

Cyclic Peptides in Pipeline: What Future for These Great Molecules?




Abstract
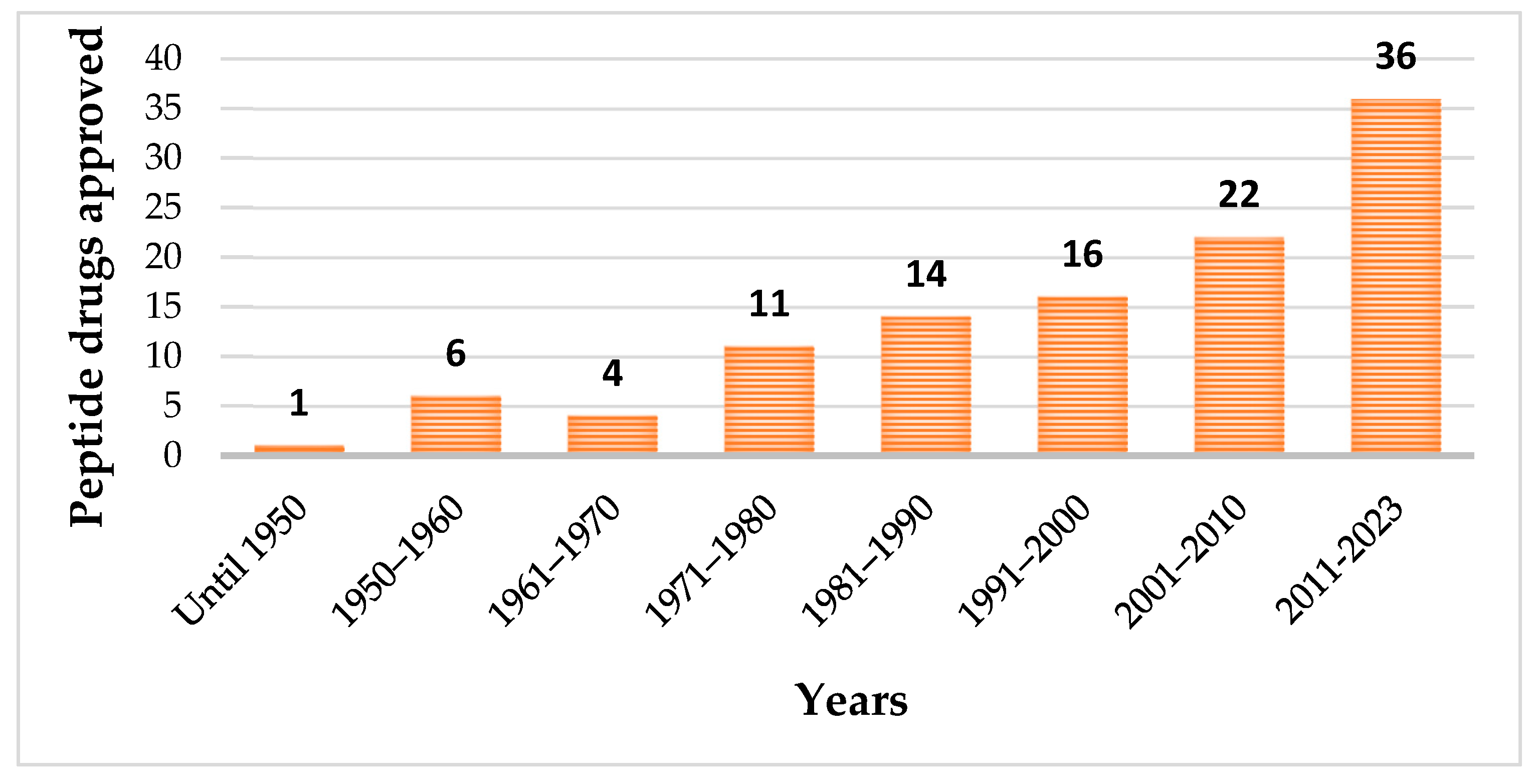
1. Introduction
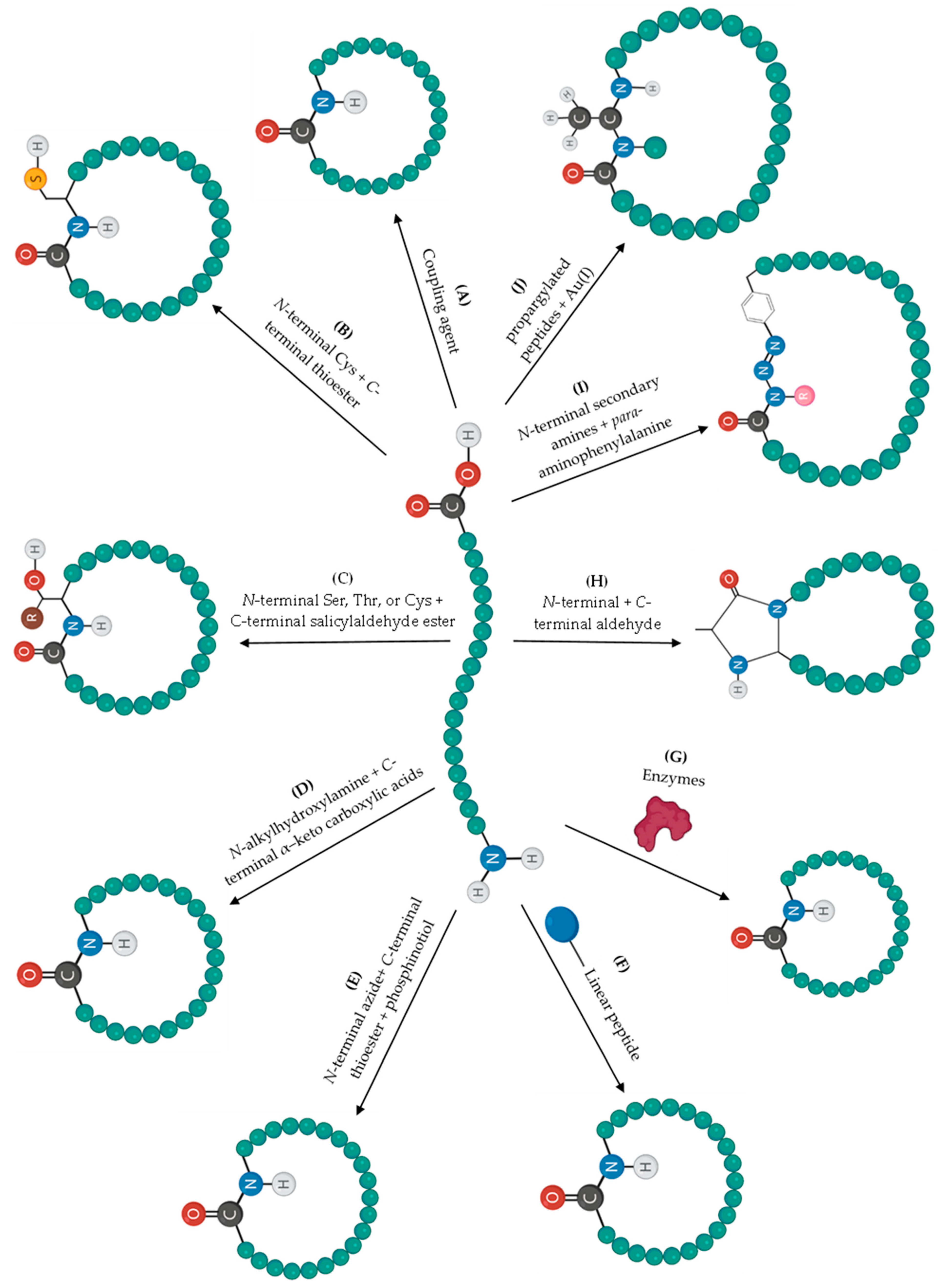
2. Cyclization Strategies
3. Strategies for Discovering and/or Optimizing Cyclic Peptides
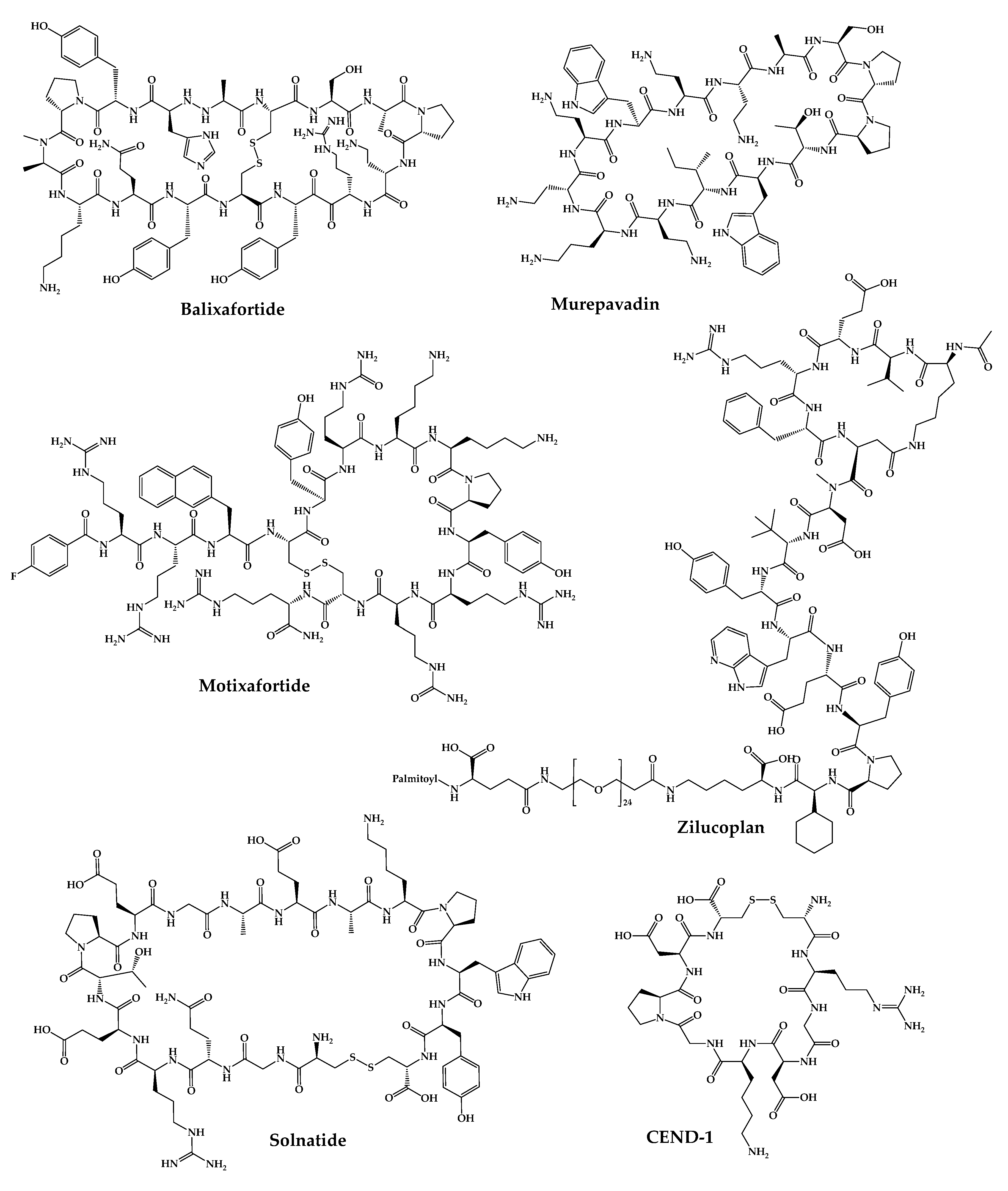
4. Cyclic Peptides in the Pipeline
4.1. POL6326—Balixafortide
4.2. BL-8040—Motixafortide
4.3. BT1718
4.4. BT8009
4.5. BT5528
4.6. VT1021
4.7. ALRN-6924
4.8. CEND-1
4.9. POL7080—Inhaled Murepavadin
4.10. Thanatin Derivatives
4.11. CD-101—Rezafungin
4.12. RA-101495—Zilucoplan
4.13. AP301—Solnatide
4.14. POL6014—Lonodelestat
4.15. THR-149
4.16. PTG-300—Rusfertide
4.17. PN-943
4.18. PL8177
4.19. PA-001
4.20. AZP-3813
4.21. PL9643
4.22. PM90001—Plitidepsin
4.23. APL-2—Pegcetacoplan
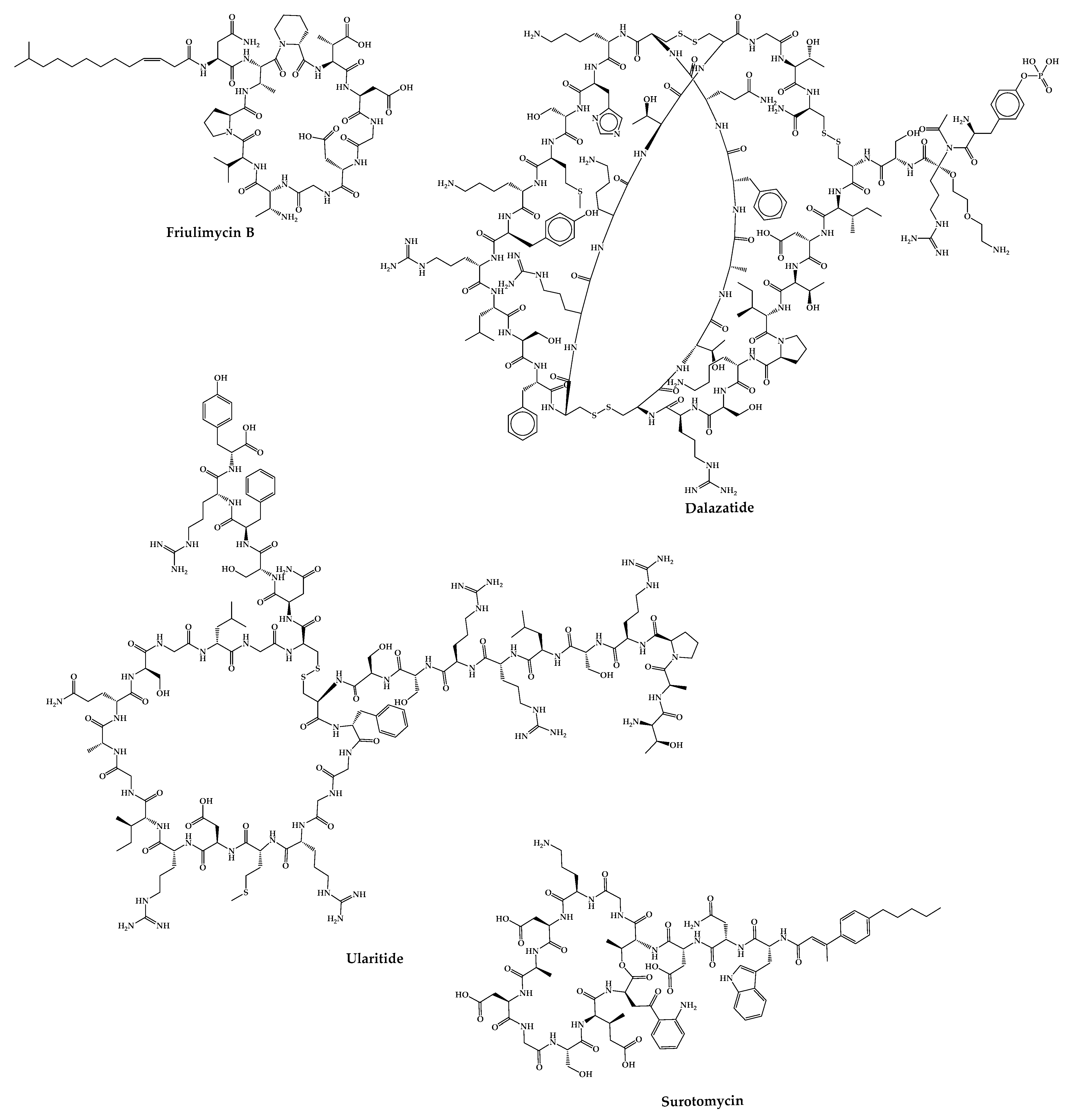
4.24. Friulimicin B
4.25. ShK-186—Dalazatide
4.26. Ularitide
4.27. CB-183,315—Surotomycin
5. Final Remarks
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- de la Torre, B.G.; Albericio, F. Peptide Therapeutics 2.0. Molecules 2020, 25, 2293. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, H.; Ghosh, D.; Williams, R.O. Just how prevalent are peptide therapeutic products? A critical review. Int. J. Pharm. 2020, 587, 119491. [Google Scholar] [CrossRef]
- Albericio, F.; Kruger, H.G. Therapeutic peptides. Future Med. Chem. 2012, 4, 1527–1531. [Google Scholar] [CrossRef]
- Akbarian, M.; Khani, A.; Eghbalpour, S.; Uversky, V.N. Bioactive Peptides: Synthesis, Sources, Applications, and Proposed Mechanisms of Action. Int. J. Mol. Sci. 2022, 23, 1445. [Google Scholar] [CrossRef]
- Zhang, H.; Chen, S. Cyclic peptide drugs approved in the last two decades (2001–2021). RSC Chem. Biol. 2022, 3, 18–31. [Google Scholar] [CrossRef] [PubMed]
- Rangel, M.; Santana, C.; Pinheiro, A.; Anjos, L.; Barth, T.; Pires Júnior, O.; Fontes, W.; S Castro, M. Marine Depsipeptides as Promising Pharmacotherapeutic Agents. Curr. Protein Pept. Sci. 2016, 17. [Google Scholar]
- Anjum, K.; Abbas, S.Q.; Akhter, N.; Shagufta, B.I.; Shah, S.A.A.; Hassan, S.S.u. Emerging biopharmaceuticals from bioactive peptides derived from marine organisms. Chem. Biol. Drug Des. 2017, 90, 12–30. [Google Scholar] [CrossRef] [PubMed]
- Venkatesan, J.; Anil, S.; Kim, S.-K.; Shim, M.S. Marine Fish Proteins and Peptides for Cosmeceuticals: A Review. Mar. Drugs 2017, 15, 143. [Google Scholar] [CrossRef]
- Charoensiddhi, S.; Conlon, M.A.; Franco, C.M.M.; Zhang, W. The development of seaweed-derived bioactive compounds for use as prebiotics and nutraceuticals using enzyme technologies. Trends Food Sci. Technol. 2017, 70, 20–33. [Google Scholar] [CrossRef]
- Ghosh, S.; Sarkar, T.; Pati, S.; Kari, Z.A.; Edinur, H.A.; Chakraborty, R. Novel Bioactive Compounds From Marine Sources as a Tool for Functional Food Development. Front. Mar. Sci. 2022, 9, 832957. [Google Scholar] [CrossRef]
- Benția, D.; Saceleanu, M.V.; Marinescu, A.A.; Ciurea, A.V. Centenary of Insulin Discovery (1921–2021): Nicolae Paulescu’s Original Contributions. Acta Endocrinol. 2021, 17, 406–411. [Google Scholar] [CrossRef]
- Ueno, H.; Zhang, W.; Nakazato, M. Regulation of feeding and therapeutic application of bioactive peptides. Pharmacol. Ther. 2022, 239, 108187. [Google Scholar] [CrossRef]
- Lau, J.L.; Dunn, M.K. Therapeutic peptides: Historical perspectives, current development trends, and future directions. Biorg. Med. Chem. 2018, 26, 2700–2707. [Google Scholar] [CrossRef]
- Muttenthaler, M.; King, G.F.; Adams, D.J.; Alewood, P.F. Trends in peptide drug discovery. Nat. Rev. Drug Discov. 2021, 20, 309–325. [Google Scholar] [CrossRef]
- Ruoslahti, E.; Pierschbacher, M.D. New Perspectives in Cell Adhesion: RGD and Integrins. Science 1987, 238, 491–497. [Google Scholar] [CrossRef]
- Storgard, C.M.; Stupack, D.G.; Jonczyk, A.; Goodman, S.L.; Fox, R.I.; Cheresh, D.A. Decreased angiogenesis and arthritic disease in rabbits treated with an αvβ3 antagonist. J. Clin. Investig. 1999, 103, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Mas-Moruno, C.; Rechenmacher, F.; Kessler, H. Cilengitide: The first anti-angiogenic small molecule drug candidate design, synthesis and clinical evaluation. Anticancer Agents Med. Chem. 2010, 10, 753–768. [Google Scholar] [CrossRef] [PubMed]
- Beer, A.J.; Haubner, R.; Goebel, M.; Luderschmidt, S.; Spilker, M.E.; Wester, H.-J.; Weber, W.A.; Schwaiger, M. Biodistribution and Pharmacokinetics of the αvβ3-Selective Tracer 18F-Galacto-RGD in Cancer Patients. J. Nucl. Med. 2005, 46, 1333–1341. [Google Scholar]
- Choi, J.S.; Joo, S.H. Recent Trends in Cyclic Peptides as Therapeutic Agents and Biochemical Tools. Biomol. Ther. 2020, 28, 18–24. [Google Scholar] [CrossRef]
- PepTherDia. Available online: http://peptherdia.herokuapp.com/ (accessed on 2 May 2023).
- D’Aloisio, V.; Dognini, P.; Hutcheon, G.A.; Coxon, C.R. PepTherDia: Database and structural composition analysis of approved peptide therapeutics and diagnostics. Drug Discov. Today 2021, 26, 1409–1419. [Google Scholar] [CrossRef] [PubMed]
- Gall, Y.M.; Konashev, M.B. The discovery of Gramicidin S: The Intellectual Transformation of G.F. Gause from Biologist to Researcher of Antibiotics and on its Meaning for the Fate of Russian Genetics. Hist. Philos. Life Sci. 2001, 23, 137–150. [Google Scholar]
- Gause, G.F.; Brazhnikova, M.G. Gramicidin S and its use in the Treatment of Infected Wounds. Nature 1944, 154, 703. [Google Scholar] [CrossRef]
- Klinker, K.P.; Borgert, S.J. Beyond Vancomycin: The Tail of the Lipoglycopeptides. Clin. Ther. 2015, 37, 2619–2636. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Zapata, D.; Petraitiene, R.; Petraitis, V. Echinocandins: The Expanding Antifungal Armamentarium. Clin. Infect. Dis. 2015, 61, S604–S611. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Zain, J. Romidepsin in the treatment of cutaneous T-cell lymphoma. J. Blood Med. 2011, 2, 37–47. [Google Scholar]
- Syed, Y.Y. Rezafungin: First Approval. Drugs 2023, 83, 833–840. [Google Scholar] [CrossRef]
- Du, Z.; Fan, B.; Dai, Q.; Wang, L.; Guo, J.; Ye, Z.; Cui, N.; Chen, J.; Tan, K.; Li, R.; et al. Supramolecular peptide nanostructures: Self-assembly and biomedical applications. Giant 2022, 9, 100082. [Google Scholar] [CrossRef]
- Panigrahi, B.; Singh, R.K.; Suryakant, U.; Mishra, S.; Potnis, A.A.; Jena, A.B.; Kerry, R.G.; Rajaram, H.; Ghosh, S.K.; Mandal, D. Cyclic peptides nanospheres: A ‘2-in-1′ self-assembled delivery system for targeting nucleus and cytoplasm. Eur. J. Pharm. Sci. 2022, 171, 106125. [Google Scholar] [CrossRef]
- Berkecz, R.; Tanács, D.; Péter, A.; Ilisz, I. Enantioselective Liquid Chromatographic Separations Using Macrocyclic Glycopeptide-Based Chiral Selectors. Molecules 2021, 26, 3380. [Google Scholar] [CrossRef]
- Teixeira, J.; Tiritan, M.E.; Pinto, M.M.M.; Fernandes, C. Chiral Stationary Phases for Liquid Chromatography: Recent Developments. Molecules 2019, 24, 865. [Google Scholar] [CrossRef]
- Armstrong, D.W.; Tang, Y.; Chen, S.; Zhou, Y.; Bagwill, C.; Chen, J.-R. Macrocyclic Antibiotics as a New Class of Chiral Selectors for Liquid Chromatography. Anal. Chem. 1994, 66, 1473–1484. [Google Scholar] [CrossRef]
- Fernandes, C.; Tiritan, M.E.; Cass, Q.; Kairys, V.; Fernandes, M.X.; Pinto, M. Enantioseparation and chiral recognition mechanism of new chiral derivatives of xanthones on macrocyclic antibiotic stationary phases. J. Chromatogr. A 2012, 1241, 60–68. [Google Scholar] [CrossRef]
- Phyo, Y.Z.; Cravo, S.; Palmeira, A.; Tiritan, M.E.; Kijjoa, A.; Pinto, M.M.M.; Fernandes, C. Enantiomeric Resolution and Docking Studies of Chiral Xanthonic Derivatives on Chirobiotic Columns. Molecules 2018, 23, 142. [Google Scholar] [CrossRef] [PubMed]
