All reactions were monitored using thin-layer chromatography (TLC). The purification of compounds via flash column chromatography (CC) and preparative TLC was performed using Macherey-Nagel silica gel 60 (0.04–0.063 mm) and Macherey-Nagel silica gel 60 (GF254) plates, respectively. Melting points were obtained under a Köfler microscope and are uncorrected. 1H and 13C NMR spectra were taken in CDCl3, DMSO, or THF at room temperature on Bruker Avance 300 and 400 instruments (300.13 MHz or 400.13 MHz for 1H, and 75.47 MHz or 100.63 MHz for 13C). Chemical shifts are expressed in δ (ppm) values relative to tetramethylsilane (TMS), used as an internal reference; 13C NMR assignments were made using 2D (HSQC and HMBC) NMR experiments (long-range 13C-1H coupling constants were optimized to 7 Hz). The spectral treatment was executed using the MestReNova v6.0.2-5475 software. The HRMS mass spectra of compounds were performed on an LTQ Orbitrap XL hybrid mass spectrometer (Thermo Fisher Scientific, Bremen, Germany), controlled by LTQ Tune Plus 2.5.5 and Xcalibur 2.1.0. at CEMUP—University of Porto, Portugal. Ions were generated using a Combi MALDI electrospray ionization (ESI) source. All reagents were purchased from Sigma Aldrich. The following materials were synthesized and purified using the described procedures. The HPLC system consisted of a Thermo Scientific SpectraSystem P4000 pump equipped with a degasser, a Thermo Scientific SpectraSystem AS3000 autosampler fitted with a maximum volume of a 100 µL loop, and a Thermo Scientific SpectraSystem UV8000 DAD detector (USA). Data acquisition was performed using ChromQuest 5.0 software, version 3.2.1. The column used in this study was ACE-C18 (150 × 4.6 mm I.D., particle size 5 µm) manufactured by Advanced Chromatography Technologies Ltd. (Aberdeen, Scotland, UK). The mobile phase composition was acetonitrile and water (70:30 v/v, for compounds 5–10, and 13–18) or acetonitrile and water (50:50 v/v; 0.1% CH3CO2H, for compounds 11 and 12, all were HPLC grade solvents obtained from Merck Life Science S.L.U. (Darmstadt, Germany). The flow rate was 1.0 mL/min and the UV detection wavelength was set at 254 nm. Analyses were performed at room temperature in an isocratic mode for 30 min. The peak purity index was determined using a total peak UV-Vis spectra of between 210 and 650 nm, with a step of 4 nm.
3.1.1. Synthesis of Chalcone-Trimethoxycinnamide Hybrid 7
Synthesis of Methyl (E)-3-(3,4,5-Trimethoxyphenyl) Acrylate (3) 3,4,5-Trimetoxycinnamic Acid (4)
Methyl (
E)-3-(3,4,5-trimethoxyphenyl) acrylate (
3) and 3,4,5-trimetoxycinnamic acid (
4) were synthesized (95% and 80% yield, respectively) and characterized according to the described procedure [
24]. An experimental description of compounds
3 and
4 is presented in the
Supplementary Materials.
Synthesis of tert-Butyl (2-(4-acetyl-3-hydroxy-2-propylphenoxy)ethyl)carbamate (5)
1-(2,4-dihydroxy-3-propylphenyl)ethan-1-one (1 g, 1 Eq, 5 mmol) was dissolved in 15 mL of anhydrous acetone. Then, K2CO3 (4 g, 5 Eq, 30 mmol) and 2-(Boc-amino)-ethyl bromide (1 g, 1.2 Eq, 6 mmol) were added. The reaction proceeded for 24 h at 70 °C with gentle stirring. After cooling, the reaction mixture was poured onto ice and neutralized with HCl. The obtained solid was filtrated, washed with water, and dried. A white solid (90% yield) corresponding to tert-butyl (2-(4-acetyl-3-hydroxy-2-propylphenoxy)ethyl)carbamate (5) was obtained.
tert-butyl (2-(4-acetyl-3-hydroxy-2-propylphenoxy)ethyl)carbamate (5): 99.3% purity; mp 175–177 °C (acetone); 1H NMR (CDCl3, 300.13 MHz) δ: 12.74 (s, 1H, 2-OH), 7.58 (d, J = 8.9 Hz, 1H, H-6), 6.42 (d, J = 8.9 Hz, 1H, H-5), 4.91 (sl, 1H, H-3″), 4.09 (t, J = 5.2 Hz, 2H, H-1″), 3.56 (q, J = 5.6 Hz, 2H, H-2″), 2.63 (t, J = 7.5 Hz, 1H, H-1′), 1.58–1.50 (m, 2H, H-2′), 2.56 (s, 3H, 1-COCH3), 1.45 (s, 9H, H-6″), 0.94 (t, J = 7.4 Hz, 3H, H-3′) ppm. 13C NMR (CDCl3. 75.47 MHz) δ: 203.2 (1-COCH3), 162.5 (C-4), 162.3 (C-2), 156.0 (C-4″), 130.2 (C-6), 118.5 (C-3), 114.6 (C-1), 102.9 (C-5), 79.8 (C-5″), 67.6 (C-1″), 40.2 (C-2″), 28.5 (C-6″), 26.5 (1-COCH3), 24.5 (C-1′), 22.1 (C-2′), 14.3 (C-3′) ppm; HRMS (ESI+): m/z Anal. Calc. for C18H27NO5 (M+Na+) 360.17814., found 360.17821.
Synthesis of tert-Butyl (E)-(2-(3-hydroxy-2-propyl-4-(3-(3,4,5-trimethoxyphenyl)acryloyl)phenoxy)ethyl)carbamate (6)
To a solution of tert-butyl (2-(4-acetyl-3-hydroxy-2-propylphenoxy)ethyl)carbamate (5, 200 mg, 1 Eq, 593 µmol) in methanol was added an aqueous solution of sodium hydroxide (40%) to pH 13–14. Then, a solution of 3,4,5-trimethoxybenzaldehyde (349 mg, 3 Eq, 1.78 mmol) in methanol was slowly added to the reaction mixture. The reaction was refluxed for 4 days. After cooling, the reaction was poured onto crushed ice, and the pH was adjusted to 6–8 with a 5 M HCl solution. The obtained solid was filtrated, washed with water, and purified via flash CC (SiO2; hexane:ethyl acetate, 7:3). A yellow solid (20% yield) corresponding to tert-butyl(E)-(2-(3-hydroxy-2-propyl-4-(3-(3,4,5-trimethoxyphenyl)acryloyl)phenoxy)ethyl)carbamate (6) was obtained.
