] [CrossRef]
- Tourlet, S.; Radjasandirane, R.; Diharce, J.; de Brevern, A.G. AlphaFold2 Update and Perspectives. BioMedInformatics 2023, 3, 378–390. [Google Scholar] [CrossRef]
- Sciacca, M.F.; Lolicato, F.; Tempra, C.; Scollo, F.; Sahoo, B.R.; Watson, M.D.; Garcia-Vinuales, S.; Milardi, D.; Raudino, A.; Lee, J.C.; et al. Lipid-Chaperone Hypothesis: A Common Molecular Mechanism of Membrane Disruption by Intrinsically Disordered Proteins. ACS Chem. Neurosci. 2020, 11, 4336–4350. [Google Scholar] [CrossRef] [PubMed]
- Fantini, J. How sphingolipids bind and shape proteins: Molecular basis of lipid-protein interactions in lipid shells, rafts and related biomembrane domains. Cell. Mol. Life Sci. 2003, 60, 1027–1032. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Su, B.H.; Tseng, Y.J. Comparative studies of AlphaFold, RoseTTAFold and Modeller: A case study involving the use of G-protein-coupled receptors. Brief Bioinform. 2022, 23, bbac308. [Google Scholar] [CrossRef]
- Tovar, A.; Eckert, H.; Bajorath, J. Comparison of 2D fingerprint methods for multiple-template similarity searching on compound activity classes of increasing structural diversity. ChemMedChem 2007, 2, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Carracedo-Reboredo, P.; Linares-Blanco, J.; Rodriguez-Fernandez, N.; Cedron, F.; Novoa, F.J.; Carballal, A.; Maojo, V.; Pazos, A.; Fernandez-Lozano, C. A review on machine learning approaches and trends in drug discovery. Comput. Struct. Biotech. J. 2021, 19, 4538–4558. [Google Scholar] [CrossRef]
- Ding, Y.; Chen, M.C.; Guo, C.; Zhang, P.; Wang, J.W. Molecular fingerprint-based machine learning assisted QSAR model development for prediction of ionic liquid properties. J. Mol. Liq. 2021, 326, 115212. [Google Scholar] [CrossRef]
- Riniker, S.; Landrum, G.A. Open-source platform to benchmark fingerprints for ligand-based virtual screening. J. Cheminform. 2013, 5, 26. [Google Scholar] [CrossRef]
- Cereto-Massague, A.; Ojeda, M.J.; Valls, C.; Mulero, M.; Garcia-Vallve, S.; Pujadas, G. Molecular fingerprint similarity search in virtual screening. Methods 2015, 71, 58–63. [Google Scholar] [CrossRef]
- Gao, K.F.; Nguyen, D.D.; Sresht, V.; Mathiowetz, A.M.; Tu, M.H.; Wei, G.W. Are 2D fingerprints still valuable for drug discovery? Phys. Chem. Chem. Phys. 2020, 22, 8373–8390. [Google Scholar] [CrossRef]
- Rogers, D.; Hahn, M. Extended-Connectivity Fingerprints. J. Chem. Inf. Model. 2010, 50, 742–754. [Google Scholar] [CrossRef]
- McGregor, M.J.; Muskal, S.M. Pharmacophore fingerprinting. 1. Application to QSAR and focused library design. J. Chem. Inf. Comput. Sci. 1999, 39, 569–574. [Google Scholar] [CrossRef]
- Schwartz, J.; Awale, M.; Reymond, J.L. SMIfp (SMILES fingerprint) Chemical Space for Virtual Screening and Visualization of Large Databases of Organic Molecules. J. Chem. Inf. Model. 2013, 53, 1979–1989. [Google Scholar] [CrossRef] [PubMed]
- Awale, M.; Jin, X.; Reymond, J.L. Stereoselective virtual screening of the ZINC database using atom pair 3D-fingerprints. J. Cheminform. 2015, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- Da, C.; Kireev, D. Structural Protein-Ligand Interaction Fingerprints (SPLIF) for Structure-Based Virtual Screening: Method and Benchmark Study. J. Chem. Inf. Model. 2014, 54, 2555–2561. [Google Scholar] [CrossRef] [PubMed]
- Durant, J.L.; Leland, B.A.; Henry, D.R.; Nourse, J.G. Reoptimization of MDL keys for use in drug discovery. J. Chem. Inf. Comput. Sci. 2002, 42, 1273–1280. [Google Scholar] [CrossRef]
- PubChem Substructure Fingerprint. Available online: https://ftp.ncbi.nlm.nih.gov/pubchem/specifications/pubchem_fingerprints.pdf (accessed on 22 July 2023).
