

Consensus Ensemble Multitarget Neural Network Model of Anxiolytic Activity of Chemical Compounds and Its Use for Multitarget Pharmacophore Design

 , ,
, ,  , and
, and
Abstract
:1. Introduction
2. Results
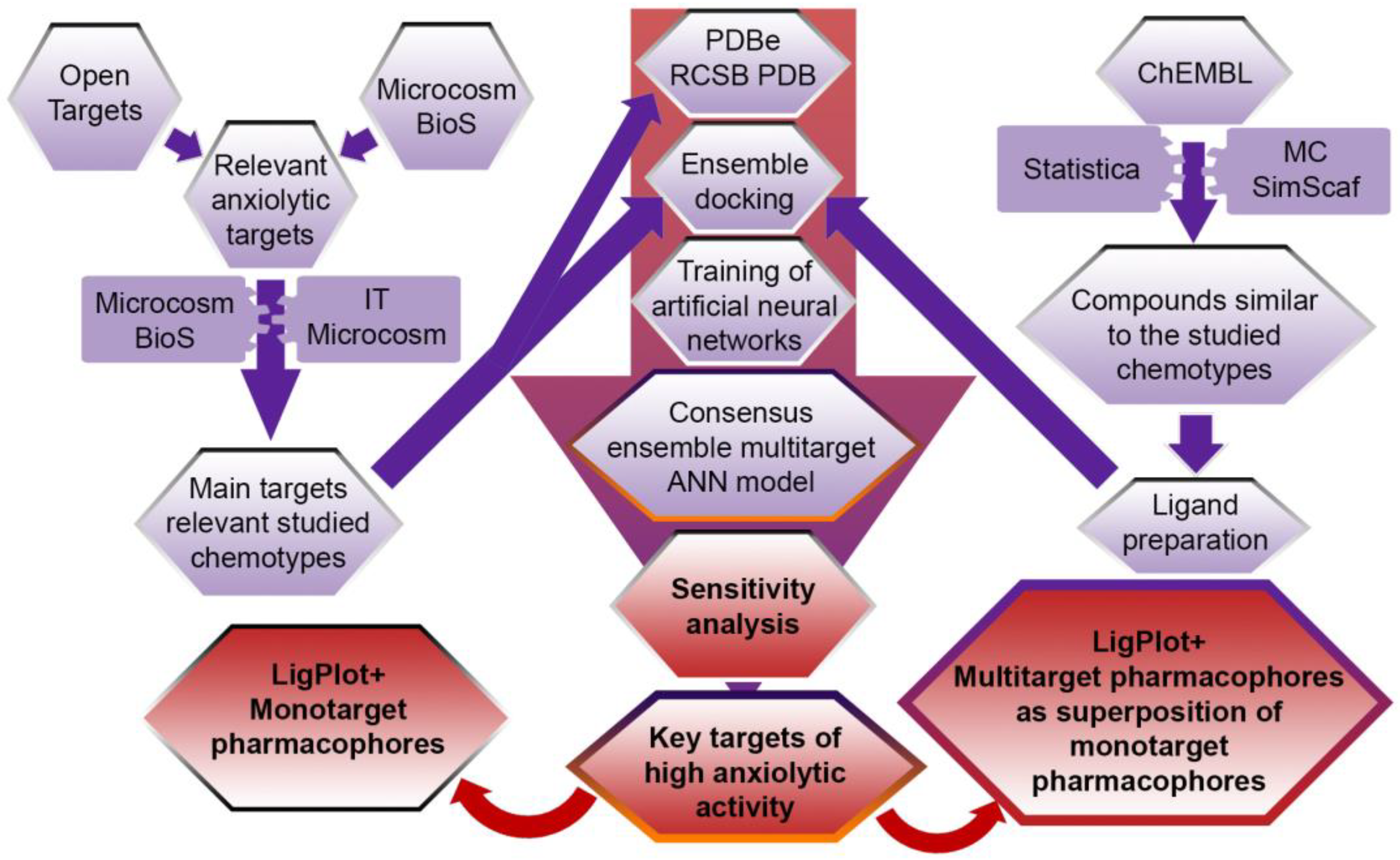
2.1. Data Preparation
2.2. Neural Network Modeling
2.3. Consensus Analysis of the Sensitivity of Neurons and Determination of Biotargets That Are the Most Significant for the Formation of High Anxiolytic Activity of Compounds Relevant to the Chemotypes of the Studied Derivatives of Nitrogen-Containing Condensed Heterocycles
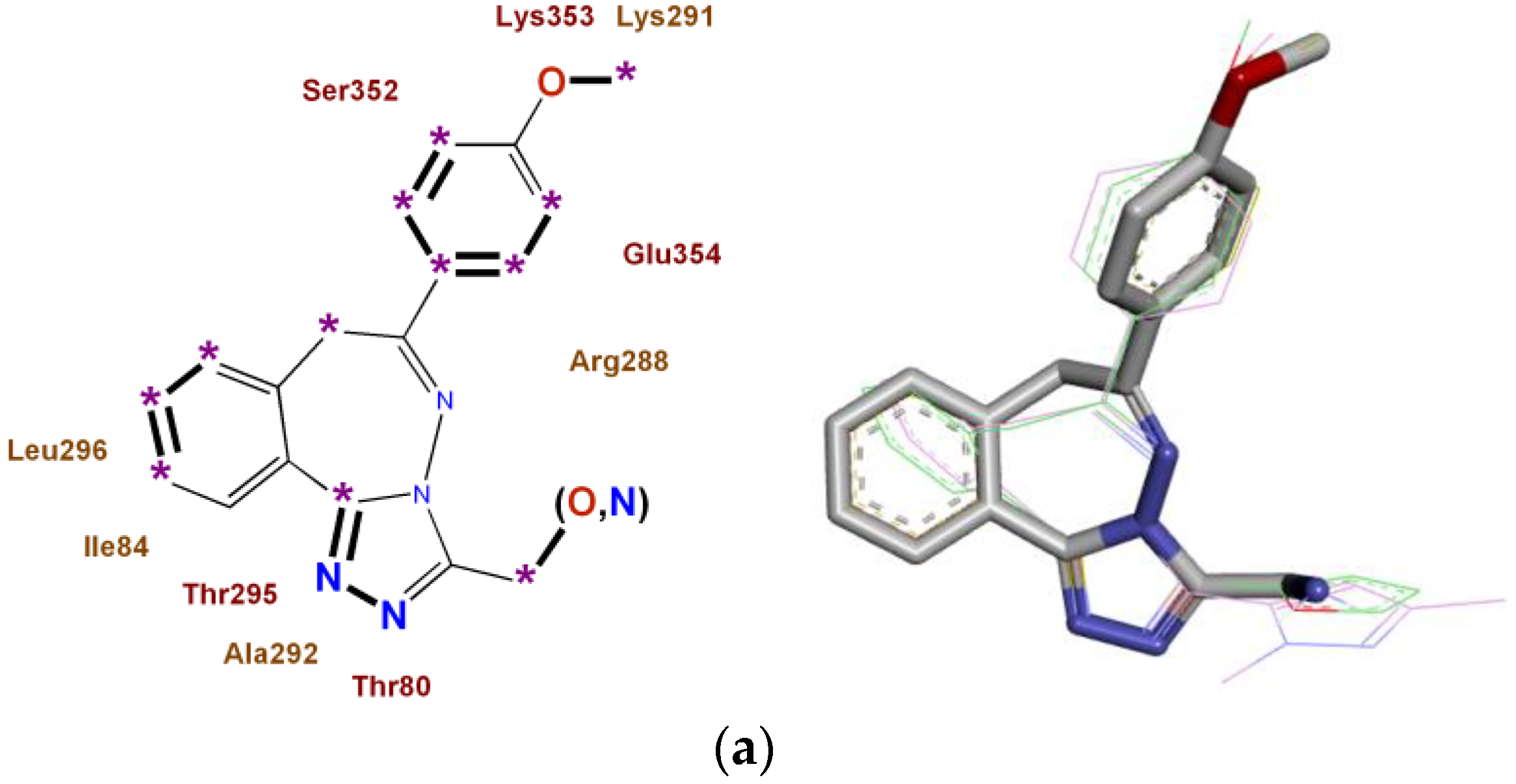
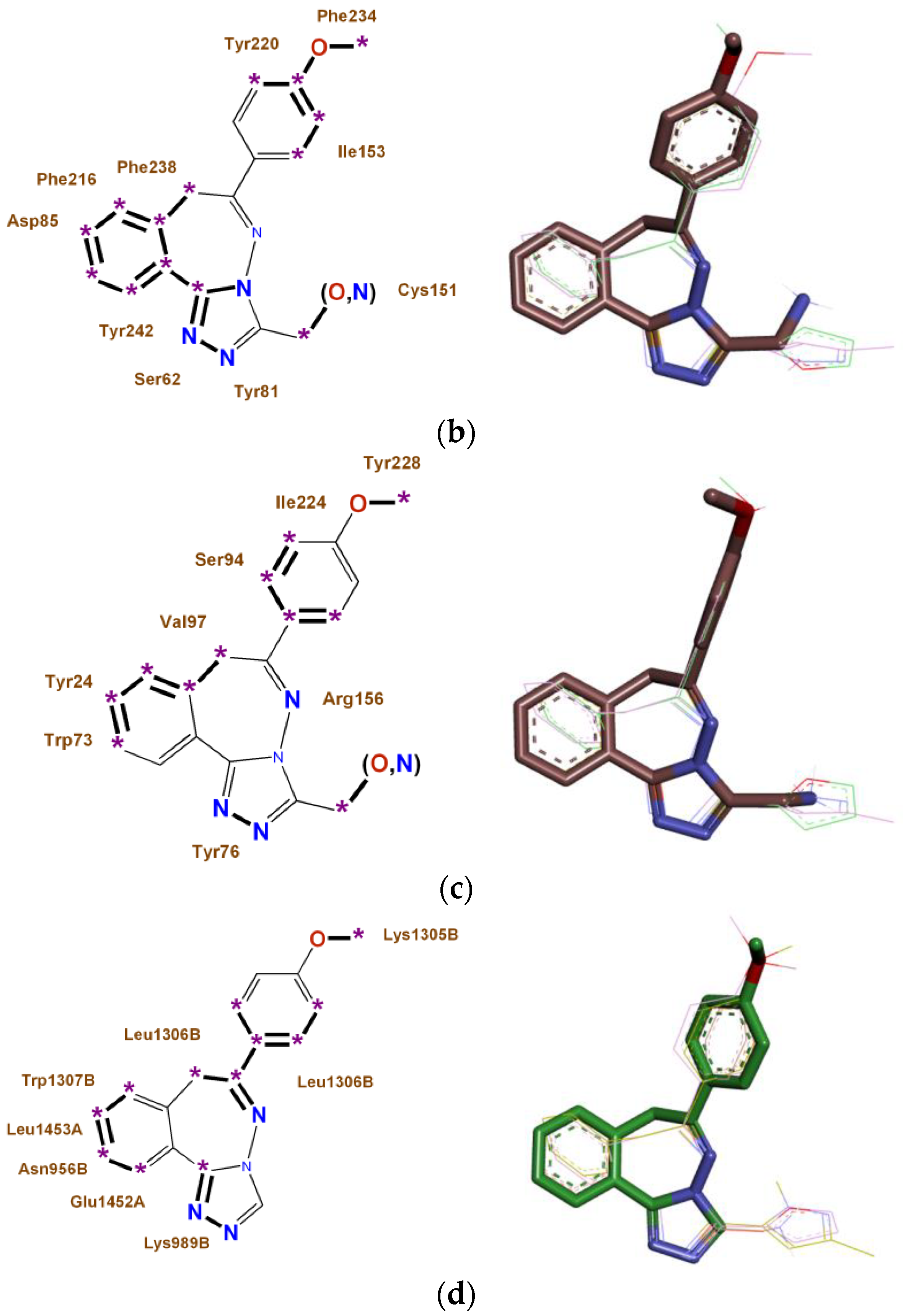
2.4. Construction of Multitarget Pharmacophores of High Anxiolytic Activity of the Studied Derivatives of Nitrogen-Containing Condensed Heterocycles
3. Discussion