tert-butyl(E)-(2-(3-hydroxy-2-propyl-4-(3-(3,4,5-trimethoxyphenyl)acryloyl)phenoxy)ethyl)carbamate (6): 98.9% purity; mp 196–198 °C (ethyl acetate); 1H NMR (DMSO, 400.13 MHz) δ: 13.68 (s, 1H, 2-OH), 8.27 (d, J = 9.2 Hz, 1H, H-6), 7.98 (d, J = 15.4 Hz, 1H, H-β), 7.79 (d, J = 15.4 Hz, 1H, H-α), 7.26 (s, 2H, H-2‴, -6‴), 7.00 (t, J = 5.7 Hz, 1H, H-3″), 6.69 (d, J = 9.2 Hz, 1H, H-5), 4.10 (t, J = 5.6 Hz, 2H, H-1″), 3.87 (s, 6H, 3‴-, 5‴-OCH3), 3.72 (s, 3H, 4‴-OCH3), 3.35 (t, J = 5.6 Hz, 2H, H-2″), 2.59 (t, J = 7.5 Hz, 1H, H-1′), 1.52–1.45 (m, 2H, H-2′), 1.39 (s, 9H, H-6″), 0.89 (t, J = 7.3 Hz, 3H, H-3″) ppm; 13C NMR (DMSO, 100.63 MHz) δ: 192.4 (C=O), 162.7 (C-2), 162.6 (C-4), 155.7 (C-4″), 153.1 (C-3‴,-5‴), 144.7 (C-β), 140.0 (C-4‴), 130.6 (C-6), 130.1 (C-1‴), 120.2 (C-α), 117.1 (C-3), 113.9 (C-1), 106.9 (C-2‴, -6‴), 103.3 (C-5), 77.7 (C-5″), 67.0 (C-1″), 60.2 (4‴-OCH3), 56.2 (3‴-, 5‴-OCH3), 40.0 (C-2″), 28.2 (C-6″), 24.3 (C-1′), 21.9 (C-2′), 14.0 (C-3′) ppm; HRMS (ESI+): m/z Anal. Calc. for C28H37NO8 (M+Na+) 538.24114., found 538.24191.
Synthesis of (E)-N-(2-(3-Hydroxy-2-propyl-4-((E)-3-(3,4,5-trimethoxyphenyl)acryloyl)phenoxy)ethyl)-3-(3,4,5-trimethoxyphenyl)acrylamide (7)
Tert-butyl(E)-(2-(3-hydroxy-2-propyl-4-(3-(3,4,5-trimethoxyphenyl)acryloyl)phenoxy)ethyl)carbamate (6, 150 mg, 1 Eq, 291 µmol) was dissolved in 5 mL of dichloromethane, and then 5 mL of TFA was added to the mixture. The reaction proceeded for 26 h with gentle stirring. After completion, the reaction mixture was diluted with dichloromethane, and then a saturated sodium hydrogen carbonate solution was added. The solution was then extracted with ethyl acetate (3 × 50 mL). The organic layers were collected, washed with water, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The crude obtained was reserved. Then, in a round-bottomed flask, (E)-3-(3,4,5-trimethoxyphenyl)acrylic acid (80 mg, 1 Eq, 0.34 mmol) was dissolved in 5 mL of anhydrous dichloromethane and N-ethyl-N-isopropylpropan-2-amine (87 mg, 0.12 mL, 2 Eq, 0.67 mmol) was added. Then, 50% of the crude material was added to the mixture. After 10 min, COMU (0.22 g, 1.5 Eq, 0.50 mmol) was also added to the mixture. The reaction proceeded for 20 h at room temperature with stirring. After completion, the reaction mixture was poured into ice. The solution was subsequently washed with dichloromethane (3 × 80 mL), with a 1 M HCl solution and saturated NaHCO3 solution. The organic phase was dried over anhydrous sodium sulfate, filtered, and solvent evaporated under reduced pressure. The reaction mixture obtained was dissolved in chloroform and purified using TLC (SiO2; chloroform:methanol, 95:5). A yellow solid (20% yield) corresponding to (E)-N-(2-(3-hydroxy-2-propyl-4-((E)-3-(3,4,5-trimethoxyphenyl)acryloyl)phenoxy)ethyl)-3-(3,4,5-trimethoxyphenyl)acrylamide (7) was obtained.
(E)-N-(2-(3-hydroxy-2-propyl-4-((E)-3-(3,4,5-trimethoxyphenyl)acryloyl)phenoxy)ethyl)-3-(3,4,5-trimethoxyphenyl)acrylamide (7): 98.9% purity; mp 205–207 °C (chloroform); 1H NMR (CDCl3, 300.13 MHz) δ: 13.68 (s, 1H, 2-OH), 8.32–8.27 (m, 1H, 3″-NH-, H-6), 7.99 (d, J = 15.4 Hz, 1H, H-β), 7.79 (d, J = 15.3 Hz, 1H, H-α), 7.28 (s, 2H, H-2‴, -6‴), 6.89 (s, 2H, H-6″, -10″), 6.87 (d, J = 15.8 Hz, 1H, H-β’), 6.62 (d, J = 15.8 Hz, 1H, H-α’), 6.73 (d, J = 9.2 Hz, 1H, H-5), 4.21 (t, J = 5.5 Hz, 2H, H-1″), 3.87 (s, 6H, 3‴-, 5‴-OCH3), 3.80 (s, 6H, 7″-, 9″-OCH3), 3.72 (s, 3H, 4‴-OCH3), 3.78 (sl, 2H, H-2″), 3.67 (s, 3H, 8″-OCH3), 2.60 (t, J = 7.5 Hz, 1H, H-1′), 1.54–1.44 (m, 2H, H-2′), 0.88 (t, J = 7.4 Hz, 3H, H-3″) ppm; 13C NMR (CDCl3, 75.47 MHz) δ: 192.5 (C=O), 165.4 (C-4″), 162.7 (C-2), 162.6 (C-4), 153.2 (C-3‴,-5‴), 153.1 (C-7″, -9″), 144.8 (C-β), 140.0 (C-4‴), 139.1 (C-4″), 130.5 (C-6), 130.5 (C-1‴), 130.1 (C-5″), 121.3 (C-α’), 120.4 (C-α), 117.2 (C-3), 114.0 (C-1), 106.9 (C-2‴, -6‴), 104.9 (C-6″, -10″), 103.6 (C-β’), 103.4 (C-5), 66.9 (C-1″), 60.2 (4‴-OCH3), 60.1 (8″-OCH3), 56.2 (3‴-, 5‴-OCH3), 56.0 (7″-, 9″-OCH3), 38.7 (C-2″), 24.0 (C-1′), 21.6 (C-2′), 14.1 (C-3′) ppm; HRMS (ESI+): m/z Anal. Calc. for C35H41NO10 (M+Na+) 658.26227., found 658.26343.
