
Overview of Cardiac Arrhythmias and Treatment Strategies

Abstract
1. Introduction
2. General Principles
2.1. Genetics
2.2. Myocardial Ischemia
2.3. Inflammation
2.4. Diet and Metabolic Disorders
3. Pharmacotherapy
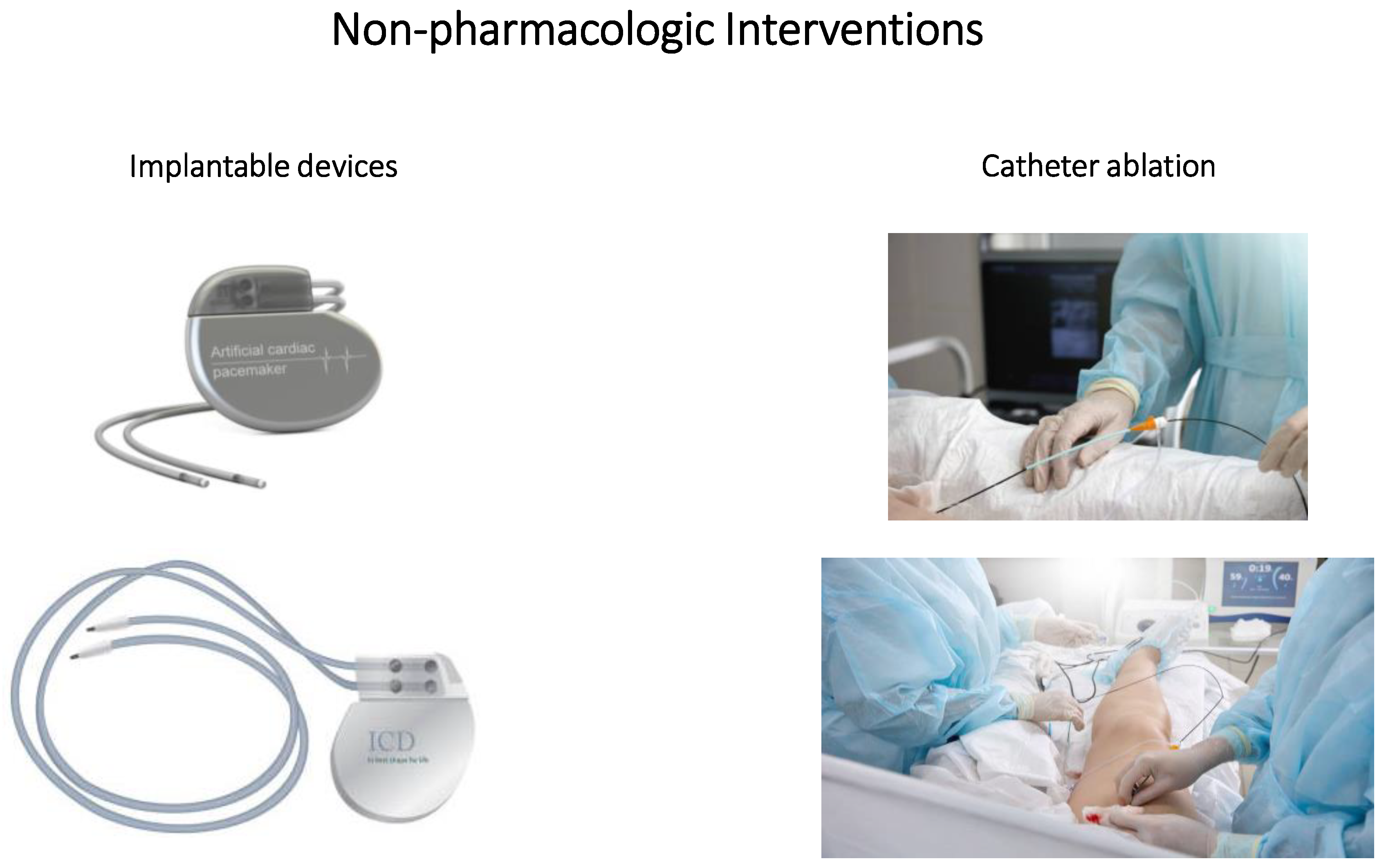
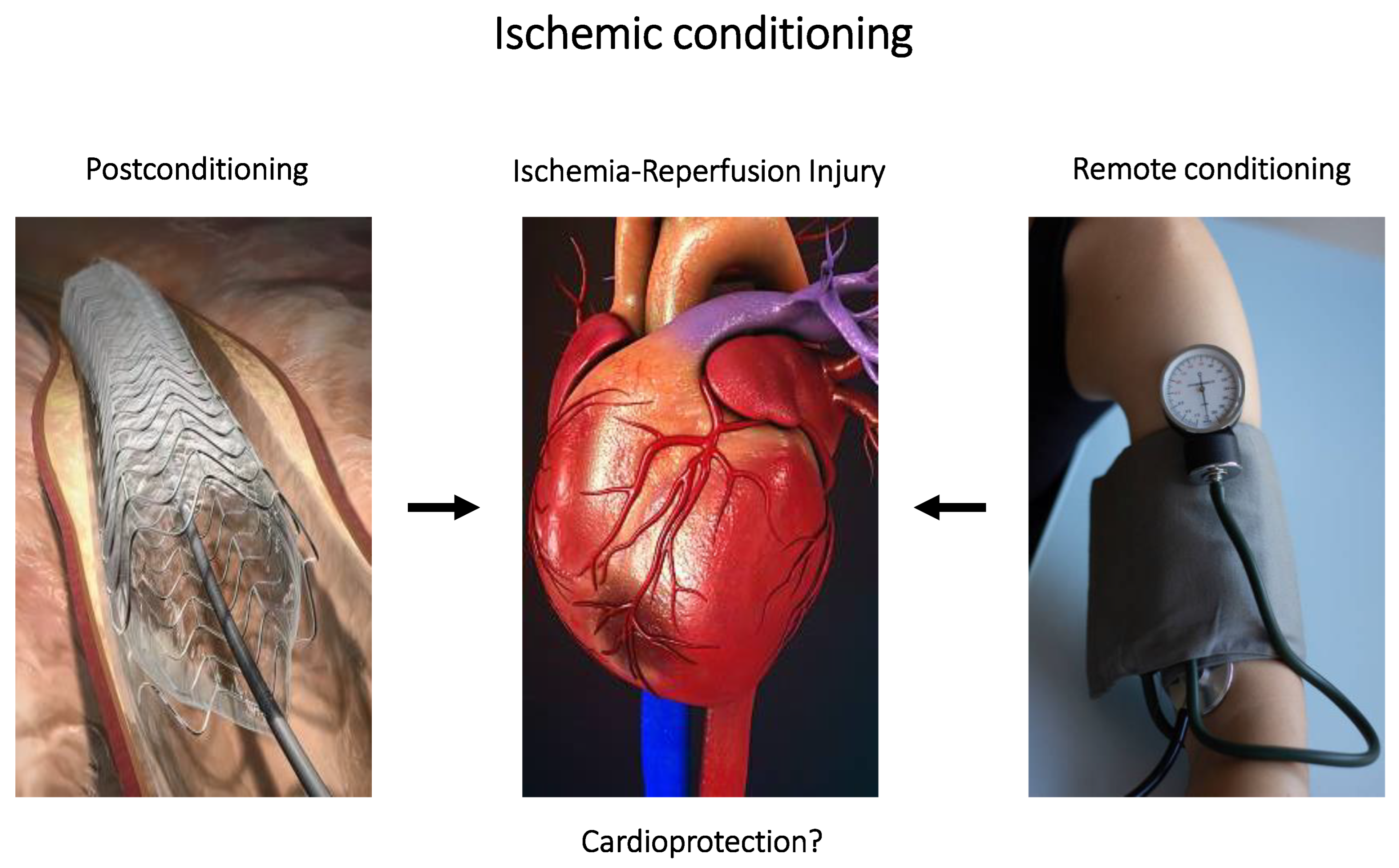
4. Non-Pharmacologic Interventions
4.1. Implantable Devices
4.2. Catheter Ablation
4.3. Ischemic Conditioning
5. Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mirchandani, S.; Phoon, C.K. Sudden cardiac death: A 2400-year-old diagnosis? Int. J. Cardiol. 2003, 90, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Luderitz, B. Historical perspectives of cardiac electrophysiology. Hell. J. Cardiol. 2009, 50, 3–16. [Google Scholar]
- O’Neill, M.; Jaïs, P.; Hocini, M.; Sacher, F.; Klein, G.J.; Clémenty, J.; Haïssaguerre, M. Catheter ablation for atrial fibrillation. Circulation 2007, 116, 1515–1523. [Google Scholar] [CrossRef] [PubMed]
- Levy, S. Factors predisposing to the development of atrial fibrillation. Pacing Clin. Electrophysiol. 1997, 20, 2670–2674. [Google Scholar] [CrossRef]
- Schotten, U.; Verheule, S.; Kirchhof, P.; Goette, A. Pathophysiological mechanisms of atrial fibrillation: A translational appraisal. Physiol. Rev. 2011, 91, 265–325. [Google Scholar] [CrossRef] [PubMed]
- Schotten, U.; Dobrev, D.; Platonov, P.G.; Kottkamp, H.; Hindricks, G. Current controversies in determining the main mechanisms of atrial fibrillation. J. Intern. Med. 2016, 279, 428–438. [Google Scholar] [CrossRef]
- Allessie, M.; Ausma, J.; Schotten, U. Electrical, contractile and structural remodeling during atrial fibrillation. Cardiovasc. Res. 2002, 54, 230–246. [Google Scholar] [CrossRef]
- Nattel, S.; Harada, M. Atrial remodeling and atrial fibrillation: Recent advances and translational perspectives. J. Am. Coll. Cardiol. 2014, 63, 2335–2345. [Google Scholar] [CrossRef]
- Iwasaki, Y.K.; Nishida, K.; Kato, T.; Nattel, S. Atrial fibrillation pathophysiology: Implications for management. Circulation 2011, 124, 2264–2274. [Google Scholar] [CrossRef]
- Lalani, G.G.; Schricker, A.; Gibson, M.; Rostamian, A.; Krummen, D.E.; Narayan, S.M. Atrial conduction slows immediately before the onset of human atrial fibrillation: A bi-atrial contact mapping study of transitions to atrial fibrillation. J. Am. Coll. Cardiol. 2012, 59, 595–606. [Google Scholar] [CrossRef]
- Skibsbye, L.; Jespersen, T.; Christ, T.; Maleckar, M.M.; van den Brink, J.; Tavi, P.; Koivumäki, J.T. Refractoriness in human atria: Time and voltage dependence of sodium channel availability. J. Mol. Cell. Cardiol. 2016, 101, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Xia, Y.; Carlson, J.; Kongstad, O.; Yuan, S. Atrial average conduction velocity in patients with and without paroxysmal atrial fibrillation. Clin. Physiol. Funct. Imaging 2017, 37, 596–601. [Google Scholar] [CrossRef] [PubMed]
- Narayan, S.M.; Franz, M.R.; Clopton, P.; Pruvot, E.J.; Krummen, D.E. Repolarization alternans reveals vulnerability to human atrial fibrillation. Circulation 2011, 123, 2922–2930. [Google Scholar] [CrossRef]
- Stătescu, C.; Sascău, R.A.; Georgescu, C.A. Functional Anatomy in Arrhythmias and Vascular Support of the Conduction System. In Cardiac Arrhythmias: From Basic Mechanism to State-of-the-Art Management; Kibos, A.S., Knight, B.P., Essebag, V., Fishberger, S.B., Slevin, M., Țintoiu, I.C., Eds.; Springer: London, UK, 2014; pp. 35–42. [Google Scholar]
- Kanno, S.; Saffitz, J.E. The role of myocardial gap junctions in electrical conduction and arrhythmogenesis. Cardiovasc. Pathol. 2001, 10, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Larson, J.; Rich, L.; Deshmukh, A.; Judge, E.C.; Liang, J.J. Pharmacologic Management for Ventricular Arrhythmias: Overview of Anti-Arrhythmic Drugs. J. Clin. Med. 2022, 11, 3233. [Google Scholar] [CrossRef]
- Tse, G. Mechanisms of cardiac arrhythmias. J. Arrhythmia 2016, 32, 75–81. [Google Scholar] [CrossRef]
- Lakatta, E.G.; Vinogradova, T.; Lyashkov, A.; Sirenko, S.; Zhu, W.; Ruknudin, A.; Maltsev, V.A. The integration of spontaneous intracellular Ca2+ cycling and surface membrane ion channel activation entrains normal automaticity in cells of the heart’s pacemaker. Ann. N. Y. Acad. Sci. 2006, 1080, 178–206. [Google Scholar] [CrossRef]
- Jalife, J.; Delmar, M.; Anumonwo, J.; Berenfeld, O.; Kalifa, J. Basic Cardiac Electrophysiology for the Clinician, 2nd ed.; Wiley-Blackwell: Hoboken, NJ, USA, 2009. [Google Scholar]
- Vagos, M.; van Herck, I.G.M.; Sundnes, J.; Arevalo, H.J.; Edwards, A.G.; Koivumaki, J.T. Computational Modeling of Electrophysiology and Pharmacotherapy of Atrial Fibrillation: Recent Advances and Future Challenges. Front. Physiol. 2018, 9, 1221. [Google Scholar] [CrossRef]
- Ravens, U.; Wettwer, E. Ultra-rapid delayed rectifier channels: Molecular basis and therapeutic implications. Cardiovasc. Res. 2011, 89, 776–785. [Google Scholar] [CrossRef]
- Skibsbye, L.; Poulet, C.; Diness, J.G.; Bentzen, B.H.; Yuan, L.; Kappert, U.; Matschke, K.; Wettwer, E.; Ravens, U.; Grunnet, M.; et al. Small-conductance calcium-activated potassium (SK) channels contribute to action potential repolarization in human atria. Cardiovasc. Res. 2014, 103, 156–167. [Google Scholar] [CrossRef]
- Liang, B.; Nissen, J.D.; Laursen, M.; Wang, X.; Skibsbye, L.; Hearing, M.C.; Andersen, M.N.; Rasmussen, H.B.; Wickman, K.; Grunnet, M.; et al. G-protein-coupled inward rectifier potassium current contributes to ventricular repolarization. Cardiovasc. Res. 2014, 101, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Tomaselli, G.F.; Marban, E. Electrophysiological remodeling in hypertrophy and heart failure. Cardiovasc. Res. 1999, 42, 270–283. [Google Scholar] [CrossRef] [PubMed]
- Nattel, S. Ionic determinants of atrial fibrillation and Ca2+ channel abnormalities: Cause, consequence, or innocent bystander? Circ. Res. 1999, 85, 473–476. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.; Akar, F.G.; Tomaselli, G.F. Molecular basis of arrhythmias. Circulation 2005, 112, 2517–2529. [Google Scholar] [CrossRef]
