3.3. Experimental Procedure for the Synthesis of Methyl Hydantoin (45)
1-(Methylamino)-4-phenylcyclohexane-1-carbonitrile hydrochloride (43). To a stirred suspension of sodium cyanide (843 mg, 17.2 mmol) and methylamine hydrochloride (1.16 g, 17.2 mmol) in 12 mL of DMSO/H2O 9:1 (v/v), a solution of 4-phenylcyclohexanone X (3.0 g, 17.2 mmol) in DMSO (24 mL) was added in one portion. The reaction mixture was stirred for 46 h at rt, poured into 130 mL of an ice-water mixture, and extracted with AcOEt (3 × 60 mL). The combined organic phases were washed with brine (2 × 60 mL) and dried with anh. Na2SO4, and the solvent were evaporated under reduced pressure. The residue was dissolved in Et2O (80 mL) and treated dropwise with an ethanolic solution saturated with gaseous hydrochloric acid to pH~2 under an ice bath. The precipitate formed was filtered off in vacuo, washed with small portions of dry Et2O, and dried over P2O5 to afford the title compound 43 as a white crystalline solid (3.2 g, 72%). This intermediate was used for the next reaction without further purification.
1-(1-Cyano-4-phenylcyclohexyl)-1-methylurea (44). To a stirred solution of the carbonitrile 43 (3.1 g, 12.4 mmol) in 20 mL acetic acid, a solution of potassium cyanate (2.01 g, 24.8 mmol) in 3 mL H2O was added. After stirring for 1 h at 35 °C, the reaction mixture was poured into 70 mL H2O and extracted with CHCl3 (3 × 50 mL). The combined organic layers were washed with H2O (3 × 50 mL) and brine (2 × 50 mL) and dried with anh. Na2SO4, and the solvents were evaporated to dryness under reduced pressure to afford the title compound 44 as a white solid (2.97 g, 93%). This intermediate was used for the next reaction without further purification.
1-Methyl-8-phenyl-1,3-diazaspiro [4.5]decane-2,4-dione (45). A stirred solution of 44 (2.9 g, 11.3 mmol) in 40 mL dry DMF was cooled in an ice bath, and sodium hydride (353 mg, 14.7 mmol, 60% dispersion in mineral oil) was added portionwise. After 4 d of stirring at 50 °C under argon, the mixture was treated with a solution of HCl 10% (96 mL), and stirring was continued for 24 h at 50 °C. After this time, the reaction mixture was poured into 400 mL of an ice-water mixture and extracted with CHCl3 (4 × 200 mL). The combined organic extracts were washed with H2O (3 × 250 mL) and brine (2 × 250 mL) and dried with anh. Na2SO4, and the solvent were evaporated in vacuo. The remaining solid was treated with Et2O and n-pentane to yield the desired compound 45 as a pale-yellow crystalline product. (2.98 g, 67%); Rf = 0.34 (CH2Cl2/AcOEt 5:1); mp 211–214 °C (AcOEt/dry Et2O-n-pentane); 1H NMR (400.13 MHz, CDCl3): δ (ppm) = 1.80–1.94 (complex m, 6H, H6, H7, H9, H10), 2.31–2.45 (complex m, 2H, H7, H9), 2.52 (tt, J1 = 11.7 Hz, J2 = 2.9 Hz, 1H, H8), 2.87 (s, 3H, CH3), 7.22 (td, J1 = 6.5 Hz, J2 = 1.9 Hz, 1H, H4′), 7.30 (d, J = 7.1 Hz, 2H, H2′, H6′), 7.32 (t, J = 7.9 Hz, 2H, H3′, H5′), 9.27 (s, 1H, H3); 13C NMR (150.9 MHz, CDCl3): δ (ppm) = 23.6 (CH3), 28.3 28.7 (C7, C9), 31.1, 31.9 (C6, C10), 40.5, 43.0 (C8), 62.8, 63.7 (C5), 126.4 (C4′), 126.8, 127.0 (C2′, C6′), 128.5, 128.7 (C3′, C5′), 145.2, 146.0 (C1′), 155.7, 156.1 (C2=O), 177.0, 177.5 (C4=O); elemental analysis calcd (%) for C15H18N2O2: C 69.74, H 7.02, N 10.84, found: C 69.78, H 7.12, N 10.77.
General experimental procedure for the synthesis of benzyl esters (7–15 and 46). Potassium bis(trimethylsilyl)amide (3.06 mmol, 1.02 eq, 95% purity) was added portionwise under ice cooling to a solution of the hydantoin (3 mmol, 1.0 eq) in dry THF (30 mL) under argon atmosphere. The reaction mixture was stirred for 20 min to 1 h at rt, and the resulting emulsion was concentrated to dryness under reduced pressure. The remaining potassium imidate salt was dissolved in dry DMF (30 mL), and benzyl bromoacetate or benzyl 2-bromopropanoate (3.15 mmol, 1.05 eq) was added dropwise. After stirring for 48–64 h at 35–38 °C under argon, the reaction mixture was diluted in ice water (300 mL) and extracted with AcOEt (3 × 150 mL). The combined organic extracts were washed with H2O (3 × 200 mL) and brine (2 × 200 mL), dried over anh. Na2SO4, and evaporated in vacuo. The crude residue was purified by silica gel chromatography to yield the title benzyl esters (7–15 and 46).
Benzyl (4′-methyl-2,5-dioxo-3′,4′-dihydro-2′H-spiro[imidazolidine-4,1′-naphthalene]-1-yl)acetate (7, 8). Hydantoin 4 (1.2 g, 5.21 mmol) was dissolved in 52 mL dry THF, and to this solution, an amount of potassium bis(trimethylsilyl)amide (1.12 g, 5.31 mmol, 95% purity) was added portionwise under ice cooling. After stirring for 1 h at rt under argon, the solvent was removed under reduced pressure, and the remaining potassium imidate salt was dissolved in dry DMF (52 mL). Benzyl bromoacetate (1.25 g, 5.47 mmol) was added dropwise. The stirring was continued at 35–38 °C for 64 h under argon, and the reaction mixture was worked up following the general procedure for the preparation of benzyl esters. The viscous oily residue was chromatographed on silica gel using CH2Cl2, CH2Cl2/AcOEt 20:1 και 10:1 as eluents to afford two pairs of enantiomers in a 70/30 ratio. (1.7 g, 86%).
((4R,4′S)/(4S,4′R)-(4′-Methyl-2,5-dioxo-3′,4′-dihydro-2′H-spiro[imidazolidine-4,1′-naphthalene]-1-yl)acetate (7). White foamy product, which was crystallized upon treatment with n-pentane under ice cooling. (1.19 g); Rf = 0.78 (CH2Cl2/AcOEt 8:1); mp 111–113 °C (AcOEt/dry Et2O-n-pentane); 1H NMR (400.13 MHz, CDCl3): δ (ppm) = 1.27, 1.28 (d + d, J = 7.1 Hz, J = 7.1 Hz, 3H, CH3), 1.45 (dddd, J1 = 13.2 Hz, J2 = 9.9 Hz, J3 = 7.3 Hz, J4 = 2.9 Hz, 0.55H, H3′), 1.84 (dtd, J1 = 11.9 Hz, J2 = 8.6 Hz, J3 = 3.8 Hz, 0.5H, H2′), 1.92 (dddd, J1 = 10.8 Hz, J2 = 9.0 Hz, J3 = 5.9 Hz, J4 = 2.7 Hz, 0.9H, H3′), 2.00 (ddd, J1 = 13.5 Hz, J2 = 8.6 Hz, J3 = 2.9 Hz, 0.5H, H2′), 2.15 (ddd, J1 = 13.0 Hz, J2 = 9.6 Hz, J3 = 3.1 Hz, 0.5H, H2′), 2.28 (ddt, J1 = 11.7 Hz, J2 = 5.5 Hz, J3 = 2.4 Hz, 0.5H, H3′), 2.33 (ddd, J1 = 13.5 Hz, J2 = 6.4 Hz, J3 = 2.8 Hz, 0.5H, H2′), 3.00 (dq, J1 = 12.9 Hz, J2 = 6.3 Hz, 1H, H4′), 4.21–4.36 (2q, AB, J1AB = J2AB = 17.4 Hz, 2H, NCH2COO), 5.07–5.17 (q, AB, JAΒ = 12.2 Hz, 2H, OCH2Ph), 5.94, 5.96 (s + s, 1H, H3), 7.02 (td, J1 = 8.3 Hz, J2 = 2.2 Hz, 1H, H7′), 7.12–7.22 (complex m, 3H, H5′, H6′, H8′), 7.23–7.34 (complex m, 5H, H2″, H3″, H4″, H5″, H6″); 13C (50.32 MHz, CDCl3): δ (ppm) = 22.4 (CH3), 26.6, 27.2 (C3′), 31.2, 31.7 (C2′), 32.0, 32.1 (C4′), 39.7 (NCH2COO), 63.7, 63.8 (C4), 67.9 (OCH2Ph), 127.1 (C7′), 127.3 (C8′), 128.6, 128.8, 129.0, 129.1 (C5′, C6′, C2″, C3″, C4″, C5″, C6″), 132.3 (C8′a), 135.0 (C1″), 143.0, 143.1 (C4′a), 155.7 (C2=O), 167.1 (COOCH2Ph), 175.7 (C5=O); elemental analysis calcd (%) for C22H22N2O4: C 69.83, H 5.86, N 7.40, found: C 69.79, H 5.89, N 7.49.
((4R,4′R)/(4S,4′S)-4′-Methyl-2,5-dioxo-3′,4′-dihydro-2′H-spiro[imidazolidine-4,1′-naphthalene]-1-yl)acetate (8). White foamy product. (510 mg); Rf = 0.70 (CH2Cl2/AcOEt 8:1); 1HNMR (400.13 MHz, CDCl3): δ (ppm) = 1.33, 1.34 (dd, J = 6.9 Hz, J = 7.0 Hz, 3H, CH3), 1.55 (ddt, J1 = 13.9 Hz, J2 = 10.1 Hz, J3 = 4.9 Hz, 0.6H, H3′), 1.88 (ddd, J1 = 13.4 Hz, J2 = 7.5 Hz, J3 = 3.6 Hz, 0.5H, H2′), 1.91–1.99 (complex m, 0.9H, H3′), 2.05 (ddd, J1 = 13.5 Hz, J2 = 8.6 Hz, J3 = 2.5 Hz, 0.45H, H2′), 2.18 (ddd, J1 = 12.7 Hz, J2 = 9.7 Hz, J3 = 2.8 Hz, 0.5H, H2′), 2.32 (ddd, J1 = 14.5 Hz, J2 = 7.1 Hz, J3 = 2.8 Hz, 0.5H, H3′), 2.36 (ddd, J1 = 13.2 Hz, J2 = 8.8 Hz, J3 = 4.4 Hz, 0.5H, H2′), 2.97 (dq, J1 = 12.0 Hz, J2 = 6.3 Hz, 1H, H4′), 4.23–4.40 (2q, AB, J1AB = J2AB = 17.4 Hz, 2H, NCH2CO), 5.11–5.24 (q, AB, JAB = 12.2 Hz, 2H, OCH2Ph), 6.84, 6.89 (s + s, 1H, H3), 7.07 (t, J = 7.2 Hz, 1H, H7′), 7.14–7.28 (complex m, 3H, H5′, H6′, H8′), 7.30–7.46 (complex m, 5H, H2″, H3″, H4″, H5″, H6″); 13C (50.32 MHz, CDCl3): δ (ppm) = 22.1 (CH3), 26.2, 26.8 (C3′), 30.7, 31.2 (C2′), 31.7, 31.9 (C4′), 39.3 (NCH2CO), 63.4, 63.5 (C4), 67.5 (OCH2Ph), 126.7 (C7′), 127.0 (C8′), 128.3, 128.4, 128.5 (C5′, C6′, C2″, C3″, C4″, C5″, C6″), 132.2 (C8′a), 134.8 (C1″), 142.76, 142.84 (C4′a), 155.8 (C2=O), 167.0 (COOCH2Ph), 175.7 (C5=O). HRMS/ESI+: m/z calcd for C22H22N2O4: 378.1580; found: 378.1586.
Benzyl 2-(8-phenyl-2,4-dioxo-1,3-diazaspiro [4.5] decan-3-yl)acetate (9). Potassium bis(trimethylsilyl)amide (527 mg, 2.51 mmol, 95% purity) was added portionwise, under ice cooling, to a solution of hydantoin 5 (600 mg, 2.46 mmol) in 30 mL dry THF. The mixture was stirred at r.t. for 20 min under argon. After removal of the solvent under reduced pressure, the residue was dissolved in 30 mL dry DMF, and benzyl bromoacetate (591 mg, 2.58 mmol) was added dropwise. The stirring was continued at 35–38 °C for 48 h under argon, and afterward, the reaction mixture was poured into 200 mL of an ice-water mixture and extracted with AcOEt (3 × 150 mL). The combined organic phases were washed with H2O (3 × 150 mL) and brine (2 × 150 mL), dried over anh. Na2SO4, and evaporated under reduced pressure affording a white crude solid product, which was purified by silica gel column chromatography using CH2Cl2 to CH2Cl2/AcOEt (9:1) as eluents. The column chromatography afforded 880 mg of 9 as a white crystalline solid (91%). Mp 199–201 °C (AcOEt/n-pentane), Rf = 0.59 (CH2Cl2/AcOEt 8:1). 1H NMR (400.13 MHz, CDCl3) δ (ppm) 1.75 (qd, 2H, J1 = 13.6 Hz, J2 = 3.2 Hz, H7e, H9a), 1.83 (d, 1.7H, J = 12.8 Hz, H6a, H10a, cis), 1.94 (dd, 1.75H, J1 = 14.0 Hz, J2 = 3.0 Hz, H7a, H9e, cis), 2.05 (td, 1.85H, J1 = 13.7 Hz, J2 = 3.9 Hz, H6e, H10e), 2.11 (d, 0.35H, J = 13.7 Hz, H6a, H10a, trans), 2.31 (qd, 0.35H, J1 = 13.2 Hz, J2 = 3.0 Hz, H7a, H9e, trans), 2.58 (tt, 0.18H, J1 = 12.0 Hz, J2 = 3.7 Hz, H8, trans), 2.63 (tt, 0.9H, J1 = 12.3 Hz, J2 = 3.6 Hz, H8, cis), 4.33 (s, 0.34H, NCH2COO, trans), 4.38 (s, 1.7H, NCH2COO, cis), 5.15 (s, 1.7H, COOCH2Ph, cis), 5.16 (s, 0.34H, COOCH2Ph, trans), 6.68 (s, 0.16H, H1, trans), 7.19 (tt, 0.88H, J1 = 7.1 Hz, J2 = 1.3 Hz, H4′, cis), 7.22 (tt, 0.18H, J1 = 7.1 Hz, J2 = 1.5 Hz, H4′, trans), 7.28 (d, 1.8H, J = 7.7 Hz, H3′, H5′), 7.30–7.39 (m, 7H, H2′, H6′, H2″, H3″, H4″, H5″, H6″), 8.64 (s, 0.8H, H1, cis); 13C NMR (150.9 MHz, CDCl3) δ (ppm) 29.2 (C7, C9), 33.5 (C6, C10, cis), 35.0 (C6, C10, trans), 39.2 (NCH2COO, trans), 39.5 (NCH2COO, cis), 42.6 (C8, trans), 42.9 (C8, cis), 60.4 (C5, trans), 62.5 (C5, cis), 67.8 (COOCH2Ph), 126.5 (C4′, trans), 126.6 (C4′, cis), 127.0 (C2′, C6′, cis), 127.1 (C2′, C6′, trans), 128.5 (C2″, C6″), 128.61 (C3′, C5′, trans), 128.65 (C3′, C5′, cis), 128.7 (C4″), 128.8 (C3″, C5″), 135.0 (C1″), 146.0 (C1′, trans), 146.1 (C1′, cis), 156.1 (C2=O, trans), 157.0 (C2=O, cis), 167.1 (COOCH2Ph), 175.8 (C4=O, trans), 176.7 (C4=O, cis). Anal. Calcd for C23H24N2O4: C, 70.39; H, 6.16; N, 7.14. Found: C, 70.35; H, 6.22; N, 7.16.
Benzyl 2-(2,4-dioxo-6-phenyl-1,3-diazaspiro [4.5]decan-3-yl) acetate (10). Using the general experimental procedure described for the preparation of benzyl esters, a solution of hydantoin 6 (1000 mg, 4.09 mmol) in 41 mL dry THF was treated with potassium bis(trimethylsilyl)amide (877 mg, 4.17 mmol, 95% purity), which was added portionwise under ice cooling. The stirring was continued at r.t. for 20 min under argon, and afterward, the solvent was evaporated under reduced pressure. The resulting potassium imidate salt was dissolved in 41 mL dry DMF, and then benzyl bromoacetate (985 mg, 4.30 mmol) was added dropwise. After being stirred at 35–38 °C for 64 h under argon, the reaction mixture was poured into 400 mL of ice-water mixture and extracted with AcOEt (3 × 200 mL). The organic phases were combined and washed with H2O (3 × 270 mL) and brine (2 × 270 mL), dried over anh. Na2SO4, and evaporated in vacuo. The residual viscous oil was chromatographed on a silica gel column, using CH2Cl2 to CH2Cl2/AcOEt (10:1), to afford the desired compound 10 as a white product (1.140 mg, 71%). Mp 182–184 °C (AcOEt/n-pentane), Rf = 0.65 (CH2Cl2/AcOEt 8:1). 1H NMR (600.11 MHz, CDCl3) δ (ppm): 1.45 (qt, 1H, J1 = 13.8 Hz, J2 = 3.1 Hz, H9), 1.49–1.56 (m, 1H, H8), 1.79–1.93 (complex m, 5H, H7, H8, H9, H10), 2.07 (td, 1H, J1 = 13.5 Hz, J2 = 4.0 Hz, H10), 3.09–3.16 (sym m, 1H, H6), 3.85–3.95 (q, AB, 2H, JAB = 17.5 Hz, NCH2COO), 5.04–5.16 (q, AB, 2H, JAB = 12.3 Hz, OCH2Ph), 7.13–7.38 (complex m, 10H, H2′, H3′, H4′, H5′, H6′, H2″, H3″, H4″, H5″, H6″), 7.98 (dd, 1H, J1 = 29.4 Hz, J2 = 8.3 Hz, H1); 13C NMR (150.9 MHz, CDCl3) δ (ppm): 21.5 (C9), 25.6 (C8), 27.7 (C7), 34.4 (C10), 39.1 (NCH2COO), 48.0 (C6), 66.9 (C5), 67.4 (OCH2Ph), 127.5 (C4″), 128.2 (C2″, C6″), 128.3 (C3″, C5″), 128.6 (C4′), 128.7 (C2′, C6′), 128.7 (C3′, C5′), 135.2 (C1″), 139.1 (C1′), 157.0 (C2=O), 166.9 (NCH2COO), 175.6 (C4=O). Anal. Calcd for C23H24N2O4: C, 70.39; H, 6.16; N, 7.14. Found: C, 70.30; H, 6.17; N, 7.08.
Benzyl 2-(1-methyl-2,4-dioxo-8-phenyl-1,3-diazaspiro [4.5]decan-3-yl)acetate (11). Sodium hydride (102 mg, 2.56 mmol, 95% purity) was added portionwise to a solution of hydantoin 45 (550 mg, 2.13 mmol) in 21 mL dry DMF under ice cooling. The mixture was stirred for 20 min at rt under argon, and then benzyl bromoacetate (586 mg, 2.56 mmol) was added dropwise. The stirring was continued for 48 h at 35–38 °C under argon, and the mixture was then worked up following the same procedure described for the synthesis of benzyl esters. The crude colorless oily residue was purified by column chromatography with CH2Cl2, CH2Cl2/AcOEt 30:1 and 20:1 to yield a colorless oily product, which was crystallized upon treatment with n-pentane and Et2O at 0 °C to afford the desired 11 as a white crystalline solid. (765 mg, 88%); Rf = 0.6, 0.37 (CH2Cl2/AcOEt 10:1); mp 92–94 °C (AcOEt/dry Et2O-n-pentane); 1H NMR (600.11 MHz, CDCl3): δ (ppm) = 1.79–1.93 (complex m, 6H, H6, H7, H9, H10), 2.37 (qd, J1 = 12.2 Hz, J2 = 3.2 Hz, 2H, H7, H9), 2.52 (tt, J1 = 12.4 Hz, J2 = 3.3 Hz, 1H, H8), 2.91 (s, 3H, CH3), 4.33 (s, 2H, NCH2COO), 5.18 (s, 2H, OCH2Ph), 7.22 (tt, J1 = 7.0 Hz, J2 = 1.7 Hz, 1H, H4′), 7.27–7.38 (complex m, 9H, H2′, H3′, H5′, H6′, H2″, H3″, H4″, H5″, H6″); 13C NMR (150.9 MHz, CDCl3): δ (ppm) = 24.0 (CH3), 29.0 (C7, C9), 31.1 (C6, C10), 39.4 (NCH2COO), 43.1 (C8), 62.2 (C5), 67.7 (OCH2Ph), 126.5 (C4′), 127.1 (C2′, C6′), 128.5 (C3′, C5′), 128.6 (C2″, C6″), 128.66 (C4″), 128.75 (C3″, C5″), 135.1 (C1″), 146.1 (C1′), 154.7 (C2=O), 167.3 (COOCH2Ph), 175.5 (C4=O); elemental analysis calcd (%) for C24H26N2O4: C 70.92, H 6.45, N 6.89, found: C 70.98, H 6.50, N 6.84.
