

Lactylation-Related Gene Signature Effectively Predicts Prognosis and Treatment Responsiveness in Hepatocellular Carcinoma



Abstract
1. Introduction
2. Results
2.1. Identifying Prognostic Lactylation-Related Genes in HCC
2.2. Constructing a Prognostic Model in the TCGA Cohort
2.3. Validating the Prognostic Lactylation-Related Signature in the ICGC Cohort
2.4. Independent Prognostic Value of the Lactylation-Related Eight-Gene Signature
2.5. Functional Annotation and Glycolysis Pathway GSEA
2.6. TIME Landscape Analysis and Treatment Responsiveness Evaluation
2.7. Signature Gene Structure Analysis and Mutation Distribution
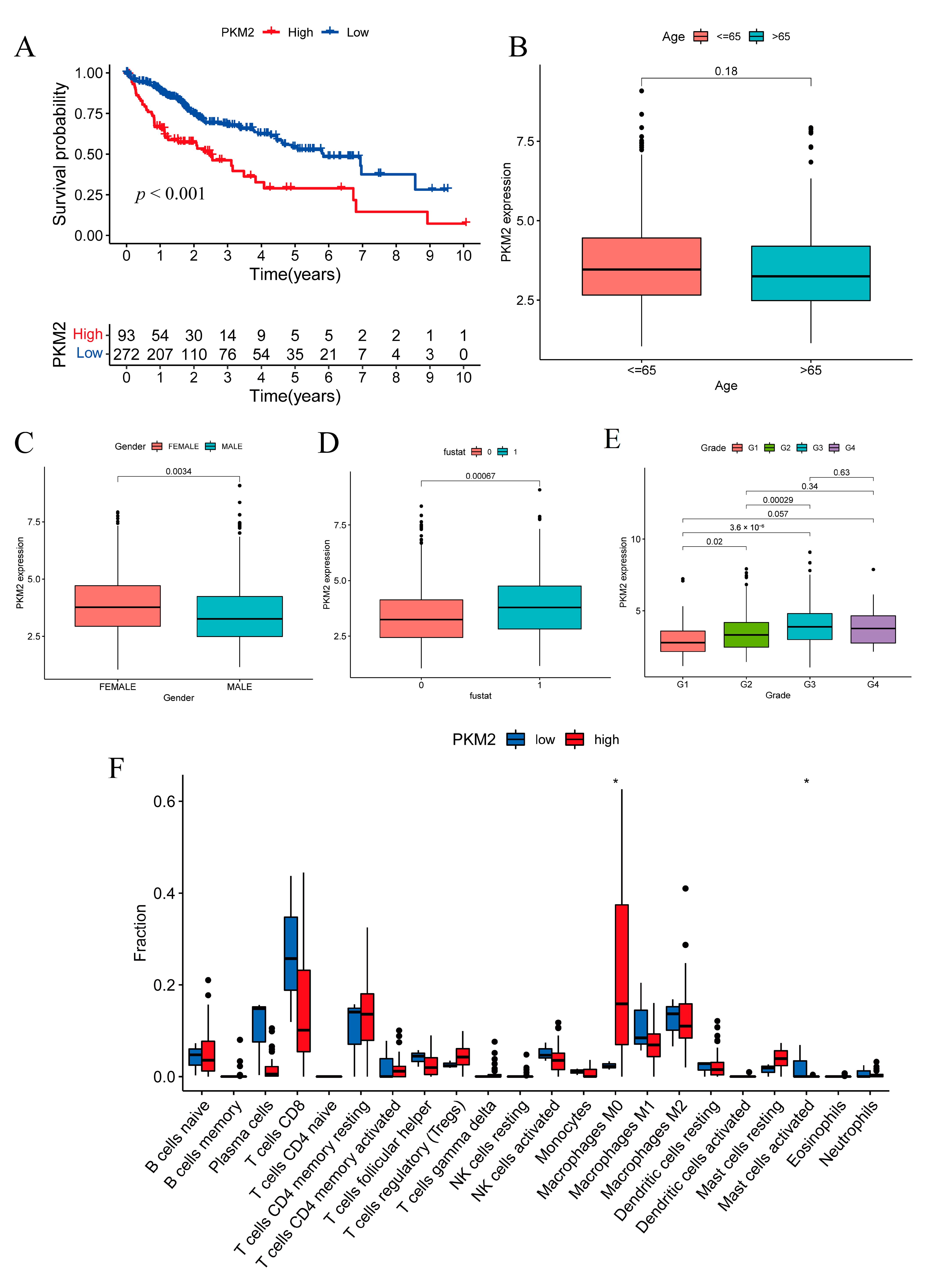
2.8. Prognostic Investigation of the Glycolysis Rate-Limiting Enzyme PKM2
3. Discussion
4. Materials and Methods
4.1. Data Acquisition
4.2. Identification of Differentially Expressed and Prognostic Genes
4.3. Analysis of Glycolysis Pathway Enrichment
4.4. Construction and Validation of a Prognostic Lactylation-Related Gene Signature
4.5. Functional Annotation Analysis
4.6. Treatment Responsiveness Evaluation
4.7. Mutation Analysis of Signature Genes
4.8. Survival Analysis and Clinical Relevance Analysis for Single Gene
4.9. Tumor-Infiltrating Immune Cell Profiling
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Wang, H.C.; Haung, L.Y.; Wang, C.J.; Chao, Y.J.; Hou, Y.C.; Yen, C.J.; Shan, Y.S. Tumor-associated macrophages promote resistance of hepatocellular carcinoma cells against sorafenib by activating CXCR2 signaling. J. Biomed. Sci. 2022, 29, 99. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.D.; Fan, T.C.; Lai, M.W.; Yeh, C.T. GALNT14-mediated O-glycosylation on PHB2 serine-161 enhances cell growth, migration and drug resistance by activating IGF1R cascade in hepatoma cells. Cell Death Dis. 2022, 13, 956. [Google Scholar] [CrossRef]
- Liu, P.H.; Hsu, C.Y.; Hsia, C.Y.; Lee, Y.H.; Su, C.W.; Huang, Y.H.; Lee, F.Y.; Lin, H.C.; Huo, T.I. Prognosis of hepatocellular carcinoma: Assessment of eleven staging systems. J. Hepatol. 2016, 64, 601–608. [Google Scholar] [CrossRef]
- Zhang, D.; Tang, Z.; Huang, H.; Zhou, G.; Cui, C.; Weng, Y.; Liu, W.; Kim, S.; Lee, S.; Perez-Neut, M.; et al. Metabolic regulation of gene expression by histone lactylation. Nature 2019, 574, 575–580. [Google Scholar] [CrossRef] [PubMed]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Guzman, G.; Chennuri, R.; Chan, A.; Rea, B.; Quintana, A.; Patel, R.; Xu, P.Z.; Xie, H.; Hay, N. Evidence for heightened hexokinase II immunoexpression in hepatocyte dysplasia and hepatocellular carcinoma. Dig. Dis. Sci. 2015, 60, 420–426. [Google Scholar] [CrossRef]