- Opie, L.H.; Coetzee, W.A. Role of calcium ions in reperfusion arrhythmias: Relevance to pharmacologic intervention. Cardiovasc. Drugs Ther. 1988, 2, 623–636. [Google Scholar] [CrossRef]
- Cascio, W.E.; Yang, H.; Johnson, T.A.; Muller-Borer, B.J.; Lemasters, J.J. Electrical properties and conduction in reperfused papillary muscle. Circ. Res. 2001, 89, 807–814. [Google Scholar] [CrossRef]
- Manning, A.S.; Hearse, D.J. Reperfusion-induced arrhythmias: Mechanisms and prevention. J. Mol. Cell. Cardiol. 1984, 16, 497–518. [Google Scholar] [CrossRef]
- Tanaka, K.; Hearse, D.J. Reperfusion-induced arrhythmias in the isolated rabbit heart: Characterization of the influence of the duration of regional ischemia and the extracellular potassium concentration. J. Mol. Cell. Cardiol. 1988, 20, 201–211. [Google Scholar] [CrossRef]
- Noble, D. A modification of the Hodgkin–Huxley equations applicable to Purkinje fibre action and pace-maker potentials. J. Physiol. 1962, 160, 317–352. [Google Scholar] [CrossRef]
- Passini, E.; Britton, O.J.; Lu, H.R.; Rohrbacher, J.; Hermans, A.N.; Gallacher, D.J.; Greig, R.J.H.; Bueno-Orovio, A.; Rodriguez, B. Human In Silico Drug Trials Demonstrate Higher Accuracy than Animal Models in Predicting Clinical Pro-Arrhythmic Cardiotoxicity. Front. Physiol. 2017, 8, 668. [Google Scholar] [CrossRef]
- Colatsky, T.; Fermini, B.; Gintant, G.; Pierson, J.B.; Sager, P.; Sekino, Y.; Strauss, D.G.; Stockbridge, N. The Comprehensive in Vitro Proarrhythmia Assay (CiPA) initiative—Update on progress. J. Pharmacol. Toxicol. Method. 2016, 81, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Miragoli, M.; Salvarani, N.; Rohr, S. Myofibroblasts induce ectopic activity in cardiac tissue. Circ. Res. 2007, 101, 755–758. [Google Scholar] [CrossRef] [PubMed]
- Pastore, J.M.; Rosenbaum, D.S. Role of structural barriers in the mechanism of alternans-induced reentry. Circ. Res. 2000, 87, 1157–1163. [Google Scholar] [CrossRef] [PubMed]
- Gaeta, S.A.; Bub, G.; Abbott, G.W.; Christini, D.J. Dynamical mechanism for subcellular alternans in cardiac myocytes. Circ. Res. 2009, 105, 335–342. [Google Scholar] [CrossRef]
- Gaeta, S.A.; Krogh-Madsen, T.; Christini, D.J. Feedback-control induced pattern formation in cardiac myocytes: A mathematical modeling study. J. Theor. Biol. 2010, 266, 408–418. [Google Scholar] [CrossRef]
- Gaeta, S.A.; Christini, D.J. Non-linear dynamics of cardiac alternans: Subcellular to tissue-level mechanisms of arrhythmia. Front. Physiol. 2012, 3, 157. [Google Scholar] [CrossRef]
- Ortmans, S.; Daval, C.; Aguilar, M.; Compagno, P.; Cadrin-Tourigny, J.; Dyrda, K.; Rivard, L.; Tadros, R. Pharmacotherapy in inherited and acquired ventricular arrhythmia in structurally normal adult hearts. Expert. Opin. Pharmacother. 2019, 20, 2101–2114. [Google Scholar] [CrossRef]
- Prystowsky, E.N.; Padanilam, B.J.; Joshi, S.; Fogel, R.I. Ventricular arrhythmias in the absence of structural heart disease. J. Am. Coll. Cardiol. 2012, 59, 1733–1744. [Google Scholar] [CrossRef]
- Killu, A.M.; Stevenson, W.G. Ventricular tachycardia in the absence of structural heart disease. Heart 2019, 105, 645–656. [Google Scholar] [CrossRef]
- Wang, M.; Tu, X. The Genetics and Epigenetics of Ventricular Arrhythmias in Patients without Structural Heart Disease. Front. Cardiovasc. Med. 2022, 9, 891399. [Google Scholar] [CrossRef]
- Bermúdez-Jiménez, F.J.; Carriel, V.; Brodehl, A.; Alaminos, M.; Campos, A.; Schirmer, I.; Milting, H.; Abril, B.; Álvarez, M.; López-Fernández, S.; et al. Novel Desmin Mutation p.Glu401Asp Impairs Filament Formation, Disrupts Cell Membrane Integrity, and Causes Severe Arrhythmogenic Left Ventricular Cardiomyopathy/Dysplasia. Circulation 2018, 137, 1595–1610. [Google Scholar] [CrossRef] [PubMed]
- Van der Heijden, J.F.; Hassink, R.J. The phospholamban p.Arg14del founder mutation in Dutch patients with arrhythmogenic cardiomyopathy. Neth. Hear. J. 2013, 21, 284–285. [Google Scholar] [CrossRef] [PubMed]
- Forleo, C.; Carmosino, M.; Resta, N.; Rampazzo, A.; Valecce, R.; Sorrentino, S.; Iacoviello, M.; Pisani, F.; Procino, G.; Gerbino, A.; et al. Clinical and functional characterization of a novel mutation in lamin a/c gene in a multigenerational family with arrhythmogenic cardiac laminopathy. PLoS ONE 2015, 10, e0121723. [Google Scholar] [CrossRef]
- Austin, K.M.; Trembley, M.A.; Chandler, S.F.; Sanders, S.P.; Saffitz, J.E.; Abrams, D.J.; Pu, W.T. Molecular mechanisms of arrhythmogenic cardiomyopathy. Nat. Rev. Cardiol. 2019, 16, 519–537. [Google Scholar] [CrossRef]
- Roselli, C.; Chaffin, M.D.; Weng, L.-C.; Aeschbacher, S.; Ahlberg, G.; Albert, C.M.; Almgren, P.; Alonso, A.; Anderson, C.D.; Aragam, K.G.; et al. Multi-ethnic genome-wide association study for atrial fibrillation. Nat. Genet. 2018, 50, 1225–1233. [Google Scholar] [CrossRef]
- Lozano-Velasco, E.; Franco, D.; Aranega, A.; Daimi, H. Genetics and Epigenetics of Atrial Fibrillation. Int. J. Mol. Sci. 2020, 21, 5717. [Google Scholar] [CrossRef] [PubMed]
- Cao, F.; Li, Z.; Ding, W.M.; Yan, L.; Zhao, Q.Y. LncRNA PVT1 regulates atrial fibrosis via miR-128-3p-SP1-TGF-beta1-Smad axis in atrial fibrillation. Mol. Med. 2019, 25, 7. [Google Scholar] [CrossRef]
- Zhao, J.B.; Zhu, N.; Lei, Y.H.; Zhang, C.J.; Li, Y.H. Modulative effects of lncRNA TCONS_00202959 on autonomic neural function and myocardial functions in atrial fibrillation rat model. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 8891–8897. [Google Scholar] [CrossRef]
- Yao, L.; Zhou, B.; You, L.; Hu, H.; Xie, R. LncRNA MIAT/miR-133a-3p axis regulates atrial fibrillation and atrial fibrillation-induced myocardial fibrosis. Mol. Biol. Rep. 2020, 47, 2605–2617. [Google Scholar] [CrossRef]
- Shen, K.; Tu, T.; Yuan, Z.; Yi, J.; Zhou, Y.; Liao, X.; Liu, Q.; Zhou, X. DNA methylation dysregulations in valvular atrial fibrillation. Clin. Cardiol. 2017, 40, 686–691. [Google Scholar] [CrossRef]