Benzyl 2-bromopropanoate. A two-necked round-bottom flask equipped with a magnetic stirrer and a dropping funnel was charged with N,N′-dicyclohexylcarbodiimide (6.19 g, 30.0 mmol) and Et2O (75 mL). To this suspension, 2-bromopropionic acid (3.82 g, 25.0 mmol) was added. The dropping funnel was charged with benzyl alcohol (3.24 g, 30 mmol), a catalytic amount of 4-dimethylaminopyridine (183 mg, 1.5 mmol), and Et2O (15 mL), and this solution was added dropwise to the suspension. After stirring at ambient temperature for 3 h, the reaction mixture was evaporated to half of its volume under reduced pressure and poured into 130 mL n-hexane. The precipitate formed was filtered off through a pad of Celite, and the filtrate was concentrated to dryness. The viscous oily residue was purified by column chromatography on silica gel with n-hexane/AcOEt 80:1, 40:1, and 20:1 as eluents to afford 4.83 g (79%) of pure benzyl 2-bromopropanoate as a colorless oil. 1H NMR spectrum in CDCl3 identical to that reported in the literature.
Benzyl 2-(2,4-dioxo-8-phenyl-1,3-diazaspiro [4.5]decan-3-yl)propanoate (46). To a solution of the hydantoin 5 (750 mg, 3.07 mmol) in dry THF (30 mL), potassium bis(trimethylsilyl)amide (657 mg, 3.13 mmol, 95% purity) was added portionwise at 0 °C. After stirring for 15 min at ambient temperature under argon, the solvent was removed under reduced pressure, and the remaining potassium imidate salt was dissolved in dry DMF (30 mL). Benzyl 2-bromopropanoate (783 mg, 3.22 mmol) was then added dropwise, and the stirring was continued for 62 h at 42 °C under argon. The reaction mixture was worked up following the general procedure described for the synthesis of benzyl esters. The resulting yellowish oil was chromatographed on silica gel using n-hexane/AcOEt 10:1, 6:1, and 4:1 as eluents to afford the title compound 46 as a white crystalline solid. (985 mg, 79%); Rf = 0.21 (n-hexane/AcOEt 4:1); mp 177–179 °C (AcOEt/n-pentane); 1H NMR (600.11 MHz, CDCl3): δ (ppm) = 1.66–1.84 (complex m, 4H, H6, H7, H9, H10), 1.76 (d, J = 7.4 Hz, 3H, NCH(CH3)COO), 1.87–1.93 (complex m, 1.9H, H7, H9), 1.99 (td, J1 = 13.8 Hz, J1 = 4.1 Hz, 2H, H6, H10), 2.27 (dtdd, J1 = 16.3 Hz, J2 = 13.1 Hz, J3 = 8.1 Hz, J4 = 3.3 Hz, 0.2H, H7, H9), 2.55 (tt, J1 = 12.1 Hz, J2 = 3.5 Hz, 0.1H, H8), 2.60 (tt, J1 = 12.2 Hz, J2 = 3.5 Hz, 0.9H, H8), 4.82 (q, J = 7.3 Hz, 0.1H, NCH(CH3)COO), 4.88 (q, J = 7.3 Hz, 0.8H, NCH(CH3)COO), 5.02–5.23 (q, AX, JAX = 12.3 Hz, 1.8H, OCH2Ph), 5.04–5.27 (q, AX, JAX = 12.3 Hz, 0.2H, OCH2Ph), 6.42 (s, 0.1H, H1), 7.22 (td, J1 = 7.0 Hz, J2 = 1.6 Hz, 1H, H4′), 7.25–7.37 (complex m, 9H, H2′, H3′, H5′, H6′, H2″, H3″, H4″, H5″, H6″), 8.67 (s, 0.9H, H1); 13C NMR (50.32 MHz, CDCl3): δ (ppm) = 14.9 (NCH(CH3)COO), 29.2 (C7, C9), 33.4 (C6, C10), 43.1 (C8), 48.2 (NCH(CH3)COO), 62.0 (C5), 67.7 (OCH2Ph), 126.6 (C4′), 127.0 (C2′, C6′), 128.4 (C2″, C6″), 128.5 (C4″), 128.6 (C3′, C5′), 128.7 (C3″, C5″), 135.3 (C1″), 146.2 (C1′), 157.3 (C2=O), 169.5 (COOCH2Ph), 176.7 (C4=O); elemental analysis calcd (%) for C24H26N2O4: C 70.92, H 6.45, N 6.89, found: C 71.00, H 6.49, N 6.82.
General N-alkylation procedure for the preparation of analogs (12–15 and 50). To a well-stirred and ice-cooled solution of the benzyl ester (3.25 mmol, 1.0 eq) in dry DMF (15 mL), NaH (3.9 mmol, 1.2 eq, 60% dispersion in mineral oil) was slowly added in small portions. After 15 min of stirring at ambient temperature under argon, the mixture was treated dropwise with methyl iodide/ethyl iodide/benzyl bromide (3.9 mmol, 1.2 eq), and it was allowed to react for 7 days at 60–65 °C under argon. The reaction mixture was poured into ice water (150 mL) with AcOEt (3 × 120 mL). The combined organic extracts were washed with H2O (3 × 180 mL) and brine (2 × 180 mL) and dried over anh. Na2SO4, and the solvent were concentrated under reduced pressure to afford a viscous oily residue. The crude product was chromatographed on silica gel to yield the respective N-alkylated products.
Benzyl 2-(1-methyl-2,4-dioxo-6-phenyl-1,3-diazaspiro [4.5]decan-3-yl)acetate (12). A stirred solution of 10 (500 mg, 1.27 mmol) in dry DMF (5.9 mL) was treated with sodium hydride (61 mg, 1.53 mmol, 60% dispersion in mineral oil), which was added portionwise under ice cooling. After stirring at r.t. for 15 min under argon, methyl iodide (217 mg, 1.53 mmol) was added dropwise. The reaction mixture was stirred at 60 °C for 7 days under argon, and then it was poured into an ice-water mixture (60 mL) and extracted with AcOEt (3 × 45 mL). The combined organic phases were washed with H2O (3 × 70 mL) and brine (2 × 70 mL), dried over anh. Na2SO4, and evaporated under reduced pressure. The yellowish crude oily product was purified by silica gel column chromatography, using CH2Cl2/AcOEt 15:1 as eluent to afford the target compound 12 (280 mg, 54%) as a colorless highly viscous oil, which, after being left under vacuum, was converted to semisolid. Low mp, Rf = 0.85 (CH2Cl2/AcOEt 8:1). 1H NMR (600.11 MHz, CDCl3) δ (ppm): 1.65 (qt, 1H, J1 = 13.2 Hz, J2 = 3.7 Hz, H8), 1.83 (qt, 1H, J1 = 14.0 Hz, J2 = 4.1 Hz, H9), 1.92 (dq, 2H, J1 = 14.3 Hz, J2 = 4.1 Hz, H7, H9), 1.97 (dp, 1H, J1 = 14.5 Hz, J2 = 1.7 Hz, H10), 2.05–2.09 (m, 1H, H8), 2.13 (ddd, 1H, J1 = 13.8 Hz, J2 = 12.8 Hz, J3 = 3.6 Hz, H7), 2.22 (td, 1H, J1 = 14.4 Hz, J2 = 5.4 Hz, H10), 3.23 (dd, 1H, J1 = 13.9 Hz, J2 = 4.1 Hz, H6), 3.34 (s, 3H, NCH3), 3.86–3.98 (q, AB, 2H, JAB = 17.4 Hz, NCH2COO), 5.05–5.13 (q, AB, 2H, JAB = 12.3 Hz, OCH2Ph), 7.06–7.11 (m, 2H, H2′, H6′), 7.17–7.20 (m, 1H, H4′), 7.21–7.24 (m, 2H, H3′, H5′), 7.26–7.29 (m, 2H, H2″, H6″), 7.30–7.37 (complex m, 3H, H3″, H4″, H5″); 13C NMR (150.9 MHz, CDCl3) δ (ppm): 22.0 (C9), 25.4 (C8), 27.8 (C7), 30.5 (NCH3), 32.9 (C10), 39.7 (NCH2COO), 49.4 (C6), 67.5 (OCH2Ph), 67.8 (C5), 127.6 (C4′), 128.1 (C2′, C6′), 128.4 (C2″, C6″), 128.5 (C3′, C5′), 128.6 (C4″), 128.7 (C3″, C5″), 135.2 (C1″), 139.2 (C1′), 155.3 (C2=O), 166.9 (NCH2COO), 175.5 (C4=O). Anal. Calcd for C24H26N2O4: C, 70.92; H, 6.45; N, 6.89. Found: C, 70.93; H, 6.39; N, 6.86.
Benzyl 2-(1-ethyl-8-phenyl-2,4-dioxo-1,3-diazaspiro [4.5]decan-3-yl)acetate (13). To a stirred solution of 9 (660 mg, 1.68 mmol) in dry DMF (12 mL), sodium hydride (81 mg, 2.02 mmol, 60% dispersion in mineral oil) was added portionwise under ice cooling. After stirring at ambient temperature for 15 min under argon, ethyl iodide (393 mg, 2.52 mmol) was added dropwise. Stirring was continued at 60 °C for 7 days under argon, and the reaction mixture was diluted with 120 mL ice-water mixture and extracted with AcOEt (3 × 80 mL). The combined organic extracts were washed with H2O (3 × 100 mL) and brine (2 × 100 mL), dried with anh. Na2SO4, and evaporated in vacuo. The yellow viscous oil was purified by column chromatography on silica gel, with CH2Cl2 to CH2Cl2/AcOEt 30:1 to afford the desired compound 13 (310 mg, 44%) as a colorless oil, which solidified on cooling. A mixture of CH2Cl2/AcOEt 20:1 was then used to elute the transesterification product 54 (80 mg, 13%) as a viscous oil, which was crystallized upon treatment with n-pentane. Mp 84–86 °C (AcOEt/n-pentane), Rf = 0.87 (CH2Cl2/AcOEt 8:1). 1H NMR (600.11 MHz, CDCl3) δ (ppm) 1.25 (t, 3H, J = 7.1 Hz, NCH2CH3), 1.88–1.95 (m, 2H, H6a, H10a), 1.98 (ddd, 2H, J1 = 13.2 Hz, J2 = 9.5 Hz, J3 = 3.3 Hz, H7a, H9e), 2.02 (ddd, 2H, J1 = 10.5 Hz, J2 = 8.7 Hz, J3 = 3.2 Hz, H6e, H10e), 2.20 (ddd, 2H, J1 = 10.2 Hz, J2 = 8.4 Hz, J3 = 3.7 Hz, H7e, H9e), 2.87 (ddd, 1H, J1 = 13.2 Hz, J2 = 9.0 Hz, J3 = 4.2 Hz, H8), 3.58 (q, 2H, J = 7.1 Hz, NCH2CH3), 4.31 (s, 2H, NCH2COO), 5.17 (s, 2H, COOCH2Ph), 7.24 (tt, 1H, J1 = 7.3 Hz, J2 = 1.1 Hz, H4′), 7.28 (d, 2H, J = 7.3 Hz, H2′, H6′), 7.32–7.39 (m, 7H, H3′, H5′, H2″, H3″, H4″, H5″, H6″); 13C NMR (150.9 MHz, CDCl3) δ (ppm) 15.0 (NCH2CH3), 28.0 (C7, C9), 31.9 (C6, C10), 37.0 (NCH2CH3), 39.6 (C8), 39.8 (NCH2COO), 63.4 (C5), 67.7 (COOCH2Ph), 126.5 (C4′), 126.9 (C2′, C6′), 128.5 (C2″, C6″), 128.7 (C4″), 128.8 (C3′, C5′, C3″, C5″), 135.1 (C1″), 144.9 (C1′), 154.7 (C2=O), 167.2 (COOCH2Ph), 176.3 (C4=O). Anal. Calcd for C25H28N2O4: C, 71.41; H, 6.71; N, 6.66. Found: C, 71.45; H, 6.77; N, 6.60.
Ethyl 2-(1-ethyl-8-phenyl-2,4-dioxo-1,3-diazaspiro [4.5]decan-3-yl)acetate (54). Mp 91–93 °C (AcOEt/n-pentane), Rf = 0.76 (CH2Cl2/AcOEt 8:1). 1H NMR (600.11 MHz, CDCl3) δ (ppm) 1.27 (t, 3H, J = 7.1 Hz, NCH2CH3), 1.28 (t, 3H, J = 7.1 Hz, COOCH2CH3), 1.97 (ddd, 2H, J1 = 16.7 Hz, J2 = 8.9 Hz, J3 = 3.8 Hz, H6a, H10a), 2.00 (ddd, 2H, J1 = 13.5 Hz, J2 = 9.6 Hz, J3 = 3.6 Hz, H7a, H9e), 2.04–2.11 (m, 2H, H7e, H9a), 2.89 (ddd, 1H, J1 = 13.1 Hz, J2 = 9.0 Hz, J3 = 4.2 Hz, H8), 3.60 (q, 2H, J = 7.1 Hz, NCH2CH3), 4.21 (q, 2H, J = 7.1 Hz, COOCH2CH3), 4.25 (s, 2H, NCH2COO), 7.24 (tt, 1H, J1 = 7.3 Hz, J2 = 1.1 Hz, H4′), 7.28 (d, 2H, J = 7.3 Hz, H2′, H6′), 7.35 (tt, 2H, J1 = 7.7 Hz, J2 = 1.1 Hz, H3′, H5′); 13C NMR (150.9 MHz, CDCl3) δ (ppm) 14.2 (COOCH2CH3), 15.0 (NCH2CH3), 28.0 (C7, C9), 32.0 (C6, C10), 37.0 (NCH2CH3), 39.7 (NCH2COO), 39.8 (C8), 61.9 (COOCH2CH3), 63.4 (C5), 126.5 (C4′), 126.9 (C2′, C6′), 128.8 (C3′, C5′), 144.9 (C1′), 154.8 (C2=O), 167.3 (COOCH2Ph), 176.4 (C4=O). Anal. Calcd for C20H26N2O4: C, 67.02; H, 7.31; N, 7.82. Found: C, 67.00; H, 7.42; N, 7.90.
Benzyl 2-(1-benzyl-2,4-dioxo-8-phenyl-1,3-diazaspiro [4.5]decan-3-yl)acetate (14). To a stirred solution of 9 (800 mg, 2.04 mmol) in 9 mL dry DMF, sodium hydride (98 mg, 2.45 mmol, 60% dispersion in mineral oil) was added portionwise under ice cooling. The mixture was stirred at ambient temperature for 30 min under argon, and then benzyl bromide (419 mg, 2.45 mmol) was added dropwise. After 7 days of stirring at 65 °C under argon, the reaction mixture was worked up according to the general N-benzylation procedure described. The crude yellowish oily residue was purified by column chromatography on silica gel with CH2Cl2 and then CH2Cl2/AcOEt 20:1 to yield an off-yellow oily product, which was crystallized after treatment with hexane to afford the title compound 14 as a white crystalline solid. (330 mg, 33%); Rf = 0.80 (CH2Cl2/AcOEt 8:1); mp 120–122 °C (AcOEt/dry Et2O-n-pentane); 1H NMR (600.11 MHz, CDCl3): δ (ppm) = 1.72 (dtd, J1 = 13.6 Hz, J2 = 9.9 Hz, J3 = 3.9 Hz, 2H, H7, H9), 1.87 (ddd, J1 = 14.8 Hz, J2 = 10.3 Hz, J3 = 4.9 Hz, 2H, H6, H10), 1.91 (ddd, J1 = 14.5 Hz, J2 = 11.3 Hz, J3 = 4.4 Hz, 2H, H6, H10), 2.02 (dq, J1 = 14.3 Hz, J2 = 5.3, 4.9 Hz, 2H, H7, H9), 2.77 (tt, J1 = 8.8 Hz, J2 = 4.0 Hz, 1H, H8), 4.39 (s, 2H, NCH2COO), 4.75 (s, 2H, NCH2Ph), 5.21 (s, 2H, OCH2Ph), 7.13 (d, J = 7.6 Hz, 2H, H2′, H6′), 7.20 (d, J = 7.4 Hz, 2H, H2Bz, H6Bz), 7.23 (t, J = 7.2 Hz, 1H, H4′), 7.27 (d, J = 7.5 Hz, 2H, H2″, H6″), 7.28 (complex m, 8H, H3′, H5′, H3″, H4″, H5″, H3Bz, H4Bz, H5Bz); 13C NMR (150.9 MHz, CDCl3): δ (ppm) = 27.8 (C7, C9), 31.4 (C6, C10), 39.3 (C8), 40.0 (NCH2COO), 44.9 (NCH2Ph), 64.1 (C5), 67.8 (OCH2Ph), 126.4 (C4′), 127.0 (C2Bz, C6Bz), 127.1 (C2′, C6′), 127.7 (C4Bz), 128.6 (C2″, C6″), 128.67 (C3′, C5′), 128.73 (C4″), 128.8 (C3″, C5″), 128.9 (C3Bz, C5Bz), 135.1 (C1″), 137.8 (C1Bz), 144.8 (C1′), 155.9 (C2=O), 167.2 (COOCH2Ph), 176.3 (C4=O); elemental analysis calcd (%) for C30H30N2O4: C 74.67, H 6.27, N 5.81, found: C 74.63, H 6.33, N 5.85.
Benzyl 2-(1-benzyl-2,4-dioxo-6-phenyl-1,3-diazaspiro [4.5] decan-3-yl)acetate (15). Prepared by N-alkylation of benzyl ester 10 (500 mg, 1.27 mmol) in dry DMF (5.9 mL) by using NaH (61 mg, 1.53 mmol, 60% dispersion in mineral oil) and benzyl bromide (262 mg, 1.53 mmol) as described in the main manuscript for the preparation of N-benzylated derivatives. After stirring at 65 °C for 7 days under argon, the reaction mixture was diluted with 60 mL ice water and extracted with AcOEt (3 × 45 mL). The combined organic extracts were washed with H2O (3 × 70 mL) and brine (2 × 70 mL), dried with anh. Na2SO4, and evaporated in vacuo. The crude oily residue was purified by column chromatography on silica gel using CH2Cl2 to afford the title compound 15 as a colorless oil, which was further crystallized to a white crystalline solid upon treatment with n-pentane/Et2O (393 mg, 64%). Mp 98–100 °C (AcOEt/n-pentane, Et2O), Rf = 0.41 (n-hexane/AcOEt 3:1). 1H NMR (400.13 MHz, CDCl3) δ (ppm): 1.29 (qt, 1H, J1 = 14.0 Hz, J2 = 4.2 Hz, H9), 1.42–1.58 (complex m, 2H, H8, H9), 1.77 (dp, 1H, J1 = 14.7 Hz, J2 = 1.9 Hz, H10), 1.81–1.96 (m, 2H, H7, H8), 2.03 (td, 1H, J1 = 14.3 Hz, J2 = 4.7 Hz, H10), 2.13 (td, 1H, J1 = 13.3 Hz, J2 = 3.4 Hz, H7), 3.15 (dd, 1H, J1 = 13.9 Hz, J2 = 4.2 Hz, H6), 3.79–4.01 (q, AB, 2H, JAB = 17.3 Hz, NCH2COO), 4.63–5.31 (q, AX, 2H, JAX = 16.7 Hz, NCH2Ph), 5.02–5.12 (q, AB, 2H, JAB = 12.2 Hz, OCH2Ph), 7.03 (~dd, 2H, J1 = 7.8 Hz, J2 = 1.8 Hz, H2′, H6′), 7.11–7.31 (complex m, 13H, H3′, H4′, H5′, H2″, H3″, H4″, H5″, H6″, H2Bz, H3Bz, H4Bz, H5Bz, H6Bz); 13C NMR (50.32 ΜHz, CDCl3) δ (ppm): 20.9 (C9), 25.2 (C8), 28.0 (C7), 32.2 (C10), 39.7 (NCH2COO), 47.0 (NCH2Ph), 50.2 (C6), 67.6 (OCH2Ph), 68.8 (C5), 126.5 (C2Bz, C6Bz), 127.4 (C4Bz), 127.8 (C4′), 128.2 (C2″, C6″), 128.3 (C2′, C6′), 128.5 (C4″, C3Bz, C5Bz), 128.7 (C3″, C5″), 128.8 (C3′, C5′), 135.1 (C1″), 137.4 (C1Bz), 138.9 (C1′), 155.9 (C2=O), 167.0 (NCH2COO), 175.5 (C4=O). Anal. Calcd for C30H30N2O4: C, 74.67; H, 6.27; N, 5.81. Found: C, 74.75; H, 6.30; N, 5.89.