- Mustafa, K.; Kanwal, J.; Musaddiq, S.; Khakwani, S. Bioactive Peptides and Their Natural Sources. In Functional Foods and Nutraceuticals: Bioactive Components, Formulations and Innovations; Egbuna, C., Dable Tupas, G., Eds.; Springer International Publishing: Cham, Germany, 2020; pp. 75–97. [Google Scholar]
- Ahmed, I.; Asgher, M.; Sher, F.; Hussain, S.M.; Nazish, N.; Joshi, N.; Sharma, A.; Parra-Saldívar, R.; Bilal, M.; Iqbal, H.M.N. Exploring Marine as a Rich Source of Bioactive Peptides: Challenges and Opportunities from Marine Pharmacology. Mar. Drugs 2022, 20, 208. [Google Scholar] [CrossRef]
- Bhat, Z.F.; Kumar, S.; Bhat, H.F. Bioactive peptides of animal origin: A review. J. Food Sci. Technol. 2015, 52, 5377–5392. [Google Scholar] [CrossRef]
- Robinson, S.D.; Undheim, E.A.B.; Ueberheide, B.; King, G.F. Venom peptides as therapeutics: Advances, challenges and the future of venom-peptide discovery. Expert Rev. Proteom. 2017, 14, 931–939. [Google Scholar] [CrossRef]
- Lewis, R.J.; Garcia, M.L. Therapeutic potential of venom peptides. Nat. Rev. Drug Discov. 2003, 2, 790–802. [Google Scholar] [CrossRef] [PubMed]
- Daly, N.L.; Wilson, D.T. Plant derived cyclic peptides. Biochem. Soc. Trans. 2021, 49, 1279–1285. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lin, M.; Xu, D.; Lai, D.; Zhou, L. Structural Diversity and Biological Activities of Fungal Cyclic Peptides, Excluding Cyclodipeptides. Molecules 2017, 22, 2069. [Google Scholar] [CrossRef] [PubMed]
- Kisil, O.V.; Efimenko, T.A.; Efremenkova, O.V. Looking Back to Amycolatopsis: History of the Antibiotic Discovery and Future Prospects. Antibiotics 2021, 10, 1254. [Google Scholar] [CrossRef]
- Debono, M.; Barnhart, M.; Carrell, C.B.; Hoffmann, J.A.; Occolowitz, J.L.; Abbott, B.J.; Fukuda, D.S.; Hamill, R.L.; Biemann, K.; Herlihy, W.C. A21978C, a complex of new acidic peptide antibiotics: Isolation, chemistry, and mass spectral structure elucidation. J. Antibiot. 1987, 40, 761–777. [Google Scholar] [CrossRef]
- Ling, L.L.; Schneider, T.; Peoples, A.J.; Spoering, A.L.; Engels, I.; Conlon, B.P.; Mueller, A.; Schäberle, T.F.; Hughes, D.E.; Epstein, S.; et al. A new antibiotic kills pathogens without detectable resistance. Nature 2015, 517, 455–459. [Google Scholar] [CrossRef]
- Gu, H.; Han, S.M.; Park, K.-K. Therapeutic Effects of Apamin as a Bee Venom Component for Non-Neoplastic Disease. Toxins 2020, 12, 195. [Google Scholar] [CrossRef]
- White, K.M.; Rosales, R.; Yildiz, S.; Kehrer, T.; Miorin, L.; Moreno, E.; Jangra, S.; Uccellini, M.B.; Rathnasinghe, R.; Coughlan, L.; et al. Plitidepsin has potent preclinical efficacy against SARS-CoV-2 by targeting the host protein eEF1A. Science 2021, 371, 926–931. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, S.; Adholeya, A.; Deshmukh, S.K. The Pharmacological Potential of Non-ribosomal Peptides from Marine Sponge and Tunicates. Front. Pharmacol. 2016, 7, 333. [Google Scholar] [CrossRef] [PubMed]
- Daletos, G.; Kalscheuer, R.; Koliwer-Brandl, H.; Hartmann, R.; de Voogd, N.J.; Wray, V.; Lin, W.; Proksch, P. Callyaerins from the Marine Sponge Callyspongia aerizusa: Cyclic Peptides with Antitubercular Activity. J. Nat. Prod. 2015, 78, 1910–1925. [Google Scholar] [CrossRef]
- Xu, W.-J.; Liao, X.-J.; Xu, S.-H.; Diao, J.-Z.; Du, B.; Zhou, X.-L.; Pan, S.-S. Isolation, Structure Determination, and Synthesis of Galaxamide, A Rare Cytotoxic Cyclic Pentapeptide from a Marine Algae Galaxaura filamentosa. Org. Lett. 2008, 10, 4569–4572. [Google Scholar] [CrossRef]
- Wibowo, J.T.; Bayu, A.; Aryati, W.D.; Fernandes, C.; Yanuar, A.; Kijjoa, A.; Putra, M.Y. Secondary Metabolites from Marine-Derived Bacteria with Antibiotic and Antibiofilm Activities against Drug-Resistant Pathogens. Mar. Drugs 2023, 21, 50. [Google Scholar] [CrossRef] [PubMed]
- Pesic, A.; Baumann, H.I.; Kleinschmidt, K.; Ensle, P.; Wiese, J.; Süssmuth, R.D.; Imhoff, J.F. Champacyclin, a New Cyclic Octapeptide from Streptomyces Strain C42 Isolated from the Baltic Sea. Mar. Drugs 2013, 11, 4834–4857. [Google Scholar] [CrossRef] [PubMed]
- Linington, R.G.; González, J.; Ureña, L.-D.; Romero, L.I.; Ortega-Barría, E.; Gerwick, W.H. Venturamides A and B: Antimalarial Constituents of the Panamanian Marine Cyanobacterium Oscillatoria sp. J. Nat. Prod. 2007, 70, 397–401. [Google Scholar] [CrossRef] [PubMed]
- Linington, R.G.; Edwards, D.J.; Shuman, C.F.; McPhail, K.L.; Matainaho, T.; Gerwick, W.H. Symplocamide A, a Potent Cytotoxin and Chymotrypsin Inhibitor from the Marine Cyanobacterium Symploca sp. J. Nat. Prod. 2008, 71, 22–27. [Google Scholar] [CrossRef] [PubMed]
- de Sá, J.D.M.; Kumla, D.; Dethoup, T.; Kijjoa, A. Bioactive Compounds from Terrestrial and Marine-Derived Fungi of the Genus Neosartorya &dagger. Molecules 2022, 27, 2351. [Google Scholar] [PubMed]
- Du, F.-Y.; Zhang, P.; Li, X.-M.; Li, C.-S.; Cui, C.-M.; Wang, B.-G. Cyclohexadepsipeptides of the Isaridin Class from the Marine-Derived Fungus Beauveria felina EN-135. J. Nat. Prod. 2014, 77, 1164–1169. [Google Scholar] [CrossRef] [PubMed]
- Wyche, T.P.; Hou, Y.; Vazquez-Rivera, E.; Braun, D.; Bugni, T.S. Peptidolipins B–F, Antibacterial Lipopeptides from an Ascidian-Derived Nocardia sp. J. Nat. Prod. 2012, 75, 735–740. [Google Scholar] [CrossRef] [PubMed]
- Amelia, T.S.M.; Suaberon, F.A.C.; Vad, J.; Fahmi, A.D.M.; Saludes, J.P.; Bhubalan, K. Recent Advances of Marine Sponge-Associated Microorganisms as a Source of Commercially Viable Natural Products. Mar. Biotechnol. 2022, 24, 492–512. [Google Scholar] [CrossRef] [PubMed]
- Prompanya, C.; Fernandes, C.; Cravo, S.; Pinto, M.M.M.; Dethoup, T.; Silva, A.M.S.; Kijjoa, A. A New Cyclic Hexapeptide and a New Isocoumarin Derivative from the Marine Sponge-Associated Fungus Aspergillus similanensis KUFA 0013. Mar. Drugs 2015, 13, 1432–1450. [Google Scholar] [CrossRef]
- May Zin, W.W.; Buttachon, S.; Dethoup, T.; Fernandes, C.; Cravo, S.; Pinto, M.M.M.; Gales, L.; Pereira, J.A.; Silva, A.M.S.; Sekeroglu, N.; et al. New Cyclotetrapeptides and a New Diketopiperzine Derivative from the Marine Sponge-Associated Fungus Neosartorya glabra KUFA 0702. Mar. Drugs 2016, 14, 136. [Google Scholar] [CrossRef]
- Phyo, Y.Z.; Ribeiro, J.; Fernandes, C.; Kijjoa, A.; Pinto, M.M.M. Marine Natural Peptides: Determination of Absolute Configuration Using Liquid Chromatography Methods and Evaluation of Bioactivities. Molecules 2018, 23, 306. [Google Scholar] [CrossRef]
- Fernandes, C.; Ribeiro, R.; Pinto, M.; Kijjoa, A. Absolute Stereochemistry Determination of Bioactive Marine-Derived Cyclopeptides by Liquid Chromatography Methods: An Update Review. Molecules 2023, 28, 615. [Google Scholar] [CrossRef]
- Kang, H.K.; Choi, M.-C.; Seo, C.H.; Park, Y. Therapeutic Properties and Biological Benefits of Marine-Derived Anticancer Peptides. Int. J. Mol. Sci. 2018, 19, 919. [Google Scholar] [CrossRef]
- Ribeiro, R.; Pinto, E.; Fernandes, C.; Sousa, E. Marine Cyclic Peptides: Antimicrobial Activity and Synthetic Strategies. Mar. Drugs 2022, 20, 397. [Google Scholar] [CrossRef] [PubMed]
- Sridhar, K.; Inbaraj, B.S.; Chen, B.-H. Recent developments on production, purification and biological activity of marine peptides. Food Res. Int. 2021, 147, 110468. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, M.; Cruz, L.J.; Hunkapiller, M.W.; Gray, W.R.; Olivera, B.M. Isolation and structure of a peptide toxin from the marine snail Conus magus. Arch. Biochem. Biophys. 1982, 218, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Kohli, R.M.; Walsh, C.T.; Burkart, M.D. Biomimetic synthesis and optimization of cyclic peptide antibiotics. Nature 2002, 418, 658–661. [Google Scholar] [CrossRef] [PubMed]
