

Inhibition of the hERG Potassium Channel by a Methanesulphonate-Free E-4031 Analogue

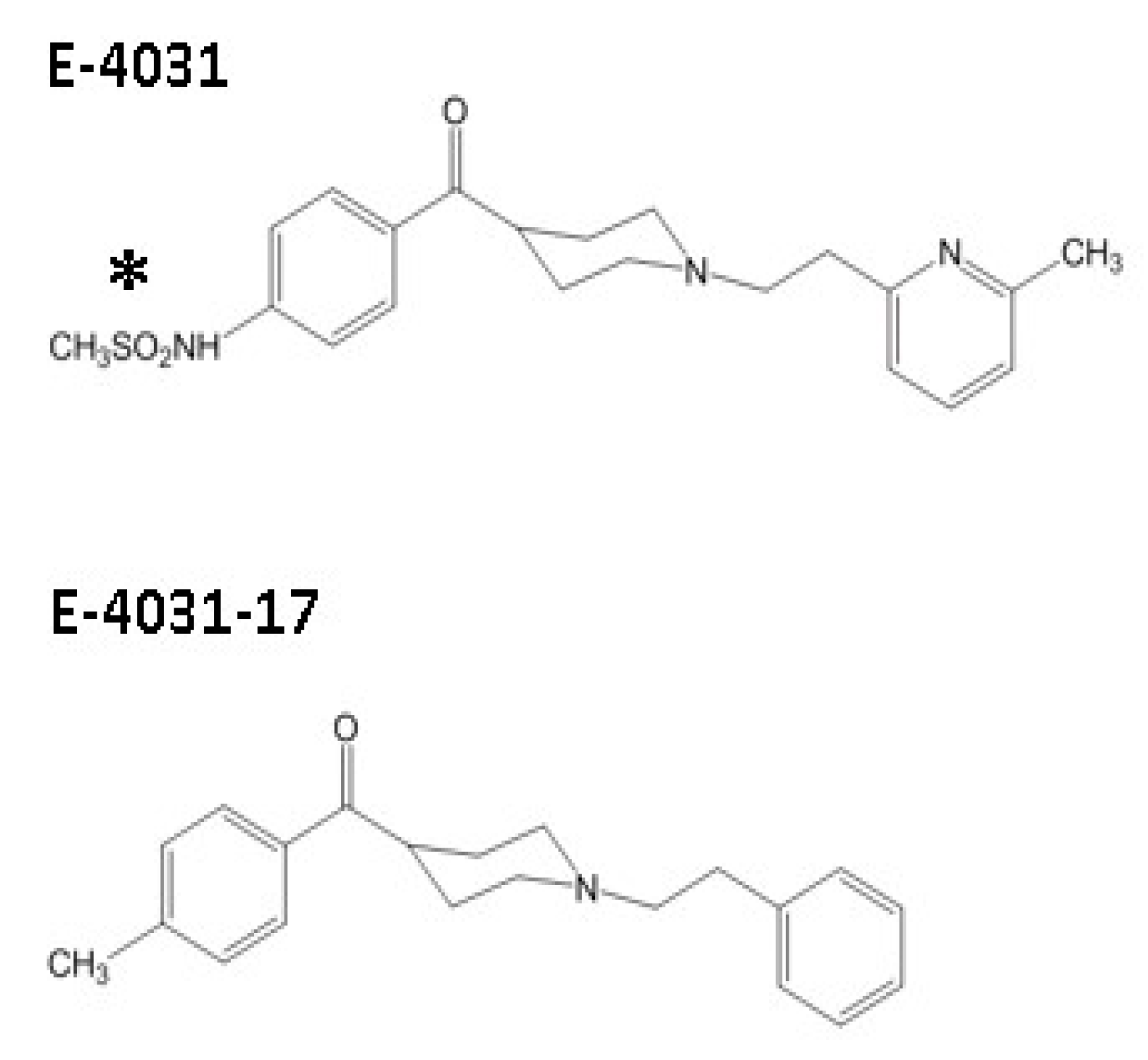{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Concentration Dependence of IhERG Inhibition by E-4031 and Its Analogue E-4031-17
2.2. IhERG Inhibition by E-4031 and E-4031-17 during a Sustained Depolarisation
2.3. Effects of E-4031 and E-4031-17 on IhERG Elicited at Different Test Voltages
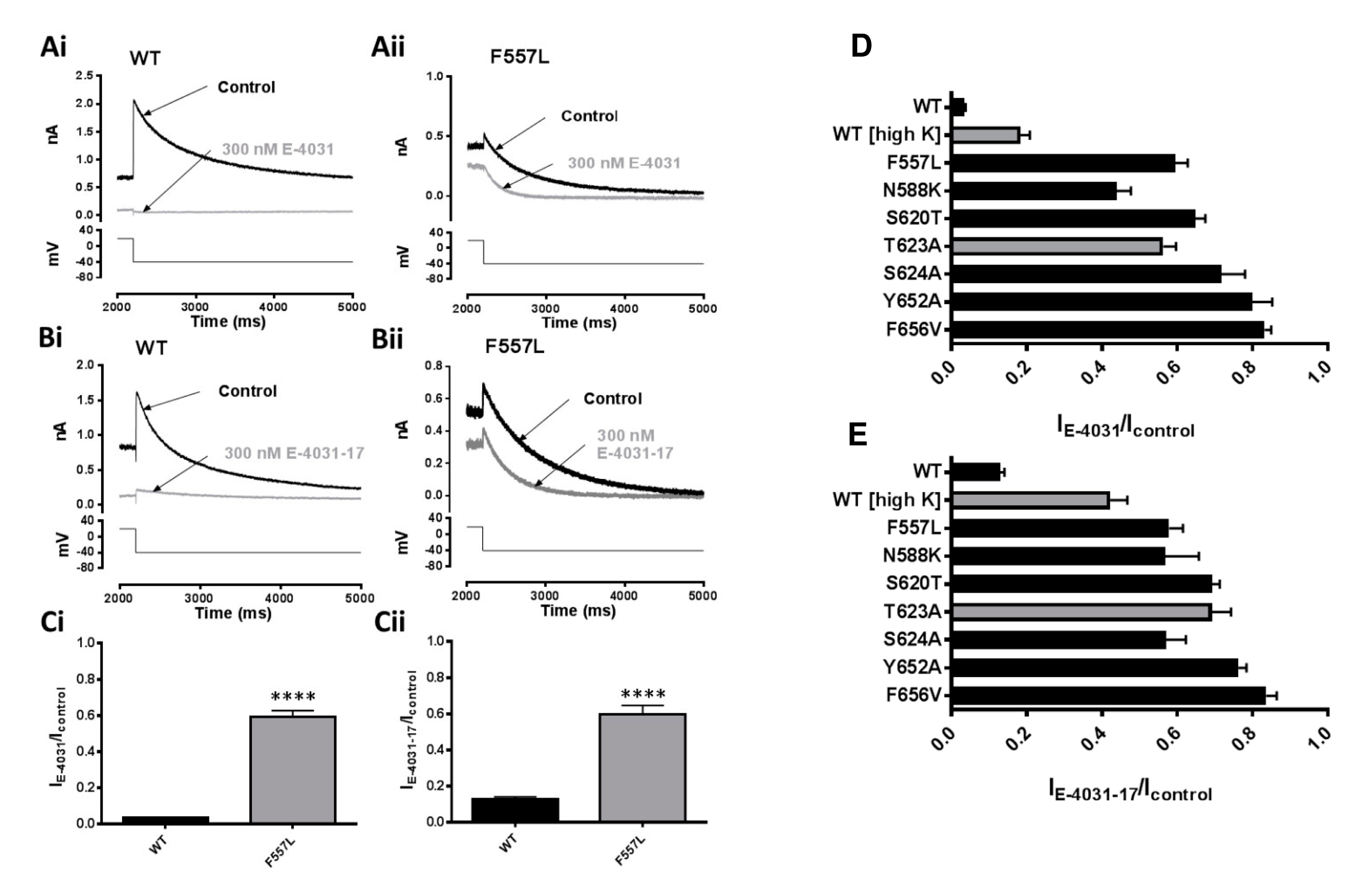
2.4. IhERG Inactivation and the Inhibitory Actions of E-4031 and E-4031-17
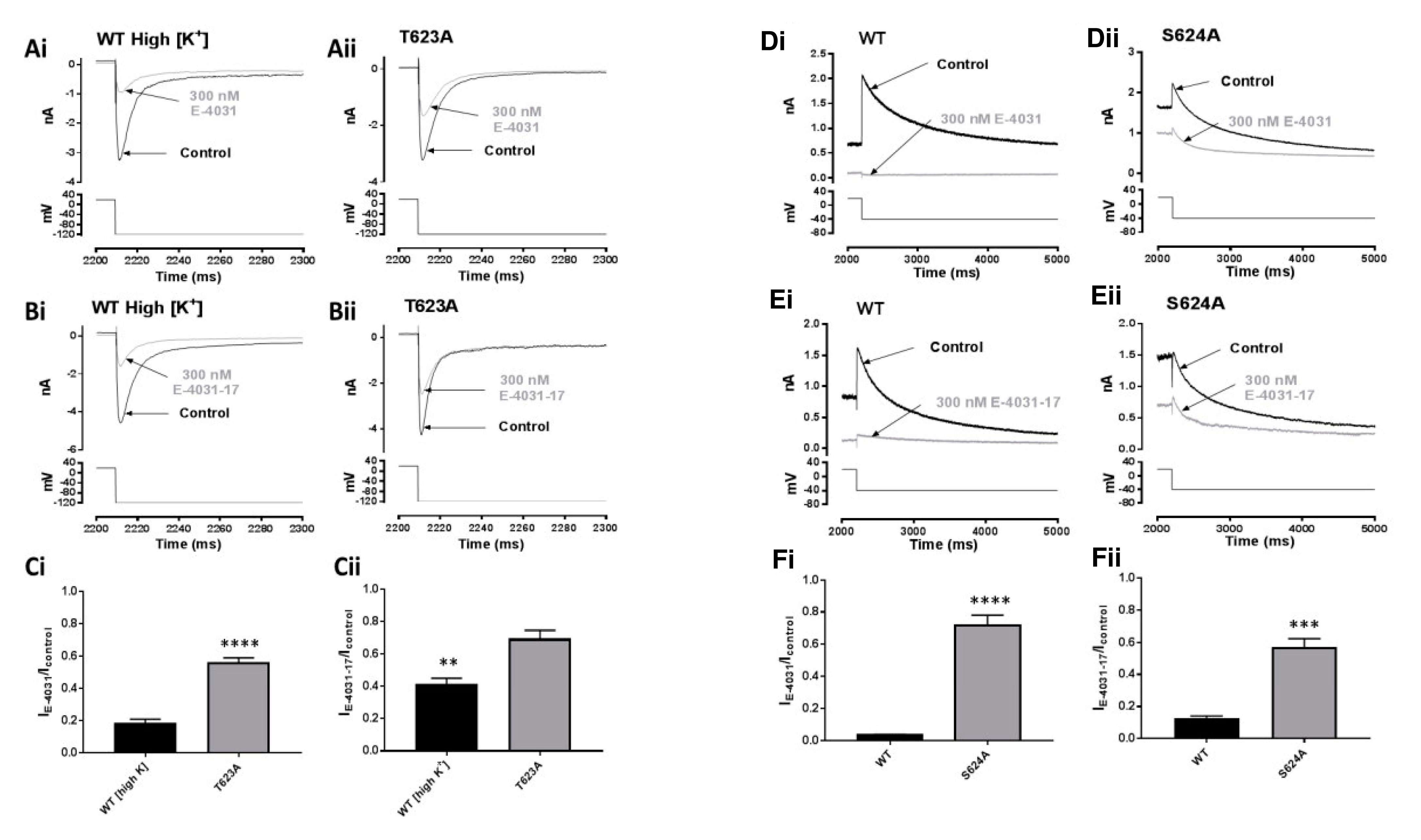
2.5. Effects of Mutations at the Base of the Pore Helix on E-4031 and E-4031-17
2.6. Effects of Mutation of S6 and S5 Aromatic Residues on Actions of E-4031 and E-4031-17
3. Discussion
3.1. Contextualizing the Inhibitory Potency of E-4031-17
3.2. E-4031—Comparison with Prior Experimental Studies
3.3. E-4031-17 Electrophysiology Data in Context
3.4. Blocking Determinants for E-4031 and E-4031-17
3.5. Conclusions
4. Materials and Methods
4.1. hERG Expressing Cell Lines and Mutants Employed
4.2. Maintenance of Mammalian Cell Lines and Transfection