Benzyl 2-(1-methyl-2,4-dioxo-8-phenyl-1,3-diazaspiro [4.5]decan-3-yl)propanoate (50). Potassium bis(trimethylsilyl)amide (664 mg, 3.16 mmol, 95% purity) was added portionwise, under ice cooling, to a solution of methyl hydantoin 45 (800 mg, 3.10 mmol) in 31 mL dry THF. The mixture was stirred for 15 min at rt under argon. After removal of the solvent under vacuum, the residue was dissolved in 31 mL dry DMF, and benzyl 2-bromopropanoate (793 mg, 3.26 mmol) was added dropwise. After stirring for another 70 h at 45 °C under argon, the reaction mixture was worked up following the general procedure previously described. The crude yellow oil was purified by column chromatography on silica gel eluting with n-hexane/AcOEt 8:1, 5:1, and 4:1 to yield the desired benzyl ester 50 as a colorless oil. Crystallization upon treatment with n-pentane under ice cooling yielded 50 as a white solid. (900 mg, 69%); Rf = 0.12 (n-hexane/AcOEt 4:1); mp 76–78 °C (AcOEt/n-pentane); 1H NMR (400.13 MHz, CDCl3): δ (ppm) = 1.60–1.90 (complex m, 6H, H6, H7, H9, H10), 1.68 (d, J = 7.3 Hz, 3H, NCH(CH3)COO), 2.32 (qd, J1 = 13.4 Hz, J2 = 4.0 Hz, 2H, H7, H9), 2.48 (tt, J1 = 12.3 Hz, J2 = 3.3 Hz, 1H, H8), 2.86 (s, 3H, NCH3), 4.82 (q, J = 7.3 Hz, 1H, NCH(CH3)COO), 5.03–5.30 (q, AX, JAX = 12.2 Hz, 2H, OCH2Ph), 7.22 (tt, J1 = 7.0 Hz, J2 = 1.9 Hz, 1H, H4), 7.25–7.36 (complex m, 9H, H2′, H3′, H5′, H6′, H2″, H3″, H4″, H5″, H6″); 13C NMR (150.9 MHz, CDCl3): δ (ppm) = 14.9 (NCH(CH3)COO), 23.9 (NCH3), 28.86, 28.89 (C7, C9), 30.9 (C6, C10), 43.1 (C8), 48.1 (NCH(CH3)COO), 61.4 (C5), 67.6 (OCH2Ph), 126.4 (C4′), 127.1 (C2′, C6′), 128.5 (C2″, C6″), 128.57, 128.60 (C3′, C5′, C3″, C4″, C5″), 135.4 (C1″), 146.1 (C1′), 154.8 (C2=O), 169.7 (COOCH2Ph), 175.3 (C4=O); elemental analysis calcd (%) for C25H28N2O4: C 71.41, H 6.71, N 6.66, found: C 71.44, H 6.80, N 6.60.
General experimental procedure for the preparation of carboxylic acids (16–24, 47, and 51). A solution of the benzyl ester (2.0 mmol) was hydrogenated over Pd/C (10 wt.%) catalyst for 3 h at 44–46 °C under 50–55 psi pressure in a mixture of EtOH/AcOEt 3:1 (40 mL). The solution was filtered off in vacuo to remove the catalyst, the filtration pad was washed with portions of hot EtOH (3 × 15 mL), and the combined filtrates were evaporated under reduced pressure to afford the desired carboxylic acid.
((4R,4′S)/(4S,4′R)-4′-Methyl-2,5-dioxo-3′,4′-dihydro-2′H-spiro[imidazolidine-4,1′-naphthalene]-1-yl)acetic acid (16). A solution of benzyl ester 7 (1.0 g, 2.64 mmol) in 53 mL abs EtOH/AcOEt (4:1) was hydrogenated in the presence of Pd/C (120 mg) according to the general procedure described for the synthesis of carboxylic acids to yield a white foamy product. Removal of the entrapped solvents upon drying under high vacuum yielded the desired compound 16 as a glass solid, which was crystallized upon treatment with dry Et2O under ice cooling (760 mg, almost quantitative yield); Rf = 0.06 (CH2Cl2/AcOEt 4:1); mp 200–202 °C (AcOEt/dry Et2O-n-pentane); 1H NMR (400.13 MHz, [D6]DMSO): δ (ppm) = 1.27, 1.29 (d + d, J = 7.1 Hz, J2 = 7.0 Hz, 3H, CH3), 1.56–1.67 (m, 0.45 H, H3′), 1.77–1.90 (m, 1H, H2′, H3′), 1.94–2.10 (m, 1.45H, H2′, H3′), 2.12–2.29 (m, 1H, H2′, H3′), 2.94 (dq, J1 = 13.4 Hz, J2 = 7.9 Hz, 1H, H4′), 4.05–4.21 (2q, AB, J1AB = J2AB = 17.6 Hz, 2H, NCH2COOH), 7.18 (t, J = 7.6 Hz, 1H, H7′), 7.22 (dd, 1H, J1 = 7.8 Hz, J2 = 2.0 Hz, H8′), 7.26–7.31 (m, 1H, H6′), 7.33 (d, J = 7.6 Hz, 1H, H5′), 8.96, 8.97 (s + s, 1H, H3′), 13.26 (s, 1H, NCH2COOH); 13C (50.32 MHz, [D6]DMSO): δ (ppm) = 22.0, 22.3 (CH3), 25.6, 26.4 (C3′), 30.1, 31.0 (C2′), 31.2, 31.6 (C4′), 39.2, 39.3 (NCH2COOH), 62.5, 62.6 (C4), 126.4 (C7′), 127.1 (C8′), 128.0, 128.3, 128.5 (C5′, C6′), 133.5, 133.6 (C8′a), 142.65, 142.74 (C4′a), 155.1 (C2=O), 168.9 (NCH2COOH), 175.9 (C5=O); elemental analysis calcd (%) for C15H16N2O4: C 62.49, H 5.59, N 9.72; found: C 62.56, H 5.63, N 9.69.
((4R,4′R)/(4S,4′S)-4′-Methyl-2,5-dioxo-3′,4′-dihydro-2′H-spiro[imidazolidine-4,1′-naphthalene]-1-yl)acetic acid (17). A total of 10 wt% Pd (52 mg) on charcoal was added to a solution of the benzyl ester 8 (430 mg, 1.14 mmol) in a mixture of EtOH/AcOEt 3:1 (23 mL), and the mixture was hydrogenated following the procedure previously described. The white foamy solid obtained strongly binds the aforementioned solvents. Removal of the entrapped solvents upon drying under high vacuum yielded the target compound 17 as a glass solid, which was crystallized upon treatment with dry Et2O under ice cooling (325 mg, almost quantitative yield); Rf = 0.02 (CH2Cl2/AcOEt 4:1); mp 154–156 °C (AcOEt/dry Et2O-n-pentane); 1H NMR (400.13 MHz, CDCl3) δ (ppm): 1.35 (~t, J = 6.5 Hz, 3H, CH3), 1.54 (ddt, J1 = 14.0 Hz, J2 = 9.7 Hz, J3 = 4.8 Hz, 0.6H, H3′), 1.89–2.04 (complex m, 1.2H, H2′, H3′), 2.11 (ddd, J1 = 13.2 Hz, J2 = 8.7 Hz, J3 = 2.7 Hz, 0.6H, H2′), 2.24 (ddd, J1 = 12.4 Hz, J2 = 9.7 Hz, J3 = 2.6 Hz, 0.6H, H2′), 2.31–2.46 (complex m, 1H, H2′, H3′), 2.94–3.06 (dt, J1 = 12.9 Hz, J2 = 6.3 Hz, 1H, H4′), 4.26–4.42 (2q, AB, J1AB = 17.6 Hz, J2AB = 18.0 Hz, 2H, NCH2COOH), 6.58, 6.60 (s + s, 1H, H3), 7.11 (td, J1 = 8.0 Hz, J2 = 2.2 Hz, 1H, H7′), 7.21 (d, J = 7.7 Hz, 1H, H8′), 7.23–7.33 (m, 2H, H5′, H6′); 13C (50.32 MHz, CDCl3): δ (ppm) = 22.4 (CH3), 26.6, 27.2 (C3′), 31.1, 31.6 (C2′), 32.0, 32.1 (C4′), 39.4 (NCH2COOH), 63.9, 64.0 (C4), 127.1 (C7′), 127.2 (C8′), 128.6, 128.9 (C6′), 129.2 (C5′), 132.0 (C8′a), 143.1 (C4′a), 156.6 (C2=O), 171.2 (NCH2COOH), 175.7 (C5=O); elemental analysis calcd (%) for C15H16N2O4: C 62.49, H 5.59, N 9.72; found: C 62.55, H 5.62, N 9.66.
2-(2,4-Dioxo-8-phenyl-1,3-diazaspiro [4.5]decan-3-yl)acetic acid (18). A mixture of benzyl ester 9 (400 mg, 1.02 mmol) and 10% Pd on charcoal (48 mg) in EtOH/AcOEt 3:1 (20 mL) was hydrogenated following the general procedure described for the preparation of carboxylic acids to afford the title compound 18 as a white crystalline solid. (308 mg, almost quantitative yield); Rf = 0.05 (CH2Cl2/AcOEt 5:1); mp >250 °C (MeOH/dry Et2O-n-pentane); 1H NMR (600.11 MHz, [D6]DMSO): δ (ppm) = 1.65 (dd, J1 = 11.5 Hz, J2 = 2.4 Hz, 1.7H, H6a, H10a, cis), 1.70–1.82 (complex m, 3.7H, H7, H9, cis), 1.86 (qd, J1 = 14.2 Hz, J2 = 3.7 Hz, 2H, H6e, H10e), 1.92 (tt, J1 = 14.0 Hz, J2 = 3.3 Hz, 0.3H, H6a, H10a, trans), 2.12 (qd, J1 = 13.6 Hz, J2 = 3.7 Hz, 0.3H, H7, H9, trans), 2.59 (tt, J1 = 11.4 Hz, J2 = 4.2 Hz, 1H, H8), 4.02 (s, 0.35H, NCH2COOH, trans), 4.06 (s, 1.65H, NCH2COOH, cis), 7.18 (tt, J1 = 7.3 Hz, J2 = 1.5 Hz, 1H, H4′), 7.26 (dd, J1 = 8.2 Hz, J2 = 1.6 Hz, 0.3H, H2′, H6′, trans), 7.29 (t, J = 7.6 Hz, 2H, H3′, H5′), 7.33 (dd, J1 = 8.3 Hz, J2 = 1.6 Hz, 1.7H, H2′, H6′, cis), 8.31 (s, 0.15H, H1, trans), 9.07 (s, 0.8H, H1, cis), 13.08 (v br s, 1H, NCH2COOH); 13C NMR (100.61 MHz, [D6]DMSO): δ (ppm) = 28.4 (C7, C9, cis), 28.7 (C7, C9, trans), 33.4 (C6, C10, cis), 33.9 (C6, C10, trans), 38.7 (NCH2COOH, trans), 39.0 (NCH2COOH, cis), 41.3 (C8, trans), 42.2 (C8, cis), 59.0 (C5, trans), 61.1 (C5, cis), 126.0 (C4′), 126.7 (C2′, C6′, trans), 126.9 (C2′, C6′, cis), 128.2 (C3′, C5′, cis),128.4 (C3′, C5′, trans), 146.1 (C1′, trans), 146.6 (C1′, cis), 154.8 (C2=O, trans), 155.2 (C2=O, cis), 168.8 (NCH2COOH, cis), 168.9 (NCH2COOH, trans), 176.1 (C4=O, trans), 176.5 (C4=O, cis); elemental analysis calcd (%) for C16H18N2O4: C 63.56, H 6.00, N 9.27, found: C 63.64, H 6.07, N 9.20.
2-(2,4-Dioxo-6-phenyl-1,3-diazaspiro [4.5]decan-3-yl)acetic acid (19). Following the general hydrogenolysis procedure for the preparation of carboxylic acids described in the main manuscript, benzyl ester 10 (400 mg, 1.02 mmol) in a mixture of 20 mL EtOH/AcOEt (3:1) provided the target compound 19 as a white crystalline solid (300 mg, almost quantitative yield). Mp > 250 °C (AcOEt/n-pentane), Rf = 0.14 (AcOEt). 1H NMR (600.11 MHz, DMSO-d6) δ (ppm): 1.46 (qt, 1H, J1 = 13.0 Hz, J2 = 3.6 Hz, H8), 1.56 (tt, 1H, J1 = 13.4 Hz, J2 = 3.5 Hz, H9), 1.59–1.69 (m, 2H, H7, H10), 1.72 (dt, 1H, J1 = 13.4 Hz, J2 = 3.6 Hz, H9), 1.80 (dt, 1H, J1 = 13.0 Hz, J2 = 3.6 Hz, H8), 1.88 (td, 1H, J1 = 13.5 Hz, J2 = 4.0 Hz, H10), 2.01 (qd, 1H, J1 = 13.2 Hz, J2 = 3.6 Hz, H7), 2.98 (dd, 1H, J1 = 13.4 Hz, J2 = 3.6 Hz, H6), 3.50–3.66 (q, AB, 2H, JAB = 17.4 Hz, NCH2COOH), 7.08–7.12 (m, 2H, H3′, H5′), 7.17 (~tt, 1H, J1 = 7.2 Hz, J2≈2.0 Hz, H4′), 7.19–7.24 (m, 2H, H2′, H6′), 8.98 (s, 1H, H1), 12.90 (brs, 1H, NCH2COOH); 13C NMR (150.9 MHz, DMSO-d6) δ (ppm): 20.5 (C9), 25.1 (C8), 27.0 (C7), 34.6 (C10), 38.4 (NCH2COOH), 47.2 (C6), 65.7 (C5), 126.9 (C4′), 127.7 (C2′, C6′), 128.3 (C3′, C5′), 139.5 (C1′), 155.1 (C2=O), 168.4 (NCH2COOH), 175.2 (C4=O). Anal. Calcd for C16H18N2O4: C, 63.56; H, 6.00; N, 9.27. Found: C, 63.61; H, 6.03; N, 9.26.
2-(1-Methyl-2,4-dioxo-8-phenyl-1,3-diazaspiro [4.5]decan-3-yl)acetic acid (20). Following the general hydrogenolysis procedure for the synthesis of carboxylic acids previously described, benzyl ester 11 (630 mg, 1.55 mmol) in a mixture of 31 mL EtOH/AcOEt (3:1) in the presence of Pd/C (76 mg) yielded the target compound 20 as a white crystalline solid (490 mg, almost quantitative yield); Rf = 0.03 (CH2Cl2/AcOEt 4:1); mp 234–236 °C (MeOH/n-pentane); 1H NMR (400.13 MHz, [D6]DMSO): δ (ppm) = 1.71 (d, J = 12.9 Hz, 2H, H6, H10), 1.76 (d, J = 12.4 Hz, 2H, H7, H9), 2.02 (td, J1 = 13.2 Hz, J2 = 3.5 Hz, 2H, H6, H10), 2.16 (qd, J1 = 12.6 Hz, J2 = 3.1 Hz, 2H, H7, H9), 2.62 (tt, J1 = 12.3 Hz, J2 = 3.6 Hz, 1H, H8), 2.83 (s, 3H, CH3), 4.08 (s, 2H, NCH2COOH), 7.20 (t, J = 7.1 Hz, 1H, H4′), 7.25 (d, J = 7.3 Hz, 2H, H2′, H6′), 7.31 (t, J = 7.4 Hz, 2H, H3′, H5′), 13.14 (br s, 1H, NCH2COOH); 13C NMR (150.9 MHz, [D6]DMSO): δ (ppm) = 23.6 (CH3), 28.6 (C7, C9), 30.0 (C6, C10), 39.0 (NCH2COOH), 41.4 (C8), 61.3 (C5), 126.1 (C4′), 126.6 (C2′, C6′), 128.4 (C3′, C5′), 146.3 (C1′), 154.2 (C2=O), 168.8 (NCH2COOH), 175.2 (C4=O); elemental analysis calcd (%) for C17H20N2O4: C 64.54, H 6.37, N 8.86, found: C 64.50, H 6.45, N 8.88.
2-(1-Methyl-2,4-dioxo-6-phenyl-1,3-diazaspiro [4.5]decan-3-yl)acetic acid (21). It was prepared by hydrogenolysis of benzyl ester 12 (250 mg, 0.62 mmol) in a mixture of EtOH/AcOEt 3:1 (12.5 mL) following the general procedure previously described. Evaporation of the solvents yielded the corresponding carboxylic acid 21 as a shiny white crystalline solid (190 mg, almost quantitative yield). Mp 184–186 °C (AcOEt/n-pentane, Et2O), Rf = 0.14 (AcOEt). 1H NMR (600.11 MHz, DMSO-d6) δ (ppm): 1.52–1.61 (sym m, 1H, H8), 1.74 (dq, 1H, J1 = 13.7 Hz, J2 = 3.7 Hz, H7), 1.79–1.86 (m, 2H, H9), 1.89–1.95 (m, 1H, H10), 1.95–2.01 (m, 1H, H8), 2.02–2.09 (m, 1H, H10), 2.20 (qd, 1H, J1 = 13.6 Hz, J2 = 3.6 Hz, H7), 3.11 (dd, 1H, J1 = 13.9 Hz, J2 = 4.1 Hz, H6), 3.23 (s, 3H, NCH3), 3.59–3.77 (q, AB, 2H, JAB = 17.3 Hz, NCH2COOH), 7.05–7.09 (m, 2H, H2′, H6′), 7.19 (~tt, 1H, J1 = 7.3 Hz, J2 ≈ 1.7 Hz, H4′), 7.21–7.26 (m, 2H, H3′, H5′), 13.05 (vbs, 1H, NCH2COOH); 13C NMR (150.9 MHz, DMSO-d6) δ (ppm): 21.2 (C9), 24.9 (C8), 27.0 (C7), 29.7 (NCH3), 32.5 (C10), 39.8 (NCH2COOH), 48.5 (C6), 66.8 (C5), 127.1 (C4′), 127.9 (C2′, C6′), 128.0 (C3′, C5′), 139.3 (C1′), 154.7 (C2=O), 168.2 (NCH2COOH), 174.9 (C4=O). Anal. Calcd for C17H20N2O4: C, 64.54; H, 6.37; N, 8.86. Found: C, 64.50; H, 6.31; N, 8.89.
2-(1-Ethyl-2,4-dioxo-8-phenyl-1,3-diazaspiro [4.5]decan-3-yl)acetic acid (22). A mixture of benzyl ester 13 (480 mg, 1.14 mmol) and 10% Pd on charcoal (58 mg) in EtOH/AcOEt 3:1 (22.8 mL) was subjected to catalytic hydrogenolysis following the general procedure previously described to yield the title compound 22 as a glass solid. Crystallization upon treatment with n-pentane (0 °C) yielded 22 as a white crystalline solid (375 mg, almost quantitative yield); Rf = 0.07 (AcOEt); mp 153–155 °C (AcOEt/dry Et2O-n-pentane); 1H NMR (600.11 MHz, [D6]DMSO): δ (ppm) = 1.16 (t, J = 7.1 Hz, 3H, NCH2CH3), 1.75 (dq, J1 = 7.8 Hz, J2 = 3.8 Hz, 0.4H, H6, H10, trans), 1.84–1.98 (m, 5.6H, H6, H10, cis, H7, H9), 2.04 (dd, J1 = 8.4 Hz, J2 = 6.0 Hz, 1.8H, H7, H9, cis), 2.18 (qd, J1 = 13.3 Hz, J2 = 4.0 Hz, 0.2H, H7, H9, trans), 2.65 (tt, J1 = 12.6 Hz, J2 = 3.6 Hz, 0.1H, H8, trans), 2.81 (dt, J1 = 9.4 Hz, J2 = 4.9 Hz, 0.9H, H8, cis), 3.54 (q, J = 7.0 Hz, 2H, NCH2CH3), 4.07 (s, 0.2H, NCH2COOH, trans), 4.08 (s, 1.8H, NCH2COOH, cis), 7.19 (tt, J1 = 7.2 Hz, J2 = 1.6 Hz, 0.1H, H4′, trans), 7.21 (tt, J1 = 6.4 Hz, J2 = 2.1 Hz, 0.9H, H4′, cis), 7.24 (dd, J1 = 8.5 Hz, J2 = 1.6 Hz, 0.2H, H2′, H6′, trans), 7.31 (td, J1 = 7.5 Hz, J2 = 1.9 Hz, 0.2H, H3′, H5′, trans), 7.21–7.38 (m, 3.8H, H2′, H6′, cis, H3′, H5′, cis), 13.15 (br s, 1H, NCH2COOH); 13C NMR (150.9 MHz, [D6]DMSO): δ (ppm) = 14.5 (NCH2CH3), 27.6 (C7, C9, cis), 28.6 (C7, C9, trans), 31.0 (C6, C10, trans), 31.7 (C6, C10, cis), 36.4 (NCH2CH3), 39.1 (NCH2COOH), 39.4 (C8), 62.3 (C5), 120.0 (C4′), 126.5 (C2′, C6′, trans), 126.8 (C2′, C6′, cis), 128.4 (C3′, C5′), 145.2 (C1′), 154.1 (C2=O), 168.6 (NCH2COOH), 175.7 (C4=O); elemental analysis calcd (%) for C18H22N2O4: C 65.44, H 6.71, N 8.48, found: C 65.52, H 6.69, N 8.55.