- Albericio, F. Developments in peptide and amide synthesis. Curr. Opin. Chem. Biol. 2004, 8, 211–221. [Google Scholar] [CrossRef]
- Jiang, S.; Li, Z.; Ding, K.; Roller, P.P. Recent Progress of Synthetic Studies to Peptide and Peptidomimetic Cyclization. Curr. Org. Chem. 2008, 12, 1502–1542. [Google Scholar] [CrossRef]
- Hamada, Y.; Shioiri, T. Recent Progress of the Synthetic Studies of Biologically Active Marine Cyclic Peptides and Depsipeptides. Chem. Rev. 2005, 105, 4441–4482. [Google Scholar] [CrossRef]
- Anand, M.; Alagar, M.; Ranjitha, J.; Selvaraj, V. Total synthesis and anticancer activity of a cyclic heptapeptide from marine sponge using water soluble peptide coupling agent EDC. Arab. J. Chem. 2019, 12, 2782–2787. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Moon, D.K. Enantioselective Total Synthesis of (+)-Jasplakinolide. Org. Lett. 2007, 9, 2425–2427. [Google Scholar] [CrossRef]
- Jing, X.; Jin, K. A gold mine for drug discovery: Strategies to develop cyclic peptides into therapies. Med. Res. Rev. 2020, 40, 753–810. [Google Scholar] [CrossRef]
- Gentilucci, L.; De Marco, R.; Cerisoli, L. Chemical modifications designed to improve peptide stability: Incorporation of non-natural amino acids, pseudo-peptide bonds, and cyclization. Curr. Pharm. Des. 2010, 16, 3185–3203. [Google Scholar] [CrossRef] [PubMed]
- Wallace, R.J. Acetylation of peptides inhibits their degradation by rumen micro-organisms. Br. J. Nutr. 1992, 68, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Moradi, S.V.; Hussein, W.M.; Varamini, P.; Simerska, P.; Toth, I. Glycosylation, an effective synthetic strategy to improve the bioavailability of therapeutic peptides. Chem. Sci. 2016, 7, 2492–2500. [Google Scholar] [CrossRef]
- Lau, J.; Bloch, P.; Schäffer, L.; Pettersson, I.; Spetzler, J.; Kofoed, J.; Madsen, K.; Knudsen, L.B.; McGuire, J.; Steensgaard, D.B.; et al. Discovery of the Once-Weekly Glucagon-Like Peptide-1 (GLP-1) Analogue Semaglutide. J. Med. Chem. 2015, 58, 7370–7380. [Google Scholar] [CrossRef]
- Trier, S.; Linderoth, L.; Bjerregaard, S.; Strauss, H.M.; Rahbek, U.L.; Andresen, T.L. Acylation of salmon calcitonin modulates in vitro intestinal peptide flux through membrane permeability enhancement. Eur. J. Pharm. Biopharm. 2015, 96, 329–337. [Google Scholar] [CrossRef]
- Li, W.; O’Brien-Simpson, N.M.; Hossain, M.A.; Wade, J.D. The 9-Fluorenylmethoxycarbonyl (Fmoc) Group in Chemical Peptide Synthesis – Its Past, Present, and Future. Aust. J. Chem. 2020, 73, 271–276. [Google Scholar] [CrossRef]
- Amblard, M.; Fehrentz, J.-A.; Martinez, J.; Subra, G. Methods and protocols of modern solid phase peptide synthesis. Mol. Biotechnol. 2006, 33, 239–254. [Google Scholar] [CrossRef]
- Ferrazzano, L.; Catani, M.; Cavazzini, A.; Martelli, G.; Corbisiero, D.; Cantelmi, P.; Fantoni, T.; Mattellone, A.; De Luca, C.; Felletti, S.; et al. Sustainability in peptide chemistry: Current synthesis and purification technologies and future challenges. Green Chem. 2022, 24, 975–1020. [Google Scholar] [CrossRef]
- Al Musaimi, O.; de la Torre, B.G.; Albericio, F. Greening Fmoc/tBu solid-phase peptide synthesis. Green Chem. 2020, 22, 996–1018. [Google Scholar] [CrossRef]
- Lamers, C. Overcoming the shortcomings of peptide-based therapeutics. Future Drug Discov. 2022, 4, FDD75. [Google Scholar] [CrossRef]
- White, C.J.; Yudin, A.K. Contemporary strategies for peptide macrocyclization. Nat. Chem. 2011, 3, 509–524. [Google Scholar] [CrossRef]
- Bechtler, C.; Lamers, C. Macrocyclization strategies for cyclic peptides and peptidomimetics. RSC Med. Chem. 2021, 12, 1325–1351. [Google Scholar] [CrossRef]
- Chow, H.Y.; Zhang, Y.; Matheson, E.; Li, X. Ligation Technologies for the Synthesis of Cyclic Peptides. Chem. Rev. 2019, 119, 9971–10001. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.-M.; Liu, S.-Z.; Cheng, X.-Z.; Ding, W.-Z.; Zhu, T.; Chen, B. Recent progress of on-resin cyclization for the synthesis of clycopeptidomimetics. Chin. Chem. Lett. 2016, 27, 1731–1739. [Google Scholar] [CrossRef]
- Dawson, P.E.; Muir, T.W.; Clark-Lewis, I.; Kent, S.B.H. Synthesis of Proteins by Native Chemical Ligation. Science 1994, 266, 776–779. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.-S.; Chang, H.-N.; Shi, J.; Liu, L. Chemical synthesis of a cyclotide via intramolecular cyclization of peptide O-esters. Sci. China Chem. 2012, 55, 64–69. [Google Scholar] [CrossRef]
- Yan, L.Z.; Dawson, P.E. Synthesis of Peptides and Proteins without Cysteine Residues by Native Chemical Ligation Combined with Desulfurization. J. Am. Chem. Soc. 2001, 123, 526–533. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.F.; Tam, J.P. Peptide segment ligation strategy without use of protecting groups. Proc. Natl. Acad. Sci. USA 1994, 91, 6584–6588. [Google Scholar] [CrossRef]
- Li, X.; Lam, H.Y.; Zhang, Y.; Chan, C.K. Salicylaldehyde Ester-Induced Chemoselective Peptide Ligations: Enabling Generation of Natural Peptidic Linkages at the Serine/Threonine Sites. Org. Lett. 2010, 12, 1724–1727. [Google Scholar] [CrossRef]
- Lam, H.Y.; Zhang, Y.; Liu, H.; Xu, J.; Wong, C.T.T.; Xu, C.; Li, X. Total Synthesis of Daptomycin by Cyclization via a Chemoselective Serine Ligation. J. Am. Chem. Soc. 2013, 135, 6272–6279. [Google Scholar] [CrossRef]
- Wong, C.T.T.; Lam, H.Y.; Li, X. Effective synthesis of kynurenine-containing peptides via on-resin ozonolysis of tryptophan residues: Synthesis of cyclomontanin B. Org. Biomol. Chem. 2013, 11, 7616–7620. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.T.T.; Lam, H.Y.; Li, X. Effective synthesis of cyclic peptide yunnanin C and analogues via Ser/Thr ligation (STL)-mediated peptide cyclization. Tetrahedron 2014, 70, 7770–7773. [Google Scholar] [CrossRef]
- Zhao, J.-F.; Zhang, X.-H.; Ding, Y.-J.; Yang, Y.-S.; Bi, X.-B.; Liu, C.-F. Facile Synthesis of Peptidyl Salicylaldehyde Esters and Its Use in Cyclic Peptide Synthesis. Org. Lett. 2013, 15, 5182–5185. [Google Scholar] [CrossRef] [PubMed]
- Bode, J.W.; Fox, R.M.; Baucom, K.D. Chemoselective Amide Ligations by Decarboxylative Condensations of N-Alkylhydroxylamines and α-Ketoacids. Angew. Chem. Int. Ed. 2006, 45, 1248–1252. [Google Scholar] [CrossRef]
- Fukuzumi, T.; Ju, L.; Bode, J.W. Chemoselective cyclization of unprotected linear peptides by α-ketoacid–hydroxylamine amide-ligation. Org. Biomol. Chem. 2012, 10, 5837–5844. [Google Scholar] [CrossRef]
- Nilsson, B.L.; Kiessling, L.L.; Raines, R.T. Staudinger Ligation: A Peptide from a Thioester and Azide. Org. Lett. 2000, 2, 1939–1941. [Google Scholar] [CrossRef]
- Saxon, E.; Armstrong, J.I.; Bertozzi, C.R. A “Traceless” Staudinger Ligation for the Chemoselective Synthesis of Amide Bonds. Org. Lett. 2000, 2, 2141–2143. [Google Scholar] [CrossRef]
- Lambert, J.N.; Mitchell, J.P.; Roberts, K.D. The synthesis of cyclic peptides. J. Chem. Soc. Perkin Trans. 1 2001, 5, 471–484. [Google Scholar] [CrossRef]
- Rosenbaum, C.; Waldmann, H. Solid phase synthesis of cyclic peptides by oxidative cyclative cleavage of an aryl hydrazide linker—Synthesis of stylostatin 1. Tetrahedron Lett. 2001, 42, 5677–5680. [Google Scholar] [CrossRef]
- Abdel Monaim, S.A.H.; Ramchuran, E.J.; El-Faham, A.; Albericio, F.; de la Torre, B.G. Converting Teixobactin into a Cationic Antimicrobial Peptide (AMP). J. Med. Chem. 2017, 60, 7476–7482. [Google Scholar] [CrossRef]
- Wills, R.; Adebomi, V.; Raj, M. Site-Selective Peptide Macrocyclization. ChemBioChem 2021, 22, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, G.K.T.; Wang, S.; Qiu, Y.; Hemu, X.; Lian, Y.; Tam, J.P. Butelase 1 is an Asx-specific ligase enabling peptide macrocyclization and synthesis. Nat. Chem. Biol. 2014, 10, 732–738. [Google Scholar] [CrossRef] [PubMed]
