
Indol-3-ylglyoxylamide as Privileged Scaffold in Medicinal Chemistry
 , , , and
, , , and
Abstract
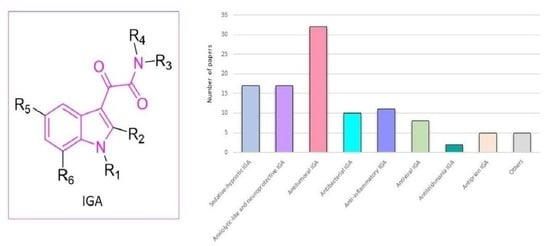
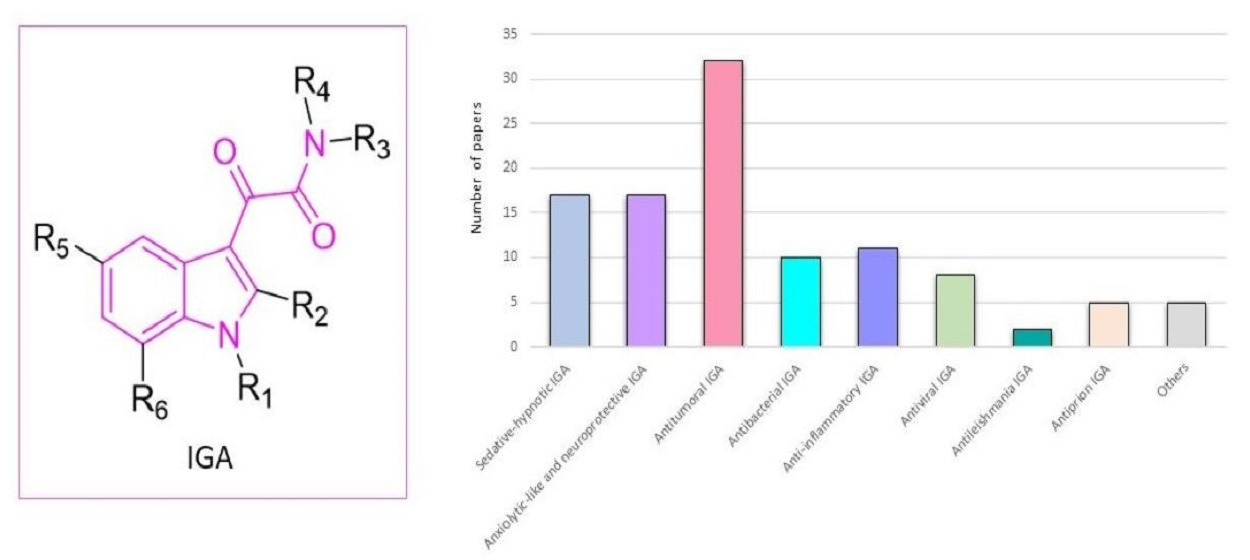
1. Introduction
2. Indolylglyoxylamides with Sedative–Hypnotic Properties
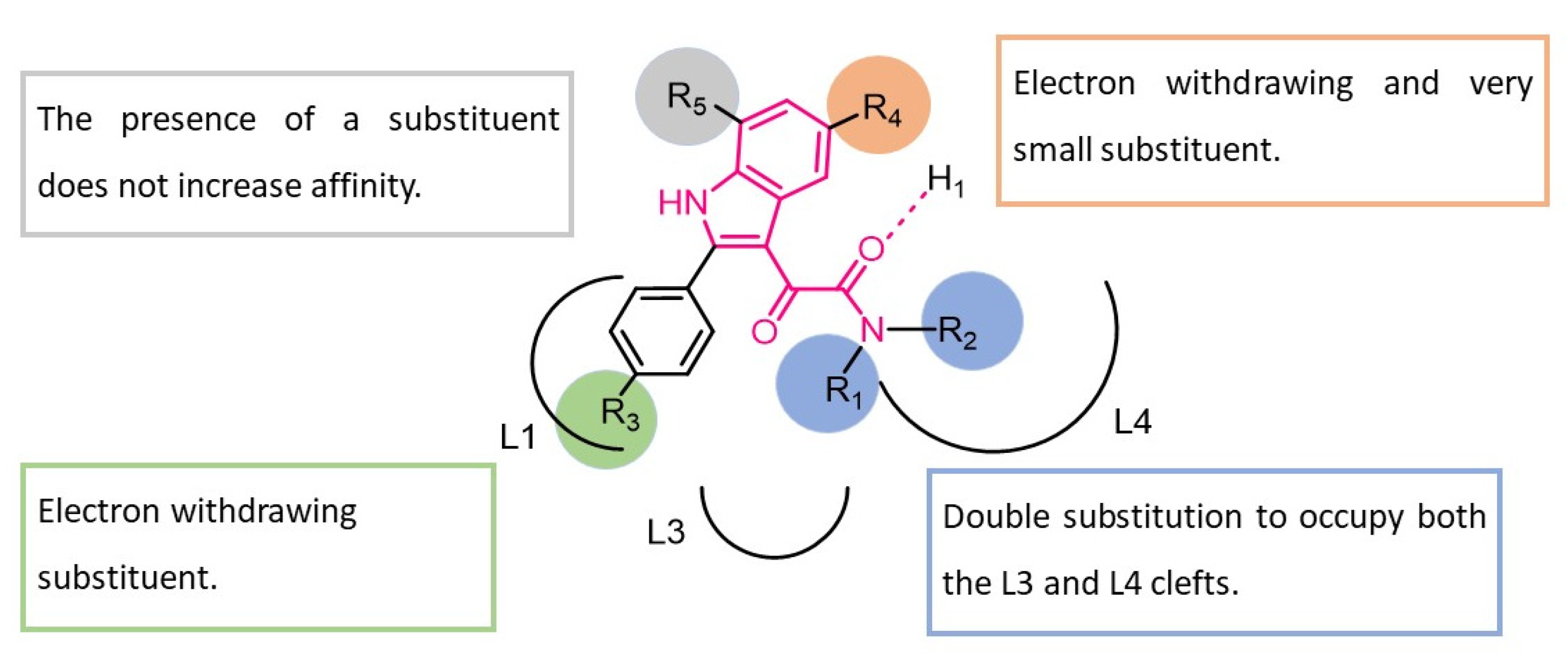
3. Indolylglyoxylamides with Anxiolytic-Like and Neuroprotective Properties
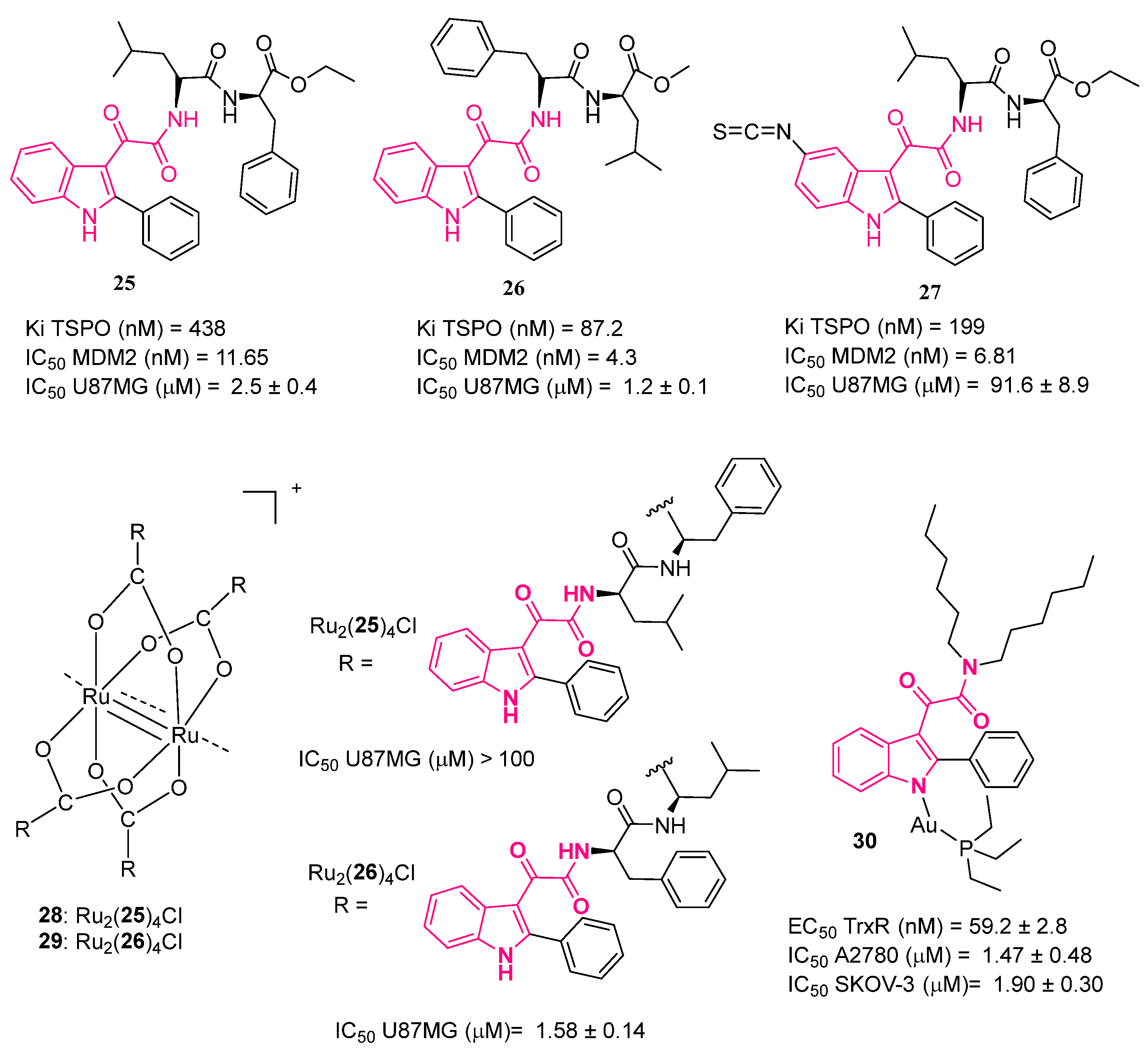
4. Indolylglyoxylamides with Antitumoral Properties
5. Indolylglyoxylamides with Antibacterial Properties
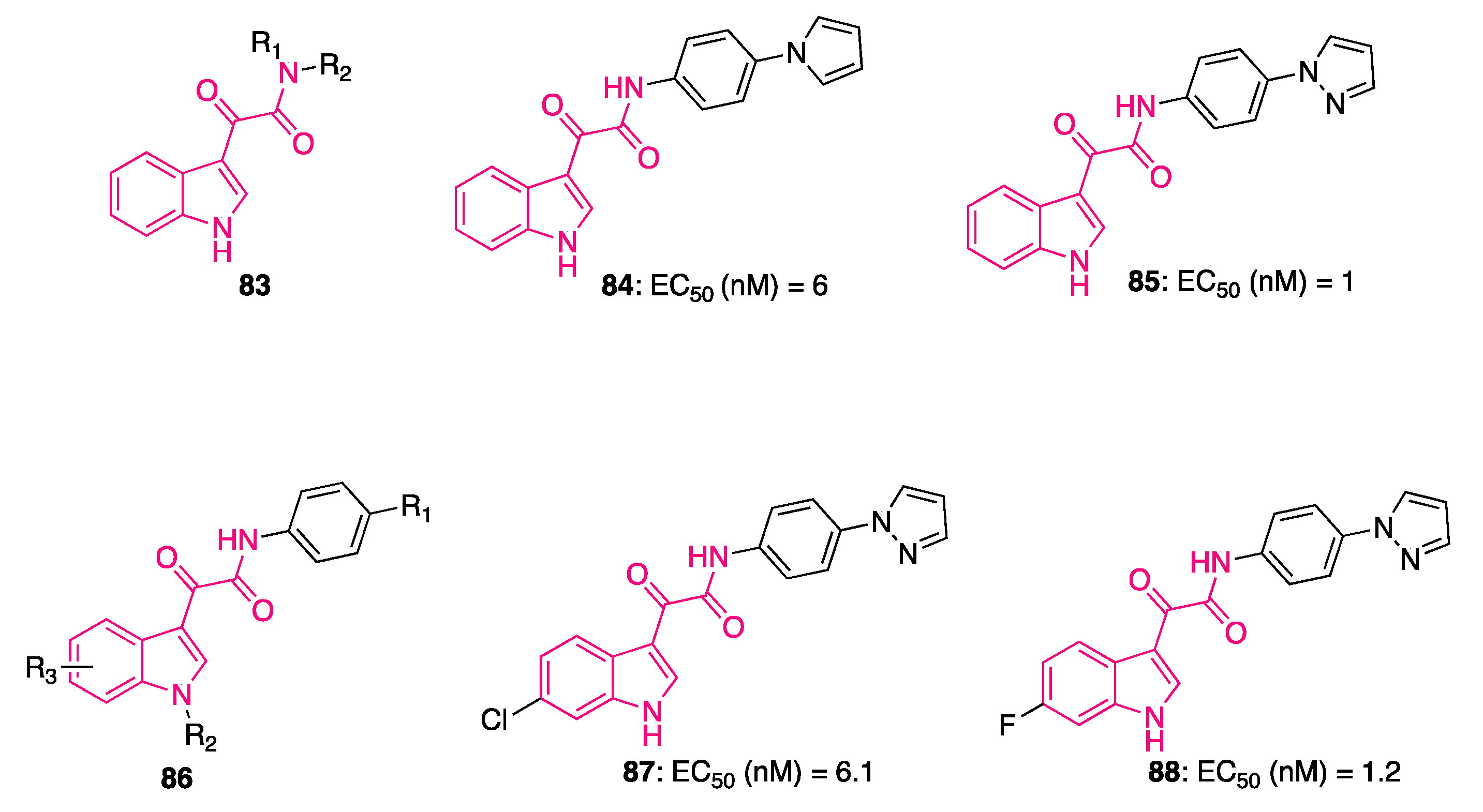
6. Indolylglyoxylamides with Anti-Inflammatory Properties
6.1. Phosphodiesterase PDE4 Inhibitors
6.2. Phospholipase A2 Inhibitors
6.3. p38α Inhibitors
7. Indolylglyoxylamides with Antiviral Properties
7.1. Viral NS2B/NS3 Protease Inhibitors
7.2. HIV-1 Inhibitors
8. Indolylglyoxylamides with Antileishmania Properties
9. Indolylglyoxylamides with Antiprion Properties
10. Others
10.1. A2B Allosteric Modulators
10.2. Carbonic Anhydrase Activators
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Evans, B.E.; Rittle, K.E.; Bock, M.G.; DiPardo, R.M.; Freidinger, R.M.; Whitter, W.L.; Lundell, G.F.; Veber, D.F.; Anderson, P.S.; Chang, R.S.L.; et al. Methods for Drug Discovery: Development of Potent, Selective, Orally Effective Cholecystokinin Antagonists. J. Med. Chem. 1988, 31, 2235–2246. [Google Scholar] [CrossRef] [PubMed]
- de Sa Alves, F.; Barreiro, E.; Manssour Fraga, C. From Nature to Drug Discovery: The Indole Scaffold as a “Privileged Structure”. Mini Rev. Med. Chem. 2009, 9, 782–793. [Google Scholar] [CrossRef] [PubMed]
- Salerno, S.; Barresi, E.; Baglini, E.; Poggetti, V.; Da Settimo, F.; Taliani, S. Target-Based Anticancer Indole Derivatives for the Development of Anti-Glioblastoma Agents. Molecules 2023, 28, 2587. [Google Scholar] [CrossRef] [PubMed]
- Taliani, S.; Da Settimo, F.; Martini, C.; Laneri, S.; Novellino, E.; Greco, G. Exploiting the Indole Scaffold to Design Compounds Binding to Different Pharmacological Targets. Molecules 2020, 25, 2331. [Google Scholar] [CrossRef]
- Shimazaki, Y.; Yajima, T.; Takani, M.; Yamauchi, O. Metal Complexes Involving Indole Rings: Structures and Effects of Metal–Indole Interactions. Coord. Chem. Rev. 2009, 253, 479–492. [Google Scholar] [CrossRef]
- Da Settimo, F.; Lucacchini, A.; Marini, A.M.; Martini, C.; Primofiore, G.; Senatore, G.; Taliani, S. Isosteric Replacement of the Indole Nucleus by Benzothiophene and Benzofuran in a Series of Indolylglyoxylylamine Derivatives with Partial Agonist Activity at the Benzodiazepine Receptor. Eur. J. Med. Chem. 1996, 31, 951–956. [Google Scholar] [CrossRef]
- Bianucci, A.M.; Da Settimo, A.; Da Settimo, F.; Primofiore, G.; Martini, C.; Giannaccini, G.; Lucacchini, A. Benzodiazepine Receptor Affinity and Interaction of Some N-(Indol-3-Ylglyoxylyl)Amine Derivatives. J. Med. Chem. 1992, 35, 2214–2220. [Google Scholar] [CrossRef]
- Bondensgaard, K.; Ankersen, M.; Thøgersen, H.; Hansen, B.S.; Wulff, B.S.; Bywater, R.P. Recognition of Privileged Structures by G-Protein Coupled Receptors. J. Med. Chem. 2004, 47, 888–899. [Google Scholar] [CrossRef]
- Rad, R.; Mracec, M.; Mracec, M.; Oprea, T. The Privileged Structures Hypothesis for G Protein-Coupled Receptors—Some Preliminary Results. Rev. Roum. Chim. 2007, 52, 853–858. [Google Scholar]
- Desiraju, G.R. Supramolecular Synthons in Crystal Engineering—A New Organic Synthesis. Angew. Chemie Int. Ed. English 1995, 34, 2311–2327. [Google Scholar] [CrossRef]
- Robello, M.; Barresi, E.; Baglini, E.; Salerno, S.; Taliani, S.; Settimo, F. Da The Alpha Keto Amide Moiety as a Privileged Motif in Medicinal Chemistry: Current Insights and Emerging Opportunities. J. Med. Chem. 2021, 64, 3508–3545. [Google Scholar] [CrossRef]
- Etter, M.C. Encoding and Decoding Hydrogen-Bond Patterns of Organic Compounds. Acc. Chem. Res. 1990, 23, 120–126. [Google Scholar] [CrossRef]
- Morin, C.M.; Jarrin, D.C. Epidemiology of Insomnia: Prevalence, Course, Risk Factors, and Public Health Burden. Sleep Med. Clin. 2022, 17, 173–191. [Google Scholar] [CrossRef]
- Mitler, M.M. Nonselective and Selective Benzodiazepine Receptor Agonists—Where Are We Today? Sleep 2000, 23, S39–S47. [Google Scholar]
- Barbera, J.; Shaprio, C. Benefit-Risk Assessment of Zaleplon in the Treatment of Insomnia. Drug Saf. 2005, 28, 301–318. [Google Scholar] [CrossRef]
- Sanger, D.J.; Benavides, J.; Perrault, G.; Morel, E.; Cohen, C.; Joly, D.; Zivkovic, B. Recent Developments in the Behavioral Pharmacology of Benzodiazepine (ω) Receptors: Evidence for the Functional Significance of Receptor Subtypes. Neurosci. Biobehav. Rev. 1994, 18, 355–372. [Google Scholar] [CrossRef]
- Terzano, M.G.; Rossi, M.; Palomba, V.; Smerieri, A.; Parrino, L. New Drugs for Insomnia: Comparative Tolerability of Zopiclone, Zolpidem and Zaleplon. Drug Saf. 2003, 26, 261–282. [Google Scholar] [CrossRef]
- Krystal, A.D.; Walsh, J.K.; Laska, E.; Caron, J.; Amato, D.A.; Wessel, T.C.; Roth, T. Sustained Efficacy of Eszopiclone Over 6 Months of Nightly Treatment: Results of a Randomized, Double-Blind, Placebo-Controlled Study in Adults with Chronic Insomnia. Sleep 2003, 26, 793–799. [Google Scholar] [CrossRef]
- Roth, T.; Soubrane, C.; Titeux, L.; Walsh, J.K.; on behalf of the Zoladult Study Group. Efficacy and Safety of Zolpidem-MR: A Double-Blind, Placebo-Controlled Study in Adults with Primary Insomnia. Sleep Med. 2006, 7, 397–406. [Google Scholar] [CrossRef]
- Farber, R.H.; Burke, P.J. Post-Bedtime Dosing with Indiplon in Adults and the Elderly: Results from Two Placebo-Controlled, Active Comparator Crossover Studies in Healthy Volunteers. Curr. Med. Res. Opin. 2008, 24, 837–846. [Google Scholar] [CrossRef]
- Sieghart, W.; Sperk, G. Subunit Composition, Distribution and Function of GABA-A Receptor Subtypes. Curr. Top. Med. Chem. 2005, 2, 795–816. [Google Scholar] [CrossRef] [PubMed]
- Möhler, H.; Fritschy, J.M.; Rudolph, U. A New Benzodiazepine Pharmacology. J. Pharmacol. Exp. Ther. 2002, 300, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Whiting, P.J. GABA-A Receptor Subtypes in the Brain: A Paradigm for CNS Drug Discovery? Drug Discov. Today 2003, 8, 445–450. [Google Scholar] [CrossRef] [PubMed]
- Da Settimo, F.; Taliani, S.; Trincavelli, M.; Montali, M.; Martini, C. GABAA/Bz Receptor Subtypes as Targets for Selective Drugs. Curr. Med. Chem. 2007, 14, 2680–2701. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Koehler, K.F.; Zhang, P.; Cook, J.M. Development of a Comprehensive Pharmacophore Model for the Benzodiazepine Receptor. Drug Des. Discov. 1995, 12, 193–248. [Google Scholar]
- Da Settimo, A.; Primofiore, G.; Da Settimo, F.; Marini, A.M.; Novellino, E.; Greco, G.; Martini, C.; Giannaccini, G.; Lucacchini, A. Synthesis, Structure-Activity Relationships, and Molecular Modeling Studies of N-(Indol-3-Ylglyoxylyl)Benzylamine Derivatives Acting at the Benzodiazepine Receptor. J. Med. Chem. 1996, 39, 5083–5091. [Google Scholar] [CrossRef]
- Primofiore, G.; Da Settimo, F.; Taliani, S.; Marini, A.M.; Novellino, E.; Greco, G.; Lavecchia, A.; Besnard, F.; Trincavelli, L.; Costa, B.; et al. Novel N-(Arylalkyl)Indol-3-Ylglyoxylylamides Targeted as Ligands of the Benzodiazepine Receptor: Synthesis, Biological Evaluation, and Molecular Modeling Analysis of the Structure-Activity Relationships. J. Med. Chem. 2001, 44, 2286–2297. [Google Scholar] [CrossRef]
- Primofiore, G.; Taliani, S.; Da Settimo, F.; Marini, A.M.; La Motta, C.; Simorini, F.; Patrizi, M.P.; Sergianni, V.; Novellino, E.; Greco, G.; et al. Novel N-Substituted Indol-3-Ylglyoxylamides Probing the LDi and L1/L2 Lipophilic Regions of the Benzodiazepine Receptor Site in Search for Subtype-Selective Ligands. J. Med. Chem. 2007, 50, 1627–1634. [Google Scholar] [CrossRef]