4.3. Electrophysiological Recordings
4.4. Equations Used for Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Varro, A.; Tomek, J.; Nagy, N.; Virag, L.; Passini, E.; Rodriguez, B.; Baczko, I. Cardiac Transmembrane Ion Channels and Action Potentials: Cellular Physiology and Arrhythmogenic Behavior. Physiol. Rev. 2021, 101, 1083–1176. [Google Scholar] [PubMed]
- Sanguinetti, M.C.; Jiang, C.; Curran, M.E.; Keating, M.T. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell 1995, 81, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Trudeau, M.C.; Warmke, J.W.; Ganetzky, B.; Robertson, G.A. HERG, an inward rectifier in the voltage-gated potassium channel family. Science 1995, 269, 92–95. [Google Scholar] [CrossRef] [PubMed]
- Sanguinetti, M.C.; Tristani-Firouzi, M. hERG potassium channels and cardiac arrhythmia. Nature 2006, 440, 463–469. [Google Scholar] [CrossRef]
- Modell, S.M.; Lehmann, M.H. The long QT syndrome family of cardiac ion channelopathies: A HuGE review. Genet. Med. 2006, 8, 143–155. [Google Scholar] [CrossRef]
- Brugada, R.; Hong, K.; Dumaine, R.; Cordeiro, J.; Gaita, F.; Borggrefe, M.; Menendez, T.M.; Brugada, J.; Pollevick, G.D.; Wolpert, C.; et al. Sudden death associated with short-QT syndrome linked to mutations in HERG. Circulation 2004, 109, 30–35. [Google Scholar] [CrossRef]
- Maury, P.; Extramiana, F.; Sbragia, P.; Giustetto, C.; Schimpf, R.; Duparc, A.; Wolpert, C.; Denjoy, I.; Delay, M.; Borggrefe, M.; et al. Short QT syndrome. Update on a recent entity. Arch. Cardiovasc. Dis. 2008, 101, 779–786. [Google Scholar] [CrossRef]
- Hu, D.; Li, Y.; Zhang, J.; Pfeiffer, R.; Gollob, M.H.; Healey, J.; Harrell, D.T.; Makita, N.; Abe, H.; Sun, Y.; et al. The Phenotypic Spectrum of a Mutation Hotspot Responsible for the Short QT Syndrome. JACC Clin. Electrophysiol. 2017, 3, 727–743. [Google Scholar] [CrossRef]
- Campuzano, O.; Fernandez-Falgueras, A.; Lemus, X.; Sarquella-Brugada, G.; Cesar, S.; Coll, M.; Mates, J.; Arbelo, E.; Jorda, P.; Perez-Serra, A.; et al. Short QT Syndrome: A Comprehensive Genetic Interpretation and Clinical Translation of Rare Variants. J. Clin. Med. 2019, 8, 1035. [Google Scholar] [CrossRef]
- Tamargo, J.; Caballero, R.; Gomez, R.; Valenzuela, C.; Delpon, E. Pharmacology of cardiac potassium channels. Cardiovasc. Res. 2004, 62, 9–33. [Google Scholar] [CrossRef]
- TeBay, C.; Hill, A.P.; Windley, M.J. Metabolic and electrolyte abnormalities as risk factors in drug-induced long QT syndrome. Biophys. Rev. 2022, 14, 353–367. [Google Scholar] [CrossRef] [PubMed]
- Gintant, G.A. Preclinical Torsades-de-Pointes screens: Advantages and limitations of surrogate and direct approaches in evaluating proarrhythmic risk. Pharmacol. Ther. 2008, 119, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Vandenberg, J.I.; Walker, B.D.; Campbell, T.J. HERG K+ channels: Friend and foe. TIPS 2001, 22, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Anonymous. ICH S7B Note for guidance on the nonclinical evaluation of the potential for delayed ventricular repolarization (QT interval prolongation) by human pharmaceuticals. 2005. London, 25 May. Reference CHMP/ICH/423/02. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/ich-s-7-b-nonclinical-evaluation-potential-delayed-ventricular-repolarization-qt-interval_en.pdf (accessed on 27 July 2023).
