


Tryptophan Substitution in CJ-15,208 (cyclo[Phe-D-Pro-Phe-Trp]) Introduces δ-Opioid Receptor Antagonism, Preventing Antinociceptive Tolerance and Stress-Induced Reinstatement of Extinguished Cocaine-Conditioned Place Preference

 , ,
, ,  and
and
Abstract
1. Introduction
2. Results
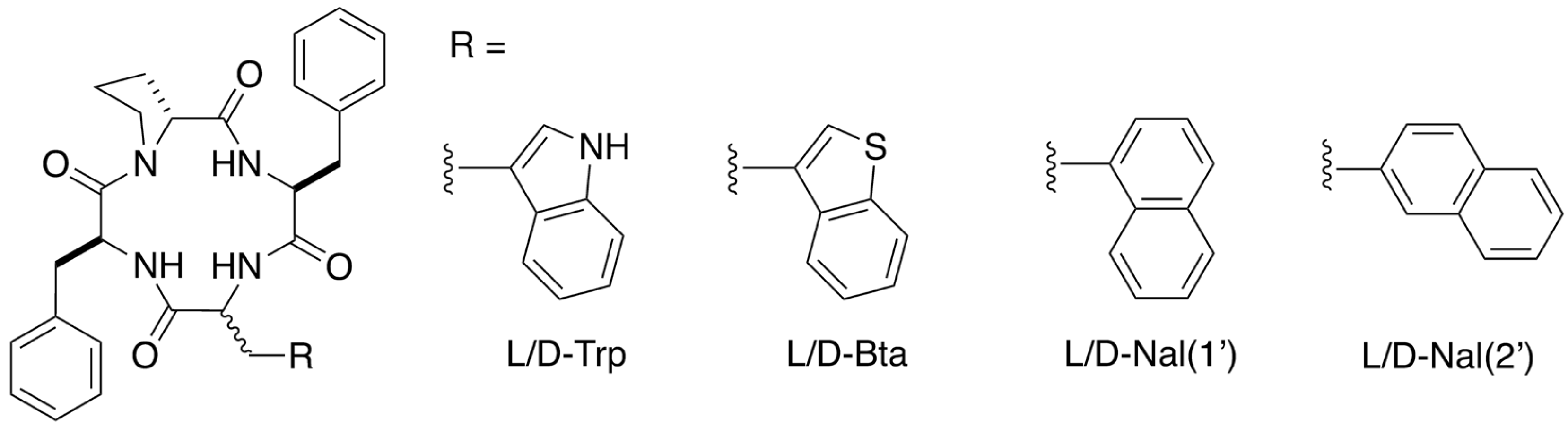
2.1. Design and Synthesis
2.2. In Vitro Pharmacological Evaluation
2.3. Metabolic Stability
2.4. In Vivo Pharmacological Evaluation
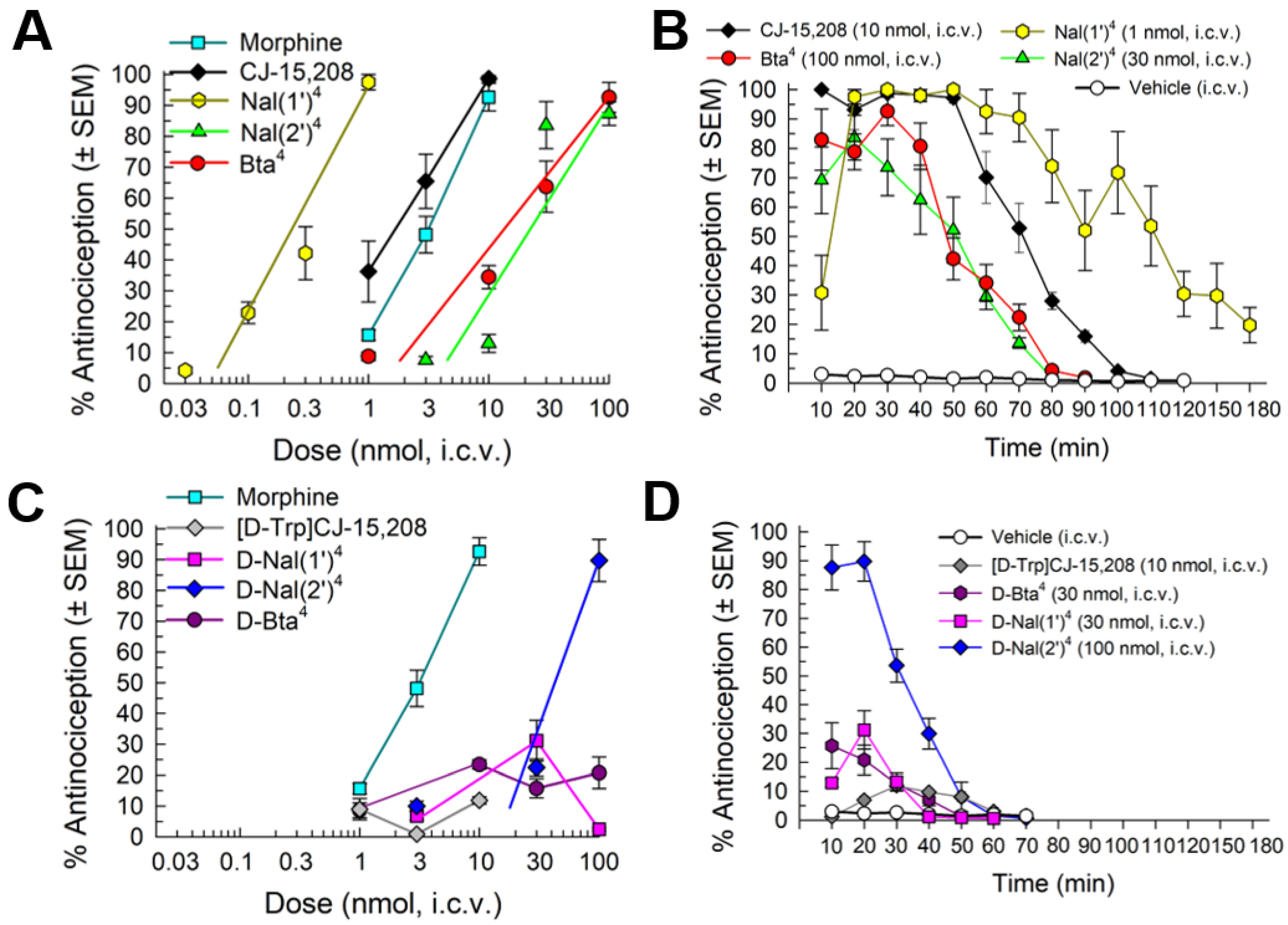
2.4.1. Antinociception
2.4.2. Opioid Receptor Involvement in Analog Antinociception
2.4.3. Evaluation of Opioid-Receptor-Selective Antagonist Activity Mediated by the Analog
2.5. In Vivo Assessment of Potential Opioid-Related Liabilities of [Nal(2′)4]CJ-15,208
2.5.1. Assessment of Acute Antinociceptive Tolerance Development
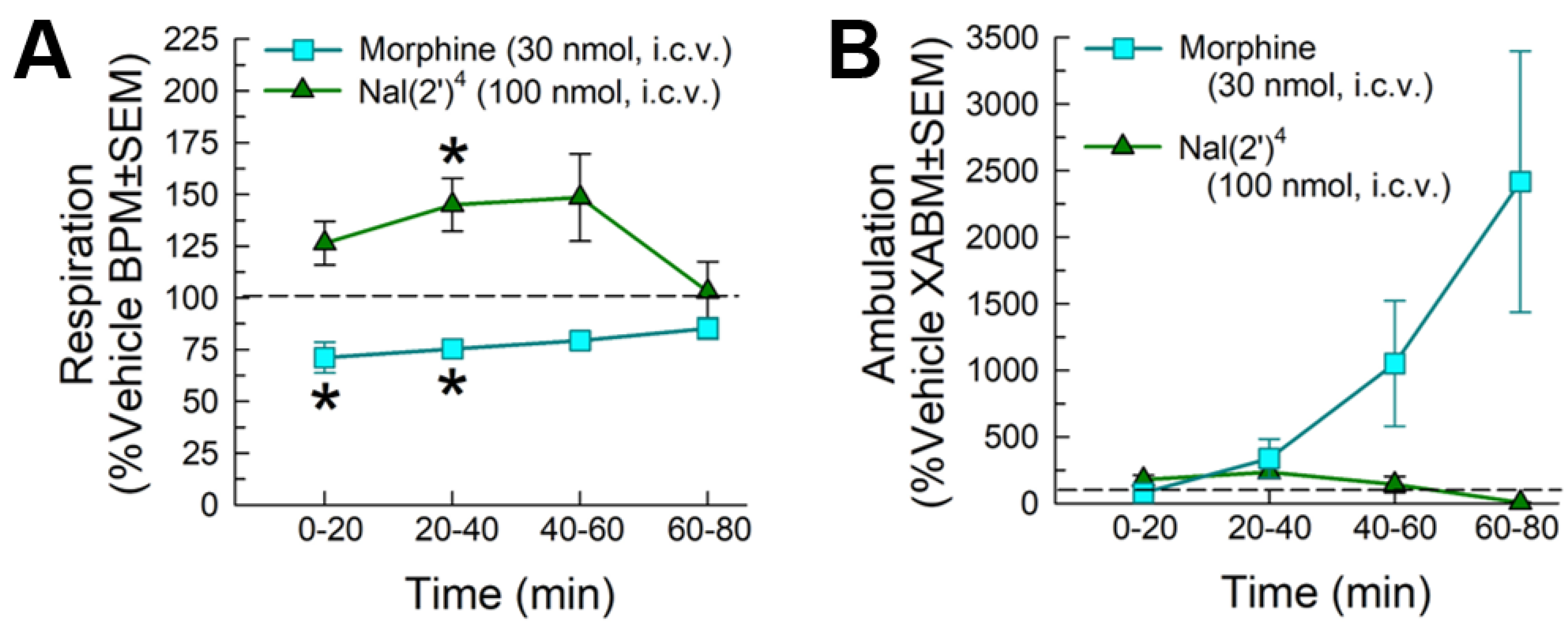
2.5.2. Evaluation of Respiratory and Spontaneous Locomotor Effects
2.5.3. Evaluation of Potential Reinforcing or Aversive Properties
2.5.4. Evaluation of [Nal(2′)4]CJ-15,208 in the Prevention of Reinstatement of Extinguished Cocaine-Conditioned Place Preference
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Instruments
4.3. Peptide Synthesis and Purification
4.3.1. [Bta4]CJ-15,208
4.3.2. [Nal(1′)4]CJ-15,208
4.3.3. [Nal(2′)4]CJ-15,208
4.3.4. [D-Bta4]CJ-15,208
4.3.5. [D-Nal(1′)4]CJ-15,208
4.3.6. [D-Nal(2′)4]CJ-15,208
4.4. Metabolism by Mouse Liver Microsomes
4.5. In Vitro Pharmacological Evaluation
4.6. In Vivo Testing
4.6.1. Animals and Drug Administration
4.6.2. Antinociceptive Testing
4.6.3. Acute Antinociceptive Tolerance Determination
4.6.4. Respiration and Ambulation
4.6.5. Evaluation of Potential Conditioned Place Preference (CPP) and Conditioned Place Aversion (CPA)
4.6.6. Cocaine-Conditioned Place Preference, Extinction, and Reinstatement Testing
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Savage, S.R.; Kirsh, K.L.; Passik, S.D. Challenges in using opioids to treat pain in persons with substance use disorders. Addict. Sci. Clin. Pract. 2008, 4, 4–25. [Google Scholar] [CrossRef]
- Cahill, C.M.; Holdridge, S.V.; Morinville, A. Trafficking of delta-opioid receptors and other G-protein-coupled receptors: Implications for pain and analgesia. Trends Pharmacol. Sci. 2007, 28, 23–31. [Google Scholar] [CrossRef]
- Fujita, W.; Gomes, I.; Devi, L.A. Heteromers of mu-delta opioid receptors: New pharmacology and novel therapeutic possibilities. Br. J. Pharmacol. 2015, 172, 375–387. [Google Scholar] [CrossRef]
- Vaught, J.L.; Takemori, A.E. Differential effects of leucine and methionine enkephalin on morphine-induced analgesia, acute tolerance and dependence. J. Pharmacol. Exp. Ther. 1979, 208, 86–90. [Google Scholar]