- He, X.; Huang, Q.; Ma, C.; Yu, S.; McKernan, R.; Cook, J.M. Pharmacophore/Receptor Models for GABA(A)/BzR Alpha2beta3gamma2, Alpha3beta3gamma2 and Alpha4beta3gamma2 Recombinant Subtypes. Included Volume Analysis and Comparison to Alpha1beta3gamma2, Alpha5beta3gamma2, and Alpha6beta3gamma2 Subtypes. Drug Des. Discov. 2000, 17, 131–171. [Google Scholar]
- Atack, J.R. Anxioselective Compounds Acting at the GABA(A) Receptor Benzodiazepine Binding Site. Curr. Drug Targets CNS Neurol. Disord. 2003, 2, 213–232. [Google Scholar] [CrossRef]
- Atack, J.R. The Benzodiazepine Binding Site of GABAA Receptors as a Target for the Development of Novel Anxiolytics. Expert Opin. Investig. Drugs 2005, 14, 601–618. [Google Scholar] [CrossRef]
- Sigel, E.; Ernst, M. The Benzodiazepine Binding Sites of GABAA Receptors. Trends Pharmacol. Sci. 2018, 39, 659–671. [Google Scholar] [CrossRef]
- Skolnick, P. Anxioselective Anxiolytics: On a Quest for the Holy Grail. Trends Pharmacol. Sci. 2012, 33, 611–620. [Google Scholar] [CrossRef]
- Primofiore, G.; Da Settimo, F.; Marini, A.M.; Taliani, S.; La Motta, C.; Simorini, F.; Novellino, E.; Greco, G.; Cosimelli, B.; Ehlardo, M.; et al. Refinement of the Benzodiazepine Receptor Site Topology by Structure-Activity Relationships of New N-(Heteroarylmethyl)Indol-3- Ylglyoxylamides. J. Med. Chem. 2006, 49, 2489–2495. [Google Scholar] [CrossRef]
- Taliani, S.; Cosimelli, B.; Da Settimo, F.; Marini, A.M.; La Motta, C.; Simorini, F.; Salerno, S.; Novellino, E.; Greco, G.; Cosconati, S.; et al. Identification of Anxiolytic/Nonsedative Agents among Indol-3- Ylglyoxylamides Acting as Functionally Selective Agonists at the γ-Aminobutyric Acid-A (GABAA) A2 Benzodiazepine Receptor. J. Med. Chem. 2009, 52, 3723–3734. [Google Scholar] [CrossRef]
- Bourin, M.; Hascoët, M. The Mouse Light/Dark Box Test. Eur. J. Pharmacol. 2003, 463, 55–65. [Google Scholar] [CrossRef]
- Olsen, R.W. GABAA Receptor: Positive and Negative Allosteric Modulators. Neuropharmacology 2018, 136, 10–22. [Google Scholar] [CrossRef]
- Papadopoulos, V.; Baraldi, M.; Guilarte, T.R.; Knudsen, T.B.; Lacapère, J.J.; Lindemann, P.; Norenberg, M.D.; Nutt, D.; Weizman, A.; Zhang, M.R.; et al. Translocator Protein (18 KDa): New Nomenclature for the Peripheral-Type Benzodiazepine Receptor Based on Its Structure and Molecular Function. Trends Pharmacol. Sci. 2006, 27, 402–409. [Google Scholar] [CrossRef]
- Taliani, S.; Da Settimo, F.; Da Pozzo, E.; Chelli, B.; Martini, C. Translocator Protein Ligands as Promising Therapeutic Tools for Anxiety Disorders. Curr. Med. Chem. 2009, 16, 3359–3380. [Google Scholar] [CrossRef]
- Le Fur, G.; Vaucher, N.; Perrier, M.L.; Flamier, A.; Benavides, J.; Renault, C.; Dubroeucq, M.C.; Guérémy, C.; Uzan, A. Differentiation between Two Ligands for Peripheral Benzodiazepine Binding Sites, [3H]R05-4864 and [3H]PK 11195, by Thermodynamic Studies. Life Sci. 1983, 33, 449–457. [Google Scholar] [CrossRef]
- Romeo, E.; Auta, J.; Kozikowski, A.P.; Ma, D.; Papadopoulos, V.; Puia, G.; Costa, E.; Guidotti, A. 2-Aryl-3-Indoleacetamides (FGIN-1): A New Class of Potent and Specific Ligands for the Mitochondrial DBI Receptor (MDR). J. Pharmacol. Exp. Ther. 1992, 262, 971–978. [Google Scholar] [PubMed]
- Kozikowski, A.P.; Ma, D.; Brewer, J.; Sun, S.; Costa, E.; Romeo, E.; Guidotti, A. Chemistry, Binding Affinities, and Behavioral Properties of a New Class of “Antineophobic” Mitochondrial DBI Receptor Complex (MDRC) Ligands. J. Med. Chem. 1993, 36, 2908–2920. [Google Scholar] [CrossRef] [PubMed]
- Costa, B.; Da Pozzo, E.; Martini, C. Translocator Protein and Steroidogenesis. Biochem. J. 2018, 475, 901–904. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, V.; Lecanu, L.; Brown, R.C.; Han, Z.; Yao, Z.X. Peripheral-Type Benzodiazepine Receptor in Neurosteroid Biosynthesis, Neuropathology and Neurological Disorders. Neuroscience 2006, 138, 749–756. [Google Scholar] [CrossRef] [PubMed]
- Nothdurfter, C.; Rammes, G.; Baghai, T.C.; Schüle, C.; Schumacher, M.; Papadopoulos, V.; Rupprecht, R. Translocator Protein (18 KDa) as a Target for Novel Anxiolytics with a Favourable Side-Effect Profile. J. Neuroendocrinol. 2012, 24, 82–92. [Google Scholar] [CrossRef]
- Nothdurfter, C.; Rupprecht, R.; Rammes, G. Recent Developments in Potential Anxiolytic Agents Targeting GABAA/BzR Complex or the Translocator Protein (18 kDa) (TSPO). Curr. Top. Med. Chem. 2012, 12, 360–370. [Google Scholar] [CrossRef]
- Primofiore, G.; Da Settimo, F.; Taliani, S.; Simorini, F.; Patrizi, M.P.; Novellino, E.; Greco, G.; Abignente, E.; Costa, B.; Chelli, B.; et al. N,N-Dialkyl-2-Phenylindol-3-Ylglyoxylamides. A New Class of Potent and Selective Ligands at the Peripheral Renzodiazepine Receptor. J. Med. Chem. 2004, 47, 1852–1855. [Google Scholar] [CrossRef]
- Da Settimo, F.; Simorini, F.; Taliani, S.; La Motta, C.; Marini, A.M.; Salerno, S.; Bellandi, M.; Novellino, E.; Greco, G.; Cosimelli, B.; et al. Anxiolytic-like Effects of N,N-Dialkyl-2-Phenylindol-3-Ylglyoxylamides by Modulation of Translocator Protein Promoting Neurosteroid Biosynthesis. J. Med. Chem. 2008, 51, 5798–5806. [Google Scholar] [CrossRef]
- Barresi, E.; Bruno, A.; Taliani, S.; Cosconati, S.; Da Pozzo, E.; Salerno, S.; Simorini, F.; Daniele, S.; Giacomelli, C.; Marini, A.M.; et al. Deepening the Topology of the Translocator Protein Binding Site by Novel N,N-Dialkyl-2-Arylindol-3-Ylglyoxylamides. J. Med. Chem. 2015, 58, 6081–6092. [Google Scholar] [CrossRef]
- Barresi, E.; Robello, M.; Costa, B.; Da Pozzo, E.; Baglini, E.; Salerno, S.; Da Settimo, F.; Martini, C.; Taliani, S. An Update into the Medicinal Chemistry of Translocator Protein (TSPO) Ligands. Eur. J. Med. Chem. 2021, 209, 112924. [Google Scholar] [CrossRef]
- Da Pozzo, E.; Giacomelli, C.; Costa, B.; Cavallini, C.; Taliani, S.; Barresi, E.; Da Settimo, F.; Martini, C. TSPO PIGA Ligands Promote Neurosteroidogenesis and Human Astrocyte Well-Being. Int. J. Mol. Sci. 2016, 17, 1028. [Google Scholar] [CrossRef]
- Costa, B.; Da Pozzo, E.; Chelli, B.; Simola, N.; Morelli, M.; Luisi, M.; Maccheroni, M.; Taliani, S.; Simorini, F.; Da Settimo, F.; et al. Anxiolytic Properties of a 2-Phenylindolglyoxylamide TSPO Ligand: Stimulation of In Vitro Neurosteroid Production Affecting GABAA Receptor Activity. Psychoneuroendocrinology 2011, 36, 463–472. [Google Scholar] [CrossRef]
- Tu, L.N.; Morohaku, K.; Manna, P.R.; Pelton, S.H.; Butler, W.R.; Stocco, D.M.; Selvaraj, V. Peripheral Benzodiazepine Receptor/Translocator Protein Global Knock-out Mice Are Viable with No Effects on Steroid Hormone Biosynthesis. J. Biol. Chem. 2014, 289, 27444–27454. [Google Scholar] [CrossRef]
- Morohaku, K.; Pelton, S.H.; Daugherty, D.J.; Butler, W.R.; Deng, W.; Selvaraj, V. Translocator Protein/Peripheral Benzodiazepine Receptor Is Not Required for Steroid Hormone Biosynthesis. Endocrinology 2014, 155, 89–97. [Google Scholar] [CrossRef]
- M Scarf, A.; M Auman, K.; Kassiou, M. Is There Any Correlation between Binding and Functional Effects at the Translocator Protein (TSPO) (18 KDa)? Curr. Mol. Med. 2012, 12, 387–397. [Google Scholar] [CrossRef]
- Costa, B.; Da Pozzo, E.; Giacomelli, C.; Barresi, E.; Taliani, S.; Da Settimo, F.; Martini, C. TSPO Ligand Residence Time: A New Parameter to Predict Compound Neurosteroidogenic Efficacy. Sci. Rep. 2016, 6, 18164. [Google Scholar] [CrossRef]
- Bruno, A.; Barresi, E.; Simola, N.; Da Pozzo, E.; Costa, B.; Novellino, E.; Da Settimo, F.; Martini, C.; Taliani, S.; Cosconati, S. Unbinding of Translocator Protein 18 KDa (TSPO) Ligands: From In Vitro Residence Time to In Vivo Efficacy via in Silico Simulations. ACS Chem. Neurosci. 2019, 10, 3805–3814. [Google Scholar] [CrossRef]