- Sanguinetti, M.C.; Jurkiewicz, N.K. Two components of cardiac delayed rectifier K+ current. J. Gen. Physiol. 1990, 96, 195–215. [Google Scholar] [CrossRef] [PubMed]
- Mitcheson, J.S.; Chen, J.; Lin, M.; Culberson, C.; Sanguinetti, M.C. A structural basis for drug-induced long QT syndrome. Proc. Natl. Acad. Sci. USA 2000, 97, 12329–12333. [Google Scholar] [CrossRef] [PubMed]
- Lees-Miller, J.P.; Duan, Y.; Teng, G.Q.; Duff, H.J. Molecular determinant of high affinity dofetilide binding to HERG1 expressed in Xenopus oocytes: Involvement of S6 sites. Mol. Pharmacol. 2000, 57, 367–374. [Google Scholar]
- Kamiya, K.; Niwa, R.; Mitcheson, J.S.; Sanguinetti, M.C. Molecular determinants of HERG channel block. Mol. Pharmacol. 2006, 69, 1709–1716. [Google Scholar] [CrossRef]
- Saxena, P.; Zangerl-Plessl, E.M.; Linder, T.; Windisch, A.; Hohaus, A.; Timin, E.; Hering, S.; Stary-Weinzinger, A. New potential binding determinant for hERG channel inhibitors. Sci. Rep. 2016, 6, 24182. [Google Scholar] [CrossRef]
- Helliwell, M.V.; Zhang, Y.; El Harchi, A.; Du, C.; Hancox, J.C.; Dempsey, C.E. Structural implications of hERG K+ channel block by a high affinity minimally-structured blocker. J. Biol. Chem. 2018, 293, 7040–7057. [Google Scholar] [CrossRef]
- Cheng, H.; Du, C.; Zhang, Y.; James, A.F.; Dempsey, C.E.; Abdala, A.P.; Hancox, J.C. Potent hERG channel inhibition by sarizotan, an investigative treatment for Rett Syndrome. J. Mol. Cell. Cardiol. 2019, 135, 22–30. [Google Scholar] [CrossRef]
- Kamiya, K.; Niwa, R.; Morishima, M.; Honjo, H.; Sanguinetti, M.C. Molecular determinants of HERG channel block by terfenadine and cisapride. J. Pharmacol. Sci. 2008, 108, 301–307. [Google Scholar] [CrossRef]
- Witchel, H.J.; Dempsey, C.E.; Sessions, R.B.; Perry, M.; Milnes, J.T.; Hancox, J.C.; Mitcheson, J.S. The low-potency, voltage-dependent HERG channel blocker propafenone—Molecular determinants and drug trapping. Mol. Pharmacol. 2004, 66, 1201–1212. [Google Scholar] [CrossRef]
- Mitcheson, J.S.; Perry, S. Molecular determinants of high affinity drug binding to HERG channels. Curr. Opin. Drug. Discov. Dev. 2004, 6, 667–674. [Google Scholar]
- Perry, M.; Stansfeld, P.J.; Leaney, J.; Wood, C.; De Groot, M.J.; Leishman, D.; Sutcliffe, M.J.; Mitcheson, J.S. Drug binding interactions in the inner cavity of HERG channels: Molecular insights from structure-activity relationships of clofilium and ibutilide analogs. Mol. Pharmacol. 2006, 69, 509–519. [Google Scholar] [CrossRef]
- Vilums, M.; Overman, J.; Klaasse, E.; Scheel, O.; Brussee, J.; IJzerman, A.P. Understanding of molecular substructures that contribute to hERG K+ channel blockade: Synthesis and biological evaluation of E-4031 analogues. ChemMedChem 2012, 7, 107–113. [Google Scholar] [CrossRef]
- Helliwell, M.V. Structural Requirements for High-Affinity Block of hERG K+ Channels Using Electrophysiology and Mutagenesis with Simplified Blockers. Ph.D. Thesis, University of Bristol, Bristol, UK, 2019. Available online: https://research-information.bris.ac.uk/en/studentTheses/structural-requirements-for-high-affinity-block-of-herg-k-channel (accessed on 1 September 2022).