2-(1-Benzyl-2,4-dioxo-8-phenyl-1,3-diazaspiro [4.5]decan-3-yl)acetic acid (23). It was prepared from benzyl ester 14 (280 mg, 0.58 mmol) by catalytic hydrogenolysis (H2/10% Pd-C, 34 mg) in a mixture of EtOH/AcOEt 3:1 (12 mL) following the general procedure previously described. The glass solid formed was crystallized upon treatment with n-pentane and some drops of Et2O under ice cooling to yield the carboxylic acid 23 as a white crystalline solid. (225 mg, almost quantitative yield); Rf = 0.10 (AcOEt); mp 129–131 °C (AcOEt/n-pentane); 1H NMR (600.11 MHz, [D6]DMSO): δ (ppm) = 1.62 (qd, J1 = 10.5 Hz, J2 = 5.6 Hz, 1.7H, H7, H9), 1.70 (d, J = 10.9 Hz, 0.8H, H6, H10), 1.75–1.91 (complex m, 4.7H, H6, H7, H9, H10), 1.94 (td, J1 = 13.2 Hz, J2 = 3.6 Hz, 0.4H, H6, H10), 2.14 (qd, J1 = 12.4 Hz, J2 = 2.6 Hz, 0.4H, H7, H9), 2.57 (t, J = 12.5 Hz, 0.2H, H8, cis), 2.67 (tt, J1 = 10.3, J2 = 3.8 Hz, 0.8H, H8, trans), 4.14 (s, 0.4H, NCH2COOH, cis), 4.17 (s, 1.6H, NCH2COOH, trans), 4.59 (s, 0.35H, NCH2Ph, cis), 4.81 (s, 1.6H, NCH2Ph, trans), 7.14–7.19 (m, 2H, H2′, H6′), 7.21 (td, J1 = 7.3 Hz, J2 = 1.3 Hz, 1H, H4′), 7.26 (tt, J1 = 7.2 Hz, J2 = 1.6 Hz, 1H, H4Bz), 7.27–7.31 (m, 4H, H2Bz, H3Bz, H5Bz, H6Bz 7.31–7.36 (m, 2H, H3′, H5′), 13.21 (v br s, 1H, NCH2COOH); 13C NMR (150.9 MHz, [D6]DMSO): δ (ppm) = 27.7 (C7, C9), 31.6 (C6, C10), 39.1 (NCH2COOH, cis), 39.4 (C8), 39.8 (NCH2COOH, trans), 41.2 (low) (NCH2Ph, cis), 44.2 (NCH2Ph, trans), 63.0 (C5), 126.0 (C4′), 126.5 (C2Bz, C6Bz), 126.8 (C2′, C6′), 128.3 (C3′, C5′, C4Bz), 128.5 (C3Bz, C5Bz), 138.2 (C1Bz), 145.3 (C1′), 155.6 (C2=O), 168.7 (NCH2COOH), 175.9 (C4=O); elemental analysis calcd (%) for C23H24N2O4: C 70.39, H 6.16, N 7.14, found: C 70.46, H 6.19, N 7.02.
2-(1-Benzyl-2,4-dioxo-6-phenyl-1,3-diazaspiro [4.5]decan-3-yl)acetic acid (24). A mixture of benzyl ester 15 (350 mg, 0.73 mmol) and 10% Pd on charcoal (42 mg) in EtOH/AcOEt 3:1 (14.5 mL) was hydrogenated following the general procedure for the preparation of carboxylic acids to yield the title compound 24 as a foamy solid. Crystallization upon treatment with n-pentane (0 °C) afforded a white crystalline solid (276 mg, almost quantitative yield). Mp 119–123 °C (AcOEt/n-pentane, Et2O), Rf = 0.29 (AcOEt). 1H NMR (600.11 MHz, DMSO-d6) δ (ppm): 1.26 (qt, 1H, J1 = 13.9 Hz, J2 = 3.9 Hz, H9), 1.41 (dt, 1H, J1 = 13.9 Hz, J2 = 3.8 Hz, H9), 1.49 (qt, 1H, J1 = 13.1 Hz, J2 = 4.0 Hz, H8), 1.69–1.78 (m, 2H, H7, H10), 1.88 (dt, 1H, J1 = 13.3 Hz, J2 = 3.5 Hz, H8), 1.95 (td, 1H, J1 = 14.3 Hz, J2 = 4.9 Hz, H10), 2.36 (qd, 1H, J1 = 13.7 Hz, J2 = 3.8 Hz, H7), 3.11 (dd, 1H, J1 = 14.0 Hz, J2 = 4.3 Hz, H6), 3.61 (brs, 1H, NCH2COOH, under DMSO-water peak), 3.61–3.90 (q, AB, 2H, JAB = 17.3 Hz, NCH2COOH), 4.91–5.10 (q, AB, 2H, JAB = 17.2 Hz, NCH2Ph), 7.13 (~dd, 2H, J1 = 7.0 Hz, J2 = 1.6 Hz, H2′, H6′), 7.20–7.35 (complex m, 8H, H3′, H4′, H5′, H2Bz, H3Bz, H4Bz, H5Bz, H6Bz); 13C NMR (50.32 ΜHz, DMSO-d6) δ (ppm): 20.0 (C9), 24.6 (C8), 26.9 (C7), 31.9 (C10), 39.5 (NCH2COOH), 45.7 (NCH2Ph), 49.3 (C6), 67.8 (C5), 126.1 (C2Bz, C6Bz), 126.9 (C4Bz), 127.2 (C4′), 128.1 (C2′, C3′, C5′, C6′), 128.3 (C3Bz, C5Bz), 137.9 (C1Bz), 139.1 (C1′), 155.3 (C2=O), 168.4 (NCH2COOH), 175.0 (C4=O). Anal. Calcd for C23H24N2O4: C, 70.39; H, 6.16; N, 7.14. Found: C, 70.32; H, 6.14; N, 7.09.
2-(2,4-Dioxo-8-phenyl-1,3-diazaspiro [4.5]decan-3-yl)propanoic acid (47). A mixture of benzyl ester 46 (900 mg, 2.21 mmol) and 10% Pd on charcoal (108 mg) in EtOH/AcOEt 3:1 (44 mL) was hydrogenated following the general procedure described above for the preparation of carboxylic acids to yield the desired compound 47 as a white crystalline solid. (695 mg, almost quantitative yield); Rf = 0.13 (AcOEt); mp 236–238 °C (MeOH/n-pentane); 1H NMR (600.11 MHz, [D6]DMSO): δ (ppm) = 1.46 (d, J = 7.3 Hz, 3H, NCH(CH3)COOH), 1.62 (d, J = 12.3 Hz, 1.8H, H6, H10), 1.67–1.80 (complex m, 3.8H, H7, H9), 1.84 (tq, J1 = 13.4 Hz, J1 = 4.4 Hz, 2H, H6, H10), 2.12 (qd, J1 = 13.7 Hz, J2 = 4.3 Hz, 0.2H, H7, H9), 2.58 (tt, J1 = 11.2 Hz, J2 = 4.2 Hz, 1H, H8), 4.55 (q, J = 7.4 Hz, 0.1H, NCH(CH3)COOH), 4.58 (q, J = 7.3 Hz, 0.8H, NCH(CH3)COOH), 7.18 (td, J1 = 7.1 Hz, J2 = 1.8 Hz, 1H, H4′), 7.26 (dd, J1 = 7.0 Hz, J2 = 1.5 Hz, 0.2H, H2′, H6′), 7.29 (t, J = 7.5 Hz, 2H, H3′, H5′), 7.33 (dd, J1 = 7.1 Hz, J2 = 1.5 Hz, 1.8H, H2′, H6′), 8.29 (s, 0.1H, H1), 9.06 (s, 0.9H, H1), 12.92 (v br s, 1H, NCH(CH3)COOH); 13C NMR (150.9 MHz, [D6]DMSO): δ (ppm) = 14.5 (NCH(CH3)COOH), 28.4, 28.7 (C7, C9), 33.3, 33.9 (C6, C10), 41.3, 42.2 (C8), 46.7, 47.0 (NCH(CH3)COOH), 58.4, 60.6 (C5), 126.0 (C4′), 126.7, 126.9 (C2′, C6′), 128.2, 128.3 (C3′, C5′), 146.2, 146.6 (C1′), 154.8, 155.1 (C2=O), 171.0 (NCH(CH3)COOH), 175.9, 176.3 (C4=O); elemental analysis calcd (%) for C17H20N2O4: C 64.54, H 6.37, N 8.86, found: C 64.50, H 6.48, N 8.88.
2-(1-Methyl-2,4-dioxo-8-phenyl-1,3-diazaspiro [4.5]decan-3-yl)propanoic acid (51). Benzyl ester 50 (800 mg, 1.90 mmol) in EtOH/AcOEt 3:1 (38 mL) was subjected to catalytic hydrogenolysis in the presence of 10% Pd on charcoal (96 mg) following the general procedure previously to yield the corresponding carboxylic acid 51 as a white crystalline solid. (625 mg, almost quantitative yield); Rf = 0.24 (AcOEt); mp 217–219 °C (AcOEt/n-pentane); 1H NMR (400.13 MHz, [D6]DMSO): δ (ppm) = 1.46 (d, J = 7.3 Hz, 3H, NCH(CH3)COOH), 1.68 (d, J = 13.0 Hz, 2H, H6, H10), 1.75 (d, J = 12.5 Hz, 2H, H7, H9), 2.00 (tt, J1 = 13.2 Hz, J2 = 4.0 Hz, 2H, H6, H10), 2.17 (qd, J1 = 12.9 Hz, J2 = 2.8 Hz, 2H, H7, H9), 2.60 (tt, J1 = 12.5 Hz, J2 = 3.8 Hz, 1H, H8), 2.81 (s, 3H, NCH3), 4.61 (q, J = 7.2 Hz, 1H, NCH(CH3)COOH), 7.19 (td, J1 = 7.2 Hz, J2 = 1.8 Hz, 1H, H4′), 7.24 (dd, J1 = 7.1 Hz, J2 = 1.7 Hz, 2H, H2′, H6′), 7.31 (t, J = 7.4 Hz, 2H, H3′, H5′), 13.03 (v br s, 1H, NCH(CH3)COOH); 13C NMR (150.9 MHz, [D6]DMSO): δ (ppm) = 14.5 (NCH(CH3)COOH), 23.6 (NCH3), 28.5, 28.6 (C7, C9), 29.9 (C6, C10), 41.4 (C8), 47.1 (NCH(CH3)COOH), 60.7 (C5), 126.1 (C4′), 126.6 (C2′, C6′), 128.4 (C3′, C5′), 146.3 (C1′), 154.1 (C2=O), 171.0 (NCH(CH3)COOH), 175.0 (C4=O); elemental analysis calcd (%) for C18H22N2O4: C 65.44, H 6.71, N 8.48, found: C 65.53, H 6.78, N 8.45.
General experimental procedure for the preparation of N-(phelylmethoxy)acetamides (25–33, 48, and 52). Method A: To a solution of the carboxylic acid (1.5 mmol, 1.0 eq) in a mixture of dry CH2Cl2/dry DMF 4:1 (15 mL), EDCI·HCl (1.8 mmol, 1.2 eq) and HOBt (1.8 mmol, 1.2 eq, monohydrate, 97%) were added, followed by O-benzylhydroxylamine hydrochloride (1.8 mmol, 1.2 eq) and TEA (8.7 mmol, 5.8 eq). After stirring for 40–45 h at 30–35 °C under argon, CH2Cl2 was evaporated under vacuum. The reaction mixture was quenched with water (30 mL) and extracted with AcOEt (4 × 30 mL). The combined organic phases were washed with H2O (3 × 50 mL) and brine (2 × 50 mL), dried over anh. Na2SO4, and concentrated under reduced pressure. The crude product was purified by column chromatography on silica gel to yield the corresponding acetamides or propanamides. Method B: The carboxylic acid (1.5 mmol, 1.0 eq) was dissolved in 20 mL dry THF, and to this solution, CDI (1.8 mmol, 1.2 eq) was added. After stirring for 1 h at 28 °C under argon, O-benzylhydroxylamine hydrochloride (1.8 mmol, 1.2 eq) and TEA (2.7 mmol, 1.8 eq) were added sequentially, and the stirring was continued for 24 h at 28 °C under argon and for 1 h at 40°C. Then, THF was evaporated under reduced pressure. The reaction mixture was poured into ice water (30 mL) and extracted with AcOEt (4 × 30 mL). The combined organic layers were washed with H2O (3 × 50 mL) and brine (2 × 50 mL), dried with anh. Na2SO4, and concentrated under vacuum. The crude residue was purified by column chromatography on silica gel to yield the title acetamides or propanamides.
N-(Phenylmethoxy)-2-((4R,4′S)/(4S,4′R)-4′-methyl-2,5-dioxo-3′,4′-dihydro-2′H- spiro[imidazolidine-4,1′-naphthalene]-1-yl)acetamide (25). To a stirred solution of carboxylic acid 16 (650 mg, 2.29 mmol) in 23 mL dry CH2Cl2/dry DMF (4:1), EDCI·HCl (527 mg, 2.75 mmol), HOBt (434 mg, 2.75 mmol, monohydrate, 97%), O-benzylhydroxylamine hydrochloride (439 mg, 2.75 mmol), and TEA (1.35 g, 13.3 mmol) were added successively, and the mixture was allowed to react for 41 h at 30 °C under argon. Following the general procedure described for the preparation of N-(phenylmethoxy)acetamides (Method A), the off-yellow oily residue was chromatographed on silica gel with CH2Cl2/AcOEt 50:1, 30:1, 3:1 και AcOEt as eluents. The white foamy product obtained was dried under high vacuum to yield 25 as a glass solid. (500 mg, 55%), Rf = 0.08 (CH2Cl2/AcOEt 8:1); 1H NMR (400.13 MHz, [D6]DMSO): δ (ppm) = 1.27, 1.30 (d +d, J = 7.0 Hz, J = 6.9 Hz, 3H, CH3), 1.62 (ddd, J1 = 13.9 Hz, J2 = 10.9 Hz, J3 = 5.2 Hz, 0.45H, H3′), 1.77–1.90 (m, 1H, H2′, H3′), 1.97–2.11 (m, 1.3H, H2′, H3′), 2.12–2.29 (m, 1H, H2′, H3′), 2.94 (td, J1 = 12.8 Hz, J2 = 6.1 Hz, 1H, H4′), 3.91–4.08 (2q, AB, 1.6H, J1AΒ = 16.1 Hz, J2AΒ = 16.2 Hz, NCH2CO), 4.27 (s, 0.35H, NCH2CO), 4.81, 4.87 (s + s, 2H, OCH2Ph), 7.21 (t, J = 7.6 Hz, 1H, H7′), 7.25–7.31 (m, 2H, H6′, H8′), 7.33 (d, J = 7.2 Hz, 1H, H5′), 7.35–7.53 (complex m, 5H, H2″, H3″, H4″, H5″, H6″), 8.94 (s, 1H, H3), 11.06 (s, 0.12H, CONHOCH2Ph), 11.44 (s, 0.72H, CONHOCH2Ph); 13C (50.32 MHz, [D6]DMSO): δ (ppm) = 21.9, 22.4 (CH3), 25.6, 26.5 (C3′), 30.0, 31.1 (C2′), 31.3, 31.7 (C4′), 38.0, 38.3 (NCH2CO), 62.6, 62.7 (C4), 77.0, 78.6 (low) (OCH2Ph), 126.4 (C7′), 127.4 (C8′), 127.9 (C5′), 128.4 (C6′, C3″, C4″, C5″), 128.8 (C2″, C6″), 133.6, 133.7 (C8′a), 135.8 (C1″), 142.6, 142.7 (C4′a), 155.3 (C2=O), 163.7 (CONHOCH2Ph), 176.1 (C5=O). HRMS/ESI+: m/z calcd for C22H23N3O4: 393.1689; found: 393.1691.
Ν-(Phenylmethoxy)-2-((4R,4′R)/(4S,4′S)-4′-methyl-2,5-dioxo-3′,4′-dihydro-2′H- spiro[imidazolidine-4,1′-naphthalene]-1-yl)acetamide (26). Using the general procedure described for the preparation of N-(benzyloxy)acetamides (Method A), the carboxylic acid precursor 17 (280 mg, 0.97 mmol) was treated successively with EDCI·HCl (222 mg, 1.16 mmol), HOBt (183 mg, 1.16 mmol, monohydrate, 97%), O-benzylhydroxylamine hydrochloride (185 mg, 1.16 mmol), and TEA (570 mg, 5.63 mmol) in dry CH2Cl2/dry DMF 4:1 (10 mL). The reaction mixture was then worked up following the general synthetic procedure previously described, and the yellowish oily residue was purified by column chromatography on silica gel eluting with CH2Cl2/AcOEt 50:1, 30:1, 10:1 and then AcOEt. The obtained foamy product was dried under high vacuum to afford 26 as a glass solid. (230 mg, 60%); Rf = 0.09 (CH2Cl2/AcOEt 8:1); 1H NMR (400.13 MHz, [D6]DMSO): δ (ppm) = 1.27, 1.30 (d + d, J = 7.0 Hz, J = 6.9 Hz, 3H, CH3), 1.57–1.68 (m, 0.55H, H3′), 1.78–1.90 (m, 0.8H, H2′, H3′), 1.95–2.11 (m, 1.5H, H2′, H3′), 2.12–2.29 (m, 1H, H2′, H3′), 2.94 (tt, J1 = 7.0 Hz, J2 = 6.9 Hz, 1H, H4′), 3.90–4.07 (2q, AB, J1AΒ = J2AΒ = 16.2 Hz, 1.5H, NCH2CO), 4.27 (s, 0.35H, NCH2CO), 4.81, 4.87 (s + s, 2H, OCH2Ph), 7.21 (t, J = 7.4 Hz, 1H, H7′), 7.25–7.35 (m, 3H, H5′, H6′, H8′), 7.36–7.52 (m, 5H, H2″, H3″, H4″, H5″, H6″), 8.94 (s, 1H, H3), 11.06 (s, 0.12H, CONHOCH2Ph), 11.44 (s, 0.75H, CONHOCH2Ph); 13C (50.32 MHz, [D6]DMSO): δ (ppm) = 21.9, 22.4 (CH3), 25.6, 26.5 (C3′), 30.0, 31.1 (C2′), 31.2, 31.7 (C4′), 38.0 (NCH2CO), 62.5, 62.7 (C4), 77.0, 78.5 (OCH2Ph), 126.4 (C7′), 127.4 (C8′), 127.9 (C5′), 128.4 (C6′, C3″, C4″, C5″), 128.8 (C2″, C6″), 133.6, 133.7 (C8′a), 135.8 (C1″), 142.6 (C4′a), 155.3 (C2=O), 163.7 (CONHOCH2Ph), 176.1 (C5=O). HRMS/ESI+: m/z calcd for C22H23N3O4: 393.1689; found: 393.1687.