- Zhang, D.; Hu, X.; Li, J.; Hoogstra-Berends, F.; Zhuang, Q.; Esteban, M.A.; de Groot, N.; Henning, R.H.; Brundel, B.J. Converse role of class I and class IIa HDACs in the progression of atrial fibrillation. J. Mol. Cell. Cardiol. 2018, 125, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Wu, C.T.; Qi, X.; Meijering, R.A.; Hoogstra-Berends, F.; Tadevosyan, A.; Deniz, G.C.; Durdu, S.; Akar, A.R.; Sibon, O.C.; et al. Activation of histone deacetylase-6 induces contractile dysfunction through derailment of alpha-tubulin proteostasis in experimental and human atrial fibrillation. Circulation 2014, 129, 346–358. [Google Scholar] [CrossRef]
- Egger, G.; Liang, G.; Aparicio, A.; Jones, P.A. Epigenetics in human disease and prospects for epigenetic therapy. Nature 2004, 429, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, G.; Heard, E. Advances in epigenetics link genetics to the environment and disease. Nature 2019, 571, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Cao, J. The functional role of long non-coding RNAs and epigenetics. Biol. Proced. Online 2014, 16, 11. [Google Scholar] [CrossRef] [PubMed]
- Priori, S.G.; Blomström-Lundqvist, C.; Mazzanti, A.; Blom, N.; Borggrefe, M.; Camm, J.; Elliott, P.M.; Fitzsimons, D.; Hatala, R.; Hindricks, G.; et al. 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: The Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC). Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Eur. Heart J. 2015, 36, 2793–2867. [Google Scholar] [CrossRef]
- Sridharan, A.; Bradfield, J.S.; Shivkumar, K.; Ajijola, O.A. Autonomic nervous system and arrhythmias in structural heart disease. Auton. Neurosci. 2022, 243, 103037. [Google Scholar] [CrossRef]
- De Azevodo, I.M.; Watanabe, Y.; Dreifus, L.S. Atrioventricular junctional rhythm: Classification and clinical significance. Chest 1973, 64, 732–740. [Google Scholar] [CrossRef]
- Jalife, J.; Antzelevitch, C.; Moe, G.K. The case for modulated parasystole. Pacing Clin. Electrophysiol. 1982, 5, 911–926. [Google Scholar] [CrossRef]
- Gussak, I.; Antzelevitch, C.; Hammill, S.C.; Shen, W.K.; Bjerregaard, P. Cardiac Repolarization: Bridging Basic and Clinical Science (Contemporary Cardiology); Humana Press: Totowa, NJ, USA, 2003. [Google Scholar]
- Wu, T.-J.; Ong, J.J.; Hwang, C.; Lee, J.J.; Fishbein, M.C.; Czer, L.; Trento, A.; Blanche, C.; Kass, R.M.; Mandel, W.J.; et al. Characteristics of wave fronts during ventricular fibrillation in human hearts with dilated cardiomyopathy: Role of increased fibrosis in the generation of reentry. J. Am. Coll. Cardiol. 1998, 32, 187–196. [Google Scholar] [CrossRef]
- Kawara, T.; Derksen, R.; de Groot, J.R.; Coronel, R.; Tasseron, S.; Linnenbank, A.C.; Hauer, R.N.; Kirkels, H.; Janse, M.J.; de Bakker, J.M. Activation delay after premature stimulation in chronically diseased human myocardium relates to the architecture of interstitial fibrosis. Circulation 2001, 104, 3069–3075. [Google Scholar] [CrossRef] [PubMed]
- Braunwald, E.; Kloner, R.A. Myocardial reperfusion: A double-edged sword? J. Clin. Investig. 1985, 76, 1713–1719. [Google Scholar] [CrossRef] [PubMed]
- Yellon, D.M.; Hausenloy, D.J. Myocardial reperfusion injury. N. Engl. J. Med. 2007, 357, 1121–1135. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, R.; Balla, C.; Malagù, M.; Guardigli, G.; Morciano, G.; Bertini, M.; Biscaglia, S.; Campo, G. Reperfusion Damage- A Story of Success, Failure, and Hope. Circ. J. 2017, 81, 131–141. [Google Scholar] [CrossRef]
- Hausenloy, D.J.; Garcia-Dorado, D.; Bøtker, H.E.; Davidson, S.M.; Downey, J.; Engel, F.B.; Jennings, R.; Lecour, S.; Leor, J.; Madonna, R.; et al. Novel targets and future strategies for acute cardioprotection: Position Paper of the European Society of Cardiology Working Group on Cellular Biology of the Heart. Cardiovasc. Res. 2017, 113, 564–585. [Google Scholar] [CrossRef]
- Spannbauer, A.; Traxler, D.; Lukovic, D.; Zlabinger, K.; Winkler, J.; Gugerell, A.; Ferdinandy, P.; Hausenloy, D.J.; Pavo, N.; Emmert, M.Y.; et al. Effect of Ischemic Preconditioning and Postconditioning on Exosome-Rich Fraction microRNA Levels, in Relation with Electrophysiological Parameters and Ventricular Arrhythmia in Experimental Closed-Chest Reperfused Myocardial Infarction. Int. J. Mol. Sci. 2019, 20, 2140. [Google Scholar] [CrossRef]
- Wang, Y.; Hill, J.A. Electrophysiological remodeling in heart failure. J. Mol. Cell. Cardiol. 2010, 48, 619–632. [Google Scholar] [CrossRef]
- Inoue, H.; Zipes, D.P. Conduction over an isthmus of atrial myocardium in vivo: A possible model of Wolff-Parkinson-White syndrome. Circulation 1987, 76, 637–647. [Google Scholar] [CrossRef] [PubMed]
- Tomaselli, G.F.; Zipes, D.P. What causes sudden death in heart failure? Circ. Res. 2004, 95, 754–763. [Google Scholar] [CrossRef]
- Yue, L.; Xie, J.; Nattel, S. Molecular determinants of cardiac fibroblast electrical function and therapeutic implications for atrial fibrillation. Cardiovasc. Res. 2011, 89, 744–753. [Google Scholar] [CrossRef]
- Wijffels, M.C.; Kirchhof, C.J.; Dorland, R.; Allessie, M.A. Atrial fibrillation begets atrial fibrillation. A study in awake chronically instrumented goats. Circulation 1995, 92, 1954–1968. [Google Scholar] [CrossRef] [PubMed]
- Moe, G.K.; Rheinboldt, W.C.; Abildskov, J.A. A Computer Model of Atrial Fibrillation. Am. Heart J. 1964, 67, 200–220. [Google Scholar] [CrossRef] [PubMed]
- Roden, D.M. Repolarization reserve: A moving target. Circulation 2008, 118, 981–982. [Google Scholar] [CrossRef] [PubMed]
- Varro, A.; Baczko, I. Cardiac ventricular repolarization reserve: A principle for understanding drug-related proarrhythmic risk. Br. J. Pharmacol. 2011, 164, 14–36. [Google Scholar] [CrossRef]
- Pu, J.; Boyden, P.A. Alterations of Na+ currents in myocytes from epicardial border zone of the infarcted heart. A possible ionic mechanism for reduced excitability and postrepolarization refractoriness. Circ. Res. 1997, 81, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Ursell, P.C.; Gardner, P.I.; Albala, A.; Fenoglio, J.J., Jr.; Wit, A.L. Structural and electrophysiological changes in the epicardial border zone of canine myocardial infarcts during infarct healing. Circ. Res. 1985, 56, 436–451. [Google Scholar] [CrossRef]
- Maltsev, V.A.; Sabbab, H.N.; Undrovinas, A.I. Down-regulation of sodium current in chronic heart failure: Effect of long-term therapy with carvedilol. Cell. Mol. Life Sci. 2002, 59, 1561–1568. [Google Scholar] [CrossRef]