- Milnes, J.T.; Witchel, H.J.; Leaney, J.L.; Leishman, D.J.; Hancox, J.C. Investigating dynamic protocol-dependence of hERG potassium channel inhibition at 37 °C: Cisapride versus dofetilide. J. Pharmacol. Toxicol. Methods 2010, 61, 178–191. [Google Scholar] [CrossRef] [PubMed]
- Melgari, D.; Brack, K.E.; Zhang, C.; Zhang, Y.; El Harchi, A.; Mitcheson, J.S.; Dempsey, C.E.; Ng, G.A.; Hancox, J.C. hERG potassium channel blockade by the HCN channel inhibitor bradycardic agent ivabradine. J. Am. Heart Assoc. 2015, 4, e001813. [Google Scholar] [CrossRef]
- Zhou, Z.; Gong, Q.; Ye, B.; Fan, Z.; Makielski, J.C.; Robertson, G.A.; January, C.T. Properties of HERG channels stably expressed in HEK 293 cells studied at physiological temperature. Biophys. J. 1998, 74, 230–241. [Google Scholar] [CrossRef] [PubMed]
- McPate, M.J.; Duncan, R.S.; Witchel, H.J.; Hancox, J.C. Disopyramide is an effective inhibitor of mutant HERG K+ channels involved in variant 1 short QT syndrome. J. Mol. Cell. Cardiol. 2006, 41, 563–566. [Google Scholar] [CrossRef] [PubMed]
- Windley, M.J.; Farr, J.; TeBay, C.; Vandenberg, J.I.; Hill, A.P. High throuput measurement of hERG drug block kinetics using the CiPA dynamic protocol. J. Pharmacol. Toxicol. Methods 2022, 22, 107192. [Google Scholar] [CrossRef]
- Spector, P.S.; Curran, M.E.; Keating, M.T.; Sanguinetti, M.C. Class III antiarrhythmic drugs block HERG, a human cardiac delayed rectifier channel: Open channel block by methanesulfonanilides. Circ. Res. 1996, 78, 499–503. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Seebohm, G.; Sanguinetti, M.C. Position of aromatic residues in the S6 domain, not inactivation, dictates cisapride sensitivity of HERG and eag potassium channels. Proc. Natl. Acad. Sci. USA 2002, 99, 12329–12333. [Google Scholar] [CrossRef] [PubMed]
- Weerapura, M.; Hebert, T.E.; Nattel, S. Dofetilide block involves interactions with open and inactivated states of HERG channels. Pflugers Arch. 2002, 443, 520–531. [Google Scholar] [CrossRef] [PubMed]
- Perrin, M.J.; Kuchel, P.W.; Campbell, T.J.; Vandenberg, J.I. Drug binding to the inactivated state is necessary but not sufficient for high-affinity binding to human ether-à-go-go-related gene channels. Mol. Pharmacol. 2008, 74, 1443–1452. [Google Scholar] [CrossRef] [PubMed]
- Herzberg, I.M.; Trudeau, M.C.; Robertson, G.A. Transfer of rapid inactivation and sensitivity to the class III antiarrhythmic drug E-4031 from HERG to M-eag channels. J. Physiol. 1998, 511, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Cordeiro, J.M.; Brugada, R.; Wu, Y.S.; Hong, K.; Dumaine, R. Modulation of IKr inactivation by mutation N588K in KCNH2: A link to arrhythmogenesis in short QT syndrome. Cardiovas. Res. 2005, 67, 498–509. [Google Scholar] [CrossRef]
- McPate, M.J.; Duncan, R.S.; Milnes, J.T.; Witchel, H.J.; Hancox, J.C. The N588K-HERG K+ channel mutation in the “short QT syndrome”: Mechanism of gain-in-function determined at 37 °C. Biochem. Biophys. Res. Comm. 2005, 334, 441–449. [Google Scholar] [CrossRef]
- Ficker, E.; Jarolimek, W.; Kiehn, J.; Baumann, A.; Brown, A.M. Molecular determinants of dofetilide block of HERG K channels. Circ. Res. 1998, 82, 386–395. [Google Scholar]
- Kamiya, K.; Mitcheson, J.S.; Yasui, K.; Kodama, I.; Sanguinetti, M.C. Open channel block of HERG K+ channels by vesnarinone. Mol. Pharmacol. 2001, 60, 244–253. [Google Scholar] [CrossRef]
- Perry, M.D.; deGroot, M.J.; Helliwell, R.; Leishman, D.; Sanguinetti, M.C. Structural determinants of HERG channel block by clofilium and ibutilide. Mol. Pharmacol. 2004, 66, 240–249. [Google Scholar] [CrossRef]