N-(Phenylmethoxy)-2-(2,4-dioxo-8-phenyl-1,3-diazaspiro [4.5]decan-3-yl)acetamide (27). The N-benzyloxy precursor 27 was prepared from carboxylic acid 18 (280 mg, 0.93 mmol) in dry THF (12 mL) upon treatment with CDI (182 mg, 1.12 mmol), O-benzylhydroxylamine hydrochloride (177 mg, 1.12 mmol), and TEA (169 mg, 1.67 mmol) successively following the same procedure described for the preparation of O-benzyl hydroxamates (Method B). The crude glass solid was purified by column chromatography on silica gel eluting with CH2Cl2/AcOEt 20:1, 10:1, 4:1, and then AcOEt to yield the corresponding O-benzyl hydroxamate 27 as a white semisolid, which was crystallized under cooling (white crystals, 280 mg, 74%); Rf = 0.29 (CH2Cl2/AcOEt 8:1); mp 193–195 °C (AcOEt/n-pentane); 1H NMR (600.11 MHz, [D6]DMSO): δ (ppm) = 1.70 (d, J = 12.6 Hz, 2H, H6, H10), 1.73–1.83 (m, 3.8H, H7, H9, cis), 1.86 (td, J1 = 12.6 Hz, J2 = 4.3 Hz, 1.7H, H6, H10, cis), 1.96 (d, J = 13.5 Hz, 0.25H, H6, H10, trans), 2.14 (qd, J1 = 12.0 Hz, J2 = 3.4 Hz, 0.15H, H7, H9, trans), 2.60 (dt, J1 = 11.2 Hz, J2 = 4.1 Hz, 1H, H8), 3.89 (low), 3.92 (s + s, 1.55H, NCH2CO), 4.20 (s, 0.30H, NCH2CO), 4.80, 4.86 (low) (s + s, 2H, OCH2Ph), 7.19 (tt, J1 = 7.2 Hz, J2 = 1.6 Hz, 1H, H4′, cis), 7.27 (d, J = 7.5 Hz, 0.2H, H2′, H6′, trans), 7.31 (t, J = 7.5 Hz, 2H, H3′, H5′), 7.35 (d, J1 = 7.3 Hz, J2 = 1.6 Hz, 1.8H, H2′, H6′, cis), 7.35–7.50 (m, 5H, H2″, H3″, H4″, H5″, H6″), 8.28 (low) (s, 0.05H, H1, trans), 9.04 (s, 0.82H, H1, cis), 10.99 (low) (s, 0.1H, CONHOCH2Ph), 11.35 (s, 0.7H, CONHOCH2Ph); 13C NMR (150.9 MHz, [D6]DMSO): δ (ppm) = 28.4 (C7, C9, cis), 28.7 (low) (C7, C9, trans), 33.4 (C6, C10, cis), 34.0 (low) (C6, C10, trans), 37.6 (low), 37.9 (NCH2CO), 41.3 (low) (C8, trans), 42.2 (C8, cis), 61.0 (low) (C5, trans), 61.1 (C5, cis), 77.1, 78.6 (low) (OCH2Ph), 126.0 (C4′), 126.7 (low) (C2′, C6′, trans), 126.9 (C2′, C6′, cis), 128.2 (C3′, C5′), 128.3 (C3″, C4″, C5″), 128.9, 129.3 (low) (C2″, C6″), 135.8 (C1″), 146.2 (low) (C1′, trans), 146.6 (C1′, cis), 155.0 (low) (C2=O, trans), 155.4 (C2=O, cis), 163.6, 168.8 (low) (CONHOCH2Ph), 176.3 (low) (C4=O, trans), 176.7 (C4=O, cis); elemental analysis calcd (%) for C23H25N3O4: C 67.80, H 6.18, N 10.31, found: C 67.88, H 6.14, N 10.33.
N-(Phenylmethoxy)-2-(2,4-dioxo-6-phenyl-1,3-diazaspiro [4.5]decan-3-yl)acetamide (28). The N-benzyloxy analog 28 was prepared from carboxylic acid 19 (250 mg, 0.83 mmol) in a mixture of dry CH2Cl2/dry DMF (8.5 mL) upon treatment with EDCI·HCl (192 mg, 1.00 mmol), HOBt (139 mg, 1.00 mmol, monohydrate, 97%), O-benzylhydroxylamine hydrochloride (160 mg, 1.00 mmol), and TEA (486 mg, 4.8 mmol) following the amidation procedure described in the main manuscript (Method A). After removal of the CH2Cl2 under vacuum, the reaction mixture was poured into 17 mL ice water and extracted with AcOEt (4 × 17 mL). The combined organic phases were washed with H2O (3 × 28 mL) and brine (2 × 28 mL), dried with anh. Na2SO4, and evaporated under reduced pressure. The resulting almost-colorless viscous oil was chromatographed on silica gel using CH2Cl2/AcOEt 20:1, 10:1, 5:1, and finally, 100% AcOEt as eluents to afford the corresponding O-benzyl hydroxamate 28 as a glass solid. The product was treated with n-pentane under ice cooling to yield a white crystalline solid (158 mg, 47%). Mp 189–191 °C (AcOEt/n-pentane), Rf = 0.31 (CH2Cl2/AcOEt 6:1). 1H NMR (600.11 MHz, DMSO-d6) δ (ppm): 1.47 (qt, 1H, J1 = 13.2 Hz, J2 = 3.3 Hz, H8), 1.58 (tt, 1H, J1 = 14.1 Hz, J2 = 3.8 Hz, H9), 1.63 (dq, 1H, J1 = 12.6 Hz, J2 = 3.5 Hz, H7), 1.73 (d, 2H, J = 11.9 Hz, H9, H10), 1.81 (~dq, 1H, J1 = 12.3 Hz, J2 = 3.0 Hz, H8), 1.89 (td, 1H, J1 = 13.0 Hz, J2 = 3.7 Hz, H10), 2.02 (qd, 1H, J1 = 13.2 Hz, J2 = 3.6 Hz, H7), 2.97 (dd, 1H, J1 = 13.4 Hz, J2 = 3.5 Hz, H6), 3.31–3.49 (q, AB, 1.7H, JAB = 16.0 Hz, NCH2CONH, E/Z-isomer), 3.62–3.80 (low) (q, AB, 0.3H, JAB = 15.5 Hz, NCH2CONH, E/Z-isomer), 4.72, 4.74 (low) (s + brs, 2H, OCH2Ph, E/Z isomers), 7.09–7.13 (m, 2H, H3′, H5′), 7.16–7.25 (complex m, 3H, H2′, H4′, H6′), 7.30–7.41 (sym m, 5H, H2″, H3″, H4″, H5″, H6″), 8.93 (s, 1H, H1), 10.86 (low), 11.10 (brs + s, 2H, NCH2CONH, E/Z isomers); 13C NMR (100.61 MHz, DMSO-d6) δ (ppm): 20.6 (C9), 25.1 (C8), 26.9 (C7), 34.3 (C10), 37.3 (NCH2CONH), 47.4 (C6), 65.7 (C5), 77.0 (OCH2Ph), 126.9 (C4′), 127.7 (C3′, C5′), 128.3 (C2′, C6′, C3″, C4″, C5″), 128.8 (C2″, C6″), 135.7 (C1″), 139.6 (C1′), 155.3 (C2=O), 163.2 (NCH2CONH), 175.3 (C4=O). Anal. Calcd for C23H25N3O4: C, 67.80; H, 6.18; N, 10.31. Found: C, 67.88; H, 6.23; N, 10.33.
N-(Phenylmethoxy)-2-(1-methyl-2,4-dioxo-8-phenyl-1,3-diazaspiro [4.5]decan-3-yl)acetamide (29). Using the general procedure described for the preparation of N-(benzyloxy)acetamides (Method A), the carboxylic acid precursor 20 (420 mg, 1.33 mmol) was treated successively with EDCI·HCl (307 mg, 1.60 mmol), HOBt (253 mg, 1.60 mmol, monohydrate, 97%), O-benzylhydroxylamine hydrochloride (255 mg, 1.60 mmol), and TEA (780 mg, 7.71 mmol) in dry CH2Cl2/dry DMF 4:1 (13.3 mL). The reaction mixture was then worked up following the general synthetic procedure described above, and the yellowish oily residue was chromatographed on a silica gel column with CH2Cl2/AcOEt 20:1, 10:1, and then AcOEt, as eluents. The obtained glass solid was solidified upon treatment with n-pentane and Et2O to afford 260 mg of 29 as white crystals (46%); Rf = 0.27 (CH2Cl2/AcOEt 7:1); mp 153–155 °C (AcOEt/dry Et2O-n-pentane); 1H NMR (400.13 MHz, CDCl3): δ (ppm) = 1.77–1.99 (m, 6H, H6, H7, H9, H10), 2.27–2.44 (m, 2H, H7, H9), 2.52 (t, J = 12.3 Hz, 1H, H8), 2.89 (s, 3H, CH3), 4.09 (s, 0.8H, NCH2CO), 4.40 (s, 0.8H, NCH2CO), 4.92 (s, 2H, OCH2Ph), 7.21 (td, 1H, J1 = 6.5 Hz, J2 = 2.3 Hz, H4′), 7.26–7.33 (m, 4H, H2′, H3′, H5′, H6′), 7.33–7.45 (m, 5H, H2″, H3″, H4″, H5″, H6″), 8.22 (s, 0.35H, CONHOCH2Ph), 8.94 (s, 0.4H, CONHOCH2Ph); 13C NMR (150.9 MHz, CDCl3): δ (ppm) = 24.0 (CH3), 28.6, 28.9 (C7, C9), 31.1, 31.8 (C6, C10), 38.9, 39.2 (NCH2CO), 43.1 (C8), 62.3 (C5), 78.4, 79.8 (OCH2Ph), 126.5 (C4′), 126.9, 127.1 (C2′, C6′), 128.6, 128.7 (C3′, C5′, C3″, C4″, C5″), 129.4 (C2″, C6″), 134.3 (C1″), 146.1 (C1′), 155.1 (C2=O), 164.5 (CONHOCH2Ph), 175.6 (C4=O); elemental analysis calcd (%) for C24H27N3O4: C 68.39, H 6.46, N 9.97, found: C 68.44, H 6.48, N 10.09.
N-(Phenylmethoxy)-2-(1-methyl-2,4-dioxo-6-phenyl-1,3-diazaspiro [4.5]decan-3-yl)acetamide (30). Using the general procedure described for the preparation of N-(phenylmethoxy)acetamides (Method A), the carboxylic acid precursor 21 (160 mg, 0.51 mmol) was treated with EDCI·HCl (116 mg, 0.61 mmol), HOBt (85 mg, 0.61 mmol, monohydrate, 97%), O-benzylhydroxylamine hydrochloride (97 mg, 0.61 mmol), and TEA (297 mg, 2.93 mmol) in dry CH2Cl2/dry DMF 4:1 (5 mL). After removal of the CH2Cl2 under reduced pressure, the reaction mixture was quenched with 10 mL ice water and extracted with AcOEt (4 × 10 mL). The combined organic phases were washed with H2O (3 × 17 mL) and brine (2 × 17 mL), dried with anh. Na2SO4, and evaporated in vacuo. The resulting yellowish crude product was chromatographed on silica gel using CH2Cl2/AcOEt 20:1, 10:1, and AcOEt as eluents to afford the corresponding O-benzyl hydroxamate 30 as an off-white glass solid. The title compound was crystallized to a white crystalline solid after treatment with n-pentane under ice cooling (106 mg, 50%). Mp 83–85 °C (AcOEt/n-pentane), Rf = 0.25 (CH2Cl2/AcOEt 6:11H NMR (600.11 MHz, DMSO-d6) δ (ppm): 1.52–1.62 (sym m, 1H, H8), 1.75 (dq, 1H, J1 = 13.7 Hz, J2 = 3.7 Hz, H7), 1.80–1.88 (m, 2H, H9), 1.92–2.02 (m, 2H, H8, H10), 2.02–2.11 (m, 1H, H10), 2.20 (qd, 1H, J1 = 13.6 Hz, J2 = 3.6 Hz, H7), 3.09 (dd, 1H, J1 = 13.9 Hz, J2 = 4.1 Hz, H6), 3.23 (s, 3H, NCH3), 3.39–3.62 (q, AB, 2H, JAB = 16.0 Hz, NCH2CONH), 4.73, 4.77 (low) (s + brs, 2H, OCH2Ph, E/Z isomers), 7.05–7.09 (m, 2H, H2′, H6′), 7.18–7.22 (m, 1H, H4′), 7.22–7.26 (m, 2H, H3′, H5′), 7.31–7.42 (m, 5H, H2″, H3″, H4″, H5″, H6″), 10.89 (low), 11.16 (vbs + s, 1H, NCH2CONH, E/Z isomers); 13C NMR (100.61 MHz, DMSO-d6) δ (ppm): 21.2 (C9), 24.9 (C8), 26.9 (C7), 29.7 (NCH3), 32.1 (C10), 38.2 (NCH2CONH), 48.8 (C6), 66.8 (C5), 77.0 (OCH2Ph), 127.1 (C4′), 127.9 (C2′, C6′), 128.0 (C3′, C5′), 128.3 (C3″, C4″, C5″), 128.8 (C2″, C6″), 135.7 (C1″), 139.3 (C1′), 154.7 (C2=O), 163.0 (NCH2CONH), 175.0 (C4=O). Anal. Calcd for C24H27N3O4: C, 68.39; H, 6.46; N, 9.97. Found: C, 68.38; H, 6.51; N, 9.99.
N-(Phenylmethoxy)-2-(1-ethyl-2,4-dioxo-8-phenyl-1,3-diazaspiro [4.5]decan-3-yl)acetamide (31). Carboxylic acid 22 (290 mg, 0.88 mmol) was treated with CDI (172 mg, 1.06 mmol), O-benzylhydroxylamine hydrochloride (169 mg, 1.06 mmol), and TEA (160 mg, 1.58 mmol) in 12 mL dry THF as described for the synthesis of N-(phenylmethoxy)acetamides (Method B). The reaction was worked up as previously described, and the resulting oily residue was purified by column chromatography on silica gel using CH2Cl2/AcOEt 20:1, 5:1, and AcOEt as eluents to afford the title compound 31 as a white foamy solid, which strongly binds the elution solvents. Removal of the entrapped solvents yielded 31 as a glass solid. (250 mg, 65%); Rf = 0.12 (CH2Cl2/AcOEt 8:1); 1H NMR (400.13 MHz, [D6]DMSO): δ (ppm) = 1.16 (t, J = 7.0 Hz, 3H, NCH2CH3), 1.56 (qd, J1 = 6.6 Hz, J2 = 2.7 Hz, 0.2H, H7, H9), 1.77 (t, J = 12.5 Hz, 0.5H, H7, H9), 1.84–2.10 (complex m, 7H, H6, H7, H9, H10), 2.16 (dt, J1 = 13.2 Hz, J2 = 6.6 Hz, 0.2H, H7, H9), 2.31 (td, J1 = 7.5 Hz, J2 = 5.3 Hz, 0.1H, H6, H10), 2.64 (t, J = 11.5 Hz, 0.1H, H8, trans), 2.74–2.86 (m, 0.9H, H8, cis), 3.40 (qd, J1 = 6.4 Hz, J2 = 2.3 Hz, 0.2H, NCH2NCH3), 3.53 (q, J = 7.1 Hz, 1.8H, NCH2NCH3), 3.95 (s, 1.6H, NCH2CO), 4.22 (s, 0.4H, NCH2CO), 4.80, 4.86 (s + s, 2H, OCH2Ph), 7.21 (ddd, J1 = 8.7 Hz, J2 = 5.9 Hz, J3 = 2.5 Hz, 1H, H4′), 7.28–7.50 (m, 9H, H2′, H3′, H5′, H6′, H2″, H3″, H4″, H5″, H6″), 11.03 (s, 0.18H, CONHOCH2Ph), 11.40 (s, 0.82H, CONHOCH2Ph); 13C NMR (150.9 MHz, [D6]DMSO): δ (ppm) = 14.6, 15.0 (NCH2CH3), 25.8, 27.7, 28.7, 28.9, 29.4 (C7, C9), 31.1, 31.7 (C6, C10), 36.4 (NCH2CH3), 37.9, 38.4 (NCH2CO), 39.5 (C8), 62.2, 62.4 (C5), 77.1, 78.6 (OCH2Ph), 126.1 (C4′), 126.6, 126.9 (C2′, C6′), 128.3 (C3′, C5′), 128.4 (C3″, C4″, C5″), 128.9, 129.3 (C2″, C6″), 135.8 (C1″), 145.2, 146.4 (C1′), 154.1, 154.3 (C2=O), 163.5, 168.6 (CONHOCH2Ph), 175.3, 175.9 (C4=O).
N-(Phenylmethoxy)-2-(1-benzyl-2,4-dioxo-8-phenyl-1,3-diazaspiro [4.5]decan-3-yl)acetamide (32). The N-benzyloxy precursor 32 was prepared from carboxylic acid 23 (190 mg, 0.48 mmol) in dry CH2Cl2/dry DMF 4:1 (5 mL) upon treatment with EDCI·HCl (111 mg, 0.58 mmol), HOBt (92 mg, 0.58 mmol, monohydrate, 97%), O-benzylhydroxylamine hydrochloride (93 mg, 0.58 mmol), and TEA (281 mg, 2.78 mmol) sequentially, following the general procedure described for the preparation of N-(phenylmethoxy)acetamides (Method A). The yellowish oily residue was purified by column chromatography on silica gel with CH2Cl2/AcOEt 30:1, 20:1, and then AcOEt as eluents to afford the N-benzyloxy precursor 32 as a white foamy solid, which strongly binds the elution solvents. Removal of the entrapped solvents upon drying under high vacuum yielded 32 as a glass solid, which was solidified upon treatment with n-pentane (0 °C) to afford white crystals. (110 mg, 46%); Rf = 0.30 (CH2Cl2/AcOEt 8:1); mp 61–63 °C (AcOEt/n-pentane); 1H NMR (600.11 MHz, [D6]DMSO): δ (ppm) = 1.62 (qd, J1 = 10.3 Hz, J2 = 5.1 Hz, 1.8H, H7, H9, trans), 1.67–1.90 (complex m, 6H, H7, H9, H6, H10, trans), 1.93 (td, J1 = 13.8 Hz, J2 = 4.0 Hz, 0.6H, H6, H10, cis), 2.15 (qd, J1 = 12.7 Hz, J2 = 2.4 Hz, 0.4H, H7, H9, cis), 2.57 (tt, J1 = 12.4 Hz, J2 = 3.1 Hz, 0.1H, H8, cis), 2.67 (td, J1 = 8.8 Hz, J2 = 3.8 Hz, 0.9H, H8, trans), 4.00, 4.04 (s + s, 1.6H, NCH2CO), 4.31 (s, 0.35H, NCH2CO), 4.59 (s, 0.35H, NCH2Ph), 4.80 (s, 1.6H, NCH2Ph), 4.82, 4.88 (s + s, 2H, OCH2Ph), 7.15–7.50 (complex m, 15H, HAr), 11.05 (s, 0.15H, CONHOCH2Ph), 11.41 (s, 0.72H, CONHOCH2Ph); 13C NMR (100.61 MHz, [D6]DMSO): δ (ppm) = 27.8, 28.6 (C7, C9), 31.3, 31.7 (C6, C10), 38.7, 38.9 (NCH2CO), 39.1 (C8), 44.2 (NCH2Ph), 62.6, 63.0 (C5), 77.1, 78.6 (OCH2Ph), 126.0 (C4′), 126.5 (C2Bz, C6Bz), 126.8 (C2′, C6′) 127.0 (C2Bz, C6Bz), 127.2 (C4Bz), 128.4 (C3Bz, C5Bz, C3′, C5′, C4″), 128.5 (C3″, C5″),128.9, 129.3 (C2″, C6″), 135.8 (C1″), 138.3, 138.6 (C1Bz), 145.4, 146.3 (C1′), 155.1, 155.7 (C2=O), 163.5, 163.6 (CONHOCH2Ph), 175.2, 176.1 (C4=O); elemental analysis calcd (%) for C30H31N3O4: C 72.41, H 6.28, N 8.44, found: C 72.49, H 6.24, N 8.55.