3.1.2. Synthesis of Compound 7 Analogues with a Chalcone Scaffold (8 and 13)
Synthesis of (E)-N-(2-(3-Hydroxy-2-propyl-4-(3-(3,4,5-trimethoxyphenyl)acryloyl)phenoxy)ethyl)-3,4,5-trimethoxybenzamide (8)
Tert-butyl(E)-(2-(3-hydroxy-2-propyl-4-(3-(3,4,5-trimethoxyphenyl)acryloyl)phenoxy)ethyl)carbamate (6, 150 mg, 1 Eq, 291 µmol) was dissolved in 5 mL of dichloromethane. Then, 5 mL of TFA was added to the mixture. The reaction proceeded for 26 h with gentle stirring. After completion, the reaction mixture was diluted with dichloromethane, and then a saturated sodium hydrogen carbonate solution was added. The solution was then extracted with ethyl acetate (3 × 50 mL). The organic layers were collected, washed with water, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The crude obtained was reserved. Then, in a round-bottomed flask, 3,4,5-trimethoxybenzoic acid (85 mg, 1 Eq, 0.40 mmol) was dissolved in 5 mL of anhydrous dichloromethane and N-ethyl-N-isopropylpropan-2-amine (0.10 g, 0.14 mL, 2 Eq, 0.80 mmol) was added. Then, 50% of the crude material was added to the mixture. After 10 min, COMU (0.26 g, 1.5 Eq, 0.60 mmol) was also added to the mixture. The reaction proceeded for 3 h at room temperature with stirring. After completion, the reaction mixture was poured onto ice. The solution was subsequently washed with 1M HCl solution and saturated NaHCO3 solution. The organic phase was dried over anhydrous sodium sulfate, filtered, and solvent evaporated under reduced pressure. The reaction mixture obtained was purified via TLC (SiO2; dichloromethane:methanol; 97:3). A light-yellow solid (20% yield) corresponding to (E)-N-(2-(3-hydroxy-2-propyl-4-(3-(3,4,5-trimethoxyphenyl)acryloyl)phenoxy)ethyl)-3,4,5-trimethoxybenzamide (8) was obtained.
(E)-N-(2-(3-hydroxy-2-propyl-4-(3-(3,4,5-trimethoxyphenyl)acryloyl)phenoxy)ethyl)-3,4,5-trimethoxybenzamide (8): 97.8% purity; mp 201–203 °C (chloroform); 1H NMR (DMSO, 400.13 MHz) δ: 13.67 (s, 1H, 2-OH), 8.63 (t, J = 5.5 Hz, 1H, 3″-NH-), 8.28 (d, J = 9.1 Hz, 1H, H-6), 7.98 (d, J = 15.4 Hz, 1H, H-β), 7.29 (d, J = 15.3 Hz, 1H, H-α), 7.26 (s, 2H, H-2‴, -6‴), 7.20 (s, 2H, H-6″, -10″), 6.76 (d, J = 9.0 Hz, 1H, H-5), 4.28 (t, J = 5.6 Hz, 2H, H-1″), 3.87 (s, 6H, 3‴-, 5‴-OCH3), 3.82 (s, 6H, 7″-, 9″-OCH3), 3.72 (s, 3H, 4‴-OCH3), 3.69–3.66 (m, 2H, H-2″), 3.68 (s, 3H, 8″-OCH3), 2.58 (t, J = 7.3 Hz, 1H, H-1′), 1.49–1.40 (m, 2H, H-2′), 0.82 (t, J = 7.3 Hz, 3H, H-3′) ppm; 13C NMR (DMSO, 100.63) δ: 192.4 (C=O), 166.0 (C-4″), 162.7 (C-2), 162.6 (C-4), 153.1 (C-3‴,-5‴), 152.5 (C-7″, -9″), 144.7 (C-β), 140.0 (C-4‴), 140.0 (C-4″), 130.6 (C-6), 130.6 (C-1‴), 129.5 (C-5″), 120.2 (C-α), 117.1 (C-3), 113.9 (C-1), 106.9 (C-2‴, -6‴), 104.8 (C-6″, -10″), 103.4 (C-5), 66.5 (C-1″), 60.2 (4‴-OCH3), 60.1 (8″-OCH3), 56.2 (3‴-, 5‴-OCH3), 56.0 (7″-, 9″-OCH3), 38.9 (C-2″), 23.9 (C-1′), 21.0 (C-2′), 14.0 (C-3′) ppm; HRMS (ESI+): m/z Anal. Calc. for C33H39NO10 (M+Na+) 632.24662, found 632.24594.
Synthesis of Methyl 2-(4-Acetyl-3-hydroxy-2-propylphenoxy)acetate (9) and Dimethyl 2,2’-((4-acetyl-2-propyl-1,3-phenylene)bis(oxy))diacetate (10)
1-(2,4-dihydroxy-3-propylphenyl)ethan-1-one (1 g, 1 Eq, 5.149 mmol) was dissolved in 5 mL of anhydrous acetone. Then, K2CO3 (711.5 mg, 1 Eq, 5.149 mmol) and methyl 2-bromoacetate (945.1 mg, 584.9 µL, 1.2 Eq, 6.178 mmol) were added. The reaction proceeded for 24 h at 70 °C with gentle stirring. After cooling, the reaction mixture was poured onto ice and neutralized with HCl. The obtained solid was filtrated, washed with water, and dried. A white solid (45% yield) corresponding to methyl 2-(4-acetyl-3-hydroxy-2-propylphenoxy)acetate (9) was obtained. The aqueous phase was then extracted with dichloromethane (3 × 50 mL). The organic layers were collected, washed with water, dried over anhydrous sodium sulfate, concentrated under reduced pressure, and purified using flash CC (SiO2; n-hexane:ethyl acetate, 7:3). A gum (25% yield) corresponding to dimethyl 2,2’-((4-acetyl-2-propyl-1,3-phenylene)bis(oxy))diacetate (10) was obtained.