- Shealy, R.T.; Murphy, A.D.; Ramarathnam, R.; Jakobsson, E.; Subramaniam, S. Sequence-function analysis of the K+-selective family of ion channels using a comprehensive alignment and the KcsA channel structure. Biophys. J. 2003, 84, 2929–2942. [Google Scholar] [CrossRef]
- Asai, T.; Adachi, N.; Moriya, T.; Oki, H.; Maru, T.; Kawasaki, M.; Suzuki, K.; Chen, S.; Ishii, R.; Yonemori, K.; et al. Cryo-EM Structure of K+ Bound hERG Channel Complexed with the Blocker Astemizole. Structure 2021, 29, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Chiu, P.J.; Marcoe, K.F.; Bounds, S.E.; Lin, C.H.; Feng, J.J.; Lin, A.; Cheng, F.C.; Crumb, W.J.; Mitchell, R. Validation of a [3H] astemizole binding assay in HEK293 cells expressing HERG K+ channels. J. Pharmacol. Sci. 2004, 95, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Baron, C.A.; Thiebaud, N.; Ren, M.; Viatchenko-Karpinski, S.; Indapurkar, A.; King, T.; Matta, M.K.; Ismaiel, O.A.; Patel, V.; Mashaee, M.; et al. hERG block potencies for 5 positive control drugs obtained per ICH E14/S7B Q&As best practices: Impact of recording temperature and drug loss. J. Pharmacol. Toxicol. Methods 2022, 117, 107193. [Google Scholar]
- Wang, S.; Morales, M.J.; Liu, S.; Strauss, H.C.; Rasmusson, R.L. Modulation of HERG affinity for E-4031 by [K]o and C-type inactivation. FEBS Lett. 1997, 417, 43–47. [Google Scholar] [CrossRef]
- Wu, W.; Gardner, A.; Sanguinetti, M.C. The Link between Inactivation and High-Affinity Block of hERG1 Channels. Mol. Pharmacol. 2015, 87, 1042–1050. [Google Scholar] [CrossRef]
- Carmeliet, E. Voltage- and time-dependent block of the delayed rectifier K+ current in cardiac myocytes by dofetilide. J. Pharmacol. Exp. Ther. 1992, 262, 809–817. [Google Scholar]
- Ohler, A.; Ravens, U. Effects of E-4031, almokalant and tedisamil on postrest action potential duration of human papillary muscles. J. Pharmacol. Exp. Ther. 1994, 270, 460–465. [Google Scholar]
- Mitcheson, J.S.; Chen, J.; Sanguinetti, M.C. Trapping of a methanesufonanlide by closure of the HERG potassium channel activation gate. J. Gen. Physiol. 2000, 115, 229–240. [Google Scholar] [CrossRef]
- Pareja, K.; Chu, E.; Dodyk, K.; Richter, K.; Miller, A. Role of the activation gate in determining the extracellular potassium dependency of block of HERG by trapped drugs. Channels 2013, 7, 23–33. [Google Scholar] [CrossRef][Green Version]
- Thouta, S.; Lo, G.; Grajauskas, L.; Claydon, T. Investigating the state dependence of drug binding in hERG channels using a trapped-open channel phenotype. Sci. Rep. 2018, 8, 4962. [Google Scholar]
- Imai, Y.N.; Ryu, S.; Oiki, S. Docking model of drug binding to the human ether-a-go-go potassium channel guided by tandem dimer mutant patch-clamp data: A synergic approach. J. Med. Chem. 2009, 52, 1630–1638. [Google Scholar] [CrossRef]
- Wang, W.; MacKinnon, R. Cryo-EM Structure of the Open Human Ether-a-go-go-Related K+ Channel hERG. Cell 2017, 169, 422–430. [Google Scholar] [CrossRef] [PubMed]
- Dempsey, C.E.; Wright, D.; Colenso, C.K.; Sessions, R.B.; Hancox, J.C. Assessing HERG pore models as templates for drug docking using published experimental constraints: The inactivated state in the context of drug block. J. Chem. Inf. Model 2014, 54, 601–612. [Google Scholar] [CrossRef]
- Farid, R.; Day, T.; Friesner, R.A.; Pearlstein, R.A. New insights about HERG blockade obtained from protein modeling, potential energy mapping, and docking studies. Bioorg. Med. Chem. 2006, 14, 3160–3173. [Google Scholar] [CrossRef] [PubMed]
- Maly, J.; Emigh, A.M.; DeMarco, K.R.; Furutani, K.; Sack, J.T.; Clancy, C.E.; Vorobyov, I.; Yarov-Yarovoy, V. Structural modeling of the hERG potassium channel and associated drug interactions. Front. Pharmacol. 2022, 13, 966463. [Google Scholar] [CrossRef]