- A verification database was created on the structure and level of anxiolytic activity of 663 experimentally studied known chemical compounds;
- A data set was formed on the structure and level of anxiolytic activity of 216 known compounds structurally similar to 15 studied chemotypes of nitrogen-containing heterocycles;
- Seventeen biotargets were identified that determine anxiolytic activity and are relevant to 15 studied chemotypes of nitrogen-containing heterocycles;
- A consensus ensemble multitarget neural network model of the dependence of the anxiolytic activity of chemical compounds on their docking energies in 17 relevant biotargets was built;
- The high recognition and prediction ability of the obtained consensus model is shown, and on average, according to the ROCanalysis, it is 98.9%;
- A consensus sensitive analysis of neurons of the obtained model was performed and four key biotargets were identified ADRA1B, ADRA2A, AGTR1, and NMDA-Glut, which are the most significant for the formation of anxiolytic activity;
- For 2,3,4,5-tetrahydro-11H-[1,3]diazepino[1,2-a]benzimidazole and [1,2,4]triazolo[3,4-a][2,3]benzodiazepine derivatives with high anxiolytic activity, eight monotarget pharmacophores were built, which determine the affinity for each of the most significant biotargets ADRA1B, ADRA2A, AGTR1, and NMDA-Glut;
- For 2,3,4,5-tetrahydro-11H-[1,3]diazepino[1,2-a]benzimidazole and [1,2,4]triazolo[3,4-a][2,3]benzodiazepine derivatives, two multitarget pharmacophores were constructed to determine the overall affinity for the most important biotargets ADRA1B, ADRA2A, AGTR1, and NMDA-Glut, which together, taking into account their mutual influence on each other, providehigh anxiolytic activity.