- Mazurek, S.; Boschek, C.B.; Hugo, F.; Eigenbrodt, E. Pyruvate kinase type M2 and its role in tumor growth and spreading. Semin. Cancer Biol. 2005, 15, 300–308. [Google Scholar] [CrossRef] [PubMed]
- Du, D.; Liu, C.; Qin, M.; Zhang, X.; Xi, T.; Yuan, S.; Hao, H.; Xiong, J. Metabolic dysregulation and emerging therapeutical targets for hepatocellular carcinoma. Acta Pharm. Sin. B 2022, 12, 558–580. [Google Scholar] [CrossRef]
- Tong, X.; Zhao, F.; Thompson, C.B. The molecular determinants of de novo nucleotide biosynthesis in cancer cells. Curr. Opin. Genet. Dev. 2009, 19, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Langbein, S.; Zerilli, M.; Zur Hausen, A.; Staiger, W.; Rensch-Boschert, K.; Lukan, N.; Popa, J.; Ternullo, M.P.; Steidler, A.; Weiss, C.; et al. Expression of transketolase TKTL1 predicts colon and urothelial cancer patient survival: Warburg effect reinterpreted. Br. J. Cancer 2006, 94, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Wan, N.; Wang, N.; Yu, S.; Zhang, H.; Tang, S.; Wang, D.; Lu, W.; Li, H.; Delafield, D.G.; Kong, Y.; et al. Cyclic immonium ion of lactyllysine reveals widespread lactylation in the human proteome. Nat. Methods 2022, 19, 854–864. [Google Scholar] [CrossRef]
- Yang, Z.; Yan, C.; Ma, J.; Peng, P.; Ren, X.; Cai, S.; Shen, X.; Wu, Y.; Zhang, S.; Wang, X.; et al. Lactylome analysis suggests lactylation-dependent mechanisms of metabolic adaptation in hepatocellular carcinoma. Nat. Metab. 2023, 5, 61–79. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Yin, J.; Shan, L.; Yi, X.; Zhang, W.; Ding, Y. Identification of lysine-lactylated substrates in gastric cancer cells. iScience 2022, 25, 104630. [Google Scholar] [CrossRef]
- Wang, J.; Yang, P.; Yu, T.; Gao, M.; Liu, D.; Zhang, J.; Lu, C.; Chen, X.; Zhang, X.; Liu, Y. Lactylation of PKM2 Suppresses Inflammatory Metabolic Adaptation in Pro-inflammatory Macrophages. Int. J. Biol. Sci. 2022, 18, 6210–6225. [Google Scholar] [CrossRef]
- Rizvi, S.; Wang, J.; El-Khoueiry, A.B. Liver Cancer Immunity. Hepatology 2021, 73 (Suppl. 1), 86–103. [Google Scholar] [CrossRef]
- Torres, R.; Judson-Torres, R.L. Research Techniques Made Simple: Feature Selection for Biomarker Discovery. J. Investig. Dermatol. 2019, 139, 2068–2074.e2061. [Google Scholar] [CrossRef]