- Sato, T.; Ohkusa, T.; Honjo, H.; Suzuki, S.; Yoshida, M.-A.; Ishiguro, Y.S.; Nakagawa, H.; Yamazaki, M.; Yano, M.; Kodama, I.; et al. Altered expression of connexin43 contributes to the arrhythmogenic substrate during the development of heart failure in cardiomyopathic hamster. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H1164–H1173. [Google Scholar] [CrossRef]
- Rodriguez-Sinovas, A. Cx43 phosphorylation and cardioprotection. Cardiovasc. Res. 2009, 83, 613–614. [Google Scholar] [CrossRef]
- Peters, N.S.; Green, C.R.; Poole-Wilson, P.A.; Severs, N.J. Reduced content of Connexin43 gap junctions in ventricular myocardium from hypertrophied and ischemic human hearts. Circulation 1993, 88, 864–875. [Google Scholar] [CrossRef]
- Akar, F.G.; Rosenbaum, D.S. Transmural electrophysiological heterogeneities underlying arrhythmogenesis in heart failure. Circ. Res. 2003, 93, 638–645. [Google Scholar] [CrossRef] [PubMed]
- Yan, G.-X.; Rials, S.J.; Wu, Y.; Liu, T.; Xu, X.; Marinchak, R.A.; Kowey, P.R. Ventricular hypertrophy amplifies transmural repolarization dispersion and induces early afterdepolarization. Am. J. Physiol. Heart Circ. Physiol. 2001, 281, H1968–H1975. [Google Scholar] [CrossRef] [PubMed]
- Wetzel, U.; Boldt, A.; Lauschke, J.; Weigl, J.; Schirdewahn, P.; Dorszewski, A.; Doll, N.; Hindricks, G.; Dhein, S.; Kottkamp, H. Expression of connexins 40 and 43 in human left atrium in atrial fibrillation of different aetiologies. Heart 2005, 91, 166–170. [Google Scholar] [CrossRef] [PubMed]
- Polontchouk, L.; Haefliger, J.-A.; Ebelt, B.; Schaefer, T.; Stuhlmann, D.; Mehlhorn, U.; Kuhn-Regnier, F.; De Vivie, E.; Dhein, S. Effects of chronic atrial fibrillation on gap junction distribution in human and rat atria. J. Am. Coll. Cardiol. 2001, 38, 883–891. [Google Scholar] [CrossRef]
- Sarrazin, J.-F.; Comeau, G.; Daleau, P.; Kingma, J.; Plante, I.; Fournier, D.; Molin, F. Reduced incidence of vagally induced atrial fibrillation and expression levels of connexins by n-3 polyunsaturated fatty acids in dogs. J. Am. Coll. Cardiol. 2007, 50, 1505–1512. [Google Scholar] [CrossRef]
- Daleau, P.; Boudriau, S.; Michaud, M.; Jolicoeur, C.; Kingma, J.G., Jr. Preconditioning in the absence or presence of sustained ischemia modulates myocardial Cx43 protein levels and gap junction distribution. Can. J. Physiol. Pharmacol. 2001, 79, 371–378. [Google Scholar] [CrossRef]
- Chung, M.K.; Martin, D.O.; Sprecher, D.; Wazni, O.; Kanderian, A.; Carnes, C.A.; Bauer, J.A.; Tchou, P.J.; Niebauer, M.J.; Natale, A.; et al. C-reactive protein elevation in patients with atrial arrhythmias: Inflammatory mechanisms and persistence of atrial fibrillation. Circulation 2001, 104, 2886–2891. [Google Scholar] [CrossRef]
- Psychari, S.N.; Apostolou, T.S.; Sinos, L.; Hamodraka, E.; Liakos, G.; Kremastinos, D.T. Relation of elevated C-reactive protein and interleukin-6 levels to left atrial size and duration of episodes in patients with atrial fibrillation. Am. J. Cardiol. 2005, 95, 764–767. [Google Scholar] [CrossRef]
- Issac, T.T.; Dokainish, H.; Lakkis, N.M. Role of inflammation in initiation and perpetuation of atrial fibrillation: A systematic review of the published data. J. Am. Coll. Cardiol. 2007, 50, 2021–2028. [Google Scholar] [CrossRef]
- Pohl, D.; Benseler, S. Systemic inflammatory and autoimmune disorders. Handb. Clin. Neurol. 2013, 112, 1243–1252. [Google Scholar] [CrossRef]
- Pasquié, J.L.; Sanders, P.; Hocini, M.; Hsu, L.F.; Scavée, C.; Jais, P.; Takahashi, Y.; Rotter, M.; Sacher, F.; Victor, J.; et al. Fever as a precipitant of idiopathic ventricular fibrillation in patients with normal hearts. J. Cardiovasc. Electrophysiol. 2004, 15, 1271–1276. [Google Scholar] [CrossRef] [PubMed]
- Lazzerini, P.E.; Capecchi, P.L.; Laghi-Pasini, F. Systemic inflammation and arrhythmic risk: Lessons from rheumatoid arthritis. Eur. Heart J. 2017, 38, 1717–1727. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, K.; Nakashima, H.; Saku, K. The HMG-CoA reductase inhibitor atorvastatin prevents atrial fibrillation by inhibiting inflammation in a canine sterile pericarditis model. Cardiovasc. Res. 2004, 62, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Tveit, A.; Grundtvig, M.; Gundersen, T.; Vanberg, P.; Semb, A.G.; Holt, E.; Gullestad, L. Analysis of pravastatin to prevent recurrence of atrial fibrillation after electrical cardioversion. Am. J. Cardiol. 2004, 93, 780–782. [Google Scholar] [CrossRef] [PubMed]
- Lazzerini, P.E.; Laghi-Pasini, F.; Boutjdir, M.; Capecchi, P.L. Inflammatory cytokines and cardiac arrhythmias: The lesson from COVID-19. Nat. Rev. Immunol. 2022, 22, 270–272. [Google Scholar] [CrossRef]
- Coromilas, E.J.; Kochav, S.; Goldenthal, I.; Biviano, A.; Garan, H.; Goldbarg, S.; Kim, J.-H.; Yeo, I.; Tracy, C.; Ayanian, S.; et al. Worldwide Survey of COVID-19-Associated Arrhythmias. Circ. Arrhythmia Electrophysiol. 2021, 14, e009458. [Google Scholar] [CrossRef]
- Wang, Y.; Zheng, Y.; Tong, Q.; Wang, L.; Lv, G.; Xi, Z.; Liu, W. Cardiac Injury and Clinical Course of Patients with Coronavirus Disease 2019. Front. Cardiovasc. Med. 2020, 7, 147. [Google Scholar] [CrossRef]
- Group WHOREAfC-TW; Shankar-Hari, M.; Vale, C.L.; Godolphin, P.J.; Fisher, D.; Higgins, J.P.T.; Spiga, F.; Savović, J.; Tierney, J.; Baron, G.; et al. Association between Administration of IL-6 Antagonists and Mortality among Patients Hospitalized for COVID-19: A Meta-analysis. JAMA 2021, 326, 499–518. [Google Scholar] [CrossRef]
- Sharma, S.; Adrogue, J.V.; Golfman, L.; Uray, I.; Lemm, J.; Youker, K.; Noon, G.P.; Frazier, O.H.; Taegtmeyer, H. Intramyocardial lipid accumulation in the failing human heart resembles the lipotoxic rat heart. FASEB J. 2004, 18, 1692–1700. [Google Scholar] [CrossRef]
- Aromolaran, A.S.; Colecraft, H.M.; Boutjdir, M. High-fat diet-dependent modulation of the delayed rectifier K(+) current in adult guinea pig atrial myocytes. Biochem. Biophys. Res. Commun. 2016, 474, 554–559. [Google Scholar] [CrossRef]
- Aromolaran, A.S.; Boutjdir, M. Cardiac Ion Channel Regulation in Obesity and the Metabolic Syndrome: Relevance to Long QT Syndrome and Atrial Fibrillation. Front. Physiol. 2017, 8, 431. [Google Scholar] [CrossRef] [PubMed]