- Porreca, F.; Heyman, J.S.; Mosberg, H.I.; Omnaas, J.R.; Vaught, J.L. Role of mu and delta receptors in the supraspinal and spinal analgesic effects of [D-Pen2, D-Pen5]enkephalin in the mouse. J. Pharmacol. Exp. Ther. 1987, 241, 393–400. [Google Scholar] [PubMed]
- Abdelhamid, E.E.; Sultana, M.; Portoghese, P.S.; Takemori, A.E. Selective blockage of delta-opioid receptors prevents the development of morphine-tolerance and dependence in mice. J. Pharmacol. Exp. Ther. 1991, 258, 299–303. [Google Scholar]
- Fundytus, M.E.; Schiller, P.W.; Shapiro, M.; Weltrowska, G.; Coderre, T.J. Attenuation of Morphine Tolerance and Dependence with the Highly Selective Delta-Opioid Receptor Antagonist TIPPΨ. Eur. J. Pharmacol. 1995, 286, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Hepburn, M.J.; Little, P.J.; Gingras, J.; Kuhn, C.M. Differential effects of naltrindole on morphine-induced tolerance and physical dependence in rats. J. Pharmacol. Exp. Ther. 1997, 281, 1350–1356. [Google Scholar] [PubMed]
- Schiller, P.W. Bi- or multifunctional opioid peptide drugs. Life Sci. 2010, 86, 598–603. [Google Scholar] [CrossRef]
- Schiller, P.W.; Fundytus, M.E.; Merovitz, L.; Weltrowska, G.; Nguyen, T.M.; Lemieux, C.; Chung, N.N.; Coderre, T.J. The Opioid µ Agonist/δ Antagonist DIPP-NH2[Ψ] Produces a Potent Analgesic Effect, No Physical Dependence, and Less Tolerance than Morphine in Rats. J. Med. Chem. 1999, 42, 3520–3526. [Google Scholar] [CrossRef] [PubMed]
- Daniels, D.J.; Lenard, N.R.; Etienne, C.L.; Law, P.Y.; Roerig, S.C.; Portoghese, P.S. Opioid-induced tolerance and dependence in mice is modulated by the distance between pharmacophores in a bivalent ligand series. Proc. Natl. Acad. Sci. USA 2005, 102, 19208–19213. [Google Scholar] [CrossRef]
- Lenard, N.R.; Daniels, D.J.; Portoghese, P.S.; Roerig, S.C. Absence of conditioned place preference or reinstatement with bivalent ligands containing mu-opioid receptor agonist and delta-opioid receptor antagonist pharmacophores. Eur. J. Pharmacol. 2007, 566, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Ananthan, S.; Saini, S.K.; Dersch, C.M.; Xu, H.; McGlinchey, N.; Giuvelis, D.; Bilsky, E.J.; Rothman, R.B. 14-Alkoxy- and 14-acyloxypyridomorphinans: Mu agonist/delta antagonist opioid analgesics with diminished tolerance and dependence side effects. J. Med. Chem. 2012, 55, 8350–8363. [Google Scholar] [CrossRef]
- Healy, J.R.; Bezawada, P.; Shim, J.; Jones, J.W.; Kane, M.A.; MacKerell, A.D., Jr.; Coop, A.; Matsumoto, R.R. Synthesis, modeling, and pharmacological evaluation of UMB 425, a mixed mu agonist/delta antagonist opioid analgesic with reduced tolerance liabilities. ACS Chem. Neurosci. 2013, 4, 1256–1266. [Google Scholar] [CrossRef]