- Fitzmaurice, C.; Abate, D.; Abbasi, N.; Abbastabar, H.; Abd-Allah, F.; Abdel-Rahman, O.; Abdelalim, A.; Abdoli, A.; Abdollahpour, I.; Abdulle, A.S.M.; et al. Global, Regional, and National Cancer Incidence, Mortality, Years of Life Lost, Years Lived With Disability, and Disability-Adjusted Life-Years for 29 Cancer Groups, 1990 to 2017: A Systematic Analysis for the Global Burden of Disease Study. JAMA Oncol. 2019, 5, 1749–1768. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Zhu, J.; Thompson, C.B. The hallmarks of cancer metabolism: Still emerging. Cell Metab. 2022, 34, 355–377. [Google Scholar] [CrossRef]
- Wu, D.; Yang, Y.; Hou, Y.; Zhao, Z.; Liang, N.; Yuan, P.; Yang, T.; Xing, J.; Li, J. Increased mitochondrial fission drives the reprogramming of fatty acid metabolism in hepatocellular carcinoma cells through suppression of Sirtuin 1. Cancer Commun. 2022, 42, 37–55. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.; Xu, L.; Yu, X.; Zhang, G.; Guo, H.; Liu, H.; Song, G.; Weng, S.; Dong, L.; Zhu, J.; et al. OGDHL silencing promotes hepatocellular carcinoma by reprogramming glutamine metabolism. J. Hepatol. 2020, 72, 909–923. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Yu, M.; Ma, X.; Sun, J.; Liu, C.; Wang, C.; Wu, S.; Fu, P.; Yang, Z.; He, Y.; et al. IFNα Potentiates Anti-PD-1 Efficacy by Remodeling Glucose Metabolism in the Hepatocellular Carcinoma Microenvironment. Cancer Discov. 2022, 12, 1718–1741. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Huang, D.; Jiang, Y.; Hou, J.; Tian, M.; Li, J.; Sun, L.; Zhang, Y.; Zhang, T.; Li, Z.; et al. Lactate Modulates Cellular Metabolism Through Histone Lactylation-Mediated Gene Expression in Non-Small Cell Lung Cancer. Front. Oncol. 2021, 11, 647559. [Google Scholar] [CrossRef]
- Hudson, T.J.; Anderson, W.; Artez, A.; Barker, A.D.; Bell, C.; Bernabé, R.R.; Bhan, M.K.; Calvo, F.; Eerola, I.; Gerhard, D.S.; et al. International network of cancer genome projects. Nature 2010, 464, 993–998. [Google Scholar] [CrossRef]
- Pitt, J.M.; Marabelle, A.; Eggermont, A.; Soria, J.C.; Kroemer, G.; Zitvogel, L. Targeting the tumor microenvironment: Removing obstruction to anticancer immune responses and immunotherapy. Ann. Oncol. 2016, 27, 1482–1492. [Google Scholar] [CrossRef]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef]
- Galon, J.; Bruni, D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 2019, 18, 197–218. [Google Scholar] [CrossRef]