N-(Phenylmethoxy)-2-(1-benzyl-2,4-dioxo-6-phenyl-1,3-diazaspiro [4.5]decan-3-yl)acetamide (33). Carboxylic acid 24 (235 mg, 0.60 mmol) was treated with CDI (117 mg, 0.72 mmol), O-benzylhydroxylamine hydrochloride (115 mg, 0.72 mmol), and TEA (109 mg, 1.08 mmol) in 8 mL dry THF as described for the synthesis of O-benzyl hydroxamates (Method B). The reaction was worked up in exactly the same way described in the main manuscript, and the resulting oily residue was purified by column chromatography on silica gel using CH2Cl2, CH2Cl2/AcOEt 20:1, 5:1, and AcOEt as eluents to yield the corresponding O-benzyl hydroxamate 33 as a colorless oily product. Crystallization upon treatment with n-pentane/Et2O afforded a white crystalline solid (131 mg, 44%). Mp 157–159 °C (AcOEt/n-pentane), Rf = 0.42 (CH2Cl2/AcOEt 8:1). 1H NMR (600.11 MHz, DMSO-d6) δ (ppm): 1.27 (qt, 1H, J1 = 17.6 Hz, J2 = 4.0 Hz, H9), 1.42 (d, 1H, J = 14.0 Hz, H9), 1.49 (qt, 1H, J1 = 13.7 Hz, J2 = 4.0 Hz, H8), 1.75 (td, 2H, J1 = 13.6 Hz, J2 = 6.7 Hz, H7, H10), 1.89 (d, 1H, J = 13.3 Hz, H8), 1.96 (td, 1H, J1 = 14.3 Hz, J2 = 4.7 Hz, H10), 2.35 (qd, 1H, J1 = 13.8 Hz, J2 = 3.7 Hz, H7), 3.09 (dd, 1H, J1 = 14.0 Hz, J2 = 4.3 Hz, H6), 3.40–3.74 (q, AB, 2H, JAB = 16.1 Hz, NCH2CONH), 4.76, 4.79 (low) (s + brs, 2H, OCH2Ph, E/Z isomers), 4.90–5.11 (q, AB, 2H, JAB = 17.3 Hz, NCH2Ph), 7.09–7.14 (m, 2H, H2′, H6′), 7.20–7.43 (complex m, 13H, H3′, H4′, H5′, H2″, H3″, H4″, H5″, H6″, H2Bz, H3Bz, H4Bz, H5Bz, H6Bz), 11.22 (vbs, 1H, NCH2CONH); 13C NMR (50.32 ΜHz, DMSO-d6) δ (ppm): 20.0 (C9), 24.6 (C8), 26.9 (C7), 31.7 (C10), 38.4 (NCH2CONH), 45.7 (ΝCH2Ph), 49.5 (C6), 67.7 (C5), 77.0 (OCH2Ph), 126.1 (C2Bz, C6Bz), 126.9 (C4Bz), 127.2 (C4′), 128.0 (C3′, C5′, C3Bz, C5Bz), 128.3 (C2′, C6′, C3″, C4″, C5″), 128.8 (C2″, C6″), 135.7 (C1″), 137.9 (C1Bz), 139.1 (C1′), 155.4 (C2=O), 163.0 (NCH2CONH), 175.1 (C4=O). Anal. Calcd for C30H31N3O4: C, 72.41; H, 6.28; N, 8.44. Found: C, 72.38; H, 6.24; N, 8.49.
N-(Phenylmethoxy)-2-(2,4-dioxo-8-phenyl-1,3-diazaspiro [4.5]decan-3-yl)propanamide (48). The N-benzyloxy precursor 48 was prepared from carboxylic acid 47 (600 mg, 1.90 mmol) in a mixture of dry CH2Cl2/dry DMF 4:1 (19 mL) upon treatment with EDCI·HCl (437 mg, 2.28 mmol), HOBt (360 mg, 2.28 mmol, monohydrate, 97%), O-benzylhydroxylamine hydrochloride (364 mg, 2.28 mmol), and TEA (1.12 g, 11.02 mmol) according to the procedure described for the preparation of acetamides (Method A). The crude colorless oily residue was chromatographed on silica gel with CH2Cl2/AcOEt 20:1, 7:1, and then AcOEt to afford the corresponding O-benzyl hydroxamate 48 as a glass solid, which was crystallized upon treatment with n-pentane under ice cooling. (240 mg, 30%); Rf = 0.21 (CH2Cl2/AcOEt 8:1); mp 157–159 °C (AcOEt/n-pentane); 1H NMR (600.11 MHz, [D6]DMSO): δ (ppm) = 1.45 (d, J = 7.2 Hz, 3H, NCH(CH3)CO), 1.59–1.90 (complex m, 8H, H6, H7, H9, H10), 2.55–2.63 (complex m, 1H, H8), 4.42 (q, J = 7.1 Hz, 0.1H, NCH(CH3)CO), 4.46 (q, J = 7.1 Hz, 0.8H, NCH(CH3)CO), 5.02–5.23 (q, AB, JAB = 10.8 Hz, 2H, OCH2Ph), 7.19 (tt, J1 = 7.2 Hz, J2 = 1.4 Hz, 1H, H4′), 7.25 (d, J = 7.4 Hz, 0.2H, H2′, H6′), 7.29 (t, J = 7.5 Hz, 2H, H3′, H5′), 7.33 (dd, J1 = 7.7 Hz, J2 = 1.2 Hz, 1.8H, H2′, H6′), 7.34–7.43 (complex m, 5H, H2″, H3″, H4″, H5″, H6″), 8.22 (s, 0.05H, H1), 8.98, 9.02 (s + s, 0.9H, H1), 11.27 (s, 0.85H, CONHOCH2Ph); 13C NMR (150.9 MHz, [D6]DMSO): δ (ppm) = 14.5 (NCH(CH3)CO), 28.3 (C7, C9), 33.2 (C6, C10), 42.2 (C8), 46.7 (NCH(CH3)CO), 60.4 (C5), 76.8 (OCH2Ph), 126.0 (C4′), 126.9 (C2′, C6′), 128.15 (C3′, C5′), 128.21 (C3″, C4″, C5″), 128.8 (C2″, C6″), 135.8 (C1″), 146.6 (C1′), 155.1 (C2=O), 166.0 (CONHOCH2Ph), 176.4 (C4=O); elemental analysis calcd (%) for C24H27N3O4: C 68.39, H 6.46, N 9.97, found: C 68.45, H 6.57, N 10.02.
N-(Phenylmethoxy)-2-(1-methyl-2,4-dioxo-8-phenyl-1,3-diazaspiro [4.5]decan-3-yl)propanamide (52). Prepared from carboxylic acid 51 (500 mg, 1.51 mmol) upon treatment with CDI (293 mg, 1.81 mmol), O-benzylhydroxylamine hydrochloride (289 mg, 1.81 mmol), and TEA (275 mg, 2.72 mmol) sequentially in dry THF (20 mL) following the general procedure for the preparation of O-benzyl hydroxamates (Method B). The colorless oil obtained was chromatographed on silica gel with CH2Cl2/AcOEt 15:1, 8:1, and then AcOEt to afford the title compound 52 as a glass solid. The product was crystallized upon treatment with n-pentane under ice cooling. (470 mg, 71%); Rf = 0.30 (CH2Cl2/AcOEt 8:1); mp 103–106 °C (AcOEt/n-pentane); 1H NMR (600.11 MHz, [D6]DMSO): δ (ppm) = 1.46 (d, J = 7.3 Hz, 3H, NCH(CH3)CO), 1.69–1.82 (m, 4H, H6, H7, H9, H10), 1.96 (tt, J1 = 13.2 Hz, J2 = 4.8 Hz, 2H, H6, H10), 2.17 (q, J = 12.9 Hz, 2H, H7, H9), 2.60 (t, J = 12.4 Hz, 1H, H8), 2.80 (s, 3H, NCH3), 4.50 (q, J = 7.1 Hz, 1H, NCH(CH3)CO), 4.71–4.83 (q, AB, JAB = 10.8 Hz, 2H, OCH2Ph), 7.19 (t, J = 7.3 Hz, 1H, H4′), 7.25 (d, J = 7.6 Hz, 2H, H2′, H6′), 7.31 (t, J = 7.5 Hz, 2H, H3′, H5′), 7.32–7.44 (complex m, 5H, H2″, H3″, H4″, H5″, H6″), 9.81 (s, 0.07H, CONHOCH2Ph), 10.79 (s, 0.03H, CONHOCH2Ph), 11.29 (s, 0.9H, CONHOCH2Ph); 13C NMR (150.9 MHz, [D6]DMSO): δ (ppm) = 14.4 (NCH(CH3)CO), 23.5 (NCH3), 28.5, 28.6 (C7, C9), 29.78, 29.85 (C6, C10), 41.5 (C8), 46.8 (NCH(CH3)CO), 60.6 (C5), 76.8, 77.3 (OCH2Ph), 126.0 (C4′), 126.6 (C2′, C6′), 128.1 (C3′, C5′), 128.2 (C3′, C5′, C4″), 128.3 (C3″, C5″), 128.6, 128.8 (C2″, C6″), 135.8 (C1″), 146.4 (C1′), 154.1 (C2=O), 166.0 (CONHOCH2Ph), 175.0 (C4=O); elemental analysis calcd (%) for C25H29N3O4: C 68.95, H 6.71, N 9.65, found: C 69.00, H 6.75, N 9.67.
General experimental procedure for the preparation of N-(hydroxy)acetamides (34–42, 49, and 53). A mixture of the N-benzyloxy precursor (1.0 mmol) and 10 wt.% Pd on charcoal in EtOH/AcOEt 3:1 (40 mL) was subjected to catalytic hydrogenolysis for 3 h under an atmosphere of 50–55 psi hydrogen at 44–46 °C. The catalyst was removed by filtration and washed with portions of hot MeOH (3 × 15 mL). The combined filtrates were concentrated to dryness under reduced pressure to obtain the desired acetohydroxamic acid analogs.
Ν-(hydroxy)-2-((4R,4′S)/(4S,4′R)-4′-methyl-2,5-dioxo-3′,4′-dihydro-2′H- spiro[imidazolidine-4,1′-naphthalene]-1-yl)acetamide (34). A mixture of O-benzyl hydroxamate 25 (400 mg, 1.02 mmol) and 10% Pd on charcoal (48 mg) in EtOH/AcOEt 3:1 (41 mL) was subjected to catalytic hydrogenolysis following the general procedure previously described to afford the title compound 34 as a glass solid. The product was chromatographed on silica gel eluting with CH2Cl2/AcOEt 5:1, AcOEt, and AcOEt/MeOH 20:1 to yield 34 as a white foamy product, which strongly binds the elution solvents. Removal of the entrapped solvents yielded 34 as a glass solid, which was crystallized upon treatment with dry Et2O to afford a white semisolid. (375 mg, almost quantitative yield); Rf = 0.38 (AcOEt); mp melted gradually from 95 °C (AcOEt/dry Et2O); 1H NMR (600.11 MHz, [D6]DMSO): δ (ppm) = 1.27, 1.30 (d + d, J = 7.1 Hz, J = 6.9 Hz, 3H, CH3), 1.62 (tdd, J1 = 13.2 Hz, J2 = 9.0 Hz, J3 = 3.0 Hz, 0.5H, H3′), 1.79–1.89 (m, 1H, H2′, H3′), 2.01 (ddd, J1 = 12.6 Hz, J2 = 7.9 Hz, J3 = 3.3 Hz, 0.5H, H3′), 2.05 (td, J1 = 10.5 Hz, J2 = 3.1 Hz, 1H, H2′), 2.16 (qd, J1 = 8.7 Hz, J2 = 3.4 Hz, 0.5H, H3′), 2.24 (~t, J = 11.2 Hz, 0.5H, H2′), 2.94 (dq, J1 = 13.4 Hz, J2 = 6.6 Hz, 1H, H4′), 3.91–4.03 (2q, AB, J1A = J2AB = 16.0 Hz, 1.5H, NCH2CO, E-isomer), 4.20–4.32 (2q, AB, J1AB = J2AB = 17.2 Hz, 0.5H, NCH2CO, Z-isomer), 7.18 (t, J = 7.9 Hz, 1H, H7′), 7.24–7.41 (complex m, 3H, H5′, H6′, H8′), 8.86 (s, 1H, H3), 8.96 (s, 0.68H, NCH2CONHOH, E-isomer), 9.34 (s, 0.2H, NCH2CONHOH, Z-isomer), 10.28 (s, 0.2H, NCH2CONHOH, Z-isomer), 10.72 (s, 0.66H, NCH2CONHOH, E-isomer); 13C NMR (150.9 MHz, [D6]DMSO): δ (ppm) = 21.9, 22.3 (CH3), 25.6, 26.5 (C3′), 29.9, 31.1 (C2′), 31.2, 31.6 (C4′), 37.9 (NCH2CO, E-isomer), 38.5 (NCH2CO, Z-isomer), 62.4, 62.6 (C4), 126.3 (C7′), 127.4, 127.8 (C8′), 128.2, 128.4 (C5′, C6′), 133.6, 133.7 (C8′a), 142.5, 142.6 (C4′a), 155.3 (C2=O, E-isomer), 155.5 (C2=O, Z-isomer), 163.4 (NCH2CONHOH, E-isomer), 168.8 (NCH2CONHOH, Z-isomer), 176.1 (C5=O, E-isomer), 176.3 (C5=O, Z-isomer); elemental analysis calcd (%) for C15H17N3O4: C 59.40, H 5.65, N 13.85; found: C 59.45, H 5.61, N 13.93.
Ν-(Hydroxy)-2-((4R,4′R)/(4S,4′S)-4′-methyl-2,5-dioxo-3′,4′-dihydro-2′H- spiro[imidazolidine-4,1′-naphthalene]-1-yl)acetamide (35). A solution of the N-phenylmethoxy acetamide 26 (180 mg, 0.46 mmol) in a mixture of EtOH/AcOEt (3:1, 18 mL) was subjected to hydrogenolysis over Pd/C (22 mg) according to the procedure described for the preparation of hydroxamate analogs. Evaporation of the solvents in vacuo yielded the title compound 35 as a white foamy product, which strongly binds the solvents. Removal of the entrapped solvents under high vacuum afforded 35 as a glass solid. Crystallization upon treatment with n-pentane and dry Et2O yielded 35 as a white semisolid. (139 mg, almost quantitative yield); Rf = 0.40 (AcOEt); mp melted gradually from 83 °C (AcOEt/dry Et2O-n-pentane); 1H NMR (400.13 MHz, [D6]DMSO): δ (ppm) = 1.26, 1.29 (d + d, J = 7.0 Hz, J = 6.9 Hz, 3H, CH3), 1.56–1.67 (m, 0.55H, H3′), 1.77–1.89 (m, 0.8H, H2′, H3′), 2.03 (td, J1 = 10.1 Hz, J2 = 2.8 Hz, 1.6H, H2′, H3′), 2.11–2.29 (m, 1H, H2′, H3′), 2.86–3.00 (dt, J1 = 13.5 Hz, J2 = 7.0 Hz, 1H, H4′), 3.89–4.08 (2q, AB, J1AΒ = 16.1 Hz, J2AΒ = 16.5 Hz, 1.5H, NCH2CO, E-isomer), 4.19–4.32 (2q, AB, J1AΒ = 17.5 Hz, J2AΒ = 17.2 Hz, 0.5H, NCH2CO, Z-isomer), 7.18 (t, J = 7.7 Hz, 1H, H7′), 7.23–7.42 (complex m, 3H, H5′, H6′, H8′), 8.86, 8.91 (s + s, 1H, H3), 9.02 (s, 0.6H, NCH2CONHOH, E-isomer), 9.39 (s, 0.2H, NCH2CONHOH, Z-isomer), 10.34 (s, 0.2H, NCH2CONHOH, Z-isomer), 10.76 (s, 0.6H, NCH2CONHOH, E-isomer); 13C NMR (50.32 MHz, [D6]DMSO): δ (ppm) = 21.9, 22.4 (CH3), 25.6, 26.5 (C3′), 29.9 (C2′), 31.2, 31.7 (C4′), 38.3 (NCH2CO, E-isomer), 39.9 (NCH2CO, Z-isomer), 62.5, 62.7 (C4), 126.4 (C7′), 127.5, 127.8 (C6′, C8′), 128.3, 128.5 (C5′), 133.7, 133.8 (C8′a), 142.6 (C4′a), 155.4 (C2=O), 163.4 (NCH2CONHOH, E-isomer), 168.9 (NCH2CONHOH, Z-isomer), 176.2 (C5=O); elemental analysis calcd (%) for C15H17N3O4: C 59.40, H 5.65, N 13.85; found: C 59.45, H 5.60, N 13.89.
N-Hydroxy-2-(2,4-dioxo-8-phenyl-1,3-diazaspiro [4.5]decan-3-yl) acetamide (36). A solution of the N-phenylmethoxy acetamide 27 (220 mg, 0.54 mmol) in a mixture of EtOH/AcOEt (3:1, 22 mL) was subjected to hydrogenolysis in the presence of Pd/C (26 mg) according to the procedure described for the preparation of hydroxamate analogs. Evaporation of the solvents in vacuo yielded the title compound 36 as a white crystalline solid. (170 mg, almost quantitative yield); Rf = 0.62 (AcOEt); mp 217–219 °C (MeOH/dry Et2O-n-pentane); 1H NMR (600.11 MHz, [D6]DMSO): δ (ppm) = 1.68 (d, J = 11.3 Hz, 2H, H6, H10), 1.71–1.83 (complex m, 3.9H, H7, H9, cis), 1.85 (td, J1 = 12.3 Hz, J2 = 4.0 Hz, 1.9H, H6, H10), 1.95 (dt, J1 = 13.9 Hz, J2 = 4.6 Hz, 0.1H, H6, H10, trans), 2.10 (qd, J1 = 13.1 Hz, J2 = 3.9 Hz, 0.1H, H7, H9, trans), 2.58 (tt, J1 = 11.2 Hz, J2 = 4.1 Hz, 1H, H8), 3.86 (low), 3.89 (s + s, 1.5H, NCH2CO, E-isomer), 4.16 (low), 4.19 (s + s, 0.45H, NCH2CO, Z-isomer), 7.18 (tt, J1 = 7.2 Hz, J2 = 1.4 Hz, 1H, H4′), 7.25 (dd, J1 = 7.0 Hz, J2 = 1.6 Hz, 0.2H, H2′, H6′, trans), 7.29 (td J1 = 7.6 Hz, J2 = 1.6 Hz, 2H, H3′, H5′), 7.33 (dd, J1 = 7.2 Hz, J2 = 1.6 Hz, 1.8H, H2′, H6′, cis), 8.22, 8.24 (s + s, 0.05H, H1, trans), 8.91 (s, 0.6H, NCH2CONHOH, E-isomer), 8.99, 9.00 (s + s, 0.95H, H1, cis), 9.31 (s, 0.2H, NCH2CONHOH, Z-isomer), 10.25 (s, 0.2H, NCH2CONHOH, Z-isomer), 10.67 (s, 0.6H, NCH2CONHOH, E-isomer); 13C NMR (150.9 MHz, [D6]DMSO): δ (ppm) = 28.4 (C7, C9, cis), 28.7 (low) (C7, C9, trans), 33.3 (C6, C10, cis), 33.9 (low) (C6, C10, trans), 37.9 (NCH2CO, E-isomer), 38.2 (NCH2CO, Z-isomer), 41.3 (low) (C8, trans), 42.2 (C8, cis), 58.8 (low) (C5, trans), 60.9 (C5, cis), 126.0 (C4′), 126.6 (low) (C2′, C6′, trans), 126.9 (C2′, C6′, cis), 128.2 (C3′, C5′, cis), 128.3 (low) (C3′, C5′, trans), 146.2 (low) (C1′, trans), 146.6 (C1′, cis), 155.4 (C2=O, E-isomer), 155.6 (low) (C2=O, Z-isomer), 163.3 (NCH2CONHOH, E-isomer), 168.8 (NCH2CONHOH, Z-isomer), 176.7 (C4=O, E-isomer), 177.0 (low) (C4=O, Z-isomer); elemental analysis calcd (%) for C16H19N3O4: C 60.56, H 6.04, N 13.24, found: C 60.60, H 6.09, N 13.32.