4. Materials and Methods
4.1. Biotargets Relevant to the Anxiolytic Activity of the Studied Derivatives of Nitrogen-Containing Condensed Heterocycles
4.2. A Verification Database on the Structure and Anxiolytic Activity of Known Compounds Relevant to the Chemotypes of the Studied Derivatives of Nitrogen-Containing Condensed Heterocycles
4.3. Ensemble Docking of Known Compounds Tested for Anxiolytic Activity, Relevant to the Chemotypes of the Studied Derivatives of Nitrogen-Containing Condensed Heterocycles
4.4. Neural Network Modeling of the Dependence of the Level of Anxiolytic Activity of Chemical Compounds on Their Affinity for Biotargets Relevant to the Chemotypes of the Studied Derivatives of Nitrogen-Containing Condensed Heterocycles
4.5. Consensus Sensitive Analysis of Neurons and Determination of Biotargets Most Significant for the Formation of High Anxiolytic Activity of Compounds Relevant to the Chemotypes of the Studied Derivatives of Nitrogen-Containing Condensed Heterocycles
4.6. Construction of Multitarget Pharmacophores of High Anxiolytic Activity of the Studied Derivatives of Nitrogen-Containing Condensed Heterocycles
5. Conclusions
- Seventeen relevant biotargets were identified that determine the anxiolytic activity of the studied 2,3,4,5-tetrahydro-11H-[1,3]diazepino[1,2-a]benzimidazole and [1,2,4]triazolo[3,4-a][2,3]benzodiazepine derivatives;
- Using the method of artificial neural networks, a consensus ensemble multitarget model of the dependence of the anxiolytic activity of chemical compounds on their docking energies in 17 relevant biotargets was built, the accuracy of which was 98.9%;
- Based on the consensus analysis of the sensitivity of neurons in the obtained model, four key biotargets ADRA1B, ADRA2A, AGTR1, and NMDA-Glut were identified, which are most significant for the formation of the anxiolytic activity of the studied compounds;
- For these biotargets ADRA1B, ADRA2A, AGTR1, and NMDA-Glut, eight monotargeted pharmacophores of the studied 2,3,4,5-tetrahydro-11H-[1,3]diazepino[1,2-a]benzimidazole and [1,2,4]triazolo[3,4-a][2,3]benzodiazepine derivatives were constructed;
- For the studied 2,3,4,5-tetrahydro-11H-[1,3]diazepino[1,2-a]benzimidazole and [1,2,4]triazolo[3,4-a][2,3]benzodiazepine derivatives by superposition of monotarget pharmacophores, two multitarget pharmacophores were constructed that determine the overall affinity for biotargets ADRA1B, ADRA2A, AGTR1, and NMDA-Glut in combination, taking into account their mutual influence on each other, which provide high anxiolytic activity;
- The formed consensus ensemble multitarget neural network model of anxiolytic activity of chemical compounds and the pharmacophores built on its basis are used in the directed search for new 2,3,4,5-tetrahydro-11H-[1,3]diazepino[1,2-a]benzimidazole and [1,2,4]triazolo[3,4-a][2,3]benzodiazepine derivatives with high anxiolytic activity.