- Alejo-Valle, O.; Weigert, K.; Bhayadia, R.; Ng, M.; Issa, H.; Beyer, C.; Emmrich, S.; Schuschel, K.; Ihling, C.; Sinz, A.; et al. The megakaryocytic transcription factor ARID3A suppresses leukemia pathogenesis. Blood 2022, 139, 651–665. [Google Scholar] [CrossRef]
- Shen, M.; Li, S.; Zhao, Y.; Liu, Y.; Liu, Z.; Huan, L.; Qiao, Y.; Wang, L.; Han, L.; Chen, Z.; et al. Hepatic ARID3A facilitates liver cancer malignancy by cooperating with CEP131 to regulate an embryonic stem cell-like gene signature. Cell Death Dis. 2022, 13, 732. [Google Scholar] [CrossRef]
- Chen, Q.; Shen, P.; Ge, W.L.; Yang, T.Y.; Wang, W.J.; Meng, L.D.; Huang, X.M.; Zhang, Y.H.; Cao, S.J.; Miao, Y.; et al. Roundabout homolog 1 inhibits proliferation via the YY1-ROBO1-CCNA2-CDK2 axis in human pancreatic cancer. Oncogene 2021, 40, 2772–2784. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Gabola, M.; Lattanzio, R.; Paul, C.; Pinet, V.; Tang, R.; Turali, H.; Bremond, J.; Longobardi, C.; Maurizy, C.; et al. Cyclin A2 maintains colon homeostasis and is a prognostic factor in colorectal cancer. J. Clin. Investig. 2021, 131, e131517. [Google Scholar] [CrossRef] [PubMed]
- Honnappa, S.; Gouveia, S.M.; Weisbrich, A.; Damberger, F.F.; Bhavesh, N.S.; Jawhari, H.; Grigoriev, I.; van Rijssel, F.J.; Buey, R.M.; Lawera, A.; et al. An EB1-binding motif acts as a microtubule tip localization signal. Cell 2009, 138, 366–376. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Dai, M.; Zhang, C.; Teng, K.; Wang, F.; Li, H.; Sun, W.; Feng, Z.; Kang, T.; Guan, X.; et al. KIF2C: A novel link between Wnt/β-catenin and mTORC1 signaling in the pathogenesis of hepatocellular carcinoma. Protein Cell 2021, 12, 788–809. [Google Scholar] [CrossRef]
- Gómez-Manzo, S.; Marcial-Quino, J.; Vanoye-Carlo, A.; Serrano-Posada, H.; Ortega-Cuellar, D.; González-Valdez, A.; Castillo-Rodríguez, R.A.; Hernández-Ochoa, B.; Sierra-Palacios, E.; Rodríguez-Bustamante, E.; et al. Glucose-6-Phosphate Dehydrogenase: Update and Analysis of New Mutations around the World. Int. J. Mol. Sci. 2016, 17, 2069. [Google Scholar] [CrossRef]
- Luo, M.; Fu, A.; Wu, R.; Wei, N.; Song, K.; Lim, S.; Luo, K.Q. High Expression of G6PD Increases Doxorubicin Resistance in Triple Negative Breast Cancer Cells by Maintaining GSH Level. Int. J. Biol. Sci. 2022, 18, 1120–1133. [Google Scholar] [CrossRef]