- Germelli, L.; Da Pozzo, E.; Giacomelli, C.; Tremolanti, C.; Marchetti, L.; Wetzel, C.H.; Barresi, E.; Taliani, S.; Settimo, F.D.; Martini, C.; et al. De Novo Neurosteroidogenesis in Human Microglia: Involvement of the 18 Kda Translocator Protein. Int. J. Mol. Sci. 2021, 22, 3115. [Google Scholar] [CrossRef]
- Corsi, F.; Baglini, E.; Barresi, E.; Salerno, S.; Cerri, C.; Martini, C.; Da Settimo Passetti, F.; Taliani, S.; Gargini, C.; Piano, I. Targeting TSPO Reduces Inflammation and Apoptosis in an In Vitro Photoreceptor-Like Model of Retinal Degeneration. ACS Chem. Neurosci. 2022, 13, 3188–3197. [Google Scholar] [CrossRef]
- Tremolanti, C.; Cavallini, C.; Meyer, L.; Klein, C.; Da Pozzo, E.; Costa, B.; Germelli, L.; Taliani, S.; Patte-Mensah, C.; Mensah-Nyagan, A.G. Translocator Protein Ligand PIGA1138 Reduces Disease Symptoms and Severity in Experimental Autoimmune Encephalomyelitis Model of Primary Progressive Multiple Sclerosis. Mol. Neurobiol. 2022, 59, 1744–1765. [Google Scholar] [CrossRef]
- Angeloni, E.; Germelli, L.; Marchetti, L.; Da Pozzo, E.; Tremolanti, C.; Wetzel, C.H.; Baglini, E.; Taliani, S.; Da Settimo, F.; Martini, C.; et al. The Human Microglial Surveillant Phenotype Is Preserved by de Novo Neurosteroidogenesis through the Control of Cholesterol Homeostasis: Crucial Role of 18 KDa Translocator Protein. Biochim. Biophys. Acta Mol. Basis Dis. 2023, 1869, 166751. [Google Scholar] [CrossRef] [PubMed]
- Chhikara, B.S.; Parang, K. Global Cancer Statistics 2022: The Trends Projection Analysis. Chem. Biol. Lett. 2023, 10, 451. [Google Scholar]
- Ganesh, K.; Massagué, J. Targeting Metastatic Cancer. Nat. Med. 2021, 27, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Mothersill, C.; Seymour, C.B. Mechanisms and Implications of Genomic Instability and Other Delayed Effects of Ionizing Radiation Exposure. Mutagenesis 1998, 13, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Charmsaz, S.; Collins, D.M.; Perry, A.S.; Prencipe, M. Novel Strategies for Cancer Treatment: Highlights from the 55th IACR Annual Conference. Cancers 2019, 11, 1125. [Google Scholar] [CrossRef]
- Shams, M.; Owczarczak, B.; Manderscheid-Kern, P.; Bellnier, D.A.; Gollnick, S.O. Development of Photodynamic Therapy Regimens That Control Primary Tumor Growth and Inhibit Secondary Disease. Cancer Immunol. Immunother. 2015, 64, 287–297. [Google Scholar] [CrossRef]
- Allen, C.; Her, S.; Jaffray, D.A. Radiotherapy for Cancer: Present and Future. Adv. Drug Deliv. Rev. 2017, 109, 1–2. [Google Scholar] [CrossRef]
- Yahya, E.B.; Alqadhi, A.M. Recent Trends in Cancer Therapy: A Review on the Current State of Gene Delivery. Life Sci. 2021, 269, 119087. [Google Scholar] [CrossRef]
- Bukowski, K.; Kciuk, M.; Kontek, R. Mechanisms of Multidrug Resistance in Cancer Chemotherapy. Int. J. Mol. Sci. 2020, 21, 3233. [Google Scholar] [CrossRef]
- Jordan, M.A. Mechanism of Action of Antitumor Drugs That Interact with Microtubules and Tubulin. Curr. Med. Chem. Anticancer Agents 2002, 2, 1–17. [Google Scholar] [CrossRef]
- Bacher, G.; Nickel, B.; Emig, P.; Vanhoefer, U.; Seeber, S.; Shandra, A.; Klenner, T.; Beckers, T. D-24851, a Novel Synthetic Microtubule Inhibitor, Exerts Curative Antitumoral Activity In Vivo, Shows Efficacy toward Multidrug-Resistant Tumor Cells, and Lacks Neurotoxicity. Cancer Res. 2001, 61, 392–399. [Google Scholar]
- Windebank, A.J. Chemotherapeutic Neuropathy: Current Opinion in Neurology. Curr. Opin. Neurol. 1999, 12, 565–571. [Google Scholar] [CrossRef]
- Rowinsky, E.K. The Development and Clinical Utility of the Taxane Class of Antimicrotubule Chemotherapy Agents. Annu. Rev. Med. 1997, 48, 353–374. [Google Scholar] [CrossRef]
- Kuppens, I.E.L.M.; Witteveen, P.O.; Schot, M.; Schuessler, V.M.; Daehling, A.; Beijnen, J.H.; Voest, E.E.; Schellens, J.H.M. Phase I Dose-Finding and Pharmacokinetic Trial of Orally Administered Indibulin (D-24851) to Patients with Solid Tumors. Invest. New Drugs 2007, 25, 227–235. [Google Scholar] [CrossRef]
- Oostendorp, R.L.; Witteveen, P.O.; Schwartz, B.; Vainchtein, L.D.; Schot, M.; Nol, A.; Rosing, H.; Beijnen, J.H.; Voest, E.E.; Schellens, J.H.M. Dose-Finding and Pharmacokinetic Study of Orally Administered Indibulin (D-24851) to Patients with Advanced Solid Tumors. Invest. New Drugs 2010, 28, 163–170. [Google Scholar] [CrossRef]
- Sorgel, F.; Kinzig, M. Pharmacokinetics of Gyrase Inhibitors, Part 2: Renal and Hepatic Elimination Pathways and Drug Interactions. Am. J. Med. 1993, 94, 56S–69S. [Google Scholar] [CrossRef]
- Hagen, S.E.; Domagala, J.; Gajda, C.; Lovdahl, M.; Tait, B.D.; Wise, E.; Holler, T.; Hupe, D.; Nouhan, C.; Urumov, A.; et al. 4-Hydroxy-5,6-Dihydropyrones as Inhibitors of HIV Protease: The Effect of Heterocyclic Substituents at C-6 on Antiviral Potency and Pharmacokinetic Parameters. J. Med. Chem. 2001, 44, 2319–2332. [Google Scholar] [CrossRef]
- Li, W.T.; Hwang, D.R.; Chen, C.P.; Shen, C.W.; Huang, C.L.; Chen, T.W.; Lin, C.H.; Chang, Y.L.; Chang, Y.Y.; Lo, Y.K.; et al. Synthesis and Biological Evaluation of N-Heterocyclic Indolyl Glyoxylamides as Orally Active Anticancer Agents. J. Med. Chem. 2003, 46, 1706–1715. [Google Scholar] [CrossRef]
- Hu, C.B.; Chen, C.P.; Yeh, T.K.; Song, J.S.; Chang, C.Y.; Chuu, J.J.; Tung, F.F.; Ho, P.Y.; Chen, T.W.; Lin, C.H.; et al. BPR0C261 Is a Novel Orally Active Antitumor Agent with Antimitotic and Anti-Angiogenic Activities. Cancer Sci. 2011, 102, 182–191. [Google Scholar] [CrossRef]
- Leu, J.D.; Lin, S.T.; Chen, C.T.; Chang, C.A.; Lee, Y.J. BPR0C261, An Analogous of Microtubule Disrupting Agent D-24851 Enhances the Radiosensitivity of Human Non-Small Cell Lung Cancer Cells via P53-Dependent and P53-Independent Pathways. Int. J. Mol. Sci. 2022, 23, 14083. [Google Scholar] [CrossRef]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the P53 Network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Choy, H. Taxanes in Combined Modality Therapy for Solid Tumors. Crit. Rev. Oncol. Hematol. 2001, 37, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Li, W.T.; Yeh, T.K.; Song, J.S.; Yang, Y.N.; Chen, T.W.; Lin, C.H.; Chen, C.P.; Shen, C.C.; Hsieh, C.C.; Lin, H.L.; et al. BPR0C305, an Orally Active Microtubule-Disrupting Anticancer Agent. Anticancer. Drugs 2013, 24, 1047–1057. [Google Scholar] [CrossRef] [PubMed]
- Colley, H.E.; Muthana, M.; Danson, S.J.; Jackson, L.V.; Brett, M.L.; Harrison, J.; Coole, S.F.; Mason, D.P.; Jennings, L.R.; Wong, M.; et al. An Orally Bioavailable, Indole-3-Glyoxylamide Based Series of Tubulin Polymerization Inhibitors Showing Tumor Growth Inhibition in a Mouse Xenograft Model of Head and Neck Cancer. J. Med. Chem. 2015, 58, 9309–9333. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.J.; Huang, W.C.; Wei, Y.L.; Hsu, S.C.; Yuan, P.; Lin, H.Y.; Wistuba, I.I.; Lee, J.J.; Yen, C.J.; Su, W.C.; et al. Elevated BCRP/ABCG2 Expression Confers Acquired Resistance to Gefitinib in Wild-Type EGFR-Expressing Cells. PLoS ONE 2011, 6, e21428. [Google Scholar] [CrossRef] [PubMed]
- Guggilapu, S.D.; Lalita, G.; Reddy, T.S.; Prajapti, S.K.; Nagarsenkar, A.; Ramu, S.; Brahma, U.R.; Lakshmi, U.J.; Vegi, G.M.N.; Bhargava, S.K.; et al. Synthesis of C5-Tethered Indolyl-3-Glyoxylamide Derivatives as Tubulin Polymerization Inhibitors. Eur. J. Med. Chem. 2017, 128, 1–12. [Google Scholar] [CrossRef]
- Guggilapu, S.D.; Guntuku, L.; Reddy, T.S.; Nagarsenkar, A.; Sigalapalli, D.K.; Naidu, V.G.M.; Bhargava, S.K.; Bathini, N.B. Synthesis of Thiazole Linked Indolyl-3-Glyoxylamide Derivatives as Tubulin Polymerization Inhibitors. Eur. J. Med. Chem. 2017, 138, 83–95. [Google Scholar] [CrossRef]
- Brel, V.K.; Artyushin, O.I.; Chuprov-Netochin, R.N.; Leonov, S.V.; Semenova, M.N.; Semenov, V.V. Synthesis and Biological Evaluation of Indolylglyoxylamide Bisphosphonates, Antimitotic Microtubule-Targeting Derivatives of Indibulin with Improved Aqueous Solubility. Bioorg. Med. Chem. Lett. 2020, 30, 127635. [Google Scholar] [CrossRef]