- Mosberg, H.I.; Yeomans, L.; Harland, A.A.; Bender, A.M.; Sobczyk-Kojiro, K.; Anand, J.P.; Clark, M.J.; Jutkiewicz, E.M.; Traynor, J.R. Opioid peptidomimetics: Leads for the design of bioavailable mixed efficacy mu opioid receptor (MOR) agonist/delta opioid receptor (DOR) antagonist ligands. J. Med. Chem. 2013, 56, 2139–2149. [Google Scholar] [CrossRef]
- Mosberg, H.I.; Yeomans, L.; Anand, J.P.; Porter, V.; Sobczyk-Kojiro, K.; Traynor, J.R.; Jutkiewicz, E.M. Development of a bioavailable mu opioid receptor (MOPr) agonist, delta opioid receptor (DOPr) antagonist peptide that evokes antinociception without development of acute tolerance. J. Med. Chem. 2014, 57, 3148–3153. [Google Scholar] [CrossRef]
- Bender, A.M.; Clark, M.J.; Agius, M.P.; Traynor, J.R.; Mosberg, H.I. Synthesis and evaluation of 4-substituted piperidines and piperazines as balanced affinity mu opioid receptor (MOR) agonist/delta opioid receptor (DOR) antagonist ligands. Bioorg. Med. Chem. Lett. 2014, 24, 548–551. [Google Scholar] [CrossRef] [PubMed]
- Bender, A.M.; Griggs, N.W.; Anand, J.P.; Traynor, J.R.; Jutkiewicz, E.M.; Mosberg, H.I. Asymmetric synthesis and in vitro and in vivo activity of tetrahydroquinolines featuring a diverse set of polar substitutions at the 6 position as mixed-efficacy mu opioid receptor/delta opioid receptor ligands. ACS Chem. Neurosci. 2015, 6, 1428–1435. [Google Scholar] [CrossRef] [PubMed]
- Aceto, M.D.; Harris, L.S.; Negus, S.S.; Banks, M.L.; Hughes, L.D.; Akgun, E.; Portoghese, P.S. MDAN-21: A Bivalent Opioid Ligand Containing mu-Agonist and Delta-Antagonist Pharmacophores and Its Effects in Rhesus Monkeys. Int. J. Med. Chem. 2012, 2012, 327257. [Google Scholar] [CrossRef] [PubMed]
- Aldrich, J.V.; Senadheera, S.N.; Ross, N.C.; Ganno, M.L.; Eans, S.O.; McLaughlin, J.P. The macrocyclic peptide natural product CJ-15,208 is orally active and prevents reinstatement of extinguished cocaine-seeking behavior. J. Nat. Prod. 2013, 76, 433–438. [Google Scholar] [CrossRef]
- Eans, S.O.; Ganno, M.L.; Reilley, K.J.; Patkar, K.A.; Senadheera, S.N.; Aldrich, J.V.; McLaughlin, J.P. The macrocyclic tetrapeptide [D-Trp]CJ-15,208 produces short acting κ opioid receptor antagonism in the CNS after oral administration. Br. J. Pharmacol. 2013, 169, 426–436. [Google Scholar] [CrossRef] [PubMed]
- Senadheera, S.N.; Kulkarni, S.S.; McLaughlin, J.P.; Aldrich, J.V. Improved Synthesis of CJ-15,208 Isomers and Their Pharmacological Activity at Opioid Receptors. In Peptides: Building Bridge; Lebl, M., Ed.; American Peptide Society: San Diego, CA, USA, 2011; pp. 346–347. [Google Scholar]