Dimethyl 2,2’-((4-acetyl-2-propyl-1,3-phenylene)bis(oxy))diacetate (10): 99.7% purity; mp N.D. (gum); 1H NMR (CDCl3, 300.13 MHz) δ: 7.50 (d, J = 8.7 Hz, 1H, H-6), 6.53 (d, J = 8.7 Hz, 1H, H-5), 4.69 (s, 2H, H-1″), 4.47 (s, 2H, H-1‴), 3.81 (s, 3H, H-3″), 3.79 (s, 3H, H-3‴), 2.69 (t, J = 7.8 Hz, 2H, H-1′), 1.63–1.53 (m, 2H, H-2′), 2.56 (s, 3H, 1-COCH3), 0.97 (t, J = 7.4 Hz, 3H, H-3′) ppm; 13C NMR (CDCl3. 75.47 MHz) δ: 198.9 (1-COCH3), 169.2 (C-2‴), 168.9 (C-2″), 160.1 (C-4), 156.8 (C-2), 129.2 (C-6), 126.8 (C-1), 126.4 (C-3), 107.1 (C-5), 71.8 (C-1‴), 65.3 (C-1″), 52.4 (C-3‴), 52.2 (C-3″), 30.0 (1-COCH3), 26.0 (C-1′), 22.9 (C-2′), 14.5 (C-3′) ppm; HRMS (ESI+): m/z Anal. Calc. for C17H22O7 (M+Na+) 361.12577, found 361.12594.
Synthesis of (E)-2-(3-Hydroxy-2-propyl-4-(3-(3,4,5-trimethoxyphenyl)acryloyl)phenoxy)acetic Acid (11)
To a solution of methyl 2-(4-acetyl-3-hydroxy-2-propylphenoxy)acetate (9, 200 mg, 1 Eq, 751 µmol) in methanol was added an aqueous solution of 40% sodium hydroxide to pH 13–14. Then, a solution of 3,4,5-trimethoxybenzaldehyde (442 mg, 3 Eq, 2.25 mmol) in methanol was slowly added to the reaction mixture. The reaction was refluxed for 2 days. After cooling, the reaction was poured into crushed ice and neutralized with a 5 M HCl solution. The solution was then extracted with ethyl acetate (3 × 50 mL). The organic layers were collected, washed with water, and dried over anhydrous sodium sulfate. The solvent was evaporated to dryness, and the obtained reaction mixture was purified via flash CC (SiO2; chloroform:methanol, 95:5). A yellow solid (30% yield) corresponding to (E)-2-(3-hydroxy-2-propyl-4-(3-(3,4,5-trimethoxyphenyl)acryloyl)phenoxy)acetic acid (11) was obtained.
(E)-2-(3-hydroxy-2-propyl-4-(3-(3,4,5-trimethoxyphenyl)acryloyl)phenoxy)acetic acid (11): 98.7% purity; mp 200–202 °C (chloroform); 1H NMR (DMSO, 400.13 MHz) δ: 13.39 (s, 1H, 2-OH), 12.73 (s, 1H, 2″-OH), 7.79 (d, J = 15.6 Hz, 1H, H-β), 7.75 (d, J = 9.2 Hz, 1H, H-6), 7.56 (d, J = 8.8 Hz, 1H, H-5), 7.45 (d, J = 15.4 Hz, 1H, H-α), 6.86 (s, 2H, H-2‴, -6‴), 4.72 (sl, 2H, H-1″), 3.92 (s, 6H, 3‴-, 5‴-OCH3), 3.90 (s, 3H, 4‴-OCH3), 2.73–2.66 (m, 2H, H-1′), 1.63–1.53 (m, 2H, H-2′), 0.94 (t, J = 7.7 Hz, 3H, H-3′) ppm; 13C NMR (DMSO, 100.63 MHz) δ: 192.4 (C=O), 172.8 (C-2″), 163.7 (C-2), 162.5 (C-4), 153.7 (C-3‴,-5‴), 145.0 (C-β), 140.8 (C-4‴), 130.1 (C-5), 130.4 (C-1‴), 129.0 (C-6), 119.7 (C-α), 119.4 (C-3), 115.3 (C-1), 106.1 (C-2‴, -6‴), 65.1 (C-1″), 61.2 (4‴-OCH3), 56.4 (3‴-, 5‴-OCH3), 24.7 (C-1′), 22.0 (C-2′), 14.4 (C-3′) ppm; HRMS (ESI+): m/z Anal. Calc. for C23H26O10 (M+H+) 431.17004, found 431.16961.
Synthesis of (E)-2,2’-((2-Propyl-4-(3-(3,4,5-trimethoxyphenyl)acryloyl)-1,3-phenylene)bis(oxy))diacetic Acid (12)
To a solution of methyl 2-(4-acetyl-3-hydroxy-2-propylphenoxy)acetate (10, 200 mg, 1 Eq, 751 µmol) in methanol was added an aqueous solution of 40% sodium hydroxide to pH 13–14. Then, a solution of 3,4,5-trimethoxybenzaldehyde (442 mg, 3 Eq, 2.25 mmol) in methanol was slowly added to the reaction mixture. The reaction was refluxed for 4 days. After cooling, the reaction was poured onto crushed ice and the pH was adjusted to 6–8, with a 5 M HCl solution. The obtained solid was filtrated, washed with water, dried, and purified via flash CC (SiO2; chloroform:methanol, 9:1). A yellow solid (35% yield) corresponding to (E)-2,2’-((2-propyl-4-(3-(3,4,5-trimethoxyphenyl)acryloyl)-1,3-phenylene)bis(oxy))diacetic acid (12) was obtained.
(E)-2,2’-((2-propyl-4-(3-(3,4,5-trimethoxyphenyl)acryloyl)-1,3-phenylene)bis(oxy))diacetic acid (12): 98.7% purity; mp 212–214 °C (chloroform); 1H NMR (DMSO, 400.13 MHz) δ: 7.55–7.52 (m, 3H, H-α,-β, -6), 7.10 (s, 2H, H-2‴’, -6‴’), 6.79 (d, J = 8.8 Hz, 1H, H-5), 4.80 (s, 2H, H-1″), 4.35 (s, 2H, H-1‴), 3.84 (s, 6H, 3‴’-, 5‴’-OCH3), 3.70 (s, 3H, 4‴’-OCH3), 2.69 (t, J = 7.4 Hz, 1H, H-1′), 1.60–1.51 (m, 2H, H-2′), 0.93 (t, J = 7.4 Hz, 3H, H-3′) ppm; 13C NMR (DMSO, 100.63 MHz) δ: 190.3 (C=O), 170.0 (C-2″), 169.5 (C-2‴), 159.6 (C-4), 156.3 (C-2), 153.1 (C-3‴’, -5‴’), 144.0 (C-β), 139.6 (C-4‴’), 130.1 (C-1‴’), 129.0 (C-6), 125.4 (C-α), 125.1 (C-1), 124.5 (C-3), 107.4 (C-5), 106.2 (C-2‴’, -6‴’), 71.4 (C-1‴), 65.0 (C-1″), 60.1 (4‴’-OCH3), 56.0 (3‴’-, 5‴’-OCH3), 25.4 (C-1′), 22.3 (C-2′), 14.3 (C-3′) ppm; HRMS (ESI+): m/z Anal. Calc. for C25H28O10 (M+Na+) 511.15747, found 511.15885.