N-Hydroxy-2-(2,4-dioxo-6-phenyl-1,3-diazaspiro [4.5]decan-3-yl) acetamide (37). The N-benzyloxy precursor 28 (120 mg, 0.29 mmol) was subjected to catalytic hydrogenation in a mixture of EtOH/AcOEt 3:1 (12 mL) according to the procedure described in the main manuscript for the synthesis of hydroxamate analogs. Concentration to dryness under reduced pressure afforded the title compound 37 as a white solid (90 mg, almost quantitative yield). Mp 196–199 °C (AcOEt/n-pentane), Rf = 0.28 (AcOEt). This compound appeared in the 1H and 13C NMR spectra as a mixture of E/Z conformers. 1H NMR (600.11 MHz, DMSO-d6) δ (ppm): 1.46 (qt, 1H, J1 = 12.7 Hz, J2 = 3.2 Hz, H8), 1.56 (tt, 1H, J1 = 14.8 Hz, J2 = 3.9 Hz, H9), 1.62 (dq, 1H, J1 = 17.6 Hz, J2 = 4.0 Hz, H7), 1.67–1.76 (m, 2H, H9, H10), 1.80 (d, 1H, J = 12.3 Hz, H8), 1.86 (td, 1H, J1 = 13.6 Hz, J2 = 4.2 Hz, H10), 2.01 (qd, 1H, J1 = 13.1 Hz, J2 = 3.6 Hz, H7), 2.96 (dd, 1H, J1 = 13.4 Hz, J2 = 3.6 Hz, H6), 3.29–3.43 (q, AB, 1.6H, JAB = 15.9 Hz, NCH2CO, E-isomer), 3.59–3.75 (q, AB, 0.4H, JAB = 17.2 Hz, NCH2CO, Z-isomer), 7.08–7.13 (m, 2H, H2′, H6′), 7.16–7.24 (complex m, 3H, H3′, H4′, H5′), 8.86 (s, 0.6H, CH2CONHOH, E-isomer), 8.89 (s, 0.3H, H1, Z-isomer), 8.92 (s, 0.7H, H1, E-isomer), 9.16 (s, 0.2H, CH2CONHOH, Z-isomer), 10.16 (s, 0.2H, CH2CONHOH, Z-isomer), 10.43 (s, 0.6H, CH2CONHOH, E-isomer); 13C NMR (150.9 MHz, DMSO-d6) δ (ppm): 20.6 (C9), 25.1 (C8), 26.9 (C7), 34.2 (C10, E-isomer), 34.4 (C10, Z-isomer), 37.3 (NCH2CO, E-isomer), 37.5 (NCH2CO, Z-isomer), 47.5 (C6), 65.6 (C5), 127.0 (C4′), 127.7 (C3′, C5′), 128.3 (C2′, C6′), 139.6 (C1′), 155.4 (C2=O, E-isomer), 155.6 (C2=O, Z-isomer), 163.0 (NCH2CO, E-isomer), 168.5 (NCH2CO, Z-isomer), 175.4 (C4=O, E-isomer), 175.6 (C4=O, Z-isomer). Anal. Calcd for C16H19N3O4: C, 60.56; H, 6.04; N, 13.24. Found: C, 60.58; H, 6.08; N, 13.25.
N-Hydroxy-2-(1-methyl-2,4-dioxo-8-phenyl-1,3-diazaspiro [4.5]decan-3-yl)acetamide (38). A mixture of the O-benzyl hydroxamate 29 (200 mg, 0.47 mmol) and 10% Pd on charcoal (24 mg) in EtOH/AcOEt 3:1 (18.8 mL) was hydrogenated following the general procedure for the preparation of acetohydroxamic acids previously described to afford the title compound 38 as a white crystalline solid. (155 mg, almost quantitative yield); Rf = 0.50 (AcOEt); mp 197–200 °C (MeOH/dry Et2O-n-pentane); 1H NMR (400.13 MHz, [D6]DMSO): δ (ppm) = 1.75 (d, J = 9.9 Hz, 4H, H6, H7, H9, H10), 2.00 (dt, J1 = 12.7 Hz, J2 = 6.7 Hz, 2H, H6, H10), 2.16 (qd, J1 = 13.2 Hz, J2 = 3.8 Hz, 2H, H7, H9), 2.61 (t, J = 12.1 Hz, 1H, H8), 2.81 (s, 3H, NCH3), 3.91, 3.94 (s + s, 1.5H, NCH2CO, E-isomer), 4.20 (s, 0.45H, NCH2CO, Z-isomer), 7.19 (t, J = 7.1 Hz, 1H, H4′), 7.24 (d, J = 7.5 Hz, 2H, H2′, H6′), 7.31 (t, J = 7.5 Hz, 2H, H3′, H5′), 8.96 (s, 0.69H, ΝCH2CONHOH, E-isomer), 9.35 (s, 0.19H, ΝCH2CONHOH, Z-isomer), 10.31 (s, 0.19H, ΝCH2CONHOH, Z-isomer), 10.72 (s, 0.69H, ΝCH2CONHOH, E-isomer); 13C NMR (150.9 MHz, [D6]DMSO): δ (ppm) = 23.6 (CH3), 28.6 (C7, C9), 30.0 (C6, C10), 38.1 (NCH2CO, E-isomer), 38.3 (NCH2CO, Z-isomer), 41.4 (C8), 61.2 (C5), 126.1 (C4′), 126.6 (C2′, C6′), 128.4 (C3′, C5′), 146.4 (C1′), 154.4 (C2=O, E-isomer), 154.6 (C2=O, Z-isomer), 163.3 (NCH2CONHOH, E-isomer), 168.7 (NCH2CONHOH, Z-isomer), 175.4 (C4=O, E-isomer), 175.7 (C4=O, Z-isomer); elemental analysis calcd (%) for C17H21N3O4: C 61.62, H 6.39, N 12.68, found: C 61.64, H 6.47, N 12.64.
N-Hydroxy-2-(1-methyl-2,4-dioxo-6-phenyl-1,3-diazaspiro [4.5]decan-3-yl)acetamide (39). A solution of the O-benzyl hydroxamate 30 (85 mg, 0.20 mmol) in EtOH/AcOEt 3:1 (8 mL) was hydrogenated as described in the main manuscript. Evaporation of the solvents in vacuo yielded the title compound 39 as a glass solid, which is crystallized with n-pentane/Et2O at 0 °C to a white solid (62 mg, almost quantitative yield). Mp 195–197 °C (AcOEt, EtOH/n-pentane), Rf = 0.49 (AcOEt). This compound exhibited distinct peaks attributed to each of the two E/Z conformers in the 1H and 13C NMR spectra. 1H NMR (600.11 MHz, MeOD) δ (ppm): 1.66 (1H, J1 = 12.9 Hz, J2 = 4.3 Hz, H8), 1. 85 (dq, 1H, J1 = 13.5 Hz, J2 = 3.6 Hz, H7), 1.89–2.00 (complex m, 2H, H9), 2.07–2.15 (m, 2H, H8, H10), 2.18 (dd, 1H, J1 = 13.3 Hz, J2 = 6.4 Hz, H10), 2.24 (qd, 1H, J1 = 13.6 Hz, J2 = 3.6 Hz), 3.16 (dd, 1H, J = 13.9, 4.1 Hz), 3.34 (s, 3H, NCH3), 3.50–3.74 (q, AB, 1.6H, JAB = 16.0 Hz, NCH2CO, E/Z-isomer), 3.85–4.10 (q, AB, 0.4H, JAB = 17.4 Hz, NCH2CO, E/Z-isomer), 4.83 (s, 2H, CH2COΝHOH, under MeOD-water peak), 7.08–7.14 (m, 2H, H2′, H6′), 7.18–7.27 (m, 3H, H3′, H4′, H5′); 13C NMR (150.9 MHz, MeOD) δ (ppm): 22.8 (C9), 26.5 (C8), 28.7 (C7), 30.8 (NCH3), 33.0 (C10, E/Z-isomer), 33.3 (C10, E/Z-isomer), 39.5 (NCH2CO, E/Z-isomer), 39.6 (NCH2CO, E/Z-isomer), 51.1 (C6, E/Z-isomer), 51.2 (C6, E/Z-isomer), 69.2 (C5), 128.5 (C4′), 129.2 (C2′, C6′), 129.3 (C3′, C5′), 140.6 (C1′), 157.2 (C2=O), 165.8 (NCH2CO, E/Z-isomer), 169.2 (NCH2CO, E/Z-isomer), 177.3 (C4=O). Anal. Calcd for C17H21N3O4: C, 61.62; H, 6.39; N, 12.68. Found: C, 61.67; H, 6.43; N, 12.66.
N-Hydroxy-2-(1-ethyl-2,4-dioxo-8-phenyl-1,3-diazaspiro [4.5]decan-3-yl)acetamide (40). A total of 10 wt% Pd (23 mg) on charcoal was added to a solution of the O-benzyl hydroxamate 31 (190 mg, 0.44 mmol) in a mixture of EtOH/AcOEt 3:1 (17.6 mL), and the mixture was hydrogenated following the procedure described for the synthesis of hydroxamate analogs. The white foamy solid obtained strongly binds the aforementioned solvents. Removal of the entrapped solvents upon drying under high vacuum yielded the target compound 40 as a glass solid, which was crystallized upon treatment under ice cooling (150 mg, almost quantitative yield); Rf = 0.25 (AcOEt); mp 147–150 °C (AcOEt/dry Et2O-n-pentane); 1H NMR (600.11 MHz, [D6]DMSO): δ (ppm) = 1.16 (t, J = 7.1 Hz, 3H, NCH2CH3), 1.75 (dq, J1 = 7.8 Hz, J2 = 3.8 Hz, 0.4H, H6, H10, trans), 1.81–1.98 (m, 5.6H, H6, H10, cis, H7, H9), 2.04 (dd, J1 = 8.6 Hz, J2 = 4.0 Hz, 1.8H, H7, H9, cis), 2.18 (qd, J1 = 13.1 Hz, J2 = 2.5 Hz, 0.2H, H7, H9, trans), 2.65 (tt, J1 = 12.6 Hz, J2 = 3.6 Hz, 0.1H, H8, trans), 2.81 (dt, J1 = 9.4 Hz, J2 = 4.9 Hz, 0.9H, H8, cis), 3.53 (q, J = 6.9 Hz, 2H, NCH2CH3), 3.91, 3.95 (s + s, 1.55H, NCH2CO, E-isomer), 4.21 (s, 0.45H, NCH2CO, Z-isomer), 7.19 (tt, J1 = 6.8 Hz, J2 = 1.4 Hz, 0.1H, H4′, trans), 7.21 (tt, J1 = 6.5 Hz, J2 = 2.0 Hz, 0.9H, H4′, cis), 7.24 (dd, J1 = 8.1 Hz, J2 = 1.5 Hz, 0.2H, H2′, H6′, trans), 7.31 (td, J1 = 7.5 Hz, J2 = 1.9 Hz, 0.2H, H3′, H5′, trans), 7.31–7.37 (m, 3.8H, H2′, H6′, cis, H3′, H5′, cis), 8.93 (s, 0.6H, ΝCH2CONHOH, E-isomer), 9.31 (s, 0.2H, ΝCH2CONHOH, Z-isomer), 10.26 (s, 0.2H, ΝCH2CONHOH, Z-isomer), 10.66 (br s, 0.4H, ΝCH2CONHOH, E-isomer); 13C NMR (150.9 MHz, [D6]DMSO): δ (ppm) = 14.6 (NCH2CH3), 27.6 (C7, C9, cis), 28.6 (C7, C9, trans), 31.6 (C6, C10, cis), 31.7 (C6, C10, trans), 36.3 (NCH2CH3), 38.4 (NCH2CO, E-isomer), 38.7 (NCH2CO, Z-isomer), 39.4 (C8), 62.3 (C5), 126.0 (C4′), 126.6 (C2′, C6′, trans), 126.8 (C2′, C6′, cis), 128.4 (C3′, C5′), 145.2 (C1′), 154.3 (C2=O, E-isomer), 154.5 (C2=O, Z-isomer), 163.1 (NCH2CONHOH, E-isomer), 168.6 (NCH2CONHOH, Z-isomer), 175.9 (C4=O, E-isomer), 176.2 (C4=O, Z-isomer); elemental analysis calcd (%) for C18H23N3O4: C 62.59, H 6.71, N 12.17, found: C 62.54, H 6.78, N 12.23.
N-Hydroxy-2-(1-benzyl-2,4-dioxo-8-phenyl-1,3-diazaspiro [4.5]decan-3-yl)acetamide (41). A mixture of the O-benzyl hydroxamate 32 (85 mg, 0.17 mmol) and 10% Pd on charcoal (10 mg) in EtOH/AcOEt 3:1 (7 mL) was hydrogenated following the general procedure for the synthesis of acetohydroxamic acids previously described to afford the title compound 41 a white foamy solid, which strongly binds the aforementioned solvents. Removal of the entrapped solvents upon drying under high vacuum yielded the desired compound 41 as a glass solid, which was crystallized upon treatment with n-pentane under ice cooling. (69 mg, almost quantitative yield); Rf = 0.48 (AcOEt); mp 105–107 °C (AcOEt/n-pentane); 1H NMR (400.13 MHz, [D6]Acetone): δ (ppm) = 1.60–2.07 (complex m, 7.3H, H6, H7, H9, H10), 2.25 (qd, J1 = 13.8 Hz, J2 = 3.6 Hz, 0.3H, H7, H9, cis), 2.50 (td, J1 = 11.8 Hz, J2 = 6.1 Hz, 0.15H, H8, cis), 2.66 (td, J1 = 10.0 Hz, J2 = 4.0 Hz, 0.7H, H8, trans), 4.11, 4.17 (s + s, 1.2H, NCH2CO, E-isomer), 4.42 (s, 0.55H, NCH2CO, Z-isomer), 4.59 (s, 0.35H, NCH2Ph), 4.82 (s, 1.5H, NCH2Ph), 7.06–7.40 (complex m, 10H, HAr), 8.18 (s, 0.25H, ΝCH2CONHOH, E-isomer), 9.68 (s, 0.12H, ΝCH2CONHOH, Z-isomer), 9.50 (s, 0.15H, ΝCH2CONHOH, Z-isomer), 10.15 (s, 0.25H, ΝCH2CONHOH, E-isomer); 13C NMR (50.32 MHz, [D6]Acetone): δ (ppm) = 29.1 (C7, C9), 32.7, 32.8 (C6, C10), 39.7 (NCH2CO, E-isomer), 40.0 (NCH2CO, Z-isomer), 41.0 (C8, trans), 42.4 (NCH2Ph), 43.2 (C8, cis), 45.3 (NCH2Ph), 63.9, 64.4 (C5), 126.9 (C4′), 127.6 (C2′, C6′), 127.8 (C2′, C6′, C2Bz, C6Bz), 128.0 (C4Bz), 128.1 (C2Bz, C6Bz), 129.2 (C3′, C5′), 129.4 (C3Bz, C5Bz), 139.7, 139.9 (C1Bz), 146.4, 147.5 (C1′), 157.1 (C2=O), 164.8 (NCH2CONHOH, E-isomer), 170.7 (NCH2CONHOH, Z-isomer), 177.1 (C4=O); elemental analysis calcd (%) for C23H25N3O4: C 67.80, H 6.18; N 10.31, found: C 67.85, H 6.15; N 10.33.
N-Hydroxy-2-(1-benzyl-2,4-dioxo-6-phenyl-1,3-diazaspiro [4.5] decan-3-yl)acetamide (42). A solution of the O-benzyl hydroxamate 33 (100 mg, 0.20 mmol) in EtOH/AcOEt 3:1 (8 mL) was hydrogenated in the presence of Pd/C (12 mg) as described for the preparation of N-(hydroxy)acetamides. Concentration to dryness yielded acetohydroxamic acid 42 as a colorless glass solid. Treatment with n-pentane/Et2O at 0 °C afforded a white crystalline solid (78 mg, almost quantitative yield). Mp 110–111 °C (AcOEt/n-pentane), Rf = 0.64 (AcOEt). Ιn the 1H and 13C NMR spectra of this compound, a double set of characteristic peaks are distinguished for each of the two E/Z conformers. 1H NMR (600.11 MHz, DMSO-d6) δ (ppm): 1.26 (qt, 1H, J1 = 14.1 Hz, J2 = 3.2 Hz, H9), 1.36–1.44 (m, 1H, H9), 1.49 (qt, 1H, J1 = 13.1 Hz, J2 = 3.9 Hz, H8), 1.69–1.79 (m, 2H, H7, H10), 1.85–1.91 (m, 1H, H8), 1.94 (td, 1H, J1 = 14.2 Hz, J2 = 4.7 Hz, H10), 2.37 (qd, 1H, J1 = 12.5 Hz, J2 = 3.8 Hz, H7), 3.09 (dd, 1H, J1 = 14.1 Hz, J2 = 4.2 Hz, H6), 3.34 (brs, 2H, CH2COΝHOH, under DMSO-water peak), 3.41–3.71 (q, AB, 1.6H, JAB = 15.9 Hz, NCH2CO, E-isomer), 3.71–4.00 (q, AB, 0.4H, JAB = 17.2 Hz, NCH2CO, Z-isomer), 4.92–5.11 (q, AB, 2H, JAB = 17.3 Hz, NCH2Ph), 7.08–7.18 (m, 2H, H2′, H6′), 7.19–7.43 (complex m, 8H, H3′, H4′, H5′, H2Bz, H3Bz, H4Bz, H5Bz, H6Bz); 13C NMR (100.61 ΜHz, DMSO-d6) δ (ppm): 20.1 (C9), 24.7 (C8), 26.9 (C7), 31.7 (C10, E-isomer), 31.9 (C10, Z-isomer), 38.1 (NCH2CO), 45.7 (NCH2Ph), 49.6 (C6), 67.7 (C5), 126.2 (C2Bz, C6Bz), 126.9 (C4Bz), 127.3 (C4′), 128.1 (C2′, C6′, C3Bz, C5Bz), 128.3 (C3′, C5′), 138.0 (C1Bz), 139.2 (C1′), 155.5 (C2=O, E-isomer), 155.7 (C2=O, Z-isomer), 162.8 (NCH2CO, E-isomer), 168.2 (NCH2CO, Z-isomer), 175.2 (C4=O, E-isomer), 175.3 (C4=O, Z-isomer). Anal. Calcd for C23H25N3O4: C, 67.80; H, 6.18; N, 10.31. Found: C, 67.89; H, 6.24; N, 10.35.
N-(Hydroxy)-2-(2,4-dioxo-8-phenyl-1,3-diazaspiro [4.5]decan-3-yl)propanamide (49). The N-benzyloxy precursor 48 (200 mg, 0.47 mmol) was subjected to catalytic hydrogenation in a mixture of EtOH/AcOEt 3:1 (19 mL) according to the procedure described for the synthesis of hydroxamate analogs to afford the title compound 49 as a white crystalline solid. (155 mg, almost quantitative yield); Rf = 0.34 (AcOEt); mp 184–186 °C (MeOH/n-pentane); 1H NMR (600.11 MHz, [D6]DMSO): δ (ppm) = 1.43 (d, J = 7.1 Hz, 0.1H, NCH(CH3)CO), 1.49 (d, J = 7.3 Hz, 2.8H, NCH(CH3)CO, 1.66–1.88 (complex m, 7.8H, H6, H7, H9, H10), 2.11 (q, J1 = 12.0 Hz, 0.15H, H7, H9), 2.57 (tt, J1 = 11.0 Hz, J2 = 5.8 Hz, 1H, H8), 4.42 (q, J = 7.4 Hz, 0.1H, NCH(CH3)CO), 4.45 (q, J = 7.4 Hz, 0.8H, NCH(CH3)CO), 7.18 (td, J1 = 7.1 Hz, J2 = 1.4 Hz, 1H, H4′), 7.25 (d, J = 7.6 Hz, 0.2H, H2′, H6′), 7.29 (t, J = 7.5 Hz, 2H, H3′, H5′), 7.33 (d, J = 7.6 Hz, 1.8H, H2′, H6′), 8.18 (s, 0.05H, H1), 8.81 (s, 0.8H, NCH(CH3)CONHOH, E-isomer), 8.95, 9.02 (s + s, 0.85H, H1), 9.16 (s, 0.05H, NCH(CH3)CONHOH, Z-isomer), 10.00 (s, 0.05H, NCH(CH3)CONHOH, Z-isomer), 10.60 (s, 0.75H, NCH(CH3)CONHOH, E-isomer); 13C NMR (150.9 MHz, [D6]DMSO): δ (ppm) = 15.1 (NCH(CH3)CO), 28.9 (C7, C9), 33.7 (C6, C10), 42.7 (C8), 47.4 (NCH(CH3)CO), 60.8 (C5), 126.5 (C4′), 127.4 (C2′, C6′), 128.7 (C3′, C5′), 147.1 (C1′), 155.8 (C2=O), 166.4 (NCH(CH3)CONHOH, E-isomer), 168.7 (NCH(CH3)CONHOH, Z-isomer), 177.1 (C4=O); elemental analysis calcd (%) for C17H21N3O4: C 61.62, H 6.39, N 12.68, found: C 61.62, H 6.39, N 12.68.