- Brice-Tutt, A.C.; Senadheera, S.N.; Ganno, M.L.; Eans, S.O.; Khaliq, T.; Murray, T.F.; McLaughlin, J.P.; Aldrich, J.V. Phenylalanine stereoisomers of CJ-15,208 and [D-Trp]CJ-15,208 exhibit distinctly different opioid activity profiles. Molecules 2020, 25, 3999. [Google Scholar] [CrossRef] [PubMed]
- Ross, N.C.; Reilley, K.J.; Murray, T.F.; Aldrich, J.V.; McLaughlin, J.P. Novel opioid cyclic tetrapeptides: Trp isomers of CJ-15,208 exhibit distinct opioid receptor agonism and short-acting kappa opioid receptor antagonism. Br. J. Pharmacol. 2012, 165, 1097–1108. [Google Scholar] [CrossRef]
- Wtorek, K.; Piekielna-Ciesielska, J.; Janecki, T.; Janecka, A. The search for opioid analgesics with limited tolerance liability. Peptides 2020, 130, 170331. [Google Scholar] [CrossRef]
- Sanchez-Blazquez, P.; Garcia-Espana, A.; Garzon, J. Antisense oligodeoxynucleotides to opioid mu and delta receptors reduced morphine dependence in mice: Role of delta-2 opioid receptors. J. Pharmacol. Exp. Ther. 1997, 280, 1423–1431. [Google Scholar] [PubMed]
- Zhu, Y.; King, M.A.; Schuller, A.G.P.; Nitsche, J.F.; Reidl, M.; Elde, R.P.; Unterwald, E.; Pasternak, G.W.; Pintar, J.E. Retention of Supraspinal Delta-like Analgesia and Loss of Morphine Tolerance in δ Opioid Receptor Knockout Mice. Neuron 1999, 24, 243–252. [Google Scholar] [CrossRef]
- Hruby, V.J. Multivalent peptide and peptidomimetic ligands for the treatment of pain without toxicities and addiction. Peptides 2019, 116, 63–67. [Google Scholar] [CrossRef]
- Nielsen, C.K.; Simms, J.A.; Bito-Onon, J.J.; Li, R.; Ananthan, S.; Bartlett, S.E. The delta opioid receptor antagonist, SoRI-9409, decreases yohimbine stress-induced reinstatement of ethanol-seeking. Addict. Biol. 2012, 17, 224–234. [Google Scholar] [CrossRef]
- Nielsen, C.K.; Simms, J.A.; Pierson, H.B.; Li, R.; Saini, S.K.; Ananthan, S.; Bartlett, S.E. A novel delta opioid receptor antagonist, SoRI-9409, produces a selective and long-lasting decrease in ethanol consumption in heavy-drinking rats. Biol. Psychiatry 2008, 64, 974–981. [Google Scholar] [CrossRef]
- Ciccocioppo, R.; Martin-Fardon, R.; Weiss, F. Effect of selective blockade of μ1 or delta opioid receptors on reinstatement of alcohol-seeking behavior by drug-associated stimuli in rats. Neuropsychopharmacology 2002, 27, 391–399. [Google Scholar] [CrossRef]
- Marinelli, P.W.; Funk, D.; Harding, S.; Li, Z.; Juzytsch, W.; Le, A.D. Roles of opioid receptor subtypes in mediating alcohol-seeking induced by discrete cues and context. Eur. J. Neurosci. 2009, 30, 671–678. [Google Scholar] [CrossRef]