- Tantak, M.P.; Gupta, V.; Nikhil, K.; Arun, V.; Singh, R.P.; Jha, P.N.; Shah, K.; Kumar, D. Sequential One-Pot Synthesis of Bis(Indolyl)Glyoxylamides: Evaluation of Antibacterial and Anticancer Activities. Bioorg. Med. Chem. Lett. 2016, 26, 3167–3171. [Google Scholar] [CrossRef]
- Tantak, M.P.; Wang, J.; Singh, R.P.; Kumar, A.; Shah, K.; Kumar, D. 2-(3′-Indolyl)-N-Arylthiazole-4-Carboxamides: Synthesis and Evaluation of Antibacterial and Anticancer Activities. Bioorg. Med. Chem. Lett. 2015, 25, 4225–4231. [Google Scholar] [CrossRef]
- Venkataramana Reddy, P.O.; Tantak, M.P.; Valdez, R.; Singh, R.P.; Singh, O.M.; Sadana, R.; Kumar, D. Synthesis and Biological Evaluation of Novel Carbazolyl Glyoxamides as Anticancer and Antibacterial Agents. RSC Adv. 2016, 6, 9379–9386. [Google Scholar] [CrossRef]
- Soni, J.P.; Nikitha Reddy, G.; Rahman, Z.; Sharma, A.; Spandana, A.; Phanindranath, R.; Dandekar, M.P.; Nagesh, N.; Shankaraiah, N. Synthesis and Cytotoxicity Evaluation of DNA-Interactive β-Carboline Indolyl-3-Glyoxamide Derivatives: Topo-II Inhibition and in Silico Modelling Studies. Bioorg. Chem. 2023, 131, 106313. [Google Scholar] [CrossRef]
- Tokala, R.; Thatikonda, S.; Vanteddu, U.S.; Sana, S.; Godugu, C.; Shankaraiah, N. Design and Synthesis of DNA-Interactive β-Carboline–Oxindole Hybrids as Cytotoxic and Apoptosis-Inducing Agents. ChemMedChem 2018, 13, 1909–1922. [Google Scholar] [CrossRef]
- Jadala, C.; Sathish, M.; Reddy, T.S.; Reddy, V.G.; Tokala, R.; Bhargava, S.K.; Shankaraiah, N.; Nagesh, N.; Kamal, A. Synthesis and In Vitro Cytotoxicity Evaluation of β-Carboline-Combretastatin Carboxamides as Apoptosis Inducing Agents: DNA Intercalation and Topoisomerase-II Inhibition. Bioorg. Med. Chem. 2019, 27, 3285–3298. [Google Scholar] [CrossRef]
- M O’Boyle, N.; J Meegan, M. Designed Multiple Ligands for Cancer Therapy. Curr. Med. Chem. 2011, 18, 4722–4737. [Google Scholar] [CrossRef]
- Villalonga-Planells, R.; Coll-Mulet, L.; Martínez-Soler, F.; Castaño, E.; Acebes, J.J.; Giménez-Bonafé, P.; Gil, J.; Tortosa, A. Activation of P53 by Nutlin-3a Induces Apoptosis and Cellular Senescence in Human Glioblastoma Multiforme. PLoS ONE 2011, 6, e18588. [Google Scholar] [CrossRef]
- Shoukrun, R.; Veenman, L.; Shandalov, Y.; Leschiner, S.; Spanier, I.; Karry, R.; Katz, Y.; Weisinger, G.; Weizman, A.; Gavish, M. The 18-KDa Translocator Protein, Formerly Known as the Peripheral-Type Benzodiazepine Receptor, Confers Proapoptotic and Antineoplastic Effects in a Human Colorectal Cancer Cell Line. Pharmacogenet. Genom. 2008, 18, 977–988. [Google Scholar] [CrossRef]
- Daniele, S.; Taliani, S.; Da Pozzo, E.; Giacomelli, C.; Costa, B.; Trincavelli, M.L.; Rossi, L.; La Pietra, V.; Barresi, E.; Carotenuto, A.; et al. Apoptosis Therapy in Cancer: The First Single-Molecule Co-Activating P53 and the Translocator Protein in Glioblastoma. Sci. Rep. 2014, 4, 4749. [Google Scholar] [CrossRef]
- Daniele, S.; La Pietra, V.; Barresi, E.; Di Maro, S.; Da Pozzo, E.; Robello, M.; La Motta, C.; Cosconati, S.; Taliani, S.; Marinelli, L.; et al. Lead Optimization of 2-Phenylindolylglyoxylyldipeptide Murine Double Minute (MDM)2/Translocator Protein (TSPO) Dual Inhibitors for the Treatment of Gliomas. J. Med. Chem. 2016, 59, 4526–4538. [Google Scholar] [CrossRef]
- Juchum, M.; Günther, M.; Laufer, S.A. Fighting Cancer Drug Resistance: Opportunities and Challenges for Mutation-Specific EGFR Inhibitors. Drug Resist. Updat. 2015, 20, 12–28. [Google Scholar] [CrossRef]
- Daniele, S.; Barresi, E.; Zappelli, E.; Marinelli, L.; Novellino, E.; Settimo, F.D.; Taliani, S.; Trincavelli, M.L.; Martini, C.; Daniele, S.; et al. Long Lasting MDM2/Translocator Protein Modulator: A New Strategy for Irreversible Apoptosis of Human Glioblastoma Cells. Oncotarget 2016, 7, 7866–7884. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Lippard, S.J. Cellular Processing of Platinum Anticancer Drugs. Nat. Rev. Drug Discov. 2005, 4, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Oun, R.; Moussa, Y.E.; Wheate, N.J. The Side Effects of Platinum-Based Chemotherapy Drugs: A Review for Chemists. Dalt. Trans. 2018, 47, 6645–6653. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, T.C.; Suntharalingam, K.; Lippard, S.J. The Next Generation of Platinum Drugs: Targeted Pt(II) Agents, Nanoparticle Delivery, and Pt(IV) Prodrugs. Chem. Rev. 2016, 116, 3436–3486. [Google Scholar] [CrossRef]
- Aquino, M.A.S. Diruthenium and Diosmium Tetracarboxylates: Synthesis, Physical Properties and Applications. Coord. Chem. Rev. 1998, 170, 141–202. [Google Scholar] [CrossRef]
- Barresi, E.; Tolbatov, I.; Pratesi, A.; Notarstefano, V.; Baglini, E.; Daniele, S.; Taliani, S.; Re, N.; Giorgini, E.; Martini, C.; et al. A Mixed-Valence Diruthenium(Ii,Iii) Complex Endowed with High Stability: From Experimental Evidence to Theoretical Interpretation. Dalt. Trans. 2020, 49, 14520–14527. [Google Scholar] [CrossRef]
- Barresi, E.; Tolbatov, I.; Marzo, T.; Zappelli, E.; Marrone, A.; Re, N.; Pratesi, A.; Martini, C.; Taliani, S.; Da Settimo, F.; et al. Two Mixed Valence Diruthenium(II,III) Isomeric Complexes Show Different Anticancer Properties. Dalt. Trans. 2021, 50, 9643–9647. [Google Scholar] [CrossRef]
- Chiaverini, L.; Baglini, E.; Mannelli, M.; Poggetti, V.; Da Settimo, F.; Taliani, S.; Gamberi, T.; Barresi, E.; La Mendola, D.; Marzo, T. A Complex Bearing TSPO PIGA Ligand Coordinated to the [Au(PEt3)]+ Pharmacophore Is Highly Cytotoxic against Ovarian Cancer Cells. BioMetals 2023, 36, 1–8. [Google Scholar] [CrossRef]
- Landini, I.; Massai, L.; Cirri, D.; Gamberi, T.; Paoli, P.; Messori, L.; Mini, E.; Nobili, S. Structure-Activity Relationships in a Series of Auranofin Analogues Showing Remarkable Antiproliferative Properties. J. Inorg. Biochem. 2020, 208, 111079. [Google Scholar] [CrossRef]
- Nutma, E.; Ceyzériat, K.; Amor, S.; Tsartsalis, S.; Millet, P.; Owen, D.R.; Papadopoulos, V.; Tournier, B.B. Cellular Sources of TSPO Expression in Healthy and Diseased Brain. Eur. J. Nucl. Med. Mol. Imaging 2021, 49, 146–163. [Google Scholar] [CrossRef]
- Takhi, M.; Singh, G.; Murugan, C.; Thaplyyal, N.; Maitra, S.; Bhaskarreddy, K.M.; Amarnath, P.V.S.; Mallik, A.; Harisudan, T.; Trivedi, R.K.; et al. Novel and Potent Oxazolidinone Antibacterials Featuring 3-Indolylglyoxamide Substituents. Bioorg. Med. Chem. Lett. 2008, 18, 5150–5155. [Google Scholar] [CrossRef]
- Clemett, D.; Markham, A. Linezolid. Drugs 2000, 59, 815–827. [Google Scholar] [CrossRef]
- Shinabarger, D.L.; Marotti, K.R.; Murray, R.W.; Lin, A.H.; Melchior, E.P.; Swaney, S.M.; Dunyak, D.S.; Demyan, W.F.; Buysse, J.M. Mechanism of Action of Oxazolidinones: Effects of Linezolid and Eperezolid on Translation Reactions. Antimicrob. Agents Chemother. 1997, 41, 2132–2136. [Google Scholar] [CrossRef]
- Kloss, P.; Xiong, L.; Shinabarger, D.L.; Mankin, A.S. Resistance Mutations in 23 S RRNA Identify the Site of Action of the Protein Synthesis Inhibitor Linezolid in the Ribosomal Peptidyl Transferase Center. J. Mol. Biol. 1999, 294, 93–101. [Google Scholar] [CrossRef]
- Johnson, A.P.; Tysall, L.; Stockdale, M.W.; Woodford, N.; Kaufmann, M.E.; Warner, M.; Livermore, D.M.; Asboth, F.; Allerberger, F.J. Emerging Linezolid-Resistant Enterococcus Faecalis and Enterococcus Faecium Isolated from Two Austrian Patients in the Same Intensive Care Unit. Eur. J. Clin. Microbiol. Infect. Dis. 2002, 21, 751–754. [Google Scholar]
- Singh, P.; Verma, P.; Yadav, B.; Komath, S.S. Synthesis and Evaluation of Indole-Based New Scaffolds for Antimicrobial Activities—Identification of Promising Candidates. Bioorg. Med. Chem. Lett. 2011, 21, 3367–3372. [Google Scholar] [CrossRef]