- Hof, W.v.t.; Maňásková, S.H.; Veerman, E.C.I.; Bolscher, J.G.M. Sortase-mediated backbone cyclization of proteins and peptides. Biol. Chem. 2015, 396, 283–293. [Google Scholar]
- Jackson, D.Y.; Burnier, J.P.; Wells, J.A. Enzymic Cyclization of Linear Peptide Esters Using Subtiligase. J. Am. Chem. Soc. 1995, 117, 819–820. [Google Scholar] [CrossRef]
- Touati, J.; Angelini, A.; Hinner, M.J.; Heinis, C. Enzymatic Cyclisation of Peptides with a Transglutaminase. ChemBioChem 2011, 12, 38–42. [Google Scholar] [CrossRef]
- Hemu, X.; Qiu, Y.; Nguyen, G.K.T.; Tam, J.P. Total Synthesis of Circular Bacteriocins by Butelase 1. J. Am. Chem. Soc. 2016, 138, 6968–6971. [Google Scholar] [CrossRef]
- Hayes, H.C.; Luk, L.Y.P.; Tsai, Y.-H. Approaches for peptide and protein cyclisation. Org. Biomol. Chem. 2021, 19, 3983–4001. [Google Scholar] [CrossRef]
- Wills, R.; Adebomi, V.; Spancake, C.; Cohen, R.D.; Raj, M. Synthesis of L-cyclic tetrapeptides by backbone amide activation CyClick strategy. Tetrahedron 2022, 126, 133071. [Google Scholar] [CrossRef]
- Adebomi, V.; Cohen, R.D.; Wills, R.; Chavers, H.A.H.; Martin, G.E.; Raj, M. CyClick Chemistry for the Synthesis of Cyclic Peptides. Angew. Chem. Int. Ed. 2019, 58, 19073–19080. [Google Scholar] [CrossRef]
- Nwajiobi, O.; Verma, A.K.; Raj, M. Rapid Arene Triazene Chemistry for Macrocyclization. J. Am. Chem. Soc. 2022, 144, 4633–4641. [Google Scholar] [CrossRef]
- Vanjari, R.; Panda, D.; Mandal, S.; Vamisetti, G.B.; Brik, A. Gold(I)-Mediated Rapid Cyclization of Propargylated Peptides via Imine Formation. J. Am. Chem. Soc. 2022, 144, 4966–4976. [Google Scholar] [CrossRef]
- Jin, K. Developing cyclic peptide-based drug candidates: An overview. Future Med. Chem. 2020, 12, 1687–1690. [Google Scholar] [CrossRef]
- Di, L. Strategic Approaches to Optimizing Peptide ADME Properties. AAPS J. 2015, 17, 134–143. [Google Scholar] [CrossRef]
- Craik, D.J.; Adams, D.J. Chemical Modification of Conotoxins to Improve Stability and Activity. ACS Chem. Biol. 2007, 2, 457–468. [Google Scholar] [CrossRef]
- Hilpert, K.; Volkmer-Engert, R.; Walter, T.; Hancock, R.E.W. High-throughput generation of small antibacterial peptides with improved activity. Nat. Biotechnol. 2005, 23, 1008–1012. [Google Scholar] [CrossRef]
- Domhan, C.; Uhl, P.; Kleist, C.; Zimmermann, S.; Umstätter, F.; Leotta, K.; Mier, W.; Wink, M. Replacement of l-Amino Acids by d-Amino Acids in the Antimicrobial Peptide Ranalexin and Its Consequences for Antimicrobial Activity and Biodistribution. Molecules 2019, 24, 2987. [Google Scholar] [CrossRef]
- Harris, A.G. Somatostatin and somatostatin analogues: Pharmacokinetics and pharmacodynamic effects. Gut 1994, 35, S1–S4. [Google Scholar] [CrossRef] [PubMed]
- Yuen, K.C.J.; Samson, S.L. Oral Octreotide: A Review of Recent Clinical Trials and Practical Recommendations for Its Use in the Treatment of Patients with Acromegaly. Endocr. Pract. 2022, 28, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Kumar, A.; Abdel Monaim, S.A.H.; Jad, Y.E.; El-Faham, A.; de la Torre, B.G.; Albericio, F. N-methylation in amino acids and peptides: Scope and limitations. Biopolymers 2018, 109, e23110. [Google Scholar] [CrossRef]
- Gui, Q.; Jiang, Z.; Zhang, L. Insights into the modulatory role of cyclosporine A and its research advances in acute inflammation. Int. Immunopharmacol. 2021, 93, 107420. [Google Scholar] [CrossRef] [PubMed]
- Biron, E.; Chatterjee, J.; Ovadia, O.; Langenegger, D.; Brueggen, J.; Hoyer, D.; Schmid, H.A.; Jelinek, R.; Gilon, C.; Hoffman, A.; et al. Improving Oral Bioavailability of Peptides by Multiple N-Methylation: Somatostatin Analogues. Angew. Chem. Int. Ed. 2008, 47, 2595–2599. [Google Scholar] [CrossRef] [PubMed]
- Cabrele, C.; Martinek, T.A.; Reiser, O.; Berlicki, Ł. Peptides Containing β-Amino Acid Patterns: Challenges and Successes in Medicinal Chemistry. J. Med. Chem. 2014, 57, 9718–9739. [Google Scholar] [CrossRef] [PubMed]
- Gademann, K.; Ernst, M.; Hoyer, D.; Seebach, D. Synthesis and Biological Evaluation of a Cyclo-β-tetrapeptide as a Somatostatin Analogue. Angew. Chem. Int. Ed. 1999, 38, 1223–1226. [Google Scholar] [CrossRef]
- Proulx, C.; Sabatino, D.; Hopewell, R.; Spiegel, J.; Ramos, Y.G.; Lubell, W.D. Azapeptides and their therapeutic potential. Future Med. Chem. 2011, 3, 1139–1164. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Walker, D.; Sun, B.; Hu, Y.; Walker, S.; Kahne, D. Vancomycin analogues active against vanA-resistant strains inhibit bacterial transglycosylase without binding substrate. Proc. Natl. Acad. Sci. USA 2003, 100, 5658–5663. [Google Scholar] [CrossRef] [PubMed]
- Devi, R.V.; Sathya, S.S.; Coumar, M.S. Evolutionary algorithms for de novo drug design—A survey. Appl. Soft Comput. 2015, 27, 543–552. [Google Scholar] [CrossRef]
- Duffy, F.J.; Devocelle, M.; Shields, D.C. Computational Approaches to Developing Short Cyclic Peptide Modulators of Protein–Protein Interactions. In Computational Peptidology; Zhou, P., Huang, J., Eds.; Humana Press: New York, NY, USA, 2015; pp. 241–271. [Google Scholar]
- Frecer, V.; Ho, B.; Ding, J.L. De Novo Design of Potent Antimicrobial Peptides. Antimicrob. Agents Chemother. 2004, 48, 3349–3357. [Google Scholar] [CrossRef]
- Hosseinzadeh, P.; Watson, P.R.; Craven, T.W.; Li, X.; Rettie, S.; Pardo-Avila, F.; Bera, A.K.; Mulligan, V.K.; Lu, P.; Ford, A.S.; et al. Anchor extension: A structure-guided approach to design cyclic peptides targeting enzyme active sites. Nat. Commun. 2021, 12, 3384. [Google Scholar] [CrossRef]
- Guillen Schlippe, Y.V.; Hartman, M.C.T.; Josephson, K.; Szostak, J.W. In Vitro Selection of Highly Modified Cyclic Peptides That Act as Tight Binding Inhibitors. J. Am. Chem. Soc. 2012, 134, 10469–10477. [Google Scholar] [CrossRef]
- Smith, G.P. Filamentous Fusion Phage: Novel Expression Vectors That Display Cloned Antigens on the Virion Surface. Science 1985, 228, 1315–1317. [Google Scholar] [CrossRef]
- Marintcheva, B. Chapter 5—Phage Display. In Harnessing the Power of Viruses; Marintcheva, B., Ed.; Academic Press: Cambridge, MA, USA, 2018; pp. 133–160. [Google Scholar]
- Nixon, A.E.; Sexton, D.J.; Ladner, R.C. Drugs derived from phage display. MAbs 2014, 6, 73–85. [Google Scholar] [CrossRef]
- Huang, Y.; Wiedmann, M.M.; Suga, H. RNA Display Methods for the Discovery of Bioactive Macrocycles. Chem. Rev. 2019, 119, 10360–10391. [Google Scholar] [CrossRef]
- Smith, G.P.; Petrenko, V.A. Phage Display. Chem. Rev. 1997, 97, 391–410. [Google Scholar] [CrossRef]
- Ercan, I.; Tufekci, K.U.; Karaca, E.; Genc, S.; Genc, K. Chapter Nine—Peptide Derivatives of Erythropoietin in the Treatment of Neuroinflammation and Neurodegeneration. In Advances in Protein Chemistry and Structural Biology; Donev, R., Ed.; Academic Press: Cambridge, MA, USA, 2018; Volume 112, pp. 309–357. [Google Scholar]
- Deyle, K.; Kong, X.-D.; Heinis, C. Phage Selection of Cyclic Peptides for Application in Research and Drug Development. Acc. Chem. Res. 2017, 50, 1866–1874. [Google Scholar] [CrossRef]
- Macdougall, I.C.; Provenzano, R.; Sharma, A.; Spinowitz, B.S.; Schmidt, R.J.; Pergola, P.E.; Zabaneh, R.I.; Tong-Starksen, S.; Mayo, M.R.; Tang, H.; et al. Peginesatide for Anemia in Patients with Chronic Kidney Disease Not Receiving Dialysis. N. Engl. J. Med. 2013, 368, 320–332. [Google Scholar] [CrossRef] [PubMed]
- Molek, P.; Strukelj, B.; Bratkovic, T. Peptide phage display as a tool for drug discovery: Targeting membrane receptors. Molecules 2011, 16, 857–887. [Google Scholar] [CrossRef] [PubMed]
