
Molecular Multi-Target Approach for Human Acetylcholinesterase, Butyrylcholinesterase and β-Secretase 1: Next Generation for Alzheimer’s Disease Treatment

 ,
,  , , , and
, , , and
Abstract
1. Introduction
2. Methods
2.1. Docking-Based Virtual Screening
2.2. Molecular Hybridization
2.3. Physicochemical Filters
2.4. Pharmacophore-Based Virtual Screening
2.5. Molecular Docking
2.6. Molecular Dynamics
2.6.1 Parameterization of the Ligand
2.6.2 MD Simulations
3. Discussion and Results
3.1. Docking-Based Virtual Screening
3.2. Molecular Hybridization
3.3. Physicochemical and Toxicological Filters
3.4. Pharmacophore-Based Virtual Screening
3.5. Molecular Docking
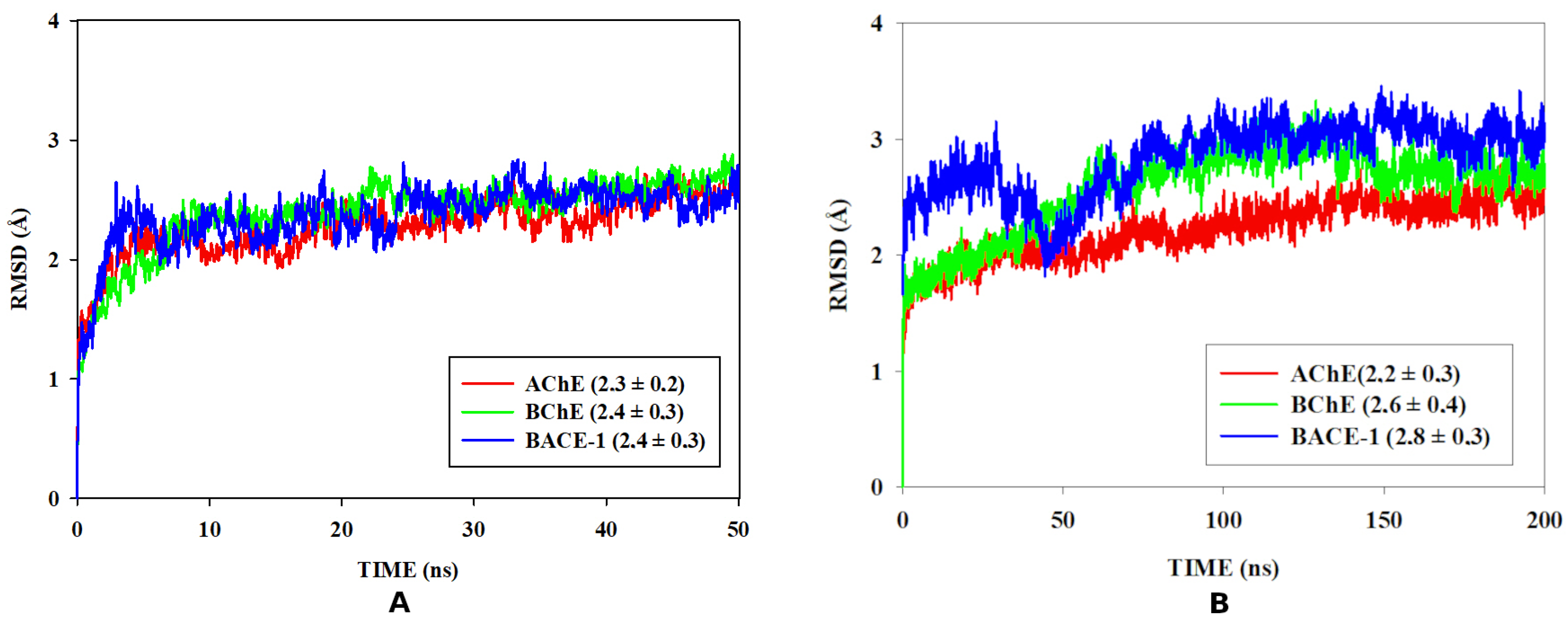
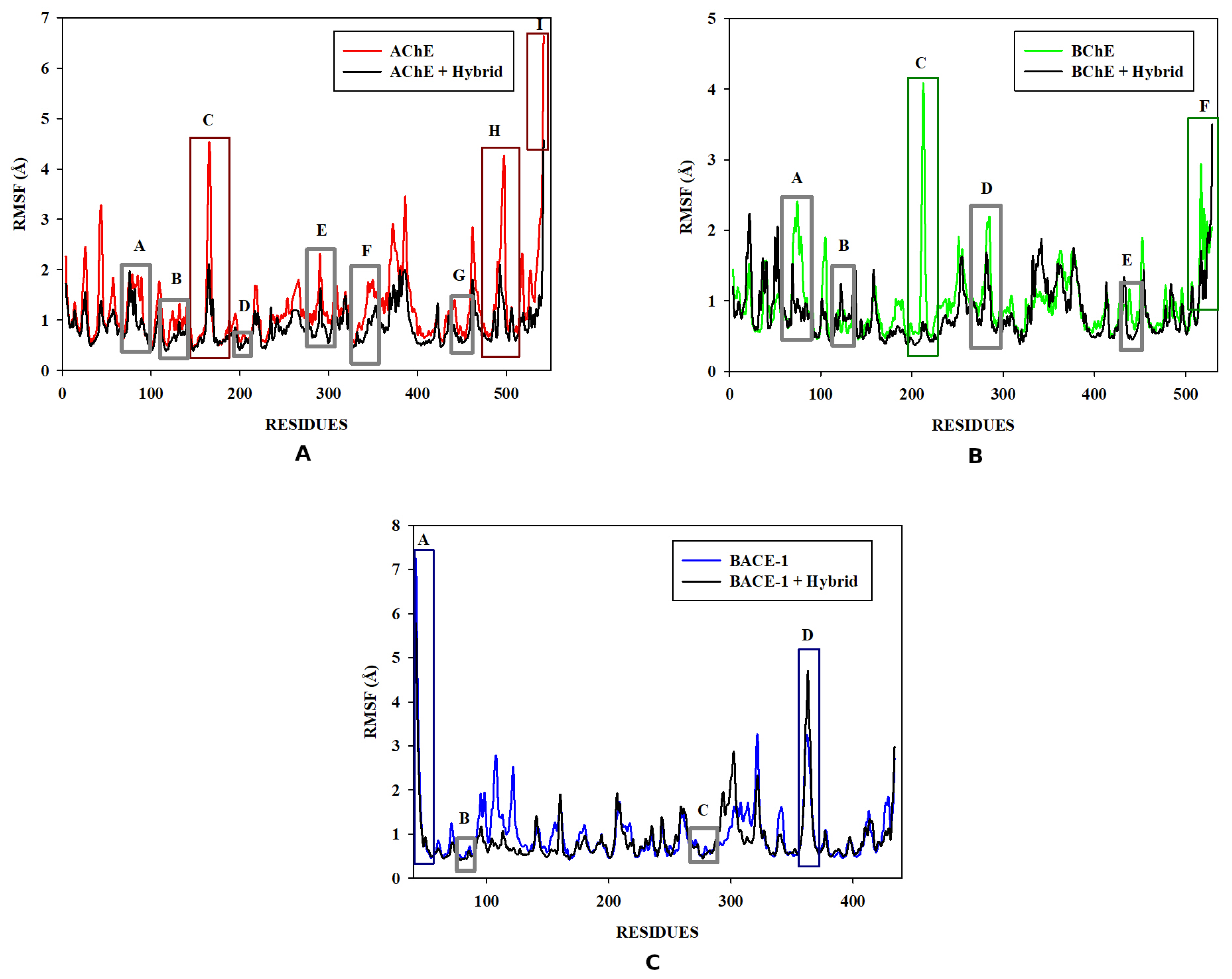
3.6. Molecular Dynamics
4. Main Limitations of This Work
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACh | Acetylcholine |
| AChE | Acetylcholinesterase |
| BChE | Butyrylcholinesterase |
| AD | Alzheimer’s disease |
| APP | Amyloid precursor protein |
| BASE-1 | -secretase 1 |
| CADD | Computer-aided drug design |
| CG | Conjugate gradient |
| CNS | Central nervous system |
| MD | Molecular dynamics |