N-(hydroxy)-2-(1-methyl-2,4-dioxo-8-phenyl-1,3-diazaspiro [4.5]decan-3-yl)propanamide (53). A mixture of the O-benzyl hydroxamate 52 (350 mg, 0.80 mmol) and 10% Pd on charcoal (42 mg) in EtOH/AcOEt 3:1 (32 mL) was hydrogenated following the general procedure for the preparation of acetohydroxamic acids previously described to afford the title compound 53 as a white foamy solid, which strongly binds the aforementioned solvents. Removal of the entrapped solvents upon drying under high vacuum yielded 53 as a glass solid. Purification of this by column chromatography on silica gel using CH2Cl2/AcOEt 4:1 and AcOEt provided the pure hydroxamic acid as a white crystalline solid. (275 mg, almost quantitative yield); Rf = 0.37 (AcOEt); mp 143–145 °C (MeOH/n-pentane); 1H NMR (600.11 MHz, [D6]DMSO): δ (ppm) = 1.45 (d, J = 7.2 Hz, 0.1H, NCH(CH3)CO), 1.49 (d, J = 7.4 Hz, 2.9H, NCH(CH3)CO), 1.70–1.82 (complex m, 4H, H6, H7, H9, H10), 1.96 (tt, J1 = 12.9 Hz, J2 = 3.9 Hz, 2H, H6, H10), 2.17 (qd, J1 = 12.3 Hz, J2 = 2.6 Hz, 2H, H7, H9), 2.59 (tt, J1 = 12.3 Hz, J2 = 3.5 Hz, 1H, H8), 2.80 (s, 3H, NCH3), 4.45 (q, J = 7.4 Hz, 0.05H, NCH(CH3)CO), 4.49 (q, J = 7.2 Hz, 0.8H, NCH(CH3)CO), 7.19 (td, J1 = 7.3 Hz, J2 = 1.5 Hz, 1H, H4′), 7.24 (dd, J1 = 7.6 Hz, J2 = 1.6 Hz, 2H, H2′, H6′), 7.31 (td, J1 = 7.6 Hz, J2 = 1.8 Hz, 2H, H3′, H5′), 8.85 (s, 0.8H, NCH(CH3)CONHOH, E-isomer), 9.20 (s, 0.05H, NCH(CH3)CONHOH, Z-isomer), 10.05 (s, 0.05H, NCH(CH3)CONHOH, Z-isomer), 10.64 (s, 0.8H, NCH(CH3)CONHOH, E-isomer); 13C NMR (50.32 MHz, [D6]DMSO): δ (ppm) = 14.6 (NCH(CH3)CO), 23.6 (NCH3), 28.6, 28.7 (C7, C9), 29.9 (C6, C10), 41.6 (C8), 47.2 (NCH(CH3)CO), 60.6 (C5), 126.1 (C4′), 126.7 (C2′, C6′), 128.5 (C3′, C5′), 146.5 (C1′), 154.4 (C2=O), 166.0 (NCH(CH3)CONHOH, E-isomer), 170.4 (NCH(CH3)CONHOH, Z-isomer), 175.4 (C4=O); elemental analysis calcd (%) for C18H23N3O4: C 62.59, H 6.71, N 12.17, found: C 62.66, H 6.74, N 12.11.



 ,
, 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
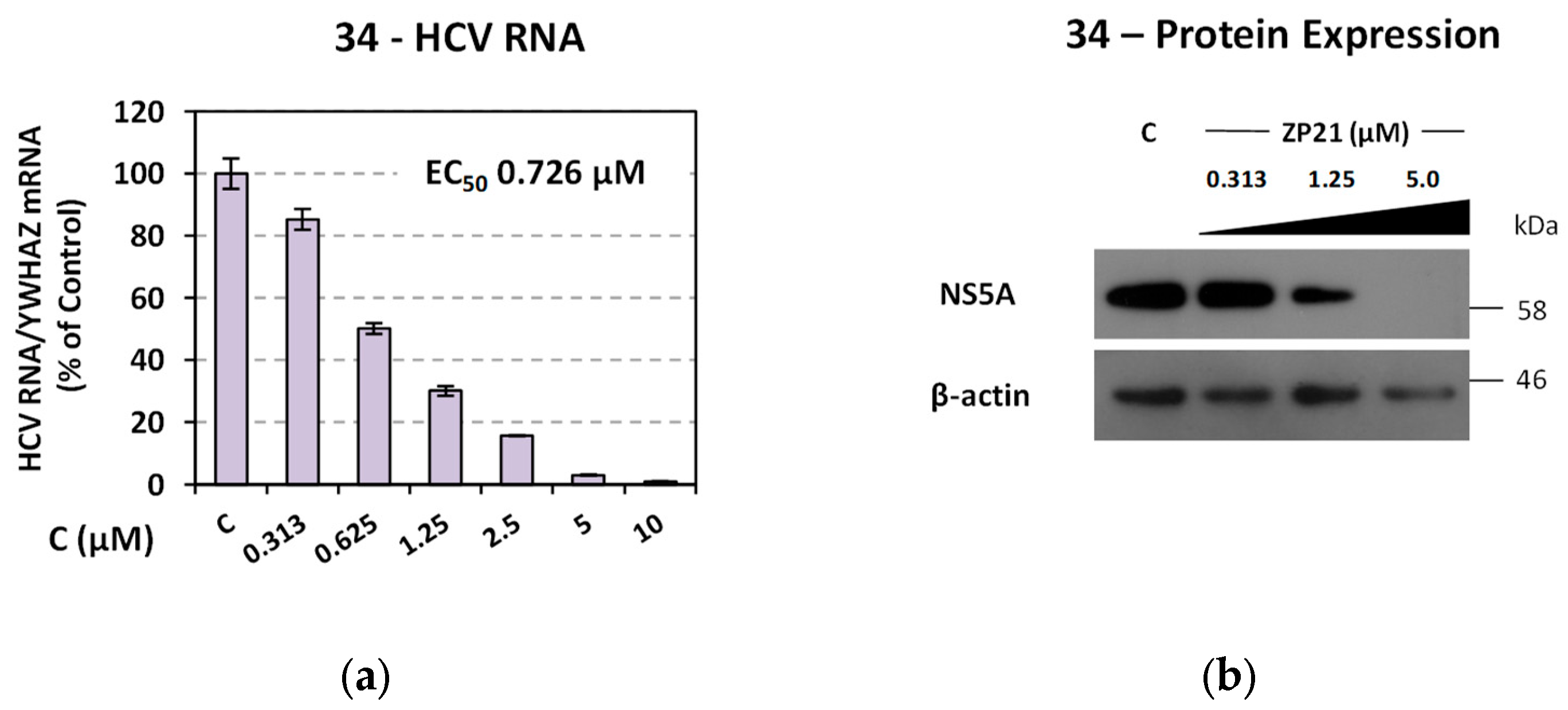{kind=link}
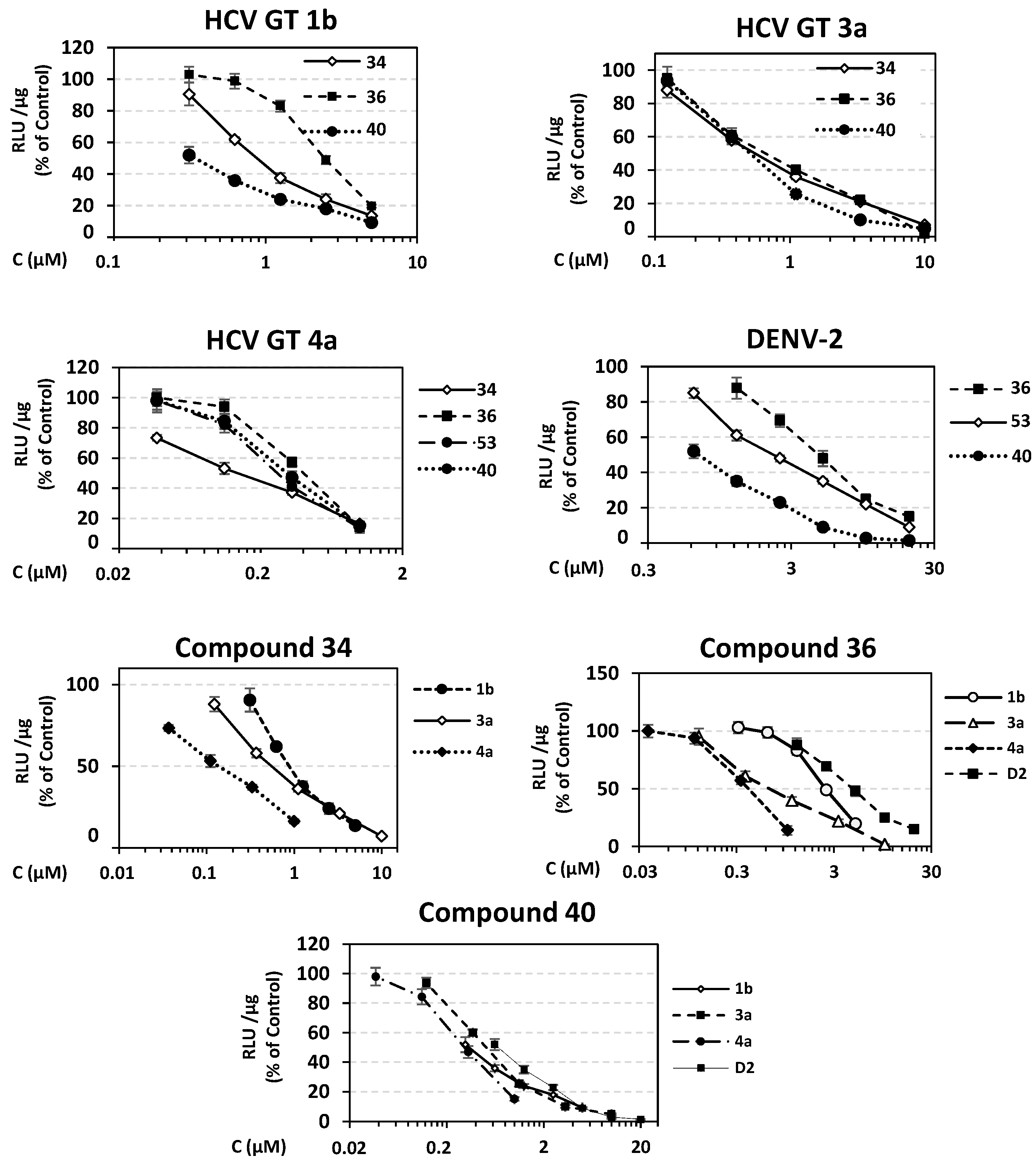{kind=link}
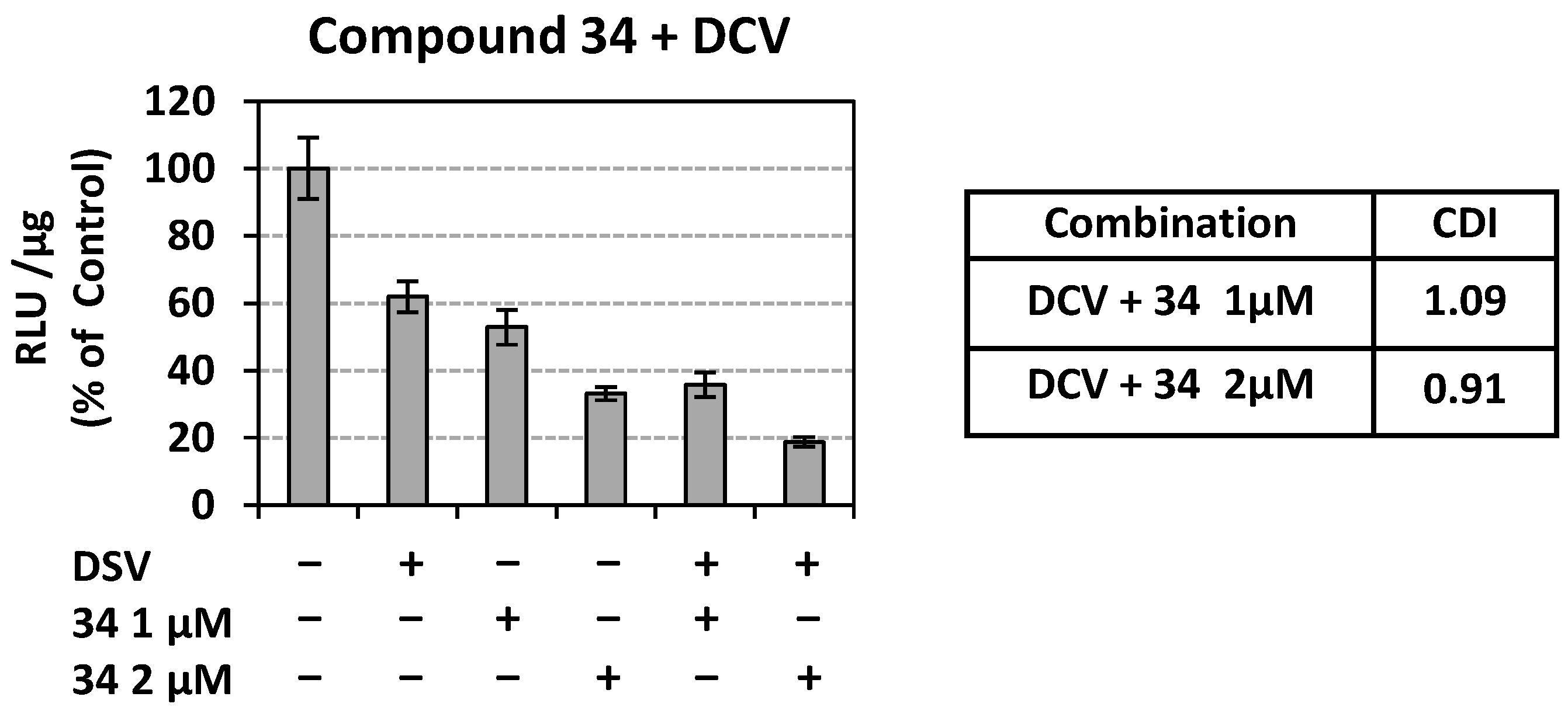{kind=link}
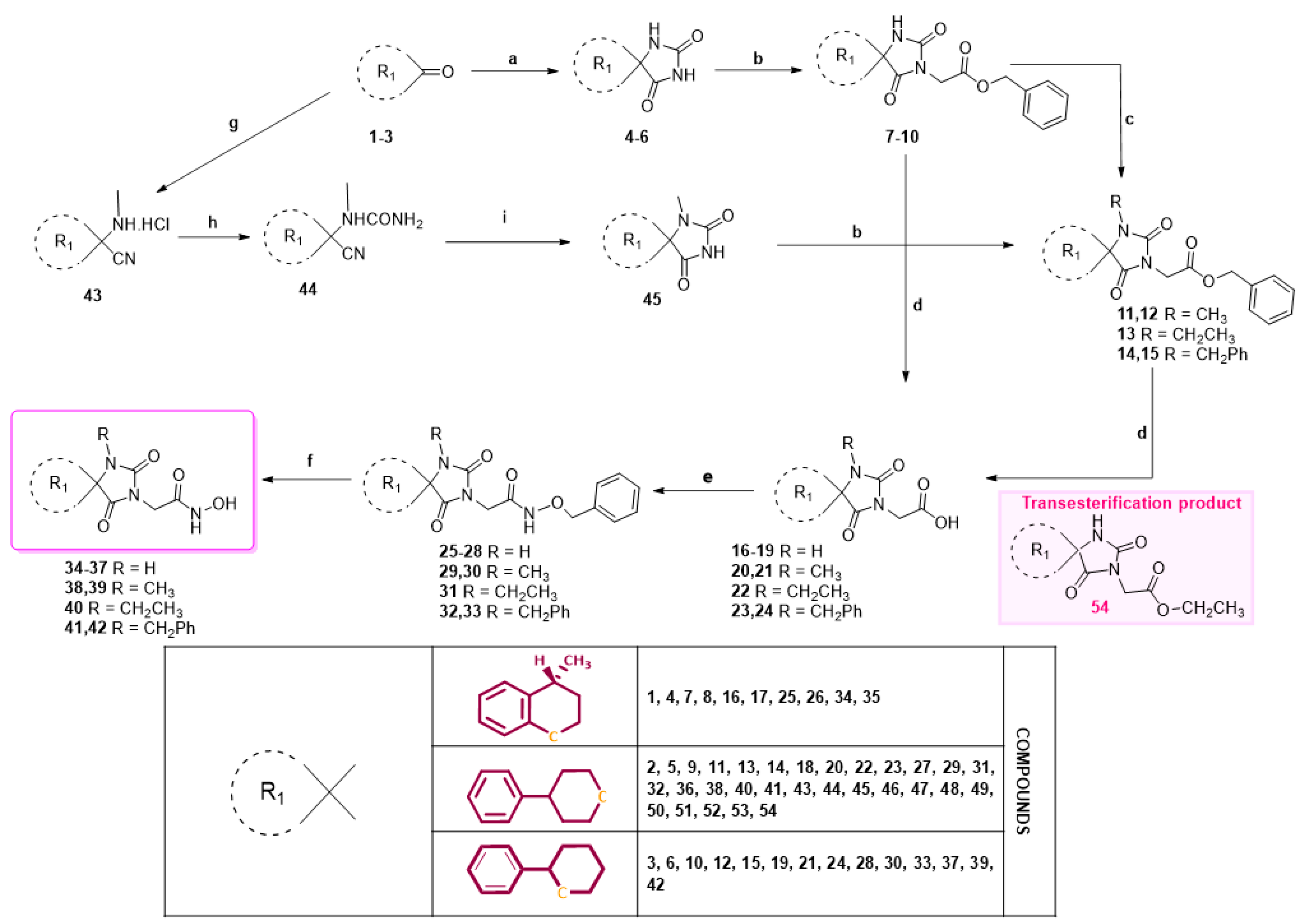{kind=link}
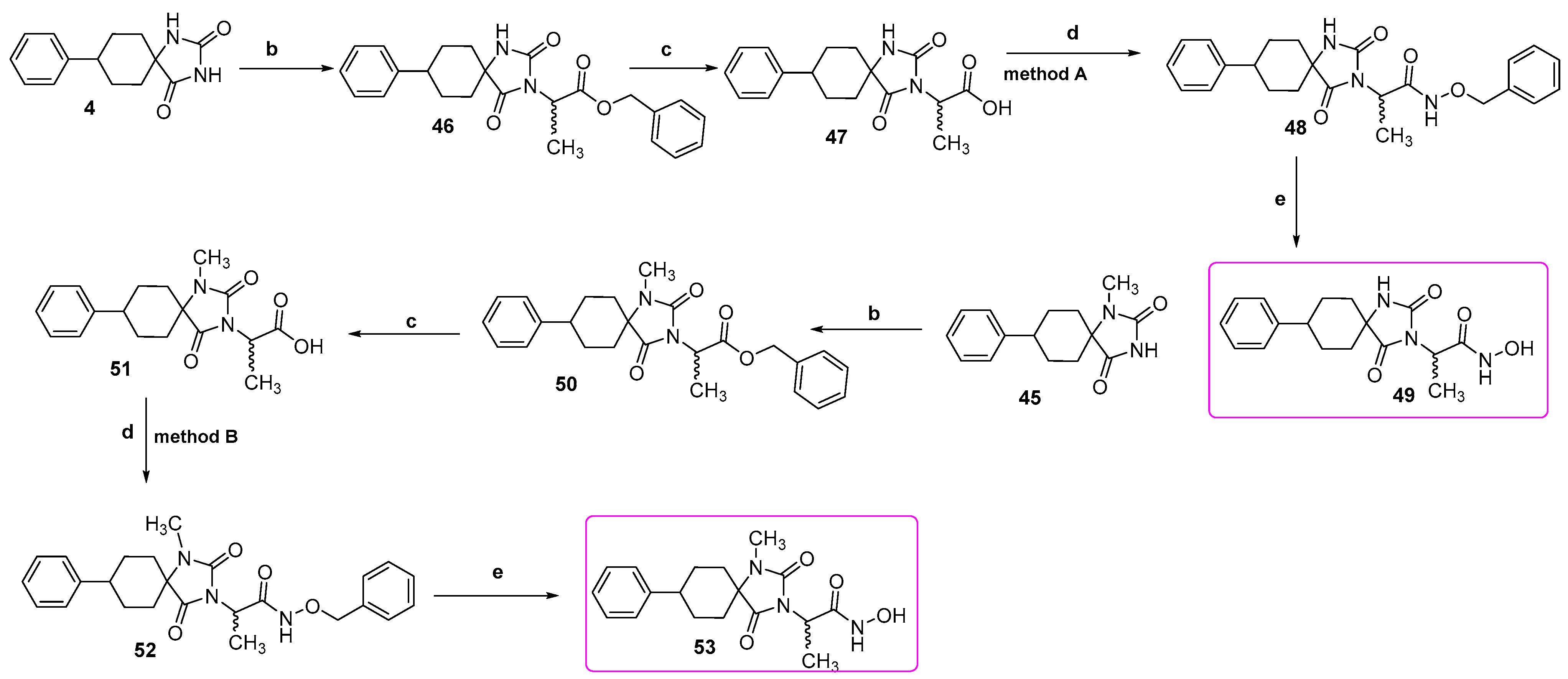{kind=link}