- Heinis, C.; Rutherford, T.; Freund, S.; Winter, G. Phage-encoded combinatorial chemical libraries based on bicyclic peptides. Nat. Chem. Biol. 2009, 5, 502–507. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, J.; Klimkait, T.; Briand, F.; Obrecht, D. Dual anti-viral and immunomodulatory activity of the CXCR4 inhibitor Balixafortide (POL6326) in preclinical in vitro and in vivo SARS-CoV2 infection models. Swiss Med. Wkly. 2021, 151, 14S. [Google Scholar]
- Motixafortide (BL-8040). Available online: https://www.biolinerx.com/pipeline/bl-8040/overview (accessed on 12 February 2023).
- Harrison, H.; Bennett, G.; Blakeley, D.M.; Brown, A.; Campbell, S.; Chen, L.; Lutz, R.J.; Pavan, S.; Rietschoten, K.v.; Teufel, D.P.; et al. Abstract 5144: BT1718, a novel bicyclic peptide-maytansinoid conjugate targeting MT1-MMP for the treatment of solid tumors: Design of bicyclic peptide and linker selection. Cancer Res. 2017, 77, 5144. [Google Scholar] [CrossRef]
- Cook, N.; Banerji, U.; Evans, J.; Biondo, A.; Germetaki, T.; Randhawa, M.; Godfrey, L.; Leslie, S.; Jeffrey, P.; Rigby, M.; et al. 464P—Pharmacokinetic (PK) assessment of BT1718: A phase I/II a study of BT1718, a first in class bicycle toxin conjugate (BTC), in patients (pts) with advanced solid tumours. Ann. Oncol. 2019, 30, v174. [Google Scholar] [CrossRef]
- Bennett, G.; Lutz, R.; Park, P.; Harrison, H.; Lee, K. Abstract 1167: Development of BT1718, a novel Bicycle Drug Conjugate for the treatment of lung cancer. Cancer Res. 2017, 77, 1167. [Google Scholar] [CrossRef]
- Rigby, M.; Bennett, G.; Chen, L.; Mudd, G.E.; Harrison, H.; Beswick, P.J.; Van Rietschoten, K.; Watcham, S.M.; Scott, H.S.; Brown, A.N.; et al. BT8009; A Nectin-4 Targeting Bicycle Toxin Conjugate for Treatment of Solid Tumors. Mol. Cancer Ther. 2022, 21, 1747–1756. [Google Scholar] [CrossRef] [PubMed]
- Mudd, G.E.; Scott, H.; Chen, L.; van Rietschoten, K.; Ivanova-Berndt, G.; Dzionek, K.; Brown, A.; Watcham, S.; White, L.; Park, P.U.; et al. Discovery of BT8009: A Nectin-4 Targeting Bicycle Toxin Conjugate for the Treatment of Cancer. J. Med. Chem. 2022, 65, 14337–14347. [Google Scholar] [CrossRef]
- Bennett, G.; Brown, A.; Mudd, G.; Huxley, P.; Van Rietschoten, K.; Pavan, S.; Chen, L.; Watcham, S.; Lahdenranta, J.; Keen, N. MMAE Delivery Using the Bicycle Toxin Conjugate BT5528. Mol. Cancer Ther. 2020, 19, 1385–1394. [Google Scholar] [CrossRef] [PubMed]
- Mahalingam, D.; Harb, W.; Patnaik, A.; Ulahannan, S.; Mahdi, H.; Ahluwalia, M.; Patel, M.; Dowlati, A.; Bullock, A.; Wen, P.; et al. A first-in-human Phase 1/2 open label trial evaluating the safety, pharmacology, and preliminary efficacy of VT1021 in subjects with advanced solid tumors. J. Immunother. Cancer 2020, 8, A228. [Google Scholar]
- Pairawan, S.; Zhao, M.; Yuca, E.; Annis, A.; Evans, K.; Sutton, D.; Carvajal, L.; Ren, J.G.; Santiago, S.; Guerlavais, V.; et al. First in class dual MDM2/MDMX inhibitor ALRN-6924 enhances antitumor efficacy of chemotherapy in TP53 wild-type hormone receptor-positive breast cancer models. Breast Cancer Res. 2021, 23, 29. [Google Scholar] [CrossRef]
- Hurtado de Mendoza, T.; Mose, E.S.; Botta, G.P.; Braun, G.B.; Kotamraju, V.R.; French, R.P.; Suzuki, K.; Miyamura, N.; Teesalu, T.; Ruoslahti, E.; et al. Tumor-penetrating therapy for β5 integrin-rich pancreas cancer. Nat. Commun. 2021, 12, 1541. [Google Scholar] [CrossRef]
- Díez-Aguilar, M.; Hernández-García, M.; Morosini, M.-I.; Fluit, A.; Tunney, M.M.; Huertas, N.; del Campo, R.; Obrecht, D.; Bernardini, F.; Ekkelenkamp, M.; et al. Murepavadin antimicrobial activity against and resistance development in cystic fibrosis Pseudomonas aeruginosa isolates. J. Antimicrob. Chemother. 2020, 76, 984–992. [Google Scholar] [CrossRef]
- Spexis Achieves First CARB-X Milestone for Its Thanatin Derivatives Program and Receives Funding of up to USD 1.9 Million to Initiate Lead Optimization. Available online: https://spexisbio.com/news-adhoc/news-detail/?newsid=2206705 (accessed on 8 March 2023).
- Krishnan, B.R.; James, K.D.; Polowy, K.; Bryant, B.J.; Vaidya, A.; Smith, S.; Laudeman, C.P. CD101, a novel echinocandin with exceptional stability properties and enhanced aqueous solubility. J. Antibiot. 2017, 70, 130–135. [Google Scholar] [CrossRef]
- Ricardo, A.; Arata, M.; DeMarco, S.; Dhamnaskar, K.; Hammer, R.; Fridkis-Hareli, M.; Rajagopal, V.; Seyb, K.; Tang, G.-Q.; Tobe, S.; et al. Preclinical Evaluation of RA101495, a Potent Cyclic Peptide Inhibitor of C5 for the Treatment of Paroxysmal Nocturnal Hemoglobinuria. Blood 2015, 126, 939. [Google Scholar] [CrossRef]
- Zhou, Q.; Wang, D.; Liu, Y.; Yang, X.; Lucas, R.; Fischer, B. Solnatide Demonstrates Profound Therapeutic Activity in a Rat Model of Pulmonary Edema Induced by Acute Hypobaric Hypoxia and Exercise. Chest 2017, 151, 658–667. [Google Scholar] [CrossRef] [PubMed]
- Barth, P.; Bruijnzeel, P.; Wach, A.; Sellier Kessler, O.; Hooftman, L.; Zimmermann, J.; Naue, N.; Huber, B.; Heimbeck, I.; Kappeler, D.; et al. Single dose escalation studies with inhaled POL6014, a potent novel selective reversible inhibitor of human neutrophil elastase, in healthy volunteers and subjects with cystic fibrosis. J. Cyst. Fibros. 2020, 19, 299–304. [Google Scholar] [CrossRef]
- Teufel, D.P.; Bennett, G.; Harrison, H.; van Rietschoten, K.; Pavan, S.; Stace, C.; Le Floch, F.; Van Bergen, T.; Vermassen, E.; Barbeaux, P.; et al. Stable and Long-Lasting, Novel Bicyclic Peptide Plasma Kallikrein Inhibitors for the Treatment of Diabetic Macular Edema. J. Med. Chem. 2018, 61, 2823–2836. [Google Scholar] [CrossRef] [PubMed]
- Van Bergen, T.; Hu, T.T.; Little, K.; De Groef, L.; Moons, L.; Stitt, A.W.; Vermassen, E.; Feyen, J.H.M. Targeting Plasma Kallikrein with a Novel Bicyclic Peptide Inhibitor (THR-149) Reduces Retinal Thickening in a Diabetic Rat Model. Investig. Ophthalmol. Vis. Sci. 2021, 62, 18. [Google Scholar] [CrossRef] [PubMed]
- Ginzburg, Y.; Kremyanskaya, M.; Kuykendall, A.T.; Yacoub, A.; Yang, J.; Gupta, S.K.; Valone, F.; Khanna, S.; Hoffman, R.; Verstovsek, S. Hepcidin Mimetic (PTG-300) Reverses Iron Deficiency While Controlling Hematocrit in Polycythemia Vera Patients. Blood 2020, 136, 40–41. [Google Scholar] [CrossRef]
- Kowdley, K.V.; Modi, N.B.; Valone, F.; Priego, V.M.; Ferris, C.; Cole, F.; Gupta, S.K. Rusfertide (PTG-300), a Hepcidin Mimetic, Maintains Liver Iron Concentration in the Absence of Phlebotomies in Patients with Hereditary Hemochromatosis. Blood 2021, 138, 943. [Google Scholar] [CrossRef]
- CHENG, L.; Venkataraman, S.; Zhao, L.; Lee, L.; Tang, T.; Liu, D.; Mattheakis, L. P004 PN-943, an oral α4β7 integrin antagonist, inhibits MAdCAM1-mediated proliferation and cytokine release from CD4+ T cells independent of trafficking. J. Crohn’s Colitis 2020, 14, S131. [Google Scholar] [CrossRef]
- Modi, N.B.; Cheng, X.; Mattheakis, L.; Hwang, C.C.; Nawabi, R.; Liu, D.; Gupta, S. Single- and Multiple-Dose Pharmacokinetics and Pharmacodynamics of PN-943, a Gastrointestinal-Restricted Oral Peptide Antagonist of α4β7, in Healthy Volunteers. Clin. Pharmacol. Drug Dev. 2021, 10, 1263–1278. [Google Scholar] [CrossRef]
- Dodd, J.; Jordan, R.; Makhlina, M.; Pesco Koplowitz, L.; Koplowitz, B.; Barnett, K.; Yang, W.H.; Spana, C. Pharmacokinetics of the Melanocortin Type 1 Receptor Agonist PL8177 After Subcutaneous Administration. Drugs RD 2021, 21, 431–443. [Google Scholar] [CrossRef]
- PeptiDream Affiliated Company, PeptiAID Inc., Completes Preclinical Studies of PA-001 Candidate Compound for COVID-19 Therapeutics and Announces Future Plans. Available online: https://contents.xj-storage.jp/xcontents/45870/bfc69946/cf52/42a1/ab2f/c8908a52f8f8/20211111150641447s.pdf (accessed on 6 April 2023).