- Carhart, R.E.; Smith, D.H.; Venkataraghavan, R. Atom Pairs as Molecular-Features in Structure Activity Studies—Definition and Applications. J. Chem. Inf. Comput. Sci. 1985, 25, 64–73. [Google Scholar] [CrossRef]
- Bender, A.; Mussa, H.Y.; Glen, R.C.; Reiling, S. Similarity searching of chemical databases using atom environment descriptors (MOLPRINT 2D): Evaluation of performance. J. Chem. Inf. Comput. Sci. 2004, 44, 1708–1718. [Google Scholar] [CrossRef]
- Schneider, P.; Schneider, G. De Novo Design at the Edge of Chaos. J. Med. Chem. 2016, 59, 4077–4086. [Google Scholar] [CrossRef]
- Mouchlis, V.D.; Afantitis, A.; Serra, A.; Fratello, M.; Papadiamantis, A.G.; Aidinis, V.; Lynch, I.; Greco, D.; Melagraki, G. Advances in De Novo Drug Design: From Conventional to Machine Learning Methods. Int. J. Mol. Sci. 2021, 22, 1676. [Google Scholar] [CrossRef]
- Girin, L.; Leglaive, S.; Bie, X.Y.; Diard, J.; Hueber, T.; Alameda-Pineda, X. Dynamical Variational Autoencoders: A Comprehensive Review. Found. Trends Mach. Learn. 2021, 15, 1–175. [Google Scholar] [CrossRef]
- Prykhodko, O.; Johansson, S.V.; Kotsias, P.C.; Arus-Pous, J.; Bjerrum, E.J.; Engkvist, O.; Chen, H.M. A de novo molecular generation method using latent vector based generative adversarial network. J. Cheminform. 2019, 11, 74. [Google Scholar] [CrossRef] [PubMed]
- Sachdev, K.; Gupta, M.K. A comprehensive review of feature based methods for drug target interaction prediction. J. Biomed. Inform. 2019, 93, 103159. [Google Scholar] [CrossRef]
- Dhakal, A.; McKay, C.; Tanner, J.J.; Cheng, J.L. Artificial intelligence in the prediction of protein-ligand interactions: Recent advances and future directions. Brief Bioinform. 2022, 23, bbab476. [Google Scholar] [CrossRef] [PubMed]
- Bissantz, C.; Kuhn, B.; Stahl, M. A Medicinal Chemist’s Guide to Molecular Interactions. J. Med. Chem. 2010, 53, 6241. [Google Scholar] [CrossRef]
- Chen, D.L.; Oezguen, N.; Urvil, P.; Ferguson, C.; Dann, S.M.; Savidge, T.C. Regulation of protein-ligand binding affinity by hydrogen bond pairing. Sci. Adv. 2016, 2, e1501240. [Google Scholar] [CrossRef] [PubMed]
- Anusuya, S.; Kesherwani, M.; Priya, K.V.; Vimala, A.; Shanmugam, G.; Velmurugan, D.; Gromiha, M.M. Drug-Target Interactions: Prediction Methods and Applications. Curr. Protein Pept. Sci. 2018, 19, 537–561. [Google Scholar] [CrossRef]
- Chen, X.; Yan, C.C.; Zhang, X.T.; Zhang, X.; Dai, F.; Yin, J.; Zhang, Y.D. Drug-target interaction prediction: Databases, web servers and computational models. Brief Bioinform. 2016, 17, 696–712. [Google Scholar] [CrossRef]
- Bagherian, M.; Sabeti, E.; Wang, K.; Sartor, M.A.; Nikolovska-Coleska, Z.; Najarian, K. Machine learning approaches and databases for prediction of drug-target interaction: A survey paper. Brief Bioinform. 2021, 22, 247–269. [Google Scholar] [CrossRef]
- Xu, L.; Ru, X.Q.; Song, R. Application of Machine Learning for Drug-Target Interaction Prediction. Front. Genet. 2021, 12, 680117. [Google Scholar] [CrossRef]
- Yamanishi, Y.; Araki, M.; Gutteridge, A.; Honda, W.; Kanehisa, M. Prediction of drug-target interaction networks from the integration of chemical and genomic spaces. Bioinformatics 2008, 24, I232–I240. [Google Scholar] [CrossRef]
- Thafar, M.A.; Olayan, R.S.; Ashoor, H.; Albaradei, S.; Bajic, V.B.; Gao, X.; Gojobori, T.; Essack, M. DTiGEMS plus: Drug-target interaction prediction using graph embedding, graph mining, and similarity-based techniques. J. Cheminform. 2020, 12, 44. [Google Scholar] [CrossRef]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of Useful Decoys, Enhanced (DUD-E): Better Ligands and Decoys for Better Benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef]
- Liu, H.; Sun, J.J.; Guan, J.H.; Zheng, J.; Zhou, S.G. Improving compound-protein interaction prediction by building up highly credible negative samples. Bioinformatics 2015, 31, 221–229. [Google Scholar] [CrossRef]
- Tsubaki, M.; Tomii, K.; Sese, J. Compound-protein interaction prediction with end-to-end learning of Neural Netw. for graphs and sequences. Bioinformatics 2019, 35, 309–318. [Google Scholar] [CrossRef]
- Wang, S.Y.; Shan, P.; Zhao, Y.L.; Zuo, L. GanDTI: A multi-task neural network for drug-target interaction prediction. Comput. Biol. Chem. 2021, 92, 107476. [Google Scholar] [CrossRef]
- Hecker, N.; Ahmed, J.; Eichborn, J.; Dunkel, M.; Macha, K.; Eckert, A.; Gilson, M.K.; Bourne, P.E.; Preissner, R. SuperTarget goes quantitative: Update on drug-target interactions. Nucleic Acids Res. 2012, 40, D1113–D1117. [Google Scholar] [CrossRef]
- Ding, Y.J.; Tang, J.J.; Guo, F.; Zou, Q. Identification of drug-target interactions via multiple kernel-based triple collaborative matrix factorization. Brief Bioinform. 2022, 23, bbab582. [Google Scholar] [CrossRef] [PubMed]
- Zitnik, M.; Sosic, R.; Leskovec, J. BioSNAP Datasets: Stanford Biomedical Network Dataset Collection. Available online: http://snap.stanford.edu/biodata/ (accessed on 25 July 2023).