- Lundman, P.; Boquist, S.; Samnegård, A.; Bennermo, M.; Held, C.; Ericsson, C.-G.; Silveira, A.; Hamsten, A.; Tornvall, P. A high-fat meal is accompanied by increased plasma interleukin-6 concentrations. Nutr. Metab. Cardiovasc. Dis. 2007, 17, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Rennison, J.H.; Van Wagoner, D.R. Impact of dietary fatty acids on cardiac arrhythmogenesis. Circ. Arrhythmia Electrophysiol. 2009, 2, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Park, T.S.; Goldberg, I.J. Sphingolipids, lipotoxic cardiomyopathy, and cardiac failure. Heart Fail. Clin. 2012, 8, 633–641. [Google Scholar] [CrossRef]
- Ali, A.; Boutjdir, M.; Aromolaran, A.S. Cardiolipotoxicity, Inflammation, and Arrhythmias: Role for Interleukin-6 Molecular Mechanisms. Front. Physiol. 2018, 9, 1866. [Google Scholar] [CrossRef]
- Billman, G.E.; Kang, J.X.; Leaf, A. Prevention of sudden cardiac death by dietary pure omega-3 polyunsaturated fatty acids in dogs. Circulation 1999, 99, 2452–2457. [Google Scholar] [CrossRef]
- Leaf, A.; Xiao, Y.F.; Kang, J.X.; Billman, G.E. Prevention of sudden cardiac death by n-3 polyunsaturated fatty acids. Pharmacol. Ther. 2003, 98, 355–377. [Google Scholar] [CrossRef]
- Grandi, E.; Ripplinger, C.M. Antiarrhythmic mechanisms of beta blocker therapy. Pharmacol. Res. 2019, 146, 104274. [Google Scholar] [CrossRef]
- Al-Gobari, M.; El Khatib, C.; Pillon, F.; Gueyffier, F. beta-Blockers for the prevention of sudden cardiac death in heart failure patients: A meta-analysis of randomized controlled trials. BMC Cardiovasc. Disord. 2013, 13, 52. [Google Scholar] [CrossRef]
- McMurray, J.; Køber, L.; Robertson, M.; Dargie, H.; Colucci, W.; Lopez-Sendon, J.; Remme, W.; Sharpe, D.N.; Ford, I. Antiarrhythmic effect of carvedilol after acute myocardial infarction: Results of the Carvedilol Post-Infarct Survival Control in Left Ventricular Dysfunction (CAPRICORN) trial. J. Am. Coll. Cardiol. 2005, 45, 525–530. [Google Scholar] [CrossRef]
- Exner, D.V.; Reiffel, J.A.; Epstein, A.E.; Ledingham, R.; Reiter, M.J.; Yao, Q.; Duff, H.; Follmann, D.; Schron, E.; Greene, H.; et al. Beta-blocker use and survival in patients with ventricular fibrillation or symptomatic ventricular tachycardia: The Antiarrhythmics Versus Implantable Defibrillators (AVID) trial. J. Am. Coll. Cardiol. 1999, 34, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Puljevic, M.; Velagic, V.; Puljevic, D.; Milicic, D. Propranolol efficiency in prevention of sustained ventricular tachycardia in patients with implanted cardioverter-defibrillator: A case series. Croat. Med. J. 2014, 55, 75–76. [Google Scholar] [CrossRef] [PubMed]
- Connolly, S.J.; Dorian, P.; Roberts, R.S.; Gent, M.; Bailin, S.; Fain, E.S.; Thorpe, K.; Champagne, J.; Talajic, M.; Coutu, B.; et al. Comparison of beta-blockers, amiodarone plus beta-blockers, or sotalol for prevention of shocks from implantable cardioverter defibrillators: The OPTIC Study: A randomized trial. JAMA 2006, 295, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Boutitie, F.; Boissel, J.-P.; Connolly, S.J.; Camm, A.J.; Cairns, J.A.; Julian, D.G.; Gent, M.; Janse, M.J.; Dorian, P.; Frangin, G. Amiodarone interaction with beta-blockers: Analysis of the merged EMIAT (European Myocardial Infarct Amiodarone Trial) and CAMIAT (Canadian Amiodarone Myocardial Infarction Trial) databases. The EMIAT and CAMIAT Investigators. Circulation 1999, 99, 2268–2275. [Google Scholar] [CrossRef]
- Bashir, Y.; Thomsen, P.-E.B.; Kingma, J.; Møller, M.; Wong, C.; Cobbe, S.M.; Jordaens, L.; Campbell, R.W.; Rasmussen, H.S.; Camm, A. Electrophysiologic profile and efficacy of intravenous dofetilide (UK-68,798), a new class III antiarrhythmic drug, in patients with sustained monomorphic ventricular tachycardia. Dofetilide Arrhythmia Study Group. Am. J. Cardiol. 1995, 76, 1040–1044. [Google Scholar] [CrossRef]
- Mounsey, J.P.; DiMarco, J.P. Cardiovascular drugs. Dofetilide. Circulation 2000, 102, 2665–2670. [Google Scholar] [CrossRef]
- Baquero, G.A.; Banchs, J.E.; De Palma, S.; Young, S.K.; Penny-Peterson, E.D.; Samii, S.M.; Wolbrette, D.L.; Naccarelli, G.V.; Gonzalez, M.D. Dofetilide reduces the frequency of ventricular arrhythmias and implantable cardioverter defibrillator therapies. J. Cardiovasc. Electrophysiol. 2012, 23, 296–301. [Google Scholar] [CrossRef]
- Singh, B.N.; Nademanee, K. Use of calcium antagonists for cardiac arrhythmias. Am. J. Cardiol. 1987, 59, 153B–162B. [Google Scholar] [CrossRef]
- Singh, B.N. Comparative mechanisms of action of antiarrhythmic agents: Significance of lengthening repolarization. In Control of Cardiac Arrhythmias by Lengthening Repolarization; Singh, B.N., Ed.; Futura Publishing Company: Mount Kisco, NY, USA, 1988; pp. 53–127. [Google Scholar]
- Van der Werf, C.; Zwinderman, A.H.; Wilde, A.A. Therapeutic approach for patients with catecholaminergic polymorphic ventricular tachycardia: State of the art and future developments. Europace 2012, 14, 175–183. [Google Scholar] [CrossRef]
- Antzelevitch, C.; Belardinelli, L.; Zygmunt, A.C.; Burashnikov, A.; Di Diego, J.M.; Fish, J.M.; Cordeiro, J.; Thomas, G. Electrophysiological effects of ranolazine, a novel antianginal agent with antiarrhythmic properties. Circulation 2004, 110, 904–910. [Google Scholar] [CrossRef]
- Olivotto, I.; Camici, P.G.; Merlini, P.A.; Rapezzi, C.; Patten, M.; Climent, V.; Sinagra, G.; Tomberli, B.; Marin, F.; Ehlermann, P.; et al. Efficacy of Ranolazine in Patients with Symptomatic Hypertrophic Cardiomyopathy: The RESTYLE-HCM Randomized, Double-Blind, Placebo-Controlled Study. Circ. Heart Fail. 2018, 11, e004124. [Google Scholar] [CrossRef] [PubMed]
- Antzelevitch, C.; Burashnikov, A.; Sicouri, S.; Belardinelli, L. Electrophysiologic basis for the antiarrhythmic actions of ranolazine. Heart Rhythm. 2011, 8, 1281–1290. [Google Scholar] [CrossRef] [PubMed]
- Morrow, D.A.; Scirica, B.M.; Karwatowska-Prokopczuk, E.; Murphy, S.A.; Budaj, A.; Varshavsky, S.; Wolff, A.A.; Skene, A.; McCabe, C.H.; Braunwald, E.; et al. Effects of ranolazine on recurrent cardiovascular events in patients with non-ST-elevation acute coronary syndromes: The MERLIN-TIMI 36 randomized trial. JAMA 2007, 297, 1775–1783. [Google Scholar] [CrossRef] [PubMed]