- Mielczarek, M.; Devakaram, R.V.; Ma, C.; Yang, X.; Kandemir, H.; Purwono, B.; Black, D.S.; Griffith, R.; Lewis, P.J.; Kumar, N. Synthesis and Biological Activity of Novel Bis-Indole Inhibitors of Bacterial Transcription Initiation Complex Formation. Org. Biomol. Chem. 2014, 12, 2882–2894. [Google Scholar] [CrossRef]
- Li, S.A.; Cadelis, M.M.; Sue, K.; Blanchet, M.; Vidal, N.; Brunel, J.M.; Bourguet-Kondracki, M.L.; Copp, B.R. 6-Bromoindolglyoxylamido Derivatives as Antimicrobial Agents and Antibiotic Enhancers. Bioorganic Med. Chem. 2019, 27, 2090–2099. [Google Scholar] [CrossRef]
- Pieri, C.; Borselli, D.; Di Giorgio, C.; De Méo, M.; Bolla, J.M.; Vidal, N.; Combes, S.; Brunel, J.M. New Ianthelliformisamine Derivatives as Antibiotic Enhancers against Resistant Gram-Negative Bacteria. J. Med. Chem. 2014, 57, 4263–4272. [Google Scholar] [CrossRef]
- Cadelis, M.M.; Pike, E.I.W.; Kang, W.; Wu, Z.; Bourguet-Kondracki, M.L.; Blanchet, M.; Vidal, N.; Brunel, J.M.; Copp, B.R. Exploration of the Antibiotic Potentiating Activity of Indolglyoxylpolyamines. Eur. J. Med. Chem. 2019, 183, 111708. [Google Scholar] [CrossRef]
- Conti, M.; Jin, S.L. The Molecular Biology of Cyclic Nucleotide Phosphodiesterases. Prog. Nucleic Acid Res. Mol. Biol. 1999, 63, 1–38. [Google Scholar] [CrossRef] [PubMed]
- Torphy, T.J.; Stadel, J.M.; Burman, M.; Cieslinski, L.B.; McLaughlin, M.M.; White, J.R.; Livi, G.P. Coexpression of Human CAMP-Specific Phosphodiesterase Activity and High Affinity Rolipram Binding in Yeast. J. Biol. Chem. 1992, 267, 1798–1804. [Google Scholar] [CrossRef] [PubMed]
- Compton, C.H.; Gubb, J.; Nieman, R.; Edelson, J.; Amit, O.; Bakst, A.; Ayres, J.G.; Creemers, J.P.H.M.; Schultze-Werninghaus, G.; Brambilla, C.; et al. Cilomilast, a Selective Phosphodiesterase-4 Inhibitor for Treatment of Patients with Chronic Obstructive Pulmonary Disease: A Randomised, Dose-Ranging Study. Lancet 2001, 358, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Cave, A.; Arlett, P.; Lee, E. Inhaled and Nasal Corticosteroids: Factors Affecting the Risks of Systemic Adverse Effects. Pharmacol. Ther. 1999, 83, 153–179. [Google Scholar] [CrossRef]
- Marx, D.; Tassabehji, M.; Heer, S.; Hüttenbrink, K.B.; Szelenyi, I. Modulation of TNF and GM-CSF Release from Dispersed Human Nasal Polyp Cells and Human Whole Blood by Inhibitors of Different PDE Isoenzymes and Glucocorticoids. Pulm. Pharmacol. Ther. 2002, 15, 7–15. [Google Scholar] [CrossRef]
- Kuss, H.; Hoefgen, N.; Johanssen, S.; Kronbach, T.; Rundfeldt, C. In Vivo Efficacy in Airway Disease Models of N-(3,5-Dichloropyrid-4-Yl)-[1-(4-Fluorobenzyl)-5-Hydroxy-Indole-3-Yl]-Glyoxylic Acid Amide (AWD 12-281), a Selective Phosphodiesterase 4 Inhibitor for Inhaled Administration. J. Pharmacol. Exp. Ther. 2003, 307, 373–385. [Google Scholar] [CrossRef]
- Burnouf, C.; Pruniaux, M.-P. Recent Advances in PDE4 Inhibitors as Immunoregulators and Anti- Inflammatory Drugs. Curr. Pharm. Des. 2005, 8, 1255–1296. [Google Scholar] [CrossRef]
- Barnette, M.S. Phosphodiesterase 4 (PDE4) Inhibitors in Asthma and Chronic Obstructive Pulmonary Disease (COPD). Prog. Drug Res. 1999, 53, 193–229. [Google Scholar] [CrossRef]
- Phillips, J.E. Inhaled Phosphodiesterase 4 (PDE4) Inhibitors for Inflammatory Respiratory Diseases. Front. Pharmacol. 2020, 11, 259. [Google Scholar] [CrossRef]
- Dennis, E.A.; Cao, J.; Hsu, Y.H.; Magrioti, V.; Kokotos, G. Phospholipase A2 Enzymes: Physical Structure, Biological Function, Disease Implication, Chemical Inhibition, and Therapeutic Intervention. Chem. Rev. 2011, 111, 6130–6185. [Google Scholar] [CrossRef]
- Dillard, R.D.; Bach, N.J.; Draheim, S.E.; Berry, D.R.; Carlson, D.G.; Chirgadze, N.Y.; Clawson, D.K.; Hartley, L.W.; Johnson, L.M.; Jones, N.D.; et al. Indole Inhibitors of Human Nonpancreatic Secretory Phospholipase A2. 1. Indole-3-Acetamides. J. Med. Chem. 1996, 39, 5119–5136. [Google Scholar] [CrossRef]
- Dillard, R.D.; Bach, N.J.; Draheim, S.E.; Berry, D.R.; Carlson, D.G.; Chirgadze, N.Y.; Clawson, D.K.; Hartley, L.W.; Johnson, L.M.; Jones, N.D.; et al. Indole Inhibitors of Human Nonpancreatic Secretory Phospholipase A2. 2. Indole-3-Acetamides with Additional Functionality. J. Med. Chem. 1996, 39, 5137–5158. [Google Scholar] [CrossRef]
- Draheim, S.E.; Bach, N.J.; Dillard, R.D.; Berry, D.R.; Carlson, D.G.; Chirgadze, N.Y.; Clawson, D.K.; Hartley, L.W.; Johnson, L.M.; Jones, N.D.; et al. Indole Inhibitors of Human Nonpancreatic Secretory Phospholipase A2. 3. Indole-3-Glyoxamides. J. Med. Chem. 1996, 39, 5159–5175. [Google Scholar] [CrossRef]
- Mihelich, E.D.; Schevitz, R.W. Structure-Based Design of a New Class of Anti-Inflammatory Drugs: Secretory Phospholipase A2 Inhibitors, SPI. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1999, 1441, 223–228. [Google Scholar] [CrossRef]
- Abraham, E.; Naum, C.; Bandi, V.; Gervich, D.; Lowry, S.F.; Wunderink, R.; Schein, R.M.; Macias, W.; Skerjanec, S.; Dmitrienko, A.; et al. Efficacy and Safety of LY315920Na/S-5920, a Selective Inhibitor of 14-KDa Group IIA Secretory Phospholipase A2, in Patients with Suspected Sepsis and Organ Failure. Crit. Care Med. 2003, 31, 718–728. [Google Scholar] [CrossRef]
- Snyder, D.W.; Bach, N.J.; Dillard, R.D.; Draheim, S.E.; Carlson, D.G.; Fox, N.; Roehm, N.W.; Armstrong, C.T.; Chang, C.H.; Hartley, L.W.; et al. Pharmacology of LY315920/S-5920, [[3-(Aminooxoacetyl)-2-Ethyl-1-(Phenylmethyl)-1H-Indol-4-Yl]Oxy]Acetate, a Potent and Selective Secretory Phospholipase A2 Inhibitor: A New Class of Anti-Inflammatory Drugs, SPI. J. Pharmacol. Exp. Ther. 1999, 288, 1117–1124. [Google Scholar]
- Rosenson, R.S.; Fraser, H.; Trias, J.; Hislop, C. Varespladib Methyl in Cardiovascular Disease. Expert Opin. Investig. Drugs 2010, 19, 1245–1255. [Google Scholar] [CrossRef]
- Bradley, J.D.; Dmitrienko, A.A.; Kivitz, A.J.; Gluck, O.S.; Weaver, A.L.; Wiesenhutter, C.; Myers, S.L.; Sides, G.D. A Randomized, Double-Blinded, Placebo-Controlled Clinical Trial of LY333013, a Selective Inhibitor of Group II Secretory Phospholipase A 2, in the Treatment of Rheumatoid Arthritis. J. Rheumatol. 2005, 32, 417–423. [Google Scholar]
- Bowton, D.L.; Dmitrienko, A.A.; Israel, E.; Zeiher, B.G.; Sides, G.D. Impact of a Soluble Phospholipase A2 Inhibitor on Inhaled Allergen Challenge in Subjects with Asthma. J. Asthma 2005, 42, 65–71. [Google Scholar] [CrossRef]
- Rosenson, R.S.; Hislop, C.; McConnell, D.; Elliott, M.; Stasiv, Y.; Wang, N.; Waters, D.D. Effects of 1-H-Indole-3-Glyoxamide (A-002) on Concentration of Secretory Phospholipase A2 (PLASMA Study): A Phase II Double-Blind, Randomised, Placebo-Controlled Trial. Lancet 2009, 373, 649–658. [Google Scholar] [CrossRef]
- Nicholls, S.J.; Kastelein, J.J.P.; Schwartz, G.G.; Bash, D.; Rosenson, R.S.; Cavender, M.A.; Brennan, D.M.; Koenig, W.; Jukema, J.W.; Nambi, V.; et al. Varespladib and Cardiovascular Events in Patients With an Acute Coronary Syndrome: The VISTA-16 Randomized Clinical Trial. JAMA 2014, 311, 252–262. [Google Scholar] [CrossRef] [PubMed]
- Schett, G.; Zwerina, J.; Firestein, G. The P38 Mitogen-Activated Protein Kinase (MAPK) Pathway in Rheumatoid Arthritis. Ann. Rheum. Dis. 2008, 67, 909–916. [Google Scholar] [CrossRef] [PubMed]
- Westra, J.; C Limburg, P. P38 Mitogen-Activated Protein Kinase (MAPK) in Rheumatoid Arthritis. Mini-Reviews Med. Chem. 2006, 6, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Schett, G.; Stach, C.; Zwerina, J.; Voll, R.; Manger, B. How Antirheumatic Drugs Protect Joints from Damage in Rheumatoid Arthritis. Arthritis Rheum. 2008, 58, 2936–2948. [Google Scholar] [CrossRef]
- Korb, A.; Tohidast-Akrad, M.; Cetin, E.; Axmann, R.; Smolen, J.; Schett, G. Differential Tissue Expression and Activation of P38 MAPK α, β, γ, and δ Isoforms in Rheumatoid Arthritis. Arthritis Rheum. 2006, 54, 2745–2756. [Google Scholar] [CrossRef]