- Davis, M.I.; Hunt, J.P.; Herrgard, S.; Ciceri, P.; Wodicka, L.M.; Pallares, G.; Hocker, M.; Treiber, D.K.; Zarrinkar, P.P. Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2011, 29, 1046–1051. [Google Scholar] [CrossRef] [PubMed]
- Song, T.; Zhang, X.D.; Ding, M.; Rodriguez-Paton, A.; Wang, S.D.; Wang, G. DeepFusion: A deep learning based multi-scale feature fusion method for predicting drug-target interactions. Methods 2022, 204, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Yazdani-Jahromi, M.; Yousefi, N.; Tayebi, A.; Kolanthai, E.; Neal, C.J.; Seal, S.; Garibay, O.O. AttentionSiteDTI: An interpretable graph-based model for drug-target interaction prediction using NLP sentence-level relation classification. Brief Bioinform. 2022, 23, bbac272. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Zhang, Z.Q.; Guan, J.H.; Zhou, S.G. Effective drug-target interaction prediction with mutual interaction neural network. Bioinformatics 2022, 38, 3582–3589. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Zhu, C.; Zhong, F.; Liu, L. MDTips: A multimodal-data-based drug-target interaction prediction system fusing knowledge, gene expression profile, and structural data. Bioinformatics 2023, 39, btad411. [Google Scholar] [CrossRef] [PubMed]
- Richard, A.M.; Huang, R.L.; Waidyanatha, S.; Shinn, P.; Collins, B.J.; Thillainadarajah, I.; Grulke, C.M.; Williams, A.J.; Lougee, R.R.; Judson, R.S.; et al. The Tox21 10K Compound Library: Collaborative Chemistry Advancing Toxicology. Chem. Res. Toxicol. 2021, 34, 189–216. [Google Scholar] [CrossRef] [PubMed]
- Thomas, R.S.; Paules, R.S.; Simeonov, A.; Fitzpatrick, S.C.; Crofton, K.M.; Casey, W.M.; Mendrick, D.L. The US Federal Tox21 Program: A Strategic and Operational Plan for Continued Leadership. Altex 2018, 35, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Mayr, A.; Klambauer, G.; Unterthiner, T.; Hochreiter, S. DeepTox: Toxicity Prediction using Deep Learning. Front. Environ. Sci. 2016, 3, 80. [Google Scholar] [CrossRef]
- Richard, A.M.; Judson, R.S.; Houck, K.A.; Grulke, C.M.; Volarath, P.; Thillainadarajah, I.; Yang, C.H.; Rathman, J.; Martin, M.T.; Wambaugh, J.F.; et al. ToxCast Chemical Landscape: Paving the Road to 21st Century Toxicology. Chem. Res. Toxicol. 2016, 29, 1225–1251. [Google Scholar] [CrossRef]
- Dix, D.J.; Houck, K.A.; Martin, M.T.; Richard, A.M.; Setzer, R.W.; Kavlock, R.J. The ToxCast program for prioritizing toxicity testing of environmental chemicals. Toxicol. Sci. 2007, 95, 5–12. [Google Scholar] [CrossRef]
- Duran-Iturbide, N.A.; Diaz-Eufracio, B.I.; Medina-Franco, J.L. In Silico ADME/Tox Profiling of Natural Products: A Focus on BIOFACQUIM. ACS Omega 2020, 5, 16076–16084. [Google Scholar] [CrossRef]
- Negus, S.S.; Banks, M.L. Pharmacokinetic-Pharmacodynamic (PKPD) Analysis with Drug Discrimination. Curr. Top Behav. Neurosci. 2018, 39, 245–259. [Google Scholar] [CrossRef]
- Maltarollo, V.G.; Gertrudes, J.C.; Oliveira, P.R.; Honorio, K.M. Applying machine learning techniques for ADME-Tox prediction: A review. Expert Opin. Drug Met. 2015, 11, 259–271. [Google Scholar] [CrossRef]
- Wenzel, J.; Matter, H.; Schmidt, F. Predictive Multitask Deep Neural Network Models for ADME-Tox Properties: Learning from Large Data Sets. J. Chem. Inf. Model. 2019, 59, 1253–1268. [Google Scholar] [CrossRef] [PubMed]
- Almazroo, O.A.; Miah, M.K.; Venkataramanan, R. Drug Metabolism in the Liver. Clin. Liver Dis. 2017, 21, 1–20. [Google Scholar] [CrossRef]
- Xu, Z.Y.; Kang, Q.J.; Yu, Z.H.; Tian, L.C.; Zhang, J.X.; Wang, T. Research on the Species Difference of the Hepatotoxicity of Medicine Based on Transcriptome. Front. Pharmacol. 2021, 12, 647084. [Google Scholar] [CrossRef] [PubMed]
- Bjornsson, E.S. Drug-induced liver injury: An overview over the most critical compounds. Arch. Toxicol. 2015, 89, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Walker, P.A.; Ryder, S.; Lavado, A.; Dilworth, C.; Riley, R.J. The evolution of strategies to minimise the risk of human drug-induced liver injury (DILI) in drug discovery and development. Arch. Toxicol. 2020, 94, 2559–2585. [Google Scholar] [CrossRef]
- Takebe, T.; Imai, R.; Ono, S. The Current Status of Drug Discovery and Development as Originated in United States Academia: The Influence of Industrial and Academic Collaboration on Drug Discovery and Development. Clin. Transl. Sci. 2018, 11, 597–606. [Google Scholar] [CrossRef]
- Clinton, J.W.; Kiparizoska, S.; Aggarwal, S.; Woo, S.; Davis, W.; Lewis, J.H. Drug-Induced Liver Injury: Highlights and Controversies in the Recent Literature. Drug Saf. 2021, 44, 1125–1149. [Google Scholar] [CrossRef]