Synthesis of (E)-2-(3-Hydroxy-2-propyl-4-(3-(3,4,5-trimethoxyphenyl)acryloyl)phenoxy)-N-(3,4,5-trimethoxyphenyl)acetamide (13)
(E)-2-(3-hydroxy-2-propyl-4-(3-(3,4,5-trimethoxyphenyl)acryloyl)phenoxy)acetic acid (11, 85 mg, 1 Eq, 0.20 mmol) was dissolved in 5 mL of anhydrous dichloromethane and N-ethyl-N-isopropylpropan-2-amine (51 mg, 69 µL, 2 Eq, 0.39 mmol) was added. Then, 3,4,5-trimethoxyaniline (43 mg, 1.2 Eq, 0.24 mmol) was added to the mixture. After 10 min, COMU (0.13 g, 1.5 Eq, 0.30 mmol) was also added to the mixture. The reaction proceeded for 3 h at room temperature with stirring. After completion, the reaction mixture was poured onto ice. The solution was subsequently washed with a 1 M HCl solution and a saturated NaHCO3 solution, and dried over anhydrous sodium sulfate. The solvent was evaporated to dryness, and the obtained reaction mixture was purified via TLC (SiO2; dichloromethane:methanol; 97:3). A yellow solid (30% yield) corresponding to (E)-2-(3-hydroxy-2-propyl-4-(3-(3,4,5-trimethoxyphenyl)acryloyl)phenoxy)-N-(3,4,5-trimethoxyphenyl)acetamide (13) was obtained.
(E)-2-(3-hydroxy-2-propyl-4-(3-(3,4,5-trimethoxyphenyl)acryloyl)phenoxy)-N-(3,4,5-trimethoxyphenyl)acetamide (13): 95.9% purity; mp 199–201 °C (chloroform); 1H NMR (DMSO, 400.13 MHz) δ: 13.66 (s, 1H, 2-OH), 10.18 (s, 1H, 3″-NH-), 8.27 (d, J = 9.2 Hz, 1H, H-6), 7.95 (d, J = 15.4 Hz, 1H, H-β), 7.80 (d, J = 15.3 Hz, 1H, H-α), 7.25 (s, 2H, H-2‴, -6‴), 7.01 (s, 2H, H-5″, -9″), 6.61 (d, J = 9.2 Hz, 1H, H-5), 4.90 (s, 2H, H-1″), 3.86 (s, 6H, 3‴-, 5‴-OCH3), 3.74 (s, 6H, 6″-, 8″-OCH3), 3.72 (s, 3H, 4‴-OCH3), 3.62 (s, 3H, 7″-OCH3), 2.66 (t, J = 7.6 Hz, 1H, H-1′), 1.58–1.51 (m, 2H, H-2′), 0.94 (t, J = 7.4 Hz, 3H, H-3″) ppm; 13C NMR (DMSO, 100.63 MHz) δ: 192.4 (C=O), 165.8 (C-2″), 162.6 (C-2), 162.1 (C-4), 153.1 (C-3‴,-5‴), 152.8 (C-6″, -8″), 145.0 (C-β), 140.0 (C-4‴), 134.6 (C-4″), 133.7 (C-7″), 130.5 (C-6), 130.5 (C-1‴), 120.2 (C-α), 117.4 (C-3), 114.2 (C-1), 106.8 (C-2‴, -6‴), 103.4 (C-5), 97.0 (C-5″, -9″), 67.2 (C-1″), 60.1 (4″-, 4‴-OCH3), 56.2 (3‴-, 5‴-OCH3), 55.7 (6″-, 8″-OCH3), 24.2 (C-1′), 21.4 (C-2′), 14.2 (C-3′) ppm; HRMS (ESI+): m/z Anal. Calc. for C32H37NO10 (M+Na+) 618.23097, found 618.23023.
3.1.3. Synthesis of Compound 7 Analogues with a Flavone Scaffold (15, 16, and 18)
Synthesis of tert-Butyl (2-((4-oxo-8-propyl-2-(3,4,5-trimethoxyphenyl)-4H-chromen-7-yl)oxy)ethyl)carbamate (14)
To a solution of tert-butyl (E)-(2-(3-hydroxy-2-propyl-4-(3-(3,4,5-trimethoxyphenyl)acryloyl)phenoxy)ethyl)carbamate (6, 190 mg, 1 Eq, 369 µmol) in DMSO (10mL/mmol of chalcone), I2 (935 µg, 0.01 Eq, 3.69 µmol) was added. The mixture was heated at reflux for 24 h. Then, the mixture was poured into ice and a sodium thiosulfate solution (10%) was added. The obtained mixture was extracted with ethyl acetate, washed with water, and then dried over anhydrous sodium sulfate. The solvent was evaporated to dryness, and the obtained reaction mixture was purified via TLC (SiO2, chloroform:methanol; 95:5). A white solid (30% yield) corresponding to tert-butyl (2-((4-oxo-8-propyl-2-(3,4,5-trimethoxyphenyl)-4H-chromen-7-yl)oxy)ethyl)carbamate (14) was obtained.