- Ward, S.J.; Roberts, D.C. Microinjection of the delta-opioid receptor selective antagonist naltrindole 5′-isothiocyanate site specifically affects cocaine self-administration in rats responding under a progressive ratio schedule of reinforcement. Behav. Brain Res. 2007, 182, 140–144. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Menkens, K.; Bilsky, E.J.; Wild, K.D.; Portoghese, P.S.; Reid, L.D.; Porreca, F. Cocaine place preference is blocked by the delta-opioid receptor antagonist, naltrindole. Eur. J. Pharmacol. 1992, 219, 345–346. [Google Scholar] [CrossRef]
- Suzuki, T.; Mori, T.; Tsuji, M.; Misawa, M.; Nagase, H. The role of delta-opioid receptor subtypes in cocaine- and methamphetamine-induced place preferences. Life Sci. 1994, 55, PL339–PL344. [Google Scholar] [CrossRef]
- de Vries, T.J.; Babovic-Vuksanovic, D.; Elmer, G.; Shippenberg, T.S. Lack of involvement of delta-opioid receptors in mediating the rewarding effects of cocaine. Psychopharmacology 1995, 120, 442–448. [Google Scholar] [CrossRef] [PubMed]
- Reid, L.D.; Glick, S.D.; Menkens, K.A.; French, E.D.; Bilsky, E.J.; Porreca, F. Cocaine self-administration and naltrindole, a delta-selective opioid antagonist. Neuroreport 1995, 6, 1409–1412. [Google Scholar] [CrossRef]
- Commons, K.G. Translocation of presynaptic delta opioid receptors in the ventrolateral periaqueductal gray after swim stress. J. Comp. Neurol. 2003, 464, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Cahill, C.M.; Morinville, A.; Hoffert, C.; O’Donnell, D.; Beaudet, A. Up-regulation and trafficking of delta opioid receptor in a model of chronic inflammation: Implications for pain control. Pain 2003, 101, 199–208. [Google Scholar] [CrossRef]
- Rothman, R.B.; Danks, J.A.; Jacobson, A.E.; Burke, T.R., Jr.; Rice, K.C.; Tortella, F.C.; Holaday, J.W. Morphine tolerance increases mu-noncompetitive delta binding sites. Eur. J. Pharmacol. 1986, 124, 113–119. [Google Scholar] [CrossRef]
- Morinville, A.; Cahill, C.M.; Aibak, H.; Rymar, V.V.; Pradhan, A.; Hoffert, C.; Mennicken, F.; Stroh, T.; Sadikot, A.F.; O’Donnell, D.; et al. Morphine-induced changes in delta opioid receptor trafficking are linked to somatosensory processing in the rat spinal cord. J. Neurosci. 2004, 24, 5549–5559. [Google Scholar] [CrossRef]
- van Rijn, R.M.; Brissett, D.I.; Whistler, J.L. Emergence of functional spinal delta opioid receptors after chronic ethanol exposure. Biol. Psychiatry 2012, 71, 232–238. [Google Scholar] [CrossRef]
- Kotlinska, J.H.; Gibula-Bruzda, E.; Pachuta, A.; Kunce, D.; Witkowska, E.; Chung, N.N.; Schiller, P.W.; Izdebski, J. Influence of new deltorphin analogues on reinstatement of cocaine-induced conditioned place preference in rats. Behav. Pharmacol. 2010, 21, 638–648. [Google Scholar] [CrossRef]