- Zareba, W.; Daubert, J.P.; Beck, C.A.; Huang, D.T.; Alexis, J.D.; Brown, M.W.; Pyykkonen, K.; McNitt, S.; Oakes, D.; Feng, C.; et al. Ranolazine in High-Risk Patients with Implanted Cardioverter-Defibrillators: The RAID Trial. J. Am. Coll. Cardiol. 2018, 72, 636–645. [Google Scholar] [CrossRef]
- Fox, K.; Ford, I.; Steg, P.G.; Tardif, J.C.; Tendera, M.; Ferrari, R. Ivabradine in stable coronary artery disease without clinical heart failure. N. Engl. J. Med. 2014, 371, 1091–1099. [Google Scholar] [CrossRef]
- Fox, K.; Ford, I.; Steg, P.G.; Tendera, M.; Ferrari, R.; BEAUTIFUL Investigators. Ivabradine for patients with stable coronary artery disease and left-ventricular systolic dysfunction (BEAUTIFUL): A randomised, double-blind, placebo-controlled trial. Lancet 2008, 372, 807–816. [Google Scholar] [CrossRef] [PubMed]
- Swedberg, K.; Komajda, M.; Böhm, M.; Borer, J.S.; Ford, I.; Dubost-Brama, A.; Lerebours, G.; Tavazzi, L.; on behalf of the SHIFT Investigators. Ivabradine and outcomes in chronic heart failure (SHIFT): A randomised placebo-controlled study. Lancet 2010, 376, 875–885. [Google Scholar] [CrossRef] [PubMed]
- Cocco, G.; Jerie, P. Torsades de pointes induced by the concomitant use of ivabradine and azithromycin: An unexpected dangerous interaction. Cardiovasc. Toxicol. 2015, 15, 104–106. [Google Scholar] [CrossRef]
- Lerman, B.B.; Ip, J.E.; Shah, B.K.; Thomas, G.; Liu, C.F.; Ciaccio, E.J.; Wit, A.L.; Cheung, J.W.; Markowitz, S.M. Mechanism-specific effects of adenosine on ventricular tachycardia. J. Cardiovasc. Electrophysiol. 2014, 25, 1350–1358. [Google Scholar] [CrossRef]
- Lerman, B.B.; Stein, K.M.; Markowitz, S.M.; Mittal, S.; Slotwiner, D.J. Right ventricular outflow tract tachycardia: An update. Card. Electrophysiol. Rev. 2002, 6, 68–71. [Google Scholar] [CrossRef]
- Markowitz, S.M.; Litvak, B.L.; Ramirez de Arellano, E.A.; Markisz, J.A.; Stein, K.M.; Lerman, B.B. Adenosine-sensitive ventricular tachycardia: Right ventricular abnormalities delineated by magnetic resonance imaging. Circulation 1997, 96, 1192–1200. [Google Scholar] [CrossRef] [PubMed]
- Ziff, O.J.; A Lane, D.; Samra, M.; Griffith, M.; Kirchhof, P.; Lip, G.Y.; Steeds, R.P.; Townend, J.; Kotecha, D. Safety and efficacy of digoxin: Systematic review and meta-analysis of observational and controlled trial data. BMJ 2015, 351, h4451. [Google Scholar] [CrossRef] [PubMed]
- Vamos, M.; Erath, J.W.; Hohnloser, S.H. Digoxin-associated mortality: A systematic review and meta-analysis of the literature. Eur. Heart J. 2015, 36, 1831–1838. [Google Scholar] [CrossRef] [PubMed]
- Williams, E.S.; Viswanathan, M.N. Current and emerging antiarrhythmic drug therapy for ventricular tachycardia. Cardiol. Ther. 2013, 2, 27–46. [Google Scholar] [CrossRef] [PubMed]
- Greene, H.L.; Roden, D.M.; Katz, R.J.; Woosley, R.L.; Salerno, D.M.; Henthorn, R.W. The Cardiac Arrhythmia Suppression Trial: First CAST... then CAST-II. J. Am. Coll. Cardiol. 1992, 19, 894–898. [Google Scholar] [CrossRef]
- Epstein, A.E.; Dimarco, J.P.; Ellenbogen, K.A.; Estes, N.A., 3rd; Freedman, R.A.; Gettes, L.S.; Gillinov, A.M.; Gregoratos, G.; Hammill, S.C.; Hayes, D.L.; et al. ACC/AHA/HRS 2008 Guidelines for device-based therapy of cardiac rhythm abnormalities. Heart Rhythm. 2008, 5, e1–e62. [Google Scholar] [CrossRef]
- Mulpuru, S.K.; Madhavan, M.; McLeod, C.J.; Cha, Y.M.; Friedman, P.A. Cardiac Pacemakers: Function, Troubleshooting, and Management: Part 1 of a 2-Part Series. J. Am. Coll. Cardiol. 2017, 69, 189–210. [Google Scholar] [CrossRef]
- Madhavan, M.; Mulpuru, S.K.; McLeod, C.J.; Cha, Y.M.; Friedman, P.A. Advances and Future Directions in Cardiac Pacemakers: Part 2 of a 2-Part Series. J. Am. Coll. Cardiol. 2017, 69, 211–235. [Google Scholar] [CrossRef]
- Marban, E.; Cho, H.C. Biological pacemakers as a therapy for cardiac arrhythmias. Curr. Opin. Cardiol. 2008, 23, 46–54. [Google Scholar] [CrossRef]
- Ambesh, P.; Kapoor, A. Biological pacemakers: Concepts and techniques. Natl. Med. J. India 2017, 30, 324–326. [Google Scholar] [CrossRef]
- Cingolani, E.; Goldhaber, J.I.; Marban, E. Next-generation pacemakers: From small devices to biological pacemakers. Nat. Rev. Cardiol. 2018, 15, 139–150. [Google Scholar] [CrossRef]
- Farraha, M.; Kumar, S.; Chong, J.; Cho, H.C.; Kizana, E. Gene Therapy Approaches to Biological Pacemakers. J. Cardiovasc. Dev. Dis. 2018, 5, 50. [Google Scholar] [CrossRef] [PubMed]
- Antiarrhythmics versus Implantable Defibrillators (AVID) Investigators. A comparison of antiarrhythmic-drug therapy with implantable defibrillators in patients resuscitated from near-fatal ventricular arrhythmias. N. Engl. J. Med. 1997, 337, 1576–1583. [Google Scholar] [CrossRef] [PubMed]
- Kuck, K.H.; Cappato, R.; Siebels, J.; Ruppel, R. Randomized comparison of antiarrhythmic drug therapy with implantable defibrillators in patients resuscitated from cardiac arrest: The Cardiac Arrest Study Hamburg (CASH). Circulation 2000, 102, 748–754. [Google Scholar] [CrossRef] [PubMed]
- Connolly, S.J.; Gent, M.; Roberts, R.S.; Dorian, P.; Roy, D.; Sheldon, R.S.; Mitchell, L.B.; Green, M.S.; Klein, G.J.; O’brien, B. Canadian implantable defibrillator study (CIDS): A randomized trial of the implantable cardioverter defibrillator against amiodarone. Circulation 2000, 101, 1297–1302. [Google Scholar] [CrossRef]
- Ferreira-Gonzalez, I.; Dos-Subira, L.; Guyatt, G.H. Adjunctive antiarrhythmic drug therapy in patients with implantable cardioverter defibrillators: A systematic review. Eur. Heart J. 2007, 28, 469–477. [Google Scholar] [CrossRef]
- Schron, E.B.; Exner, D.V.; Yao, Q.; Jenkins, L.S.; Steinberg, J.S.; Cook, J.R.; Kutalek, S.P.; Friedman, P.L.; Bubien, R.S.; Page, R.L.; et al. Quality of life in the antiarrhythmics versus implantable defibrillators trial: Impact of therapy and influence of adverse symptoms and defibrillator shocks. Circulation 2002, 105, 589–594. [Google Scholar] [CrossRef]