| RMSD | Root mean square deviation |
| RMSF | Root mean square fluctuation |
| ROC | Receiver operating characteristic |
| SD | Steepest descent |
References
- Yusufzai, S.K.; Khan, M.S.; Sulaiman, O.; Osman, H. Molecular docking studies of coumarin hybrids as potential acetylcholinesterase, butyrylcholinesterase, monoamine oxidase A/B and β-amyloid inhibitors for Alzheimer’s disease. Chem. Cent. J. 2018, 12, 128. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization Dementia. Available online: https://www.who.int/es/news-room/fact-sheets/detail/dementia/ (accessed on 1 April 2020).
- Falco, A.D.; Cukierman, D.S.; Hauser-Davis, R.A.; Rey, N.A. Doença de Alzheimer: Hipóteses etiológicas e perspectivas de tratamento. Química Nova 2016, 39, 63–80. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. Embo Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Association, A. 2019 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2019, 15, 321–387. [Google Scholar] [CrossRef]
- Tan, W.N.; Khairuddean, M.; Wong, K.C.; Khaw, K.Y.; Vikneswaran, M. New cholinesterase inhibitors from Garcinia atroviridis. Fitoterapia 2014, 97, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Lima, E.P.; Esmeraldo, A.R.A.A.; Costa, C.E.T.L.; Virgulino, R.R.; Franca, R.J.; de Lima, A.M. Níveis de colinesterase como marcador de risco de distúrbios neurológicos em agentes de endemias. J. Health Biol. Sci. 2015, 3, 73–76. [Google Scholar] [CrossRef]
- Menichini, F.; Tundis, R.; Loizzo, M.R.; Bonesi, M.; Marrelli, M.; Statti, G.A.; Menichini, F.; Conforti, F. Acetylcholinesterase and butyrylcholinesterase inhibition of ethanolic extract and monoterpenes from Pimpinella anisoides V Brig.(Apiaceae). Fitoterapia 2009, 80, 297–300. [Google Scholar] [CrossRef]
- Pohanka, M. Cholinesterases, a target of pharmacology and toxicology. Biomed. Pap. Med. Fac. Palacky Univ. Olomouc 2011, 155, 219–230. [Google Scholar] [CrossRef]
- Sabbah, D.A.; Zhong, H.A. Modeling the protonation states of β-secretase binding pocket by molecular dynamics simulations and docking studies. J. Mol. Graph. Model. 2016, 68, 206–215. [Google Scholar] [CrossRef]
- Food, F.; Administration, D. FDA Grants Accelerated Approval for Alzheimer’s Drug. Available online: https://www.fda.gov/news-events/press-announcements/fda-grants-accelerated-approval-alzheimers-drug (accessed on 1 April 2020).