- Milano, S.; Kurasaki, H.; Tomiyama, T.; Reid, P.; Jan, V.; Culler, M. AZP-3813, a bicyclic 16-amino acid peptide antagonist of the human growth hormone receptor as a potential new treatment for acromegaly. Endocr. Abstr. 2022, 81, P148. [Google Scholar] [CrossRef]
- Kenyon, K.; Ousler, G.W.; Watson, M.; Torkildsen, G.; Vollmer, P.; McLaurin, E.B.; Evans, D.; Winters, J.; Dodd, J.; Jordan, R.; et al. Efficacy and Safety of the Melanocortin Agonist PL9643 in a Phase 2 Study of Subjects with Dry Eye Disease. Investig. Ophthalmol. Vis. Sci. 2021, 62, 1333. [Google Scholar]
- Alonso-Álvarez, S.; Pardal, E.; Sánchez-Nieto, D.; Navarro, M.; Caballero, M.D.; Mateos, M.V.; Martín, A. Plitidepsin: Design, development, and potential place in therapy. Drug Des. Devel. Ther. 2017, 11, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Rehan, S.T.; Hashmi, M.R.; Asghar, M.S.; Tahir, M.J.; Yousaf, Z. Pegcetacoplan—A novel C3 inhibitor for paroxysmal nocturnal hemoglobinuria. Health Sci. Rep. 2022, 5, e512. [Google Scholar] [CrossRef]
- Vértesy, L.; Ehlers, E.; Kogler, H.; Kurz, M.; Meiwes, J.; Seibert, G.; Vogel, M.; Hammann, P. Friulimicins: Novel lipopeptide antibiotics with peptidoglycan synthesis inhibiting activity from Actinoplanes friuliensis sp. nov. II. Isolation and structural characterization. J. Antibiot. 2000, 53, 816–827. [Google Scholar] [CrossRef] [PubMed]
- Schneider, T.; Gries, K.; Josten, M.; Wiedemann, I.; Pelzer, S.; Labischinski, H.; Sahl, H.-G. The Lipopeptide Antibiotic Friulimicin B Inhibits Cell Wall Biosynthesis through Complex Formation with Bactoprenol Phosphate. Antimicrob. Agents Chemother. 2009, 53, 1610–1618. [Google Scholar] [CrossRef] [PubMed]
- Daley, P.; Louie, T.; Lutz, J.E.; Khanna, S.; Stoutenburgh, U.; Jin, M.; Adedoyin, A.; Chesnel, L.; Guris, D.; Larson, K.B.; et al. Surotomycin versus vancomycin in adults with Clostridium difficile infection: Primary clinical outcomes from the second pivotal, randomized, double-blind, Phase 3 trial. J. Antimicrob. Chemother. 2017, 72, 3462–3470. [Google Scholar] [CrossRef]
- Tarcha, E.J.; Olsen, C.M.; Probst, P.; Peckham, D.; Muñoz-Elías, E.J.; Kruger, J.G.; Iadonato, S.P. Safety and pharmacodynamics of dalazatide, a Kv1.3 channel inhibitor, in the treatment of plaque psoriasis: A randomized phase 1b trial. PLoS ONE 2017, 12, e0180762. [Google Scholar] [CrossRef]
- Anker, S.D.; Ponikowski, P.; Mitrovic, V.; Peacock, W.F.; Filippatos, G. Ularitide for the treatment of acute decompensated heart failure: From preclinical to clinical studies. Eur. Heart J. 2015, 36, 715–723. [Google Scholar] [CrossRef]
- Emani, S.; Meyer, M.; Palm, D.; Holzmeister, J.; Haas, G.J. Ularitide: A natriuretic peptide candidate for the treatment of acutely decompensated heart failure. Future Cardiol. 2015, 11, 531–546. [Google Scholar] [CrossRef]
- Kaufman, P.A.; Pernas, S.; Martin, M.; Gil-Martin, M.; Gomez Pardo, P.; Sara, L.T.; Manso, L.; Ciruelos, E.; Perez-Fidalgo, J.A.; Hernando, C.; et al. Balixafortide (a CXCR4 antagonist) plus eribulin in HER2-negative metastatic breast cancer (MBC): A Phase I open-label trial. Eur. J. Cancer 2018, 92, S117–S118. [Google Scholar] [CrossRef]
- Zimmermann, J.; Obrecht, D.; Remus, T. Abstract A003: Anti-angiogenic activity of the CXCR4 antagonist balixafortide. Mol. Cancer Ther. 2019, 18, A003. [Google Scholar] [CrossRef]
- Schmitt, S.; Weinhold, N.; Dembowsky, K.; Neben, K.; Witzens-Harig, M.; Braun, M.; Klemmer, J.; Wuchter, P.; Ludin, C.; Ho, A.D.; et al. First Results of a Phase-II Study with the New CXCR4 Antagonist POL6326 to Mobilize Hematopoietic Stem Cells (HSC) In Multiple Myeloma (MM). Blood 2010, 116, 824. [Google Scholar] [CrossRef]
- Tsomaia, N. Peptide therapeutics: Targeting the undruggable space. Eur. J. Med. Chem. 2015, 94, 459–470. [Google Scholar] [CrossRef]
- Pernas, S.; Martin, M.; Kaufman, P.A.; Gil-Martin, M.; Gomez Pardo, P.; Lopez-Tarruella, S.; Manso, L.; Ciruelos, E.; Perez-Fidalgo, J.A.; Hernando, C.; et al. Balixafortide plus eribulin in HER2-negative metastatic breast cancer: A phase 1, single-arm, dose-escalation trial. Lancet Oncol. 2018, 19, 812–824. [Google Scholar] [CrossRef]
- Kaufman, P.A.; Simon, S.P.; Martin, M.; Gil-Martin, M.; Pardo, P.G.; Lopez-Tarruella, S.; Manso, L.; Ciruelos, E.; Perez-Fidalgo, J.A.; Hernando, C.; et al. Balixafortide (a CXCR4 antagonist) plus eribulin in HER2 negative metastatic breast cancer: Dose-response analysis of efficacy from phase I single-arm trial. J. Clin. Oncol. 2020, 38, e15209. [Google Scholar] [CrossRef]
- Kaufman, P.A.; Martin, M.; Mayer, I.; Vahdat, L.T.; Simon, S.P.; Schmid, P.; McArthur, H.L.; Dent, R.; Rugo, H.S.; Barrios, C.; et al. 359TiP International phase III trial: Balixafortide (a CXCR4 antagonist) + eribulin versus eribulin alone in patients with HER2-negative, locally recurrent or metastatic breast cancer (FORTRESS). Ann. Oncol. 2020, 31, S394–S395. [Google Scholar] [CrossRef]
- Polyphor Provides Final Update on the Phase III FORTRESS Study of Balixafortide in Patients with Advanced HER2 Negative Breast Cancer. Available online: https://spexisbio.com/news-adhoc/news-detail/?newsid=2139995 (accessed on 12 February 2023).
- Spexis’ CXCR4 Inhibitor Balixafortide Demonstrates Synergistic Efficacy in Combination with Docetaxel in a Metastatic Prostate Cancer Preclinical Model. Available online: https://spexisbio.com/news/corporate-news-details/?newsid=2295967 (accessed on 12 February 2023).
- Abraham, M.; Pereg, Y.; Bulvik, B.; Klein, S.; Mishalian, I.; Wald, H.; Eizenberg, O.; Beider, K.; Nagler, A.; Golan, R.; et al. Single Dose of the CXCR4 Antagonist BL-8040 Induces Rapid Mobilization for the Collection of Human CD34+ Cells in Healthy Volunteers. Clin. Cancer Res. 2017, 23, 6790–6801. [Google Scholar] [CrossRef] [PubMed]
- Beider, K.; Begin, M.; Abraham, M.; Wald, H.; Weiss, I.D.; Wald, O.; Pikarsky, E.; Zeira, E.; Eizenberg, O.; Galun, E.; et al. CXCR4 antagonist 4F-benzoyl-TN14003 inhibits leukemia and multiple myeloma tumor growth. Exp. Hematol. 2011, 39, 282–292. [Google Scholar] [CrossRef]
- Burger, J.A.; Stewart, D.J.; Wald, O.; Peled, A. Potential of CXCR4 antagonists for the treatment of metastatic lung cancer. Expert Rev. Anticancer Ther. 2011, 11, 621–630. [Google Scholar] [CrossRef]
- Bockorny, B.; Semenisty, V.; Macarulla, T.; Borazanci, E.; Wolpin, B.M.; Stemmer, S.M.; Golan, T.; Geva, R.; Borad, M.J.; Pedersen, K.S.; et al. BL-8040, a CXCR4 antagonist, in combination with pembrolizumab and chemotherapy for pancreatic cancer: The COMBAT trial. Nat. Med. 2020, 26, 878–885. [Google Scholar] [CrossRef]
- Tamamura, H.; Xu, Y.; Hattori, T.; Zhang, X.; Arakaki, R.; Kanbara, K.; Omagari, A.; Otaka, A.; Ibuka, T.; Yamamoto, N.; et al. A Low-Molecular-Weight Inhibitor against the Chemokine Receptor CXCR4: A Strong Anti-HIV Peptide T140. Biochem. Biophys. Res. Commun. 1998, 253, 877–882. [Google Scholar] [CrossRef]
- Gaur, P.; Verma, V.; Gupta, S.; Sorani, E.; Haras, A.V.; Oberkovitz, G.; Peled, A.; Khleif, S. CXCR4 antagonist (BL-8040) to enhance antitumor effects by increasing tumor infiltration of antigen-specific effector T-cells. J. Clin. Oncol. 2018, 36, 73. [Google Scholar] [CrossRef]
- Gowland, C.; Berry, P.; Errington, J.; Jeffrey, P.; Bennett, G.; Godfrey, L.; Pittman, M.; Niewiarowski, A.; Symeonides, S.N.; Veal, G.J. Development of a LC–MS/MS method for the quantification of toxic payload DM1 cleaved from BT1718 in a Phase I study. Bioanalysis 2021, 13, 101–113. [Google Scholar] [CrossRef]
- Mudd, G.E.; Brown, A.; Chen, L.; van Rietschoten, K.; Watcham, S.; Teufel, D.P.; Pavan, S.; Lani, R.; Huxley, P.; Bennett, G.S. Identification and Optimization of EphA2-Selective Bicycles for the Delivery of Cytotoxic Payloads. J. Med. Chem. 2020, 63, 4107–4116. [Google Scholar] [CrossRef]
- Bendell, J.C.; Wang, J.S.-Z.; Bashir, B.; Richardson, D.L.; Bennett, G.; Campbell, C.; Hennessy, M.G.; Jeffrey, P.; Kirui, J.; Mahnke, L.; et al. BT5528-100 phase I/II study of the safety, pharmacokinetics, and preliminary clinical activity of BT5528 in patients with advanced malignancies associated with EphA2 expression. J. Clin. Oncol. 2020, 38, TPS3655. [Google Scholar] [CrossRef]
- Mahalingam, D.; Mulcahy, M.; Juric, D.; Patel, M.; Pant, S.; Ulahannan, S.; Dowlati, A.; Bullock, A.; Vaickus, L.; Fyfe, S.; et al. Clinical update of VT1021, a first-in-class CD36 and CD47 targeting immunomodulating agent, in subjects with pancreatic cancer and other solid tumors stratified by novel biomarkers. J. Immunother. Cancer 2021, 9, A397. [Google Scholar] [CrossRef]
- Harb, W.; Patnaik, A.; Mahalingam, D.; Liu, J.; Wen, P.Y.; Shapiro, G.I.; Bullock, A.J.; Juric, D.; Zheng, L.; Moore, K.; et al. 465P—A phase I open label dose escalation trial evaluating VT1021 in patients with advanced solid tumours. Ann. Oncol. 2019, 30, v175. [Google Scholar] [CrossRef]
- Tanase, C.; Enciu, A.M.; Codrici, E.; Popescu, I.D.; Dudau, M.; Dobri, A.M.; Pop, S.; Mihai, S.; Gheorghișan-Gălățeanu, A.A.; Hinescu, M.E. Fatty Acids, CD36, Thrombospondin-1, and CD47 in Glioblastoma: Together and/or Separately? Int. J. Mol. Sci. 2022, 23, 604. [Google Scholar] [CrossRef] [PubMed]
- OUR APPROACH. Available online: https://vigeotherapeutics.com/our-approach/overview/ (accessed on 12 February 2023).