- Qin, Z.; Xiang, C.; Zhong, F.; Liu, Y.; Dong, Q.; Li, K.; Shi, W.; Ding, C.; Qin, L.; He, F. Transketolase (TKT) activity and nuclear localization promote hepatocellular carcinoma in a metabolic and a non-metabolic manner. J. Exp. Clin. Cancer Res. 2019, 38, 154. [Google Scholar] [CrossRef]
- Shukla, S.K.; Purohit, V.; Mehla, K.; Gunda, V.; Chaika, N.V.; Vernucci, E.; King, R.J.; Abrego, J.; Goode, G.D.; Dasgupta, A.; et al. MUC1 and HIF-1alpha Signaling Crosstalk Induces Anabolic Glucose Metabolism to Impart Gemcitabine Resistance to Pancreatic Cancer. Cancer Cell 2017, 32, 71–87.e77. [Google Scholar] [CrossRef]
- Hsu, P.P.; Sabatini, D.M. Cancer cell metabolism: Warburg and beyond. Cell 2008, 134, 703–707. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Wei, H.; Wang, H.; Wang, Z.; Li, J.; Ou, Y.; Xiao, X.; Wang, W.; Chang, A.; Sun, W.; et al. Zeb1-induced metabolic reprogramming of glycolysis is essential for macrophage polarization in breast cancer. Cell Death Dis. 2022, 13, 206. [Google Scholar] [CrossRef]
- Qing, Y.; Dong, L.; Gao, L.; Li, C.; Li, Y.; Han, L.; Prince, E.; Tan, B.; Deng, X.; Wetzel, C.; et al. R-2-hydroxyglutarate attenuates aerobic glycolysis in leukemia by targeting the FTO/m(6)A/PFKP/LDHB axis. Mol. Cell 2021, 81, 922–939.e929. [Google Scholar] [CrossRef]
- Shen, J.; Jin, Z.; Lv, H.; Jin, K.; Jonas, K.; Zhu, C.; Chen, B. PFKP is highly expressed in lung cancer and regulates glucose metabolism. Cell. Oncol. (Dordr.) 2020, 43, 617–629. [Google Scholar] [CrossRef]
- Park, Y.Y.; Kim, S.B.; Han, H.D.; Sohn, B.H.; Kim, J.H.; Liang, J.; Lu, Y.; Rodriguez-Aguayo, C.; Lopez-Berestein, G.; Mills, G.B.; et al. Tat-activating regulatory DNA-binding protein regulates glycolysis in hepatocellular carcinoma by regulating the platelet isoform of phosphofructokinase through microRNA 520. Hepatology 2013, 58, 182–191. [Google Scholar] [CrossRef]
- Zhang, Z.; Deng, X.; Liu, Y.; Liu, Y.; Sun, L.; Chen, F. PKM2, function and expression and regulation. Cell Biosci. 2019, 9, 52. [Google Scholar] [CrossRef]
- Moreno-Yruela, C.; Zhang, D.; Wei, W.; Bæk, M.; Liu, W.; Gao, J.; Danková, D.; Nielsen, A.L.; Bolding, J.E.; Yang, L.; et al. Class I histone deacetylases (HDAC1-3) are histone lysine delactylases. Sci. Adv. 2022, 8, eabi6696. [Google Scholar] [CrossRef]