- Blennow, K.; Mattsson, N.; Schöll, M.; Hansson, O.; Zetterberg, H. Amyloid biomarkers in Alzheimer’s disease. Trends Pharmacol. Sci. 2015, 36, 297–309. [Google Scholar] [CrossRef]
- Ramos, R.S.; Macêdo, W.J.; Costa, J.S.; da Silva, C.H.d.P.; Rosa, J.M.; da Cruz, J.N.; de Oliveira, M.S.; de Aguiar Andrade, E.H.; e Silva, R.B.; Souto, R.N.; et al. Potential inhibitors of the enzyme acetylcholinesterase and juvenile hormone with insecticidal activity: Study of the binding mode via docking and molecular dynamics simulations. J. Biomol. Struct. Dyn. 2020, 38, 4687–4709. [Google Scholar] [CrossRef]
- Santana, I.B.; Leite, F.H.A.; Santos Junior, M.C. Identification of Lutzomyia longipalpis odorant binding protein modulators by comparative modeling, hierarchical virtual screening, and molecular dynamics. J. Chem. 2018, 2018, 4173479. [Google Scholar] [CrossRef]
- Zhou, J.; Jiang, X.; He, S.; Jiang, H.; Feng, F.; Liu, W.; Qu, W.; Sun, H. Rational design of multitarget-directed ligands: Strategies and emerging paradigms. J. Med. Chem. 2019, 62, 8881–8914. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, D.B. Identificação de novos Inibidores Triplos Frente Acetilcolinesterase, Butirilcolinesterase e Beta-Secretase 1 Humana. Ph. D. Thesis, Estadual University of Feira de Santana, Novo Horizonte, Brazil, 2021. [Google Scholar]
- Verli, H. Dinâmica molecular. Bioinform. Biol. Flexibilidade Mol. 2014, 8, 173–187. [Google Scholar]
- Durrant, J.D.; McCammon, J.A. Molecular dynamics simulations and drug discovery. BMC Biol. 2011, 9, 71. [Google Scholar] [CrossRef]
- Morphy, R.; Rankovic, Z. Designed multiple ligands. An emerging drug discovery paradigm. J. Med. Chem. 2005, 48, 6523–6543. [Google Scholar] [CrossRef]
- Oliveira Pedrosa, M.D.; Duarte da Cruz, R.M.; Oliveira Viana, J.D.; de Moura, R.O.; Ishiki, H.M.; Barbosa Filho, J.M.; Diniz, M.F.; Scotti, M.T.; Scotti, L.; Bezerra Mendonca, F.J. Hybrid compounds as direct multitarget ligands: A review. Curr. Top. Med. Chem. 2017, 17, 1044–1079. [Google Scholar] [CrossRef]
- Sterling, T.; Irwin, J.J. ZINC 15–ligand discovery for everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef]
- TRIPOS. SYBYL-X 2.0. Available online: https://sybyl-x.software.informer.com/2.0/ (accessed on 1 April 2020).
- Søndergaard, C.R.; Olsson, M.H.; Rostkowski, M.; Jensen, J.H. Improved treatment of ligands and coupling effects in empirical calculation and rationalization of p K a values. J. Chem. Theory Comput. 2011, 7, 2284–2295. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Mascarenhas, A.M.S.; de Almeida, R.B.M.; de Araujo Neto, M.F.; Mendes, G.O.; da Cruz, J.N.; Dos Santos, C.B.R.; Botura, M.B.; Leite, F.H.A. Pharmacophore-based virtual screening and molecular docking to identify promising dual inhibitors of human acetylcholinesterase and butyrylcholinesterase. J. Biomol. Struct. Dyn. 2021, 39, 6021–6030. [Google Scholar] [CrossRef] [PubMed]
- Domíngues, J.L.; Christopeit, T.; Villaverde, M.C.; Gossas, T.; Otero, J.M.; Nyström, S.; Baraznenok, V.; Lindström, E.; Danielson, U.H.; Sussman, F. Effect of the protonation state of the titratable residues on the inhibitor affinity to BACE-1. Biochemistry 2010, 49, 7255–7263. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed]
- Database, C.C. GOLD 4.0. Available online: https://www.ccdc.cam.ac.uk/solutions/software/gold/ (accessed on 1 April 2020).