- Margolis, E.B.; Fields, H.L.; Hjelmstad, G.O.; Mitchell, J.M. Delta-opioid receptor expression in the ventral tegmental area protects against elevated alcohol consumption. J. Neurosci. 2008, 28, 12672–12681. [Google Scholar] [CrossRef]
- Bie, B.; Zhu, W.; Pan, Z.Z. Ethanol-induced delta-opioid receptor modulation of glutamate synaptic transmission and conditioned place preference in central amygdala. Neuroscience 2009, 160, 348–358. [Google Scholar] [CrossRef]
- Nielsen, C.K.; Simms, J.A.; Li, R.; Mill, D.; Yi, H.; Feduccia, A.A.; Santos, N.; Bartlett, S.E. delta-opioid receptor function in the dorsal striatum plays a role in high levels of ethanol consumption in rats. J. Neurosci. 2012, 32, 4540–4552. [Google Scholar] [CrossRef]
- Mendez, M.; Morales-Mulia, M.; Leriche, M. [3H]DPDPE binding to delta opioid receptors in the rat mesocorticolimbic and nigrostriatal pathways is transiently increased by acute ethanol administration. Brain Res. 2004, 1028, 180–190. [Google Scholar] [CrossRef]
- Froehlich, J.C.; Zweifel, M.; Harts, J.; Lumeng, L.; Li, T.K. Importance of delta opioid receptors in maintaining high alcohol drinking. Psychopharmacology 1991, 103, 467–472. [Google Scholar] [CrossRef]
- Herz, A. Endogenous opioid systems and alcohol addiction. Psychopharmacology 1997, 129, 99–111. [Google Scholar] [CrossRef]
- Borg, P.J.; Taylor, D.A. Involvement of mu- and delta-opioid receptors in the effects of systemic and locally perfused morphine on extracellular levels of dopamine, DOPAC and HVA in the nucleus accumbens of the halothane-anaesthetized rat. Naunyn-Schmiedeberg’s Arch. Pharmacol. 1997, 355, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Chu Sin Chung, P.; Kieffer, B.L. Delta opioid receptors in brain function and diseases. Pharmacol. Ther. 2013, 140, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Ross, N.C.; Kulkarni, S.S.; McLaughlin, J.P.; Aldrich, J.V. Synthesis of CJ-15,208, a novel κ-opioid receptor antagonist. Tetrahedron Lett. 2010, 51, 5020–5023. [Google Scholar] [CrossRef] [PubMed]
- Aldrich, J.V.; Senadheera, S.N.; Ross, N.C.; Reilley, K.J.; Ganno, M.L.; Eans, S.O.; Murray, T.F.; McLaughlin, J.P. Alanine analogues of [D-Trp]CJ-15,208: Novel opioid activity profiles and prevention of drug- and stress-induced reintatement of cocaine-seeking behaviour. Br. J. Pharmacol. 2014, 171, 3212–3222. [Google Scholar] [CrossRef] [PubMed]
- Nomenclature Committee of IUB (NC-IUB) and IUPAC-IUB Joint Commission on Biochemical Nomenclature (JCBN). Eur. J. Biochem. 1984, 138, 9–37.