- Ai, H.X.; Chen, W.; Zhang, L.; Huang, L.C.; Yin, Z.M.; Hu, H.; Zhao, Q.; Zhao, J.; Liu, H.S. Predicting Drug-Induced Liver Injury Using Ensemble Learning Methods and Molecular Fingerprints. Toxicol. Sci. 2018, 165, 100–107. [Google Scholar] [CrossRef]
- Li, M.Y.; Peng, L.M.; Chen, X.P. Pharmacogenomics in drug-induced cardiotoxicity: Current status and the future. Front. Cardiovasc. Med. 2022, 9, 966261. [Google Scholar] [CrossRef]
- Food and Drug Administration. International Conference on Harmonisation; guidance on S7B Nonclinical Evaluation of the Potential for Delayed Ventricular Repolarization (QT Interval Prolongation) by Human Pharmaceuticals. Fed. Regist. 2005, 70, 61133–61134. [Google Scholar]
- Lamothe, S.M.; Guo, J.; Li, W.T.; Yang, T.H.; Zhang, S.T. The Human Ether-a-go-go-related Gene (hERG) Potassium Channel Represents an Unusual Target for Protease-mediated Damage. J. Biol. Chem. 2016, 291, 20387–20401. [Google Scholar] [CrossRef] [PubMed]
- Babcock, J.J.; Li, M. hERG channel function: Beyond long QT. Acta Pharmacol. Sin. 2013, 34, 329–335. [Google Scholar] [CrossRef] [PubMed]
- De Bruin, M.L.; Pettersson, M.; Meyboom, R.H.B.; Hoes, A.W.; Leufkens, H.G.M. Anti-HERG activity and the risk of drug-induced arrhythmias and sudden death. Eur. Heart J. 2005, 26, 590–597. [Google Scholar] [CrossRef]
- Thomas, D.; Karle, C.A.; Kiehn, J. The cardiac hERG/I-Kr potassium channel as pharmacological target: Structure, function, regulation, and clinical applications. Curr. Pharm. Des. 2006, 12, 2271–2283. [Google Scholar] [CrossRef]
- Stergiopoulos, C.; Tsopelas, F.; Valko, K. Prediction of hERG inhibition of drug discovery compounds using biomimetic HPLC measurements. ADMET DMPK 2021, 9, 191–207. [Google Scholar] [CrossRef] [PubMed]
- Honma, M. An assessment of mutagenicity of chemical substances by (quantitative) structure-activity relationship. Genes Environ. 2020, 42, 23. [Google Scholar] [CrossRef]
- Zhang, L.; Ai, H.X.; Chen, W.; Yin, Z.M.; Hu, H.; Zhu, J.F.; Zhao, J.; Zhao, Q.; Liu, H.S. CarcinoPred-EL: Novel models for predicting the carcinogenicity of chemicals using molecular fingerprints and ensemble learning methods. Sci. Rep. 2017, 7, 2118. [Google Scholar] [CrossRef]
- Basu, A.K. DNA Damage, Mutagenesis and Cancer. Int. J. Mol. Sci. 2018, 19, 970. [Google Scholar] [CrossRef]
- Drevon, C.; Piccoli, C.; Montesano, R. Mutagenicity Assays of Estrogenic Hormones in Mammalian Cells. Mutat. Res. 1981, 89, 83–90. [Google Scholar] [CrossRef]
- Ferguson, L.R. Chronic inflammation and mutagenesis. Mutat. Res. Fund. Mol. Mech. Mutagenes. 2010, 690, 3–11. [Google Scholar] [CrossRef]
- Barnes, J.L.; Zubair, M.; John, K.; Poirier, M.C.; Martin, F.L. Carcinogens and DNA damage. Biochem. Soc. T 2018, 46, 1213–1224. [Google Scholar] [CrossRef] [PubMed]
- Fradkin, P.; Young, A.; Atanackovic, L.; Frey, B.; Lee, L.J.; Wang, B. A graph neural network approach for molecule carcinogenicity prediction. Bioinformatics 2022, 38, i84–i91. [Google Scholar] [CrossRef] [PubMed]
- Bartsch, H.; Tomatis, L. Comparison between Carcinogenicity and Mutagenicity Based on Chemicals Evaluated in the IARC Monographs. Environ. Health Persp. 1983, 47, 305–317. [Google Scholar] [CrossRef] [PubMed]
- Hughes, J.P.; Rees, S.; Kalindjian, S.B.; Philpott, K.L. Principles of early drug discovery. Brit. J. Pharmacol. 2011, 162, 1239–1249. [Google Scholar] [CrossRef]
- Knuiman, M.W.; Laird, N.M.; Louis, T.A. Inter-Laboratory Variability in Ames Assay Results. Mutat. Res. 1987, 180, 171–182. [Google Scholar] [CrossRef]
- Galloway, S.M. International Regulatory Requirements for Genotoxicity Testing for Pharmaceuticals Used in Human Medicine, and Their Impurities and Metabolites. Environ. Mol. Mutagen. 2017, 58, 296–324. [Google Scholar] [CrossRef]
- Li, T.; Tong, W.D.; Roberts, R.; Liu, Z.C.; Thakkar, S. DeepCarc: Deep learning-powered carcinogenicity prediction using model-level representation. Front. Artif. Intell. 2022, 4, 757780. [Google Scholar] [CrossRef]
- Zaslayskiy, M.; Jegou, S.; Tramel, E.W.; Wainrib, G. ToxicBlend: Virtual screening of toxic compound with ensemble predictors. Comput. Toxicol. 2019, 10, 81–88. [Google Scholar] [CrossRef]
- Wu, Z.Q.; Ramsundar, B.; Feinberg, E.N.; Gomes, J.; Geniesse, C.; Pappu, A.S.; Leswing, K.; Pande, V. MoleculeNet: A benchmark for molecular machine learning. Chem. Sci. 2018, 9, 513–530. [Google Scholar] [CrossRef]
- Registry of Toxic Effects of Chemical Substances (RTECS). Available online: https://www.3ds.com/ko/products-services/biovia/ (accessed on 25 July 2023).