- Schett, G.; Tohidast-Akrad, M.; Smolen, J.S.; Schmid, B.J.; Steiner, C.W.; Bitzan, P.; Zenz, P.; Redlich, K.; Xu, Q.; Steiner, G. Activation, Differential Localization, and Regulation of the Stress-Activated Protein Kinases, Extracellular Signal-Regulated Kinase, c-Jun N-Terminal Kinase, and P38 Mitogen-Activated Protein Kinase, in Synovial Tissue and Cells in Rheumatoid Arthritis. Arthritis Rheum. 2000, 43, 2501–2512. [Google Scholar] [CrossRef]
- Pargellis, C.; Tong, L.; Churchill, L.; Cirillo, P.F.; Gilmore, T.; Graham, A.G.; Grob, P.M.; Hickey, E.R.; Moss, N.; Pav, S.; et al. Inhibition of P38 MAP Kinase by Utilizing a Novel Allosteric Binding Site. Nat. Struct. Biol. 2002, 9, 268–272. [Google Scholar] [CrossRef]
- Mavunkel, B.J.; Chakravarty, S.; Perumattam, J.J.; Dugar, S.; Lu, Q.; Liang, X. Indole-Type Derivatives as Inhibitors of P38 Kinase; Scios LLC.: Titusville, NJ, USA, 2000. [Google Scholar]
- Snoonian, J.R.; Oliver-Shaffer, P.-A. Processes for the Preparation Of N-Heteroaryl-N-Aryl-Amines by Reacting an N-Aryl Carbamic Acid Ester with a Halo-Heteroaryl And Analogous Processes; Vertex Pharmaceuticals Incorporated: Boston, MA, USA, 2004. [Google Scholar]
- Hill, R.J.; Dabbagh, K.; Phippard, D.; Li, C.; Suttmann, R.T.; Welch, M.; Papp, E.; Song, K.W.; Chang, K.C.; Leaffer, D.; et al. Pamapimod, a Novel P38 Mitogen-Activated Protein Kinase Inhibitor: Preclinical Analysis of Efficacy and Selectivity. J. Pharmacol. Exp. Ther. 2008, 327, 610–619. [Google Scholar] [CrossRef]
- Kuma, Y.; Sabio, G.; Bain, J.; Shpiro, N.; Márquez, R.; Cuenda, A. BIRB796 Inhibits All P38 MAPK Isoforms In Vitro and In Vivo. J. Biol. Chem. 2005, 280, 19472–19479. [Google Scholar] [CrossRef]
- Goldstein, D.M.; Kuglstatter, A.; Lou, Y.; Soth, M.J. Selective P38α Inhibitors Clinically Evaluated for the Treatment of Chronic Inflammatory Disorders. J. Med. Chem. 2010, 53, 2345–2353. [Google Scholar] [CrossRef]
- Nikas, S.N.; Drosos, A.A. SCIO-469 Scios Inc. Curr. Opin. Investig. Drugs 2004, 5, 1205–1212. [Google Scholar]
- Ji, R.R. Peripheral and Central Mechanisms of Inflammatory Pain, with Emphasis on MAP Kinases. Curr. Drug Targets. Inflamm. Allergy 2004, 3, 299–303. [Google Scholar] [CrossRef]
- Schindler, J.F.; Monahan, J.B.; Smith, W.G. P38 Pathway Kinases as Anti-Inflammatory Drug Targets. J. Dent. Res. 2007, 86, 800–811. [Google Scholar] [CrossRef]
- Genovese, M.C.; Cohen, S.B.; Wofsy, D.; Weinblatt, M.E.; Firestein, G.S.; Brahn, E.; Strand, V.; Baker, D.G.; Tong, S.E. A 24-Week, Randomized, Double-Blind, Placebo-Controlled, Parallel Group Study of the Efficacy of Oral SCIO-469, a P38 Mitogen-Activated Protein Kinase Inhibitor, in Patients with Active Rheumatoid Arthritis. J. Rheumatol. 2011, 38, 846–854. [Google Scholar] [CrossRef]
- Rescher, S.L.U.; Brunotte, L.; Faist, A.; Schloer, S.M. P38-Inhibitors for the Treatment of Coronavirus Infections and/or COVID-19 Cytokine Storm. International Application No. PCT/EP2022/068724, 12 January 2023. [Google Scholar]
- Leitmeyer, K.C.; Vaughn, D.W.; Watts, D.M.; Salas, R.; Villalobos, I.; de Chacon; Ramos, C.; Rico-Hesse, R. Dengue Virus Structural Differences That Correlate with Pathogenesis. J. Virol. 1999, 73, 4738–4747. [Google Scholar] [CrossRef]
- Wu, H.; Bock, S.; Snitko, M.; Berger, T.; Weidner, T.; Holloway, S.; Kanitz, M.; Diederich, W.E.; Steuber, H.; Walter, C.; et al. Novel Dengue Virus NS2B/NS3 Protease Inhibitors. Antimicrob. Agents Chemother. 2015, 59, 1100–1109. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, J.; Li, H. Flavivirus NS2B/NS3 Protease: Structure, Function, and Inhibition. In Viral Proteases and Their Inhibitors; Academic Press: Cambridge, MA, USA, 2017; pp. 163–188. [Google Scholar] [CrossRef]
- Steuer, C.; Gege, C.; Fischl, W.; Heinonen, K.H.; Bartenschlager, R.; Klein, C.D. Synthesis and Biological Evaluation of α-Ketoamides as Inhibitors of the Dengue Virus Protease with Antiviral Activity in Cell-Culture. Bioorg. Med. Chem. 2011, 19, 4067–4074. [Google Scholar] [CrossRef]
- Nie, S.; Yao, Y.; Wu, F.; Wu, X.; Zhao, J.; Hua, Y.; Wu, J.; Huo, T.; Lin, Y.L.; Kneubehl, A.R.; et al. Synthesis, Structure-Activity Relationships, and Antiviral Activity of Allosteric Inhibitors of Flavivirus NS2B-NS3 Protease. J. Med. Chem. 2021, 64, 2777–2800. [Google Scholar] [CrossRef]
- Mehand, M.S.; Al-Shorbaji, F.; Millett, P.; Murgue, B. The WHO R&D Blueprint: 2018 Review of Emerging Infectious Diseases Requiring Urgent Research and Development Efforts. Antiviral Res. 2018, 159, 63–67. [Google Scholar] [CrossRef]
- del Rosario García-Lozano, M.; Dragoni, F.; Gallego, P.; Mazzotta, S.; López-Gómez, A.; Boccuto, A.; Martínez-Cortés, C.; Rodríguez-Martínez, A.; Pérez-Sánchez, H.; Manuel Vega-Pérez, J.; et al. Piperazine-Derived Small Molecules as Potential Flaviviridae NS3 Protease Inhibitors. In Vitro Antiviral Activity Evaluation against Zika and Dengue Viruses. Bioorg. Chem. 2023, 133, 106408. [Google Scholar] [CrossRef]
- Kling, A.; Jantos, K.; Mack, H.; Hornberger, W.; Drescher, K.; Nimmrich, V.; Relo, A.; Wicke, K.; Hutchins, C.W.; Lao, Y.; et al. Discovery of Novel and Highly Selective Inhibitors of Calpain for the Treatment of Alzheimer’s Disease: 2-(3-Phenyl-1H-Pyrazol-1-Yl)-Nicotinamides. J. Med. Chem. 2017, 60, 7123–7138. [Google Scholar] [CrossRef] [PubMed]
- Dorababu, A. Indole—A Promising Pharmacophore in Recent Antiviral Drug Discovery. RSC Med. Chem. 2020, 11, 1335–1353. [Google Scholar] [CrossRef] [PubMed]
- Meanwell, N.A.; Krystal, M.R.; Nowicka-Sans, B.; Langley, D.R.; Conlon, D.A.; Eastgate, M.D.; Grasela, D.M.; Timmins, P.; Wang, T.; Kadow, J.F. Inhibitors of HIV-1 Attachment: The Discovery and Development of Temsavir and Its Prodrug Fostemsavir. J. Med. Chem. 2018, 61, 62–80. [Google Scholar] [CrossRef] [PubMed]
- Ruwizhi, N.; Aderibigbe, B.A. Cinnamic Acid Derivatives and Their Biological Efficacy. Int. J. Mol. Sci. 2020, 21, 5712. [Google Scholar] [CrossRef]
- Abd El-Raouf, O.M.; El-Sayed, E.S.M.; Manie, M.F. Cinnamic Acid and Cinnamaldehyde Ameliorate Cisplatin-Induced Splenotoxicity in Rats. J. Biochem. Mol. Toxicol. 2015, 29, 426–431. [Google Scholar] [CrossRef]
- Wang, R.; Yang, W.; Fan, Y.; Dehaen, W.; Li, Y.; Li, H.; Wang, W.; Zheng, Q.; Huai, Q. Design and Synthesis of the Novel Oleanolic Acid-Cinnamic Acid Ester Derivatives and Glycyrrhetinic Acid-Cinnamic Acid Ester Derivatives with Cytotoxic Properties. Bioorg. Chem. 2019, 88, 102951. [Google Scholar] [CrossRef]
- Amano, R.; Yamashita, A.; Kasai, H.; Hori, T.; Miyasato, S.; Saito, S.; Yokoe, H.; Takahashi, K.; Tanaka, T.; Otoguro, T.; et al. Cinnamic Acid Derivatives Inhibit Hepatitis C Virus Replication via the Induction of Oxidative Stress. Antivir. Res. 2017, 145, 123–130. [Google Scholar] [CrossRef]
- Chen, Y.; Li, Z.; Pan, P.; Lao, Z.; Xu, J.; Li, Z.; Zhan, S.; Liu, X.; Wu, Y.; Wang, W.; et al. Cinnamic Acid Inhibits Zika Virus by Inhibiting RdRp Activity. Antiviral Res. 2021, 192, 105117. [Google Scholar] [CrossRef]
- Nitsche, C.; Behnam, M.A.M.; Steuer, C.; Klein, C.D. Retro Peptide-Hybrids as Selective Inhibitors of the Dengue Virus NS2B-NS3 Protease. Antivir. Res. 2012, 94, 72–79. [Google Scholar] [CrossRef]
- Nitsche, C.; Steuer, C.; Klein, C.D. Arylcyanoacrylamides as Inhibitors of the Dengue and West Nile Virus Proteases. Bioorg. Med. Chem. 2011, 19, 7318–7337. [Google Scholar] [CrossRef]
- Deng, J.; Li, N.; Liu, H.; Zuo, Z.; Liew, O.W.; Xu, W.; Chen, G.; Tong, X.; Tang, W.; Zhu, J.; et al. Discovery of Novel Small Molecule Inhibitors of Dengue Viral NS2B-NS3 Protease Using Virtual Screening and Scaffold Hopping. J. Med. Chem. 2012, 55, 6278–6293. [Google Scholar] [CrossRef]
- Guo, S.; Zhen, Y.; Zhu, Z.; Zhou, G.; Zheng, X. Cinnamic Acid Rescues Behavioral Deficits in a Mouse Model of Traumatic Brain Injury by Targeting MiR-455-3p/HDAC2. Life Sci. 2019, 235, 116819. [Google Scholar] [CrossRef]