- Khaliq, T.; Williams, T.D.; Senadheera, S.N.; Aldrich, J.V. Development of a robust, sensitive and selective liquid chromatography-tandem mass spectrometry assay for the quantification of the novel macrocyclic peptide kappa opioid receptor antagonist [D-Trp]CJ-15,208 in plasma and application to an initial pharmacokinetic study. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2016, 1028, 11–15. [Google Scholar] [CrossRef]
- Arttamangkul, S.; Ishmael, J.E.; Murray, T.F.; Grandy, D.K.; DeLander, G.E.; Kieffer, B.L.; Aldrich, J.V. Synthesis and opioid activity of conformationally constrained dynorphin A analogues. 2. Conformational constraint in the “address” sequence. J. Med. Chem. 1997, 40, 1211–1218. [Google Scholar] [CrossRef]
- Cheng, Y.C.; Prusoff, W.H. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 percent inhibition (IC50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar] [CrossRef]
- McGrath, J.C.; Drummond, G.B.; McLachlan, E.M.; Kilkenny, C.; Wainwright, C.L. Guidelines for reporting experiments involving animals: The ARRIVE guidelines. Br. J. Pharmacol. 2010, 160, 1573–1576. [Google Scholar] [CrossRef]
- McLaughlin, J.P.; Hill, K.P.; Jiang, Q.; Sebastian, A.; Archer, S.; Bidlack, J.M. Nitrocinnamoyl and chlorocinnamoyl derivatives of dihydrocodeinone: In vivo and in vitro characterization of mu-selective agonist and antagonist activity. J. Pharmacol. Exp. Ther. 1999, 289, 304–311. [Google Scholar]
- Brice-Tutt, A.C.; Wilson, L.L.; Eans, S.O.; Stacy, H.M.; Simons, C.A.; Simpson, G.G.; Coleman, J.S.; Ferracane, M.J.; Aldrich, J.V.; McLaughlin, J.P. Multifunctional opioid receptor agonism and antagonism by a novel cyclic tetrapeptide prevents reinstatement of morphine-seeking behavior. Br. J. Pharmacol. 2020, 177, 4209–4222. [Google Scholar] [CrossRef]
- Jiang, Q.; Seyed-Mozaffari, A.; Sebastian, A.; Archer, S.; Bidlack, J.M. Preventing morphine antinociceptive tolerance by irreversible mu opioid antagonists before the onset of their antagonism. J. Pharmacol. Exp. Ther. 1995, 273, 680–688. [Google Scholar] [CrossRef]
- Mathews, J.L.; Smrcka, A.V.; Bidlack, J.M. A novel Gβγ-subunit inhibitor selectively modulates mu-opioid-dependent antinociception and attenuates acute morphine-induced antinociceptive tolerance and dependence. J. Neurosci. 2008, 28, 12183–12189. [Google Scholar] [CrossRef] [PubMed]
- Hoot, M.R.; Sypek, E.I.; Reilley, K.J.; Carey, A.N.; Bidlack, J.M.; McLaughlin, J.P. Inhibition of Gβγ-subunit signaling potentiates morphine-induced antinociception but not respiratory depression, constipation, locomotion, and reward. Behav. Pharmacol. 2013, 24, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Way, E.L.; Loh, H.H.; Shen, F.H. Simultaneous quantitative assessment of morphine tolerance and physical dependence. J. Pharmacol. Exp. Ther. 1969, 167, 1–8. [Google Scholar] [PubMed]
- Spetea, M.; Eans, S.O.; Ganno, M.L.; Lantero, A.; Mairegger, M.; Toll, L.; Schmidhammer, H.; McLaughlin, J.P. Selective kappa receptor partial agonist HS666 produces potent antinociception without inducing aversion after i.c.v. administration in mice. Br. J. Pharmacol. 2017, 174, 2444–2456. [Google Scholar] [CrossRef]
- Ferracane, M.J.; Brice-Tutt, A.; Coleman, J.; Simpson, G.; Wilson, L.; Eans, S.O.; Stacy, H.; Murray, T.F.; McLaughlin, J.P.; Aldrich, J.V. Design, synthesis, and characterization of the macrocyclic tetrapeptide cyclo[Pro-Sar-Phe-D-Phe]: A mixed opioid receptor agonist-antagonist following oral administration. ACS Chem. Neurosci. 2020, 11, 1324–1336. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ki (nM ± SEM) | |||