- Sharma, B.; Chenthamarakshan, V.; Dhurandhar, A.; Pereira, S.; Hendler, J.A.; Dordick, J.S.; Das, P. Accurate clinical toxicity prediction using multi-task deep neural nets and contrastive molecular explanations. Sci. Rep. 2023, 13, 4908. [Google Scholar] [CrossRef]
- Gold, L.S.; Manley, N.B.; Slone, T.H.; Rohrbach, L.; Garfinkel, G.B. Supplement to the Carcinogenic Potency Database (CPDB): Results of animal bioassays published in the general literature through 1997 and by the National Toxicology Program in 1997–1998. Toxicol. Sci. 2005, 85, 747–808. [Google Scholar] [CrossRef] [PubMed]
- Veith, H.; Southall, N.; Huang, R.L.; James, T.; Fayne, D.; Artemenko, N.; Shen, M.; Inglese, J.; Austin, C.P.; Lloyd, D.G.; et al. Comprehensive characterization of cytochrome P450 isozyme selectivity across chemical libraries. Nat. Biotechnol. 2009, 27, 1050–1055. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.B.; Lou, C.F.; Sun, L.X.; Li, J.; Cai, Y.C.; Wang, Z.; Li, W.H.; Liu, G.X.; Tang, Y. admetSAR 2.0: Web-service for prediction and optimization of chemical ADMET properties. Bioinformatics 2019, 35, 1067–1069. [Google Scholar] [CrossRef]
- Wei, Y.; Li, S.S.; Li, Z.L.; Wan, Z.W.; Lin, J.P. Interpretable-ADMET: A web service for ADMET prediction and optimization based on deep neural representation. Bioinformatics 2022, 38, 2863–2871. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.Z.; Yan, Z.Y.; Huang, Y.Y.; Liu, L.H.; He, D.L.; Wang, W.; Fang, X.M.; Zhang, X.N.; Wang, F.; Wu, H.; et al. HelixADMET: A robust and endpoint extensible ADMET system incorporating self-supervised knowledge transfer. Bioinformatics 2022, 38, 3444–3453. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.J.; Suzuki, A.; Thakkar, S.; Yu, K.; Hu, C.C.; Tong, W.D. DILIrank: The largest reference drug list ranked by the risk for developing drug-induced liver injury in humans. Drug Discov. Today 2016, 21, 648–653. [Google Scholar] [CrossRef]
- Liu, A.; Walter, M.; Wright, P.; Bartosik, A.; Dolciami, D.; Elbasir, A.; Yang, H.B.; Bender, A. Prediction and mechanistic analysis of drug-induced liver injury (DILI) based on chemical structure. Biol. Direct 2021, 16, 6. [Google Scholar] [CrossRef]
- Thakkar, S.; Li, T.; Liu, Z.C.; Wu, L.H.; Roberts, R.; Tong, W.D. Drug-induced liver injury severity and toxicity (DILIst): Binary classification of 1279 drugs by human hepatotoxicity. Drug Discov. Today 2020, 25, 201–208. [Google Scholar] [CrossRef]
- TDC Benckmark Dataset. Available online: https://tdcommons.ai/single_pred_tasks/tox/#dili-drug-induced-liver-injury (accessed on 25 July 2023).
- Lim, S.; Kim, Y.; Gu, J.; Lee, S.; Shin, W.; Kim, S. Supervised chemical graph mining improves drug-induced liver injury prediction. iScience 2023, 26, 105677. [Google Scholar] [CrossRef]
- Kadioglu, O.; Klauck, S.M.; Fleischer, E.; Shan, L.T.; Efferth, T. Selection of safe artemisinin derivatives using a Mach. Learn.-based cardiotoxicity platform and in vitro and in vivo validation. Arch. Toxicol. 2021, 95, 2485–2495. [Google Scholar] [CrossRef]
- Kazius, J.; McGuire, R.; Bursi, R. Derivation and validation of toxicophores for mutagenicity prediction. J. Med. Chem. 2005, 48, 312–320. [Google Scholar] [CrossRef] [PubMed]
- Hansen, K.; Mika, S.; Schroeter, T.; Sutter, A.; ter Laak, A.; Steger-Hartmann, T.; Heinrich, N.; Muller, K.R. Benchmark Data Set for in Silico Prediction of Ames Mutagenicity. J. Chem. Inf. Model. 2009, 49, 2077–2081. [Google Scholar] [CrossRef] [PubMed]
- Mattocks, A.R. Chemistry and Toxicology of Pyrrolizidine Alkaloids; Academic Press: Cambridge, MA, USA, 1986. [Google Scholar]
- European Food Safety Authority (EFSA) Dataset. Available online: https://data.europa.eu/data/datasets/database-pesticide-genotoxicity-endpoints?locale=data (accessed on 25 July 2023).