- Geeleher, P.; Cox, N.; Huang, R.S. pRRophetic: An R package for prediction of clinical chemotherapeutic response from tumor gene expression levels. PLoS ONE 2014, 9, e107468. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Gu, S.; Pan, D.; Fu, J.; Sahu, A.; Hu, X.; Li, Z.; Traugh, N.; Bu, X.; Li, B.; et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat. Med. 2018, 24, 1550–1558. [Google Scholar] [CrossRef]
- Fu, J.; Li, K.; Zhang, W.; Wan, C.; Zhang, J.; Jiang, P.; Liu, X.S. Large-scale public data reuse to model immunotherapy response and resistance. Genome Med. 2020, 12, 21. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | TCGA Cohort | ICGC LIRI-JP Cohort | |
|---|---|---|---|
| Total number of patients | 365 | 231 | |
| Survival status | Survival | 235 (64.38%) | 189 (81.82%) |
| Death | 130 (35.62%) | 42 (18.18%) | |
| Age | ≤65 years | 227 (62.19%) | 89 (38.53%) |
| >65 years | 138 (37.81%) | 142 (61.47%) | |
| Sex | Male | 246 (67.40%) | 170 (73.59%) |
| Female | 119 (32.60%) | 61 (26.41%) | |
| Stage | I | 170 (46.58%) | 36 (15.58%) |
| II | 84 (23.01%) | 105 (45.45%) | |
| III | 83 (22.74%) | 71 (30.74%) | |
| IV | 4 (1.10%) | 19 (8.23%) | |
| Unknow | 24 (6.57%) | 0 | |
| Pathological grade (Edmondson) | G1 | 55 (15.07%) | 20 (8.66%) |
| G2 | 175 (47.94%) | 134 (58.01%) | |
| G3 | 118 (32.33%) | 56 (24.24%) | |
| G4 | 12 (3.29%) | 1 (0.43%) | |
| Unknow | 5 (1.37%) | 20 (8.66%) | |
| Metastasis | Primary HCC | 365 | 201 (87.01%) |
| Metastatic HCC | 0 | 30 (12.99%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, Z.; Huang, H.; Li, M.; Liang, X.; Tan, Y.; Chen, Y. Lactylation-Related Gene Signature Effectively Predicts Prognosis and Treatment Responsiveness in Hepatocellular Carcinoma. Pharmaceuticals 2023, 16, 644. https://doi.org/10.3390/ph16050644
Cheng Z, Huang H, Li M, Liang X, Tan Y, Chen Y. Lactylation-Related Gene Signature Effectively Predicts Prognosis and Treatment Responsiveness in Hepatocellular Carcinoma. Pharmaceuticals. 2023; 16(5):644. https://doi.org/10.3390/ph16050644
Chicago/Turabian StyleCheng, Zhe, Huichao Huang, Maoyu Li, Xujun Liang, Yuying Tan, and Yongheng Chen. 2023. "Lactylation-Related Gene Signature Effectively Predicts Prognosis and Treatment Responsiveness in Hepatocellular Carcinoma" Pharmaceuticals 16, no. 5: 644. https://doi.org/10.3390/ph16050644
APA StyleCheng, Z., Huang, H., Li, M., Liang, X., Tan, Y., & Chen, Y. (2023). Lactylation-Related Gene Signature Effectively Predicts Prognosis and Treatment Responsiveness in Hepatocellular Carcinoma. Pharmaceuticals, 16(5), 644. https://doi.org/10.3390/ph16050644