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization. Mental Health Atlas 2020; World Health Organization: Geneva, Switzerland, 2021; pp. 1–126. ISBN 978-92-4-003670-3. Available online: https://www.who.int/publications/i/item/9789240036703 (accessed on 25 August 2022).
- DeMartini, J.; Patel, G.; Fancher, T.L. Generalized Anxiety Disorder. Ann. Intern. Med. 2019, 170, ITC49–ITC64. [Google Scholar] [CrossRef] [PubMed]
- Bandelow, B.; Michaelis, S.; Wedekind, D. Treatment of anxiety disorders. Dialogues Clin. Neurosci. 2017, 19, 93–107. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.R.; Prabhavalkar, K. Combination therapy with neuropeptides for the treatment of anxiety disorder. Neuropeptides 2021, 86, 102127. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, R.; Singh, A.; Chaudhary, K.K.; Yadav, M.K. Pharmacophore modeling and its applications. In Bioinformatics: Methods and Applications; Academic Press: London, UK, 2022; pp. 269–289. ISBN 978-0-323-89775-4. [Google Scholar] [CrossRef]
- Wermuth, C. Glossary of Terms Used in Medicinal Chemistry, IUPAC Recommendations. Pure Appl. Chem. 1998, 70, 1129–1143. [Google Scholar] [CrossRef]
- Zhang, W.; Pei, J.; Lai, L. Computational Multitarget Drug Design. J. Chem. Inf. Model. 2017, 57, 403–412. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Mulgaonkar, N.; Pérez, L.M.; Fernando, S. ELIXIR-A: An Interactive Visualization Tool for Multi-Target Pharmacophore Refinement. ACS Omega 2022, 7, 12707–12715. [Google Scholar] [CrossRef] [PubMed]
- Patel, L.; Shukla, T.; Huang, X.; Ussery, D.W.; Wang, S. Machine Learning Methods in Drug Discovery. Molecules 2020, 25, 5277. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Zhou, R.; Tang, J.; Li, M. PGMG: A Pharmacophore-Guided Deep Learning Approach for Bioactive Molecular Generation. Res. Sq. 2022; preprint. [Google Scholar] [CrossRef]
- Open Targets. Available online: https://www.opentargets.org/ (accessed on 29 November 2021).
- Vassiliev, P.M.; Spasov, A.A.; Kosolapov, V.A.; Kucheryavenko, A.F.; Gurova, N.A.; Anisimova, V.A. Consensus Drug Design Using IT Microcosm. In Application of Computational Techniques in Pharmacy and Medicine; Gorb, L., Kuz’min, V., Muratov, E., Eds.; Challenges and Advances in Computational Chemistry and Physics; Springer Science + Business Media: Dordrecht, The Netherlands, 2014; Volume 17, pp. 369–431. ISBN 978-94-017-9256-1. [Google Scholar] [CrossRef]
- UniProt. Available online: https://www.uniprot.org/ (accessed on 29 November 2021).
- PDBe. Available online: https://www.ebi.ac.uk/pdbe/ (accessed on 29 November 2021).
- RCSB PDB. Available online: https://www.rcsb.org/ (accessed on 29 November 2021).
- AlphaFold. Available online: https://alphafold.ebi.ac.uk/ (accessed on 29 November 2021).
- ChEMBL. Available online: https://www.ebi.ac.uk/chembl/ (accessed on 29 November 2021).
- Hilbe, J.M. Statistica 7: An overview. Am. Stat. 2007, 61, 91–94. [Google Scholar] [CrossRef]
- Kubinyi, H. Similarity and Dissimilarity: A Medicinal Chemist’s View. Perspect. Drug Discov. Des. 1998, 9, 225–252. [Google Scholar] [CrossRef]
- ChemAxon: Marvin. Available online: https://chemaxon.com/products/marvin (accessed on 29 November 2021).
- MOPAC2012. Available online: http://www.openmopac.net/MOPAC2012.html (accessed on 29 November 2021).
- LigPlot+. Available online: https://www.ebi.ac.uk/thornton-srv/software/LigPlus/ (accessed on 29 November 2021).
- PyRx. Available online: https://sourceforge.net/projects/pyrx/ (accessed on 29 November 2021).
- AutoDock Vina. Available online: https://vina.scripps.edu/ (accessed on 29 November 2021).