|---|---|---|---|
| Analog | KOR | MOR | DOR |
| CJ-15,208 2 | 27.4 ± 4.6 | 451 ± 114 | 1720 ± 350 |
| Bta4 | 51.9 ± 4.8 | 90.3 ± 38.5 | 1600 ± 360 |
| Nal(1′)4 | 103 ± 6 | 110 ± 16 | 1920 ± 120 |
| Nal(2′)4 | 358 ± 137 | 583 ± 59 | 3300 ± 360 |
| [d-Trp]CJ-15,208 2 | 21.8 ± 4.8 | 259 ± 29 | 4190 ± 860 |
| d-Bta4 | 4.29 ± 0.38 | 84.2 ± 11.8 | >10,000 |
| d-Nal(1′)4 | 21.4 ± 0.7 | 101 ± 25 | >10,000 3 |
| d-Nal(2′)4 | 12.3 ± 1.8 | 63.8 ± 5 | >10,000 |
| ED50 (and 95% C.I. 1) Values | ||
|---|---|---|
| Compound | i.c.v. (nmol) | Receptors Involved |
| Morphine | 2.35 (1.13–5.03) | MOR |
| CJ-15,208 | 1.74 (0.62–4.82) | KOR, MOR |
| Bta4 | 13.2 (9.62–18.2) | KOR, MOR, DOR |
| Nal(1′)4 | 0.24 (0.19–0.31) | KOR, MOR, DOR |
| Nal(2′)4 | 19.5 (15.8–24.0) | MOR |
| [d-Trp]CJ-15,208 | ~ | - |
| d-Bta4 | ~ | KOR, MOR |
| d-Nal(1′)4 | ~ | - |
| d-Nal(2′)4 | 31.2 (22.5–41.9) | MOR |
| Compound | Naïve ED50 (95% C.I.) | ED50 (95% C.I.) Pretreated Mice | Fold-Shift, Naïve ED50 vs. Second ED50 |
|---|---|---|---|
| [Nal(2′)4]CJ-15,208 | 19.5 (15.8–24.0) | 7.26 * (5.57–9.71) | 0.37 |
| Morphine | 2.35 (1.13–5.03) | 18.1 * (13.7–23.7) | 7.70 |
| ESI-MS m/z | TLC | HPLC | HPLC | ||
|---|---|---|---|---|---|
| Analog | Observed 1 | Calc 1 | Rf (EtOAc) | System 1 2 | System 2 3 |
| Bta4 | 617.3 4 | 617.2 | 0.23 | 24.3 | 29.4 |
| Nal(1′)4 | 611.3 | 611.3 | 0.27 5 | 15.1 6 | 27.4 7 |
| Nal(2′)4 | 611.3 | 611.3 | 0.18 8 | 24.6 | 28.9 |
| D-Bta4 | 617.3 4 | 617.2 | 0.67 | 32.4 | 37.4 |
| D-Nal(1′)4 | 611.3 | 611.3 | 0.66 | 32.8 | 37.4 |
| D-Nal(2′)4 | 611.3 | 611.3 | 0.68 | 32.8 | 36.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scherrer, K.H.; Eans, S.O.; Medina, J.M.; Senadheera, S.N.; Khaliq, T.; Murray, T.F.; McLaughlin, J.P.; Aldrich, J.V. Tryptophan Substitution in CJ-15,208 (cyclo[Phe-D-Pro-Phe-Trp]) Introduces δ-Opioid Receptor Antagonism, Preventing Antinociceptive Tolerance and Stress-Induced Reinstatement of Extinguished Cocaine-Conditioned Place Preference. Pharmaceuticals 2023, 16, 1218. https://doi.org/10.3390/ph16091218
Scherrer KH, Eans SO, Medina JM, Senadheera SN, Khaliq T, Murray TF, McLaughlin JP, Aldrich JV. Tryptophan Substitution in CJ-15,208 (cyclo[Phe-D-Pro-Phe-Trp]) Introduces δ-Opioid Receptor Antagonism, Preventing Antinociceptive Tolerance and Stress-Induced Reinstatement of Extinguished Cocaine-Conditioned Place Preference. Pharmaceuticals. 2023; 16(9):1218. https://doi.org/10.3390/ph16091218
Chicago/Turabian StyleScherrer, Kristen H., Shainnel O. Eans, Jessica M. Medina, Sanjeewa N. Senadheera, Tanvir Khaliq, Thomas F. Murray, Jay P. McLaughlin, and Jane V. Aldrich. 2023. "Tryptophan Substitution in CJ-15,208 (cyclo[Phe-D-Pro-Phe-Trp]) Introduces δ-Opioid Receptor Antagonism, Preventing Antinociceptive Tolerance and Stress-Induced Reinstatement of Extinguished Cocaine-Conditioned Place Preference" Pharmaceuticals 16, no. 9: 1218. https://doi.org/10.3390/ph16091218
APA StyleScherrer, K. H., Eans, S. O., Medina, J. M., Senadheera, S. N., Khaliq, T., Murray, T. F., McLaughlin, J. P., & Aldrich, J. V. (2023). Tryptophan Substitution in CJ-15,208 (cyclo[Phe-D-Pro-Phe-Trp]) Introduces δ-Opioid Receptor Antagonism, Preventing Antinociceptive Tolerance and Stress-Induced Reinstatement of Extinguished Cocaine-Conditioned Place Preference. Pharmaceuticals, 16(9), 1218. https://doi.org/10.3390/ph16091218