- Faucher, A.M.; Bailey, M.D.; Beaulieu, P.L.; Brochu, C.; Duceppe, J.S.; Ferland, J.M.; Ghiro, E.; Gorys, V.; Halmos, T.; Kawai, S.H.; et al. Synthesis of BILN 2061, an HCV NS3 Protease Inhibitor with Proven Antiviral Effect in Humans. Org. Lett. 2004, 6, 2901–2904. [Google Scholar] [CrossRef]
- Li, F.; Lee, E.M.; Sun, X.; Wang, D.; Tang, H.; Zhou, G.C. Design, Synthesis and Discovery of Andrographolide Derivatives against Zika Virus Infection. Eur. J. Med. Chem. 2020, 187, 111925. [Google Scholar] [CrossRef]
- Barbosa-Lima, G.; Moraes, A.M.; da Araújo, A.S.; da Silva, E.T.; de Freitas, C.S.; Vieira, Y.R.; Marttorelli, A.; Neto, J.C.; Bozza, P.T.; de Souza, M.V.N.; et al. 2,8-Bis(Trifluoromethyl)Quinoline Analogs Show Improved Anti-Zika Virus Activity, Compared to Mefloquine. Eur. J. Med. Chem. 2017, 127, 334–340. [Google Scholar] [CrossRef]
- Balasubramanian, A.; Teramoto, T.; Kulkarni, A.A.; Bhattacharjee, A.K.; Padmanabhan, R. Antiviral Activities of Selected Antimalarials against Dengue Virus Type 2 and Zika Virus. Antiviral Res. 2017, 137, 141–150. [Google Scholar] [CrossRef]
- de la Guardia, C.; Stephens, D.E.; Dang, H.T.; Quijada, M.; Larionov, O.V.; Lleonart, R. Antiviral Activity of Novel Quinoline Derivatives against Dengue Virus Serotype 2. Molecules 2018, 23, 672. [Google Scholar] [CrossRef]
- Kaptein, S.J.F.; Vincetti, P.; Crespan, E.; Rivera, J.I.A.; Costantino, G.; Maga, G.; Neyts, J.; Radi, M. Identification of Broad-Spectrum Dengue/Zika Virus Replication Inhibitors by Functionalization of Quinoline and 2,6-Diaminopurine Scaffolds. ChemMedChem 2018, 13, 1371–1376. [Google Scholar] [CrossRef]
- Beesetti, H.; Tyagi, P.; Medapi, B.; Krishna, V.S.; Sriram, D.; Khanna, N.; Swaminathan, S. A Quinoline Compound Inhibits the Replication of Dengue Virus Serotypes 1-4 in Vero Cells. Antivir. Ther. 2018, 23, 385–394. [Google Scholar] [CrossRef]
- Zhao, J.; Zhang, Y.; Wang, M.; Liu, Q.; Lei, X.; Wu, M.; Guo, S.; Yi, D.; Li, Q.; Ma, L.; et al. Quinoline and Quinazoline Derivatives Inhibit Viral RNA Synthesis by SARS-CoV-2 RdRp. ACS Infect. Dis. 2021, 7, 1535–1544. [Google Scholar] [CrossRef]
- Vella, S.; Palmisano, L. The Global Status of Resistance to Antiretroviral Drugs. Clin. Infect. Dis. 2005, 41, S239–S246. [Google Scholar] [CrossRef] [PubMed]
- Qian, K.; Morris-Natschke, S.L.; Lee, K.H. HIV Entry Inhibitors and Their Potential in HIV Therapy. Med. Res. Rev. 2009, 29, 369–393. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Zhang, Z.; Wallace, O.B.; Deshpande, M.; Fang, H.; Yang, Z.; Zadjura, L.M.; Tweedie, D.L.; Huang, S.; Zhao, F.; et al. Discovery of 4-Benzoyl-1-[(4-Methoxy-1H-Pyrrolo [2,3-b]Pyridin-3-Yl)Oxoacetyl]-2-(R) -Methylpiperazine (BMS-378806): A Novel HIV-1 Attachment Inhibitor That Interferes with CD4-Gp120 Interactions. J. Med. Chem. 2003, 46, 4236–4239. [Google Scholar] [CrossRef] [PubMed]
- Meanwell, N.A.; Wallace, O.B.; Fang, H.; Wang, H.; Deshpande, M.; Wang, T.; Yin, Z.; Zhang, Z.; Pearce, B.C.; James, J.; et al. Inhibitors of HIV-1 Attachment. Part 2: An Initial Survey of Indole Substitution Patterns. Bioorg. Med. Chem. Lett. 2009, 19, 1977–1981. [Google Scholar] [CrossRef]
- Meanwell, N.A.; Wallace, O.B.; Wang, H.; Deshpande, M.; Pearce, B.C.; Trehan, A.; Yeung, K.S.; Qiu, Z.; Wright, J.J.K.; Robinson, B.A.; et al. Inhibitors of HIV-1 Attachment. Part 3: A Preliminary Survey of the Effect of Structural Variation of the Benzamide Moiety on Antiviral Activity. Bioorg. Med. Chem. Lett. 2009, 19, 5136–5139. [Google Scholar] [CrossRef]
- Wang, T.; Kadow, J.F.; Zhang, Z.; Yin, Z.; Gao, Q.; Wu, D.; Parker, D.D.G.; Yang, Z.; Zadjura, L.; Robinson, B.A.; et al. Inhibitors of HIV-1 Attachment. Part 4: A Study of the Effect of Piperazine Substitution Patterns on Antiviral Potency in the Context of Indole-Based Derivatives. Bioorg. Med. Chem. Lett. 2009, 19, 5140–5145. [Google Scholar] [CrossRef]
- Wang, T.; Yin, Z.; Zhang, Z.; Bender, J.A.; Yang, Z.; Johnson, G.; Yang, Z.; Zadjura, L.M.; D’Arienzo, C.J.; Parker, D.D.G.; et al. Inhibitors of Human Immunodeficiency Virus Type 1 (HIV-1) Attachment. 5. An Evolution from Indole to Azaindoles Leading to the Discovery of 1-(4-Benzoylpiperazin-1-Yl)-2-(4,7-Dimethoxy-1H-Pyrrolo[2,3-c]-Pyridin-3-Yl) Ethane-1,2-Dione (BMS-488043), a Drug. J. Med. Chem. 2009, 52, 7778–7787. [Google Scholar] [CrossRef]
- Lin, P.F.; Blair, W.; Wang, T.; Spicer, T.; Guo, Q.; Zhou, N.; Gong, Y.F.; Wang, H.G.H.; Rose, R.; Yamanaka, G.; et al. A Small Molecule HIV-1 Inhibitor That Targets the HIV-1 Envelope and Inhibits CD4 Receptor Binding. Proc. Natl. Acad. Sci. USA 2003, 100, 11013–11018. [Google Scholar] [CrossRef]
- Ho, H.-T.; Fan, L.; Nowicka-Sans, B.; McAuliffe, B.; Li, C.-B.; Yamanaka, G.; Zhou, N.; Fang, H.; Dicker, I.; Dalterio, R.; et al. Envelope Conformational Changes Induced by Human Immunodeficiency Virus Type 1 Attachment Inhibitors Prevent CD4 Binding and Downstream Entry Events. J. Virol. 2006, 80, 4017. [Google Scholar] [CrossRef]
- Yang, Z.; Zadjura, L.; D’Arienzo, C.; Marino, A.; Santone, K.; Klunk, L.; Greene, D.; Lin, P.F.; Colonno, R.; Wang, T.; et al. Preclinical Pharmacokinetics of a Novel HIV-1 Attachment Inhibitor BMS-378806 and Prediction of Its Human Pharmacokinetics. Biopharm. Drug Dispos. 2005, 26, 387–402. [Google Scholar] [CrossRef]
- Hanna, G.J.; Lalezari, J.; Hellinger, J.A.; Wohl, D.A.; Nettles, R.; Persson, A.; Krystal, M.; Lin, P.; Colonno, R.; Grasela, D.M. Antiviral Activity, Pharmacokinetics, and Safety of BMS-488043, a Novel Oral Small-Molecule HIV-1 Attachment Inhibitor, in HIV-1-Infected Subjects. Antimicrob. Agents Chemother. 2011, 55, 722–728. [Google Scholar] [CrossRef]
- Pancera, M.; Lai, Y.T.; Bylund, T.; Druz, A.; Narpala, S.; O’Dell, S.; Schön, A.; Bailer, R.T.; Chuang, G.Y.; Geng, H.; et al. Crystal Structures of Trimeric HIV Env with Entry Inhibitors BMS-378806 and BMS-626529. Nat. Chem. Biol. 2017, 13, 1115. [Google Scholar] [CrossRef]
- Kozal, M.; Aberg, J.; Pialoux, G.; Cahn, P.; Thompson, M.; Molina, J.-M.; Grinsztejn, B.; Diaz, R.; Castagna, A.; Kumar, P.; et al. Fostemsavir in Adults with Multidrug-Resistant HIV-1 Infection. N. Engl. J. Med. 2020, 382, 1232–1243. [Google Scholar] [CrossRef]
- Akopyants, N.S.; Kimblin, N.; Secundino, N.; Patrick, R.; Peters, N.; Lawyer, P.; Dobson, D.E.; Beverley, S.M.; Sacks, D.L. Demonstration of Genetic Exchange during Cyclical Development of Leishmania in the Sand Fly Vector. Science 2009, 324, 265–268. [Google Scholar] [CrossRef]
- Rogers, M.; Kropf, P.; Choi, B.S.; Dillon, R.; Podinovskaia, M.; Bates, P.; Müller, I. Proteophosophoglycans Regurgitated by Leishmania-Infected Sand Flies Target the L-Arginine Metabolism of Host Macrophages to Promote Parasite Survival. PLoS Pathog. 2009, 5, e1000555. [Google Scholar] [CrossRef]
- Gupta, L.; Talwar, A.; Nishi; Palne, S.; Gupta, S.; Chauhan, P.M.S. Synthesis of Marine Alkaloid: 8,9-Dihydrocoscinamide B and Its Analogues as Novel Class of Antileishmanial Agents. Bioorg. Med. Chem. Lett. 2007, 17, 4075–4079. [Google Scholar] [CrossRef]
- Kumar, A.; Katiyar, S.B.; Gupta, S.; Chauhan, P.M.S. Syntheses of New Substituted Triazino Tetrahydroisoquinolines and β-Carbolines as Novel Antileishmanial Agents. Eur. J. Med. Chem. 2006, 41, 106–113. [Google Scholar] [CrossRef]
- Costa, E.V.; Pinheiro, M.L.B.; Xavier, C.M.; Silva, J.R.A.; Amaral, A.C.F.; Souza, A.D.L.; Barison, A.; Campos, F.R.; Ferreira, A.G.; Machado, G.M.C.; et al. A Pyrimidine-β-Carboline and Other Alkaloids from Annona Foetida with Antileishmanial Activity. J. Nat. Prod. 2006, 69, 292–294. [Google Scholar] [CrossRef]