tert-butyl (2-((4-oxo-8-propyl-2-(3,4,5-trimethoxyphenyl)-4H-chromen-7-yl)oxy)ethyl)carbamate (14): 98.9% purity; mp 191–193 °C (ethyl acetate); 1H NMR (DMSO, 400.14 MHz) δ: 7.88 (d, J = 8.9 Hz, 1H, H-5), 7.34 (s, 2H, H-2‴, -6‴), 7.20 (d, J = 8.9 Hz, 1H, H-6), 7.07 (s, 1H, H-3), 7.02 (t, J = 5.8 Hz, 1H, 3″-NH-), 4.18 (t, J = 5.5 Hz, 2H, H-1″), 3.91 (s, 6H, 3‴-, 5‴-OCH3), 3.90–3.67 (m, 2H, H-2″), 3.76 (s, 3H, 4‴-OCH3), 2.74 (t, J = 7.1 Hz, 2H, H-1′), 1.68–1.61 (m, 2H, H-2′), 1.38 (s, 9H, H-6″), 0.97 (t, J = 7.3 Hz, 3H, H-3′) ppm; 13C NMR (DMSO, 100.63 MHz) δ: 177.0 (C=O), 161.8 (C-2), 160.6 (C-7), 155.7 (C-4″), 154.4 (C-8a), 153.3 (C-3‴, -5‴), 140.4 (C-4‴), 126.8 (C-1‴), 123.8 (C-5), 118.2 (C-4a), 117.2 (C-8), 110.4 (C-6), 106.1 (C-3), 103.5 (C-2‴, -6‴), 77.7 (C-5″), 67.6 (C-1″), 60.2 (4‴-OCH3), 56.3 (C-2″), 56.1 (3‴-, 5‴-OCH3), 28.2 (6″-CH3), 24.8 (C-1′), 22.1 (C-2′), 14.2 (C-3′) ppm; HRMS (ESI+): m/z Anal. Calc. for C28H35NO8 (M+H+) 514.24354, found 514.24311.
Synthesis of (E)-N-(2-((4-oxo-8-Propyl-2-(3,4,5-trimethoxyphenyl)-4H-chromen-7-yl)oxy)ethyl)-3-(3,4,5-trimethoxyphenyl)acrylamide (15)
To a solution of (E)-N-(2-(3-hydroxy-2-propyl-4-((E)-3-(3,4,5-trimethoxyphenyl)acryloyl)phenoxy)ethyl)-3-(3,4,5-trimethoxyphenyl)acrylamide (7, 10 mg, 1 Eq, 16 μmol) in DMSO (10mL/mmol of chalcone), I2 (40 μg, 0.01 Eq, 0.16 μmol) was added. The mixture was heated at reflux for 2 h. Then, the mixture was poured into ice and a sodium thiosulfate solution (10%) was added. The obtained mixture was extracted with ethyl acetate, washed with water, and then dried over anhydrous sodium sulfate. The solvent was evaporated to dryness, and the obtained reaction mixture was purified via TLC (SiO2, chloroform:methanol; 95:5). A white solid (35% yield) corresponding to (E)-N-(2-((4-oxo-8-propyl-2-(3,4,5-trimethoxyphenyl)-4H-chromen-7-yl)oxy)ethyl)-3-(3,4,5-trimethoxyphenyl)acrylamide (15) was obtained.
(E)-N-(2-((4-oxo-8-propyl-2-(3,4,5-trimethoxyphenyl)-4H-chromen-7-yl)oxy)ethyl)-3-(3,4,5-trimethoxyphenyl)acrylamide (15): 98.4% purity; mp 198–199 °C (chloroform); 1H NMR (CDCl3, 400.13 MHz) δ: 8.09 (d, J = 8.9 Hz, 1H, H-5), 7.59 (d, J = 15.8 Hz, 1H, H-β), 7.15 (s, 2H, H-2‴, -6‴), 7.02 (d, J = 8.9 Hz, 1H, H-6), 6.73 (s, 2H, H-6″, -10″), 6.72 (s, 1H, H-3), 6.32 (d, J = 15.5 Hz, 1H, H-α), 5.97 (t, J = 5.7 Hz, 1H, 3″-NH-), 4.28 (t, J = 5.2 Hz, 2H, H-1″), 3.95 (s, 6H, 3‴-, 5‴-OCH3), 3.94 (s, 3H, 4‴-OCH3), 3.88 (s, 6H, 7″-, 9″-OCH3), 3.87 (s, 3H, 8″-OCH3), 3.95–3.90 (m, 2H, H-2″), 2.98 (t, J = 7.5 Hz, 1H, H-1′), 1.78–1.72 (m, 2H, H-2′), 1.06 (t, J = 7.4 Hz, 3H, H-3′) ppm; 13C NMR (CDCl3, 100.63 MHz) δ: 178.9 (C=O), 166.2 (C-4″), 162.9 (C-2), 160.6 (C-7), 155.4 (C-8a), 153.8 (C-3‴,-5‴), 153.6 (C-7″, -9″), 142.1 (C-β), 140.0 (C-4‴), 139.2 (C-8″), 130.4 (C-5″), 127.4 (C-1‴), 124.7 (C-5), 119.4 (C-4a), 119.3 (C-α), 118.7 (C-8), 109.9 (C-6), 106.6 (C-3), 105.2 (C-2‴, -6‴), 103.5 (C-6″, -10″), 67.8 (C-1″), 61.2 (4‴-OCH3), 61.1 (4″-OCH3), 56.4 (3‴-, 5‴-OCH3), 56.3 (7″-, 9″-OCH3), 39.5 (C-2″), 25.7 (C-1′), 22.8 (C-2′), 14.7 (C-3′) ppm; HRMS (ESI+): m/z Anal. Calc. for C35H39NO10 (M+CH3OHH+) 666.29089, found 666.29022.
Synthesis of 3,4,5-Trimethoxy-N-(2-((4-oxo-8-propyl-2-(3,4,5-trimethoxyphenyl)-4H-chromen-7-yl)oxy)ethyl)benzamide (16)
To a solution of (E)-N-(2-(3-hydroxy-2-propyl-4-(3-(3,4,5-trimethoxyphenyl)acryloyl)phenoxy)ethyl)-3,4,5-trimethoxybenzamide (8, 10 mg, 1 Eq, 16 μmol) in DMSO (10mL/mmol of chalcone), I2 (42 μg, 0.01 Eq, 0.16 μmol) was added. The mixture was heated at reflux for 2 h. Then, the mixture was poured into ice and a sodium thiosulfate solution (10%) was added. The obtained mixture was extracted with ethyl acetate, washed with water, and then dried over anhydrous sodium sulfate. The solvent was evaporated to dryness and the reaction mixture was purified via TLC (SiO2, chloroform:methanol; 95:5). A white solid (33% yield) corresponding to 3,4,5-trimethoxy-N-(2-((4-oxo-8-propyl-2-(3,4,5-trimethoxyphenyl)-4H-chromen-7-yl)oxy)ethyl)benzamide (16) was obtained.