- Helma, C.; Schoning, V.; Drewe, J.; Boss, P. A Comparison of Nine Machine Learning Mutagenicity Models and Their Application for Predicting Pyrrolizidine Alkaloids. Front. Pharmacol. 2021, 12, 708050. [Google Scholar] [CrossRef] [PubMed]
- Inventory of Hazardous Chemicals. Available online: https://www.mem.gov.cn/fw/cxfw/ (accessed on 25 July 2023).
- The Globally Harmonized System of Classification and Labeling of Chemicals (GHS). Available online: https://unece.org/# (accessed on 25 July 2023).
- Hao, N.; Sun, P.X.; Zhao, W.J.; Li, X.X. Application of a developed triple-classification machine learning model for carcinogenic prediction of hazardous organic chemicals to the US, EU, and WHO based on Chinese database. Ecotoxicol. Environ. Safe 2023, 255, 114806. [Google Scholar] [CrossRef] [PubMed]
| Database | URL * | Description | Ref. |
|---|---|---|---|
| Compound and Drug Databases | |||
| PubChem | https://pubchem.ncbi.nlm.nih.gov/ | Launched in 2004 as part of the Molecular Libraries Roadmap Initiatives by the US National Institutes of Health (NIH), PubChem is a public database for information regarding chemical substances and their biological activities. | [86] |
| ChEMBL | https://www.ebi.ac.uk/chembl/ | ChEMBL is a well-curated database of bioactive molecules with drug-like properties, integrating chemical, bioactivity, and genomic data to aid in the transformation of genomic information into effective new drugs. | [87] |
| ZINC | https://zinc.docking.org/ | ZINC is a free database of over 230 million commercially available compounds in 3D formats, suitable for virtual screening, provided by the Irwin and Shoichet Lab at UCSF. | [88] |
| ChemSpider | http://www.chemspider.com | ChemSpider is a free-access website that serves as a chemical database and a structure-centric community for chemists, aiming to aggregate and index accessible information on chemical structures and related data from various online sources. It includes analytical data, synthesis reactions, experimental properties, and more. | [89] |
| DrugBank | http://www.drugbank.ca | DrugBank is a robust online database that provides wide-ranging biochemical and pharmacological data about drugs, including their mechanisms of action and targets. | [90] |
| DrugCentral | http://drugcentral.org/ | DrugCentral is a publicly accessible online compendium that consolidates information on the structure, bioactivity, regulatory, and pharmacological actions, and indications of active pharmaceutical ingredients approved by the FDA and other regulatory bodies. | [91] |
| Drugs@FDA | https://www.accessdata.fda.gov/scripts/cder/daf/ | Drugs@FDA is a comprehensive database that contains information about FDA-approved prescription and over-the-counter drug products, including brand-name and generic drugs, as well as many therapeutic biological products, with majority of data dating back to 1998 and some extending to 1939. | [92] |
| Metabolic and Biomolecular Pathway Databases | |||
| KEGG | https://www.kegg.jp | KEGG is a database designed to provide insights into the high-level biological functions of cells, organisms, and ecosystems using molecular-level data, particularly from large-scale genome sequencing and other high-throughput experiments. | [93] |
| BioCyc | https://biocyc.org/ | BioCyc is a comprehensive collection of pathway/genome databases and a suite of bioinformatics tools that offer insights into the genomes, metabolic pathways, and regulatory networks of numerous sequenced organisms, helping to accelerate scientific research. | [94] |
| Reactome | https://reactome.org | Reactome is an open-access, peer-reviewed pathway database that aims to offer user-friendly bioinformatics resources for visualizing, interpreting, and analyzing pathway information. These resources aid various fields, including basic research, genome examination, modeling, systems biology, and education. | [95] |
| HMDB | http://www.hmdb.ca | The Human Metabolome Database (HMDB) is an open-access online database that provides comprehensive information regarding small molecule metabolites identified in the human body. | [96] |
| Protein–Protein Interaction and Network Databases | |||
| IntAct | http://www.ebi.ac.uk/intact/ | IntAct is a freely accessible database that houses molecular interaction data, obtained either directly from data submissions or curated from scholarly publications. | [97] |
| BioGRID | https://thebiogrid.org | BioGRID is an online repository that meticulously compiles and hosts extensive data on protein and genetic interactions, chemical associations, and post-translational modifications from major model organisms. | [98] |
| STRING | https://string-db.org/ | STRING is a comprehensive repository that comprises both acknowledged and projected protein associations. These interactions encompass both direct interactions, which involve physical contact, and indirect ones, which imply functional relationships. | [99] |
| STITCH | http://stitch.embl.de/ | STITCH is a platform used to investigate established and anticipated connections between proteins and chemicals, with connections supported by experimental data, databases, and the academic literature. | [100] |
| Drug–Target Interaction Databases | |||
| BindingDB | http://www.bindingdb.org/bind/index.jsp | BindingDB is an open, web-based database dedicated primarily to measuring binding affinities between proteins, viewed as drug targets, and small, drug-like molecules. | [101] |
| TTD | http://db.idrblab.net/ttd/ | The Therapeutic Target Database (TTD) is a resource that offers details regarding established and potential therapeutic protein and nucleic acid targets, the diseases they target, associated pathway information, and the specific drugs designed to interact with these targets. | [102] |