- Kumar, R.; Khan, S.; Verma, A.; Srivastava, S.; Viswakarma, P.; Gupta, S.; Meena, S.; Singh, N.; Sarkar, J.; Chauhan, P.M.S. Synthesis of 2-(Pyrimidin-2-Yl)-1-Phenyl-2,3,4,9-Tetrahydro-1H-β-Carbolines as Antileishmanial Agents. Eur. J. Med. Chem. 2010, 45, 3274–3280. [Google Scholar] [CrossRef]
- Chauhan, S.S.; Gupta, L.; Mittal, M.; Vishwakarma, P.; Gupta, S.; Chauhan, P.M.S. Synthesis and Biological Evaluation of Indolyl Glyoxylamides as a New Class of Antileishmanial Agents. Bioorganic Med. Chem. Lett. 2010, 20, 6191–6194. [Google Scholar] [CrossRef]
- Singh, A.; Mohan, M.L.; Isaac, A.O.; Luo, X.; Petrak, J.; Vyoral, D.; Singh, N. Prion Protein Modulates Cellular Iron Uptake: A Novel Function with Implications for Prion Disease Pathogenesis. PLoS ONE 2009, 4, e4468. [Google Scholar] [CrossRef]
- Hu, W.; Kieseier, B.; Frohman, E.; Eagar, T.N.; Rosenberg, R.N.; Hartung, H.P.; Stüve, O. Prion Proteins: Physiological Functions and Role in Neurological Disorders. J. Neurol. Sci. 2008, 264, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.J.; Borsenberger, V.; Louth, J.C.; Judd, K.E.; Chen, B. Design, Synthesis, and Structure—Activity Relationship of Indole-3-Glyoxylamide Libraries Possessing Highly Potent Activity in a Cell Line Model of Prion Disease. J. Med. Chem. 2009, 52, 7503–7511. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.J.; Louth, J.C.; Ferrara, S.; Jackson, M.P.; Sorrell, F.J.; Cochrane, E.J.; Gever, J.; Baxendale, S.; Silber, B.M.; Roehl, H.H.; et al. Discovery of 6-Substituted Indole-3-Glyoxylamides as Lead Antiprion Agents with Enhanced Cell Line Activity, Improved Microsomal Stability and Low Toxicity. Eur. J. Med. Chem. 2011, 46, 4125–4132. [Google Scholar] [CrossRef]
- Thompson, M.J.; Louth, J.C.; Ferrara, S.; Sorrell, F.J.; Irving, B.J.; Cochrane, E.J.; Meijer, A.J.H.M.; Chen, B. Structure–Activity Relationship Refinement and Further Assessment of Indole-3-Glyoxylamides as a Lead Series against Prion Disease. ChemMedChem 2011, 6, 115–130. [Google Scholar] [CrossRef]
- Taliani, S.; Trincavelli, M.L.; Cosimelli, B.; Laneri, S.; Severi, E.; Barresi, E.; Pugliesi, I.; Daniele, S.; Giacomelli, C.; Greco, G.; et al. Modulation of A2B Adenosine Receptor by 1-Benzyl-3-Ketoindole Derivatives. Eur. J. Med. Chem. 2013, 69, 331–337. [Google Scholar] [CrossRef]
- Trincavelli, M.L.; Giacomelli, C.; Daniele, S.; Taliani, S.; Cosimelli, B.; Laneri, S.; Severi, E.; Barresi, E.; Pugliesi, I.; Greco, G.; et al. Allosteric Modulators of Human A2B Adenosine Receptor. Biochim. Biophys. Acta Gen. Subj. 2014, 1840, 1194–1203. [Google Scholar] [CrossRef]
- Trincavelli, M.L.; Daniele, S.; Giacomelli, C.; Taliani, S.; Da Settimo, F.; Cosimelli, B.; Greco, G.; Novellino, E.; Martini, C. Osteoblast Differentiation and Survival: A Role for A2B Adenosine Receptor Allosteric Modulators. Biochim. Biophys. Acta Mol. Cell Res. 2014, 1843, 2957–2966. [Google Scholar] [CrossRef]
- Barresi, E.; Giacomelli, C.; Marchetti, L.; Baglini, E.; Salerno, S.; Greco, G.; Da Settimo, F.; Martini, C.; Trincavelli, M.L.; Taliani, S. Novel Positive Allosteric Modulators of A2B Adenosine Receptor Acting as Bone Mineralisation Promoters. J. Enzyme Inhib. Med. Chem. 2021, 36, 286–294. [Google Scholar] [CrossRef]
- Supuran, C.T. Carbonic Anhydrases: Novel Therapeutic Applications for Inhibitors and Activators. Nat. Rev. Drug Discov. 2008, 7, 168–181. [Google Scholar] [CrossRef]
- Supuran, C.T. Carbonic Anhydrase Activators. Future Med. Chem. 2018, 10, 561–573. [Google Scholar] [CrossRef]
- Barresi, E.; Ravichandran, R.; Germelli, L.; Angeli, A.; Baglini, E.; Salerno, S.; Marini, A.M.; Costa, B.; Da Pozzo, E.; Martini, C.; et al. Carbonic Anhydrase Activation Profile of Indole-Based Derivatives. J. Enzyme Inhib. Med. Chem. 2021, 36, 1783–1797. [Google Scholar] [CrossRef]
- Santoro, A.; Mattace Raso, G.; Taliani, S.; Da Pozzo, E.; Simorini, F.; Costa, B.; Martini, C.; Laneri, S.; Sacchi, A.; Cosimelli, B.; et al. TSPO-Ligands Prevent Oxidative Damage and Inflammatory Response in C6 Glioma Cells by Neurosteroid Synthesis. Eur. J. Pharm. Sci. 2016, 88, 124–131. [Google Scholar] [CrossRef]
- Ruusuvuori, E.; Huebner, A.K.; Kirilkin, I.; Yukin, A.Y.; Blaesse, P.; Helmy, M.; Jung Kang, H.; El Muayed, M.; Christopher Hennings, J.; Voipio, J.; et al. Neuronal Carbonic Anhydrase VII Provides GABAergic Excitatory Drive to Exacerbate Febrile Seizures. EMBO J. 2013, 32, 2275–2286. [Google Scholar] [CrossRef]
- Matthews, T.A.; Abel, A.; Demme, C.; Sherman, T.; Pan, P.W.; Halterman, M.W.; Parkkila, S.; Nehrke, K. Expression of the CHOP-Inducible Carbonic Anhydrase CAVI-b Is Required for BDNF-Mediated Protection from Hypoxia. Brain Res. 2014, 1543, 28–37. [Google Scholar] [CrossRef]
- Primofiore, G.; Marini, A.M.; Settimo, F.D.; Martini, C.; Bardellini, A.; Giannaccini, G.; Lucacchini, A. Specific Inhibition of Benzodiazepine Receptor Binding by Some N-(Indo1-3-Ylglyoxylyl)Amino Acid Derivatives: Stereoselective Interactions. J. Med. Chem. 1989, 32, 2514–2518. [Google Scholar] [CrossRef]




























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
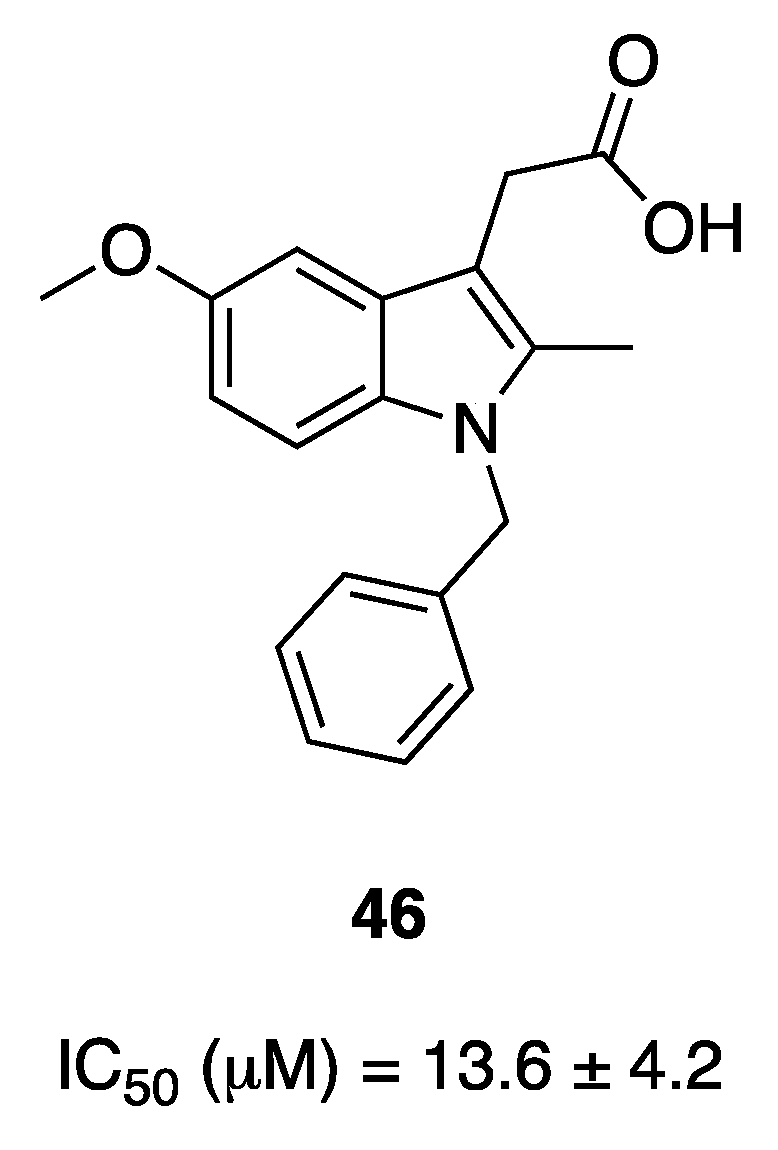{kind=link}
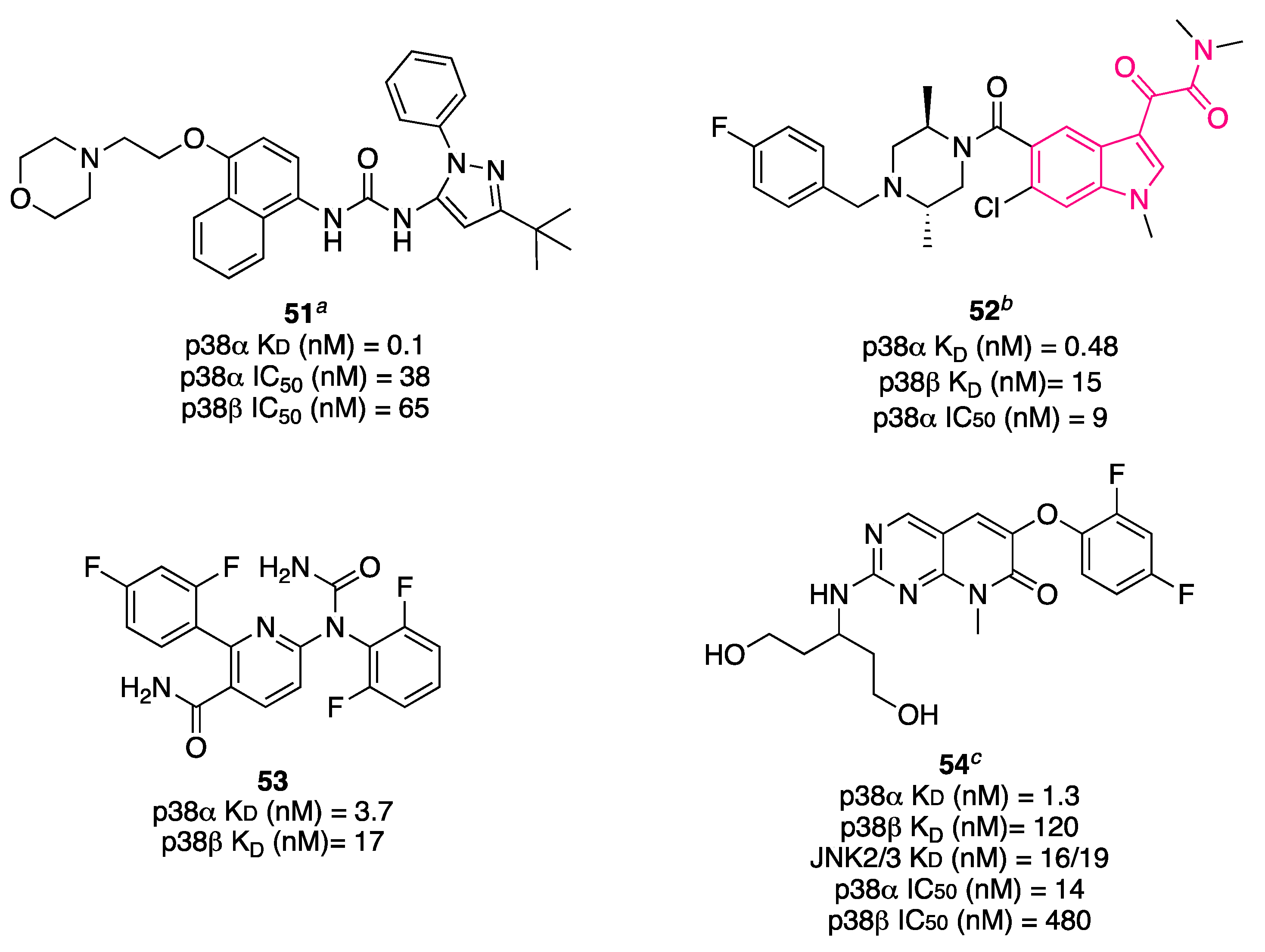{kind=link}
{kind=link}
{kind=link}
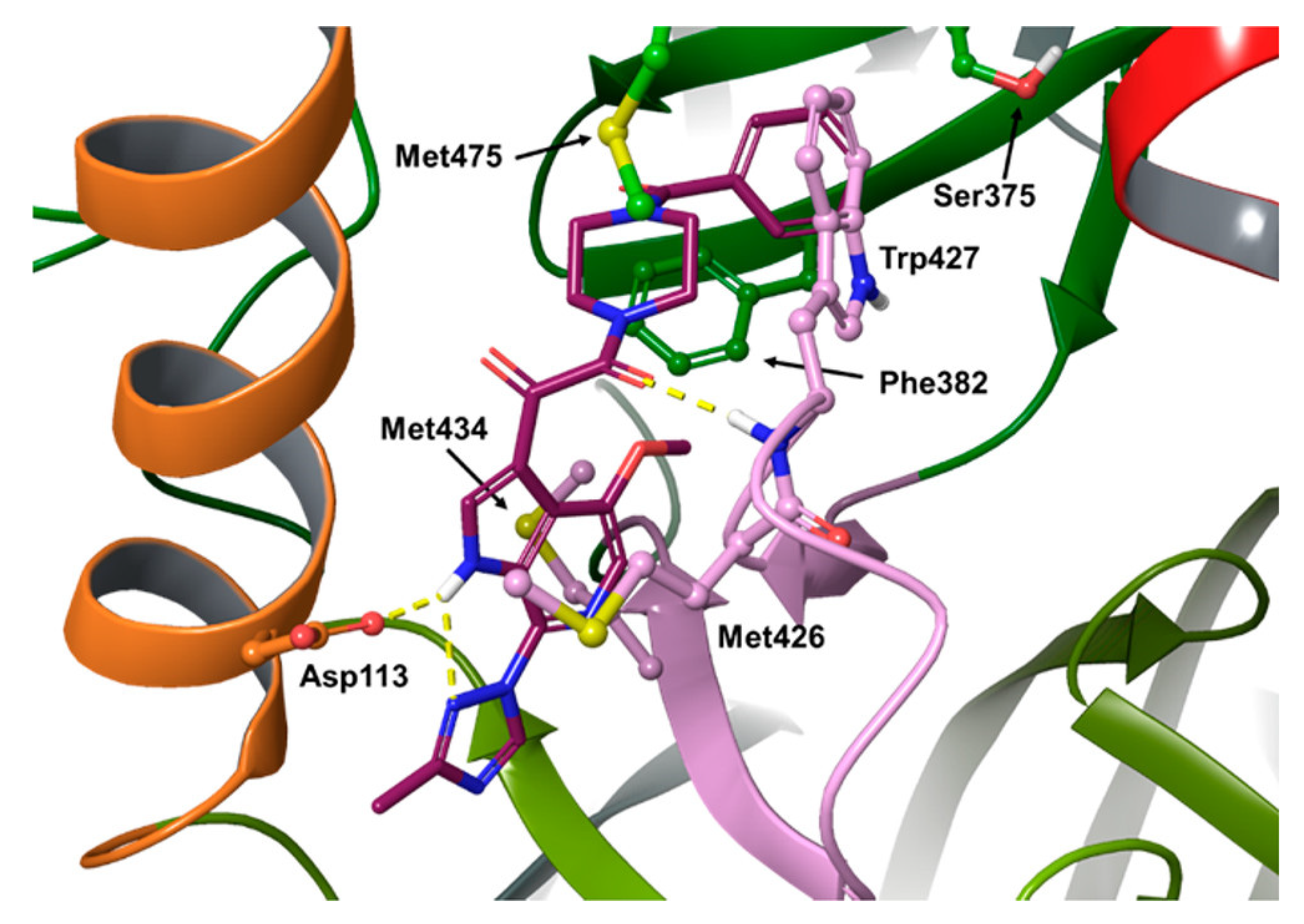{kind=link}
{kind=link}
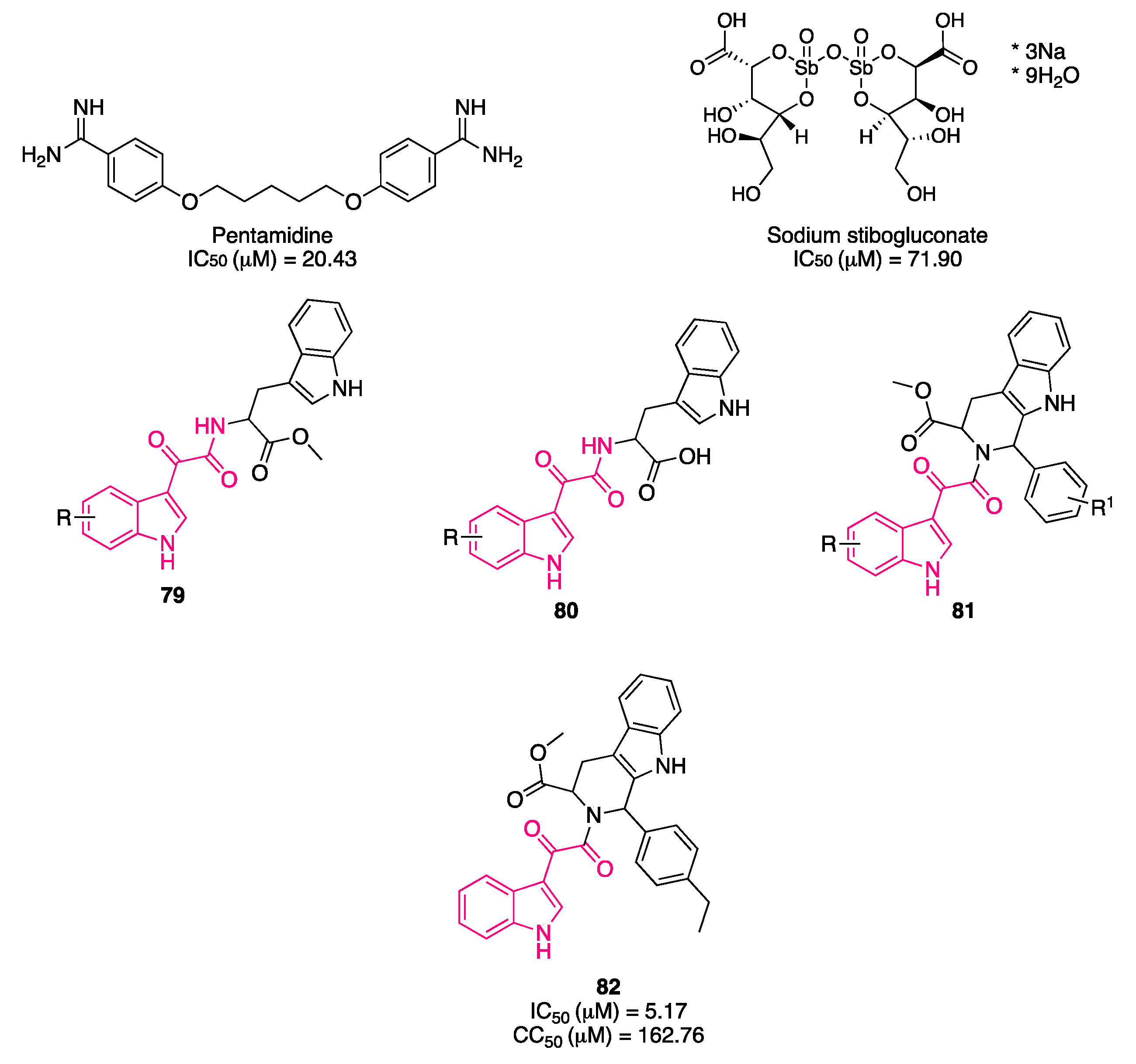{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||
|---|---|---|
| Activity | Cell Line/Animal Model | Reference |
| Immunomodulation | Human C20 microglial cells | [58] |
| Modulation of inflammatory-based retinal neurodegeneration | LPS-induced degeneration in 661W cells | [59] |
| Neuroprotection | Female mouse model of primary progressive multiple sclerosis | [60] |
| Maintenance of the correct functionality of microglia in neuroinflammation | Human microglia C20 and HMC3 cells | [61] |
 | |||||
|---|---|---|---|---|---|
| cpd | R1 | R2 | hGIIA PLA2 (μM) | hGIB PLA2 (μM) | pGIB PLA2 (μM) |
| 47 | 2-(C6H5)C6H4CH2 | CH3 | 0.006 ± 0.001 | 0.364 | 0.097 |
| 48 | 3-(C6H5)C6H4CH2 | CH3 | 0.009 ± 0.001 | 0.57 | 0.007 |
| 49 | C6H5CH2 | CH3 | 0.011 ± 0.004 | 0.761 | 0.015 |
| 50 | -- | -- | 0.009 ± 0.001 | 0.228 | 0.048 |
 | ||
|---|---|---|
| cpd | EC50 (nM) | CC50 (μM) |
| 65 | 86 ± 24 (LAI) | 145 ± 23 |
| 66 | 4.0 (JR-FL) 4.9 (LAI) | 200 |
| 67 | 1.52 (JR-FL) | >300 (n = 2) |
| 68 | 575.9 (JR-FL) | >300 (n = 2) |
| 69 | 21.6 (JR-FL) | >300 (n = 2) |
| 70 | 1.7 ± 1.6 (JR-FL, n = 11) | 280 |
| 71 | 1.47 ± 0.63 (JR-FL) 2.68 ± 1.64 (LAI) | >300 |
| 72 | 0.88 ± 0.46 (JR-FL, n = 56) 1.15 (LAI) | >300 |
 | ||
|---|---|---|
| Co-Receptor Tropism | Virus | EC50 (nM) |
| CCR5 | JR-FL | 0.4 ± 0.1 |
| SF-162 | 0.5 ± 0.2 | |
| Bal | 1.7 ± 0.5 | |
| CXCR4 | LAI | 0.7 ± 0.4 |
| NL4−3 | 2.2 ± 0.6 | |
| MN | 14.8 ± 5.2 | |
| IIIb | 16.2 ± 1.7 | |
| RF | >2000 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barresi, E.; Robello, M.; Baglini, E.; Poggetti, V.; Viviano, M.; Salerno, S.; Da Settimo, F.; Taliani, S. Indol-3-ylglyoxylamide as Privileged Scaffold in Medicinal Chemistry. Pharmaceuticals 2023, 16, 997. https://doi.org/10.3390/ph16070997
Barresi E, Robello M, Baglini E, Poggetti V, Viviano M, Salerno S, Da Settimo F, Taliani S. Indol-3-ylglyoxylamide as Privileged Scaffold in Medicinal Chemistry. Pharmaceuticals. 2023; 16(7):997. https://doi.org/10.3390/ph16070997
Chicago/Turabian StyleBarresi, Elisabetta, Marco Robello, Emma Baglini, Valeria Poggetti, Monica Viviano, Silvia Salerno, Federico Da Settimo, and Sabrina Taliani. 2023. "Indol-3-ylglyoxylamide as Privileged Scaffold in Medicinal Chemistry" Pharmaceuticals 16, no. 7: 997. https://doi.org/10.3390/ph16070997
APA StyleBarresi, E., Robello, M., Baglini, E., Poggetti, V., Viviano, M., Salerno, S., Da Settimo, F., & Taliani, S. (2023). Indol-3-ylglyoxylamide as Privileged Scaffold in Medicinal Chemistry. Pharmaceuticals, 16(7), 997. https://doi.org/10.3390/ph16070997