3,4,5-trimethoxy-N-(2-((4-oxo-8-propyl-2-(3,4,5-trimethoxyphenyl)-4H-chromen-7-yl)oxy)ethyl)benzamide (16): 95.8% purity; mp 196–199 °C (chloroform); 1H NMR (DMSO, 400.13 MHz) δ: 8.64 (t, J = 5.6 Hz, 1H, 3″-NH-), 7.90 (d, J = 8.8 Hz, 1H, H-5), 7.33 (s, 2H, H-2‴, -6‴), 7.27 (d, J = 8.9 Hz, 1H, H-6), 7.07 (s, 2H, H-6″, -10″), 7.07 (s, 1H, H-3), 4.02 (t, J = 5.2 Hz, 2H, H-1″), 3.90 (s, 6H, 3‴-, 5‴-OCH3), 3.81 (s, 6H, 7″-, 9″-OCH3), 3.75 (s, 3H, 4‴-OCH3), 3.69 (s, 3H, 8″-OCH3), 3.51–3.46 (m, 2H, H-2″), 2.89 (t, J = 7.4 Hz, 2H, H-1′), 1.68–1.59 (m, 2H, H-2′), 0.89 (t, J = 7.4 Hz, 3H, H-3′) ppm; 13C NMR (DMSO, 100.63 MHz) δ: 177.4 (C=O), 166.1 (C-4″), 162.5 (C-2), 160.2 (C-7), 155.9 (C-8a), 154.5 (C-3‴,-5‴), 154.4 (C-7″, -9″), 140.4 (C-4‴), 140.2 (C-8″), 129.8 (C-5″), 126.8 (C-1‴), 124.3 (C-5), 119.8 (C-4a), 117.3 (C-8), 109.7 (C-6), 106.0 (C-3), 104.7 (C-6″, -10″), 103.5 (C-2‴, -6‴), 67.6 (C-1″), 61.4 (4‴-OCH3), 60.0 (8″-OCH3), 56.1 (3‴-, 5‴-OCH3), 55.9 (7″-, 9″-OCH3), 38.9 (C-2″), 30.7 (C-1′), 22.1 (C-2′), 14.1 (C-3′) ppm; HRMS (ESI+): m/z Anal. Calc. for C33H37NO10 (M+H+) 608.24902, found 608.24725.
Synthesis of 2-((4-oxo-8-Propyl-2-(3,4,5-trimethoxyphenyl)-4H-chromen-7-yl)oxy)-N-(3,4,5-trimethoxyphenyl)acetamide (17)
Method A: To a solution of (E)-2-(3-hydroxy-2-propyl-4-(3-(3,4,5-trimethoxyphenyl)acryloyl)phenoxy)-N-(3,4,5-trimethoxyphenyl)acetamide (13, 100 mg, 1 Eq, 168 µmol) in DMSO (10 mL/mmol of chalcone), I2 (427 µg, 0.01 Eq, 1.68 µmol) was added. The mixture was heated at reflux for 24 h. Then, the mixture was poured into ice and a sodium thiosulfate solution (10%) was added. The obtained mixture was extracted with ethyl acetate, washed with water, and then dried over anhydrous sodium sulfate. The solvent was evaporated to dryness and the obtained mixture was purified via TLC (SiO2; chloroform:methanol; 95:5). A yellow solid (30% yield) corresponding to (2-((4-oxo-8-propyl-2-(3,4,5-trimethoxyphenyl)-4H-chromen-7-yl)oxy)-N-(3,4,5-trimethoxyphenyl)acetamide (17) was obtained.
Method B: 2-((4-oxo-8-propyl-2-(3,4,5-trimethoxyphenyl)-4H-chromen-7-yl)oxy)acetic acid (18, 100 mg, 1 Eq, 233 µmol) was dissolved in 5 mL of anhydrous dichloromethane and N-ethyl-N-isopropylpropan-2-amine (60.3 mg, 81.3 µL, 2 Eq, 467 µmol), and 3,4,5-trimethoxyaniline (51.3 mg, 1.2 Eq, 280 µmol) was added to the mixture. After 10 min, COMU (150 mg, 1.5 Eq, 350 µmol) was also added to the mixture. The reaction proceeded for 5 h at room temperature with stirring. After completion, the reaction mixture was poured onto ice. The solution was subsequently washed with 1M HCl solution and saturated NaHCO3 solution, and dried over anhydrous sodium sulfate. The solvent was evaporated to dryness and the obtained reaction mixture was purified via TLC (SiO2; chloroform:methanol; 95:5). A yellow solid (17.5% yield) corresponding to (2-((4-oxo-8-propyl-2-(3,4,5-trimethoxyphenyl)-4H-chromen-7-yl)oxy)-N-(3,4,5-trimethoxyphenyl)acetamide (17) was obtained.
(2-((4-oxo-8-propyl-2-(3,4,5-trimethoxyphenyl)-4H-chromen-7-yl)oxy)-N-(3,4,5-trimethoxyphenyl)acetamide (17): 96.4% purity; mp 195–197 °C (chloroform); 1H NMR (DMSO, 400.13 MHz) δ: 10.15 (s,1H, 3″-NH-), 7.90 (d, J = 9.1 Hz, 1H, H-5), 7.36 (s, 2H, H-2‴, -6‴), 7.00 (s, 2H, H-5″, -9″), 7.14 (d, J = 9.0 Hz, 1H, H-6), 7.10 (s, 1H, H-3), 4.94 (s, 2H, H-1″), 3.92 (s, 6H, 3‴-, 5‴-OCH3), 3.76 (s, 3H, 4‴-OCH3), 3.76 (s, 6H, 6″-, 8″-OCH3), 3.62 (s, 3H, 7″-OCH3), 3.07 (t, J = 7.3 Hz, 1H, H-1′), 1.71–1.67 (m, 2H, H-2′), 1.01 (t, J = 7.3 Hz, 3H, H-3′) ppm; 13C NMR (DMSO, 100.63 MHz) δ: 177.0 (C=O), 166.9 (C-2″), 162.9 (C-2), 160.1 (C-7), 154.6 (C-8a), 153.0 (C-3‴, -5‴), 152.8 (C-6″, -8″), 140.0 (C-4‴), 134.6 (C-7″), 133.8 (C-4″), 126.7 (C-1‴), 123.7 (C-5), 119.5 (C-8), 118.5 (C-4a), 110.4 (C-6), 106.1 (C-3), 103.8 (C-2‴, -6‴), 97.0 (C-5″, -9″), 67.9 (C-1″), 60.3 (4‴-OCH3), 60.1 (7″-OCH3), 56.1 (3‴-, 5‴-OCH3), 55.7 (6″-, 8″-OCH3), 24.1 (C-1′), 22.3 (C-2′), 14.5 (C-3′) ppm; HRMS (ESI+): m/z Anal. Calc. for C33H35NO10 (M+K+) 632.18925, found 632.18769.