| IUPHAR/BPS Guide to PHARMACOLOGY | https://www.guidetopharmacology.org/ | The IUPHAR/BPS Guide to PHARMACOLOGY is an expert-curated database offering comprehensive information on drug targets, prescription medicines, and experimental drugs, enriched with links to other databases, aiming to be a centralized resource for pharmacology and drug discovery. | [103] |
| DGIdb | http://www.dgidb.org | The Drug–Gene Interaction Database is an online tool that amalgamates various datasets detailing interactions between drugs and genes, and the druggability of genes. It presents a user-friendly visual interface and a well-documented API for data queries. | [104] |
| Toxicity and Side Effect Databases | |||
| CTD | http://ctdbase.org/ | CTD is a comprehensive, public database that collates data from various sources on the impacts of environmental exposures on human health, including chemical genes, chemical disease, and chemical–exposure interactions across all species, offering analytical tools for hypothesis generation. | [105] |
| DrugMatrix/ToxFX | https://ntp.niehs.nih.gov/data/drugmatrix | DrugMatrix, accompanied by its reporting system ToxFX, serves as one of the largest toxicogenomic reference databases, providing comprehensive profiles for over 600 compounds, aimed at enhancing the efficiency of toxicological assessments and understanding of the potential toxicity of xenobiotics. | [106] |
| OECD eChemPortal | https://www.echemportal.org/echemportal/ | eChemPortal is a free public global database that collects and provides direct links to chemical characteristics data and safety information from various national, regional, and international government programs. | [107] |
| SIDER | http://sideeffects.embl.de/ | SIDER is a database that provides information about marketed drugs and their documented adverse reactions, including side effect frequency, drug classifications, and additional resources such as drug–target relations. | [108] |
| Protein and Gene Databases | |||
| UniProt | https://www.uniprot.org | UniProt offers the scientific community a thorough, superior, and freely accessible database of protein sequences and functional data. | [109] |
| InterPro | https://www.ebi.ac.uk/interpro/ | InterPro facilitates the functional examination of proteins by grouping them into families and forecasting the presence of domains and significant sites. | [110] |
| GenBank | http://www.ncbi.nlm.nih.gov/genbank/ | GenBank is the NIH’s genetic sequence database, a comprehensive, annotated collection of all publicly accessible DNA sequences, participating in the International Nucleotide Sequence Database Collaboration, with data updates every two months. | [111] |
| RCSB PDB | http://rcsb.org/ | RCSB PDB is a resource-driven by the Protein Data Bank archive, offering detailed information about 3D structures of proteins, nucleic acids, and complex assemblies, aiding students and researchers in exploring biomedicine, agriculture, protein synthesis, and various health and disease conditions. | [112] |
| Ligand Expo | http://ligand-expo.rcsb.org/ | Ligand Expo is a resource offering chemical and structural information about small molecules found within the Protein Data Bank entries, along with tools for searching, identifying entries with specific molecules, downloading 3D molecule structures, and creating new chemical definitions. | [113] |
| Databases offering diverse types of information | |||
| LINCS | https://lincsproject.org/ | The LINCS Consortium is a project that provides public data on cellular responses to various genetic and environmental stressors, aiming to deepen our understanding of cellular pathways and aid in the development of therapies to normalize disturbed pathways and networks, with their website and data portal offering comprehensive information on assays, cell types, perturbations, and related software for data analysis. | [114] |
| BRENDA | http://www.brenda-enzymes.org/ | BRENDA is a comprehensive resource that consolidates extensive information about enzymes and enzyme–ligand relationships derived from various sources and offers adaptable search systems and assessment tools. | [115] |
| COCONUT | https://coconut.naturalproducts.net | Natural Products Online is a freely accessible, open-source platform dedicated to storing, searching, and analysis of natural products (NPs). It currently features COCONUT, a comprehensive and well-documented collection of open natural products, which is one of the most significant resources available without any restrictions. | [116] |
| TDR targets | https://tdrtargets.org | TDR Targets is a website that serves two purposes. Firstly, it provides information on targets, drugs, and bioactive compounds. Secondly, it can be used to prioritize targets within whole genomes. | [117] |
| Approach | Year | Datasets | Features | Algorithms | Performance | Ref. |
|---|---|---|---|---|---|---|
| DTiGEMS+ | 2020 | The literature [173] | Similarity-based features | Graph embedding, graph mining, similarity network fusion, MLP, RF, Adaboost | AUPR of 0.88, 0.86, 0.96, and 0.97 for the NR, GPCR, IC, and E datasets | [174] |
| GanDTI | 2021 | DUD-E [175], bindingDB inhibition, the literature [176,177] | Molecule fingerprints with a radius of two, protein data encoded overlapping amino acid sequences | GNN, attention mechanism to formulate summarized protein feature vectors, MLP | AUC of 0.983, Recall of 0.933 and Precision of 0.960 | [178] |