- do Bomfim, M.R.; Barbosa, D.B.; de Carvalho, P.B.; da Silva, A.M.; de Oliveira, T.A.; Taranto, A.G.; Leite, F.H.A. Identification of potential human beta-secretase 1 inhibitors by hierarchical virtual screening and molecular dynamics. J. Biomol. Struct. Dyn. 2022, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Chemaxon. Marvin Sketch Version 15.4.20: ChemAxon. Available online: https://chemaxon.com/marvin (accessed on 1 April 2020).
- Hopkins, A.L.; Keserü, G.M.; Leeson, P.D.; Rees, D.C.; Reynolds, C.H. The role of ligand efficiency metrics in drug discovery. Nat. Rev. Drug Discov. 2014, 13, 105–121. [Google Scholar] [CrossRef] [PubMed]
- Kawai, K.; Nagata, N.; Takahashi, Y. De novo design of drug-like molecules by a fragment-based molecular evolutionary approach. J. Chem. Inf. Model. 2014, 54, 49–56. [Google Scholar] [CrossRef]
- Pires, D.E.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Barbezan, A.B.; Martins, R.; Bueno, J.B.; Villavicencio, A.L.C. Ames test to detect mutagenicity of 2-alkylcyclobutanones: A review. J. Food Sci. 2017, 82, 1518–1522. [Google Scholar] [CrossRef]
- Chen, J.; Myint, K. New QSAR Prediction Models Derived from GPCR CB2-Antagonistic Triaryl Bis-Sulfone Analogues by a Combined Molecular Morphological and Pharmacophoric Approach. SAR QSAR Environ. Res. 2012, 22, 525–544. [Google Scholar] [CrossRef]
- Stroet, M.; Caron, B.; Visscher, K.M.; Geerke, D.P.; Malde, A.K.; Mark, A.E. Automated topology builder version 3.0: Prediction of solvation free enthalpies in water and hexane. J. Chem. Theory Comput. 2018, 14, 5834–5845. [Google Scholar] [CrossRef] [PubMed]
- Schmid, N.; Eichenberger, A.P.; Choutko, A.; Riniker, S.; Winger, M.; Mark, A.E.; Van Gunsteren, W.F. Definition and testing of the GROMOS force-field versions 54A7 and 54B7. Eur. Biophys. J. 2011, 40, 843–856. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed]
- Mermelstein, D.J.; McCammon, J.A.; Walker, R.C. pH-dependent conformational dynamics of beta-secretase 1: A molecular dynamics study. J. Mol. Recognit. 2019, 32, e2765. [Google Scholar] [CrossRef]
- Berendsen, H.; Grigera, J.; Straatsma, T. The missing term in effective pair potentials. J. Phys. Chem. 2010, 53, 5061–5084. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An Nlog(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Daura, X.; van Gunsteren, W.F.; Mark, A.E. Folding–unfolding thermodynamics of a β-heptapeptide from equilibrium simulations. Proteins Struct. Funct. Bioinform. 1999, 34, 269–280. [Google Scholar] [CrossRef]
- Leite, F.H.A.; da Silva Santiago, P.B.G.; Froes, T.Q.; da Silva Filho, J.; da Silva, S.G.; Ximenes, R.M.; de Faria, A.R.; Brondani, D.J.; de Albuquerque, J.F.; Castilho, M.S. Structure-guided discovery of thiazolidine-2, 4-dione derivatives as a novel class of Leishmania major pteridine reductase 1 inhibitors. Eur. J. Med. Chem. 2016, 123, 639–648. [Google Scholar] [CrossRef]
- Ballester, P.J.; Mangold, M.; Howard, N.I.; Robinson, R.L.M.; Abell, C.; Blumberger, J.; Mitchell, J.B. Hierarchical virtual screening for the discovery of new molecular scaffolds in antibacterial hit identification. J. R. Soc. Interface 2012, 9, 3196–3207. [Google Scholar] [CrossRef]
- Kumar, A.; Zhang, K.Y. Hierarchical virtual screening approaches in small molecule drug discovery. Methods 2015, 71, 26–37. [Google Scholar] [CrossRef] [PubMed]
- SCHRÖDINGER. The PyMOL Molecular Graphics System. Available online: https://pymol.org/2/ (accessed on 1 April 2020).