- NEWS. Available online: https://vigeotherapeutics.com/news/vigeo-therapeutics-advances-vt1021-into-phase-2-3-registrational-study-for-glioblastoma/ (accessed on 12 February 2023).
- Zhang, S.; Lou, J.; Li, Y.; Zhou, F.; Yan, Z.; Lyu, X.; Zhao, Y. Recent Progress and Clinical Development of Inhibitors that Block MDM4/p53 Protein–Protein Interactions. J. Med. Chem. 2021, 64, 10621–10640. [Google Scholar] [CrossRef] [PubMed]
- Saleh, M.N.; Patel, M.R.; Bauer, T.M.; Goel, S.; Falchook, G.S.; Shapiro, G.I.; Chung, K.Y.; Infante, J.R.; Conry, R.M.; Rabinowits, G.; et al. Phase 1 Trial of ALRN-6924, a Dual Inhibitor of MDMX and MDM2, in Patients with Solid Tumors and Lymphomas Bearing Wild-type TP53. Clin. Cancer Res. 2021, 27, 5236–5247. [Google Scholar] [CrossRef]
- Sallman, D.A.; Borate, U.; Cull, E.H.; Donnellan, W.B.; Komrokji, R.S.; Steidl, U.G.; Corvez, M.M.; Payton, M.; Annis, D.A.; Pinchasik, D.; et al. Phase 1/1b Study of the Stapled Peptide ALRN-6924, a Dual Inhibitor of MDMX and MDM2, As Monotherapy or in Combination with Cytarabine for the Treatment of Relapsed/Refractory AML and Advanced MDS with TP53 Wild-Type. Blood 2018, 132, 4066. [Google Scholar] [CrossRef]
- Yang, J.; Zhu, Q.; Wu, Y.; Qu, X.; Liu, H.; Jiang, B.; Ge, D.; Song, X. Utilization of macrocyclic peptides to target protein-protein interactions in cancer. Front. Oncol. 2022, 12, 992171. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.S.; Graves, B.; Guerlavais, V.; Tovar, C.; Packman, K.; To, K.H.; Olson, K.A.; Kesavan, K.; Gangurde, P.; Mukherjee, A.; et al. Stapled α-helical peptide drug development: A potent dual inhibitor of MDM2 and MDMX for p53-dependent cancer therapy. Proc. Natl. Acad. Sci. USA 2013, 110, E3445–E3454. [Google Scholar] [CrossRef]
- Zhou, X.; Singh, M.; Sanz Santos, G.; Guerlavais, V.; Carvajal, L.A.; Aivado, M.; Zhan, Y.; Oliveira, M.M.S.; Westerberg, L.S.; Annis, D.A.; et al. Pharmacologic Activation of p53 Triggers Viral Mimicry Response Thereby Abolishing Tumor Immune Evasion and Promoting Antitumor Immunity. Cancer Discov. 2021, 11, 3090–3105. [Google Scholar] [CrossRef]
- Our Approach to Protect Against Chemotherapy-Induced Side Effects. Available online: https://aileronrx.com/our-science/#our-focus (accessed on 13 February 2023).
- Carvajal, L.A.; Neriah, D.B.; Senecal, A.; Benard, L.; Thiruthuvanathan, V.; Yatsenko, T.; Narayanagari, S.R.; Wheat, J.C.; Todorova, T.I.; Mitchell, K.; et al. Dual inhibition of MDMX and MDM2 as a therapeutic strategy in leukemia. Sci. Transl. Med. 2018, 10, eaao3003. [Google Scholar] [CrossRef]
- Clinical Development. Available online: https://aileronrx.com/clinical-development/ (accessed on 13 February 2023).
- Sugahara, K.N.; Teesalu, T.; Karmali, P.P.; Kotamraju, V.R.; Agemy, L.; Girard, O.M.; Hanahan, D.; Mattrey, R.F.; Ruoslahti, E. Tissue-Penetrating Delivery of Compounds and Nanoparticles into Tumors. Cancer Cell 2009, 16, 510–520. [Google Scholar] [CrossRef] [PubMed]
- Dean, A.; Gill, S.; McGregor, M.; Broadbridge, V.; Jarvelainen, H.A.; Price, T.J. 1528P Phase I trial of the first-in-class agent CEND-1 in combination with gemcitabine and nab-paclitaxel in patients with metastatic pancreatic cancer. Ann. Oncol. 2020, 31, S941. [Google Scholar] [CrossRef]
- Pipeline. Available online: https://cendrx.com/pipeline/ (accessed on 13 February 2023).
- Martin-Loeches, I.; Dale, G.E.; Torres, A. Murepavadin: A new antibiotic class in the pipeline. Expert Rev. Anti-Infect. Ther. 2018, 16, 259–268. [Google Scholar] [CrossRef]
- Batur, G.; Ermert, P.; Zimmermann, J.; Obrecht, D. Macrocycle therapeutics to treat life-threatening diseases. Chimia 2021, 75, 508. [Google Scholar] [CrossRef]
- Melchers, M.J.; Teague, J.; Warn, P.; Hansen, J.; Bernardini, F.; Wach, A.; Obrecht, D.; Dale, G.E.; Mouton, J.W. Pharmacokinetics and Pharmacodynamics of Murepavadin in Neutropenic Mouse Models. Antimicrob. Agents Chemother. 2019, 63, 10–1128. [Google Scholar] [CrossRef]
- Steinberg, D.A.; Hurst, M.A.; Fujii, C.A.; Kung, A.H.; Ho, J.F.; Cheng, F.C.; Loury, D.J.; Fiddes, J.C. Protegrin-1: A broad-spectrum, rapidly microbicidal peptide with in vivo activity. Antimicrob. Agents Chemother. 1997, 41, 1738–1742. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.A.; Shankaramma, S.C.; Jetter, P.; Kienzl, U.; Schwendener, R.A.; Vrijbloed, J.W.; Obrecht, D. Properties and structure–activity studies of cyclic β-hairpin peptidomimetics based on the cationic antimicrobial peptide protegrin I. Biorg. Med. Chem. 2005, 13, 2055–2064. [Google Scholar] [CrossRef] [PubMed]
- Srinivas, N.; Jetter, P.; Ueberbacher, B.J.; Werneburg, M.; Zerbe, K.; Steinmann, J.; Van der Meijden, B.; Bernardini, F.; Lederer, A.; Dias, R.L.A.; et al. Peptidomimetic Antibiotics Target Outer-Membrane Biogenesis in Pseudomonas aeruginosa. Science 2010, 327, 1010–1013. [Google Scholar] [CrossRef] [PubMed]
- Shankaramma, S.C.; Athanassiou, Z.; Zerbe, O.; Moehle, K.; Mouton, C.; Bernardini, F.; Vrijbloed, J.W.; Obrecht, D.; Robinson, J.A. Macrocyclic Hairpin Mimetics of the Cationic Antimicrobial Peptide Protegrin I: A New Family of Broad-Spectrum Antibiotics. ChemBioChem 2002, 3, 1126–1133. [Google Scholar] [CrossRef]
- Luther, A.; Moehle, K.; Chevalier, E.; Dale, G.; Obrecht, D. Protein epitope mimetic macrocycles as biopharmaceuticals. Curr. Opin. Chem. Biol. 2017, 38, 45–51. [Google Scholar] [CrossRef]
- Dash, R.; Bhattacharjya, S. Thanatin: An Emerging Host Defense Antimicrobial Peptide with Multiple Modes of Action. Int. J. Mol. Sci. 2021, 22, 1522. [Google Scholar] [CrossRef]
- Moura, E.C.C.M.; Baeta, T.; Romanelli, A.; Laguri, C.; Martorana, A.M.; Erba, E.; Simorre, J.-P.; Sperandeo, P.; Polissi, A. Thanatin Impairs Lipopolysaccharide Transport Complex Assembly by Targeting LptC–LptA Interaction and Decreasing LptA Stability. Front. Microbiol. 2020, 11, 909. [Google Scholar] [CrossRef]
- Ong, V.; James, K.D.; Smith, S.; Krishnan, B.R. Pharmacokinetics of the Novel Echinocandin CD101 in Multiple Animal Species. Antimicrob. Agents Chemother. 2017, 61, 10–1128. [Google Scholar] [CrossRef]
- Garcia-Effron, G. Rezafungin—Mechanisms of Action, Susceptibility and Resistance: Similarities and Differences with the Other Echinocandins. J. Fungi 2020, 6, 262. [Google Scholar] [CrossRef]
- James, K.D.; Laudeman, C.P.; Malkar, N.B.; Krishnan, R.; Polowy, K. Structure-Activity Relationships of a Series of Echinocandins and the Discovery of CD101, a Highly Stable and Soluble Echinocandin with Distinctive Pharmacokinetic Properties. Antimicrob. Agents Chemother. 2017, 61, 10–1128. [Google Scholar] [CrossRef]
- Howard, J.F.; Vissing, J.; Gilhus, N.E.; Leite, M.I.; Utsugisawa, K.; Duda, P.W.; Farzaneh-Far, R.; Murai, H.; Wiendl, H. Zilucoplan: An Investigational Complement C5 Inhibitor for the Treatment of Acetylcholine Receptor Autoantibody–Positive Generalized Myasthenia Gravis. Expert Opin. Investig. Drugs 2021, 30, 483–493. [Google Scholar] [CrossRef]
- Duda, P.; Farzaneh-Far, R.; Ma, Z.; Zhu, N.; Thackaberry, E.; Ricardo, A. Neurological Disease Treatment with Zilucoplan. U.S. Patent 17/287,581, 5 May 2022. [Google Scholar]
- Zilucoplan (RA101495). Available online: https://www.ucb.com/clinical-studies/Clinical-studies-index/Zilucoplan-RA101495 (accessed on 13 February 2023).
- Howard, J.F., Jr.; Nowak, R.J.; Wolfe, G.I.; Freimer, M.L.; Vu, T.H.; Hinton, J.L.; Benatar, M.; Duda, P.W.; MacDougall, J.E.; Farzaneh-Far, R.; et al. Clinical Effects of the Self-administered Subcutaneous Complement Inhibitor Zilucoplan in Patients with Moderate to Severe Generalized Myasthenia Gravis: Results of a Phase 2 Randomized, Double-Blind, Placebo-Controlled, Multicenter Clinical Trial. JAMA Neurol. 2020, 77, 582–592. [Google Scholar] [CrossRef]
- Howard, J.F., Jr.; Bresch, S.; Genge, A.; Hewamadduma, C.; Hinton, J.; Hussain, Y.; Juntas-Morales, R.; Kaminski, H.J.; Maniaol, A.; Mantegazza, R.; et al. Safety and efficacy of zilucoplan in patients with generalised myasthenia gravis (RAISE): A randomised, double-blind, placebo-controlled, phase 3 study. Lancet Neurol. 2023, 22, 395–406. [Google Scholar] [CrossRef]
- UCB Announces Positive Data in Myasthenia Gravis with Zilucoplan Phase 3 Study Results. Available online: https://www.ucb.com/stories-media/Press-Releases/article/UCB-announces-positive-data-in-myasthenia-gravis-with-zilucoplan-phase-3-study-results (accessed on 13 February 2023).