2-((4-oxo-8-Propyl-2-(3.4.5-trimethoxyphenyl)-4H-chromen-7-yl)oxy)acetic acid (18)
To a solution of (E)-2-(3-hydroxy-2-propyl-4-(3-(3,4,5-trimethoxyphenyl)acryloyl)phenoxy)acetic acid (11, 100 mg, 1 Eq, 232 µmol) in DMSO (10mL/mmol of chalcone), I2 (590 µg, 0.01 Eq, 2.32 µmol) was added. The mixture was heated at reflux for 24 h. Then, the mixture was poured into ice and a sodium thiosulfate solution (10%) was added. The obtained mixture was extracted with ethyl acetate, washed with water, and then dried over anhydrous sodium sulfate. The solvent was evaporated to dryness, and the obtained oil was purified via TLC (SiO2, n-hexane: ethyl acetate:formic acid (3:7:0.1%). A white solid (35% yield) corresponding to 2-((4-oxo-8-propyl-2-(3,4,5-trimethoxyphenyl)-4H-chromen-7-yl)oxy)acetic acid (18) was obtained.
2-((4-oxo-8-propyl-2-(3.4.5-trimethoxyphenyl)-4H-chromen-7-yl)oxy)acetic acid (18): 95.9% purity; mp 199–201 °C (chloroform); 1H NMR (THF, 400.13 MHz) δ: 7.92 (d, J = 8.8 Hz, 1H, H-5), 7.38 (s, 2H, H-2‴, -6‴), 7.10 (d, J = 8.9 Hz, 1H, H-6), 7.01 (s, 1H, H-3), 4.85 (s, 2H, H-1″), 3.94 (s, 6H, 3‴-, 5‴-OCH3), 3.79 (s, 3H, 4‴-OCH3), 3.04 (t, J = 7.7 Hz, 2H, H-1′), 1.73–1.72 (m, 2H, H-2′), 1.04 (t, J = 7.4 Hz, 3H, H-3′) ppm; 13C NMR (THF, 100.63 MHz) δ: 178.1 (C=O), 170.8 (C-2″), 163.2 (C-2), 161.1 (C-7), 156.1 (C-8a), 154.9 (C-3‴, -5‴), 142.2 (C-4‴), 128.3 (C-1‴), 124.9 (C-5), 119.8 (C-8), 119.2 (C-4a), 111.1 (C-6), 107.3 (C-3), 104.7 (C-2‴, -6‴), 66.4 (C-1″), 61.0 (4‴-OCH3), 57.0 (3‴-, 5‴-OCH3), 26.4 (C-1′), 23.5 (C-2′), 15.1 (C-3′) ppm; HRMS (ESI+): m/z Anal. Calc. for C23H24O8 (M+H+) 429.15439, found 429.15658.


 ,
, 












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
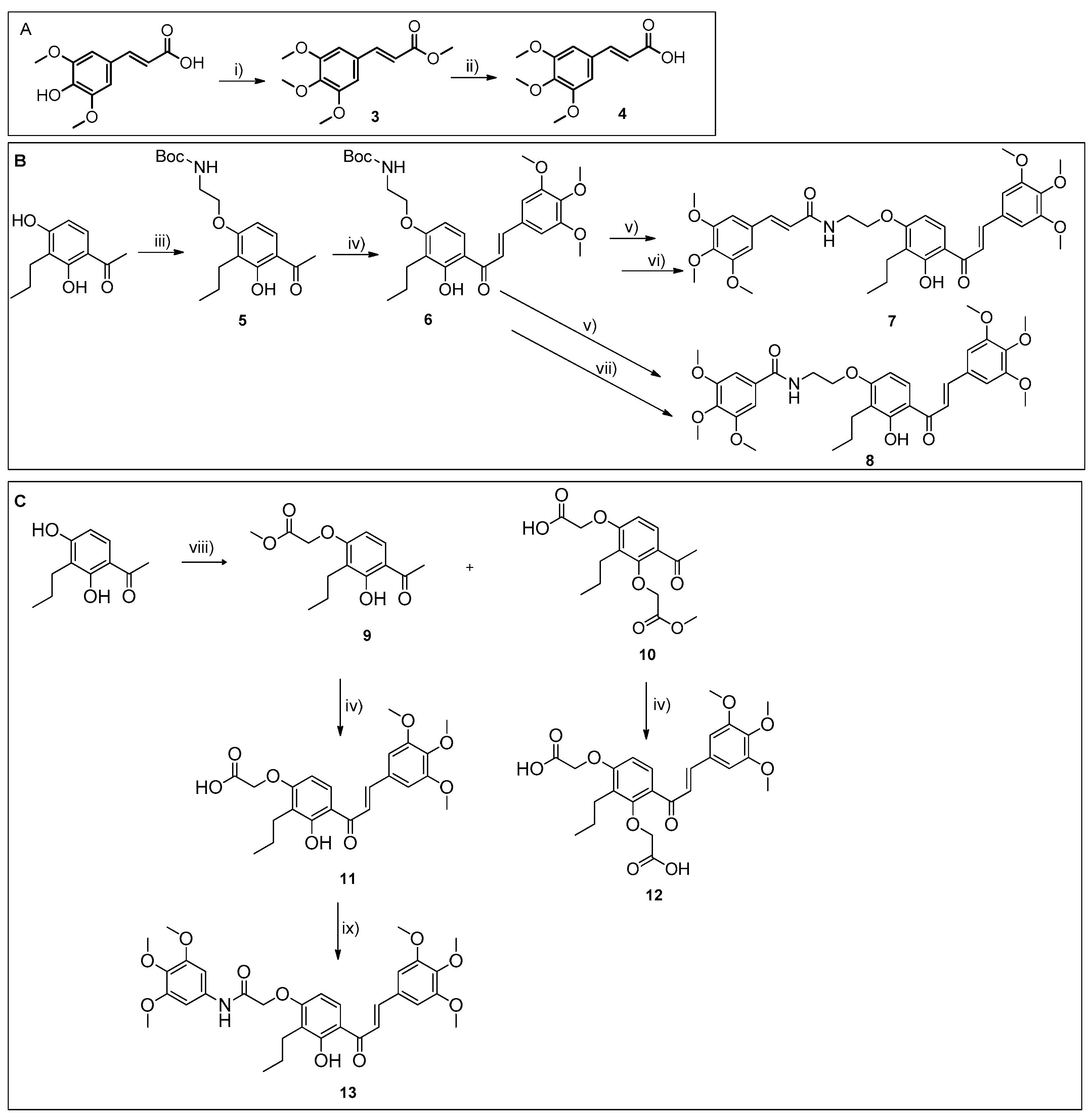{kind=link}
{kind=link}