- Munoz-Torrero, D.; Camps, P. Dimeric and hybrid anti-Alzheimer drug candidates. Curr. Med. Chem. 2006, 13, 399–422. [Google Scholar] [CrossRef] [PubMed]
- Cheung, J.; Gary, E.N.; Shiomi, K.; Rosenberry, T.L. Structures of human acetylcholinesterase bound to dihydrotanshinone I and territrem B show peripheral site flexibility. ACS Med. Chem. Lett. 2013, 4, 1091–1096. [Google Scholar] [CrossRef]
- Li, Q.; Yang, H.; Chen, Y.; Sun, H. Recent progress in the identification of selective butyrylcholinesterase inhibitors for Alzheimer’s disease. Eur. J. Med. Chem. 2017, 132, 294–309. [Google Scholar] [CrossRef] [PubMed]
- Barman, A.; Prabhakar, R. Elucidating the catalytic mechanism of β-secretase (BACE1): A quantum mechanics/molecular mechanics (QM/MM) approach. J. Mol. Graph. Model. 2013, 40, 1–9. [Google Scholar] [CrossRef]
- Paz, O.S.; Froes, T.Q.; Leite, F.H.; Castilho, M.S. Modeling of BACE-1 Inhibitors as Anti-Alzheimer’s Agents. In Computational Modeling of Drugs Against Alzheimer’s Disease; Springer: New York, NY, USA, 2018; pp. 177–206. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2012, 64, 4–17. [Google Scholar] [CrossRef]
- DeGoey, D.A.; Chen, H.J.; Cox, P.B.; Wendt, M.D. Beyond the rule of 5: Lessons learned from AbbVie’s drugs and compound collection: Miniperspective. J. Med. Chem. 2017, 61, 2636–2651. [Google Scholar] [CrossRef]
- Clark, R.D.; Abrahamian, E. Using a staged multi-objective optimization approach to find selective pharmacophore models. J. Comput. Aided Mol. Des. 2009, 23, 765. [Google Scholar] [CrossRef]
- Dorfman, R.J.; Smith, K.M.; Masek, B.B.; Clark, R.D. A knowledge-based approach to generating diverse but energetically representative ensembles of ligand conformers. J. Comput. Aided Mol. Des. 2008, 22, 681–691. [Google Scholar] [CrossRef]
- de Carvalho Gallo, J.C.; de Mattos Oliveira, L.; Araújo, J.S.C.; Santana, I.B.; dos Santos Junior, M.C. Virtual screening to identify Leishmania braziliensis N-myristoyltransferase inhibitors: Pharmacophore models, docking, and molecular dynamics. J. Mol. Model. 2018, 24, 260. [Google Scholar] [CrossRef]
- Xie, H.; Qiu, K.; Xie, X. 3D QSAR Studies, Pharmacophore Modeling and Virtual Screening on a Series of Steroidal Aromatase Inhibitors. Int. J. Mol. Sci. 2014, 15, 20927–20947. [Google Scholar] [CrossRef]
- Empereur-mot, C.; Guillemain, H.; Latouche, A.; Zagury, J.F.; Viallon, V.; Montes, M. Predictiveness curves in virtual screening. J. Cheminform. 2015, 7, 52. [Google Scholar] [CrossRef]
- Kothandan, G.; Madhavan, T.; Gadhe, C.G.; Cho, S.J. A combined 3D QSAR and pharmacophore-based virtual screening for the identification of potent p38 MAP kinase inhibitors: An in silico approach. Med. Chem. Res. 2013, 22, 1773–1787. [Google Scholar] [CrossRef]
- Prati, F.; Bottegoni, G.; Bolognesi, M.L.; Cavalli, A. BACE-1 Inhibitors: From Recent Single-Target Molecules to Multitarget Compounds for Alzheimer’s Disease. J. Med. Chem. 2018, 61, 619–637. [Google Scholar] [CrossRef]