- Vassiliev, P.M.; Spasov, A.A.; Kochetkov, A.N.; Perfilev, M.A.; Koroleva, A.R. Consensus ensemble neural network multitarget model of RAGE inhibitory activity of chemical compounds. Biochem. Suppl. Ser. B Biomed. Chem. 2021, 15, 281–289. [Google Scholar] [CrossRef]
- Kolmogorov, A.N. On the representation of continuous functions of many variables by superposition of continuous functions of one variable and addition. Proc. USSR Acad. Sci. 1957, 114, 953–956. [Google Scholar]
- Dubin, U. Cross-Entropy Method: Theory with Applications; LAMBERT Academic Publishing: Chisinau, Moldova, 2013; pp. 1–148. ISBN 978-3659477164. [Google Scholar]
- Suzuki, K. (Ed.) Artificial Neural Networks—Methodological Advances and Biomedical Applications; IntechOpen: London, UK, 2011; pp. 1–376. ISBN 978-953-307-243-2. [Google Scholar] [CrossRef]
- El-Shahat, A. (Ed.) Advanced Applications for Artificial Neural Networks; IntechOpen: London, UK, 2018; pp. 1–296. ISBN 978-953-51-3781-8. [Google Scholar] [CrossRef]
- Du, K.-L.; Swamy, M.N.S. Neural Networks and Statistical Learning; Springer: London, UK, 2019; pp. 1–988. ISBN 978-1-4471-7451-6. [Google Scholar] [CrossRef]
- MedCalc. Available online: https://www.medcalc.org/ (accessed on 29 November 2021).
- Pukelshein, F. The three sigma rule. Am. Stat. 1994, 48, 88–91. [Google Scholar]
- Perfilev, M.A.; Vassiliev, P.M.; Kochetkov, A.N. Detailing of the targeted mechanism of action of RAGE inhibitors through consensus sensitivity analysis of neural network models. In Modern Medicine: New Approaches and Current Research, Proceedings of the International Scientific and Practical Conference Dedicated to the 30th Anniversary of the Medical Institute of the Chechen State University, Grozny, Russia, 22 October 2020; Saidov, Z.A., Ed.; Chechen State University Publishing House: Grozny, Russia, 2020; pp. 361–368. ISBN 978-5-91127-280-7. [Google Scholar]
- Maltsev, D.V.; Spasov, A.A.; Miroshnikov, M.V.; Skripka, M.O.; Divaeva, L.N. Influence of Diazepino[1,2-a]benzimidazole derivative (DAB-19) on behavioral aspects of animals. Res. Results Pharmacol. 2020, 6, 9–14. [Google Scholar] [CrossRef]
- Maltsev, D.V.; Spasov, A.A.; Yakovlev, D.S.; Vassiliev, P.M.; Skripka, M.O.; Miroshnikov, M.V.; Sultanova, K.T.; Kochetkov, A.N.; Divaeva, L.N.; Kuzmenko, T.A.; et al. Searching for new anxiolytic agents among derivatives of 11-dialkylaminoethyl-2,3,4,5-tetrahydrodiazepino[1,2-a]benzimidazole. Eur. J. Pharm. Sci. 2021, 161, 105792. [Google Scholar] [CrossRef] [PubMed]
- Skripka, M.O.; Spasov, A.A.; Maltsev, D.V.; Miroshnikov, M.V.; Yakovlev, D.S.; Sultanova, K.T.; Kochergin, M.A.; Divaeva, L.N. Screening of anxiolytic properties and analysis of structure-activity relationship of new derivatives of 6-(4-methoxy)-7H-[1,2,4]triazolo[3,4-a][2,3]benzodiazepine under the code RD. Res. Results Pharmacol. 2021, 7, 31–37. [Google Scholar] [CrossRef]
- BIOVIA Discovery Studio. 2021. Available online: https://www.3ds.com/products-services/biovia/products/molecular-modeling-simulation/biovia-discovery-studio/ (accessed on 9 November 2021).