- Schmid, B.; Kredel, M.; Ullrich, R.; Krenn, K.; Lucas, R.; Markstaller, K.; Fischer, B.; Kranke, P.; Meybohm, P.; Zwißler, B.; et al. Safety and preliminary efficacy of sequential multiple ascending doses of solnatide to treat pulmonary permeability edema in patients with moderate-to-severe ARDS—A randomized, placebo-controlled, double-blind trial. Trials 2021, 22, 643. [Google Scholar] [CrossRef]
- Schmid, B.; Kranke, P.; Lucas, R.; Meybohm, P.; Zwissler, B.; Frank, S. Safety and preliminary efficacy of sequential multiple ascending doses of solnatide to treat pulmonary permeability edema in patients with moderate to severe ARDS in a randomized, placebo-controlled, double-blind trial: Preliminary evaluation of safety and feasibility in light of the COVID-19 pandemic. Trials 2022, 23, 252. [Google Scholar]
- Shabbir, W.; Scherbaum-Hazemi, P.; Tzotzos, S.; Fischer, B.; Fischer, H.; Pietschmann, H.; Lucas, R.; Lemmens-Gruber, R. Mechanism of action of novel lung edema therapeutic AP301 by activation of the epithelial sodium channel. Mol. Pharmacol. 2013, 84, 899–910. [Google Scholar] [CrossRef]
- Tzotzos, S.; Fischer, B.; Fischer, H.; Pietschmann, H.; Lucas, R.; Dupré, G.; Lemmens-Gruber, R.; Hazemi, P.; Prymaka, V.; Shabbir, W. AP301, a synthetic peptide mimicking the lectin-like domain of TNF, enhances amiloride-sensitive Na+ current in primary dog, pig and rat alveolar type II cells. Pulm. Pharmacol. Ther. 2013, 26, 356–363. [Google Scholar] [CrossRef]
- Schwameis, R.; Eder, S.; Pietschmann, H.; Fischer, B.; Mascher, H.; Tzotzos, S.; Fischer, H.; Lucas, R.; Zeitlinger, M.; Hermann, R. A FIM study to assess safety and exposure of inhaled single doses of AP301-A specific ENaC channel activator for the treatment of acute lung injury. J. Clin. Pharmacol. 2014, 54, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Krenn, K.; Lucas, R.; Croizé, A.; Boehme, S.; Klein, K.U.; Hermann, R.; Markstaller, K.; Ullrich, R. Inhaled AP301 for treatment of pulmonary edema in mechanically ventilated patients with acute respiratory distress syndrome: A phase IIa randomized placebo-controlled trial. Crit. Care 2017, 21, 194. [Google Scholar] [CrossRef] [PubMed]
- Hooftman, L.; Chevalier, E.; Wach, A.; Zimmermann, J.; Bruijnzeel, P.; Naue, N.; Heimbeck, I.; Kappeler, D.; Barth, P. WS01.4 A randomised, double-blind, placebo-controlled, parallel-group, dose-escalation study of inhaled single doses of POL6014, a potent and selective reversible inhibitor of human neutrophil elastase (NE), in cystic fibrosis (CF) patients. J. Cyst. Fibros. 2017, 16, S2. [Google Scholar] [CrossRef]
- Taranath, R.; Bourne, G.; Zhao, L.; Frederick, B.; King, C.; Liu, D. Regulation of Iron Homeostasis By PTG-300 Improves Disease Parameters in Mouse Models for Beta-Thalassemia and Hereditary Hemochromatosis. Blood 2019, 134, 3540. [Google Scholar] [CrossRef]
- Taranath, R.; Zhao, L.; Vengalam, J.; Lee, L.; Tang, T.; Dion, C.; Su, A.; Tovera, J.; Bhandari, A.; Cheng, X.; et al. Regulation of Iron Homeostasis and Efficacy of Rusfertide Analog Peptide in a Mouse Model for Polycythemia Vera. Blood 2021, 138, 2006. [Google Scholar] [CrossRef]
- Verstovsek, S.; Kuykendall, A.T.; Hoffman, R.; Ginzburg, Y.; Pemmaraju, N.; Valone, F.; Modi, N.B.; Khanna, S.; O’Connor, P.G.; Gupta, S.K.; et al. A Phase 3 Study of the Hepcidin Mimetic Rusfertide (PTG-300) in Patients with Polycythemia Vera. Blood 2021, 138, 1504. [Google Scholar] [CrossRef]
- Dodd, J.; Yang, W.H.; Makhlina, M. Fr486 ORAL ADMINISTRATION OF THE MELANOCORTIN-1 RECEPTOR AGONIST PL8177 IN A RAT MODEL OF COLITIS. Gastroenterology 2021, 160, S-327. [Google Scholar] [CrossRef]
- Available online: https://docs.publicnow.com/viewDoc?hash_primary=8E226B09BA24D896775EFE0AD9A91AB41D99129D (accessed on 5 April 2023).
- Cary, D.R.; Ohuchi, M.; Reid, P.C.; Masuya, K. Constrained Peptides in Drug Discovery and Development. J. Synth. Org. Chem Jpn. 2017, 75, 1171–1178. [Google Scholar] [CrossRef]
- Culler, M.; Milano, S.; Kurasaki, H.; Tomiyama, T.; Reid, P.; van der Lely, A.J.; Culler, M. ODP353 Sustained Suppression of IGF1 with AZP-3813, a Bicyclic 16-Amino Acid Peptide Antagonist of the Human Growth Hormone Receptor and a Potential New Treatment for Acromegaly. J. Endocr. Soc. 2022, 6, A511. [Google Scholar] [CrossRef]
- Losada, A.; Muñoz-Alonso, M.J.; García, C.; Sánchez-Murcia, P.A.; Martínez-Leal, J.F.; Domínguez, J.M.; Lillo, M.P.; Gago, F.; Galmarini, C.M. Translation Elongation Factor eEF1A2 is a Novel Anticancer Target for the Marine Natural Product Plitidepsin. Sci. Rep. 2016, 6, 35100. [Google Scholar] [CrossRef]
- Wong, R.S.M. Safety and efficacy of pegcetacoplan in paroxysmal nocturnal hemoglobinuria. Ther. Adv. Hematol. 2022, 13, 20406207221114673. [Google Scholar] [CrossRef]
- Our Pipeline. Available online: https://apellis.com/our-science/our-pipeline/ (accessed on 14 February 2023).
- Dijksteel, G.; Ulrich, M.; Middelkoop, E.; Boekema, B. Review: Lessons learned from clinical trials using antimicrobial peptides (AMPs). Front. Microbiol. 2021, 12, 616979. [Google Scholar] [CrossRef]
- Castañeda, O.; Sotolongo, V.; Amor, A.M.; Stöcklin, R.; Anderson, A.J.; Harvey, A.L.; Engström, Å.; Wernstedt, C.; Karlsson, E. Characterization of a potassium channel toxin from the Caribbean sea anemone Stichodactyla helianthus. Toxicon 1995, 33, 603–613. [Google Scholar] [CrossRef]
- Pennington, M.W.; Chang, S.C.; Chauhan, S.; Huq, R.; Tajhya, R.B.; Chhabra, S.; Norton, R.S.; Beeton, C. Development of Highly Selective Kv1.3-Blocking Peptides Based on the Sea Anemone Peptide ShK. Mar. Drugs 2015, 13, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Tarcha, E.J.; Chi, V.; Muñoz-Elías, E.J.; Bailey, D.; Londono, L.M.; Upadhyay, S.K.; Norton, K.; Banks, A.; Tjong, I.; Nguyen, H.; et al. Durable Pharmacological Responses from the Peptide ShK-186, a Specific Kv1.3 Channel Inhibitor That Suppresses T Cell Mediators of Autoimmune Disease. J. Pharmacol. Exp. Ther. 2012, 342, 642–653. [Google Scholar] [CrossRef] [PubMed]
- Dalazatide. Available online: https://www.kv13therapeutics.com/pipeline/dalazatide/ (accessed on 14 February 2023).
- Schulz-Knappe, P.; Forssmann, K.; Herbst, F.; Hock, D.; Pipkorn, R.; Forssmann, W.G. Isolation and structural analysis of “Urodilatin”, a new peptide of the cardiodilatin-(ANP)-family, extracted from human urine. Klin. Wochenschr. 1988, 66, 752–759. [Google Scholar] [CrossRef] [PubMed]
- Joseph, S.M.; Cedars, A.M.; Ewald, G.A.; Geltman, E.M.; Mann, D.L. Acute decompensated heart failure: Contemporary medical management. Tex. Heart Inst. J. 2009, 36, 510–520. [Google Scholar] [PubMed]
- KENTSCH, M.; LUDWIG, D.; DRUMMER, C.; GERZER, R.; MÜLLER-ESCH, G. Haemodynamic and renal effects of urodilatin bolus injections in patients with congestive heart failure. Eur. J. Clin. Investig. 1992, 22, 662–669. [Google Scholar] [CrossRef]
- Mitrovic, V.; Seferovic, P.M.; Simeunovic, D.; Ristic, A.D.; Miric, M.; Moiseyev, V.S.; Kobalava, Z.; Nitsche, K.; Forssmann, W.-G.; Lüss, H.; et al. Haemodynamic and clinical effects of ularitide in decompensated heart failure. Eur. Heart J. 2006, 27, 2823. [Google Scholar] [CrossRef]
- Packer, M.; O’Connor, C.; McMurray, J.J.V.; Wittes, J.; Abraham, W.T.; Anker, S.D.; Dickstein, K.; Filippatos, G.; Holcomb, R.; Krum, H.; et al. Effect of Ularitide on Cardiovascular Mortality in Acute Heart Failure. N. Engl. J. Med. 2017, 376, 1956–1964. [Google Scholar] [CrossRef]
- Mascio, C.T.; Mortin, L.I.; Howland, K.T.; Van Praagh, A.D.; Zhang, S.; Arya, A.; Chuong, C.L.; Kang, C.; Li, T.; Silverman, J.A. In vitro and in vivo characterization of CB-183,315, a novel lipopeptide antibiotic for treatment of Clostridium difficile. Antimicrob. Agents Chemother. 2012, 56, 5023–5030. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide Name | Generic Name | Indication | Highest Phase | Company | Source | References |
|---|---|---|---|---|---|---|
| POL6326 | Balixafortide | Advanced breast cancers | Spexis | Synthesis | [143] | |
| BL-8040 | Motixafortide | HSCs, solid tumors, and AML | III | Bioline RX | Synthesis | [144] |
| BT1718 |  | Cancer with MT1-MMP expression | I/II | Bicycle Therapeutics Ltd. | Synthesis | [145,146,147] |
| BT8009 | | Cancer types, where Nectin-4 is expressed | I/II | Bicycle Therapeutics Ltd. | Synthesis | [148,149] |
| BT5528 | | Advanced solid tumors associated with EphA2 expression | I/II | Bicycle Therapeutics Ltd. | Synthesis | [150] |
| VT1021 | | Antitumor | II | Vigeo Therapeutics | | [151] |
| ALRN-6924 | | Chemoprotective agent | Ib | Aileron Therapeutics | Synthesis | [152] |
| CEND-1 | | Enhance the efficacy of chemotherapy | II | Cend Therapeutics | | [153] |
| POL7080 | Inhaled murepavadin | Antibiotic to treat Pseudomonas infections in patients with cystic fibrosis | I | Spexis | Synthesis | [154] |
| Thanatin-derivative | | Pre-clinical | Spexis/University of Zurich | | [155] |
| CD-101 | Rezafungin | Treatment of candidemia and/or invasive candidiasis and invasive fungal diseases | III | Cidara Therapeutics Inc. | Semi-synthesis | [156] |
| RA-101495 | Zilucoplan | Paroxysmal nocturnal hemoglobinuria, generalized myasthenia gravis | III | UCB Pharma | Synthesis | [157] |
| AP301 | Solnatide | Pulmonary permeability edema | II | Apeptico | Synthesis | [158] |
| POL6014 | Lonodelestat | Cystic fibrosis | II | Santhera Pharmaceuticals | Synthesis | [159] |
| THR-149 | Diabetic macular edema | II | Oxurion NV | Synthesis | [160,161] | |
| PTG-300 | Rusfertide | Polycythemia vera | III | Protagonist Therapeutics, Inc. | Synthesis | [162,163] |
| PN-943 | | Ulcerative colitis | II | Protagonist Therapeutics, Inc. | | [164,165] |
| PL8177 | | Ulcerative colitis | II | Palatin Technologies Inc. | Synthesis | [166] |
| PA-001 | | SARSCov-2 infection | I | PeptiAID | | [167] |
| AZP-3813 | | Acromegaly | IND-Enabling | Amolyt Pharma | | [168] |
| PL9643 | | Dry eye disease | III | Palatin Technologies Inc. | Synthesis | [169] |
| PM90001 | Plitidepsin | SARS-CoV-2 infection | III | PharmaMar | Synthesis | [46,170] |
| APL-2 | Pegcetacoplan | ALS, IC-MPGN, and C3G, CAD, and HSCT-TMA | | Apellis Pharmaceuticals Inc. | | [171] |
| Friulimicin B | Antibacterial activity against Gram-positive bacteria | Stopped in phase I in 2008 due to unfavourable pharmacokinetic properties | MerLion Pharmaceuticals GmbH | Nature | [172,173] |
| CB-183,315 | Surotomycin | Antibacterial activity against Clostridium difficile | Stopped in phase III in 2019 (no improvement over vancomycin) | Merck | | [174] |
| ShK-186 | Dalazatide | Plaque psoriasis | I (finished in May 2015) | Kv1.3 | Synthesis | [175] |
| Ularitide | Acutely decompensated heart failure | III (finished in October 2018) | Cardiorentis | Synthesis | [176,177] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Costa, L.; Sousa, E.; Fernandes, C. Cyclic Peptides in Pipeline: What Future for These Great Molecules? Pharmaceuticals 2023, 16, 996. https://doi.org/10.3390/ph16070996
Costa L, Sousa E, Fernandes C. Cyclic Peptides in Pipeline: What Future for These Great Molecules? Pharmaceuticals. 2023; 16(7):996. https://doi.org/10.3390/ph16070996
Chicago/Turabian StyleCosta, Lia, Emília Sousa, and Carla Fernandes. 2023. "Cyclic Peptides in Pipeline: What Future for These Great Molecules?" Pharmaceuticals 16, no. 7: 996. https://doi.org/10.3390/ph16070996
APA StyleCosta, L., Sousa, E., & Fernandes, C. (2023). Cyclic Peptides in Pipeline: What Future for These Great Molecules? Pharmaceuticals, 16(7), 996. https://doi.org/10.3390/ph16070996