- Mendes, G.O. Identificação de Potenciais Inibidores Triplos Frente Acetilcolinesterase, Butirilcolinesterase e Beta-Secretase 1; Estadual University of Feira de Santana: Novo Horizonte, Brazil, 2020. [Google Scholar]
- Türkeş, C.; Arslan, M.; Demir, Y.; Çoçaj, L.; Rifati Nixha, A.; Beydemir, Ş. Synthesis, biological evaluation and in silico studies of novel N-substituted phthalazine sulfonamide compounds as potent carbonic anhydrase and acetylcholinesterase inhibitors. Bioorganic Chem. 2019, 89, 103004. [Google Scholar] [CrossRef]
- Kumar, V.; Saha, A.; Roy, K. In silico modeling for dual inhibition of acetylcholinesterase (AChE) and butyrylcholinesterase (BuChE) enzymes in Alzheimer’s disease. Comput. Biol. Chem. 2020, 88, 107355. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.A.; Ali, M.Y.; Choi, R.J.; Jeong, H.O.; Chung, H.Y.; Choi, J.S. Kinetics and molecular docking studies of fucosterol and fucoxanthin, BACE1 inhibitors from brown algae Undaria pinnatifida and Ecklonia stolonifera. Food Chem. Toxicol. 2016, 89, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Sargsyan, K.; Grauffel, C.; Lim, C. How molecular size impacts RMSD applications in molecular dynamics simulations. J. Chem. Theory Comput. 2017, 13, 1518–1524. [Google Scholar] [CrossRef]
- Hubbard, R.E.; Haider, M.K. Hydrogen bonds in proteins: Role and strength. In Encyclopedia of Life Sciences; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2010. [Google Scholar]
- Nalivaeva, N.N.; Turner, A.J. Post-translational modifications of proteins: Acetylcholinesterase as a model system. Proteomics Int. Ed. 2001, 1, 735–747. [Google Scholar] [CrossRef]
- Torda, A.E.; van Gunsteren, W.F. Algorithms for clustering molecular dynamics configurations. J. Comput. Chem. 1994, 15, 1331–1340. [Google Scholar] [CrossRef]
- Pita, S.S.d.R.; Pascutti, P.G. Análise Farmacofórica da Tripanotiona Redutase (TR) de Trypanosoma cruzi complexada com Inibidores Peptídeo miméticos. Rev. Virtual Química 2012, 4, 788–804. [Google Scholar] [CrossRef]
- Bissantz, C.; Kuhn, B.; Stahl, M. A medicinal chemist’s guide to molecular interactions. J. Med. Chem. 2010, 53, 5061–5084. [Google Scholar] [CrossRef] [PubMed]
- Nicolet, Y.; Lockridge, O.; Masson, P.; Fontecilla-Camps, J.C.; Nachon, F. Crystal structure of human butyrylcholinesterase and of its complexes with substrate and products. J. Biol. Chem. 2003, 278, 41141–41147. [Google Scholar] [CrossRef] [PubMed]
- Masson, P.; Xie, W.; Froment, M.T.; Levitsky, V.; Fortier, P.L.; Albaret, C.; Lockridge, O. Interaction between the peripheral site residues of human butyrylcholinesterase, D70 and Y332, in binding and hydrolysis of substrates. Biochim. Biophys. Acta (BBA) Protein Struct. Mol. Enzymol. 1999, 1433, 281–293. [Google Scholar] [CrossRef]
- Dhanabalan, A.K.; Kesherwani, M.; Velmurugan, D.; Gunasekaran, K. Identification of new BACE1 inhibitors using pharmacophore and molecular dynamics simulations approach. J. Mol. Graph. Model. 2017, 76, 56–69. [Google Scholar] [CrossRef]