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No 1 | Architecture 2 | Train 3 | Test 4 | Val 5 | F0 6 | Fa 7 | Fn 8 | ROC 9 |
|---|---|---|---|---|---|---|---|---|
| Activity level—High | ||||||||
| 634 | MLP 17-16-2 Tanh Softmax | 100.0 | 74.2 | 90.0 | 94.9 | 90.0 | 95.7 | 91.5 |
| 39 | MLP 17-16-2 Tanh Softmax | 100.0 | 96.7 | 71.0 | 95.4 | 86.7 | 96.8 | 90.7 |
| 417 | MLP 17-16-2 Tanh Softmax | 99.4 | 74.2 | 80.6 | 93.1 | 83.3 | 94.6 | 89.1 |
| 250 | MLP 17-16-2 Tanh Softmax | 100.0 | 87.1 | 80.6 | 95.4 | 83.3 | 97.3 | 87.9 |
| 467 | MLP 17-11-2 Tanh Softmax | 99.4 | 74.2 | 90.3 | 94.4 | 80.0 | 96.8 | 84.8 |
| 427 | MLP 17-16-2 Tanh Softmax | 100.0 | 80.6 | 77.4 | 94.0 | 80.0 | 96.2 | 83.8 |
| 215 | MLP 17-12-2 Tanh Softmax | 100.0 | 77.4 | 80.6 | 94.0 | 90.0 | 94.6 | 92.5 |
| Mean | 99.8 | 80.6 | 81.5 | 94.5 | 84.8 | 96.0 | 88.6 | |
| Consensus | — | — | — | 100.0 | 100.0 | 100.0 | 100.0 | |
| Activity level—High or Moderate | ||||||||
| 219 | MLP 17-14-2 Tanh Softmax | 94.8 | 83.9 | 73.3 | 90.3 | 91.5 | 89.3 | 91.7 |
| 119 | MLP 17-12-2 ExponentialSoftmax | 99.4 | 86.7 | 61.3 | 92.1 | 90.4 | 93.4 | 92.5 |
| 390 | MLP 17-15-2 Logistic Softmax | 98.7 | 80.6 | 51.6 | 89.4 | 88.3 | 90.2 | 91.1 |
| 8 | MLP 17-15-2 ExponentialSoftmax | 98.7 | 90.3 | 64.5 | 92.6 | 91.5 | 93.4 | 93.1 |
| 587 | MLP 17-16-2 Tanh Softmax | 100.0 | 77.4 | 67.7 | 92.1 | 89.4 | 94.3 | 92.3 |
| 259 | MLP 17-11-2 Tanh Softmax | 98.1 | 77.4 | 67.7 | 90.7 | 94.7 | 87.7 | 93.0 |
| 49 | MLP 17-17-2 Tanh Softmax | 96.8 | 90.3 | 71.0 | 92.1 | 90.4 | 93.4 | 92.3 |
| Mean | 98.1 | 83.8 | 65.3 | 91.3 | 90.9 | 91.7 | 92.3 | |
| Consensus | — | — | — | 97.2 | 96.8 | 97.5 | 97.2 | |
| Activity level—Active | ||||||||
| 67 | MLP 17-15-2 Logistic Softmax | 99.4 | 93.5 | 83.3 | 96.3 | 97.9 | 81.0 | 93.6 |
| 497 | MLP 17-7-2 Tanh Softmax | 99.4 | 80.0 | 90.3 | 95.4 | 95.9 | 90.5 | 95.6 |
| 44 | MLP 17-6-2 Tanh Softmax | 97.4 | 100.0 | 87.1 | 96.3 | 96.9 | 90.5 | 96.4 |
| 222 | MLP 17-7-2 Tanh Softmax | 99.4 | 93.5 | 87.1 | 96.8 | 98.5 | 81.0 | 97.0 |
| 47 | MLP 17-12-2 Logistic Softmax | 100.0 | 100.0 | 90.3 | 98.6 | 99.0 | 95.2 | 95.8 |
| 206 | MLP 17-11-2 Tanh Softmax | 100.0 | 77.4 | 90.3 | 95.4 | 96.9 | 81.0 | 89.2 |
| 222 | MLP 17-7-2 Tanh Softmax | 96.8 | 93.5 | 87.1 | 94.9 | 95.4 | 90.5 | 94.6 |
| Mean | 98.9 | 91.1 | 87.9 | 96.2 | 97.2 | 87.1 | 94.6 | |
| Consensus | — | — | — | 99.1 | 99.0 | 100.0 | 99.5 | |
| General mean | 98.9 | 85.2 | 78.2 | 94.0 | 91.0 | 91.6 | 91.8 | |