- Carroll, D.L.; Hamilton, G.A. Quality of life in implanted cardioverter defibrillator recipients: The impact of a device shock. Heart Lung 2005, 34, 169–178. [Google Scholar] [CrossRef]
- Borne, R.T.; Katz, D.; Betz, J.; Peterson, P.N.; Masoudi, F.A. Implantable Cardioverter-Defibrillators for Secondary Prevention of Sudden Cardiac Death: A Review. J. Am. Heart Assoc. 2017, 6, e005515. [Google Scholar] [CrossRef]
- Abboud, J.; Ehrlich, J.R.; Josefs-Hospital, W.S. Antiarrhythmic Drug Therapy to Avoid Implantable Cardioverter Defibrillator Shocks. Arrhythmia Electrophysiol. Rev. 2016, 5, 117–121. [Google Scholar] [CrossRef]
- Wilber, D.J.; Pappone, C.; Neuzil, P.; De Paola, A.; Marchlinski, F.; Natale, A.; Macle, L.; Daoud, E.G.; Calkins, H.; Hall, B.; et al. Comparison of antiarrhythmic drug therapy and radiofrequency catheter ablation in patients with paroxysmal atrial fibrillation: A randomized controlled trial. JAMA 2010, 303, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Pappone, C.; Rosanio, S.; Augello, G.; Gallus, G.; Vicedomini, G.; Mazzone, P.; Gulletta, S.; Gugliotta, F.; Pappone, A.; Santinelli, V.; et al. Mortality, morbidity, and quality of life after circumferential pulmonary vein ablation for atrial fibrillation: Outcomes from a controlled nonrandomized long-term study. J. Am. Coll. Cardiol. 2003, 42, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Fuster, V.; Rydén, L.E.; Cannom, D.S.; Crijns, H.J.; Curtis, A.B.; Ellenbogen, K.A.; Halperin, J.L.; Le Heuzey, J.-Y.; Kay, G.N.; Lowe, J.E.; et al. ACC/AHA/ESC 2006 Guidelines for the Management of Patients with Atrial Fibrillation: A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the European Society of Cardiology Committee for Practice Guidelines (Writing Committee to Revise the 2001 Guidelines for the Management of Patients With Atrial Fibrillation): Developed in collaboration with the European Heart Rhythm Association and the Heart Rhythm Society. Circulation 2006, 114, e257–e354. [Google Scholar] [CrossRef] [PubMed]
- Wazni, O.M.; Marrouche, N.F.; Martin, D.O.; Verma, A.; Bhargava, M.; Saliba, W.; Bash, D.; Schweikert, R.; Brachmann, J.; Gunther, J.; et al. Radiofrequency ablation vs antiarrhythmic drugs as first-line treatment of symptomatic atrial fibrillation: A randomized trial. JAMA 2005, 293, 2634–2640. [Google Scholar] [CrossRef]
- Pappone, C.; Augello, G.; Sala, S.; Gugliotta, F.; Vicedomini, G.; Gulletta, S.; Paglino, G.; Mazzone, P.; Sora, N.; Greiss, I.; et al. A randomized trial of circumferential pulmonary vein ablation versus antiarrhythmic drug therapy in paroxysmal atrial fibrillation: The APAF Study. J. Am. Coll. Cardiol. 2006, 48, 2340–2347. [Google Scholar] [CrossRef]
- Ardell, J.L.; Andresen, M.C.; Armour, J.A.; Billman, G.E.; Chen, P.-S.; Foreman, R.D.; Herring, N.; O’Leary, D.S.; Sabbah, H.N.; Schultz, H.; et al. Translational neurocardiology: Preclinical models and cardioneural integrative aspects. J. Physiol. 2016, 594, 3877–3909. [Google Scholar] [CrossRef]
- Goldberger, J.J.; Basu, A.; Boineau, R.; Buxton, A.E.; Cain, M.E.; Canty, J.M., Jr.; Chen, P.-S.; Chugh, S.S.; Costantini, O.; Exner, D.V.; et al. Risk stratification for sudden cardiac death: A plan for the future. Circulation 2014, 129, 516–526. [Google Scholar] [CrossRef]
- Khemani, P.; Mehdirad, A.A. Cardiovascular Disorders Mediated by Autonomic Nervous System Dysfunction. Cardiol. Rev. 2020, 28, 65–72. [Google Scholar] [CrossRef]
- Verrier, R.L.; Antzelevitch, C. Autonomic aspects of arrhythmogenesis: The enduring and the new. Curr. Opin. Cardiol. 2004, 19, 2–11. [Google Scholar] [CrossRef]
- Fukuda, K.; Kanazawa, H.; Aizawa, Y.; Ardell, J.L.; Shivkumar, K. Cardiac innervation and sudden cardiac death. Circ. Res. 2015, 116, 2005–2019. [Google Scholar] [CrossRef]
- Shen, M.J.; Zipes, D.P. Role of the autonomic nervous system in modulating cardiac arrhythmias. Circ. Res. 2014, 114, 1004–1021. [Google Scholar] [CrossRef] [PubMed]
- Haegeli, L.M.; Calkins, H. Catheter ablation of atrial fibrillation: An update. Eur. Heart J. 2014, 35, 2454–2459. [Google Scholar] [CrossRef]
- Hanna, P.; Shivkumar, K. Targeting the Cardiac Ganglionated Plexi for Atrial Fibrillation: Modulate or Destroy? JACC Clin. Electrophysiol. 2018, 4, 1359–1361. [Google Scholar] [CrossRef] [PubMed]
- Armour, J.A.; Murphy, D.A.; Yuan, B.X.; Macdonald, S.; Hopkins, D.A. Gross and microscopic anatomy of the human intrinsic cardiac nervous system. Anat. Rec. 1997, 247, 289–298. [Google Scholar] [CrossRef]
- Avazzadeh, S.; McBride, S.; O’brien, B.; Coffey, K.; Elahi, A.; O’Halloran, M.; Soo, A.; Quinlan, L.R. Ganglionated Plexi Ablation for the Treatment of Atrial Fibrillation. J. Clin. Med. 2020, 9, 3081. [Google Scholar] [CrossRef] [PubMed]
- Safaei, N.; Montazerghaem, H.; Azarfarin, R.; Alizadehasl, A.; Alikhah, H. Radiofrequency ablation for treatment of atrial fibrillation. Bioimpacts 2011, 1, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Jaïs, P.; Cauchemez, B.; Macle, L.; Daoud, E.; Khairy, P.; Subbiah, R.; Hocini, M.; Extramiana, F.; Sacher, F.; Bordachar, P.; et al. Catheter ablation versus antiarrhythmic drugs for atrial fibrillation: The A4 study. Circulation. 2008, 118, 2498–2505. [Google Scholar] [CrossRef]
- Morillo, C.A.; Verma, A.; Connolly, S.J.; Kuck, K.H.; Nair, G.M.; Champagne, J.; Sterns, L.D.; Beresh, H.; Healey, J.S.; Natale, A. Radiofrequency ablation vs antiarrhythmic drugs as first-line treatment of paroxysmal atrial fibrillation (RAAFT-2): A randomized trial. JAMA 2014, 311, 692–700. [Google Scholar] [CrossRef]
- Franciosi, S.; Perry, F.K.G.; Roston, T.M.; Armstrong, K.R.; Claydon, V.E.; Sanatani, S. The role of the autonomic nervous system in arrhythmias and sudden cardiac death. Auton. Neurosci. 2017, 205, 1–11. [Google Scholar] [CrossRef]
- Rotter, M.; Takahashi, Y.; Sanders, P.; Haïssaguerre, M.; Jaïs, P.; Hsu, L.-F.; Sacher, F.; Pasquié, J.-L.; Clementy, J.; Hocini, M. Reduction of fluoroscopy exposure and procedure duration during ablation of atrial fibrillation using a novel anatomical navigation system. Eur. Heart J. 2005, 26, 1415–1421. [Google Scholar] [CrossRef]