- Manoharan, P.; Ghoshal, N. Fragment-based virtual screening approach and molecular dynamics simulation studies for identification of BACE1 inhibitor leads. J. Biomol. Struct. Dyn. 2018, 36, 1878–1892. [Google Scholar] [CrossRef]
- Doak, B.C.; Over, B.; Giordanetto, F.; Kihlberg, J. Oral Druggable Space beyond the Rule of 5: Insights from Drugs and Clinical Candidates. Chem. Biol. 2014, 21, 1115–1142. [Google Scholar] [CrossRef]
- Marsault, E.; Peterson, M.L. Macrocycles are Great Cycles: Applications, Opportunities, and Challenges of Synthetic Macrocycles in Drug Discovery. J. Med. Chem. 2011, 54, 1961–2004. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | MW (g/mol) | HBD | HBA | cLog P | PSA (Å) | RB | HBD + HBA | AMES |
|---|---|---|---|---|---|---|---|---|
| A | 578.24 | 2 | 5 | 6.39 | 251 | 7 | 7 | YES |
| B | 545.61 | 3 | 10 | 1 | 226 | 8 | 13 | NO |
| C | 588.13 | 4 | 9 | 2.35 | 244 | 7 | 12 | YES |
| D | 550.08 | 6 | 7 | 2.94 | 238 | 9 | 13 | NO |
| E | 518.70 | 3 | 7 | 6.63 | 227 | 8 | 10 | NO |
| F | 542.60 | 2 | 10 | 5.03 | 229 | 8 | 12 | NO |
| G | 546.08 | 2 | 7 | 7.06 | 238 | 7 | 9 | NO |
| H | 567.05 | 2 | 9 | 6.15 | 240 | 8 | 11 | NO |
| I | 540.58 | 2 | 9 | 6.13 | 229 | 6 | 11 | NO |
| J | 1098.0 | 1 | 15 | 14.94 | 466 | 11 | 16 | NO |
| K | 503.56 | 1 | 10 | 2.64 | 213 | 9 | 11 | NO |
| L | 586.49 | 1 | 7 | 6.10 | 232 | 9 | 8 | NO |
| M | 585.74 | 4 | 10 | 6.37 | 254 | 5 | 14 | YES |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mendes, G.O.; Pita, S.S.d.R.; Carvalho, P.B.d.; Silva, M.P.d.; Taranto, A.G.; Leite, F.H.A. Molecular Multi-Target Approach for Human Acetylcholinesterase, Butyrylcholinesterase and β-Secretase 1: Next Generation for Alzheimer’s Disease Treatment. Pharmaceuticals 2023, 16, 880. https://doi.org/10.3390/ph16060880
Mendes GO, Pita SSdR, Carvalho PBd, Silva MPd, Taranto AG, Leite FHA. Molecular Multi-Target Approach for Human Acetylcholinesterase, Butyrylcholinesterase and β-Secretase 1: Next Generation for Alzheimer’s Disease Treatment. Pharmaceuticals. 2023; 16(6):880. https://doi.org/10.3390/ph16060880
Chicago/Turabian StyleMendes, Géssica Oliveira, Samuel Silva da Rocha Pita, Paulo Batista de Carvalho, Michel Pires da Silva, Alex Gutterres Taranto, and Franco Henrique Andrade Leite. 2023. "Molecular Multi-Target Approach for Human Acetylcholinesterase, Butyrylcholinesterase and β-Secretase 1: Next Generation for Alzheimer’s Disease Treatment" Pharmaceuticals 16, no. 6: 880. https://doi.org/10.3390/ph16060880
APA StyleMendes, G. O., Pita, S. S. d. R., Carvalho, P. B. d., Silva, M. P. d., Taranto, A. G., & Leite, F. H. A. (2023). Molecular Multi-Target Approach for Human Acetylcholinesterase, Butyrylcholinesterase and β-Secretase 1: Next Generation for Alzheimer’s Disease Treatment. Pharmaceuticals, 16(6), 880. https://doi.org/10.3390/ph16060880