| DTI prediction using multiple kernel-based triple collaborative matrix factorization | 2022 | DrugBank, BRENDA, SuperTarget [179], KEGG BRITE | Gaussian interaction profile, network of drug-side effect associations, MACCs drug substructure fingerprint, and chemical structure for drug kernels, Gaussian interaction profile for target, PPIs network of target, functional information of target and sequence information of target for target kernels | Multiple kernel-based triple collaborative matrix factorization (MKTC-MF) | AUPR of 0.933 on ion channel | [180] |
| DeepFusion | 2022 | BIOSNAP [181], DAVIS dataset [182] | Global structural similarity feature based on similarity theory and convolutional neural network for both drug and protein, local chemical sub-structure semantic feature using transformer network for both drug and protein | Deep-learning-based multi-scale feature fusion method including CNN and transformer network | Best ROC-AUC of 0.911 | [183] |
| AttentionSiteDTI | 2022 | Protein Data Bank, DUD-E, human dataset from Liu et al. [176], BindingDB | Graph-based features of proteins and drugs | Topology adaptive graph CNN (TAGCN), MLP, self-attention mechanism, bidirectional long short-term memory (LSTM) | Best AUC of 0.991 in human dataset | [184] |
| MINN-DTI | 2022 | DUD-E, human dataset from Liu et al. [176], BindingDB | A 2D distance map for the target and the 2D molecular graph for the molecule | Dynamic CNN (DyCNN), inter-CMPNN, MLP | Best AUC of 0.967 in human dataset | [185] |
| MDTips | 2023 | Drug repurposing knowledge graph (DRKG), DrugBank, UniProt | Knowledge graph, drug-structure-based feature, target amino-acid-sequence-based feature, drug perturbation signatures, gene over-expression signatures, gene knockout/knockdown signature | Attentive FP and transformer encoders, knowledge graph embedding, ConvE, GAT, GNN, CNN, GCN | AUPR: 0.951 ± 0.003 | [186] |
| Approach | Year | Datasets | Features | Algorithms | Performance | Ref. |
|---|---|---|---|---|---|---|
| ToxicBlend | 2019 | Tox21 data, ToxCast | Physical chemicals descriptors, PubChem molecular fingerprints, SMILES n-grams | Multi-task XGBoost, multi-task NNs, graph convolutional model | AUC of 0.866 in Tox21 by random splits, AUC of 0.763 in ToxCast by scaffold splits | [222] |
| CEM-DNN | 2023 | ClinTox [223], Tox21, RTECS [224] | Morgan fingerprints, SMILES embeddings (SE) | Single-task DNN, multi-task DNN | AUC-ROC: 0.991 ± 0.011, balanced accuracy: 0.963 ± 0.028 | [225] |
| admetSAR2.0 | 2019 | DrugBank, ChEMBL, CPDB [226], Tox21, CYP450 dataset [227] | RDKit, Morgan, atom pairs, torsions, MACCS, SubFP fingerprints | kNN | AUC ranging from 0.625 to 0.992, with an average of 0.842 | [228] |
| Interpretable-ADMET | 2022 | ChEMBL, PubChem, DrugBank, publications in the literature | Matched molecular pair (MMP)-processed fingerprint | Graph convolutional neural network (GCNN), graph attention network (GAT) | AUC of 0.977 in GCNN, AUC of 0.974 in GAT | [229] |
| HelixADMET | 2022 | ZINC15, DrugBank, ChEMBL, CPDB, Tox21, CYP450, PubChem assays | Subgraph (local structure) of a compound, molecular 3D conformation, molecular fingerprints | GNN, RF | AUC range of 0.803 to 0.967 | [230] |
| Prediction and mechanistic analysis of DILI based on chemical structure | 2021 | DILIrank [231], SIDER | ECFP4 fingerprints, predicted protein targets, Mordred molecular descriptors | SVM, RF | Mean balanced accuracy of 0.759 ± 0.027 | [232] |
| DILI prediction by maximizing fidelity through explicit subgraph feature mining | 2022 | DILIst [233], TDC [234] | SMILES converted to RDKit mol and networkx graph object | Supervised subgraph mining (SSM) | AUC: 0.691, F1-score: 0.784, MCC: 0.338 | [235] |
| deephERG | 2019 | ChEMBL | Mol2vec, 2D MOE descriptors | Multitask DNN | Best AUC: 0.967, 29.6% of FDA-approved drugs potentially possessed hERG inhibitory activity | [31] |
| Cardiotoxicity prediction of Artemisinin derivatives | 2021 | PubMed, PubChem, DrugBank | The calculated descriptors | RF | AUC greater than 0.830 for cardio-toxicity parameters | [236] |
| Predicting mutagenicity in pyrrolizidine alkaloids | 2021 | The literatures [237,238,239], EFSA dataset [240] | MolPrint2D fingerprints, chemistry development kit | Lazar with high confidence, all lazar predictions, RF, logistic regression (stochastic gradient descent), logistic regression (scikit), NN, SVM | Accuracies of 80–85% | [241] |
| Carcinogenic classification using a triple classification prediction model | 2023 | Inventory of Hazardous Chemicals [242], Globally Harmonized System of Classification and Labeling of Chemicals (GHS) [243] | Generated by calculation and RF feature selection, including AATSC0p and GATS1e | MLP. XGBoost. kNN, complement naïve Bayes, SVM, LR. RF | The best accuracy in IARC dataset by RF | [244] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, R.; Yoon, H.; Kim, G.; Lee, H.; Lee, Y. Revolutionizing Medicinal Chemistry: The Application of Artificial Intelligence (AI) in Early Drug Discovery. Pharmaceuticals 2023, 16, 1259. https://doi.org/10.3390/ph16091259
Han R, Yoon H, Kim G, Lee H, Lee Y. Revolutionizing Medicinal Chemistry: The Application of Artificial Intelligence (AI) in Early Drug Discovery. Pharmaceuticals. 2023; 16(9):1259. https://doi.org/10.3390/ph16091259
Chicago/Turabian StyleHan, Ri, Hongryul Yoon, Gahee Kim, Hyundo Lee, and Yoonji Lee. 2023. "Revolutionizing Medicinal Chemistry: The Application of Artificial Intelligence (AI) in Early Drug Discovery" Pharmaceuticals 16, no. 9: 1259. https://doi.org/10.3390/ph16091259
APA StyleHan, R., Yoon, H., Kim, G., Lee, H., & Lee, Y. (2023). Revolutionizing Medicinal Chemistry: The Application of Artificial Intelligence (AI) in Early Drug Discovery. Pharmaceuticals, 16(9), 1259. https://doi.org/10.3390/ph16091259