| General mean for consensus | — | — | — | 98.8 | 98.6 | 99.2 | 98.9 | |
| Target 1 | 634 2 | 39 2 | 417 2 | 250 2 | 467 2 | 427 2 | 215 2 | Sign 3 |
|---|---|---|---|---|---|---|---|---|
| ADRA1A | 5.7 | 3.3 | 3.4 | 4.6 | 6.8 | 3.7 | 5.8 | 1 |
| ADRA1B | 6.4 | 8.8 | 6.9 | 7.0 | 4.0 | 7.7 | 8.0 | 6 |
| ADRA2A | 7.6 | 12.4 | 11.0 | 6.0 | 6.7 | 7.5 | 4.1 | 5 |
| ADRA2B | 3.0 | 3.7 | 4.3 | 4.5 | 11.6 | 4.3 | 6.0 | 1 |
| AGTR1 | 6.7 | 6.5 | 7.6 | 6.1 | 4.8 | 7.7 | 11.2 | 6 |
| GABA-A-GABA | 4.3 | 3.1 | 4.2 | 6.1 | 4.2 | 3.8 | 3.0 | 1 |
| GABA-A-Benz | 6.1 | 5.5 | 3.3 | 4.2 | 5.0 | 6.7 | 7.0 | 3 |
| HTR1A | 6.0 | 4.1 | 3.6 | 5.5 | 3.9 | 4.3 | 2.6 | 0 |
| HTR2A-Spec | 4.7 | 4.7 | 5.2 | 5.8 | 8.1 | 5.6 | 7.0 | 2 |
| HTR2A-Allo | 6.5 | 14.6 | 12.3 | 4.2 | 5.9 | 2.6 | 6.4 | 4 |
| HTR4 | 10.9 | 6.8 | 2.0 | 6.1 | 5.4 | 5.0 | 5.6 | 3 |
| HTR7 | 3.7 | 4.1 | 5.8 | 8.0 | 10.7 | 6.8 | 6.6 | 4 |
| MTNR1A | 4.4 | 3.4 | 3.0 | 4.5 | 4.0 | 4.0 | 4.2 | 0 |
| MTNR1B | 4.8 | 5.0 | 4.4 | 10.5 | 4.7 | 3.6 | 3.2 | 1 |
| NMDA-Glut | 12.1 | 5.7 | 15.8 | 7.3 | 3.2 | 12.6 | 11.6 | 5 |
| NMDA-Ca | 3.5 | 5.0 | 4.0 | 5.3 | 6.4 | 10.9 | 3.7 | 2 |
| SLC18A2 | 3.4 | 3.4 | 3.3 | 4.1 | 4.5 | 3.1 | 4.0 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vassiliev, P.M.; Maltsev, D.V.; Spasov, A.A.; Perfilev, M.A.; Skripka, M.O.; Kochetkov, A.N. Consensus Ensemble Multitarget Neural Network Model of Anxiolytic Activity of Chemical Compounds and Its Use for Multitarget Pharmacophore Design. Pharmaceuticals 2023, 16, 731. https://doi.org/10.3390/ph16050731
Vassiliev PM, Maltsev DV, Spasov AA, Perfilev MA, Skripka MO, Kochetkov AN. Consensus Ensemble Multitarget Neural Network Model of Anxiolytic Activity of Chemical Compounds and Its Use for Multitarget Pharmacophore Design. Pharmaceuticals. 2023; 16(5):731. https://doi.org/10.3390/ph16050731
Chicago/Turabian StyleVassiliev, Pavel M., Dmitriy V. Maltsev, Alexander A. Spasov, Maxim A. Perfilev, Maria O. Skripka, and Andrey N. Kochetkov. 2023. "Consensus Ensemble Multitarget Neural Network Model of Anxiolytic Activity of Chemical Compounds and Its Use for Multitarget Pharmacophore Design" Pharmaceuticals 16, no. 5: 731. https://doi.org/10.3390/ph16050731
APA StyleVassiliev, P. M., Maltsev, D. V., Spasov, A. A., Perfilev, M. A., Skripka, M. O., & Kochetkov, A. N. (2023). Consensus Ensemble Multitarget Neural Network Model of Anxiolytic Activity of Chemical Compounds and Its Use for Multitarget Pharmacophore Design. Pharmaceuticals, 16(5), 731. https://doi.org/10.3390/ph16050731