- Perissinotti, L.; Guo, J.; Kudaibergenova, M.; Lees-Miller, J.; Ol’khovich, M.; Sharapova, A.; Perlovich, G.L.; Muruve, D.A.; Gerull, B.; Noskov, S.Y.; et al. The Pore-Lipid Interface: Role of Amino-Acid Determinants of Lipophilic Access by Ivabradine to the hERG1 Pore Domain. Mol. Pharmacol. 2019, 96, 259–271. [Google Scholar]
- Butler, A.; Helliwell, M.V.; Zhang, Y.; Hancox, J.C.; Dempsey, C.E. An Update on the Structure of hERG. Front. Pharmacol. 2019, 10, 1572. [Google Scholar] [CrossRef] [PubMed]
- Stansfeld, P.J.; Gedeck, P.; Gosling, M.; Cox, B.; Mitcheson, J.S. Drug block of the hERG potassium channel: Insight from modeling. Proteins 2007, 68, 568–580. [Google Scholar] [CrossRef]
- Stary, A.; Wacker, S.J.; Boukharta, L.; Zachariae, U.; Karimi-Nejad, Y.; Åqvist, J.; Vriend, G.; De Groot, B.L. Toward a consensus model of the HERG potassium channel. ChemMedChem 2010, 5, 455–467. [Google Scholar] [CrossRef]
- Hondeghem, L.M.; Snyders, D.J. Class III antiarrhythmic agents have a lot of potential but a long way to go. Circulation 1990, 81, 686–690. [Google Scholar] [CrossRef] [PubMed]
- Dorian, P.; Newman, D. Rate dependence of the effect of antiarrhythmic drugs delaying cardiac repolarization: An overview. Europace 2000, 2, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Milnes, J.T.; Crociani, O.; Arcangeli, A.; Hancox, J.C.; Witchel, H.J. Blockade of HERG potassium currents by fluvoxamine: Incomplete attenuation by S6 mutations at F656 or Y652. Br. J. Pharmacol. 2003, 139, 887–898. [Google Scholar] [CrossRef] [PubMed]
- Al-Moubarak, E.; Shiels, H.A.; Zhang, Y.; Du, C.; Hanington, O.; Harmer, S.C.; Dempsey, C.E.; Hancox, J.C. Inhibition of the hERG potassium channel by phenanthrene: A polycyclic aromatic hydrocarbon pollutant. Cell. Mol. Life Sci. 2021, 78, 7899–7914. [Google Scholar] [CrossRef]
- Wang, S.; Liu, S.; Morales, M.J.; Strauss, H.C.; Rasmusson, R.L. A quantitative analysis of the activation and inactivation kinetics of HERG expressed in Xenopus oocytes. J. Physiol. 1997, 502, 45–60. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.D.; Singleton, C.B.; Bursill, J.A.; Wyse, K.R.; Valenzuela, S.M.; Qiu, M.R.; Breit, S.N.; Campbell, T.J. Inhibition of the human ether-a-go-go related gene (HERG) potassium channel by cisapride: Affinity for open and inactivated states. Br. J. Pharmacol. 1999, 128, 444–450. [Google Scholar] [CrossRef]
- Zou, A.; Xu, Q.P.; Sanguinetti, M.C. A mutation in the pore region of HERG K channels expressed in Xenopus oocytes reduces rectification by shifting the voltage dependence of inactivation. J. Physiol. 1998, 509, 129–137. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Helliwell, M.V.; Zhang, Y.; El Harchi, A.; Dempsey, C.E.; Hancox, J.C. Inhibition of the hERG Potassium Channel by a Methanesulphonate-Free E-4031 Analogue. Pharmaceuticals 2023, 16, 1204. https://doi.org/10.3390/ph16091204
Helliwell MV, Zhang Y, El Harchi A, Dempsey CE, Hancox JC. Inhibition of the hERG Potassium Channel by a Methanesulphonate-Free E-4031 Analogue. Pharmaceuticals. 2023; 16(9):1204. https://doi.org/10.3390/ph16091204
Chicago/Turabian StyleHelliwell, Matthew V., Yihong Zhang, Aziza El Harchi, Christopher E. Dempsey, and Jules C. Hancox. 2023. "Inhibition of the hERG Potassium Channel by a Methanesulphonate-Free E-4031 Analogue" Pharmaceuticals 16, no. 9: 1204. https://doi.org/10.3390/ph16091204
APA StyleHelliwell, M. V., Zhang, Y., El Harchi, A., Dempsey, C. E., & Hancox, J. C. (2023). Inhibition of the hERG Potassium Channel by a Methanesulphonate-Free E-4031 Analogue. Pharmaceuticals, 16(9), 1204. https://doi.org/10.3390/ph16091204