- Kistler, P.; Rajappan, K.; Jahngir, M.; Earley, M.J.; Harris, S.; Abrams, D.; Gupta, D.; Liew, R.; Ellis, S.; Sporton, S.C.; et al. The impact of CT image integration into an electroanatomic mapping system on clinical outcomes of catheter ablation of atrial fibrillation. J. Cardiovasc. Electrophysiol. 2006, 17, 1093–1101. [Google Scholar] [CrossRef] [PubMed]
- Murry, C.E.; Jennings, R.B.; Reimer, K.A. Preconditioning with ischemia: A delay of lethal cell injury in ischemic myocardium. Circulation 1986, 74, 1124–1136. [Google Scholar] [CrossRef] [PubMed]
- Shiki, K.; Hearse, D.J. Preconditioning of ischemic myocardium: Reperfusion-induced arrhythmias. Am. J. Physiol. 1987, 253, H1470–H1476. [Google Scholar] [CrossRef] [PubMed]
- Kloner, R.A.; Dow, J.; Bhandari, A. Postconditioning markedly attenuates ventricular arrhythmias after ischemia-reperfusion. J. Cardiovasc. Pharmacol. Ther. 2006, 11, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Kolettis, T.M.; Vilaeti, A.D.; Tsalikakis, D.G.; Zoga, A.; Valenti, M.; Tzallas, A.T.; Papalois, A.; Iliodromitis, E.K. Effects of pre- and postconditioning on arrhythmogenesis in the in vivo rat model. J. Cardiovasc. Pharmacol. Ther. 2013, 18, 376–385. [Google Scholar] [CrossRef]
- Krogstad, L.E.; Slagsvold, K.H.; Wahba, A. Remote ischemic preconditioning and incidence of postoperative atrial fibrillation. Scand. Cardiovasc. J. 2015, 49, 117–122. [Google Scholar] [CrossRef]
- Candilio, L.; Malik, A.; Ariti, C.; Barnard, M.; Di Salvo, C.; Lawrence, D.; Hayward, M.; Yap, J.; Roberts, N.; Sheikh, A.; et al. Effect of remote ischaemic preconditioning on clinical outcomes in patients undergoing cardiac bypass surgery: A randomised controlled clinical trial. Heart 2015, 101, 185–192. [Google Scholar] [CrossRef]
- Hong, D.M.; Lee, E.H.; Kim, H.J.; Min, J.J.; Chin, J.H.; Choi, D.K.; Bahk, J.-H.; Sim, J.-Y.; Choi, I.-C.; Jeon, Y. Does remote ischaemic preconditioning with postconditioning improve clinical outcomes of patients undergoing cardiac surgery? Remote Ischaemic Preconditioning with Postconditioning Outcome Trial. Eur. Heart J. 2014, 35, 176–183. [Google Scholar] [CrossRef]
- Meybohm, P.; Bein, B.; Brosteanu, O.; Cremer, J.; Gruenewald, M.; Stoppe, C.; Coburn, M.; Schaelte, G.; Böning, A.; Niemann, B.; et al. A Multicenter Trial of Remote Ischemic Preconditioning for Heart Surgery. N. Engl. J. Med. 2015, 373, 1397–1407. [Google Scholar] [CrossRef]
- Lotfi, A.S.; Eftekhari, H.; Atreya, A.R.; Kashikar, A.; Sivalingam, S.K.; Giannoni, M.; Visintainer, P.; Engelman, D. Randomized controlled trial of remote ischemic preconditioning and atrial fibrillation in patients undergoing cardiac surgery. World J. Cardiol. 2016, 8, 615–622. [Google Scholar] [CrossRef]
- Hausenloy, D.J.; Candilio, L.; Evans, R.; Ariti, C.; Jenkins, D.P.; Kolvekar, S.; Knight, R.; Kunst, G.; Laing, C.; Nicholas, J.; et al. Remote Ischemic Preconditioning and Outcomes of Cardiac Surgery. N. Engl. J. Med. 2015, 373, 1408–1417. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Singh, H.; Shariff, M. Remote ischemic preconditioning and its role in the prevention of new onset atrial fibrillation post-cardiac surgery. A meta-analysis of randomized control trials. J. Arrhythmia 2019, 35, 789–794. [Google Scholar] [CrossRef] [PubMed]
- Galagudza, M.M.; Sonin, D.L.; Vlasov, T.D.; Kurapeev, D.I.; Shlyakhto, E.V. Remote vs. local ischaemic preconditioning in the rat heart: Infarct limitation, suppression of ischaemic arrhythmia and the role of reactive oxygen species. Int. J. Exp. Pathol. 2016, 97, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Oxman, T.; Arad, M.; Klein, R.; Avazov, N.; Rabinowitz, B. Limb ischemia preconditions the heart against reperfusion tachyarrhythmia. Am. J. Physiol. (Heart Circ. Physiol.) 1997, 273, H1707–H1712. [Google Scholar] [CrossRef]
- Ahmed, L.A.; Salem, H.A.; Attia, A.S.; Agha, A.M. Comparative study of the cardioprotective effects of local and remote preconditioning in ischemia/reperfusion injury. Life Sci. 2012, 90, 249–256. [Google Scholar] [CrossRef]
- Dow, J.; Bhandari, A.; Simkhovich, B.Z.; Hale, S.L.; Kloner, R.A. The effect of acute versus delayed remote ischemic preconditioning on reperfusion induced ventricular arrhythmias. J. Cardiovasc. Electrophysiol. 2012, 23, 1374–1383. [Google Scholar] [CrossRef]
- Wolbrette, D.; Naccarelli, G.; Curtis, A.; Lehmann, M.; Kadish, A. Gender differences in arrhythmias. Clin. Cardiol. 2002, 25, 49–56. [Google Scholar] [CrossRef]
- Westerman, S.; Wenger, N. Gender Differences in Atrial Fibrillation: A Review of Epidemiology, Management, and Outcomes. Curr. Cardiol. Rev. 2019, 15, 136–144. [Google Scholar] [CrossRef]
- Amuthan, R.; Curtis, A.B. Sex-Specific Considerations in Drug and Device Therapy of Cardiac Arrhythmias: JACC Focus Seminar 6/7. J. Am. Coll. Cardiol. 2022, 79, 1519–1529. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Class | Drugs |
|---|---|
| Class I: Sodium Channel Blockers | |
| Ia | Disopyramide, Procainamide, Quinidine |
| Ib | Lidocaine, Mexiletine |
| Ic | Flecainide, Propafenone |
| Class II: Beta-blockers | Acebutolol, Atenolol, Bisoprolol, Carvedilol, Esmolol, Metoprolol, Nadolol, Propranolol |
| Class III: Potassium Channel Blockers | Amiodarone, Bretylium, Dofetilide, Dronedarone, Ibutilide, Sotalol, Vernakalant (not available in USA) |
| Class IV: Calcium Channel Blockers | Diltiazem, Verapamil |
| Others | Adenosine, Atropine, Digoxin |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kingma, J.; Simard, C.; Drolet, B. Overview of Cardiac Arrhythmias and Treatment Strategies. Pharmaceuticals 2023, 16, 844. https://doi.org/10.3390/ph16060844
Kingma J, Simard C, Drolet B. Overview of Cardiac Arrhythmias and Treatment Strategies. Pharmaceuticals. 2023; 16(6):844. https://doi.org/10.3390/ph16060844
Chicago/Turabian StyleKingma, John, Chantale Simard, and Benoît Drolet. 2023. "Overview of Cardiac Arrhythmias and Treatment Strategies" Pharmaceuticals 16, no. 6: 844. https://doi.org/10.3390/ph16060844
APA StyleKingma, J., Simard, C., & Drolet, B. (2023). Overview of Cardiac Arrhythmias and Treatment Strategies. Pharmaceuticals, 16(6), 844. https://doi.org/10.3390/ph16060844